

Биоорганическая химия, 2021, T. 47, № 6, стр. 659-682
Азаадамантаны – новый перспективный каркасный блок для медицинской химии и фармакологии
Е. В. Суслов 1, К. Ю. Пономарев 1, К. П. Волчо 1, *, Н. Ф. Салахутдинов 1
1 ФГБУН Новосибирский институт органической химии им. Н.Н. Ворожцова СО РАН
630090 Новосибирск, просп. акад. Лаврентьева, 9, Россия
* E-mail: volcho@nioch.nsc.ru
Поступила в редакцию 25.02.2021
После доработки 10.03.2021
Принята к публикации 14.03.2021
Аннотация
Азаадамантаны – азотистые аналоги адамантана, у которых один или несколько атомов углерода замещены на азот. Подобное строение приводит к появлению ряда специфических свойств, как химических, так и физических, и обусловливает широкий спектр полезных качеств этих соединений. Производные азаадамантанов обладают меньшей липофильностью по сравнению с адамантановыми аналогами, что влияет, в частности, на их взаимодействие с биологическими мишенями и биодоступность. Значительный рост числа публикаций, посвященных изучению реакционной способности и биологической активности азаадамантанов и их производных, произошедший за последнее десятилетие (2009–2020 гг.), говорит о большом теоретическом и практическом интересе к ним. Среди производных азаадамантанов уже обнаружены соединения, обладающие выраженной биологической активностью, включая противовирусную, антибактериальную, фунгицидную, противоопухолевую, анальгетическую, ингибиторную (в отношении разных ферментов) и др. В настоящем обзоре представлены основные методы синтеза ди- и триазаадамантанов с узловым расположением атомов азота; систематизированы и обсуждены накопленные в мировой литературе данные по результатам исследований биологической активности азаадамантанов и их производных. Обсуждены перспективы использования азаадамантанов в области медицинской химии и фармакологии.
ВВЕДЕНИЕ
Азаадамантанами называют азотистые аналоги адамантана, у которых один или несколько атомов углерода замещены на азот (рис. 1). Эти соединения известны еще с 50-х гг. прошлого столетия [1, 2], однако систематическое исследование биологической активности азаадамантанов и их производных началось относительно недавно, и основные успехи были достигнуты в последние 20 лет. Частичное замещение атомов углерода на азот заметно изменяет и химические, и физические свойства, приводя, например, к более высокой растворимости в воде азаадамантанов по сравнению с адамантанами. Наличие у азаадамантанов разнообразной биологической активности [3–10] в сочетании с умеренной токсичностью и возможность синтеза различными путями из доступных реагентов делает их привлекательными для использования в качестве базового блока в дизайне новых биологически активных соединений.

Некоторые производные диазаадамантана были обнаружены в природе, например, алкалоиды акосмин (acosmine) и его ацетат, панакосмин (panacosmine), которые были выделены из экстракта семян Acosmium panamense, дасикарпумин (dasycarpumine), выделенный из экстракта Acosmium dasycarpum [9]. Акосмин, как и его производное бовдихин (bowdichine), являющийся сложным эфиром 3,4,5-триметоксибензойной кислоты, были получены из коры стебля Bowdichia virgilioides [9]. Отметим, что в настоящее время биологическая роль этих соединений не ясна, и их биологическая активность не изучалась.
В первом разделе настоящего обзора приведена краткая информация по основным методам синтеза ди- и триазаадамантанов с узловым расположением атомов азота. Далее впервые систематизированы накопленные в мировой литературе данные по результатам исследований биологической активности указанных типов азаадамантанов. Обзор структурирован в соответствии с типом проявляемой азаадамантанами биологической активности.
МЕТОДЫ СИНТЕЗА АЗААДАМАНТАНОВ
Методы синтеза азаадамантанов достаточно подробно рассмотрены в работах [3, 4, 11, 12]. В данном обзоре, для удобства читателей, мы приводим наиболее широко применяющиеся подходы к синтезу 1,3-диазаадамантанов и 1,3,5-триазаадамантанов, остовы которых наиболее часто встречаются в биологически активных производных азаадамантанов. Обычно для синтеза азаадамантанов используют методы, основанные на конденсации ациклических или моноциклических соединений с последующим замыканием трех циклов гетероадамантановой структуры в одну стадию.
Получение 1,3-диазаадамантана и его производных. Часто применяемый метод, позволяющий получать с хорошими выходами азаадамантаны, содержащие два атома азота в узловых положениях, – конденсация различных кетонов с уротропином (гексаметилентетрамином), включающая несколько последовательных реакций Манниха (схема 1 ) [11]. В качестве карбонильной компоненты могут использоваться и ароматические, и алифатические кетоны. С применением этого подхода могут быть синтезированы диазаадамантаны, содержащие как симметричные, так и несимметричные заместители в 5-м и 7-м положениях гетероадамантановой структуры. Кетогруппа в 6-м положении диазаадамантанового остова может быть восстановлена до гидроксильной [13] или метиленовой группы [14] или превращена в другую функциональную группу [15] классическими методами органического синтеза, используемыми для модификации кетонов. Аминальная метиленовая группа во 2-м положении диазаадамантана может быть относительно легко замещена на другой фрагмент раскрытием диазаадамантанового цикла под действием различных реагентов [16, 17], обычно уксусного ангидрида, с образованием соответствующего биспидина, который затем может быть превращен обратно в диазаадамантан под действием различных конденсирующих агентов, таких как кетоны, альдегиды, дигалогениды и т.п.

Схема 1 . Синтез 1,3-диазаадамантанов конденсацией кетонов с уротропином и получение их 2-замещенных производных [11].
Одним из распространенных методов синтеза биспидинов, помимо раскрытия диазаадамантанового остова (схема 1 ), является взаимодействие аминов и кетонов с формальдегидом в кислых условиях по реакции Манниха (схема 2 ) [18, 19]. Однако при этом стоит учесть, что азотсодержащая компонента должна включать хорошо уходящую защитную группу, например, бензильную, для получения в последующем N,N-незамещенного биспидина.

Схема 2 . Синтез биспидинов взаимодействием аминов и кетонов с формальдегидом [18, 19].
Еще одна модификация реакции Манниха для синтеза биспидинов – использование в качестве азотсодержащей компоненты аммиака или ацетата аммония, а вместо формальдегида – ароматических альдегидов (схема 3 ) [20–25]. При этом происходит образование в качестве промежуточных продуктов различных замещенных пиперидонов. Полученные таким образом биспидиноны могут быть превращены в соответствующие диазаадамантаны, как показано на схеме 1 .

Схема 3 . Синтез биспидинов взаимодействием аммиака с кетонами и ароматическими альдегидами [20–25].
Получение 1,3,5-триазаадамантана и его производных. Наиболее часто для синтеза производных 1,3,5-триазаадамантанов (1) применяется методика, основанная на получении триаминов (2) из трис(оксиметил)метана и его гомологов через синтез и восстановление соответствующих триазидов с последующим взаимодействием полученного триамина с различными соединениями, содержащими карбонильную группу (схема 4 ) [26].

Схема 4 . Синтез 1,3,5-триазаадамантанов из трис(оксиметил)метана и его гомологов [26].
Производные 1,3,5-триазаадамантана с различными функциональными группами в 7-м положении гетероадамантановой структуры могут быть получены на основе 7-нитро-1,3,5-триазаадамантана (3), который, в свою очередь, синтезируют конденсацией нитрометана с гексаметилентетрамином в присутствии уксусной кислоты [27] (схема 5 ). Для последующей модификации нитрогруппы применяют классические методы органической химии.

Схема 5 . Синтез 7-нитро-1,3,5-триазаадамантана [27].
Имеющиеся методы синтеза производных ди- и триазаадамантанов, в особенности основанные на конденсации гексаметилентетрамина с карбонильными и нитросоединениями, делают эти азаадамантаны доступными для изучения и использования в синтезе других производных азаадамантанов.
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ АЗААДАМАНТАНОВ
Противовирусная активность. Одними из первых противовирусных лекарств против вируса гриппа были амантадин и римантадин (рис. 2). Их противовирусный эффект связан с блокированием ионных M2-каналов вируса, предотвращающим его проникновение в клетку [28]. Однако в настоящее время практически все эпидемически важные штаммы вируса гриппа выработали резистентность к этим лекарствам [29]. Введение двух атомов азота и двух метильных групп при переходе к соединению (4) (рис. 2) позволило частично преодолеть устойчивость к римантадину штамма вируса гриппа A/California/07/09 (H1N1)pdm09, повысив индекс селективности SI (отношение концентрации, вызывающей гибель 50% клеток (CC50), к концентрация полумаксимального ингибирования (IC50), SI = CC50/IC50) до 13, в сравнении с SI = 5 для римантадина [30]. Присоединение к аминодиазаадамантану (4) фрагмента монотерпеноида цитронеллаля с последующим восстановлением привело к образованию амина (5), продемонстрировавшего высокий SI = 30 и IC50 = 8 мкM [31]. Введение остатка цитронеллаля в 1- или 2-аминоадамантаны, не содержащие узловых атомов азота (соединения (6) и (7)), также увеличивало активность в отношении вируса гриппа, но менее существенно (SI увеличился до 22) [32]. На основании результатов компьютерного моделирования было сделано предположение [30], что диазаадамантан (5) может связываться с белковым каналом M2, хотя нельзя исключить его воздействие и на другие молекулярные мишени.

Среди производных триазаадамантана наличие выраженной противовирусной активности было показано для 7-нитро-1,3,5-триазаадамантана (3) (схема 5 ) в отношении вируса гриппа A штамма Фрунзе в концентрации 1 мкМ, его солянокислой соли в отношении вирусов герпеса 1C и 9С в концентрации 100 мкM, а также для 7-бром- и 7-амино-1,3,5-триазаадамантанов (8) и (9) (рис. 3) в отношении вируса болезни Ньюкасла в концентрации 0.125 мкМ [16].
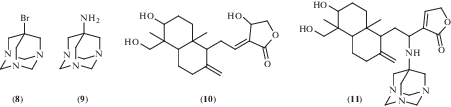
В рамках работы, направленной на синтез и исследование биологической активности новых производных природного дитерпеноида андрографолида (10) (рис. 3), обладающего широким спектром биологической активности [33, 34], было обнаружено, что соединение (11), сочетающее фрагменты 7-амино-1,3,5-триазаадамантана и андрографолида, в концентрации 1 мкг/мл ингибирует репликацию различных вирусов, включая коронавирус SARS (штамм Urbani), энтеровирус-71 (штамм Tainan/4643/98) и вирус лихорадки Рифт-Валли (штамм MP-12) [35]. В более высокой концентрации (50 мкг/мл) соединение (11) было эффективно в отношении вирусов гриппа A (H1N1/09), гепатита A (pHM175), гепатита B (02094), гепатита C (CON1), простого герпеса (тип 2), папилломы человека (тип 8), ВИЧ-1 (группа М), лихорадки Денге (тип 2, Новая Гвинея C), японского энцефалита (SA14/V1) и конского энцефалита венесуэльского происхождения (TC-83).
Таким образом, можно заключить, что хотя сами ди- и триазаадамантаны проявляют лишь умеренную ингибирующую активность в отношении ряда вирусов, их присоединение к другим биологически активным соединениям может значительно усилить потенциал последних.
Антимикробная активность. Большое внимание было уделено изучению антимикробной активности производных азаадамантанов, наиболее значительный вклад был внесен исследователями из Армении. В 1986 г. ими была опубликована работа, включающая данные по синтезу 2-замещенных 5,7-диметил-1,3-диазаадамантанов, содержащих алифатические, ароматические, гетероароматические, гетероциклические, спироциклические заместители, а также моносахариды, исходя из 1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-она (12) и разнообразных альдегидов и кетонов [36]. На модели генерализированной стафилококковой инфекции белых мышей, вызванной Staphylococcus aureus (S. aureus штамм 4-O), была исследована антибактериальная активность соединений (13–19) (рис. 4).
Рис. 4.
1,5-Диметилбиспидин-9-он и полученные из него 2-замещенные 5,7-диметил-1,3-диазаадамантан-6-оны, изученные на наличие антибактериальной активности [36].

