

Биоорганическая химия, 2020, T. 46, № 6, стр. 580-592
Перспективные молекулярные мишени сигнальных каскадов Ras-белков для создания противоопухолевых препаратов
С. Г. Клочков 1, *, М. Е. Неганова 1, Ю. Р. Александрова 1
1 Институт физиологически активных веществ РАН
142432 Московская обл., Черноголовка, Северный проезд, 1, Россия
* E-mail: klochkov@ipac.ac.ru
Поступила в редакцию 17.02.2020
После доработки 27.02.2020
Принята к публикации 28.02.2020
Аннотация
Разработка эффективных противоопухолевых препаратов считается одной из приоритетных задач современного здравоохранения. Для химиотерапии опухолей используются ингибиторы белков, ферментов, рецепторов, регулирующих сигнальные пути опухолевых клеток, индукторы апоптоза. Наиболее значимые и изучаемые сигнальные пути – это пути Raf/MEK/ERK и фосфоинозитид-3-киназы. Изменения в каскадах этих путей ведут к модуляции многих клеточных функций – роста и выживания клетки, пролиферации, дифференциации, адгезии. Эти сигнальные каскады тесно связаны с Ras-белками. Онкогенные формы Ras-белков детектируются в большом числе опухолевых заболеваний – более 30% всех форм новообразований. В настоящем обзоре рассмотрены литературные данные за последние 30 лет по исследованию ключевых этапов сигнальных каскадов, инициируемых онкобелками. Отдельно проанализированы особенности процессинга Ras-белков, рассматриваемые в настоящее время как перспективные молекулярные мишени для разработки противоопухолевых препаратов. Приведены и обсуждены литературные данные о новых стратегиях поиска эффективных антинеопластов на основе ингибиторов сигнальных путей Ras-белков.
ВВЕДЕНИЕ
Одной из наиболее значимых причин смертности населения, независимо от пола, возраста и качества жизни, являются онкологические заболевания. Естественно, эти заболевания и стратегии создания противоопухолевых лекарственных препаратов находятся в зоне пристального внимания исследователей во всем мире. В результате успешного развития современной молекулярной биологии было обнаружено, что ряд белков и генов играют критическую роль в канцерогенезе. Необратимые изменения в клеточном генетическом содержании считаются основной причиной канцерогенеза, поскольку они могут модулировать как экспрессию генов, так и функционирование трансляционных продуктов, которые связаны с регуляцией роста и дифференцировки клеток. С появлением современных технологий и благодаря непрерывным усилиям исследователей был достигнут огромный прогресс в выявлении и понимании роли мутаций и изменений генов в структуре процессов образования злокачественных опухолей человека и канцерогенеза в целом.
Для терапии разных типов опухолей используются ингибиторы белков, ферментов, рецепторов, регулирующих сигнальные пути опухолевых клеток, индукторы апоптоза, стероидные соединения и вакцины. К наиболее многообещающим и изучаемым сигнальным путям принадлежат сигнальные пути Raf/MEK/ERK и фосфоинозитид-3-киназы (PI3K) [1]. Изменения в каскадах этих сигнальных путей ведут к нарушению многих клеточных функций – функций роста и выживания клетки, пролиферации, дифференциации, адгезии. Эти сигнальные каскады тесно связаны с Ras-белками. Онкогенные формы Ras-белков детектируются в большом числе опухолевых клеток– их содержат около 30% всех форм новообразований, в том числе свыше 50% рака ободочной кишки, свыше 30% рака легких и более 90% опухолей поджелудочной железы [2, 3]. Онкогенный потенциал белков семейства Ras, таких как H-Ras, K-Ras и N-Ras, в различных опухолях убедительно доказан исследованиями нескольких десятилетий [4, 5].
Анализ образцов опухолевых клеток показал, что изменение в гене белка K-Ras наиболее часто сопровождается также изменениями генов N-Ras и H-Ras [5, 6]. Онкосопряженные изменения в гене белка K-Ras наиболее часто обнаруживаются при панкреатических карциномах, колоректальных опухолях и злокачественных новообразованиях легких, в то время как мутированный H-Ras наиболее часто встречается в дерматологических и злокачественных новообразованиях головы и шеи, гематологические злокачественные опухоли часто сопряжены с мутациями N-Ras [5]. С момента открытия онкобелков Ras в 1960-х годах в многочисленных исследованиях изучался механизм их активности и связанные с ними регуляторные пути.
Гиперэкспрессия онкогенов Ras реализует свою антиапоптотическую функцию через несколько эффекторных путей. Так, активация сигнальных путей PI3K-Akt и Raf-MEK-MAPK приводит к фосфорилированию и инактивации проапоптотического белка BAD [7], блокируя тем самым процесс апоптоза. Кроме того, белки Ras регулируют и многие транскрипционные факторы, связанные с началом и прогрессированием клеточного цикла, включая ядерный фактор κB (NF-κB) и активирующий фактор транскрипции 2 (ATF2) [5], сверхактивация которых в свою очередь способствует дисрегуляции процессов пролиферации неопластических клеток, приводя к их бесконтрольному делению [7].
Онкогенные белки Ras в опухолевых клетках могут способствовать и ангиогенезу, увеличивая транскрипционную активацию генов, контролирующих данный процесс через ряд сигнальных каскадов [8]. В частности, запуск вышеупомянутых путей PI3K-Akt и Raf-MEK-MAPK приводит к стабилизации индуцированного гипоксией фактора 1α (HIF-1α), который способен активировать промотор фактора роста эндотелия сосудов A (VEGFA) [9–12], играющего ключевую роль в индукции пролиферации эндотелиальных клеток и прорастания новых кровеносных сосудов [5]. Кроме того, посредством активации фермента циклооксигеназы 2 (COX-2) Ras могут стимулировать выработку и других проангиогенных факторов, в частности, тромбоцитарного фактора роста (PDGF) и основного фактора роста фибробластов (FGF-2) [13].
Первые успехи в создании лекарственных препаратов, ингибирующих сигнальные пути Ras, вдохновили исследователей к изучению этих белков и поиску новых потенциальных лекарственных препаратов, направленных на изменение их функций. В настоящем обзоре рассмотрены ключевые этапы сигнальных каскадов, инициируемых онкогенными белками Ras, и рассматриваемых в настоящее время как перспективные молекулярные мишени для разработки противоопухолевых препаратов.
ПРОЦЕССИНГ ОНКОГЕНОВ СЕМЕЙСТВА Ras
Онкогены семейства Ras являются генами наиболее распространенных и известных белков, которые регулируют клеточную пролиферацию, выживание, рост опухолевой клетки, а также ряд других функций, обеспечивающих процесс малигнизации клеток. К настоящему времени массив белков Ras разделяется на пять основных семейств – Ran, Ras, Rab, Rho и Arf [14]. Семейство онкобелков Ras представлено четырьмя классами: HRas, KRas4a, KRas4b, и NRas [3, 15]. Эти белки являются GTP-азами, работающими в качестве молекулярных переключателей: “включено” – когда белок связан с GTP и “выключено”, когда белок связан с GDP. Ras-GTP в свою очередь связывается с большим количеством разнообразных эффекторных белков. Взаимодействия белков Ras с эффекторами ведут к запуску каскада сигнальных путей, передающих экстраклеточный стимул внутрь клетки [16, 17]. В качестве внеклеточных сигналов выступают фактор эпидермального роста (EGF), фактор роста нервов (NGF), фактор роста тромбоцитов (PDGF), фактор роста фибробластов (FGF), которые активируют рецепторные тирозинкиназы (RTK). Данные экстраклеточные сигналы активируют рецепторные тирозинкиназы, передавая сигнал на Ras-белки и запуская два основополагающих сигнальных пути развития опухолевой клетки: Raf/MEK/ERK и PI3K [18–21]. В опухолевых клетках сигнальные пути, запускаемые белками Ras, являются ключевыми для процессов пролиферации и дифференциации, они определяют как выживание клетки, так и ее способность избегать апоптоза [22] (рис. 1).
Рис. 1.
Сигнальные пути, запускаемые белками Ras. Онкобелки Ras являются GTP-азами. Когда Ras связан с GTP – он активен, форма белка, связанная с GDP, – не активна. Ras-GTP связывается с большим количеством разнообразных эффекторных белков, которые по каскадам сигнальных путей передают стимул внутрь клетки, модулируя процессы пролиферации, дифференциации клетки и ее способность избегать апоптоза.
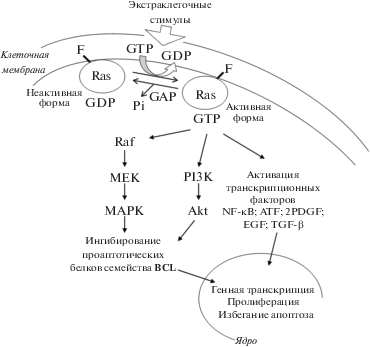
Превращение Ras-GTP в Ras-GDP является очень медленным процессом и происходит с участием регуляторных белков. Белок GAP (GTPase-activating protein) катализирует превращение Ras-GTP в Ras-GDP (включено-выключено), тогда как GEF (guanine nucleotide exchange factor) катализирует выход GDP из комплекса (выключено-включено) [23]. В дополнение к значительной роли мутированных онкобелков Ras следует заметить, что не мутированные белки Ras также присутствуют в опухолях, и это является необходимым условием малигнизации. Так, например, для развития опухоли, вызванной онкобелком K-Ras, необходимо наличие не мутированных функционирующих H-Ras или N-Ras [24–26].
Для того чтобы Ras-белки стали активными и участвовали в каскаде передачи экстраклеточного сигнала ростовых факторов, гормонов и цитокинов, необходимо их встраивание в мембрану клетки. Синтезируются белки семейства Ras в виде неактивного цитозольного пропептида – pro-p21. Все три основных белка семейства Ras H-, N-, и K-Ras проходят серию пострансляционных модификаций, которые можно разделить на два этапа [27] (рис. 2).
Рис. 2.
Процессинг онкогенов семейства Ras. Концевой участок Ras-белка CAAX алкилируется фарнезилтрансферазой или геранилгеранилтрансферазой. Концевой пептидный участок AAX отщепляется в эндоплазматическом ретикулуме эндопептидазой RCE1 и метилируется ICMT. В комплексе Гольджи, H-Ras, N-Ras, и K-Ras4a дополнительно пальмитоилируются. K-Ras4b встраивается в мембрану посредством фарнезильного остатка. H-Ras, N-Ras, и K-Ras4a – за счет пальмитоильного и фарнезильного остатка [28].
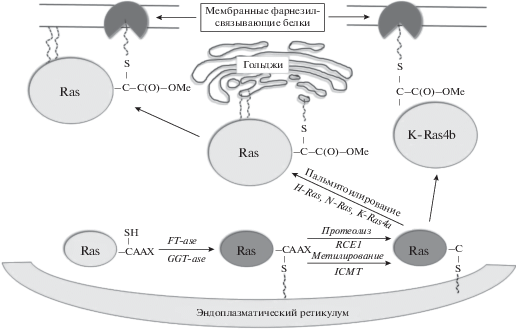
Первый этап представлен модификациями участка аминокислотной последовательности CAAX (C = цистеин, A = алифатическая аминокислота, X = любая аминокислота), который находится на С-конце всех белков Ras. Вначале остаток цистеина (Cys-188) алкилируется фарнезилтрансферазой (FT-аза) (обычно). Это превращение является критическим, так как обеспечивает присоединение белка Ras к мембране [29–37]. Затем участок AAX отщепляется протеолитически в эндоплазматическом ретикулуме эндопептидазой RCE1 [38]. H-Ras, N-Ras и K-Ras4A метилируются метилтрансферазой ICMT (isoprenylcysteine carboxyl methyltransferase) по α-карбоксильной группе нового C-концевого цистеина [39–42]. Первый этап процессинга таким образом превращает белок pro-p21 в промежуточную форму – c-p21 (cytosolic processed p21). Эта форма белка Ras значительно гидрофобнее pro-p21 и слабо ассоциируется с клеточными мембранами [41, 43].
Второй этап процессинга онкогенов Ras касается белков N-Ras, H-Ras, и K-Ras4a и происходит в комплексе Гольджи. Он представляет собой пальмитоилирование цистеина гипервариабельного участка этих белков. Пальмитоилирование достоверно увеличивает способность к встраиванию в мембрану белков H-Ras и N-Ras [41]. Таким образом, встраивание белка Ras в плазматическую мембрану является результатом комбинации двух посттрансляционных модификаций.
K-Ras4b не требует пальмитоилирования и встраивается в мембрану за счет фарнезильного остатка [44]. В результате процессинга фарнезилированный белок Ras становится гидрофобным и легко встраивается в липидный бислой плазматической мембраны [38, 41, 44]. Это позволяет ему переключаться из неактивной GDP-связанной формы в активную GTP-связанную форму в ответ на экстраклеточный сигнал. Очень важно отметить, что существуют другие пренилтрансферазы, прежде всего это геранилгеранилтрансферазы (GGT-азы), которые присоединяют один или два 20-углеродных изопреноида к белкам Ras, что позволяет им встраиваться в мембраны. Геранилгеранилированные белки более гидрофобны чем фарнезилированные, что может служить основой для различий этих двух форм в белок-белковых взаимодействиях. GGT-азы пренилируют белки, в которых в участке CAAX остаток X представлен лейцином [45]. Таким образом, GGT-аза может пренилировать K-Ras4b и другие G-белки, которые обычно фарнезилируются [46]. Важно отметить, что сигнальные G-белки могут быть и фарнезилированы, и геранилгеранилированы, например, белок RhoB (белки семейства Rho регулируют актин цитоскелета клеток, а также их адгезию) [47]. Этот потенциал пренилирования Ras-белков предполагает, что GGT-аза может восстанавливать функции этих белков, если FT-аза заингибирована [48]. Тем не менее, не все Ras-белки пренилируются GGT-азой, и до конца не выяснены функции геранилированных белков Ras и насколько они схожи с функциями фарнезилированных Ras.
МИШЕНИ СИГНАЛЬНЫХ ПУТЕЙ Ras ДЛЯ ТАРГЕТНОЙ ТЕРАПИИ ОПУХОЛЕЙ
Онкобелки семейства Ras экспрессированы в большом количестве опухолей и играют важную роль в их малигнизации. В связи с этим за последние 20–30 лет предпринимались неоднократные попытки разработки лекарственных препаратов, мишенью для которых служили бы эти онкобелки и их сигнальные пути [49]. Для этого использовали непосредственное взаимодействие ингибиторов как с FT-азой, так и с различными сайтами связывания самих белков Ras, а также их комплексами с белками-трансляторами и эффекторами [15, 50].
Ингибиторы обмена нуклеотидов
Активация каскада Ras и передача сигнала на дальнейшие его ступени происходит за счет обмена нуклеотидами между GTP- и GDP-связанными формами Ras. Регулируется этот процесс факторами GEF и GAP, однако активация самого Ras происходит без его ковалентной модификации нуклеотидами [51, 52]. Ras-мутации, которые изменяют их активность, нечувствительны к GAP, что приводит к накоплению мутантных форм Ras-белков в их активном состоянии, то есть связанными с GTP, и запускают сигнальные каскады, инициирующие рост онкогенных клеток [51, 53, 54].
Ингибиторы нуклеотидного обмена можно разделить на два класса: соединения, которые связываются в нуклеотидном сайте Ras, и соединения, связывающиеся на сайте белка GEF.
Ингибиторы связывания нуклеотидного сайта
Этот класс ингибиторов объединяет нуклеотидные аналоги и некоторые структурно не связанные c ними молекулы. Стратегии разработки этих ингибиторов были сфокусированы на синтезе веществ, способных конкурировать с GDP и GTP в сайте их связывания на Ras. С этой целью были разработаны библиотеки нуклеотидных аналогов N2-замещенных гуанозин-5'-трифосфатов [55] и пиразоло[3,4-b]хинолиновых рибофуранозидов [56] (рис. 3). Однако ни одна из рассмотренных молекул не показала достаточного ингибирования процесса нуклеотидного обмена онкогенного Ras, что легко объясняется очень высокой аффинностью нуклеотидов к сайтам связывания Ras.
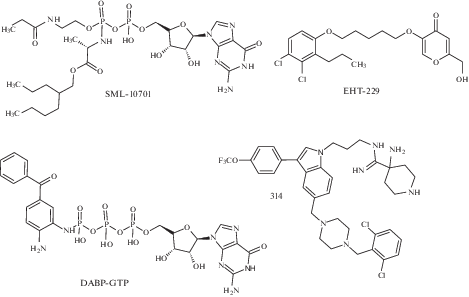
Ингибиторы комплексообразования белка Ras с белком SOS семейства GEFs
Альтернативный подход к прямому взаимодействию с нуклеотидным сайтом Ras для ингибирования связывания GTP заключается в блокировании взаимодействия с белками, которые регулируют процесс нуклеотидного обмена. Одной из перспективных молекулярных мишеней направленной Ras-терапии является комплекс белка Ras с одним из белков семейства GEFs–SOS, который отвечает за переход белка Ras в активное состояние [57]. Во время нуклеотидного обмена Ras вступает в межбелковое взаимодействие с SOS с образованием комплекса, содержащего одну SOS и две молекулы Ras. SOS является уникальным среди Ras-специфических GEF в том, что он имеет аллостерический Ras-связывающий сайт, который увеличивает его каталитическую активность. Кристаллическая структура комплекса основного каталитического домена (SOScat) показывает, что H-Ras расположен в участке Cdc25 домена SOScat и включен в комплекс фрагментом, отвечающим за обмен нуклеотидов. При присоединении SOS происходят значительные конформационные изменения в структуре Ras, что приводит к открытию его сайта связывания нуклеотидов (рис. 4) [58]. SOS стабилизирует свободную от нуклеотидов форму Ras путем формирования комплекса и приводит к открытию активного сайта Ras с высвобождением нуклеотида, переводя Ras в активную форму. Аллостерическое связывание Ras-GTP стабилизирует белок SOS на мембране, где он может катализировать другие молекулы Ras, приводя к сверхчувствительному ответу, отличному от ответа других Ras-специфических GEF [59].
Рис. 4.
Комплекс Ras-SOS. А – сайт связывания белка SOS, Б – сайт связывания, сформированный совместно белками Ras и SOS, В – сайт связывания SOS в Ras-белке, Г – сайт связывания нуклеотидов в Ras-белке.
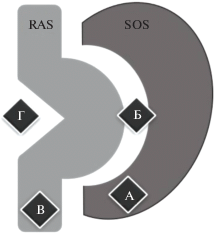
Несколькими группами исследователей с использованием скрининга на основе фрагментов известных лигандов были определены эффективные структуры, ответственные за взаимодействия с сайтами комплекса Ras-SOS [60–62] (рис. 5), и синтезирован ряд оптимизированных соединений, связывающихся с комплексом Ras-SOS.
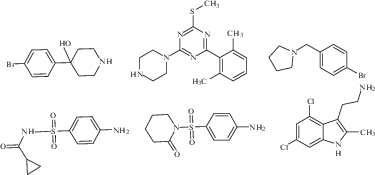
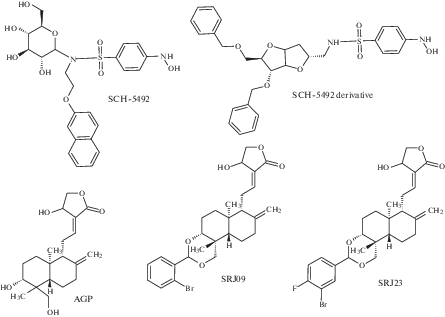
Эти соединения можно разделить на несколько классов (рис. 5): производные сахаров (соединение SCH-54292 и его аналоги), производные индола и сульфонамида, производные андрографолида и его бензилиденовые производные (соединения AGP, SRJ09, SRJ23) [61]. Было показано, что соединение SCH-54292 и его аналоги способны ингибировать активацию Ras с IC50 на микромолярном уровне [63, 64].
Как было показано в экспериментах in vitro, соединения AGP, SRJ09, SRJ23, связываясь с сайтами белка KRas4B, стабилизируют комплекс Ras-SOS, не позволяя проходить SOS-катализируемому обмену нуклеотидов и переводу Ras-белка в активную форму [65]. В совокупности эти результаты показывают, что исследованные соединения могут ингибировать процесс нуклеотидного обмена мутированных белков Ras и предлагают новый способ ингибирования SOS-опосредованной активации Ras, что открывает потенциальный путь для предотвращения развития и роста опухоли [66].
Однако, хотя эти соединения и способны влиять на нуклеотидный обмен между активной и неактивной формами белка Ras, по-видимому, для успешного применения требуются соединения, обладающие большей аффинностью. Возможно, более эффективным решением будет получение гибридных молекул, содержащих фармакофоры, соединенные рассчитанной длины линкерами, связывающихся с несколькими сайтами комплекса Ras-SOS.
Фарнезилтрансфераза (FT-аза) и ее ингибиторы
На протяжении последних 20–30 лет наиболее традиционной молекулярной мишенью потенциальных противоопухолевых препаратов остается фермент FT-аза [49].
Для посттрансляционного процессинга белков Ras FT-азы, наряду с GGT-азой-1, играют ключевую роль и обеспечивают активацию белков семейств Ras и RhoB [67, 68]. Эти ферменты локализованы в цитозоле, катализируют пренилирование белков и различаются белковыми мишенями и присоединяемым изопреноидным субстратом [69].
FT-аза катализирует присоединение 15-углеродного фрагмента фарнезилдифосфата [69] к белкам, содержащим CAAX-фрагмент (рис. 2). FT-аза представляет собой гетеродимер двух субъединиц, α и β, с молекулярной массой 48 и 46 кДа, соответственно [71, 72]. Активные центры фермента формируются пересечением центра β-субъединицы с частями α-субъединицы. FT-аза содержит в своем активном центре двухвалентный цинк (Zn2+) для связывания с фрагментом CAAX [73], для катализа требует наличия иона магния (Mg2+) [70]. Субстратная специфичность FT-аз определяется аминокислотными остатками участка CAAX, в частности аминокислотным остатком X [74]. Белки, содержащие в качестве Х метионин или серин, проявляют большую аффинность к FT-азе. Это белки N-Ras, содержащие фрагмент Cys-Val-Val-Met, K-Ras4A с Cys-Ile-Ile-Met, K-Ras4B с Cys-Val-Ile-Met, и H-Ras с Cys-Val-Leu-Ser, причем аффинность белков, содержащих метионин, в 30 раз превышает аффинность белков, содержащих серин или глицин [69, 75, 76]. Важно отметить, что FT-аза содержит места связывания как для CAAX-содержащего пептида, так и для фарнезилдифосфата (FDP) [77–79]. Сайты связывания FDP и CAAX расположены в β-субъединице, в то время как α-субъединица стабилизирует β-субъединицу и катализирует передачу фарнезила [77]. α-Субъединица после передачи фарнезила фосфорилируется, восстанавливая активность фермента [78].
GGT-аза-1, также как и FT-аза, состоит из α- и β-субъединиц. α-Субъединица обоих ферментов имеет одинаковую молекулярную массу – 48 кДа, и одинаковое строение [79]. β-Субъединицы этих ферментов различны, идентичность аминокислотных последовательностей составляет около 30% [80]. Оба фермента, FT-аза и GGT-аза-1 в соответствии со своими функциями имеют два субстрата – изопреноид и белок (Ras) [81].
Ион цинка находится в активном центре и необходим для проявления энзиматической активности. Он координируется тремя аминокислотными остатками (Aspb297, Cysb299 и Hisb362 в β-субъединице FT-азы; и Aspb269, Cysb271 и Hisb321 в GGT-азе-I [58, 71, 82 ].
В организме человека к настоящему времени идентифицировано много белков, содержащих CAAX-фрагмент: Ras, Rho, Rac, Rap [83–85]. Белки семейства Ras считаются первыми идентифицированными субстратами FT-азы. Однако было обнаружено, что в присутствии ингибиторов фарнезилтрансферазы белки N-Ras и K-Ras могут пренилироваться геранилгеранилтрансферазой-1 и только H-Ras фарнезилируется [86, 87]. Это приводит к необходимости одновременного использования ингибиторов обоих ферментов, и является одним из направлений разработки противоопухолевых препаратов.
С момента открытия онкобелков Ras и FT-азы активно использовался подход по созданию препаратов, блокирующих фарнезилирование, – ингибиторов FT-азы, – конкурирующих с FPP или CAAX-содержащим субстратом. В результате имеется огромное количество литературных данных об исследованиях in vitro, в которых показана эффективность ингибиторов FT-азы при H-Ras-опосредованной клеточной трансформации [87–89]. Эта стратегия долгое время была основной в разработке ингибиторов FT-азы. К настоящему времени синтезировано большое количество таких соединений [27, 36, 91, 92].
Ингибиторы FT-азы и GGT-азы-I можно разделить на четыре класса: 1) небольшие молекулы, конкурирующие с изопреноидным субстратом, 2) небольшие молекулы, конкурирующие с белковым субстратом (CAAX-содержащим), 3) бисубстратные аналоги, 4) соединения, не конкурирующие ни с белковым, ни с изопреноидным субстратом [91, 93, 94] (рис. 7).
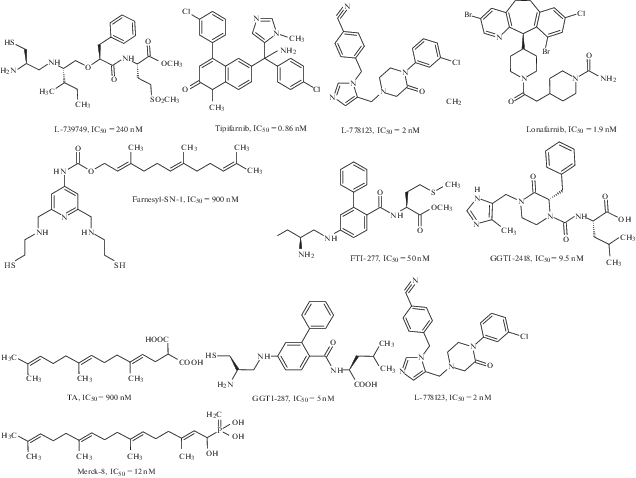
Ингибиторы FT-азы исследуются или были исследованы на разных фазах клинических испытаний. Это, прежде всего, такие препараты, как лонафарниб (Schering-Plough), типифарниб (Ortho Biotech Products), L778123 (Merck), BMS-214662 (Bristol-Myers). Высокая активность in vitro и специфические функции ингибиторов FT-азы обеспечили их интенсивное исследование на 1-й фазе клинических испытаний. Было показано, что их цитотоксические эффекты достигались в концентрациях много ниже токсических для нормальных тканей [95]. Однако клинические исследования фаз 2–3, которые являются критическими для выбора специфических опухолей и определяют дальнейшую судьбу препаратов, показали далеко не обнадеживающие результаты [96, 97].
Неутешительные результаты клинических исследований FTI привели к некоторому забвению этой терапевтической области. Тем не менее, несмотря на отсутствие эффективности против опухолей, экспрессирующих K-Ras, FTI являются мощными супрессорами H-Ras и в настоящее время испытываются в клинических исследованиях на пациентах с онкогенными мутациями H-Ras [98].
Одновременно с этим развиваются и новые подходы к использованию ингибиторов FT-азы, основанные на более глубоком понимании механизмов их действия. Основной проблемой при создании эффективных в клинике ингибиторов FT-азы является тот факт, что белки Ras являются GTP-азами, регуляция которых происходит за счет цикла присоединения GTP (активация, этот процесс стимулируется GEFs) и гидролиза GTP до GDP для дезактивации (стимулируется GAPs) (рис. 1). В результате прямого ингибирования FT-азы происходит накопление GTP-связанного Ras, что может приводить к негативным последствиям [99].
Еще одной серьезной проблемой разработки ингибиторов Ras является высокая аффинность связывания белков Ras с GTP – этот процесс происходит на пикомолярном уровне, тогда как для многих протеинкиназ такое связывание отмечается на микромолярном уровне [100]. Наличие этого феномена приводит к тому, что синтезированные аналоги нуклеотидов активно блокируют связывание ATP с киназами, но не способны блокировать связывание GTP с Ras [101].
Белки Ras для корректного встраивания в мембрану и активации требуют фарнезилирования. Однако в присутствии ингибиторов FT-азы белки K-Ras и N-Ras могут быть геранилгеранилированы GGTase-I [87, 102]. В соответствии с этим перспективным направлением разработки ингибиторов Ras является синтез гибридных молекул, одновременно ингибирующих FT-азу и GGT-азу-I. Наиболее известным представителем этого класса соединений является L-778123 (рис. 7), который в наномолярных концентрациях ингибирует пренилирование H-Ras, N-Ras и K-Ras [103] и в настоящий момент препарат проходит клинические испытания.
Интересным подходом представляется создание ингибиторов FT-азы, блокирующих возможность не только фарнезилирования, но и дальнейшего пренилирования CAAX-мотива [99]. Предыдущие поколения ингибиторов были нацелены на ингибирование функции самой FT-азой, оставляя мотив CAAX доступным для пренилирования GGT-азы, что является достаточным для последующей мембранной локализации Ras. Было показано, что с использованием нео-субстратной стратегии возможно устранить этот недостаток. Был разработан альтернативный субстрат для FT-азы, который ковалентно модифицировал концевой цистеин белка K-Ras. Это приводило не только к невозможности альтернативного (GGT-азой) пренилирования, но и к нарушению локализации K-Ras в клетке. Известно, что существенным фактором для малигнизации опухоли является необходимость присутствия не только мутированных форм онкобелков Ras, но и не подвергнувшихся мутации и функционирующих. Так, например, онкогенная активность K-Ras требует наличия функционирующих белков H-Ras или N-Ras [25, 26]. В результате применения нео-субстратной стратегии получается не только ингибирование нормального фарнезилирования и альтернативного пренилирования, но и накопление Ras белка в несвойственном ему месте – в цитоплазме, что повышает эффективность действия препаратов [99].
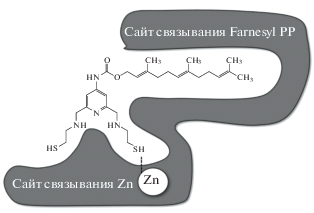
Перспективным направлением поиска ингибиторов FT-азы является применение бисубстратной стратегии. При этом в одной молекуле объединяются фрагменты, отвечающие за взаимодействие с несколькими сайтами связывания. Так, например, в работе [104] созданы новые ингибиторы FT-азы, содержащие фрагмент, связывающий ион цинка активного центра и фарнезильную группу для более точного позиционирования – например, соединение Farnesyl-SN-1 (рис. 7, 8). Полученные соединения проявляют выраженную ингибирующую активность в отношении FT-азы и обладают цитотоксичностью для ряда опухолевых клеток.
Рис. 8.
Позиционирование соединения Farnesyl-SN-1 в сайте связывания Zn2+ и фарнезилпирофосфата [94].
Следует отметить, что когда было выяснено, что мутантные формы Ras являются движущей силой образования многих видов злокачественных опухолей человека, огромные усилия и ресурсы были направлены на то, чтобы разработать их ингибиторы. К сожалению, это они не нашли успешного применения в клинике. При дальнейших исследованиях выяснилось, что функции онкобелков Ras и FT-азы более обширны и разнообразны [105]. Понимание тонких механизмов регуляции процессинга онкобелков Ras и их взаимосвязи с процессом малигнизации опухоли, а также использование новых стратегий создания лекарственных препаратов, основанных как на регуляции локализации онкобелков Ras, так и на применении новых, бифармакофорных соединений, позволяют надеяться на возрождение интереса к разработке противоопухолевых лекарственных препаратов на основе ингибиторов FT-аз.
ЗАКЛЮЧЕНИЕ
Мутантные формы Ras являются причиной образования многих видов злокачественных опухолей человека. Регулируемые ими сигнальные пути, такие, как сигнальные пути Raf/MEK/ERK и фосфоинозитид-3-киназы, регулируют важнейшие свойства малигнантных клеток – рост и выживание, пролиферацию, дифференциацию, адгезию, способность избегать апоптоза. Понимание тонких механизмов регуляции процессинга онкобелков Ras и их взаимосвязи с процессом малигнизации опухоли, а также использование новых стратегий, основанных как на регуляции локализации онкобелков Ras в клетке, так и на применении новых, бифармакофорных соединений, действующих на разные сайты связывания фермента, позволяют надеяться на возрождение интереса к ингибиторам FT-азы, как потенциальным антинеопластам.
Список литературы
Jiang K., Coppola D., Crespo N.C., Nicosia S.V., Hamilton A.D., Sebti S.M., Cheng J.Q. // Mol. Cell. Biol. 2000. V. 20. P. 139–148. https://doi.org/10.1128/mcb.20.1.139-148.2000
Bos J.L. // Cancer Res. 1989. V. 49. P. 4682–4689.
Barbacid M. // Annu. Rev. Biochem. 1987. V. 56. P. 779–827.
Samatar A.A., Poulikakos P.I. // Nat. Rev. Drug Discov. 2014. V. 13. P. 928–942. https://doi.org/10.1038/nrd4281
Pylayeva-Gupta Y., Grabocka E., Bar-Sagi D. // Nat. Rev. Cancer. 2011. V. 11. P. 761–774. https://doi.org/10.1038/nrc3106
Baines A.T., Xu D., Der C.J. // Future Med. Chem. 2011. V. 3. P. 1787–1808. https://doi.org/10.4155/fmc.11.121
Wang J., Yao X., Huang J. // Med. Chem. Comm. 2017. V. 8. P. 841–854. https://doi.org/10.1039/c7md00030h
Kranenburg O., Gebbink M., Voest E. // Biochim. Biophys. Acta. 2004. V. 1654. P. 23–37. https://doi.org/10.1016/j.bbcan.2003.09.004
Jung F. // Circ. Res. 2002. V. 91. P. 38–45.
Blancher C., Moore J.W., Robertson N., Harris A.L. // Cancer Res. 2001. V. 61. P. 7349–7355.
Rak J., Mitsuhashi Y., Sheehan C., Tamir A., Viloria-Pet-it A., Filmus J., Mansour S.J., Ahn N.G., Kerbel R.S. // Cancer Res. 2000. V. 60. P. 490–498.
Sharma V., Shaheen S.S., Dixit D., Sen E. // Inflammation. 2012. V. 35. P. 516–519. https://doi.org/10.1007/s00109-010-0683-5
Tsujii M., Kawano S., Tsuji S., Sawaoka H., Hori M., DuBois R.N. // Cell. 1998. V. 93. P. 705–716. https://doi.org/10.1016/s0092-8674(00)81433-6
Rojas A.M., Fuentes G., Rausell A., Valencia A. // Cell Biol. 2012. V. 196. P. 189–201. https://doi.org/10.1083/jcb.201103008
Marín-Ramos N.I., Ortega-Gutiérrez S., López-Rodríguez M.L. // Semin Cancer Biol. 2019. V. 54. P. 91–100. https://doi.org/10.1016/j.semcancer.2018.01.017
Mitin N., Rossman K.L., Der C.J. // Curr. Biol. CB. 2005. V. 15. P. 563–574. https://doi.org/10.1016/j.cub.2005.07.010
Vasan N., Boyer J.L., Herbst R.S. // Clin. Cancer Res. 2014. V. 2. P. 3921–3930. https://doi.org/10.1158/1078-0432.CCR-13-1762
Rodriguez-Viciana P., Warne P.H., Dhand R., Vanhaesebroeck B., Gout I., Fry M.J., Waterfield M.D., Downward J. // Nature. 1994. V. 370. P. 527–532. https://doi.org/10.1038/370527a0
Lodish H., Berk A., Zipursky S.L., Matsudaira P., Baltimore D., Darnell J. // Molecular Cell Biology (4th ed.). 2000.
Vivanco I., Sawyers C.L. // Nature Review Cancer. 2002. V. 2. P. 489–501. https://doi.org/10.1038/nrc839
Sever R., Brugge J.S. // Cold Spring Harbor Perspectives in Medicine. 2015. V. 5. https://doi.org/10.1101/cshperspect.a006098
Bourne H.R., Sanders D.A., McCormick F. // Nature. 1990. V. 348. P. 125–132. https://doi.org/10.1038/348125a0
Bos J.L., Rehmann H., Wittinghofer // Cell. 2007. V. 129. P. 865–877. https://doi.org/10.1016/j.cell.2007.05.018
Brock E.J., Ji K., Reiners J.J., Mattingly R.R. // Mini Rev. Med. Chem. 2016. V. 16. P. 358–369. https://doi.org/10.2174/1389557515666151001154002
Lim K.H., Ancrile B.B., Kashatus D.F., Counter C.M. // Nature. 2008. V. 452. P. 646–649. https://doi.org/10.1038/nature06778
Bentley C., Jurinka S.S., Kljavin N.M., Vartanian S., Ramani S.R., Gonzalez L.C., Yu K., Modrusan Z., Du P., Bourgon R., Neve R.M., Stokoe D. // Biochem. 2013. V. 452. P. 313–320. https://doi.org/10.1042/BJ20121578
Agrawal A.G., Somani R.R. // Mini-Reviews in Medicinal Chemistry. 2009. V. 9. P. 638–652. https://doi.org/10.2174/138955709788452702
Klochkov S.G., Neganova M.E., Yarla N.S., Parvathaneni M., Sharma B., Tarasov V.V., Barreto G., Bachurin S.O., Ashraf G.M., Aliev G. // Semin. Cancer. Biol. 2019. V. 56. P. 128–134. https://doi.org/10.1016/j.semcancer.2017.10.010
McCormick F. // Nature. 1993. V. 363. P. 15–16. https://doi.org/10.1038/363015a0
McCormack F. // Curr. Opin. Genet. Dev. 1994. V. 4. P. 71–76. https://doi.org/10.1016/0959-437x(94)90093-0
Kato K., Cox A.D., Hisaka M.M., Graham S.M., Bus J.E., Der C.J. // Proc. Natl. Acad. Sci. 1992. V. 89. P. 6403–6407. https://doi.org/10.1073/pnas.89.14.6403
Casey P.J. // Proc. Natl. Acad. Sci. 1989. V. 86. P. 8323–8327. https://doi.org/10.1073/pnas.86.21.8323
Gelb M.H. // Science. 1997. V. 275. P. 1750–1751. https://doi.org/10.1126/science.275.5307.1750
Cox A.D., Der C.J. // Biochim. Biophys. Acta. 1997. V. 1333. P. 51–71. https://doi.org/10.1016/s0304-419x(97)00011-5
Omer C.A., Anthony N.J., Buser-Doepner C.A., Burkhardt A.L., deSolms S.J., Dinsmore C.J., Gibbs J.B., Hartman G.D., Koblan K.S., Lobell R.B., Oliff A., Williams T.M., Kohl N.E. // Biofactors. 1997. V. 6. P. 359–366. https://doi.org/10.1002/biof.5520060306
Gibbs J.B., Oliff A. // Ann. Rev. Pharmacol. Toxicol. 1997. V. 37. P. 143–166. https://doi.org/10.1146/annurev.pharmtox.37.1.143
Schafer W.R., Kim R., Sterne R., Thorner J., Kim S., Rine J. // Science. 1989. V. 245. P. 379–385. https://doi.org/10.1126/science.2569235
Gibbs J.B. // Springer-Verlag. 1993. V. 137.
Clarke S., Vogel J.P., Deschenes R.J., Stock J. // Proc. Natl. Acad. Sci. 1988. V. 85. P. 4843–4847. https://doi.org/10.1073/pnas.85.13.4643
Fujita-Yamaguchi Y., Kathuria S., Xu Q-Y., McDonald J.M., Nakano H., Kamata T. // Proc. Nab. Acad. Sci. 1989. V. 86. P. 7306–7310.
Hancock J.F., Magee A.I., Childs J.E., Marshall C. // Cell. 1989. V. 57. P. 1167–1177. https://doi.org/10.1016/0092-8674(89)90054-8
Casey P.J., Solski P.A., Der C.J., Buss J. // Proc. Natl. Acad. Sci. 1989. V. 86. P. 1167–1177. https://doi.org/10.1073/pnas.86.21.8323
Gutierrez L., Magee A.I., Marshall C. J., Hancock J.F. // EMBO. 1989. V. 8. P. 1093–1098.
Hancock J.F., Paterson H., Marshall C.J. // Cell. 1990. V. 63. P. 133–139. https://doi.org/10.1016/0092-8674(90)90294-o
Casey P.J., Seabra M.C. // Biol. Chem. 1996. V. 271. P. 5289–5292. https://doi.org/10.1074/jbc.271.10.5289
James G.L., Goldstein J.L., Brown M.S. // Biol. Chem. 1995. V. 266. P. 14 603–14 610. https://doi.org/10.1074/jbc.270.11.6221
Armstrong S.A., Hannah V.C., Goldstein J.L., Brown M.S. // Biol. Chem. 1995. V. 270. P. 7864–7868. https://doi.org/10.1074/jbc.270.14.7864
Marks R.E., Ho A.W., Robbel C., Kuna T., Berk S., Gajewski T.F. // Blood. 2007. V. 110. P. 1982–1988. https://doi.org/10.1182/blood-2006-06-031088
Khan A.Q., Kuttikrishnan S., Siveen K.S., Prabhu K.S., Shanmugakonar M., Al-Naemi H.A., Haris M., Dermime S., Uddin S. // Seminars in Cancer Biology. 2018. https://doi.org/10.1016/j.semcancer.2018.03.001
Spencer-Smith R., O’Bryan J.P. // Seminars in Cancer Biology. 2017. https://doi.org/10.1016/j.semcancer.12.005
Vigil D., Cherfils J., Rossman K.L., Der C.J. // Nat. Rev. Cancer. 2010. V. 10. P. 842–857. https://doi.org/10.1038/nrc2960
Ahearn I.M., Haigis K., Bar-Sagi D., Philips M.R. // Nat. Rev. Mol. Cell Biol. 2012. V. 13. P. 39–51. https://doi.org/10.1038/nrm3255
Adjei A.A. // Natl. Cancer Inst. 2001. V. 93. P. 1062–1074. https://doi.org/10.1093/jnci/93.14.1062
Schubbert S., Shannon K., Bollag G. // Nat. Rev. Cancer. 2007. V. 7. P. 295–308. https://doi.org/10.1038/nrc2109
Noonan T., Brown N., Dudycz L., Wright G. // Med. Chem. 1991. V. 34. P. 1302–1307. https://doi.org/10.1021/jm00108a010
Wolin R., Wang D., Kelly J., Afonso A., James L., Kirschmeier P., McPhail A.T. // Bioorg. Med. Chem. Lett. 1996. V. 6. P. 195–200. https://doi.org/10.1016/0960-894X(95)00574-D
Wilson C.Y., Tolias P. // Drug Discov. Today. 2016. V. 21. P. 1915–1919. https://doi.org/10.1016/j.drudis.2016.08.002
Boriack-Sjodin P.A., Margarit S.M., Bar-Sagi D., Kuriyan J. // Nature. 1998. V. 394. P. 337–343. https://doi.org/10.1038/28548
Bandaru P., Kondo Y., Kuriyan J. // Cold Spring Harb. Perspect. Med. 2019. V. 9. P. a031534. https://doi.org/10.1101/cshperspect.a031534
Maurer T., Garrenton L.S., Oh A., Pitts K., Anderson D.J., Skelton N.J., Fauber B.P., Pan B., Malek S., Stokoe D., Ludlam M.J., Bowman K.K., Wu J., Giannetti A.M., Starovasnik M.A., Mellman I., Jackson P.K., Rudolph J., Wang W., Fang G. // Proc. Natl. Acad. Sci. USA. 2012. V. 109. P. 5299–5304. https://doi.org/10.1073/pnas.1116510109
Sun Q., Burke J.P., Phan J., Burns M.C., Olejniczak E.T., Waterson A.G., Lee T., Rossanese O.W., Fesik S.W. // Angew. Chem. Int. Ed. Engl. 2012. V. 51. P. 6140–6143. https://doi.org/10.1002/anie.201201358
Winter J.J., Anderson M., Blades K., Brassington C., Breeze A.L., Chresta C., Embrey K., Fairley G., Faulder P., Finlay M.R., Kettle J.G., Nowak T., Overman R., Patel S.J., Perkins P., Spadola L., Tart J., Tucker J.A., Wrigley G. // Med. Chem. 2015. V. 58. P. 2265–2274. https://doi.org/10.1021/jm501660t
Ganguly A.K., Pramanik B.N., Huang E.C., Liberles S., Heimark L., Liu Y.H., Tsarbopoulos A., Doll R.J., TaveRas A.G., Remiszewski S., Snow M.E., Wang Y.S., Vibulbhan B., Cesarz D., Brown J.E., del Rosario J., James L., Kirschmeier P., Girijavallabhan V. // Bioorg. Med. Chem. 1997. V. 5. P. 817–820. https://doi.org/10.1016/s0968-0896(97)00021-7
Taveras A.G., Remiszewski S.W., Doll R.J., Cesarz D., Huang E.C., Kirschmeier P., Pramanik B.N., Snow M.E., Wang Y.S., del Rosario J.D., Vibulbhan B., Bauer B.B., Brown J.E., Carr D., Catino J., Evans C.A., Girijavallabhan V., Heimark L., James L., Liberles S., Nash C., Perkins L., Senior M.M., Tsarbopoulos A., Webber S.E. // Bioorg. Med. Chem. 1997. V. 5. P. 125–133. https://doi.org/10.1016/S0968-0896(96)00202-7
Hocker H.J., Cho K.-J., Chen C.-Y.K., Rambahal N., Sagineedu S.R., Shaari K., Stanslas J., Hancock J.F., Gorfe A.A. Proc. Natl. Acad. Sci. USA. 2013. V. 110. P. 10 201–10 206. https://doi.org/10.1073/pnas.1300016110
Lu S., Jang H., Zhang J., Nussinov R. // Chem. Med. Chem. 2016. V. 11. P. 814–821. https://doi.org/10.1038/srep21949
Delarue F.L., Adnane J., Joshi B., Blaskovich M.A., Wang D.A., Hawker J., Bizouarn F., Ohkanda J., Zhu K., Hamilton A.D., Chellappan S., Sebti S.M. // Oncogene. 2007. V. 26. P. 633–640. https://doi.org/10.1038/sj.onc.1209819
Fritz G., Kaina B. // Curr. Cancer Drug Targets. 2006. V. 6. P. 1–14.
Shen M., Pan P., Li Y., Li D., Yu H., Hou T. // Drug Discov. Today. 2015. V. 20. P. 267–276. https://doi.org/10.1016/j.drudis.2014.10.002
Subramanian T., Liu S., Troutman J.M., Andres D.A., Spielmann H.P. // Chembiochem. 2008. V. 9. P. 2872–2882. https://doi.org/10.1002/cbic.200800248
Zhang F.L., Casey P.J. // Ann. Rev. Biochem. 1996. V. 65. P. 241–269. https://doi.org/10.1146/annurev.bi.65.070196.001325
Dunten P., Kammlott U., Crowther R., Weber D., Palermo R., Birktoft J. // Biochemistry. 1998. V. 37. P. 7907–7912. https://doi.org/10.1021/bi980531o
Sousa S.F., Fernandes P.A., Ramos M.J. // Curr. Med. Chem. 2008. V. 15. P. 1478–1492. https://doi.org/10.2174/092986708784638825
Moores S.L., Schaber M.D., Mosser S.D., Rands E., O’Hara M.B., Garsky V.M., Marshall M.S., Pompliano D.L., Gibbs J.B. // Biol. Chem. 1990. V. 266. P. 14 603–14 610.
Reiss Y., Goldstein J.L., Seabra M.C., Casey P.J., Brown M.S. // Cell. 1990. V. 62. P. 81–88.
Reiss Y., Stradley S.J., GieRasch L.M., Brown M.S., Goldstein J.L. // Proc. Natl. Acad. Sci. 1991. V. 88. P. 732–36. https://doi.org/10.1073/pnas.88.3.732
Andres D.A., Goldstein J.L., Ho Y.K., Brown M.S. // B-iol. Chem. 1993. V. 268. P. 1383–1390.
Kumar A., Beresini M.H., Dhawan P., Mehta K.D. // Biochem. Biophys. Res. Commun. 1996. V. 222. P. 445–452. https://doi.org/10.1006/bbrc.1996.0764
Seabra M.C., Reiss Y., Casey P.J., Brown M.S., Goldstein J.L. // Cell. 1991. V. 65. P. 429–434. https://doi.org/10.1016/0092-8674(91)90460-g
Long S.B., Casey P.J., Beese L.S. // Biochemistry. 1998. V. 37. P. 9612–9618. https://doi.org/10.1021/bi980708e
Park H.W., Boduluri S.R., Moomaw J.F., Casey P.J., Beese L.S. // Science. 1997. V. 275. P. 1800–2180. https://doi.org/10.1126/science.275.5307.1800
Fu H.W., Beese L.S., Casey P.J. // Biochemistry. 1998. V. 37. P. 4465–4472. https://doi.org/10.1021/bi972511c
Kinsella B.T., Erdman R.A., Maltese W.A. // Biol. Chem. 1991. V. 266. P. 9786–9794.
Nobes C.D., Lauritzen I., Mattei M.G., Paris S., Hall A., Chardin P. // Cell Biol. 1998. V. 141. P. 187–197. https://doi.org/10.1083/jcb.141.1.187
Kontani K., Tada M., Ogawa T., Okai T., Saito K., Araki Y., Katada T. // Biol. Chem. 2002. V. 277. P. 41 070–41 078. https://doi.org/10.1074/jbc.M202150200
Whyte D.B., Kirschmeier P., Hockenberry T.N., Nunez-Oliva I., James L., Catino J.J., Bishop W.R., Pai J.K. // Biol. Chem. 1997. V. 272. P. 14 459–14 464. https://doi.org/10.1074/jbc.272.22.14459
Zhang F.L., Kirschmeier P., Carr D., James L., Bond R.W., Wang L., Patton R., Windsor W.T., Syto R., Zhang R., Bishop W.R. // Biol. Chem. 1997. V. 272. P. 10 232–10 239. https://doi.org/10.1074/jbc.272.15.10232
Nagasu T., Yoshimatsu K., Rowell C., Lewis M.D., Garcia A.M. // Cancer Res. 1995. V. 55. P. 5310–5314.
Sun J., Qian Y., Hamilton A.D., Sebti S.M. // Oncogene. 1998. V. 16. P. 1467–1473. https://doi.org/10.1038/sj.onc.1201656
Brunner T.B., Hahn S.M., Gupta A.K., Muschel R.J., McKenna W.G., Bernhard E.J. // Cancer Res. 2003. V. 63. P. 5656–5668.
Strickland C.L., Weber P.C., Windsor W.T., Wu Z., Le H.V., Albanese M.M., Alvarez C.S., Cesarz D., del Rosario J., Deskus J., Mallams A.K., Njoroge F.G., Piwinski J.J., Remiszewski S., Rossman R.R., TaveRas A.G., Vibulbhan B., Doll R.J., Girijavallabhan V.M., Ganguly A.K. // Med. Chem. 1999. V. 42. P. 2125–2135. https://doi.org/10.1021/jm990030g
Клочков С.Г., Афанасьева С.В., Зефиров Н.С. // Технологии живых систем. 2008. С. 31–39.
Crul M., de Klerk G.J., Beijnen J.H, Schellens J.H. // Anticancer Drugs. 2001. V. 12. P. 163–184. https://doi.org/10.1097/00001813-200103000-00001
Reid T.S., Long S.B., Beese L.S. // Biochemistry. 2004. V. 43. P. 9000–9008. https://doi.org/10.1021/bi049280b
Midgley R.S., Kerr D.J. // Crit. Rev. Oncol. Hematol. 2002. V. 44. P. 109–120. https://doi.org/10.1016/s1040-8428(01)00189-5
Gordon L.B., Kleinman M.E., Massaro J., D’Agostino R.B. Sr., Shappell H., Gerhard-Herman M., Smoot L.B., Gordon C.M., Cleveland R.H., Nazarian A., Snyder B.D., Ullrich N.J, Silvera V.M., Liang M.G., Quinn N., Miller D.T., Huh S.Y., Dowton A.A., Littlefield K., Greer M.M., Kieran M.W. // Circulation. 2016. V. 134. P. 114–125. https://doi.org/10.1161/CIRCULATIONAHA.116.022188
Jabbour E., Kantarjian H., Cortes J. // Expert Opin. Investig. Drugs. 2007. V. 16. P. 381–392. https://doi.org/10.1517/13543784.16.3.381
Chen X., Makarewicz J.M., Knauf J.A., Johnson L.K., Fagin J.A. // Oncogene. 2013. V. 33. P. 5442–5449. https://doi.org/10.1038/onc.2013.489
Novotny C.J., Hamilton G.L., McCormick F., Shokat K.M. // ACS Chem. Biol. 2017. https://doi.org/10.1021/acschembio.7b00374
John J., Rensland H., Schlichting I., Vetter I., BoRasio G.D., Goody R.S., Wittinghofer A. // Biol. Chem. 1993. V. 268. P. 923–929.
Goekjian P.G. // Curr. Med. Chem. 1999. V. 6. P. 877–903.
Fu H.W., Casey P.J. // Rec. Prog. Hormone Res. 1999. V. 54. P. 315–342.
Morgan M.A., Onono F.O., Spielmann H.P., Subramanian T., Scherr M., Venturini L., Dallmann I., Ganser A., Reuter C.W. // Mol. Med. 2012. V. 90. P. 149–161. https://doi.org/10.1007/s00109-011-0814-7
Tanaka A., Radwan M.O., Hamasaki A., Ejima A., Obata E., Koga R., Tateishi H., Okamoto Y., Fujita M., Nakao M., Umezawa K., Tamanoi F., Otsuka M. // Bioorg. Med. Chem. Lett. 2017. V. 27. P. 3862–3866. https://doi.org/10.1016/j.bmcl.2017.06.047
Cox A.D., Der C.J. // Oncogene. 2003. V. 22. P. 8999–9006. https://doi.org/10.1038/sj.onc.1207111
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия