

Нефтехимия, 2019, T. 59, № 6-1, стр. 696-700
Аэробное жидкофазное окисление пара-трет-бутилкумола до гидропероксида
Е. М. Яркина 1, Е. А. Курганова 1, А. С. Фролов 1, Н. В. Лебедева 1, Г. Н. Кошель 1, *
1 Ярославский государственный технический университет
150000 Ярославль, Россия
* E-mail: koshelgn@ystu.ru
Поступила в редакцию 17.12.2018
После доработки 01.07.2019
Принята к публикации 02.07.2019
Аннотация
Изучена реакция аэробного жидкофазного окисления пара-трет-бутилкумола (ПТБК) в присутствии N-гидроксифталимида (N-ГФИ). Установлено, что вплоть до конверсии углеводорода (УВ) 35–50% селективность образования третичного гидропероксида ПТБК составляла более 95–98%. Показана целесообразность использования этой реакции в процессе совместного получения ПТБК и ацетона. Рассмотрен механизм процесса.
Аэробное жидкофазное окисление изопропилбензола и его метильных производных (цимолов, изопропилксилолов, триметилкумолов) до гидропероксидов (ГП) изучено достаточно подробно и составляет основу промышленных методов получения крупнотоннажных продуктов нефтехимического синтеза – фенола, крезолов, ксиленолов [1, 2].
В то же время, информация о жидкофазном окислении изопропильных производных алкилароматических УВ с большими алкильными заместителями в научно-технической литературе отсутствует. В этой связи изучение закономерностей жидкофазного окисления указанных УВ представляет научный и практический интерес, так как позволяет расширить сферу использования процессов жидкофазного окисления в синтезе высших алкилфенолов окислительным методом. Решению этой задачи в значительной степени посвящена настоящая работа.
В качестве объекта для изучения закономерностей жидкофазного окисления изопропильных производных алкилароматических УВ с большими алкильными заместителями был выбран пара-трет-бутилкумол (ПТБК). Дело в том, что в случае эффективного проведения процесса окисления ПТБК до третичного ГП (трет-ГП) открывается реальная возможность получения пара-трет-бутилфенола (ПТБФ) методом, аналогичным “Кумольному” способу синтеза фенола и ацетона. Окислительный метод получения ПТБФ состоит из трех стадий: синтез ПТБК алкилированием кумола изобутиленом или трет-бутиловым спиртом, его окисление до гидропероксида и кислотное разложение последнего в ПТБФ и ацетон по схеме:

Очевидно, что предлагаемый способ синтеза ПТБФ имеет существенные преимущества перед методом получения ПТБФ алкилированием фенола изобутиленом [2–4], так как он базируется на доступном нефтехимическом сырье (изопропилбензоле), хорошо апробированной технологии синтеза фенола и ацетона “Кумольным” методом, исключает образование м- и п-изомеров трет-бутилфенола. Сфера применения ПТБФ – ценного продукта основного органического и нефтехимического синтеза – постоянно расширяется, охватывая производство антиоксидантов, пестицидов, каучуков, лаков, красок, макроциклических соединений (каликсаренов) и, в последнее время, фармацевтических препаратов [2, 3].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовался ПТБК с содержанием основного вещества 98.5%, полученный алкилированием кумола трет-бутиловым спиртом по известной методике [4], и имеющий следующие константы: Ткип = 217°С. ЯМР 1Н (400 МГц), δ, м.д. (J, Гц): 7.28 д (2H (аром.), H-2, H-6, 3J = 8.1), 7.14 д (2H (аром.), H-3, H-5, 3J = 8.1), 2.8–2.9 септет (1H, СH (изопропил)); 1.25 с (9H, C(CH3)3), 1.18 д (6H, 2СН3 (изопропил), 3J = 6.8). Спектры записаны с использованием ЯМР-спектрометра Bruker DRX 400 (400.4 МГц). В качестве растворителя использована смесь ДМСО-d6-CCl4. В качестве внутреннего стандарта использовали тетраметилсилан. ИК-спектр ПТБК: 3000 см–1 (С–Н-связи в ароматическом кольце), 1611.94, 1513.85 см–1 (С=С-связи бензольного кольца), 827.54 см–1 (замещение в бензольном кольце), симметричные колебания 1393.87, 1362.96, и 2869.76 см–1 и антисимметричные 1462.86 см–1 (δ СН3), 829.73 см–1 (1,4 замещение), 1709.09 и 1016.02 см–1 (ν О–Н).
Окисление ПТБК проводили при атмосферном давлении на установке проточно-замкнутого типа [4]. Оксидат анализировали на содержание ГП методом иодометрического титрования [4]. В качестве катализатора использовали N-гидроксифталимид (и его производные), полученные взаимодействием фталевого ангидрида или его производного и гидроксиламина солянокислого по известной методике [4].
Полученный в работе ГП ПТБК – белое кристаллическое вещество, Tпл = 72°С. Спектр ЯМР 1Н (400 МГц), δ, м.д. (J, Гц): 7.42 м (4H, 4CH (аром.)), 7.28 с (1H, O–O–H), 1.63 д (6H, 2CH3 (изопропил), 4J = 1.7), 1.35 д (9H, 3CH3, 4J = 1.1).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Жидкофазное окисление ПТБК первоначально было изучено с использованием в качестве инициатора ГП ПТБК в интервале температур от 100 до 120°С. Из табл. 1 видно, что изменение условий проведения процесса (температуры, продолжительность, концентрации инициатора) не позволяет достичь конверсии ПТБК более 20–25% при селективности образования ГП не выше 80–85%.
Таблица 1.
Влияние различных факторов на конверсию ПТБК и селективность образования ГП ПТБК
| Катализатор/ инициатор | Содержание катализатора/ инициатора, мас. % | Время, мин | Температура, °С |
Содержание ГП ПТБК
в продуктах окисления, мас. % |
Селек-тивность образования ГП ПТБК, % | Конверсия ПТБК, % |
|---|---|---|---|---|---|---|
| ГП ПТБК | 0.07 | 60 | 100 | 6.7 | 89.0 | 7.5 |
| ГП ПТБК | 0.07 | 60 | 110 | 9.2 | 88.3 | 10.4 |
| ГП ПТБК | 0.07 | 60 | 120 | 12.9 | 87.1 | 14.8 |
| ГП ПТБК | 0.07 | 90 | 120 | 19.0 | 85.0 | 22.4 |
| ГП ПТБК | 2.0 | 60 | 120 | 20.5 | 79.3 | 25.9 |
| N-ГФИ | 2.0 | 75 | 100 | 10.0 | 97.1 | 10.3 |
| N-ГФИ | 2.0 | 75 | 110 | 21.1 | 97.7 | 21.6 |
| N-ГФИ | 2.0 | 75 | 120 | 31.9 | 95.6 | 33.4 |
| N-ГФИ | 2.0 | 75 | 130 | 36.8 | 95.2 | 39.5 |
| N-ГФИ | 2.0 | 75 | 140 | 41.4 | 94.2 | 43.9 |
| 4-метил-N-ГФИ | 2.0 | 30 | 110 | 23.1 | 98.1 | 23.5 |
| 4-метил-N-ГФИ | 2.0 | 30 | 120 | 35.1 | 93.7 | 37.4 |
| 4-метил-N-ГФИ | 2.0 | 30 | 130 | 40.1 | 92.5 | 43.4 |
| 4-фенил-N-ГФИ | 2.0 | 40 | 110 | 17.0 | 99.1 | 17.2 |
| 4-фенил-N-ГФИ | 2.0 | 40 | 120 | 33.1 | 97.5 | 30.9 |
| 4-фенил-N-ГФИ | 2.0 | 40 | 130 | 36.0 | 95.2 | 35.7 |
| N-ГФИ+ ГП ПТБК | 2.0 + 0.07 | 75 | 130 | 52.1 | 95.6 | 54.5 |
| N-ГФИ | 2.0 | 75 | 120 | 22.4 | 96.4 | 23.2 |
| N-ГФИ* | 2.0 | 75 | 120 | 22.6 | 96.7 | 23.4 |
| N-ГФИ | 1.0 | 15 | 120 | 2.4 | 98.9 | 2.4 |
| N-ГФИ | 2.0 | 15 | 120 | 4.5 | 99.0 | 4.5 |
| N-ГФИ | 3.0 | 15 | 120 | 7.0 | 98.7 | 7.1 |
| N-ГФИ | 4.0 | 15 | 120 | 10.4 | 98.5 | 10.6 |
Увеличить скорость окисления ПТБК и селективность образования трет‑ГП ПТБК до 90–95% удалось за счет применения N-гидроксифталимида (N-ГФИ), использование которого позволяет существенно повысить эффективность окисления втор-алкиларенов до ГП [1, 5–10].
Как видно из табл. 1 и рис. 1, в присутствии 2.0 мас. % N-ГФИ повышение температуры со 100 до 140°С приводит к равномерному увеличению скорости окисления ПТБК, однако при температуре 130–140°С через 1 ч реакции начинается разложение ГП, что приводит к снижению селективности образования ГП. Максимальная конверсия углеводорода (40–43%) достигается за 75 мин реакции при температуре 140°С. Селективность образования трет-ГП ПТБК во всех экспериментах была не ниже 95–97%.Таким образом, использование N-ГФИ позволило повысить конверсию ПТБК в 2–3 раза, а селективность образования ГП ПТБК – с 80–85 до 95–98%, по сравнению с окислением в присутствии инициатора ГП ПТБК.
Рис. 1.
Влияние температуры и структуры катализатора на реакцию жидкофазного окисления ПТБК в присутствии N-ГФИ. Содержание катализатора 2.0 мас.%. Катализатор: 1–5 – N-ГФИ, 6 – 4-фенил-N-ГФИ, 7 – 4-метил-N-ГФИ. Температура,°С: 1 – 100; 2 – 110; 3, 6, 7– 120; 4 – 130; 5 – 140.
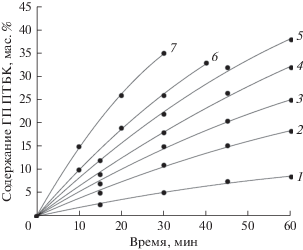
Из данных, представленных в табл. 1, видно также, что повышение концентрации катализатора с 1.0 до 4.0 мас. % при температуре 120°С позволяет увеличить скорость образования ГП ПТБК с 8–10 до 35–40% за один час. Однако использование N-ГФИ в количестве 1–1.5 мас. % вполне достаточно для эффективного окисления ПТБК в ГП.
Совместное применение катализатора N-ГФИ и ГП ПТБК в качестве инициатора, как видно из табл. 1, также оказывает положительное влияние на параметры окисления ПТБК. Так, при температуре 130°С, содержании N-ГФИ 2 мас. %, содержании ГП ПТБК 0.07 мас. % удалось за 75 мин достичь конверсии УВ свыше 50 мас. % при селективности образования целевого продукта 95.6%.
При охлаждении продуктов окисления до комнатной температуры примерно 90–95% N-ГФИ, содержащегося в оксидате, выпадает в осадок. При этом состав и качественные характеристики N-ГФИ не меняются, что позволяет использовать его в процессе окисления многократно. Так, при использовании выпавшего в осадок N-ГФИ при жидкофазном окислении ПТБК его каталитическая активность не снижается как минимум в течение 5 циклов.
Повысить эффективность окисления втор-алкилароматических УВ можно за счет использования N-ГФИ, модифицированного введением в его структуру электронодонорных заместителей [11]. Этот подход был использован и при окислении ПТБК. Из табл. 1 и рис. 1 видно, что наличие в молекуле N-ГФИ электронодонорных заместителей (метильных, фенильных групп) способствует повышению скорости окисления ПТБК. Так, например, в присутствии 4-метил-N-ГФИ при температуре 120°С удается достичь содержания ГП ПТБК в 34–40% (с селективностью выше 95%) вдвое быстрее, чем при использовании незамещенного N-ГФИ.
Достижение высокой концентрации ГП ПТБК при исключительно высокой селективности его образования по-видимому связано с протеканием комплекса превращений с участием фталимидного катализатора, который приводит к необрываемому продолжению цепи N-гидроксифталимидными радикалами, по схеме [5]:
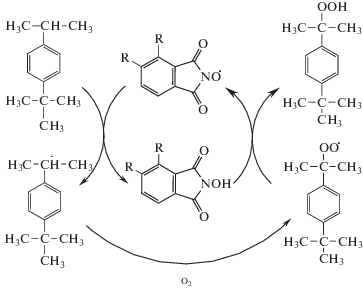
Схема. Схема реакции аэробного жидкофазного окисления пара-трет-бутилкумола в присутствии N-гидроксифталимида.
Таким образом, одновременно с инициирующим и каталитическим эффектом влияние N‑ГФИ сводится к увеличению длины цепи, что естественно приводит к уменьшению образования побочных продуктов по сравнению с образованием целевого ГП.
ЗАКЛЮЧЕНИЕ
Изучено влияние основных технологических параметров, таких как температура, концентрация и структура катализатора, продолжительность реакции на показатели жидкофазного окисления п-ара-трет-бутилкумола до гидропероксида.
Аэробное окисление пара-трет-бутилкумола проведенное в присутствии N-гидроксифталимида и его производных как индивидуально, так и совместно с гидропероксидным инициатором, позволяет достичь конверсии УВ 40–55% при селективности образования ГП на уровне 95–98% за 60–75 мин реакции.
Подтверждена возможность повторного использования N-ГФИ без снижения его каталитической активности. Рассмотрен механизм процесса.
Список литературы
Курганова Е.А., Сапунов В.Н., Кошель Г.Н., Фролов А.С. // Известия РАН. Серия хим. 2016. № 2. С. 2115.
Апостолов С.А., Бабаш С.Е., Белкина Е.И., Беренц А.Д. Новый справочник химика и технолога: сырье и продукты промышленности органических и неорганических веществ. Санкт-Петербург: АНО МПО “Профессионал”, 2002. 988 с.
Saha M., Hossain M.K., Ashaduzzama M., Afroza S.T., Galib M., Sharif N. // Bangladesh J. Sci. Ind. Res. 2009. V. 44. P. 131.
Frolov A.S., Kurganova E.A., Koshel’ G.N., Nesterova T.N. // European J. of Analytical and Applied Chemistry. 2015. V. 1. P. 16.
Ishii Y., Sakaguchi S., Iwahama T. // Adv. Synth. Catal. 2001. V. 343. № 5. P. 393
Kasperczyk K., Orlińska B., Zawadiak J. // Cent. Eur. J. Chem. 2014. V. 12. № 1. P. 1176.
Fukuda O., Sakaguchi S., Ishii Y. // Adv. Synth. Catal. 2001. V. 343. № 8. P. 809.
Melone L., Prosperini S., Ercole G., Pastor N., Punta C. // J. Chem. Technol. Biotechnol. 2014. V. 89. № 9. P. 1370.
Курганова Е.А., Фролов А.С., Кошель Г.Н., Нестерова Т.Н., Шакун В.А., Мазурин О.А. // Нефтехимия. 2018. Т. 58. № 3. С. 357.
Orlińska B., Zawadiak J. // Reac. Kinet. Mech. Cat. 2013. V. 110. P. 15.
Boring E., Geletii Y.V., Hill C.L. // J. of Molecular Catalysis A: Chemical. 2001 V. 176. P. 49.
Дополнительные материалы отсутствуют.