Наибольшую активность проявили соединения, содержащие фрагменты пиридина (15) и 2,2-диметилтетрагидропирана (16), которые продлевали жизнь зараженных животных на 20–30% при однократном введении в дозах 800 и 1500 мг/кг соответственно. Остальные изученные производные диазаадамантана не увеличивали продолжительность жизни зараженных животных по сравнению с контролем. Показано, что эти производные диазаадамантанов малотоксичны и хорошо переносятся животными при однократном введении в дозах 1500–2000 мг/кг.
В 2008 г. были представлены результаты изучения антибактериальной активности большого набора 2-замещенных диазаадамантанов, включающего различные производные, содержащие метильные, этильные, изопропильные заместители в положениях 5 и 7 молекулы диазаадамантана, а также метиленовую, карбонильную и спиртовую группу в 6-м положении каркаса. В качестве заместителей по 2-му положению использовали разнообразные ароматические и гетероароматические фрагменты (схема 6 ) [37]. Кроме того, исследовали активность и ранее описанного 6-амино-5,7-диметил-1,3-диазаадамантана (4).

Схема 6 . 5,7-Диалкилдиазаадамантаны и синтез их 2-арилзамещеных производных [37].
Антибактериальная активность соединений была изучена агар-диффузионным методом в отношении грамположительных стафилококков (S. aureus 209p, S. aureus 1, S. aureus Makarov, S. aureus 34, S. aureus 118) и грамотрицательных бактерий (Shigella dysenteriae Flexneri (Sh. dysent. Flexneri), Salmonella enterica серовар Typhimurium (E. typhi), Escherichia coli (E. coli), Proteus (Prot.). Испытуемые соединения наносили в дозе 4 мг, определяя диаметр зон отсутствия роста микроорганизмов в месте нанесения соединений (в мм) после суточного выращивания микроорганизмов. Активность считали высокой, если зона подавления роста была >20 мм, промежуточной – при зоне 15–20 мм (табл. 1).
Таблица 1.
Антибактериальная активность соединений (4), (20–27), (29), (32), (35) и (37) [37]
| Соединение | Диаметр зоны отсутствия роста, мм | |||||
|---|---|---|---|---|---|---|
| грамположительные бактерии | грамотрицательные бактерии | |||||
| S. aureus 209p | S. aureus 1 | Sh. dysent. Flexneri 6858 | E. typhi 79 | E. coli 0-55 | Prot. | |
| (4) | 9 | 7 | 7 | 7 | – | – |
| (20) | 22 | 13 | 13 | 15 | 18 | 14 |
| (21) | 5 | 5 | 5 | 6 | – | – |
| (22) | 5 | 5 | 13 | 15 | 7 | 10 |
| (23) | 0 | 0 | 7 | 7 | – | – |
| (24) | 5 | 6 | 5 | 6 | – | – |
| (25) | 6 | 0 | 0 | 6 | – | – |
| (26) | 10 | 10 | 10 | 15 | 15 | 11 |
| (27) | 7 | 7 | 7 | 10 | – | – |
| (29) | 22 | 20 | 18 | 22 | 15 | 13 |
| (32) | 10 | 10 | 13 | 14 | 10 | 5 |
| (35) | 9 | 13 | 6 | 8 | – | – |
| (37) | 9 | 7 | 9 | 10 | – | – |
| Норсульфазол | 30 | 30 | 20 | 30 | – | – |
| Сульфадимезин | 14 | 14 | 0 | 0 | – | – |
Наиболее эффективными соединениями против и грамположительных, и грамотрицательных микроорганизмов оказались 5,7-диметил-1,3-диазадамантантан (20) и его 2-пиридилзамещенный аналог (29). Эти соединения оказались более эффективными, чем сульфадимезин, но менее эффективными, чем норсульфазол, использованные в качестве препаратов сравнения. Введение в 6-е положение карбонильной, гидроксильной или аминной групп (соединения (4), (20–27)), а также пиридил-3- (32), (35) или фурильной групп (37) в положение 2 приводило к снижению антибактериальной активности соединений. При введении во 2-е положение ароматических (соединения (30), (31), (33), (36)), тиенильной (34) или индольной (38) групп наблюдалось исчезновение антибактериальных свойств.
Позже были представлены результаты исследования активности различных хинолинов (39–53) (рис. 5), содержащих диазаадамантановый фрагмент с метильными, этильными и пропильными заместителями в 5-м и 7-м положениях, а также карбонильную, спиртовую или метиленовую группу в 6-м положении каркасной структуры, в отношении грамположительных (S. aureus 209p) и грамотрицательных (Sh. dysent. Flexneri 6858, E. coli 0-55) бактерий [38]. Антибактериальная активность определяли так же, как и в работе Arutyunyan et al. [37], в качестве положительного контроля использовали 5-нитро-8-гидроксихинолин. Слабую антибактериальную активность (d = 10–13 мм) в отношении всех исследуемых видов бактерий проявили соединения (40, 41, 43, 44, 49–53), которые были значительно менее активными, чем нитрохинолин (d = 20–28 мм). Остальные соединения не продемонстрировали антибактериальных свойств.
Рис. 5.
2-Хинолинзамещенные 1,3-диазаадамантаны, изученные на наличие антибактериальной активности [37].

Была изучена антибактериальная активность азометинов (54–66) (схема 7 ), взятых в виде дигидрохлоридов [39]. Антибактериальные свойства исследовали с использованием диффузии в агаре и методом серийных разведений в отношении грамположительных (S. aureus 209p, S. aureus 1) и грамотрицательных (Sh. dysent. Flexneri 6858, E. coli 0-55) бактерий. Испытуемые соединения использовали в дозе 5 мг, помещали на чашки Петри, замеряли диаметр зон отсутствия роста микроорганизмов после инкубации в течение 20–24 ч, как и в работах [37, 38]. Для наиболее активных соединений в тестах с серийными разведениями определяли минимальную подавляющую концентрацию (МПК) для S. aureus 209p и Sh. dysent. Flexneri 6858. В качестве положительного контроля использовали фуразолидон.

Схема 7 . 6-Аминодиазаадамантаны и синтез их азометиновых производных [39].
Все изученные солянокислые азометины, за исключением соединений (55) и (61), обладали антибактериальной активностью (табл. 2). Дигидрохлориды (62–64), содержащие нитрофурильные группы, проявили наибольшую бактериостатическую активность. Они оказывали сопоставимое воздействие на грамположительные и грамотрицательные бактерии и были, как правило, более активными, чем фуразолидон. Дигидрохлориды (57) и (60) с индольными группами были эффективны в отношении грамотрицательных бактерий. МПК для гидрохлоридов (57), (60), (62–64) составила 125–250 мкг/мл, что значительно выше, чем у препарата сравнения фуразолидона (МПК = 31 мкг/мл).
Таблица 2.
Антибактериальная активность соединений (54), (56–60) и (62–66) [39]
| Соединение | Диаметр зоны отсутствия роста, мм (МПК, мг/мл) | |||
|---|---|---|---|---|
| грамположительные бактерии | грамотрицательные бактерии | |||
| S. aureus 209p | S. aureus 1 | Sh. dysent. Flexneri 6858 | E. coli 0-55 | |
| (54) | 14 | 14 | 15 | 13 |
| (56) | 13 | 13 | 15 | 13 |
| (57) | 18 | 19 | 30 (125) | 28 |
| (58) | 14 | 14 | 15 | 15 |
| (59) | 0 | 0 | 15 | 15 |
| (60) | 18 | 18 | 32 (125) | 28 |
| (62) | 30 (125) | 27 | 25 (250) | 23 |
| (63) | 32 (125) | 28 | 27 (250) | 24 |
| (64) | 29 (125) | 25 | 23 (250) | 23 |
| (65) | 14 | 14 | 14 | 12 |
| (66) | 15 | 14 | 15 | 15 |
| Фуразолидон | 25 (31) | 24 | 24 (31) | 23 |
Антимикробная активность ряда 4,8,9,10-тетрафенил-1,3-диазаадамантанов (67–72) (рис. 6) в отношении грамположительных и грамотрицательных бактерий, спорообразующих бактерий, дрожжеподобных грибов и грибов дерматофитов (Bacillus mycoides, Bacillus subtilis (B. subtilis), Bacillus anthracis, S. aureus, Bacteria carativorum, Corynebacterium, E. coli; Saccharomyces cerevisiae, Sarcina lutea, Epidermophyton rubrum, Trichophyton gypseum, Fusarium aolani и Candida albicans) была изучена Байсалбаевой с соавт. методом серийных разведений на питательной среде [40]. Активность соединений оценивали по минимальной бактериостатической или микостатической концентрации (мкг/мл).
Рис. 6.
4,8,9,10-Тетрафенил-1,3-диазаадамантаны, изученные на наличие антимикробной активности [40].

Среди исследованных соединений значительную активность проявили диазаадамантаны (67) и (71) в отношении грамположительных бактерий Corynebacterium (МПК = 7.4 и 0.27 мкг/мл соответственно) и соединение (68) в отношении Sarcina lutea (МПК = 0.5 мкг/мл). Остальные соединения были менее активными (МПК = 14–67 мкг/мл). Данных об использовании препарата сравнения в статье не приводится.
Антибактериальная активность 4,8,9,10-тетраарил-1,3-диазаадамантанов (73–95), содержащих различные заместители в ароматическом кольце, а также ранее упомянутых соединений (67), (68) и (71), имеющих незамещенные фенильные фрагменты, в отношении бактерий B. subtilis, S. aureus и E. coli была изучена Balaji et al. [41] (рис. 7, табл. 3). Исследование проводили методом диффузии в агаре с использованием микробного инокулянта, содержащего 106 кл./мл. В качестве контроля был выбран стрептомицин. Авторы определили МПК (табл. 3).
Рис. 7.
4,8,9,10-Тетраарил-1,3-диазаадамантаны, изученные на наличие антибактериальной активности [41].

Таблица 3.
Антибактериальная активность соединений (67), (68), (71), (80–83), (85), (87), (88), (93) и (95) [41]
| Соединение | МПК, мкг/мл | ||
|---|---|---|---|
| B. subtilis | S. aureus | E. coli | |
| (67) | 50 | – | 25 |
| (68) | – | 12.5 | – |
| (71) | 12.5 | 12.5 | 100 |
| (80) | 12.5 | – | 100 |
| (81) | 50 | 12.5 | 50 |
| (82) | 50 | 50 | 12.5 |
| (83) | 50 | 12.5 | 50 |
| (85) | 6.25 | 6.25 | 100 |
| (87) | 6.25 | 50 | 50 |
| (88) | 6.25 | 6.25 | – |
| (93) | 6.25 | – | 50 |
| (95) | 50 | – | 6.25 |
| Стрептомицин | 12.5 | 12.5 | 6.25 |
Диазаадамантаны (80), (85), (87), (88) и (93) показали высокую активность против бактерий B. subtilis (МПК = 6.25–12.5 мкг/мл), соединения (68), (71), (81), (83), (85) и (88) были активны в отношении S. aureus (МПК = 6.25–12.5 мкг/мл), а соединения (82) и (95) – в отношении E. coli (МПК = 12.5 и 6.25 мкг/мл соответственно). В остальных случаях соединения подавляли рост бактерий в концентрации 25–100 мкг/мл или не подавляли вовсе. Препарат сравнения, стрептомицин, подавлял рост бактерий B. subtilis и S. aureus в концентрации 12.5 мкг/мл, а бактерий E. coli – 6.25 мкг/мл. В целом фтор-, хлор-, бром- или алкоксизамещенные арилдиазаадамантаны оказались более активными в сравнении с незамещенными по ароматическому фрагменту соединениями [41].
Изучению бактерио- и микостатического действия 7-замещенных 1,3,5-триазаадамантанов было уделено значительно меньше внимания. Первые данные были получены в 70-х гг. прошлого столетия Hodge et al. [42] для 7-нитро- (3), 7‑амино- (9) и 7-гидроксиламино- (96) 1,3,5-триазаадамантанов (рис. 8). Соединения (3) и (9) при использовании метода штрихов проявили фунгистатирующее действие в отношении грибов вида Fusarium oxysporum только в высокой концентрации (500–1000 мкг/мл). Триазаадамантанамин (9) в тех же концентрациях был активен и в отношении Aspergillus fumigatus. Соединение (96) оказалось активным в отношении S. aureus и Pasteurella pseudotuberculosis (МПК = 250–500 мкг/мл), Streptococcus fecalis (МПК = 500–750 мкг/мл), а также Streptococcus hemolyticus и Sh. dysent (МПК = = 500–1000 мкг/мл). Данных об использовании препарата сравнения в работе не приводится.
Рис. 8.
7-Замещенные 1,3,5-триазаадамантаны и некоторые их производные, изученные на наличие антимикробной активности [42].

Антибактериальная активность производных 7-амино-триазаадамантана, содержащих нафтильный (97) и 5-нитрофурильный (98) заместители, взятых в виде солянокислых солей, была изучена Арутюняном с соавт. (табл. 4) [39]. Умеренную бактериостатическую активность в отношении грамположительных и грамотрицательных бактерий проявило соединение (98), содержащее нитрофурильную группу. Метод серийных разведений показал, что МПК для соединения (98) составляет 125 мкг/мл, что значительно выше, чем у препарата сравнения фуразолидона.
Таблица 4.
Антибактериальная активность соединений (97) и (98) [39]
| Соединение | Диаметр зоны отсутствия роста, мм (МПК, мкг/мл) | |||
|---|---|---|---|---|
| грамположительные бактерии | грамотрицательные бактерии | |||
| S. aureus 209p | S. aureus 1 | Sh. dysent. Flexneri 6858 | E. coli 0-55 | |
| (97) | 16 | 14 | 17 | 15 |
| (98) | 28 (125) | 26 | 30 (125) | 28 |
| Фуразолидон | 25 (31) | 24 | 24 (31) | 23 |
Наличие антипротозойной активности у четвертичных солей 5,7-диметил-1,3-диазаадамантанона было предположено в результате виртуального скрининга на модели фермента трипантионредуктазы (TryR) и изучено Perez-Pineiro et al. [43]. Ингибирование этого фермента приводит к накоплению в трипаносомах токсических кислородных продуктов, вызывающих окисление тиолов клеточных мембран и гибель паразитов. Для ряда диазаадамантанов, содержащих бромбензильную группу (рис. 9), было показано, что они способны связываться с трипанотионредуктазой, однако даже для наиболее активного N-(3-бромбензил)-производного диазаадамантана (99) IC50 была более 100 мкМ, что значительно превышает действующую концентрацию известных препаратов, например, нифуртимоксома [44].
Рис. 9.
Четвертичное аммониевое производное 5,7-диметил-1,3-диазаадамантан-6-она, обладающее антипротозойной активностью [43].

Суммируя литературные данные, можно отметить, что наибольшей антибактериальной активностью обладают производные диазаадамантанов, содержащие метильные заместители в 5-м и 7-м положениях каркаса, гетероароматический заместитель во 2-м положении и спиртовую или нитрофурановую группу, присоединенную через азометиновый фрагмент в 6-м положении молекулы. Кроме того, антибактериальные свойства были обнаружены у 4,8,9,10-тетраарил-1,3-диазаадамантанов с различными заместителями в ароматическом кольце. Несмотря на небольшое количество данных по активности производных триазаадамантанов, можно отметить, что они оказывают меньшее ингибирующее действие на микроорганизмы по сравнению с диазаадамантанами.
Противоопухолевая активность. Изучение противоопухолевой активности азаадамантанов было начато Чачояном с соавт. в 1991 г. [45]. Ими были получены и исследованы диазаадамантаны (17), (100–110) (рис. 10), содержащие фрагменты замещенных индолов с высокой противоопухолевой активностью [46].
Рис. 10.
5-(Диметиламиносульфонил)-индол-3-карбоновая кислота, 5,7-диметил-1,3-диазаадамантан-6-он и полученные из него 2-индолзамещенные производные, изученные на наличие противоопухолевой активности [45].

Противоопухолевую активность соединений изучали на крысах с перевиваемыми опухолями: саркомой 45, лимфосаркомой Плисса и лейкозом Швеца. Наиболее активные соединения изучали и на мышах с саркомами 180 и 37. Терапевтический эффект соединений оценивали по проценту торможения роста опухолей (ТРО, %), сравнивая с эффективностью ранее изученной 5-(диметиламиносульфонил)-индол-3-карбоновой кислоты (111) [46] и солянокислого 5,7-диметил-6-оксо-1,3-диазаадамантана (21). Разовую терапевтическую дозу для каждого вещества рассчитывали как ~1/20 от LD100.
Обнаружено, что производные индолил-1,3-диазаадамантана, независимо от места и характера заместителей в индольном кольце, как и соединение (111), как правило, являются нетоксичными веществами (LD100 ≥ 5000 мг/кг) (табл. 5). Несколько большей токсичностью обладают соединения (100), (101) и (105) (LD100 = 3500–4000 мг/кг), при этом сам диазаадамантан (21) существенно более токсичен (LD100 = 1100 мг/кг). Введение индольных фрагментов в структуру диазаадамантана снижает его токсичность.
Таблица 5.
| Соедине-ние | LD100, мг/кг (мыши) | ТРО, % | ТРО, % | |||||
|---|---|---|---|---|---|---|---|---|
| доза, мг/кг | саркома 45 | лимфосар-кома Плисса | лейкоз Швеца | доза, мг/кг | саркома 180 | саркома 37 | ||
| (17) | >5000 | 250 | 71.0 | – | 0 | 500 | Стим. | – |
| (100) | 4000 | 200 | 36.0 | – | 43.0 | – | – | – |
| (101) | 4000 | 200 | 47.0 | 0 | 0 | 500 | – | – |
| (102) | >5000 | 250 | 19.0 | 0 | 0 | 500 | – | 55.5 |
| (103) | >5000 | 250 | 37.0 | 0 | Стим. | – | – | – |
| (104) | 5000 | 250 | 45.0 | Стим. | 45.5 | 500 | 55.0 | 65.7 |
| (105) | 3500 | 160 | 0 | 29.0 | 21.7 | 350 | 45.8 | – |
| (106) | 5000 | 250 | 62.0 | 27.5 | 42.0 | 500 | 54.0 | 58 |
| (107) | 5000 | 250 | 34.0 | 28.0 | 34.8 | – | – | – |
| (108) | 5000 | 250 | 29.0 | 32.0 | 0 | 500 | – | – |
| (109) | 5000 | 250 | 56.5 | 36.0 | 39.0 | – | – | – |
| (110) | 5000 | 250 | 0 | 44.0 | 24.0 | 500 | 41.6 | – |
| (111) | 5000 | 250 | 65.0 | 45.0 | – | – | – | – |
| (21) | 1100 | 50 | 52.0 | 0 | 0 | – | – | – |
Незамещенный индолил-1,3-диазаадамантан (17), как и соединение (111), проявляет противоопухолевую активность против саркомы 45 (ТРО = 71%), превосходя по активности на этой линии клеток соединение (21) [45]. На моделях саркомы 180 и асцитной карциономы Эрлиха соединения (21), (102) и (112) не оказывали достоверного противоопухолевого эффекта. Замещение в положении 5 индольного кольца в индолил-1,3-диазаадамантане алкиламиносульфонильной группой (соединения (105–109) приводит к снижению (ТРО = 29.0–62.0%; для соединений (107) и (108) – недостоверно (p > 0.05)), а в случае диметилсульфонильной группы (соединение (105)) – к полной потере противоопухолевой активности в отношении саркомы 45, при сохранении терапевтической эффективности против лейкоза Швеца (ТРО = 21.7–42.0%) и саркомы 180 (ТРО = 45.8–54.0%). В отличие от веществ (103) и (105), соединения (104) и (106), имеющие метильные группы в 1-м положении или положениях 1 и 2 индольного кольца, проявили заметную противоопухолевую активность в отношении саркомы 37 (ТРО = = 65.7 и 58.0% соответственно). В то же время аналогичное изменение структуры соединения (107) при переходе к соединению (108) не оказало существенного влияния на его противоопухолевый эффект. Исходная 5-(диметиламиносульфонил)-индол-3-карбоновая кислота (111) показала умеренную активность в отношении лимфосаркомы Плисса (ТРО = 45.0%). Менее выраженный антипролиферативный эффект (ТРО = 27.5–44.0%) проявили соединения (105–110). Соединения (17), (103) и (104) оказывали стимулирующее воздействие на рост саркомы 180, лейкоза Швеца и лимфосаркомы Плисса соответственно.
Были изучены физико-химические свойства, мембранотропная и антиоксидантная активность наиболее активных соединений (104) и (106) [45]. Установлено, что в исследуемых концентрациях (0.01, 0.1, 1 и 10 мг/мл) они не влияют на резистентность мембран эритроцитов к действию 0.1 н. HCl и смеси растительных сапонинов [45]. Однако, ввиду их высокого сродства к биологическим мембранам, соединения (104) и (106) снижают резистентность мембран эритроцитов, сенсибилизируя эти мембраны, и в концентрации до 10 мг/мл оказывают гемолизирующее действие. Наибольшее гемолитическое действие проявляет соединение (104). Кроме того, установлено, что соединения (104) и (106) обладают существенной антиоксидантной активностью, ингибируя процессы Fe-индуцированного перекисного окисления липосом. Антиоксидантная активность этих соединений сопоставима с таковой для известного антиоксиданта ионола (2,6-дитретбутил-4-метилфенола) в концентрации 0.1 мг/мл.
Несколькими годами позже этим же коллективом авторов были представлены результаты изучения биологической активности нескольких серий гетероциклических и спироциклических диазаадамантанов (рис. 11, 12) [7, 47, 48]. Противоопухолевую активность соединений изучали на перевиваемых клеточных линиях опухолей: сарком 180, 37 и 45, лейкоза Швеца, карциносаркомы Уокера, асцитной и солидной форм карциномы Эрлиха. Терапевтический эффект соединений оценивали по величине TРО (%) и увеличению продолжительности жизни подопытных мышей с асцитом по сравнению с контролем. Изучаемая доза для каждого вещества была установлена как 1/20 LD100 для крыс и 1/10 LD100 для мышей.
Рис. 11.
5,7-Ди(этоксикарбонилметил)-1,3-диазаадаманан-6-он и полученный из него пиридазинон, изученные на наличие противоопухолевой активности [47].


Диазаадамантан (112) и синтезированный из него пиридазинон (113) (рис. 11) оказались малотоксичными веществами (LD100 = 2500 и >5000 мг/кг соответственно). Показано, что диазаадамантан (112) проявляет умеренную активность в отношении саркомы 45 (ТРО = 50%) в дозе 120 мг/кг, не оказывает достоверного противоопухолевого действия на лейкоз Швеца и стимулирует рост саркомы 180 в дозе 250 мг/кг. Введение соединения (112) не оказывало терапевтического воздействия на мышей с саркомой 37, карциносаркомой Уокера, асцитной и солидной карциномой Эрлиха. Пиридазинон (113) в дозе 500 мг/кг был эффективен в отношении сарком 37 (ТРО = 62%) и 180 (ТРО = 46%) и на 28% продлевал жизнь подопытных мышей с асцитной карциномой Эрлиха, не вызывая видимого токсического воздействия на организм подопытных животных. В то же время соединение (113) не воздействовало на карциносаркому Уокера, саркому 45, лейкоз Швеца и солидную форму карциномы Эрлиха [47]. Данных об использовании препарата сравнения в статье не приводится.
Результаты исследования спироциклических (16), (18), (19) (рис. 4), (114–116), 2-фосфоарил- (117), (118), 2-алкил- (119–121), 2-фосфо- (122), (123) производных и незамещенного 5,7-диметил-1,3-диазаадамантан-6-она (21) (рис. 12) представлены в табл. 6 [7, 48].
Таблица 6.
Противоопухолевая активность и острая токсичность 1,3-диазаадамантанов (16), (18), (19), (21) и (114–123) [7, 48]
| Соединение | LD100 (мыши) мг/кг | МПД (мыши) мг/кг | Доза (мг/кг) × × количество введений | Саркома 45 (крысы) | Доза (мг/кг) × × количество введений | Саркома 180 (мыши) |
|---|---|---|---|---|---|---|
| ТРО, % | ТРО, % | |||||
| (16) | 500 | 400 | 25 × 8 | 38.7 | 50 × 6 | – |
| (18) | 1400 | 1200 | 70 × 8 | 50.0 | 125 × 6 | – |
| (19) | 800 | 500 | 40 × 8 | 0 | 50 × 6 | 40.0 |
| (114) | >2500 | Н.д. | 150 × 8 | 0 | 250 × 6 | – |
| (115) | 300 | 200 | 15 × 8 | 40.3 | 30 × 6 | 76.0 |
| (116) | 550 | 400 | 25 × 8 | 18.0 | 50 × 6 | 31.0 |
| (117) | 4000 | 3000 | 150 | 30.5 | 300 × 1 | – |
| (118) | 5000 | Н.д. | 250 | – | 500 × 1 | – |
| (119) | 5000 | Н.д. | 250 | – | 500 × 1 | – |
| (120) | 1250 | 1000 | 60 | – | 125 × 1 | – |
| (121) | 1100 | 900 | 50 | – | 100 × 1 | – |
| (122) | 2500 | 2000 | 120 | 52.5 | 250 × 1 | 42.0 |
| (123) | 1000 | 850 | 50 | 35.0 | 100 × 1 | 33.6 |
| (21) | 1100 | 800 | 50 × 8 | 52.0 | 100 × 6 | 33.0 |
Токсичность и противоопухолевая активность соединений (16), (18), (19), (114–123) сравнивалась с таковой у незамещенного 5,7-диметил-6-оксо-1,3-диазаадамантана (21), взятого в виде солянокислой соли. В целом спироциклические соединения оказались умеренно токсичными (LD100 = 300–800 мг/кг для соединений (16), (19), (115) и (116)) или малотоксичными (LD100 = 1400 и >2500 мг/кг для соединений (18) и (114)) [7]. Сопоставимо низкую токсичность (LD100 = 1000–2500 мг/кг) проявили соединения (120), (122) и (123). Диазаадамантаны (117) и (118), полученные введением замещенной фосфорильной группы в соединение (21), а также 2-(4'-гидроксифенил)диазаадамантан (119) оказались нетоксичными (LD100 = 4000–5000 мг/кг) [48].
Среди исследованных соединений умеренную терапевтическую активность в отношении саркомы 45 и 180 проявили диазаадамантаны с фрагментами циклопентана (18) и циклогексана (19) (ТРО = 50 и 40% соответственно) [7]. Сопоставимой активностью обладают фосфорсодержащие диазаадамантаны (122) и (123), оказавшие воздействие на оба вида опухолей. Так, соединение (122), содержащее феноксигруппу, достоверно подавляло рост саркомы 45 на 52.5%, а саркомы 180 – на 42.0%. Соединение (123) c бис(2-хлорэтил)аминной группой в тех же условиях угнетало рост саркомы 45 на 35.0%, а саркомы 180 – на 33.6%. Для незамещенного 1,3-диазаадамантана (21) ТРО составило 52.0 и 33.0% в отношении сарком 45 и 180 соответственно [48].
Значительная противоопухолевая активность обнаружена у соединения (115) с бензилпиперидиновым кольцом. Оно ингибирует рост саркомы 180 (ТРО = 76.0%) и лейкоза Швеца (ТРО = = 69.0%), не оказывая токсического действия на подопытных животных. Среди изученных производных диазаадамантана только соединения (115) и (116) приводили к достоверному увеличению (на 26–39%) продолжительности жизни у мышей с асцитной карциномой Эрлиха [7]. Все остальные синтезированные соединения не проявили противоопухолевого действия в отношении карциносаркомы Уокера, лейкоза Швеца и асцитной карциномы Эрлиха.
Синтез и исследование противоопухолевой активности нескольких серий 2-тио- и 2-фосфозамещенных-1,3-диазаадамантанов (рис. 13) в отношении клеток меланомы мышей B16-F10 были проведены Sharabi-Ronen et al. [49]. Как отмечают авторы статьи, 2-тиозамещенные 1,3-диазаадамантаны (124) и (125) проявляли низкую активность в отношении клеток опухоли (IC50 > 200 мкМ). Более перспективные результаты были получены для фосфорсодержащих аналогов (126) и (127), показавших значительную противоопухолевую активность (IC50 = 10–60 мкМ) [49]. Кроме исследования цитотоксичности полученных соединений, для наиболее активных 5,7-ди(4-(трифторметил)бензил-1,3-диаза-2-фосфоадамантанов (126) и (127) было изучено влияние на клеточный цикл и механизм активации апоптоза. Посредством исследований изменения митохондриального мембранного потенциала, оцениваемого с помощью реагента JC-1, активации каспаз 3 и 7, окрашивания аннексином V-FITC/PI и выявления морфологических изменений было показано, что данные соединения вызывали остановку клеточного цикла в фазе G2/M, что приводило к апоптозу клеток меланомы мышей B16-F10.
Рис. 13.
2-Тио- и 2-фосфо-5,7-дизамещенные-1,3-диазаадамантан-6-оны, изученные на наличие противоопухолевой активности [49].

Менее изученной остается противоопухолевая активность триазаадамантанов. Среди производных 7-амино-1,3,5-триазаадамантана, включающих замещенные оксииндолин-, эпоксиизоиндол-, пирролидин-, ацетамид-, а также 6,6-дизамещенные триазаадамантаны, содержащие ароматические заместители, антипролиферативная активность была обнаружена у соединения (128) (рис. 14) в отношении линий клеток HT29 (рак прямой кишки, IC50 = 1 мкг/кг), MCF7 (рак молочной железы, IC50 = 2.3 мкг/кг), Panc-1 (рак поджелудочной железы, IC50 = 3.4 мкг/кг) и NCI-H460 (рак легких, IC50 = 3.4 мкг/кг) [35]. Кроме того, соединение (128) в дозе 70 мг/кг подавляло на 54% рост саркомы 180 у мышей [35].

В качестве потенциальных противоопухолевых агентов были предложены металл-органические комплексы триазаадамантана с координированными к ионам марганца молекулами CO [50]. Предполагается, что подобные соединения при введении в организм будут выделять при облучении CO. Известно, что в небольших концентрациях (<200 ppm) СО проявляет противовоспалительные и антиапоптотические свойства на здоровых клетках [51], при этом воздействие СО во время химиотерапии вызывает апоптоз клеток рака предстательной железы [52]. Наиболее перспективным авторы считают соединение (129) (рис. 14), которое имеет умеренную, зависящую от рН, растворимость в воде, что может обеспечить дополнительное преимущество в селективности действия. При физиологическом рН фрагмент триазаадамантана не несет заряда и может диффундировать через липидные мембраны. Однако в раковых клетках с относительно низким уровнем рН могут происходить протонирование и захват триазаадамантанового фрагмента. Протонированный фрагмент триазаадамантана также может приводить к повреждению ДНК. Кроме того, водные растворы комплекса (129) стабильны в темноте и нечувствительны к восстановителям, таким как дитионит натрия и глутатион, и быстро высвобождают три молекулы СО при воздействии видимого света низкой мощности. Исследования по доставке CO в раковые клетки с помощью соединения (129) были запланированы [50], но их результаты в настоящее время не опубликованы.
ВЗАИМОДЕЙСТВИЕ АЗААДАМАНТАНОВ С ДНК, А ТАКЖЕ С ФЕРМЕНТАМИ И РЕЦЕПТОРАМИ
Способность связываться с азотистыми основаниями молекулы ДНК, увеличивая скорость реакции обмена цепочек в коротких нуклеотидах, была обнаружена у 1'-бензил-5,7-диалкил-1,3-диазаспиро[адамантан-2,4'-пиперидин]-6-онов (130) и (131) (рис. 15) [53]. В экспериментах использовали ДНК тимуса теленка, олигонуклеотиды d(CAATCGGATCGAATTCGATCCGATTG) (ds26), Cy3d(CAATCGGATCGAATTCGATCCGATTG) (Cy3ds26) и Cy5d(CAATCGGATCGAATTCGATCCGATTG) (Cy5ds26). Регистрацию взаимодействия 1,3-диазаадамантанов с ДНК осуществляли с помощью флуоресцентно-контролируемого вытеснения лигандами красителя тиазолового оранжевого (ТО) из его комплекса с ДНК. Для регистрации кривых вытеснения ТО добавляли к ДНК (0.25 мкМ) в такой концентрации (СTO = 1 мкМ), чтобы все места связывания с ДНК были практически заполнены. При образовании комплекса ТО с ДНК значительно увеличивается флуоресценция красителя. Титрование 1,3-диазаадамантанами вызывало тушение флуоресценции, связанное с вытеснением ТО из комплекса с ДНК.
Рис. 15.
5,7-Диалкилзамещенные 1,3-диазаадамантаны, способные связываться с азотистыми основаниями молекулы ДНК [53].

Оказалось, что при pH 7.8 диазаадамантан (131) с н-бутильными заместителями связывается с ДНК с Kа = 180 М–1, а диазаадамантан (130) с пропильными заместителями – с Kа = 15 М–1. При pH 6.5 сродство обоих 1,3-диазаадамантанов к ДНК увеличивается, константы ассоциации 5,7-диалкилзамещенных диазаадамантанов с ДНК становятся близкими и равными 1400 и 500 М–1 соответственно. Таким образом, диазаадамантаны (130) и (131) продемонстрировали рН-зависимую аффинность к ДНК [54, 55].
О наличии ингибирующей активности в отношении некоторых ферментов у производных 5,7-диметил-1,3-диазаадамантана сообщается в работе Захаренко с соавт. [56].
У 1,3-диазаадамантанов (132) и (133) (рис. 16), содержащих фрагменты ациклических монотерпенов цитронеллаля и цитраля соответственно, обнаружена ингибирующая активность в отношении фермента репарации ДНК Tdp1 (тирозил-ДНК-фосфодиэстераза 1), который является перспективной мишенью для комплексной противоопухолевой терапии [57–59]. Действие некоторых противоопухолевых препаратов, например, камптотецина и его клинически важных производных топотекана и иринотекана [60], опосредовано ингибированием процесса отщепления топоизомеразы 1B (Top1) от 3'-конца ДНК [61, 62]. Tdp1 способна разрушать фосфодиэфирную связь между тирозином (Top1) и 3'-фосфатной группой ДНК в случаях, когда это не протекает по естественным причинам. Таким образом, соединения, способные ингибировать Tdp1, могут повышать терапевтическую эффективность ингибиторов Тор1 [63–66].
Рис. 16.
Производные 5,7-диметил-1,3-диазаадамантана, проявляющие ингибирующую активность в отношении фермента Tdp1 [56].

Захаренко с соавт. [56] использовали опубликованный ими ранее [67] метод флуоресцентной детекции активности Tdp1, основанный на способности Tdp1 отщеплять объемные заместители с 3'-конца ДНК. К 16-звенному олигонуклеотиду с 5'-конца был присоединен донор флуоресценции 5(6)-карбоксифлуоресцеин (FAM), а к 3'-концу – гаситель флуоресценции BHQ1 (Black Hole Quencher-1). Флуоресценция флуорофора гасится, т.к. BHQ1 расположен в пределах ферстеровского радиуса. Инкубация олигонуклеотида-биосенсора с Tdp1 приводит к удалению BHQ1 с 3'-конца олигонуклеотида, что приводит к разгоранию флуоресценции. Величины IC50 для соединений (132) и (133) составили 14.8 и 16.7 мкМ, что в целом является умеренной активностью, т.к. в настоящее время известны соединения, ингибирующие Tdp1 в субмикромолярном диапазоне [68–70].
Golubovskaya et al. [71] исследовали способность малых молекул, содержащих триазаадамантановый фрагмент, связываться с фокальной адгезивной киназой (FAK, или протеинтирозинкиназа 2, PTK2), нарушая ее связывание с белком p53. FAK (PTK2) – это нерецепторная тирозинкиназа, которая контролирует клеточные процессы, такие как пролиферация, адгезия, распространение, подвижность и выживаемость [72]. Белок p53 выполняет функцию супрессора образования злокачественных опухолей [73]. Активация p53 снижает жизнеспособность и клоногенность раковых клеток и ингибирует рост опухоли in vivo. Достоверное снижение жизнеспособности у клеток рака линии HCT116 p53 показали выбранные в результате молекулярного докинга N‑алкил-N-метил-1,3,5-триазаадамантаны, содержащие бутильный (134) и гексильный (135) заместители (рис. 17), однако их аффинность к FAK оказалось недостаточной, чтобы нарушить ее связывание с p53.

Eisenbarth et al. [74] изучили способность N-бутил-N-метилтриазаадамантана (134) (рис. 17) к подавлению аутоиммунного ответа, вызывающего диабет 1-го типа. Исследовался ответ клонированных Т-лимфоцитов, ограниченных DQ8, на аминокислоты В-цепи инсулина 9–23, а также на ингибирование взаимодействия этих белков с молекулами главного комплекса гистосовместимости II класса, кодируемых геном DQ8. Соединение (134) действовало в концентрациях <0.1 мкМ и >100 мкМ (IC50) в первом и во втором случаях соответственно. Продемонстрированная активность недостаточно высока, чтобы рассматривать соединение (134) как средство профилактики и лечения диабета 1-го типа.
Известно, что связывание с рецептором CCR1 приводит к предотвращению его взаимодействия c хемокинами, которые вовлечены в развитие и поддержание многочисленных воспалительных и иммунологических состояний и других нарушений [75]. Аффинность к рецептору CCR1 (IC50 = 2 мкМ) была продемонстрирована для соединения (136) (рис. 17) [76]. Следует отметить, что подобную активность можно назвать умеренной, поскольку известны соединения, связывающиеся с рецептором CCR1 в субнаномолярной концентрации [77].
ДЕЙСТВИЕ АЗААДАМАНТАНОВ НА НЕРВНУЮ СИСТЕМУ
У некоторых производных диазаадамантана была обнаружена существенная анальгетическая активность. Показано [78], что среди 8,9-замещенных диметил 5,7-карбоксил-1,3-диазаадамантанов, содержащих ароматические и гетероароматические заместители, соединение (137) (рис. 18), имеющее 2-пиридиновые заместители, на 80% снижает боль в тесте Рандалла–Селитто (сдавливание лапы) при введении перорально в дозе 1 мг/кг.

Наличие выраженной анальгетической активности было обнаружено у 2-замещенного 5,7-диметил-1,3-диазаадамантан-6-она (138), содержащего фрагмент (–)-миртеналя (рис. 18) [79]. Исследования, проводившиеся на мышах в тестах “уксусные корчи” и “горячая пластина”, показали, что при пероральном введении в дозе 20 мг/кг диазаадамантан (138) достоверно снижал болевые проявления на 61 и 82% соответственно, не уступая по эффективности препарату сравнения диклофенаку натрия. Соединение (138) показало умеренную острую токсичность на мышах, его LD50 превышала 1000 мг/кг, при этом LD50 диклофенака натрия составляет 370 мг/кг [80].
Ульцерогенную активность (действие, выражающееся в образовании дефектов слизистой оболочки желудочно-кишечного тракта) соединения (138) исследовали в сравнении с нестероидным противовоспалительным препаратом индометацином на крысах линии Wistar [81]. Во время эксперимента животным в течение трех дней вводили соединение (138) в дозе 30 мг/кг или индометацин в дозе 20 мг/кг. В группе животных, получавших соединение (138), все животные дожили до конца эксперимента, а язв не было обнаружено, тогда как среди животных, получавших индометацин, до окончания эксперимента погибла половина группы, а у остальных животных были обнаружены эрозии и язвы [79].
Важной проблемой, которая неизбежно возникает при обнаружении эффективного анальгетика, является изучение возможного механизма его действия. В отличие от (–)-миртенола, который имеет тот же монотерпеновый фрагмент, что и соединение (138), и обладает противовоспалительным действием [82], соединение (138) в дозе 60 мг/кг не проявило противовоспалительной активности на модели, основанной на введении 3%-ного формалина в качестве флогогена (от лат. phlogosis – воспаление; патогенный раздражитель, который может вызвать воспалительную реакцию) [79].
Исследование влияния соединения (138) на поведение мышей в тесте “открытое поле” показало [79], что оно снижает скорость передвижения и, как следствие, дистанцию передвижения, но не оказывает существенного влияния на исследовательскую активность животных. Подобное ингибирующее влияние на двигательную активность животных характерно для опиоидных анальгетиков [83, 84], а также анальгетиков, влияющих на каннабиноидную систему, таких как ∆9-тетрагидроканнабинол [85]. Для выявления возможного механизма действия было изучено влияние неселективного антагониста опиоидных рецепторов налоксона [86] и селективного антагониста каннабиноидных рецепторов CB1 римонабанта [87] на анальгетическую активность соединения (138) в тесте “уксусные корчи”. Анальгетическая активность соединения (138) сохранялась на фоне введения налоксона, тогда как введение римонабанта приводило к нивелированию обезболивающего эффекта [79]. Полученные результаты позволяют предположить, что анальгетический эффект соединения (138), по крайней мере частично, опосредован каннабиноидной системой с вовлечением CB1-рецепторов.
Сопоставимая с диазаадамантаном (138) анальгетическая активность была обнаружена и у вторичного амина (139), также содержащего фрагмент (–)-миртеналя, но присоединенного к гетероадамантановому остову через аминогруппу. Соединение (139), вводимое в дозе 20 мг/кг, снижало боль испытуемых животных в тестах “уксусные корчи” и “горячая пластина” на 46 и 89% соответственно [15].
Высокая анальгетическая активность была обнаружена у ранее упомянутого диазаадамантана (132) (рис. 16), содержащего фрагмент цитронеллаля. В дозе 20 мг/кг он снижал боль в тестах “уксусные корчи” и “горячая пластина” на 32 и 94% соответственно [79]. Интересно, что в случае содержащего остаток цитронеллаля соединения (5) наблюдалось исчезновение анальгетической активности в тесте “уксусные корчи”, а в тесте “горячая пластина” соединение (5) и вовсе проявляло гипералгезивные свойства (повышало чувствительность организма к болевым раздражителям). Исходный диазаадамантанамин (4) не продемонстрировал какой-либо анальгетической активности [79].
Для большинства исследованных производных диазаадамантана было показано, что они обладают низкой острой токсичностью (сотни и тысячи мг/кг). Однако некоторые его производные, в частности 5,7-дифенил-замещенные диазаадамантанолы, проявляют токсичные свойства в дозах от 1 мг/кг. Так, стрихниноподобная активность (способность вызвать сильные болезненные тетанические судороги) была обнаружена Longo et al. [88] у вводимого внутривенно 5,7-дифенил-1,3-диазаадмантан-6-ола (140) (рис. 19) с LD50 = 1.1 и 2.0 мг/кг для мышей и крыс соответственно.
Рис. 19.
Структуры 5,7-диарилзамещенных 1,3-диазаадамантанов, обладающих высокой токсичностью и стрихниноподобной активностью [88].

С целью изучения зависимости “структура–токсичность” различных 6-замещенных 5,7-дифенилдиазаадамантанов Chiavarelli et al. получили ряд 6-алкил- и 6-арил-6-гидроксипроизводных 5,7-дифенилдиазаадмантана (141) [89], а также синтезировали 6-алкил-6-гидроксидиазаадамантаны, содержащие в 5-м и 7-м положениях фенильные группы с различными заместителями, и исследовали их стрихниноподобную активность [90]. Тесты, проведенные на мышах и крысах, показали, что введение в 5,7-дифенил-1,3-диазаадамантан-6-ол алкильного заместителя (метил-, этил-, пропил-, н-бутил-, изобутил-, н-додецил- и α-нафтил-) по 6-му положению приводило к снижению токсичности (LD50 = 12.5–75.0 мг/кг) с сохранением стрихниноподобного воздействия на организм подопытных животных (повышенная возбудимость с последующими тоническими и тонико-клоническими судорогами). Введение в 6-е положение фенильной группы также снижало токсичность соединения, при этом механизм возникновения судорог несколько отличался: сначала наблюдалась мелкая дрожь, а только затем – клонические судороги [89].
Среди 6-гидроксипроизводных, содержащих замещенные фенильные группы или 6-алкоксизамещенные группы, наибольшая токсичность и стрихниноподобное действие были отмечены у 2‑метокси-, 3-метокси- и 4-метоксифенилдиазаадамантан-6-олов (142) (LD50 = 1.5–20 мг/кг) (рис. 19). 4-Метокси-замещенные 5,7-дифенилдиазаадамантаны, содержащие 6-алкоксизамещенную группу (6-метокси- и 6-этоксидиазаадамантаны), проявляли значительно меньшую токсичность (LD50 = 100–150 мг/кг) и оказывали только судорожное (стрихниноподобное) воздействие на подопытных животных. Увеличение количества метоксигрупп в фенильном кольце с сохранением незамещенной 6-гидроксигруппы приводило к резкому снижению (LD50 = 300 мг/кг для 2,3-диметоксипроизводного) или полному исчезновению (для 2,3,4-триметоксипроизводного) токсичности [90]. Таким образом, можно отметить ключевую роль наличия спиртовой группы в 6-м положении и фенильных заместителей в 5-м и 7-м положениях диазаадамантана для проявления ими стрихниноподобного действия.
Агаджанян с соавт. показали наличие α-адреноблокирующей активности у 5,7-диметилдиазаадамантана (115) (рис. 12), содержащего бензилпиперидиновый заместитель во втором положении молекулы [91]. Исследования проводили в опытах на изолированных семявыносящих протоках крыс, об эффекте судили по уменьшению сокращений, вызванных трансмуральным электрическим раздражением или норадреналином в концентрации 10 мкг/мл. В качестве препаратов сравнения использовали α-адреноблокаторы пипероксан, фентоламин и симпатолитик октадин, вводимые, как и исследуемый препарат, в концентрации 0.05 мкМ. Хотя в течение первых минут воздействия соединение (115) не оказывало существенного влияния на сокращение семявыносящего протока, вызванного норадреналином, его блокирующее действие развивалось постепенно, приводя к 50%-ному уменьшению реакции на 60-й минуте. Адреноблокирующее действие препаратов сравнения пипероксана и фентоламина проявлялось гораздо быстрее, приводя к уменьшению сокращений протока через 10 мин после введения препаратов на 30 и 55% соответственно. К 60-й минуте эффект пипероксана уменьшался наполовину, а фентоламина – держался на исходном уровне. Соединение (115) не влияло на сокращения протока, вызванные трансмуральным электрическим раздражением, не оказывая тормозящего эффекта при передаче нервного импульса и не обладая симпатолитической активностью.
Противосудорожные и психотропные свойства ряда 1,3-диазадамантанов (28) (схема 6 ) и (143–145) (рис. 20) были исследованы Арутюняном с соавт. [92] в тесте конвульсий, индуцированных коразолом, и тесте “открытое поле”. Исследованные 5-метил-7-фенил-, 5-метил-7-бензилдиазаадамантаноны (143), (144) и 2-метил, 2‑пиридил-5,7-диметилдиазаадамантаноны (145), (28) проявили противосудорожное действие в дозах ED50 = 30–35 мг/кг, в то время как для препарата сравнения, диазепама, ED50 составила 0.5 мг/кг. Большинство веществ снижали двигательную и исследовательскую активность, что может быть связано с их седативными свойствами.

ДРУГИЕ ТИПЫ АКТИВНОСТЕЙ АЗААДАМАНТАНОВ
Некоторое улучшение гипогликемической активности (способности снижать уровень глюкозы в крови) антидиабетических средств в результате присоединения фрагмента азаадамантана к биологически активным соединениям было показано Агаджаняном с соавт. [93]. Путем замены алкильной группы в 4-хлоро-N-(пропилкарбомоил)бензенсульфонамиде (хлорпропамид; препарат, применяющийся при лечении сахарного диабета 2-го типа, диабетической микроангиопатии (начальные формы) и несахарного диабета) на 1,3,5-триаазаадамантан было получено соединение (146) (рис. 20). Соединение (146) в дозах 100 и 250 мг/кг снижало содержание глюкозы в крови здоровых крыс на 13 и 18% соответственно, почти не отличаясь по активности и эффективности от хлорпропамида. В то же время у крыс с аллоксановым диабетом соединение (146) в дозе 250 мг/кг понижало содержание глюкозы на 31%, тогда как хлорпропамид – только на 23%.
Иммунодепрессивная активность 5-фенилдиазаадамантанолов, содержащих различные заместители по 7-му положению гетероадамантанового остова (рис. 21), была изучена Якушевым с соавт. [94]. Соединения (147) и (149) проявили иммунодепрессивную активность в отношении антител с лизирующими и агглютинирующими свойствами в дозах 0.5 мг/кг, при увеличении дозы до 5 и 50 мг/кг активность пропадала. В то же время соединение (148) в дозе 5 мг/кг проявило иммунодепрессивную активность в отношении обоих исследуемых антител. В дозе 50 мг/кг соединение (148) оказалось активным в отношении антител с агглютинизирующими свойствами.
Рис. 21.
Структуры 5,7-дизамещенных 1,3-диазаадамантанов, обладающих иммунодепрессивной активностью [94].

ЗАКЛЮЧЕНИЕ
Наибольшее число работ, направленных на изучение биологической активности диазаадамантанов, посвящено исследованию антимикробной и противоопухолевой активности. У тетрафенил-замещенных диазаадамантанов и производных, содержащих гетероциклические группировки по второму положению молекулы азаадамантана, были обнаружены выраженные антибактериальные свойства. Различные 2-функционализированные производные, а также спиро- и конденсированные производные, содержащие гетероатомы, продемонстрировали противоопухолевую активность, в основном в экспериментах in vivo. Среди производных диазаадамантана обнаружены соединения, обладающие выраженным противовирусным, психотропным, стрихниноподобным или анальгетическим действием. Большинство исследованных соединений показало умеренную или низкую острую токсичность in vivo, однако был выявлен ряд диазаадамантанов, обладающих крайне высокой токсичностью (LD50 < 10 мг/кг).
Производные триазаадамантанов, как и замещенные диазаадамантаны, проявляют антибактериальную и противоопухолевую активность, но, как правило, менее выраженную. Некоторые производные триазаадамантана обладают гипогликемическими свойствами. Наличие в адамантановом остове трех атомов азота приводит к увеличению растворимости в воде, что делает возможным использование триазаадамантанов в качестве фрагментов для модификации существующих, а также для дизайна новых лекарственных соединений.
Накопленный большой экспериментальный материал позволяет связать строение заместителей в азаадамантанах с влиянием на противовирусную, антибактериальную и противоопухолевую активность. Благодаря возможности синтеза различными путями из доступных реагентов и наличию разнообразной биологической активности в сочетании с низкой токсичностью, ди- и триазаадамантаны – перспективные платформы для поиска фармакологически активных производных.
Список литературы
Зефиров H.С., Рогозина С.В. // Успехи химии. 1973. Т. 42. С. 4–23. [Zefirov N.S., Rogozina S.V. // Russ. Chem. Reviews. 1973. V. 42. P. 190–198.] https://doi.org/10.1070/RC1973v042n03ABEH002574
Lukes R., Syhora K. // Chem. Listy. 1952. V. 46. P. 731–734.
Кузнецов А.И., Зефиров Н.С. // Успехи химии. 1989. Т. 58. С. 1815–1843. [Kuznetsov A.I., Zefirov N.S. // Russ. Chem. Rev. 1989. V. 58. P. 1033–1047.] https://doi.org/10.1070/RC1989v058n11ABEH003495
Izumi H., Yamagami S., Futamura S. // Curr. Med. Chem. – Cardiovasc. Hematol. Agents. 2003. V. 1. P. 99–111. https://doi.org/10.2174/1568016033477478
Yamawaki I., Bukovac S.W., Sunami A. // Chem. Pharm. Bull. 1994. V. 42. P. 2365–2369. https://doi.org/10.1248/cpb.42.2365
Brandt W., Drosihn S., Haurand M., Holzgrabe U., Nachtsheim C. // Arch. Pharm. Pharm. Med. Chem. 1996. V. 329. P. 311–323. https://doi.org/10.1002/ardp.19963290607
Arutyunyan G.L., Chachoyan A.A., Agadzhanyan T.E., Garibdzhanyan B.T. // Pharm. Chem. J. 1996. V. 30. P. 739–741. https://doi.org/10.1007/BF02218824
Becker D.P., Flynn D.L., Shone R.L., Gullikson G. // Bioorg. & Med. Chemistry. 2004. V. 14. P. 5509–5512. https://doi.org/10.1016/j.bmcl.2004.09.005
Dembitsky V.M., Gloriozova T.A., Poroikov V.V. // Biochem. Biophys. Res. Commun. 2020. V. 529. P. 1225–1241. https://doi.org/10.1016/j.bbrc.2020.06.123
Narayan S., Ramisetti S., Jaiswal A.S., Law B.K., Singh-Pillay A., Singh P., Amin S., Sharma A.K. // Eur. J. Med. Chem. 2019. V. 161. P. 456–467. https://doi.org/10.1016/j.ejmech.2018.10.052
Кузнецов А.И. // Успехи химии адамантана. Москва, 2007.
Semakin A.N., Nelyubina Y.V., Ioffe S.L., Sukhorukov A.Yu. // Eur. J. Org. Chem. 2020. V. 43. P. 6723–6735. https://doi.org/10.1002/ejoc.202000832
Kuznetsov A.I., Basargin E.B., Ba M.K., Moskovkin A.S., Miroshnichenko I.V., Botnikov M.Y. // Chem. Heterocycl. Compd. V. 25. P. 541–547. https://doi.org/10.1007/bf00482502
Palyulin V.A., Emets S.V., Potekhin K.A., Lysov A.E., Chertkov V.A., Zefirov N.S. // Dokl. Chem. 2000. V. 375. P. 289–292. https://doi.org/10.1023/a:1026651502291
Ponomarev K., Morozova E., Pavlova A., Suslov E., Korchagina D., Nefedov A., Tolstikova T., Volcho K., Salakhutdinov N. // Med. Chem. 2017. V. 13. P. 773–779. https://doi.org/10.2174/1573406413666170525124316
Вичканова С.А., Горюнова Л.В., Шипулина Л.Д., Бадаев Ф.А., Кузнецов И.А., Толкачев О.Н., Унковский Б.В. // Фармакология и токсикология. 1974. Т. 37. С. 76–79.
Агаджанян Ц.Е., Арутюнян А.Д., Арутюнян Г.Л. // ХГС. 1992. Т. 7. С. 929–932. https://doi.org/10.1007/BF00474490
Tomassoli I., Gündisch D. // Curr. Top. Med. Chem. 2016. V. 16. P. 1314–1342. https://doi.org/10.2174/1568026615666150915111434
Ruenitz P.C., Smissman E.J. // J. Heterocycl. Chem. 1976. V. 13. P. 1111–1113. https://doi.org/10.1002/jhet.5570130539
Chiavarelli S., Seilimj G., Rabagliati-Canessa F.M. // Gazz. Chim. Ital. 1960. V. 90. P. 311.
Kafka Z., Galík V., Šafář M. // Collect. Czech. Chem. Commun. 1975. V. 40. P. 174–178. https://doi.org/10.1135/cccc19750174
Baliah V., Usha R. // Indian J. Chem. 1977. V. 158. P. 684–689.
Quast H., Muller B. // Chem. Ber. 1980. V. 113. P. 2959. https://doi.org/10.1002/cber.19801130913
Quast H., Muller B., Peters E.-M. // Chem. Ber. 1982. V. 115. P. 3631. https://doi.org/10.1002/cber.19821151115
Balaji G.L., Vijayakumar V., Rajesh K. // Arab. J. Chem. 2016. V. 9. Suppl. 2. P. 1101–1104. https://doi.org/10.1016/j.arabjc.2011.12.011
Sletter H., Bochmann W. // Chem. Ber. 1951. V. 84. P. 834. https://doi.org/10.1002/cber.19510840908
Кузнецов А.И., Бурделев О.Т., Унковский Б.В. // Тр. Юбилейной конференции, посвященной 70-летию МИТХТ им. М. В. Ломоносова. Москва. 1970. С. 163.
Schnell J.R., Chou J.J. // Nature. 2008. V. 451. P. 591–595. https://doi.org/10.1038/nature06531
Fiore A.E., Fry A., Shay D., Gubareva L., Bresee J.S., Uyeki T.M. // MMWR Recomm. Rep. 2011. V. 60. P. 1–24.
Suslov E.V., Zarubaev V.V., Slita A.V., Ponomarev K.Y., Korchagina D.V., Ayine-Tora D.M., Reynisson J., Volcho K.P., Salakhutdinov N.F. // Bioorg. Med. Chem. Lett. 2017. V. 27. P. 4531–4535. https://doi.org/10.1016/j.bmcl.2017.08.062
Narayan S., Ramisetti S., Jaiswal A.S., Law B.K., Singh-Pillay A., Singh P., Amin S., Sharma A.K. // Eur. J. Med. Chem. 2019. V. 161. P. 456–467. https://doi.org/10.1016/j.ejmech.2018.10.052
Teplov G.V., Suslov E.V., Zarubaev V.V., Shtro A.A., Karpinskaya L.A., Rogachev A.D., Korchagina D.V., Volcho K.P., Salakhutdinov N.F., Kiselev O.I. // Lett. Drug. Des. Discov. 2013. V. 10. P. 477–485. https://doi.org/10.2174/1570180811310060002
Chao W.W., Lin B.F. // Chin. Med. 2010. V. 5. P. 17. https://doi.org/10.1186/1749-8546-5-17
Wiart C., Kumar K., Yusof M.Y., Hamimah H., Fauzi Z.M., Sulaiman M. // Phytother. Res. 2005. V. 19. P. 1069–1070. https://doi.org/10.1002/ptr.1765
Lifeng Xu // U.S. Pat. Appl. Publ. 2014, US 20140045779 A1.
Саакян Г.С., Арутюнян Г.Л., Агаджанян Ц.Е., Пароникян Р.В. // Арм. хим. журнал. 1986. Т. 39. С. 242–246.
Arutyunyan G.L., Paronikyan R.V., Saakyan G.S., Arutyunyan A.D., Gevorkyan K.A. // Pharm. Chem. J. 2008. V. 42. P. 18–22. https://doi.org/10.1007/s11094-008-0048-3
Arutyunyan G.L., Gevorkyan K.A., Arutyunyan A.D., Paronikyan R.V., Stepanyan G.M., Panosyan G.A. // Russ. J. Org. Chem. 2014. V. 50. P. 1480–1484. https://doi.org/10.1134/S1070428014100133
Arutyunyan G.L., Arutyunyan A.D., Gevorkyan K.A., Gasparyan S.P., Paronikyan R.V., Stepanyan G.M., Minasyan N.S. // Pharm. Chem. J. 2018. V. 52. P. 419–423. https://doi.org/10.1007/s11094-018-1834-1
Байсалбаева С.А., Омаров Т.Т., Никитина Е.Т., Казакова Г.Г. // ХФЖ. 1987. Т. 21. С. 191–195. [Baisalbaeva S.A., Omarov T.T., Nikitina E.T., Kazakova G.G. // Pharm. Chem. J. 1987. V. 21. P. 120–123.] https://doi.org/10.1007/BF00765110
Balaji G.L., Sarveswari S., Vijayakumar V. // Res. Chem. Intermed. 2015. V. 41. P. 6765–6776. https://doi.org/10.1007/s11164-014-1775-6
Hodge E.B. // U.S. Pat. Appl. Publ. 1971, US 3575974 A.
Perez-Pineiro R., Burgos As., Deuan C. Jones, Andrew L.C., Rodriguez H., Suarez M., Fairlamb A.H., Wishart D.S. // J. Med. Chem. 2009. V. 52. P. 1670–1680. https://doi.org/10.1021/jm801306g
Wyllie S., Foth B.J., Kelner A., Sokolova A.Y., Berriman M., Fairlamb A.H. // J. Antimicrob. Chemother. 2015. V. 71. P. 625–634. https://doi.org/10.1093/jac/dkv376
Чачоян А.А., Шкулев В.А., Писарский Ю.Б., Саакян Г.С., Агаджанян Ц.Е., Гарибджанян Б.Т. // ХФЖ. 1991. Т. 25. С. 45–48. [Chachoyan A.A., Shkulev V.A., Pisarskii Y.B., Saakyan G.S., Agadzhanyan T.E., Garibdzhanyan B.T. // Pharm. Chem. J. 1991. V. 25. P. 275–278.] https://doi.org/10.1007/BF00772114
Чачоян А.А., Шкулев В.А., Самвелян К.Г., Гарибджанян Б.Т., Папаян Г.Л. // ХФЖ. 1989. Т. 23. С. 166–169. [Chachoyan A.A., Shkulev V.A., Samvelyan K.G., Garibdzhanyan B.T., Papayan G.L. // Pharm. Chem. J. 1989. V. 23. P. 126–129.] https://doi.org/10.1007/BF00764459
Шкулев В.А., Адамян Г.Г., Агаджанян Ц.Е., Чачоян А.А., Гарибджанян Б.Г. // ХФЖ. 1995. Т. 29. С. 36–37. [Shkulev V.A., Adamyan G.G., Agadzhanyan T.E., Chachoyan A.A., Garibdzhanyan B.T. // Pharm. Chem. J. 1995. V. 29. P. 192–193.] https://doi.org/10.1007/BF02219065
Арутюнян Г.Л., Чачоян А.А., Шкулев В.А., Адамян Г.Г., Агаджанян Ц.Е., Гарибджанян Б.Г. // ХФЖ. 1995. Т. 29. С. 33–35. [Arutyunyan G.L., Chachoyan A.A., Shkulev V.A., Adamyan G.G., Agadzhanyan T.E., Garibdzhanyan B.T. // Pharm. Chem. J. 1995. V. 29. P. 188–191.] https://doi.org/10.1007/BF02219064
Sharabi-Ronen Y., Levinger S., Lellouche M.B, Albeck A. // Med. Chem. 2014. V. 10. P. 27–37. https://doi.org/10.2174/15734064113099900002
Jimenez J., Chakraborty I., Carrington S.J., Mascharak P.K. // ChemMedChem. 2014. V. 9. P. 1266–1274. https://doi.org/10.1039/c6dt01358a
Ryter S.W., Choi A.M.K. // Korean J. Intern. Med. 2013. V. 28. P. 123–140. https://doi.org/10.3904/kjim.2013.28.2.123
Wegiel B., Gallo D., Csizmadia E., Harris C., Belcher J., Vercellotti G.M., Penacho N., Seth P., Sukhatme V., Ahmed A., Pandolfi P.P., Helczynski L., Bjartell A., Persson J.L., Otterbien L.E. // Cancer Res. 2013. V. 73. P. 7009–7021. https://doi.org/110.1158/0008-5472
Gabrielian A., Bocharova T.N., Smirnova E.A., Volodin A.A., Harutjunyan A., Harutjunyan G. // J. Biomol. Struct. Dyn. 2011. V. 27. P. 1124–1125. https://doi.org/10.1080/073911011010524986
Gabrielian A., Bocharova T.N., Smirnova E.A., Volodin A.A., Harutjunyan A., Gevorkyan K., Harutjunyan G. // Biomol. Struct. Dyn. 2013. V. 31. P. 82–82. https://doi.org/10.1080/07391102.2013.786370
Mamaeva O.K., Gabrielyan A.G., Arutyunyan G.L., Bocharov T.N., Smirnov E.A., Volodin A.A., Shchyolkina A.K., Kaluzhnya D.N. // Mol. Biol. 2014. V. 48. P. 741–748. https://doi.org/10.1134/S0026893314050100
Захаренко А.Л., Пономарев К.Ю., Суслов Е.В., Корчагина Д.В., Волчо К.П., Васильева И.А., Салахутдинов Н.Ф., Лаврик О.И. // Биоорг. химия. 2015. Т. 41. С. 731–736. [Zakharenko A.L., Ponomarev K.U., Suslov E.V., Korchagina D.V., Volcho K.P., Vasil’eva I.A., Salakhutdinov N.F., Lavrik O.I. // Russ. J. Bioorg. Chem. 2015. V. 41. P. 657–662.] https://doi.org/10.1134/S1068162015060199
Cortes Ledesma F., El Khamisy S.F., Zuma M.C., Osborn K., Caldecott K.W. // Nature. 2009. V. 461. P. 674–678. https://doi.org/10.1038/nature08444
Laev S.S., Salakhutdinov N.F., Lavrik O.I. // Bioorg. Med. Chem. 2016. V. 24. P. 5017–5027. https://doi.org/10.1016/j.bmc.2016.09.045
Wang W., Rodriguez-Silva, M., Acanda de la Rocha A.M., Wolf A.L., Lai Y., Liu,Y., Reinhold W.C., Pommier Y., Chambers J.W., Tse-Dinh Y.-C. // Cancers. 2019. V. 11. P. 1416. https://doi.org/10.3390/cancers11101416
Beretta G., Cossa G., Gatti L., Zunino F., Perego P. // Curr. Med. Chem. 2010. V. 17. P. 1500–1508. https://doi.org/10.2174/092986710790979971
Yang S.W., Burgin A.B., Huizenga B.N., Robertson C.A., Yao K.C. Nash H.A. // Proc. Natl. Acad. Sci. 1996. V. 93. P. 11534–11539. https://doi.org/10.1073/pnas.93.21.11534
Bailly C. // Pharmacol. Res. 2019. 104398. https://doi.org/10.1016/j.phrs.2019.104398
Pommier Y., Leo E., Zhang H., Marchand C. // Chem. Biol. 2010. V. 17. P. 421–433. https://doi.org/10.1016/j.chembiol.2010.04.012
Zakharenko A.L., Luzina O.A., Sokolov D.N., Kaledin V.I., Nikolin V.P., Popova N.A., Patel J., Zakharova O.D., Chepanova A.A., Zafar A., Reynisson J., Leung E., Leung I.K.-H., Volcho K.P., Salakhutdinov N.F., Lavrik O.I. // Eur. J. Med. Chem. 2019. V. 161. P. 581–593. https://doi.org/10.1016/j.ejmech.2018.10.055
Chepanova A.A., Li-Zhulanov N.S., Sukhikh A.S., Zafar A., Reynissone J., Zakharenko A.L., Zakharova O.D., Korchagina D.V., Volcho K.P., Salakhutdinov N.F., Lavrik O.I. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 647–655. https://doi.org/10.1134/s1068162019060104
Khomenko T.M., Zakharenko A.L., Chepanova A.A., Ilina E.S., Zakharova O.D., Kaledin V.I., Nikolin V.P., Popova N.A., Korchagina D.V., Reynisson J., Chand R., Ayine-Tora D.M., Patel J., Leung I.K.H., Volcho K.P., Salakhutdinov N.F., Lavrik O.I. // Int. J. Mol. Sci. 2020. V. 21. P. 126. https://doi.org/10.3390/ijms21010126
Zakharenko A., Khomenko T., Zhukova S., Koval O., Zakharova O., Anarbaev R., Lebedeva N., Korchagina D., Komarova N., Vasil’ev V., Reynisson J., Volcho K., Salakhutdinov N., Lavrik O. // Bioorg. Med. Chem. 2015. V. 23. P. 2044–2052. https://doi.org/10.1016/j.bmc.2015.03.020
Il’ina I.V., Dyrkheeva N.S., Zakharenko A.L., Sidorenko A.Yu., Li-Zhulanov N.S., Korchagina D.V., Chand R., Ayine-Tora D.M., Chepanova A.A., Zakharova O.D., Ilina E.S., Reynisson J., Malakhova A.A., Medvedev S.P., Zakian S.M., Volcho K.P., Salakhutdinov N.F., Lavrik O.I. // Molecules. 2020. V. 25. P. 3496. https://doi.org/10.3390/molecules25153496
Gladkova E.D., Nechepurenko I.V., Bredikhin R.A., Chepanova A.A., Zakharenko A.L., Luzina O.A., Ilina E.S., Dyrkheeva N.S., Mamontova E.M., Anarbaev R.O., Reynisson J., Volcho K.P., Salakhutdinov N.F., Lavrik O.I. // Int. J. Mol. Sci. 2020. V. 21. P. 7162. https://doi.org/10.3390/ijms21197162
Salomatina O.V., Popadyuk I.I., Zakharenko A.L., Zakharova O.D., Chepanova A.A., Dyrkheeva N.S., Komarova N.I., Reynisson J., Anarbaev R.O., Salakhutdinov N.F., Lavrik O.I., Volcho K.P. // Steroids. 2021. V. 165. 108771. https://doi.org/10.1016/j.steroids.2020.108771
Golubovskaya V.M., Ho B., Zheng M., Magis A., Ostrov D., Morrison C., Cance W.G. // BMC Cancer. 2013. V. 13. P. 342–348. https://doi.org/10.1186/1471-2407-13-342
Martínez T.P., Navajas L.P., Lietha D. // Biomolecules. 2020. V. 10. P. 179. https://doi.org/10.3390/biom10020179
Sabapathy K., Lane D.P. // J. Mol. Cell. Biol. 2019. V. 11. P. 317–329. https://doi.org/10.1093/jmcb/mjz010
Eisenbarth G., Michels A., Nakayama M., Ostrov D. // U.S. Pat. Appl. Publ. 2012, US 9629848 B2.
Kasama T., Strieter R.M., Lukacs N.W., Lincoln P.M., Burdick M.D., Kunkel, S.L. // J. Clin. Invest. 1995. V. 95. P. 2868–2876. https://doi.org/10.1172/JCI117993
McMaster B. // U.S. Pat. Appl. Publ. 2003, US 6727241 B2.
Hossain N., Ivanova S., Bergare J., Eriksson T. // Bioorg. Med. Chem. Lett. 2013. V. 23. P. 1883–1886. https://doi.org/10.1016/j.bmcl.2012.12.095
Samhammer A., Holzgrabe U., Haller R. // Arch. Pharm. (Weinheim). 1989. V. 322. P. 551–555. https://doi.org/10.1002/ardp.19893220908
Ponomarev K.Y., Pavlova A.V., Suslov E.V., Ardashov O.V., Korchagina D.V., Nefedov A.V., Tolstikova T.G., Volcho K.P., Salakhutdinov N.F. // Med. Chem. Res. 2015. V. 24. P. 4146–4156. https://doi.org/10.1007/s00044-015-1464-z
Syubaev R.D., Mashkovskii M.D., Shvarts G.Y., Pokryshkin V.I. // Pharm. Chem. J. 1986. V. 20. P. 17–22. https://doi.org/10.1007/BF00766871
Scarpignato C., Hunt R.H. // Gastroenterol. Clin. N. Am. 2010. V. 39. P. 433–464. https://doi.org/10.1016/j.gtc.2010.08.010
Silva R.O., Salvadori M.S., Sousa F.B.M., Santos M.S., Carvalho N.S., Sousa D.P., Gomes B.S., Oliveira F.A., Barbosa A.L.R., Freitas R.M., de Almeida R.N., Medeiros J.-V.R. // Flavour. Fragr. J. 2014. V. 29. P. 184–192. https://doi.org/10.1002/ffj.319
Knych H.K., Steffey E.P., Casbeer H.C., Mitchell M.M. // Equine. Vet. J. 2014. V. 47. P. 592–598. https://doi.org/10.1111/evj.12318
Craft R.M., Clark J.L., Hart S.P., Pinckney M.K. // Pharmacol. Biochem. Behav. 2006. V. 85. P. 850–858. https://doi.org/10.1016/j.pbb.2006.11.022
Marshell R., Kearney-Ramos T., Brents L.K., Hyatt W.S., Tai S., Prather P.L., Fantegrossi W.E. // Pharmacol. Biochem. Behav. 2014. V. 124. P. 40–47. https://doi.org/10.1016/j.pbb.2014.05.010
Lewanowitsch T., Moller J.H., Irvine R.J. // Life Sci. 2006. V. 78. P. 682–688. https://doi.org/10.1016/j.lfs.2005.05.062
Booker L., Naidu P.S., Razdan R.K., Mahadevan A., Lichtman A.H. // Drug. Alcohol. Depend. 2009. V. 105. P. 42–47. https://doi.org/10.1016/j.drugalcdep.2009.06.009
Longo V.G., Silvestrini B., Bovet D. // J. Pharmacol. Exp. Ther. 1959. V. 126. P. 41–49.
Chiavarelli S., Fennoy L.V. // J. Org. Chem. 1961. V. 26. P. 4895–4898. https://doi.org/10.1021/jo01070a028
Chiavarelli S., Fennoy L.V., Settimj G., De Baran L. // J. Med. Pharm. Chem. 1962. V. 5. P. 1293–1297. https://doi.org/10.1021/jm01241a020
Агаджанян Ц.Е., Арутюнян Г.Л., Авакян О.М., Калтрикян А.А., Вартанян С.А. // Авторское свидетельство СССР № 1221878, 17.08.1984, SU 1221878 A1.
Arutyunyan G.L., Dzhagatspanyan I.A., Nazaryan I.M., Akopyan A.G., Arutyunyan A.D. // Pharm. Chem. J. 2007. V. 41. P. 591–593. https://doi.org/10.1007/s11094-008-0025-x
Agadzhanyan Ts.E., Arutyunyan A.D., Stepanyan N.O., Bunatyan Zh.M. // Pharm. Chem. J. 1997. V. 31. P. 15–16. https://doi.org/10.1007/BF02464677
Якушев П.Ф., Кузнецов A.И., Дуракова Л.И., Ковалев И.Е., Унковский Б.В. // Авторское свидетельство СССР № 536673, 25.10.1977, SU 536673 A1.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия