

Журнал физической химии, 2022, T. 96, № 7, стр. 991-997
Влияние добавления Al(NO3)3 на молекулярную подвижность в нитрате этиламмония по данным молекулярно-динамического моделирования
М. Убович a, *, А. В. Егоров a, В. И. Чижик a
a Санкт-Петербургский государственный университет
Санкт-Петербург, Россия
* E-mail: ubovich.milosh@yandex.ru
Поступила в редакцию 14.01.2022
После доработки 14.01.2022
Принята к публикации 18.01.2022
- EDN: OTSURE
- DOI: 10.31857/S0044453722070342
Аннотация
С помощью молекулярно-динамического моделирования предпринята попытка выявить механизмы изменения молекулярной подвижности при добавлении нитрата алюминия в нитрат этиламмония. Основной целью было не только количественно оценить кинетические характеристики компонентов исследуемой смеси при их разном соотношении, но и выявить связь между возникающей при увеличении концентрации соли перестройкой локальной структуры и процессами вращательной переориентации ионов. Рассчитаны модельные системы, функции радиального распределения, величины коэффициентов самодиффузии и времен переориентации внутримолекулярных векторов нитрат-аниона. Показано, что с увеличением концентрации нитрата алюминия в системе замедляются процессы переориентации нитрат-аниона и трансляционные движения компонент системы. С помощью функций радиального распределения продемонстрировано, что ион алюминия не проникает в ближайшее окружение катиона этиламмония. Однако появление трехзарядных ионов алюминия оказывают заметное воздействие на упорядочение нитрат-анионов, в том числе входящих в окружение катиона этиламмония. Это и приводит к замедлению вращательной переориентации нитрат-аниона.
За последние десятилетия значительно вырос интерес к изучению ионных жидкостей (ИЖ), характерная особенность которых заключается в том, что они состоят из ионов, но при этом, в отличие от “классических” солей, находятся в жидком состоянии при относительно низкой температуре (примерно до 100°C) [1–5]. ИЖ привлекают внимание исследователей своими замечательными свойствами, такими как высокая термальная стабильность, высокая температура кипения, низкое давление паров и др. Благодаря этим свойствам, ионные жидкости могут применяться в различных электрохимических приложениях (например, в ионно-литиевых батареях, суперконденсаторах, топливных ячейках и т.д.) [6–16]. Одной из актуальных проблем современной физической химии является получение ИЖ с требуемыми для конкретной задачи свойствами. Однако, многообразие возможных модификаций ИЖ (подбор аниона, добавление к катиону различных функциональных групп, добавление примесей и др.) и, соответственно, необходимость приготовления большого числа образцов и проведения множества экспериментов серьезно усложняет решение этой задачи. Несмотря на то, что в этой области проведено множество исследований, существуют заметные пробелы в понимании природы ионных жидкостей. В данном случае одним из наиболее перспективных подходов является компьютерное моделирование, позволяющее описывать ИЖ на микроуровне. Понимание механизмов формирования локальной структуры и выявление ее связи с особенностями молекулярной подвижности способствовало бы оптимизации процесса поиска новых вариантов ИЖ.
К одному из наиболее интересных классов объектов относятся системы на основе ионов аммония. Аммониевые ИЖ привлекают внимание исследователей необычным сочетанием свойств (в частности, обладают относительно невысокой стоимостью и низкой токсичностью). Характерным представителем данной группы является нитрат этиламмония (ЭАН). В работе в качестве основного объекта исследования были выбраны смеси ЭАН ($[{{{\text{C}}}_{2}}{{{\text{H}}}_{5}}{\text{NH}}_{3}^{ + }][{\text{NO}}_{3}^{ - }]$) и нитрата алюминия (Al(NO3)3). Добавление неорганических солей металлов является одним из перспективных способов получения ИЖ с нужными свойствами. Добавление соли алюминия перспективно с нескольких точек зрения. В настоящее время в качестве потенциальной замены солей на основе лития (Li+) рассматриваются соли с более высокой валентностью, такие как соли, содержащие магний (Mg2+) или алюминий (Al3+), поскольку их использование может привести к более эффективным системам хранения энергии. Для алюминия характерна гораздо более высокая теоретическая плотность энергии, чем для лития (1060 Вт ч/кг и 406 Вт ч/кг, соответственно) [17]. Кроме того, алюминий более распространен в природе по сравнению с литием, а это означает, что стоимость продукта будет более низкой [17]. Ранее смеси ЭАН и нитрата алюминия в диапазоне концентраций от 0 до 25 мол. % соли уже были исследованы в работе [17]. Авторы подробно рассмотрели влияние добавки Al(NO3)3 на особенности локальной микроструктуры смеси, однако никаких оценок величин, характеризующих молекулярную подвижность, сделано не было.
В данной работе с помощью метода молекулярной динамики (МД) предпринята попытка выявить механизмы изменения молекулярной подвижности при добавлении нитрата алюминия в ЭАН. При этом основной целью ставилось не только количественно оценить кинетические характеристики компонентов смеси при их разном соотношении, но и выявить связь между возникающей при добавлении соли перестройкой локальной структуры и процессами вращательной переориентации ионов.
МЕТОДИКА МОДЕЛИРОВАНИЯ
В работе с помощью метода МД промоделированы три системы: чистая ИЖ ЭАН без добавок и две смеси с содержанием 5 и 10 мол. % нитрата алюминия. Данные о составе модельных систем приведены в табл. 1. Моделирование проводилось в кубической ячейке с периодическими граничными условиями в NPT-ансамбле при 298 K и атмосферном давлении с помощью программного пакета MDynaMix v5.0 [18]. Температура поддерживалась постоянной с помощью термостата Нозе–Хувера [19, 20], давление – с помощью баростата Хувера [21].
Таблица 1.
Состав модельных систем
| ${{С}_{{{\text{Al(N}}{{{\text{O}}}_{{\text{3}}}}{{{\text{)}}}_{{\text{3}}}}}}}$, % | m, моль/кг | Число ионов в модельной ячейке | ||
|---|---|---|---|---|
| ЭА+ | ${\text{NO}}_{3}^{ - }$ | Al3+ | ||
| 0 | 0 | 300 | 300 | 0 |
| 5 | 0.489 | 500 | 578 | 26 |
| 10 | 1.028 | 500 | 668 | 56 |
Для описания межмолекулярного взаимодействия между катионами этиламмония (ЭА+) был выбран потенциал, предложенный в работах [22, 23]. Ранее (см. [17, 22, 24–26]) было показано, что он достаточно хорошо воспроизводит плотность и основные структурные характеристики этой ионной жидкости. Нитрат-анион рассматривался как четырехцентровая плоская структура, в центре которой находится атом азота, а на расстоянии 1.22 Å от него расположены атомы кислорода, а причем все углы O–N–O равны 120° [27]. Взаимодействие между атомами модельного нитрат-аниона и другими частицами представляет собой сумму кулоновского и леннард-джонсовского (12–6) потенциалов:
В литературе предложено несколько вариантов соответствующих параметров [22, 28, 29], но на основании опубликованных данных не представляется возможным обоснованно выбрать какой-то один из них. Поэтому для оценки влияния величин параметров модельных потенциалов для нитрат-аниона на расчетные характеристики ИЖ и смесей в работе рассмотрено три различных варианта параметров [22, 28, 29], значения которых приведены в табл. 2. Для описания иона алюминия в литературе предложено около десятка потенциалов (см., например, [30–36]), но только в двух случаях авторы рассматривали сумму кулоновского и леннард-джонсовского (12–6) потенциалов [30, 31]. Предварительное тестовое моделирование показало, что вариант, предложенный в работе [31], предсказывает завышенный размер первой сольватной оболочки катиона алюминия и в дальнейшем в расчетах использовался только потенциал [30], параметры которого приведены в табл. 2.
Таблица 2.
Параметры модельных потенциалов для нитрат-аниона и катиона алюминия
| Ион | Центр | q, |e| | σ, Å | $\mathcal{E}$, кДж/моль |
|---|---|---|---|---|
| ${\text{NO}}_{3}^{ - }$, вариант I [28] | N | +0.860 | 3.9000 | 0.8370 |
| O | –0.620 | 3.1540 | 0.6490 | |
| ${\text{NO}}_{3}^{ - }$, вариант II [22] | N | +0.905 | 3.2500 | 0.7118 |
| O | –0.635 | 2.9600 | 0.8792 | |
| ${\text{NO}}_{3}^{ - }$, вариант III [29] | N | +1.310 | 3.1000 | 0.3077 |
| O | –0.770 | 3.0000 | 0.3077 | |
| Al3+ [30] | Al | +3.000 | 1.4472 | 0.9063 |
Уравнения движения решались методом Верле с шагом 2 фс. Структура модельного нитрат-аниона сохранялась при помощи алгоритма SHAKE [37]. Потенциал электростатического взаимодействия рассчитывался методом Эвальда. Время уравновешивания системы каждой модельной системы составляло 2 нс, время последующего моделирования составило 1 нс.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Плотность модельных систем
В качестве первого критерия корректности данных моделирования были рассчитаны плотности как чистой ИЖ, так и смесей с нитратом алюминия. Соответствующие результаты приведены в табл. 3 вместе с экспериментальными данными, опубликованными в литературе [17, 24]. Очевидно, что значения потенциальных параметров нитрат-аниона оказывают заметное (до 11%) влияние на плотность моделируемой системы. На основании сопоставления расчетных данных с экспериментальными, можно сделать вывод, что все рассмотренные варианты могут быть использованы для анализа молекулярной подвижности рассматриваемых систем.
Молекулярная подвижность в смеси нитрата этиламмония и нитрата алюминия
Коэффициенты самодиффузии. Скорость трансляционного движения частиц вещества характеризуется коэффициентом самодиффузии (D). В данной работе он рассчитывался как отношение среднеквадратичного смещения молекул за определенный промежуток времени к длительности этого промежутка:
Таблица 4.
Расчетные коэффициенты самодиффузии в рассмотренных системах
| Потенциал ${\text{NO}}_{3}^{ - }$ |
${{С}_{{{\text{Al(N}}{{{\text{O}}}_{{\text{3}}}}{{{\text{)}}}_{{\text{3}}}}}}}$, % | 1011D, м2/с | ||
|---|---|---|---|---|
| Al3+ | ${\text{NO}}_{3}^{ - }$ | ЭА+ | ||
| I | 0 | – | 0.54 | 0.41 |
| 5 | 0.16 | 0.46 | 0.38 | |
| 10 | 0.15 | 0.40 | 0.37 | |
| II | 0 | – | 0.62 | 0.46 |
| 5 | 0.13 | 0.52 | 0.42 | |
| 10 | 0.15 | 0.50 | 0.32 | |
| III | 0 | – | 0.31 | 0.28 |
| 5 | 0.09 | 0.34 | 0.33 | |
| 10 | 0.09 | 0.29 | 0.26 | |
Время моделирования недостаточно велико, для того чтобы рассчитывать характеристики модельной системы с высокой точностью, но позволяет оценить основные эффекты, возникающие при добавлении нитрата алюминия в ИЖ. Отметим, что хотя значения потенциальных параметров нитрат-аниона достаточно заметно влияют на величину D, но все три значения примерно одного порядка. Как уже упоминалось выше, основной целью численного эксперимента являлось выявление связи между возникающей при добавлении соли перестройкой локальной структуры ИЖ и процессами вращательной переориентации ионов. По данным моделирования, с увеличением мольной доли Al(NO3)3, трансляционное движение в системе замедляется, но эффект выражен достаточно слабо.
Вращательная переориентация нитрат-аниона. Другой важной характеристикой молекулярной подвижности являются функции автокорреляции вращательных переориентаций различных внутримолекулярных векторов, анализ которых позволяет получить информацию о вращательном движении ионов. Для описания переориентации нитрат-аниона могут рассматриваться четыре вектора (см. рис. 1): $\overrightarrow {{\text{N}}{{{\text{O}}}_{1}}} $, $\overrightarrow {{\text{N}}{{{\text{O}}}_{2}}} $ и $\overrightarrow {{\text{N}}{{{\text{O}}}_{3}}} $ направлены вдоль трех химических связей N–O, а вектор $\overrightarrow {{{{\text{N}}}_{п}}} $ – перпендикулярен плоскости аниона. Анализ данных моделирования показал, что в случае нитрат-аниона указанные функции могут быть с удовлетворительной точностью аппроксимированы одной экспонентой вида:
где ${{\tau }_{c}}$ – время вращательной переориентации рассматриваемого вектора. Оценки величин времен вращательной переориентации рассмотренных внутримолекулярных векторов иона ${\text{NO}}_{3}^{ - }$ приведены в табл. 5.Рис. 1.
Внутримолекулярные вектора в нитрат-анионе, для которых рассчитывались функции автокорреляции вращательных переориентаций.

Таблица 5.
Времена вращательной переориентации внутримолекулярных векторов иона ${\text{NO}}_{3}^{ - }~$ (см. рис. 1)
| Потенциал | ${{С}_{{{\text{Al(N}}{{{\text{O}}}_{{\text{3}}}}{{{\text{)}}}_{{\text{3}}}}}}}$, % | Времена переориентации соответствующих векторов, пс | |||
|---|---|---|---|---|---|
| $\overrightarrow {{\text{N}}{{{\text{O}}}_{1}}} $ | $\overrightarrow {{\text{N}}{{{\text{O}}}_{2}}} $ | $\overrightarrow {{\text{N}}{{{\text{O}}}_{3}}} $ | $\overrightarrow {{{{\text{N}}}_{п}}} $ | ||
| I | 0 | 160 ± 20 | 150 ± 20 | 160 ± 20 | 510 ± 50 |
| 5 | 360 ± 30 | 360 ± 30 | 380 ± 30 | 650 ± 60 | |
| 10 | 640 ± 60 | 640 ± 60 | 570 ± 60 | 680 ± 70 | |
| II | 0 | 240 ± 20 | 240 ± 20 | 260 ± 20 | 610 ± 60 |
| 5 | 440 ± 40 | 460 ± 40 | 460 ± 40 | 830 ± 80 | |
| 10 | 820 ± 80 | 810 ± 80 | 820 ± 80 | 1100 ± 100 | |
| III | 0 | 920 ± 90 | 890 ± 90 | 910 ± 90 | 2400 ± 300 |
| 5 | 1400 ± 100 | 1300 ± 100 | 1300 ± 100 | 2900 ± 300 | |
| 10 | 1900 ± 200 | 1900 ± 200 | 1900 ± 200 | 3300 ± 300 | |
Во-первых, функции автокорреляции вращательных переориентаций векторов $\overrightarrow {{\text{N}}{{{\text{O}}}_{1}}} $, $\overrightarrow {{\text{N}}{{{\text{O}}}_{2}}} $ и $\overrightarrow {{\text{N}}{{{\text{O}}}_{3}}} $, как и можно было ожидать, практически одинаковы. Во-вторых, разница между величинами времен переориентации, полученными с использованием разных модельных потенциалов для нитрат-аниона выражена гораздо заметнее, чем в случае коэффициентов самодиффузии. Однако, характерные изменения в подвижности иона ${\text{NO}}_{3}^{ - }$ схожи: во всех случаях с увеличением мольной доли нитрата алюминия в системе наблюдается существенное замедление процессов вращательной переориентации. В-третьих, во всех случаях время вращательной переориентации вектора $\overrightarrow {{{{\text{N}}}_{п}}} $ заметно превышает таковое для векторов, направленных вдоль химической связи N–O. Это свидетельствует о том, что частота вращательных движений в плоскости нитрат-аниона выше чем, частота поворотов самой плоскости. Заметим, что этот естественный эффект получил количественную оценку.
Изменения в микроструктуре нитрата этиламмония при добавлении нитрата алюминия
Как уже упоминалось, основной целью данной работы было выявление возможной связи между возникающей при добавлении соли перестройкой локальной структуры и процессами молекулярной подвижности. Для описания микроструктуры рассматриваемых систем были рассчитаны 16 функций радиального распределения (ФРР). Необходимо отметить, что существенных отличий в виде всех рассмотренных ФРР, полученных с использованием разных модельных потенциалов для нитрат-аниона, не наблюдалось.
Анализ данных моделирования показал:
1. Вид ФРР между атомами иона ЭА+ (атом углерода групп CH2 и CH3 – атом углерода групп CH2 и CH3, атом углерода группы CH3 – атом азота группы NH3, атом азота группы NH3 – атом азота группы NH3, атом азота группы NH3 – атом углерода групп CH2 и CH3) практически не меняется с добавлением нитрата алюминия. Это говорит о том, что добавление соли в диапазоне концентраций до 10 мольных процентов не приводит к каким-либо существенным изменениям в локальной упорядоченности катионов ЭА+.
2. В случае ФРР между атомами ионов ЭА+ и нитрат-аниона (атом азота в ${\text{NO}}_{3}^{ - }$ – атом углерода группы CH3, атом кислорода в ${\text{NO}}_{3}^{ - }$ – атом углерода группы CH3, атом азота в ${\text{NO}}_{3}^{ - }$ – атом азота группы NH3, атом кислорода в ${\text{NO}}_{3}^{ - }$ – атом азота группы NH3), частично представленных на рис. 2, наблюдается уменьшение интенсивности первого пика ФРР с ростом мольной доли нитрата алюминия. Это говорит о том, что локальные изменения связаны c перераспределением нитрат-анионов. Эти данные согласуются с результатами работы [17].
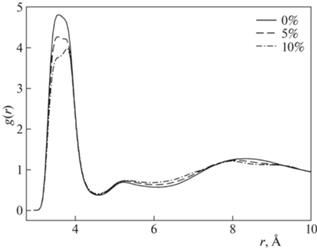
3. ФРР (атом азота в ${\text{NO}}_{3}^{ - }$ – атом азота в ${\text{NO}}_{3}^{ - }$) представленные на рис. 3, наглядно демонстрируют существенные изменения в относительном положении нитрат-анионов при добавлении соли. Наличие в системе трехзарядных ионов алюминия приводят к существенному увеличению упорядоченности нитрат-анионов в рассматриваемой системе, что проявляется в увеличении интенсивности пиков соответствующих ФРР. Наблюдаемые изменения хорошо согласуются с фактом существенного замедления вращательной переориентации нитрат-аниона.
Рис. 3.
ФРР атом азота в ${\text{NO}}_{3}^{ - }$ – атом азота в ${\text{NO}}_{3}^{ - }$. Потенциал I.

4. Расположение ионов алюминия относительно иона ЭА+, описываемое ФРР (атом алюминия – атом углерода группы CH3, атом алюминия – атом азота NH3-группы) отражено на рис. 4 и 5. Катионы Al3+ и ЭА+ испытывают электростатическое отталкивание и оказывают слабое влияние друг на друга. ФРР типа ЭА+ – Al3+ не изменяется значительно (что также продемонстрировано в работе [17]), поскольку ион алюминия не может проникнуть в ближайшее окружение катиона ЭА+. При этом многозарядный ион алюминия выстраивает около себя оболочку из нитрат-анионов, в том числе и входящих в окружение ЭА+, что приводит к замедлению вращательной переориентации ${\text{NO}}_{3}^{ - }$.


Таким образом, было промоделировано методом МД три системы: чистый ЭАН и две смеси данной ИЖ с нитратом алюминия (5 и 10 мольных процентов). Моделирование было выполнено для трех разных наборов параметров потенциала нитрат-аниона. В работе рассчитаны следующие характеристики: плотность модельных систем, различные функции радиального распределения, величины коэффициентов самодиффузии, времена переориентации внутримолекулярных векторов иона ${\text{NO}}_{3}^{ - }$.
Оценка величины коэффициентов самодиффузии показала, что с увеличением мольной доли нитрата алюминия, трансляционное движение в системе замедляется, но эффект выражен относительно слабо. Продемонстрировано, что значения рассмотренных наборов потенциальных параметров для нитрат-аниона влияют на величину коэффициента самодиффузии, но при этом для всех вариантов порядок величин тот же. Кроме того, была проведена оценка времен переориентации нитрат-аниона. Показано, что с увеличением концентрации соли замедляются процессы переориентации нитрат-аниона, что приводит к уменьшению общей диффузии в рассматриваемых системах.
Показано также, что добавление нитрат алюминия в ИЖ слабо меняет вид функций радиального распределения типа: катион ЭА+ – катион ЭА+, Al3+ – катион ЭА+. Данный факт свидетельствует о том, что ион алюминия не проникает в ближайшее окружение катиона ИЖ, т.е. присутствие Al3+ не оказывает существенного влияния на ближайших соседей катиона ЭА+. Тогда как для функций радиального распределения типа нитрат-анион – нитрат-анион при добавлении нитрата алюминия характерно появление ярко выраженного второго пика, что свидетельствует о том, что при добавлении соли Al(NO3)3 возникает упорядочение структуры.
Список литературы
Ionic Liquids – Classes and Properties / Handy, S. Ed.: InTech, 2011. 344 p.
Ionic Liquids in Synthesis / Wasserscheid P., Welton T. Ed.: Wiley-VCH: Weinheim, 2008. 776 p.
Ionic Liquids / Kirchner B., Clare B., Eds.: Topics in current chemistry; Springer Verlag: Heidelberg; New York, 2009. 345 p.
Electrochemical Aspects of Ionic Liquids, Second Edition / Ohno H., Ed.: Wiley: Hoboken, N.J, 2011. 504 p.
Ghandi K. // Green Sustain. Chem. 2014. V. 4. № 1. P. 44. https://doi.org/10.4236/gsc.2014.41008
MacFarlane D.R., Forsyth M., Howlett P.C. et al. // Acc. Chem. Res. 2007. V. 40. № 11. P. 1165. https://doi.org/10.1021/ar7000952
Armand M., Endres F., MacFarlane D.R. et al. // Nat. Mater. 2009. V. 8. № 8. P. 621. https://doi.org/10.1038/nmat2448
Lu X., Burrell G., Separovic F. et al. // J. Phys. Chem. B. 2012. V. 116. № 30. P. 9160. https://doi.org/10.1021/jp304735p
Fedorov M.V., Kornyshev A.A. // Chem. Rev. 2014. V. 114. № 5. P. 2978. https://doi.org/10.1021/cr400374x
Menne S., Pires J., Anouti M. et al. // Electrochem. Commun. 2013. V. 31. P. 39. https://doi.org/10.1016/j.elecom.2013.02.026
Mayrand-Provencher L., Lin S., Lazzerini D. et al. // J. Power Sources. 2010. V. 195. № 15. P. 5114. https://doi.org/10.1016/j.jpowsour.2010.02.073
Timperman L., Skowron P., Boisset А. et al. // Phys. Chem. Chem. Phys. 2012. V. 14. P. 8199. https://doi.org/10.1039/C2CP40315C
Timperman L., Galiano H., Lemordant D. et al. // Electrochem. Commun. 2011. V. 13. № 10. P. 1112. https://doi.org/10.1016/j.elecom.2011.07.010
Timperman L., Béguin F., Frackowiak E. et al. // J. Electrochem. Soc. 2014. V. 161. № 3. P. A228. https://doi.org/10.1149/2.016403jes
Salanne M. // Top. Curr. Chem. 2017. V. 375. № 3. P. 63. https://doi.org/10.1007/s41061-017-0150-7
Matsuoka H., Nakamoto H., Susan M.A.B.H. et al. // Electrochim. Acta. 2005. V. 50. № 19. P. 4015. https://doi.org/10.1016/j.electacta.2005.02.038
Gomez-Gonzalez V., Docampo-Alvarez B., Montes-Campos H. et al. // Phys. Chem. Chem. Phys. 2018. V. 20. № 28. P. 19071. https://doi.org/10.1039/C8CP02933D
Lyubartsev A.P., Laaksonen A. // Comput. Phys. Commun. 2000. V. 128. № 3. P. 565. https://doi.org/10.1016/S0010-4655(99)00529-9
Nose S. // Mol. Phys. 1984. V. 52. № 2. P. 255. https://doi.org/10.1080/00268978400101201
Martyna G.J., Tobias D.J., Klein M.L. // J. Chem. Phys. 1994. V. 101. № 5. P. 4177. https://doi.org/10.1063/1.467468
Martyna G.J., Tuckerman M.E., Tobias D.J. et al. // Mol. Phys. V. 87. № 5. P. 1117. https://doi.org/10.1080/00268979600100761
Umebayashi Y., Chung W.-L., Mitsugi T. et al. // J. Comput. Chem. Jpn. 2008. V. 7. № 4. P. 125. https://doi.org/10.2477/jccj.H2013
Choe J., Kim K., Chang S. // Bull. Korean Chem. Soc. 2000. V. 21. P. 200.
Mendez-Morales T., Carrete J., Cabeza O. et al. // J. Phys. Chem. B. 2014. V. 118. № 3. 761. https://doi.org/10.1021/jp410090f
Gómez-González V., Docampo-Álvarez B., Cabeza O. et al. // J. Chem. Phys. 2015. V. 143. P. 124507. https://doi.org/10.1063/1.4931656
Gómez-González V., Docampo-Álvarez B., Otero-Mato J. et al. // Phys. Chem. Chem. Phys. 2018. V. 20. P. 12767. https://doi.org/10.1039/C8CP01180J
Ebner C., Sansone R., Hengrasmee S. et al. // Int. J. Quant. Chem. 1999. V. 75. P. 805. https://doi.org/10.1002/(SICI)1097-461X(1999)75:4/5<805::AID-QUA45>3.0.CO;2-Y
Megyes T., Balint S., Peter E. et al. // J. Phys. Chem. B. 2009. V. 113. № 13. P. 4054. https://doi.org/10.1021/jp806411c
Laaksonen A., Kovacs H. // Can. J. Chem. 1994. V. 72. P. 2278. https://doi.org/10.1139/v94-290
Faro T.M.C., Thim G.P., Skaf M.S. // J. Chem. Phys. 2010. V. 132. P. 11450. https://doi.org/10.1063/1.3364110
Rappé A., Casewit C., Colwell K. et al. // J. Am. Chem. Soc. 1992. V. 114. № 25. P. 10024. https://doi.org/10.1021/ja00051a040
Martínez J.M., Pappalardo R.R., Marcos E.S. // J. Am. Chem. Soc. 1999. V. 121. № 13. P. 3175. https://doi.org/10.1021/ja9830748
Spångberg D., Hermansson K. // J. Chem. Phys. 2004. V. 120. P. 4829. https://doi.org/10.1063/1.1641191
Wasserman E., Rustad J.R., Xantheas S.S. // J. Chem. Phys. 1997. V. 106. P. 9769. https://doi.org/10.1063/1.473866
Hofer T.S., Randolf B.R., Rode B.M. // Phys. Chem. Chem. Phys. 2005. V. 7. P. 1382. https://doi.org/10.1039/B417491G
Lauenstein A., Hermansson K., Lindgren J. et al. // Int. J. Quant. Chem. 2000. V. 80. P. 892.https://doi.org/10.1002/1097-461X(2000)80:4/5<892::AID-QUA39>3.0.CO;2-Q
Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. // J. Comput. Phys. 1977. V. 23. № 3. P. 327. https://doi.org/10.1016/0021-9991(77)90098-5
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии