

Нефтехимия, 2020, T. 60, № 6, стр. 727-744
Перспективы развития катализаторов и процессов получения этиленоксида (обзор)
Л. Г. Пинаева 1, *, А. С. Носков 1
1 Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, Россия
* E-mail: pinaeva@catalysis.ru
Поступила в редакцию 24.05.2020
После доработки 03.07.2020
Принята к публикации 10.07.2020
Аннотация
Рассмотрены основные этапы развития промышленных Ag–Al2O3-катализаторов газофазного эпоксидирования этилена. Наибольшее внимание уделяется методам приготовления, направленным на повышение производительности каталитических систем последних поколений. Обобщены также результаты исследований альтернативных гетерогенных систем на основе силикатов, модифицированных Ti, Nb, W, для жидкофазного эпоксидирования этилена пероксидом водорода. Сделан анализ причин и способов решения проблемы их низкой активности/производительности и стабильности, а также оценены направления повышения эффективности использования Н2О2.
Этиленоксид (ЭО) – один из основных интермедиатов в нефтехимической промышленности, до 70% процентов которого используется далее в производстве моно-, ди- и три-этиленгликолей [1, 2]. Другие основные продукты, получаемые из ЭО, включают этоксилаты, этаноламины, гликолевые эфиры, полиэтиленгликоль и полиэфирные полиспирты. Объем ежегодного мирового производства ЭО составил 19 млн т за 2009 г. [3] с оценкой дальнейшего роста около 3% в год до 26–27 млн т в 2017–2018 гг. [4]. Однако уже в 2016 г. было произведено около 30 млн т ЭО [5], в то время как мировые мощности по его производству составили 34.5 млн т/год [6]. Известно, что в 2018 г. ЭО обеспечил около 16% переработки от 164 млн т произведенного в мире этилена [7], что соответствует значению примерно 34 млн т. В стоимостном выражении объем рынка ЭО в 2017 г. составил около 44 млрд долл. с перспективой роста к 2023 г. до примерно 58 млрд долл. (4.9%/год) [8]. Есть данные о более быстром совокупном среднегодовом темпе роста 6.3% [9], обусловленном постоянно растущим спросом на полиэфирные волокна и полиэтилентерефталат, обеспечивающий около 2/3 мирового потребления моноэтиленгликоля.
В РФ производство ЭО на основе кислородного процесса компании Scientific Design сосредоточено на “СИБУР-Нефтехим”, г. Дзержинск (266–300 тыс. т/год [10]) и ПАО “Нижнекамскнефтехим”, г. Нижнекамск (215 тыс. т/год). Дальнейшего увеличения объемов производства в ближайшие годы не планируется, поскольку имеющиеся мощности в 2014 г. были загружены на 77% и 88%, соответственно.
Весь рынок ЭО распределен примерно между 20 основными участниками, среди которых необходимо отметить как компании-производители, так и компании-разработчики процессов/катализаторов: BASF, Dow Chemical, Indorama Ventures, INEOS Group Holdings PLC, Lyondell Basell Industries, Reliance Industries Limited, Saudi Basic Industries Corporation (SABIC), Shell Global, Sinopec Corp., PetroChina.
Все современные промышленные процессы производства ЭО основаны на газофазном окислении этилена в трубчатых реакторах, проводимом при температурах 220–300°C и давлении 1–3 МПа с использованием катализаторов на основе серебра, нанесенного на Al2O3. Для увеличения выхода ЭО используется газовое промотирование этил/винил хлоридом (до 6 м.д.), схема с рециклом (конверсия этилена за один проход не превышает 20%), а также добавление в смесь метана (2–40 об. %, в зависимости от технологии) [11]. Такие технологии, изначально лицензированные компаниями Shell, Scientific Design (SD), UCC, Japan Catalytic, Snam Progetti и Hüls, очень близки по сути и отличаются только окислителем: в одном случае в качестве окислителя используется кислород воздуха, а в другом – чистый кислород. Поскольку стоимость ЭО на 80% определяется стоимостью этилена [12], в последние годы “воздушные” процессы, в которых из-за технологических особенностей реализации максимально достижимые значения выхода значительно ниже, чем в “кислородных” (80 и 87–90% соответственно), практически вышли из употребления, в том числе и на предприятиях РФ.
В настоящее время подавляющая часть производителей ЭО использует технологии, разработанные тремя компаниями: Dow Chemical, Scientific Design (приобретена в 2003 г. SABIC) и Shell Global [13–15]. Компания Dow Chemical предлагает технологию METEORTM, согласно которой производимый ЭО на следующей стадии перерабатывается в моноэтиленгликоль (МЭГ). В 2016 г. общее число проданных лицензий обеспечивало ежегодное производство до 4.5 млн т ЭО. Компания Shell Global разработала 2 технологии – Shell MASTER и Shell OMEGA – для производства только ЭО или с последующим получением МЭГ, соответственно [14]. Согласно данным, приведенным в [12], в 2010 г. до 30% ЭО производилось на заводах, построенных по лицензии Shell. Аналогично, как ЭО, так и МЭГ (более 100 заводов [16]) может производиться по разным вариантам технологии Scientific Design’s Ethylene Oxide/Ethylene Glycol (EO/EG) [15].
Объем общемирового ежегодного потребления катализаторов эпоксидирования этилена можно оценить только приблизительно, т.к. на заводах, построенных в разные годы, до настоящего времени эксплуатируются катализаторы нескольких поколений с существенно разным сроком жизни (см. ниже). Исходя из наших оценок, основанных на данных по объемам загрузок реакторов различной производительности, плотности и усредненных сроках пробега катализатора, а также данных, приведенных в [17, 18], для производства 27 млн т ЭО требуется 4.8–6.8 тыс. т катализатора в год. В стоимостном выражении мировой рынок катализаторов производства ЭО еще в 2011 г. составил 170 млн долл. [19]. Суммарная ежегодная потребность в катализаторах для всех производств в РФ составляет около 110 т/год. В случае запуска в 2022 г. в Восточной НХК производства МЭГ из ЭО мощностью 700 тыс. т [20, 21] потребность в катализаторах ЭО удвоится.
ПРОМЫШЛЕННЫЕ КАТАЛИЗАТОРЫ ЭПОКСИДИРОВАНИЯ ЭТИЛЕНА
Промышленные катализаторы, независимо от компании-производителя, представляют собой частицы серебра размером 50–100 нм, которые характеризуются максимальной активностью и селективностью в реакции эпоксидирования на единицу веса [22–25], нанесенные на α-Al2O3 и промотированные соединениями Cs и Re [11]. Анализ патентов показал, что компании Scientific Design и Shell Global используют в своих процессах катализаторы (торговые марки SynDox и CRI соответственно), содержащие ~15 мас. % Ag, в то время как в Dow Chemical (торговая марка METEOR™ EO-RETRO [26]) – c содержанием ~27–30 мас. % Ag [27–30]. При близости технологических режимов, катализаторы Shell Global можно использовать на заводах, построенных по технологии Scientific Design, и наоборот. Выбор α-Al2O3 обусловлен низкой поверхностью (<1 м2/г), благодаря чему минимизируется концентрация поверхностных гидроксильных групп, которые отвечают за вторичные превращения целевого продукта [31–36].
Хронология развития катализаторов эпоксидирования (на примере компании Shell)
Хронология возникновения и развития различных семейств катализаторов для процессов компании Shell, разрабатываемых компанией CRI/Criterion Inc., а также характеризующие их значения средней селективности по ЭО за полный цикл пробега приведены на рис. 1. Катализаторы SynDox, производимые для процессов Scientific Design компанией Clariant, развивались в таком же порядке и примерно в тех же временных интервалах. Все серии катализаторов производятся до настоящего времени, чтобы обеспечивать эксплуатацию существующих до настоящего времени установок получения ЭО [37].
Рис. 1.
Хронология возникновения и развития различных серий катализаторов торговой марки CRI для технологий компании Shell по [38].
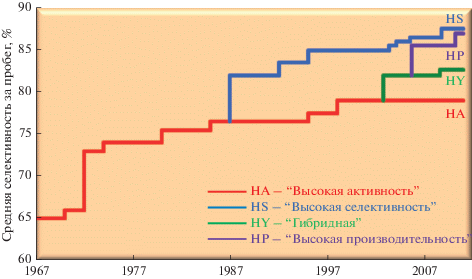
Примерно в 1972 г. Shell были разработаны концептуально новые высокоактивные катализаторы (серия “высокая активность”, HA), модификация которых впоследствии привела к увеличению селективности по ЭО примерно до 79% [38]. Такие катализаторы (серия “HAC”, в аббревиатуре Scientific Design Со.), включают Cs в качестве единственного промотора [39].
Цезий, добавляемый в количестве, не превышающем 1000 м.д., используется в качестве промотора практически во всех последующих сериях промышленных катализаторов эпоксидирования разных производителей [28, 30, 40–45]. Общепринятой является точка зрения, что его роль заключается в уменьшении концентрации кислых центров на поверхности носителя, благодаря чему замедляются реакции изомеризации ЭО [46–54] или снижается прочность его связи с поверхностью [47, 55, 56]. В некоторых работах показано, что на модифицированной Cs-поверхности возрастает дисперсность частиц Ag [57], а также достигается более однородное распределение частиц по размерам [58]. С другой стороны, результаты исследований, проведенных на монокристаллах [59] или Ag–α-Al2O3 [58, 60], свидетельствуют о том, что Cs также воздействует непосредственно на серебро, снижая прочность связи предшественника ЭО с поверхностью, что способствует десорбции последнего. Авторы работы [61] считают, что Cs локализуется преимущественно на боковых стенках ступенек поверхности Ag, которые менее селективны в отношении эпоксидирования, чем плоские участки, и таким образом блокирует диссоциативную адсорбцию кислорода на них. Кроме того, Cs поддерживает высокую степень покрытия поверхности катализатора хлором в промышленных условиях при газофазном промотировании [62]. Возможно, поэтому улучшение селективности проявляется только при совместном промотировании цезием и хлором, а в отсутствие последнего лучше протекает полное окисление этилена [63] с участием поверхностных комплексов CsOx, генерирующих кислородные вакансии. На таких вакансиях в условиях реакции стабилизируются промежуточные частицы, ответственные за образование ацетальдегида [64].
В 1986 г. была разработана серия катализаторов “Высокая селективность” (HS) [65, 66], для которой начальная селективность по ЭО была повышена сразу на 6–7%, а средняя селективность за пробег достигла величины 82% и выше (рис. 1). Более высокое значение селективности в катализаторах серий CRI-HS и Syndox-HSC достигалось посредством введения, в дополнение к цезию, рения в виде высоковалентных оксосоединений [12] в количестве до 300 м.д. Необходимым условием увеличения селективности является последовательное нанесение, сначала Re, а затем – Cs и Ag [67]. Природа промотирующего действия Re не выяснена полностью, но показано [56, 67], что он изменяет электронное состояние поверхностных атомов серебра таким образом, что на них вместо нуклеофильного кислорода, отвечающего за активацию молекулы этилена, образуется кислород с более электрофильными свойствами, необходимый для образования ЭО [24, 25]. Именно поэтому активность высокоселективных катализаторов была значительно ниже, чем у катализаторов серий HA (HAC). Необходимость реализации процесса с использованием таких катализаторов при более высоких температурах (≥240°С вместо 200°С) приводила к их быстрой дезактивации, из-за чего длительность пробега сокращалась с 3–4 лет до 1.5–2 [37], а в отдельных случаях неприемлемая потеря активности снижала срок службы даже до 12 месяцев [39]. Для увеличения длительности срока эксплуатации таких катализаторов на вновь строящихся установках была снижена объемная скорость подачи сырья, улучшена очистка от СО2 газа, подаваемого по рециклу в реактор. Кроме того, практиковалось увеличение объемов реакторов или строительство дополнительных реакторов. Все это приводило к увеличению капитальных затрат на оборудование. Впоследствии введение в катализаторы CRI-HS и Syndox-HSC сопромоторов, представляющих собой высоковалентные оксоанионы как переходных металлов (Mo, W, Cr, Ti, Hf, Zr, V, Ta, Hf, Nb), так и неметаллов (S, P, B), привели к увеличению начальных и средних по циклу значений селективности до величины ≥90 и 87%, соответственно, а также длительности его работы, обусловленной большей активностью (рис. 1, 2) [68–70].
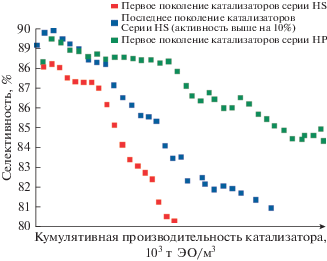
Рис. 2.
Промышленные данные, показывающие выгоду от использования новых поколений катализаторов CRI. Одна точка отвечает работе примерно в течение 1 мес (по [38]).
Гибридный катализатор (серия HY, в аббревиатуре Scientific Design Со – MSC) по сути являлся промежуточной версией с точки зрения активности и селективности между катализаторами серий HA и HS (рис. 1), обеспечивающей максимальное значение выхода и производительности в ходе пробега [37]. После проведения на старых заводах реконструкции системы очистки рецикловой смеси от СО2 [17, 18, 38] такие катализаторы демонстрируют значения начальной селективности, по меньшей мере, от 83 до 87% [39]. Известно, что для увеличения стабильности в катализаторах HS (HSC), а также HY (MSC) использовали добавки, вводимые как непосредственно на стадии формирования носителя, такие как оксиды хрома [71], Zn, так и в готовый носитель перед и одновременно с нанесением серебра, например Zn [41]. Компания Shell использует такие катализаторы на более чем 60 заводах.
Начиная c 2005 г., на рынок выведена серия катализаторов “Высокая производительность”, НР (рис. 1) [37]. Первый катализатор этой серии – S-888 – также приготовлен на базе систем Ag–Ce/Al2O3, модифицированных Re [12]. При близких к образцам серии HS начальной и средней за пробег селективности, у образцов серии НР заметно меньшая скорость дезактивации (рис. 2), которая обусловлена более высокой активностью. Это позволяет работать при более высоких расходах (или использовать реакторы с меньшим числом (на 30%) или длиной (на 20%) трубок) [38]. Производитель катализаторов гарантирует пробег в течение ~4 лет, более быстрый запуск и достижение стационарного состояния, а также бόльшую толерантность к высоким концентрациям СО2. Ниже будут проанализированы способы приготовления катализаторов, позволившие оптимизировать характеристики катализаторов последних поколений.
Для полноценной замены катализаторов Shell и Scientific Design Со в соответствующих процессах компания Sinopec, начиная с 1989 г., разработалa технологии производства более 11 катализаторов YS четырех аналогичных серий [72]:
– с высокой активностью (YS-7, средняя селективность по пробегу – 79.6%);
– c высокой селективностью (YS-8810, YS-9010, селективность по ЭО 89–91%);
– гибридные катализаторы (YS-8520, средняя селективность после 7 мес. работы 83.8%, а в целом по пробегу (3 года) – 82.8%);
– с высокой производительностью (YS-8830).
С учетом того, что подтвержденные промышленные характеристики всех серий сравнимы с показателями катализаторов CRI и Syndox [73–75], меньшая на 45% стоимость дает китайским производителям определенное преимущество при продажах. Так, заметный рост продаж катализаторов начался в 2011 г. после успешного пробега высокоселективного катализатора YS-8810 на заводе по производству МЭГ Sinopec Shanghai Petrochemical Company [74]. Всего к 2017 г. сделано более 54 загрузок катализаторов на 14 реакторах ЭО/ЭГ-заводов по всему миру [75], а общий объем продаж к 2017 г. превысил 3000 т [17, 18].
Носители для катализаторов с необходимыми свойствами производятся компанией Saint Gobain NorPro Corporation (торговая марка NorPro®) [76], являющейся правообладателем многих патентов на носители состава α-Al2O3 и катализаторы эпоксидирования на их основе [44, 77–81]. Повышенная активность в патентовании катализаторов эпоксидирования и носителей для таких катализаторов в последние 10 лет отмечается у BASF [82–87], хотя на рынке промышленные катализаторы данной компании отсутствуют.
Современное состояние производства катализаторов получения этиленоксида в Российской Федерации
На основе результатов работ, изложенных в [88, 89], в 1995 г. на ОАО “Казаньоргсинтез” было налажено производство катализатора “ЭТОКС-Ю” для воздушных процессов [90] на основе аминных комплексов серебра с добавками карбоната Cs и боратов Mg, Ca, Zn, нанесенных на мелкопористый кольцеобразный носитель. При меньшем на 14% содержании Ag, он характеризовался на 10% более высокой производительностью и селективностью (7–8%) по сравнению с ранее использовавшимся катализатором на шариковом носителе, для производства которого приходилось импортировать носитель и молочную кислоту. Более высокая устойчивость к воздействию контактных ядов, в частности сернистых соединений и ацетилена, позволила исключить из аппаратурного оформления ряд громоздких стадий очистки этилена, воздуха и увеличить срок его службы в 2–2.5 раза. Селективность по ЭО катализаторов “ЭТОКС” при несколько меньшем содержании серебра была сравнима с аналогичными образцами фирмы Shell (табл. 1). При этом их цена была на 60% ниже зарубежного аналога [91]. К 2001 г. мощность производства катализатора была увеличена почти в два раза (от 170 до 330 т в год), что обеспечивало потребность всей отрасли (ПАО “Казаньоргсинтез”, ПАО “Нижнекамскнефтехим”, ОАО “Салаватнефтеоргсинтез”). Последняя выпускаемая марка для воздушного процесса – катализатор “Этокс-111”, разработанный ООО НПП “Оксит”, который обеспечивал среднюю селективность на уровне 78–74% (первая ступень) и 70–62% (вторая ступень) при длительности эксплуатации 4–5 лет [92, 93]. После вывода из эксплуатации установок воздушных процессов к 2008 г. производство катализатора было остановлено. Была сделана попытка замены импортных катализаторов на запущенном на ПАО “Нижнекамскнефтехим” кислородном процессе. На лучших образцах в качестве промоторов, кроме традиционного Cs (0.02–0.2 мас. %), добавляли Ca, Zn, Сd или La (0.11–0.15%), а селективность по ЭО в лабораторном реакторе достигала 83.9–85% [94, 95], что было ниже, чем на разработанных к тому времени катализаторах Shell с добавками Re (86–87%) [96] (табл. 1). Кроме того, основное преимущество данного катализатора – меньшая стоимость – перекрывалось повышенным пылеобразованием и уносом серебра по сравнению с импортными образцами [92]. В настоящий момент производство катализаторов ЭО в РФ полностью отсутствует, исследования в данной области носят фрагментарный характер.
Таблица 1.
Сравнительные характеристики российских катализаторов и катализаторов Shell в одинаковом временном интервале (1988–2000 гг.)
| Содержание серебра, мас. % | Промоторы/сопромоторы/ комплексообразователь | Носитель | Cелективность по ЭО, %/Т, °С | Ссылка/год выхода патента/правообладатель |
|---|---|---|---|---|
| Воздушный процесс | ||||
| 12 | Cs (0.022%)/борат Zn (0.11%) или Cd (0.15%) или La (0.15%)/моноэтаноламин | α-Al2O3, SiC, <1 м2/г, пористость 30–60% | 80.2–80.5/219 | [89]/1992/РФ |
| Кислородный процесс | ||||
| 12 | Cs/борат Zn или Cd или La (0.15%)/моноэтаноламин | α-Al2O3, <1 м2/г, пористость 50–65% | 84–85.2/219 | [94]/1997/РФ |
| 12 | Cs (0.022%)/борат Zn (0.11%) или Ca (0.14%) или La (0.15%)/моноэтаноламин | α-Al2O3, SiC, кольца 5–8 мм, <1м2/г, пористость 30–60% | 83.5–83.9/219 | [95]/2002/РФ |
| 13–14 | Cs (0.06%) | α-Al2O3 | 82.9/217 | [96]/1995/Shell |
| Cs (0.07%) + Re (0.028%) | 87.2/249 | |||
Методы синтеза современных катализаторов
Способы получения катализаторов с частицами серебра оптимального размера 50–100 нм [22–25] хорошо известны. В подавляющем большинстве запатенованных способов приготовления используют метод пропитки распылением раствора на предварительно вакуумированный носитель с последующим удалением избыточного раствора. Может быть использована однократная пропитка раствором, содержащим исходные соли серебра и все необходимые промоторы, а также многократные пропитки (в том числе с промежуточной сушкой образцов), во время которых серебро может заводиться по частям, а введение промоторов – проводиться до, одновременно или после нанесения серебра [65, 66, 96–104]. В качестве исходных солей серебра могут использовать нитраты [105], ацетаты [106], оксалаты [107]. Однако еще в 1970-х гг. [108–110] было показано, что равномерно распределенные по поверхности носителя частицы Ag необходимых размеров получают только при использовании водных растворов комплексов на основе солей карбоновых кислот (прежде всего, оксалата) и органических аминов (прежде всего, этилендиамина, иногда – моноэтаноламина) с последующим нагреванием образцов в отсутствие водорода. Такие комплексы используют в подавляющем большинстве последующих патентов основных промышленных производителей носителей и катализаторов [42, 65, 66, 111–113], и только в отдельных случаях описывают применение водных растворов лактата серебра [114]. В свою очередь, оксалат серебра, как правило, получают непосредственно перед или одновременно с приготовлением комплексов с аминами [42] из нитрата [42, 115–117] или оксида [112, 113] серебра. Повышенная дисперсность серебра обеспечивается его восстановлением до металлического состояния в результате прокаливания в токе инертного газа [40, 41, 118] или воздуха [28, 30, 42, 56], в том числе в сложных температурных режимах.
Длительность пробега катализатора находится в прямой зависимости от его активности, определяемой концентрацией частиц серебра оптимального размера. Для ее увеличения предлагается использовать несколько способов:
– модифицирование поверхности α-Al2O3 с целью создания центров, на которых впоследствии будут формироваться и стабилизироваться наночастицы серебра;
– переход на носители с более высокой поверхностью;
– увеличение активной поверхности катализатора за счет регулирования формы гранул.
Формирование на поверхности носителя центров зародышеобразования для частиц активного компонента может достигаться как введением модифицирующей добавки на стадии его приготовления [45, 119], так и пропиткой уже готового носителя солями предшественников таких центров до стадии нанесения серебра [40, 41, 81, 118, 120, 121]. Так, повышение активности катализаторов (ΔT = = 9°С) без снижения селективности (~83.5%), а также прочности на раздавливание (более 100 Н/гранула) в 2 раза возможно при введении 0.01–3 мас. % соединений La и Si в состав носителя на стадии приготовления формовочной пасты на основе тригидрата оксида алюминия и псевдобемита [45]. В патенте [119] значимое увеличение селективности без снижения активности достигалось добавлением в формовочную массу до 1 мас. % различных соединений Cu, Zn, Ag, Au. При использовании в качестве добавок оксида меди или коллоидного золота возможно даже увеличение активности.
Положительный эффект модифицирования α-Al2O3 на активность/стабильность и селективность наблюдался при нанесении 0.56% СeО2 [120], а также Sn (из раствора SnCl2, содержащего этилендиамитетрауксусную кислоту) [121] или 150–1500 м.д. Zn (пропиткой под вакуумом водным раствором ацетата Zn c этилендиамином) [81]. В одной из самых последних заявок Scientific Design Company [122] центры зародышеобразования, представляющие собой изолированные центры (50–60%) и/или нанокластеры размером не более 1 нм, получают нанесением некаталитических (~0.1%) количеств серебра методом пропитки с последующими сушкой и прокаливанием образцов. Такие катализаторы характеризуются бóльшей (примерно на 1%) селективностью по ЭО, которая не снижается в течение как минимум 1500 ч работы при меньшей температуре (ΔТ = 5–7°С), по сравнению с образцами, на которые весь активный компонент наносится за одну пропитку. Увеличение активности и селективности возможно также при нанесении серебра двухкратной пропиткой с промежуточными сушкой и термообработкой [80, 123].
Принципиальная важность материала и пористой структуры носителя для катализатора эпоксидирования отмечалась уже 50 лет назад [124]. С одной стороны, высокая поверхность носителя способствует формированию максимального количества частиц серебра оптимального размера. Однако известно, что серебро, нанесенное на α-Al2O3 с удельной поверхностью Sуд менее 1 м2/г, более селективно превращает этилен в ЭО, чем в случае носителей на основе того же исходного материала, но характеризуемых Sуд в интервале 1.8–7.4 м2/г [125]. Дальнейшее увеличение поверхности Al2O3 от 20 м2/г до 78–104 м2/г приводило к снижение активности в реакции эпоксидирования до нуля [35, 126]. Это обусловлено увеличением концентрации на поверхности гидроксильных групп с кислыми свойствами, которые катализируют реакцию изомеризации этиленоксида в ацетальдегид. Последний, в свою очередь, быстро доокисляется до СО2 как на самих частицах серебра, так и на поверхности носителей состава γ-Al2O3, MgO, SiC, TiO2, Y2O3, CeO2, алюмосиликаты/цеолиты и ZrO2 [31, 34, 36]. Из всех носителей SiO2 и α-Al2O3 наименее активны в реакции превращения ЭО в ацетальдегид [36]. Во многих патентах особо отмечается важность контроля содержания Na в конечных катализаторах для обеспечения стабильности их работы [39]. Компания Scientific Design запатентовала способы отмывки поверхности и приповерхностных слоев уже готовых носителей от Na, которые включают обработку водой различной температуры [127].
Дополнительным фактором, ухудшающим показатели селективности катализаторов на основе носителей с высокой удельной поверхностью, является затрудненная диффузия ЭО в порах с нанометровыми характерными размерами, которая заметно увеличивает вероятность протекания его вторичных реакций, в том числе с участием локализованных в таких порах гидроксильных групп [35, 125, 128]. Для снижения их концентрации в алюмосиликатах осуществляли силанирование поверхности, что приводило к значительному увеличению скорости образования этиленоксида [49]. Также для получения однородных металлических нанесенных частиц при очень высоких концентрациях активного компонента и снижения диффузионного торможения в порах были разработаны мезопористые силикаты с бидисперсной структурой пор, сочетающие трехмерную структуру решеточных мезопор размером 3.5 нм с высокой текстурной (межчастичной) порозностью [129]. Такие катализаторы на основе носителей МСМ-41 и НМS благодаря формированию в них частиц Ag размером 20–50 нм при относительно низкой температуре (230°С) показывали более высокую активность в эпоксидировании и селективность, близкую к измеренной на непористом SiO2 и традиционном α-Al2O3. Однако при 300°С происходило резкое снижение селективности по ЭО [130]. В процессе эпоскидирования этилена необходимо поддержание примерно одинаковой конверсии за проход, и снижение активности катализатора по мере его старения компенсируется постепенным подъемом температуры во время пробега. Соответственно, сохранение необходимых значений селективности в максимально широком интервале температур является необходимым требованием к катализатору. Это ограничивает применение катализаторов на основе мезопористых силикатов.
В качестве носителей также был использован мезопористый SrTiO3, в котором, благодаря применению золь–гель метода и ПАВ-ов формировались поры с узким распределением по размерам [131]. По сравнению с другими нанокристаллическими перовскитоподобными носителями (MgTiO3, CaTiO3, SrTiO3 и BaTiO3) и α-Al2O3, при близком содержании (17.6 мас. %) и размерах частиц серебра, Ag–SrTiO3 демонстрировали заметно более высокую активность в эпоксидировании этилена, обеспечивая более высокое значение выхода ЭО (4.5%) при 99% селективности по ЭО [132, 133]. Однако во время пробега в смесях этилен/О2 = 1 : 1 на поверхности таких катализаторов происходило более интенсивное отложение углерода, чем на образцах на основе α-Al2O3, что приводило к снижению выхода (как селективности, так и конверсии) до 3.3% уже в течение 32 ч [133]. Таким образом, ни один из носителей, отличных от α-Al2O3, до настоящего момента не конкурентоспособен с точки зрения селективности или стабильности получаемых на их основе нанесенных катализаторов.
Стабилизация максимального количества частиц серебра оптимального размера при минимальном вторичном превращении ЭО достигается формированием носителя состава α-Al2O3 с высокой порозностью, обеспечиваемой бидисперсной структурой пор [42, 65, 66, 77, 97–101, 113]. Оптимальные текстурные характеристики такого носителя чаще всего обеспечиваются за счет использования:
– смеси двух порошков α-Al2O3 с различными размерами частиц и образующих их кристаллитов, которую можно получить, подбирая условия размалывания агломератов;
– варьирования природы связующего (поливиниловые спирты, клеи на основе целлюлозы, пептизирующие кислоты);
– добавок псевдобемита и гиббсита/байерита в качестве гидратированного предшественника α-Al2O3, служащих дополнительным связующим;
– различных выгорающих добавок (углеродсодержащие материалы, порошкообразные пластики, материалы на основе целлюлозы и т.д.) с варьируемыми размером частиц и количеством.
Носители с необходимыми характеристиками уже выпускаются компаниями Saint Gobain Norpro Co, Sud Chemie AG и Сeram Tec AG. В принципе, близкие значения активности и селективности по ЭО можно получать, используя носитель с близкой пористой структурой, приготовленный только из смеси гиббсита и бемита (т.е. без добавок α-Al2O3), за счет подбора особенного режима прокаливания и введения различных выгорающих добавок. Однако такие носители характеризуются гораздо меньшей прочностью [118]. Между тем, прочность гранул катализатора играет важную роль для увеличения его срока жизни. Так, у носителей с бидисперсной структурой пор частично теряется прочность и устойчивость к истиранию [80, 123]. Эта проблема решается введением в носитель дополнительного связующего. Кроме традиционных глин, в этом качестве используют различные растворимые в воде силикаты Na, К, Mg, Ca [44, 80], стабилизированные силиказоли, которые превращаются после прокаливания в алюмосиликаты со структурой муллита [39], а также непосредственно керамические частицы на основе алюмосиликатов с примесями (2–4%) алюминатов Ca(Mg) и оксидов щелочных металлов [65, 66, 97–101]. Существует также точка зрения, что введение Si дополнительно снижает концентрацию кислых центров на поверхности носителя [49], что способствует увеличению стабильности.
Однако даже при мономодальных порах размером 3.8 мкм и, соответственно, значительно меньшем объеме пор, возможны более высокие значения селективности (88.8% вместо 87.5%) и активности (ΔТ = 13°С), чем в образцах с бимодальной структурой пор, в которых до 58% пор размером 1.4–3.8 мкм формируются за счет введения порообразующей выгорающей добавки [80, 123]. Это достигается двухкратным нанесением серебра с промежуточной сушкой образцов, причем при первой пропитке наносится только серебро (9.8%), а во время второй – остальное серебро (7%) совместно с промоторами (Re, W, S, Li, K, Cs). Возможность получать высокоселективные и стабильные катализаторы на таких носителях весьма привлекательна, т.к. они характеризуются гораздо более высокой прочностью и стойкостью к истиранию по сравнению с более высокопористыми образцами с бидисперсной структурой.
В 2007 г. [134] компания Shell предложила новую форму частиц носителя – полые цилиндры. По сравнению с традиционно используемыми гранулами это позволяет увеличить активную поверхность в единице объема реактора, в т.ч. за счет нанесения большего количества активного компонента (17.8% Ag, 460 м.д. Cs, 1.5 мкмоль Re, 0.75 мкмоль W, 15 мкмоль Li на грамм катализатора). При оптимальной геометрии прочность таких катализаторов достаточно высока, а снижение селективности за время пробега, обеспечивающего производство 600 и 1400 т ЭО/м3 катализатора, не превышает величины 2.4 и 9.9%, соответственно. При этом длительность пробега можно дополнительно увеличить на образцах с повышенным содержанием серебра, достигаемым использованием двухкратной пропитки носителя. Позднее компания Saint-Gobain Ceramics запатентовала все возможные геометрические формы носителя [79], которые обеспечивают минимальный перепад давления по слою катализатора при максимальной плотности упаковки в трубчатом реакторе. Такой носитель имеет многодольную форму с неоднородным скруглением на пересечении краев и стенок носителя (рис. 3).

Другой способ увеличения активности/селективности основан на принципе модифицирования состояния серебра в катализаторе. В качестве добавок, способных изменить электронное состояние собственно серебра, пробовали использовать Сu [60, 105] и Pd [135]. Образцы Cs–Ag/Al2O3, промотированные медью (оптимальное количество – 0.2 мол. %) или Pd (100 м.д.), демонстрировали более высокие активность и селективность по ЭО. Положительный эффект сохранялся и в отсутствие Cs, а также при одновременном добавлении Cs и газофазном промотировании хлором. Такой необычный эффект одновременного увеличения активности и селективности, скорее всего, можно отнести к увеличению скорости диссоциации кислорода на поверхности в присутствии меди за счет снижения активационного барьера для адсорбции кислорода. Однако в присутствии как Cu, так и Pd происходит гораздо более быстрая агломерация частиц Ag [135–137], которая является наиболее важной причиной дезактивации катализатора [138–146].
Таким образом, анализ основных тенденций развития Ag(+Cs,Re)–α-Al2O3 катализаторов, а также выполненных исследований свидетельствует об отсутствии явных предпосылок для существенного увеличения их производительности в ближайшие годы. Несмотря на достаточно высокие значения селективности по ЭО, достигаемые на катализаторах последних поколений (ок. 85% за пробег), происходит неизбежное превращение части этилена в СО2. Это означает, что при объемах ежегодного производства ЭО около 30 млн т образуется более 10 млн т/год СО2. Кроме того, в денежном эквиваленте потеря около 3.3 млн т/год этилена составляют 4 млрд долл./год. Эта цифра будет расти по мере увеличения спроса на легкие олефины и, соответственно, стоимости этилена. Поэтому особый интерес представляют процессы и катализаторы, позволяющие перерабатывать этилен в ЭО с большей селективностью, с параллельным получением отличных от СО2 органических продуктов. При этом стабильность и производительность новых систем должна быть сравнима с Ag(+Cs,Re)–α-Al2O3 (2.2–4.1 г ЭО/ч/г активного металла).
РАЗВИТИЕ АЛЬТЕРНАТИВНЫХ СПОСОБОВ ПОЛУЧЕНИЯ ЭТИЛЕНОКСИДА
В 2010 г. в Центре экологического катализа (Канзас, CША) был запатентован процесс CEBC-EO для гомогенного жидкофазного (в среде метанола) эпоксидирования этилена Н2О2 в присутствии органического основания (пиридин N-оксид) в относительно мягких условиях (40°С, 5 МПа) [147, 148]. В данном процессе в качестве катализатора предлагалось использовать гомогенный метилтриоксорений (CH3ReO3), являющийся льюисовкой кислотой, который обеспечивает почти 100%-ную селективность по ЭО и высокую эффективность использования Н2О2. В табл. 2 приведены основные параметры процесса CEBC-EO в сравнении с традиционным газофазным процессом на серебряных катализаторах [149].
Таблица 2.
Ключевые параметры и характеристики газофазного и жидкофазного (СЕВС-ЕО) процессов эпоксидирования этилена [149]
| Характеристика | Газофазный процесс* | Процесс CEBC-EO |
|---|---|---|
| Давление, МПа | 1.0–2.0 | 50 |
| Температура, °С | 200–300 | 20–40 |
| Конверсия С2Н4 за проход, % | 10 | 50 |
| Селективность по ЭО, % | 80–90 | >99 |
| CO2, % | 10–20 | – |
| Производительность катализатора, г ЭО/ч/г активного металла |
2.2–4.1 | 1.61–4.97 |
* Взято из [150].
Существенный вклад в увеличение стоимости ЭО, производимого газофазным способом, вносят стоимость выделения и повторной компрессии этилена. К преимуществам жидкофазного процесса следует отнести отсутствие О2 в газовой фазе и нахождение ЭО преимущественно в жидкой фазе, что существенно снижает стоимость очистки этилена и затраты на рецикл. К недостаткам данного процесса следует отнести высокую стоимость катализатора, а также H2O2, используемого в качестве оксиданта, что требует развития новых технологий ее безопасного выделения и рецикла.
В работе [151] обсуждаются результаты предварительных экономических оценок обоих процессов (мощность 200 тыс. т/год). Показано, что при близких капитальных затратах, процесс CEBC может стать конкурентоспособным, если катализатор сохранит свои характеристики в течение года, а скорость уноса Re из реактора не превысит 0.11 фунта/ч (0.11 м.д. Re на выходе из реактора). Необходимо отметить, что на момент публикации были известны только принципиальные способы иммобилизации катализатора на полимерном носителе [152, 153], что необходимо для удержания катализатора наномембранами на выходе из реактора. Компания в 2013 г. анонсировала проведение работ по иммобилизации метилтриоксорения на растворимом поли(4-винил-пиридине), однако информация о результатах этой работы отсутствует. Одним из наиболее эффективных решений этой проблемы мог бы стать переход на гетерогенные катализаторы, как это реализовано в концептуально очень близком промышленном процессе Dow-BASF Hydrogen Peroxide Propylene Oxide (НРРО) получения пропиленоксида (ПО). Выход ПО в данном процессе на титансиликате TS-1 достигает 99.7% [154], а степень использования Н2О2 в среднем по циклу – 90% [155], причем последняя характеристика катализатора особенно важна, поскольку стоимость пероксида водорода вносит существенный вклад в себестоимость продукта. Очевидно, что для случая эпоксидирования этилена, как и в процессе СЕВС-ЕО, она будет иметь не меньшее значение.
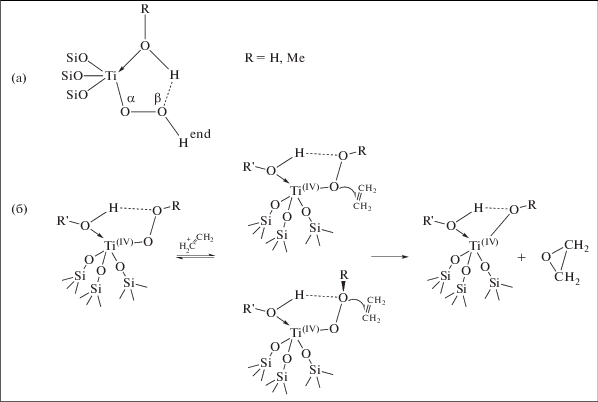
Реакционная способность титансиликатов в реакции эпоксидирования обусловлена формированием изолированных Ti4+ в тетраэдрическом окружении, которые при реакции с молекулой Н2О2 в присутствии метанола или воды образуют гидропероксо-интермедиат Ti–Oα–Oβ–Hend (рис. 4а). В таком интермедиате более электрофильный кислород Oα, непосредственно связанный с Ti, атакует двойную связь олефина с образованием эпоксида, согласно одной из предложенных схем (рис. 4б) [156].
Рис. 4.
Возможная схема образования эпоксида (б) с участием гидропероксо-интермедиата, образующегося при реакции изолированных Ti4+ в тетраэдрическом окружении, с молекулой Н2О2 (а).
В многочисленных теоретических исследованиях была показана возможность реализации такой же схемы и для этилена [157–161]. Однако TS-1 и подобные ему титансиликаты Ti–STT и Ti–CHA с мелкими порами продемонстрировали низкую активность и селективность по ЭО (табл. 3) [162–164]. Поскольку активность как на TS-1, так и на Ti–MWW в эпоксидировании более длинных линейных алкенов была заметно выше, хотя и в обоих случаях падала по мере увеличения длины цепи от пропилена к 1-гексену из-за стерических ограничений [165], авторы сделали вывод о низкой реакционной способности именно этилена, обусловленной дефицитом электронов на двойной связи в его молекуле. Снижение селективности обусловлено тем, что при использовании традиционного СН3ОН (а также трет-бутанола, ацетона или Н2О) в качестве растворителя в мелких порах происходит эффективный сольволиз или гидратация ЭО с раскрытием эпоксидного кольца с последующей конденсацией образующихся этиленгликоля и его метилового эфира (рис. 5).
Таблица 3.
Каталитические характеристики различных силикатов в жидкофазном эпоксидировании этилена Н2О2
| Катализатор | Условия | Растворитель | Выход продуктов, %1 | Селективность по ЭО, % | Эффективность использования Н2О2, % | Производительность по ЭО, 10–3 г ЭО/(ч г металла) | Ссылка | |
|---|---|---|---|---|---|---|---|---|
| навеска, мг | Т, Р, длительность цикла | |||||||
| TS-1 | 200 | 60°C, 0.4 МПа, 6 ч | СН3ОН | – | 69 | – | – | [163] |
| Ti–CHA | – | 62 | – | – | ||||
| Ti–STT | 200 | 60°C, –, 6 ч | СН3ОН | – | 49 | – | – | [162] |
| TS-1 | 150 | 2.5 МПа, 40°С, 1.5 ч | СН3ОН | 87.1 | 15.2 | 98.9 | – | [164] |
| СН3CN | 5.2 | 99.9 | 99.2 | – | ||||
| Ti–MWW | 100 | 2.5 МПа, 40°С, 1.5 ч | СН3CN | 35.5 | 98.9 | 86.3 | 91 | [165] |
| F–Ti–MWW | 89.0 | 99.6 | 95.8 | 241 | ||||
| W–KIT-6(10)2 | 500 | 5.0 МПа, 35°С, 5 ч | СН3ОН (624 ммоль) + + СН3CN (4.9 ммоль) |
– | 81.4 | 3.6 | 34 | [184] |
| W–KIT-6(100)2 | – | 80.0 | 5.0 | 153 | ||||
| Nb–KIT-6(10)2 | – | 46.8 | 17.1 | 234 | ||||
| Nb–KIT-6(100)2 | – | 73.4 | 11.2 | 794 | ||||
| Nb–TUD-1(10)2 | 500 | 5.0 МПа, 35°С, 5 ч | – | 78.5 | 21.6 | 342 | [188] | |
| Nb–TUD-1(100)2 | – | 87.8 | 12.8 | 2539 | ||||
| Nb–KIT-5(100)2 | – | 56.1 | 7.0 | 867 | ||||
| Nb–MCM-48(100)2 | – | 81.8 | 6.4 | 219 | ||||
| Nb–TUD-1(200)2 | 300 | 5.0 МПа, 35°С, 5 ч | СН3ОН (624 ммоль) + + СН3CN (7.3 ммоль) |
– | >99 | 6.6 | 1748 | [190] |
| NR4–Nb-TUD-1(200)2,3 | – | 99.5 | 89.8 | 1768 | ||||
| Nb–TUD-1(40)2 | – | 68.5 | 16.6 | 2255 | ||||
| Bn–Nb-TUD-1(40)2,4 | – | 98.7 | 59.7 | 597 | ||||

Замена СН3ОН на апротонный и более оснóвный ацетонитрил существенно увеличивала селективность, но при этом резко снижалась активность (табл. 3) [164]. Среди исследованных титансиликатов (Ti–MWW, TS-1, Ti–MOR и Ti–Beta) с относительно близкими значениями удельной поверхности (527–614 м2/г), отношением Si/Ti (38–51) и содержанием изолированных тетраэдрически координированных Ti4+ в решетке цеолита лучшие характеристики продемонстрировал Ti–MWW. В оптимальных условиях усредненный по циклу длительностью 5 ч выход продуктов эпоксидирования (включая МЭГ и метиловый эфир) и селективность по ЭО на нем составили около 75 и 97.9%, соответственно, эффективность использования Н2О2 – 77.7%. Центрифугирование и отмывка катализатора ацетоном от тяжелых продуктов (подобных этиленгликолю), отлагающихся в микропорах, а также последующее прокаливание при 550°С полностью восстанавливали эффективность использования Н2О2 как минимум в течение 10 циклов. При этом не изменялись как структурные характеристики Ti–MWW, так и содержание в нем изолированных центров Ti4+. Тем не менее, активность таких катализаторов (~90 × 10–3 г ЭО/(ч г металла) [165]) недостаточна для их использования в промышленном процессе получения ЭО. Авторы работы [165] предложили также увеличить активность Ti–MWW, повышая электроположительность Ti-центров в результате оттягивания с него электронов за счет индуктивного эффекта при появлении в непосредственной близости электроотрицательного F. Было высказано предположение, что при этом также увеличивается прочность водородной связи между Hend в интермедиате Ti–Oα–Oβ–Hend и находящихся в непосредственной близости частиц силанольных групп Si–F (рис. 4а), что увеличивает его стабильность.
Известно, однако, что при добавлении фтора возможно модифицирование кислотности и гидрофобно-гидрофильных характеристик цеолита. Это происходит благодаря реакции наиболее часто используемого для этой цели NH4F с терминальными Si–OH-группами с образованием тетраэдрических SiO3/2F [166] и ионных электроотрицательных пентакоординированных SiO4/2F− (а, в отдельных случаях, и внерешеточных ${\text{SiF}}_{6}^{{2--}}$) частиц [167, 168], которые положительно и, соответственно, отрицательно действуют на селективное окисление алкенов [169–171]. Для селективного удаления SiO4/2F− использовали обмен с хлоридами щелочных металлов, который приводил к увеличению активности фторированных Ti–MWW (F–Ti–MWW) в эпоксидировании различных алкенов [170, 171]. Однако при избытке щелочи наблюдался обратный эффект. Более предпочтительно фторирование обработкой раствором NH4F в метаноле, которое позволило избежать образования нежелательных SiO4/2F− на Ti–MOR [172] и Ti–MWW [173] и таким образом значительно увеличить активность катализаторов в окислительном аммонолизе различных кетонов и эпоксидировании пропилена соответственно.
Аналогичное пост-модифицирование Ti–MWW, сделанное авторами работы [165], привело к существенному увеличению селективности и выхода этиленоксида, а также эффективности использования Н2О2 при использовании в качестве растворителя как метанола, так и ацетонитрила во всем диапазоне отношений F/Si (0–0.4) (табл. 3, образцы Ti–MWW и F–Ti–MWW). Исследование кинетики эпоксидирования показало, что положительный эффект фторирования на активность можно объяснить модифицированием локального окружения решеточных Ti-мест. Снижение концентрации кислых центров (бора и группы Si–OH) после фторирования приводит к увеличению селективности по ЭО благодаря ингибированию реакций с раскрытием эпоксидного кольца (рис. 5), а также эффективности использования Н2О2. Авторы также полагают, что второй немаловажной причиной увеличения активности, селективности и стабильности работы таких катализаторов может быть формирование мезопор после фторирования образцов, которое облегчает доступность активных центров и диффузию продуктов. В пользу данного утверждения свидетельствовало дополнительное увеличение активности после прокаливания частично реактивированных образцов. Целесообразность поиска других титансиликатов с бóльшим размером пор для увеличения эффективности реакции также отмечается в других работах [174, 175].
В последние годы опубликовано большое число обзоров, анализирующих стратегии синтеза материалов с иерархической структурой пор [176], а также мезопористых силикатов с регулируемыми размерами и разветвленной системой транспортных пор, в том числе с использованием различных темплатов (TUD-1, KIT-5, KIT-6, MCM-48) [177, 178]. Их функционализация Nb и W позволяет получать системы, на которых в присутствии H2O2 образуются гидропероксо-комплексы [179, 180], аналогичные эффективному в эпоксидирования этилена Ti–Oα–Oβ–Hend (рис. 4а). С практической точки зрения наибольшую перспективу имеет так называемый one-pot синтез, который позволяет формирование системы металл–носитель в одну стадию. Один из вариантов такого синтеза реализуется в кислой среде с использованием в качестве темплата сополимера на основе полиалкиленоксида EOnPOmEOn Pluronic F127 и дополнительного ПАВ – н-бутанола [181]. Он более прост в исполнении, дает лучшую воспроизводимость и позволяет более равномерно распределять атомы металла по сравнению с методами пропитки и прививания, что приводит к большей относительной доли металла в высокодисперсном состоянии [182]. При этом, например, в M–KIT-6 (M = W, Zr, Nb), как удельная поверхность, так и объем пор снижались с количеством заведенного металла, в то время как распределение пор по размерам с преимущественным размером, определяемым типом металла (6–9 нм), практически не менялось [183, 184].
На таких системах с варьируемым соотношением Si/M = 10–100 (M = W, Nb) в условиях, близких к реализуемым в процессе НРРО (35°С, 50 бар, растворитель – метанол), при полном отсутствии СО2 в продуктах селективность по ЭО составила около 80% на W–KIT-6 и снижалась от 73 до 46% на Nb–KIT-6 с увеличением содержания Nb (табл. 3) [184]. Последний эффект авторы связали с изменением концентрации бренстедовских кислотных центров (БКЦ) на Nb–KIT-6 (искаженные октаэдрические структуры NbO6) [183], которые при жидкофазном окислении отвечают за хемосорбцию эпоксидов, приводящую к раскрытию эпоксидного кольца с образованием гликолей [185]. Производительность Nb–KIT-6 (234–794 г ЭО/ч/кг Nb) значительно выше, чем у образцов W–KIT-6 (34–152 г ЭО/ч/кг W), что логично объясняется присутствием в Nb–KIT-6 только изолированных и олигомерных частиц вплоть до содержания металла 10 мас. %, обеспечивающих высокую концентрацию льюисовских кислых центров (ЛКЦ) [183], в то время как в образце W–KIT-6, скорее всего, из-за большего размера иона W, даже при его содержании 3–5 мас. % регистрировали низкоактивную фазу WO3 [186]. Тем не менее, такие значения производительности заметно ниже, чем на Ag–Al2O3-катализаторах газофазного эпоксидирования этилена (700–4400 г ЭО/ч/кг Ag) [150, 187, 188]. Не менее существенные недостатки данных катализаторов связаны с:
– низкой эффективностью использования H2O2 (≤19%),
– высокой скоростью вымывания Nb из решетки KIT-6 (от 30 до 75% в течение 5 ч, в зависимости от содержания Nb).
Авторы объяснили это высокой концентрацией БКЦ, возникающих при внедрении металла в решетку носителя.
Альтернативный способ приготовления, основанный на золь-гель методологии, применяется для синтеза рентгеноаморфных силикатов TUD-1. В качестве источника Si используется тетраэтилортосиликат (TEOS). Вместо ПАВ для получения регулярной пористой структуры в качестве хелатирующих кремний реагентов применяют триэтаноламин или тетраэтиленгликоль, которые (вместе с солями, предпочтительно алкоголятами, необходимых металлов) смешиваются с TEOS и Н2О для последующего старения и термообработки [189]. В отличие от других силикатов, например, KIT-5, KIT-6, приготовленных с использованием темплатов [183, 184], увеличение содержания Nb в Nb–TUD-1 от 1.4 до 12.2% приводило к значительному увеличению среднего размера пор (от 5 до 20 нм) [188]. В ряду протестированных мезопористых систем Nb-силикат, приготовленных различными способами (Nb–KIT-5, Nb–MCM-48 и Nb–TUD-1) [188] наибольшая производительность по ЭО в случае использования СН3ОН в качестве растворителя была измерена на рентгеноаморфном Nb–TUD-1 (342–2539 г ЭО/ч/кг Nb при варьировании Si/Nb от 10 до 100) (табл. 3). Тем не менее, эффективность использования Н2О2 во всех случаях не превышала 12%, а скорость вымывания Nb за цикл длительностью 5 ч оставалась значительной (12–60.8%). Моделирование взаимодействия Н2О2 с различными модельными структурами, формирующимися в системе Nb–Si–O, с использованием теории функционала плотности показало, что разложение H2O2 протекает наиболее легко на БКЦ, в особенности Nb–OH, образующихся в силикатах ниобила с тетраэдрическим кислородным окружением Nb. Авторы показали, что снижение концентрации БКЦ при уменьшении содержания Nb до величины 0.23–0.88 мас. % приводит к резкому увеличению производительности по ЭО (1041–4304 г ЭО/ч/кг Nb). На таких мезопористых (средний размер пор 6.6–7.8 нм) образцах селективность по ЭО варьировалась в интервале 87.8–99.9%, а степень использования H2O2 возросла до 20.6–78.7%. Однако скорость вымывании Nb в цикле практически не изменилась.
Другой способ увеличения гидрофобности поверхности Nb–TUD-1, предложенный авторами работы [190], заключался в обработке образцов органическими основаниями (тетраэтиламмоний гидроксид и диизопропилэтиламин) или взаимодействием с различными реагентами, содержащими ковалентно связывающиеся группы (метил-, триметилсилицил-, трет-бутил-, бензил-) с различной гидролитической стабильностью. При использовании органических оснований (образец NR4–Nb-TUD-1, табл. 3) заметно возрастала эффективность использования Н2О2 (до 90%), однако устойчивость к вымыванию металлов улучшилась незначительно. Обработанный бензиловым спиртом образец Bn–Nb–TUD-1 сочетал высокую эффективность использования Н2О2 (~60−71%) с заметно пониженной скоростью вымывания Nb (∼3%) и высокой селективностью по ЭО (>98%). Однако это сопровождалось снижением производительности по сравнению с немодифицированным образцом из-за дезактивации/блокировки части ЛКЦ в результате процедуры пассивации БКЦ (табл. 3).
Этой же группой авторов [191] был предложен альтернативный Evaporation-Induced Self-Assembly (EISA) способ приготовления мезопористых модифицированных Nb- силикатов с использованием в качестве темплата полиалкиленоксида EO20PO70EO20 (Pluronic 123). Он позволяет избежать формирования нежелательных кристаллитов Nb2O5 вплоть до содержания Nb ~ 22 мас. %. Высокая удельная поверхность (780–980 м2/г) при наличии мезопор размером 3.9–4.9 нм, а также увеличение гидрофобности поверхности из-за неполного удаления углерода при прокаливании образцов в азоте привели к увеличению активности, селективности и степени использования H2O2 в эпоксидировании циклогексена на таких системах по сравнению с приготовленными другими методами содержащими Nb силикатами (Nb–TUD-1, Nb–SBA-16, Nb–KIT-6, Nb–KIT-5). Не исключено, что подобные образцы могут быть перспективны и для эпоксидировании этилена.
Таким образом, в настоящее время ни один из известных катализаторов жидкофазного эпоксидирования этилена Н2О2 не обладает характеристиками, достаточными для того, чтобы процесс на их основе составил конкуренцию газофазному процессу, использующему в качестве катализаторов системы Ag–Al2O3. Снижение до приемлемых для реализации процесса в непрерывном режиме значений скорости уноса из реактора высокоактивного и селективного гомогенного катализатора на основе метилтриоксорения станет возможным только после его успешной иммобилизации. Эффективное протекание при использовании СН3ОН в качестве растворителя реакций, приводящих к раскрытию эпоксидного цикла, ограничивает применение мелкопористых титансиликатов TS-1, Ti–CHA, Ti–STT, Ti–MWW. Замена растворителя на апротонный ацетонитрил решает проблему селективности, но резко снижает активность таких систем, даже в случая формирования мезопористой структуры. Для высокоактивных модифицированных Nb силикатов KIT-5(6), TUD-1 характерны высокая степень вымывания Nb, а также низкая эффективность использования Н2О2. Попытки селективного удаления БКЦ, с участием которых протекают гидролиз Н2О2, приводят к дезактивации/блокировке части ЛКЦ, отвечающих за активность.
ЗАКЛЮЧЕНИЕ
Используемый в промышленности для получения ЭО более 50 лет газофазный процесс эпоксидирования этилена кислородом характеризуется высокой степенью оптимизации. Используемые в данном процессе катализаторы Ag–α-Al2O3, модифицированные Cs и Re, на которых при конверсии С2Н4 за проход около 10% достигается средняя за пробег селективность около 85%, хорошо изучены. В последние годы увеличение длительности их пробега, связанное с увеличением концентрации частиц серебра оптимальных состава и размеров, достигалось в основном:
– модифицированием поверхности носителя, направленным на создания центров для последующего формирования и стабилизации наночастиц серебра;
– оптимизацией пористой структуры носителя, облегчающей диффузию продуктов реакции;
– увеличением активной поверхности катализатора за счет регулирования формы гранул.
Невозможность дальнейшего значительного снижения селективности по основному побочному продукту – СО2, привело к активным исследованиям жидкофазных процессов, в которых в качестве окислителя используется Н2О2. Значения производительности лучших катализаторов на основе метилтриоксорения для процесса CEBC-EO и используемых в промышленности Ag–α-Al2O3 сравнимы, а селективность по ЭО при конверсии этилена до 50% за проход на гомогенных системах превышает 99%. Их использование в непрерывных процессах станет возможным после развития технологий их иммобилизации на носителе. Выделяют несколько причин, из-за которых невозможно одновременно достичь высокой активности/производительности, селективности и эффективности использования Н2О2 на известных гетерогенных силикатах, модифицированных Ti, Nb или W. Низкая активность модифицированных W силикатов объясняется преимущественным формированием массивных оксидов W c октаэдрическим кислородным окружением. На мелкопористых силикатах эффективно протекают реакции, приводящие к раскрытию эпоксидного цикла. Высокая концентрация БКЦ в наиболее активных и селективных силикатах ниобила отвечает за высокую скорость вымывания Nb и гидролиз Н2О2. Попытки их удаления приводят к блокировке ЛКЦ, отвечающих за активность. Основные перспективы развития этих систем связывают с поиском методик синтеза, обеспечивающих формирование мезопористых систем, гидрофобные свойства поверхности которых можно регулировать полнотой удаления углерода темплата при прокаливании.
Список литературы
Электронный ресурс. URL: https://ihsmarkit.com/ products/ethylene-oxide-chemical-economics-handbook.html (дата обращения 30.04.2020).
SRI Consulting, 2009 // URL: https://hub.globalccsinstitute.com/publications/ccs-roadmap-industry- high-purity-co2-sources-sectoral-assessment-%E2% 80%93-final-draft-report-2 (дата обращения 20.04.2020).
Bernardo P., Clarizia G. // Clean Technol. Envir. 2012. V. 14. № 3. P. 475.
Ethylene Oxide (EO) (Complete Report). A Global Strategic Business Report Global Industry Analysts Inc. 2011, 346 p.
Электронный ресурс. URL: https://www.shell.com/ business-customers/global-solutions/petrochemicals-technologies-licensing/ethylene-oxide-ethylene-glycol-processes.html (дата обращения 15.10.2018).
Global and China Ethylene Oxide (EO) Industry Report. 2017– 2021. Электронный ресурс. URL: http://www.researchinchina.com/UpLoads/ArticleFreePartPath/20170714171038.pdf (дата обращения 01.07.2020).
Электронный ресурс. URL: http://rupec.ru/news/ 40737/ (дата обращения 24.04.2020).
Global Ethylene Oxide Market –Forecasts from 2018 to 2023 // Электронный ресурс. URL: https://www. researchandmarkets.com/research/rn64pq/global_ ethylene?w=4 (дата обращения 24.04.2020.).
Электронный ресурс. URL: https://www.energiasmarketresearch.com/global-ethylene-oxide-market-report/ (дата обращения 24.04.2020).
Электронный ресурс. URL: https://www.kommersant.ru/doc/2535838 (дата обращения 29.04.2020).
Rebsdat S., Mayer D. Ethylene Oxide. Ulmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2012. P. 568.
Selective Reasoning. Shell Chemicals Magazine, Autumn/Winter. 2010. P. 12.
Электронный ресурс. URL: http://www.toyo-eng.com/jp/en/products/petrochmical/eo/ (дата обращения 30.04.2020).
Электронный ресурс. URL: https://www.shell.com/ business-customers/global-solutions/petrochemicals-technologies-licensing/ethylene-oxide-ethylene-glycol-processes.html (дата обращения 30.04.2020).
Электронный ресурс. URL: http://www.scidesign.com/technologies/ethylene-oxide-and-derivatives/ethylene-oxideethylene-glycols# (дата обращения 20.11.2018).
Электронный ресурс. URL: http://www.hydrocarbonprocessing.com/news/2018/05/sabic-selects-scientific-design-company-eoeg-technology-for-complex-in-al-jubail-saudi-arabia (дата обращения 30.04.2020).
Электронный ресурс. URL: http://briciysfy.sinopec.com/briciysfy/en/ps/pro_presentation/yxhyhzhyywychj.shtml (дата обращения 30.04.2020).
Электронный ресурс. URL: http://briciysfy.sinopec.com/briciysfy/en/rd/tech_achi/20170822/ news_20170822_324012701230.shtml-2017-08-22 (дата обращения 20.11.2018).
CHM046C – Global Markets for Catalysts: Focus on Catalyst Regeneration. BCC Research. June 2015. ISBN: 1-62296-116-1. P. 455.
Электронный ресурс. URL: http://docplayer.ru/ 37465919-Plan-2030-klasternoe-razvitie.html (дата обращения 29.04.2020).
Электронный ресурс. URL: https://www.kommersant.ru/doc/3374207 (дата обращения 29.04.2020).
Lee J.K., Verykios X.E., Pitchai R. // Appl. Catal. 1989. V. 50. № 1. P. 171.
Goncharova S.N., Paukshtis E.A., Bal’zhinimaev B.S. // Appl. Catal. A Gen. 1995. V. 126. № 1. P. 67.
Mastikhin V.M., Goncharova S.N., Tapilin V.M., Terskikh V.V., Balzhinimaev B.S. // J. Mol. Catal. A Chem. 1995. V. 96. № 2. P. 175.
Bukhtiyarov V.I., Prosvirin I.P., Kvon R.I., Goncharova S.N., Bal’zhinimaev B.S. // J. Chem. Soc. Faraday Trans. 1997. V. 93. № 13. P. 2323.
Электронный ресурс. URL: https://www.dow.com/ en-us/news/press-releases/dow-meteor-technology-selected-for-new-meglobal-meg-production-facility-on-the-us-gulf-coast (дата обращения 01.11.2018).
Soo H., Chou P.-Y., Bhasin M.M. // Patent US № 5102848. 1991.
Liu A.C., Soo H. // Patent US № 8546294. 2013.
Zhang L., Tupe R.R., Phillips A.G., Hinman P.V., Soo H. // Patent. US № 8815769. 2013.
Gopal S., Murthy L.V., Basrur A.G. // Patent US № 9458121. 2016.
Rojluechai S., Chavadej S., Schwank J.W., Meeyoo V. // Catal. Commun. 2007. V. 8. № 1. P. 57.
Petitto C., Delahay G. // Catal. Letters. 2012. V. 142. № 4. P. 433.
Raharjo P., Ishizaki C., Ishizaki K.J. // Ceram. Soc. Japan. 2000. V. 108. № 1257. P. 449.
Lee J.K., Verykios X.E., Pitchai R. // Appl. Catal. 1988. V. 44(C). P. 223.
Mao C.-F., Vannice M.A. // Appl. Catal. A Gen. 1995. V. 122. № 1. P. 61.
Yong Y.S., Kennedy E.M., Cant N.W. // Appl. Catal. 1991. V. 76. P. 31.
CRI, Catalyst training material. 2014. Электронный ресурс. URL: https://www.cricatalyst.com/cricatalyst/catalysts/ethylene-oxide.html (дата обращения 13.08.2018).
van Milligen H., VanderWilp B., Wells G.J. Enhancements in Ethylene Oxide/Ethylene Glycol Manufacturing Technology. Shell Global Solutions. 2016. 12 p.
Пак С., Рокицки А., Кавабата С. // Патент РФ № 2495715. 2009.
Rizkalla N., Rokicki A. // Patent US № 8883675. 2014.
Suchanek W., Rokicki A. // Patent US № 8975424. 2015.
Gerdes W.H., Lockemeyer J.R., Remus D.J., Szymanski T., Yeates R.C. // Patent US № 8084390. 2011.
Abdallah R., Mäurer T., Rosendahl T., Rosowski F., Theis G., Mazanec T., Deshmukh S., Silva L.J. // Patent US № 8933254. 2015.
Richard M.A., Lockemeyer J.R. // Patent US № 9339798. 2016.
Li X., Li J., Sun X., Chen J., Cao S., Gao L., Wang H., Liang R., Xue Q., Zhang Z. // Patent US № 9592491. 2017.
Grant R.B., Lambert R.M. // J. Catal. 1985. V. 93. P. 92.
Tan S.A., Grant R.B., Lambert R.M. // J. Catal. 1987. V. 106. P. 54.
Macleod N., Keel J.M., Lambert R.M. // Catal. Lett. 2003. V. 86. P. 51.
Jung K.-H., Chung K.-H., Kim M.-Y., Kim J.-H., Seo G. // Korean J. Chem. Eng. 1999. V. 16. P. 396.
Mao C.-F., Vannice A.M. // Appl. Catal. A. 1995. V. 122. P. 61.
Epling W.S., Hoflund G.B., Minahan D.M. // J. Catal. 1997. V. 171. P. 490.
Liu K. // Patent US № 4897376. 1990.
Hertzauk K., Beaning K., Pryucan U., Muros V.D., Schvartzman M. // Patent Japan № H4-3177411. 1992.
Batsfam J.E., Gardes W.H., Kowareski R.M. // Patent Japan № H5-84440. 1993.
Grant R.B., Lambert R.M. // Langmuir. 1985. V. 1. P. 29.
Diao W., DiGiulio C.D., Schaal M.T., Ma S., Monnier J.R. // J. Catal. 2015. V. 322. P. 14.
Amorim de Carvalho M.C.N., Passos F.B., Schmal M. // J. Catal. 2007. V. 248. P. 124.
Ren D., Xu H., Li J., Li J., Cheng D. // Molecular Catalysis. 2017. V. 441. P. 92.
Campbell C.T. // J. Phys. Chem. 1985. V. 89. P. 5789.
Jankowiak J.T., Barteau M.A. // J. Catal. 2005. V. 236. P. 379.
Atkins M., Couves J., Hague M., Sakakini B.H., Waugh K.C. // J. Catal. 2005. V. 235. P. 103.
van Santen R.A. In: Catalysis: Theory to Practice, Proceedings of Characterization and Metal Catalysts. Ottawa Canada, 1988. P. 1152.
Grant R.B., Harbach C.A.J., Lambert R.M., Tan S.A. // J. Chem. Soc. Faraday Trans. 1987. V. 1. № 83. P. 2035.
Ozbek M.O., Onal I., van Santen R.A. // Chem. Cat. Chem. 2013. V. 5. P. 443.
Lauritzen A.M. // Patent US № 4761394. 1988.
Lauritzen A.M. // Patent US № 4766105. 1988.
Ren D., Cheng G., Li J., Li J., Dai W., Sun X.X., Cheng. D. // Catal. Lett. 2017. V. 147. P. 2920.
Evans W.E., Matusz M., McCallister P.M. // Patent WO № 078603. 2012.
Rizkalla N., Rokicki A. // Patent US № 0281724. 2011.
Lockemeyer J.R., Yeates R.C., Reinalda D. // Patent US № 0160655. 2010.
Rizkalla N., Rokicki A. // Patent US. № 9452419. 2016.
Shimin J., Jianshe C., Junjun A., Linfa C. // Petrochem. Technology. 2013. V. 42. № 2. P. 204.
Электронный ресурс. URL: http://briciysfy.sinopec.com/briciysfy/en/rd/professional_field/ (дата обращения 29.04.2020).
Электронный ресурс. URL: http://www.sinopecgroup.com/group/en/Sinopecnews/20121126/ news_20121126_479410000000.shtml (дата обращения 19.10.2018).
Электронный ресурс. URL: http://scc.sinopec.com/ scc/en/products/basic_catalyst/11YSseriesAg.shtml (дата обращения 29.04.2020).
Электронный ресурс. URL: https://www.norpro.saint-gobain.com/catalyst-carrier/materials (дата обращения 01.10.2018).
Szymanski T., Remus D.J., Lockemeyer J.R., Yeates R.C., Gerdes W.H. // Patent US № 6831037. 2004.
Szymanski T., Remus D.J., Lockemeyer J.R., Yeates R.C., Gerdes W.H. // Patent US № 0096219. 2005.
Richard M.A., Covey J.D. // Patent US № 8871677. 2014.
Richard M.A., Lockemeyer J.R. // Patent US № 9073035. 2015.
Rosendahl T., Mäurer T., Dobner C.K., Zühlke J. // Patent US № 0264953. 2012.
Rosendahl T., Mäurer T., Dobner C.K., Geiger T., Gritsch A. // Patent US № 8629079. 2014.
Rosendahl T., Mäurer T., Dobner C.K. // Patent US № 8921587. 2014.
Rosendahl T., Mäurer T., Dobner C.K., Lehr A., Wanka J. // Patent US № 8987482. 2015.
Rosendahl T., Mäurer T., Dobner C.K., Lehr A. // Patent US № 079154. 2015.
Rosendahl T., Mäurer T., Dobner C.K., Lehr A., Wanka J. // Patent US № 9199223. 2015.
Zakzeski J.J., Bosch M., Dobner C., Bartosch C., Zühlke J. // Patent US № 9714227. 2017.
Габутдинов М.С. Автореферат дис. … к.т.н. 1995. С. 26. АО “Казаньоргсинтез”, Казань.
Соколов В.С., Давыдов В.А., Парфенов А.Н., Парфенов В.Н., Чесноков Б.Б., Габутдинов М.С., Розанов И.Ю., Черевин В.Ф., Ионов Ю.В., Романов Н.В., Юсупов Н.Х., Гусев Ю.В. // Патент РФ № 2046015. 1992.
Ламберов А.А., Гильманов Х.Х., Бусыгин В.М. // Катализ в промышленности. 2006. № 3. С.12.
Электронный ресурс. URL: https://neftegaz.ru/ news/view/6195-Tehnologii-ekonomii (дата обращения 29.04.2020).
Сафин Д.Х., Петухов А.А., Швец В.Ф. // Химическая промышленность сегодня. 2005. № 8. С. 45.
Бусыгин В.М., Гильманов Х.Х., Трифонов С.В., Ламберов А.А. // Катализ в промышленности. 2005. № 3. С. 33.
Парфенов А.Н., Парфенов В.Н., Соколов В.С., Давыдов В.А., Куликов Г.Н., Ионов Ю.В., Юсупов Н.Х., Габутдинов М.С., Гусев Ю.В., Трусов А.И. // Патент РФ № 2123383. 1997.
Парфенов А.Н., Парфенов В.Н., Соколов В.С., Куликов Г.Н. // Патент РФ № 2177829. 2002.
Matusz M., Mesters C.M.A.M., Buffum J.E. // Patent US № 5380697. 1995.
Bhasin M.M. // Patent US № 4908343. 1990.
Bhasin M.M. // Patent US № 5057481. 1991.
Thorsteinson E.M., Bhasin M.M., Seyedmonir S.R. // Patent US № 5187140. 1993.
Soo H.,Chou P.-Y., Bhasin M.M. // Patent US № 5102848. 1992.
Hayden P., Pinnegar H. // Patent US № 5011807. 1991.
Hayden P., Pinnegar H. // Patent US № 5099041. 1992.
Herzog K., Boeck S., Mross W.D., Plueckhan J., Boehning K.-H., Gallei E. // Patent US № 5407888. 1995.
Lockemeyer J.R. // Patent US № 6368998. 2002.
Jankowiak J., Barteau M.A. // J. Catal. 2005. V. 236. P. 366.
Martens A. // Patent French № 1499285. 1967.
Bhasin M.M., Warner G.H. // Patent US № 4419276. 1983.
Nielsen R.P. // Patent US № 3702259. 1972.
Cavitt S.B. // Patent US № 4102820. 1978.
Cannon J.C. // Patent US № 4235757. 1980.
Zhang Z.X., Jin J.Q., Gao Z. // Patent CN № 1426836A. 2003.
Natal M.A.W., Bhasin M.M., Soo H., Liu A.C. // Patent US № 177000. 2009.
Gueckel C. // Patent US № 7977274. 2011.
Bhasin M.M., Ellgen P.C., Hendrix C.D. // Patent US № 4916243. 1990.
Pourmortazavi S.M., Hajimirsadeghi S.S., Kohsari I., Fareghi Alamdari R., Rahimi-Nasrabadi M. // Chem. Eng. Technol. 2008. V. 31. № 10. P. 532.
Boldyrev V.V. // Thermochim. Acta. 2002. V. 388. № 1–2. P. 63.
Fiorucci A.R., Saran L.M., Cavalheiro É.T.G., Neves E.A. // Thermochim. Acta. 2000. V. 356. № 1–2. P. 71.
Rizkalla N. // Patent US № 8791280. 2014.
Shima M., Takada H. // Patent US № 6153556. 2000.
Wang S., Li J., Ji S. // Petrochem. Technology. 2010. V. 39. № 7. P.729.
Chen J., Jiang J., Zhang Z., Xue Q., Wang S. // Petrochem. Technology. 2011. V. 40. № 12. P. 1292.
Pak S. // Patent US № 0201595. 2018.
Richard M.A., Lockemeyer J.R. // Patent US № 9339979. 2016.
Miller S. A. In: Ethylene and its Industrial Derivatives. London: Ernest Benn Limited, 1969. P. 546.
Kanoh H., Nishimura T., Ayame A. // J. Catal. 1979. V. 57. P. 372.
Harriott P. // J. Catal. 1971. V. 21. P. 56.
Rizkalla N., Rokicki A. // Patent US № 0369460. 2017.
Wolf R., Gotze H. // Chem. Tech. 1962. V. 14. № 10. P. 600.
Beck J.S., Vartuli J.C., Roth W.J., Leonowicz M.E., Kresge C.T., Schmitt K.D., Chu C.T.W., Olson D.H., Sheppard E.W., McCullen S.B., Higgins J.B., Schlenker J.L. // J. Am. Chem. Soc. 1992. V. 114. P. 1.
Fotopoulos A.P., Triantafyllidis K.S. // Catal. Today. 2007. V. 127. P.148.
Puangpetch T., Sreethawong T., Yoshikawa S., Chavadej S. // J. Mol. Catal. A: Chem. 2008. V. 287. P. 70.
Chongterdtoonskul A., Schwank J.W., Chavadej S. // Catal. Lett. 2012. V. 142. P. 991.
Chongterdtoonskul A., Schwank J.W., Chavadej S. // J. Molec. Catal. A: Chemical. 2012. V. 358. P. 58.
Matusz M., Richard M.A., Lockemeyer J.R., Bos A.N.R., Rekers D.M., Reinalda D., Yeates R.C. // Patent US № 7259129. 2007.
Dellamorte J.C., Lauterbach J., Barteau. M.A. // Appl. Catal. A: Gen. 2011. V. 391. P. 281.
Dellamorte J.C., Lauterbach J., Barteau. M.A. // Top. Catal. 2010. V. 53. P. 13.
Dellamorte J.C., Lauterbach J., Barteau. M.A. // Ind. Eng. Chem. Res. 2009. V. 48. P. 5943.
Montrasi G.L., Tauszik G.R., Solari M., Leofanti G. // Appl. Catal. 1983. V. 5. P. 359.
Schouten E.P.S., Borman P.C., Westerterp K.R. // Chem. Eng. Process. 1996. V. 35. P. 43.
Murakami Y., Komai S., Hattori T. // Stud. Surf. Sci. Catal. 1991. V. 68. P. 645.
Presland A.E.B., Price G.L., Trimm D.L. // J. Catal. 1972. V. 26. P. 313.
Verykios X.E., Stein F.P., Coughlin R.W. // J. Catal. 1980. V. 66. P. 368.
Hoflund G.B., Minahan D.M. // J. Catal. 1996. V. 162. P. 48.
Jiang W., Zhang Z., Chen J. // Petrochem. Industry Technology. 2004-02.
Boskovic G., Wolf D., Brückner A., Baerns M. // J. Catal. 2004. V. 224. P. 187.
Wu J.C., Harriot P. // J. Catal. 1975. V. 39. P. 395.
Subramaniam B., Busch D.H., Lee H.-J., Ghanta M., Shi T-P. // US Patent 8080677. 2011.
Lee H.-J., Ghanta M., Busch D.H., Subramaniam B. // Chem. Eng. Sci. 2010. V. 65. P. 128.
Ghanta M., Subramaniam B., Lee H.-J., Busch D.H. // AIChEJ. 2013. V. 59. № 1. P. 180.
Buffum J.E., Gerdes W.H., Kowaleski R.M. // Patent US № 5145824. 1992.
Ghanta M., Ruddy T., Fahey D., Busch D., Subramaniam B. // Ind. Eng. Chem. Res. 2013. V. 52. P. 18.
Vezzosi S., Ferre A.G., Crucianelli M., Crestini C., Saladino R. // J. Catal. 2008. V. 257. № 2. P. 262.
Bracco L.L.B., Juliarena M.P., Ruiz G.T., Feliz M.R., Ferraudi G.J., Wolcan E. // J. Phys. Chem. B. 2008. V. 112. № 37. P. 11506.
Pujado P.R., Hammerman J.I. // Patent US № 5599956. 1997.
Ghanta M., Fahey D.R., Busch D.H., Subramaniam B. // ACS Sustainable Chem. Eng. 2013. V. 1. P. 268.
Clerici M.G., Ingallina P. // J. Catal. 1993. V. 140. P. 71.
Vayssilov G.N., van Santen R.A. // J. Catal. 1998. V. 175. P. 170.
Karlsen E., Schöffel. K. // Catal. Today. 1996. V. 32. P. 107.
Neurock M., Manzer L.E. // Chem. Commun. 1996. P. 1133.
Munakata H., Oumi Y., Miyamoto A. // J. Phys. Chem. B. 2001. V. 105. P. 3493.
Sinclair P.E., Catlow C.R.A. // J. Phys. Chem. B. 1999. V. 103. P. 1084.
Eilertsen E.A., Giordanino F., Lamberti C., Bordiga S., Damin A., Bonino F., Olsbye U., Lillerud K.P. // Chem. Commun. 2011. P. 11 867.
Eilertsen E.A., Bordiga S., Lamberti C., Damin A., Bonino F., Arstad B., Svelle S., Olsbye U., Lillerud K.P. // ChemCatChem. 2011. V. 3. P. 1869.
Lu X.Q., Zhou W.J., Wu H.H., Liebens A., Wu P. // Ap-pl. Catal. A: Gen. 2016. V. 515. P. 51.
Lu X., Zhou W.-J., Guan Y., Liebens A., Wu P. // Catal. Sci. Technol. 2017. V. 7. P. 2624.
Youngman R.E., Sen S.J. // J. Non-Cryst. Solids. 2004. V. 349. P. 10.
Ogino I., Nigra M.M., Hwang S.J., Ha J.M., Rea T., Zones S.I., Katz A. // J. Am. Chem. Soc. 2011. V. 133. P. 3288.
Louis B., Kiwi-Minsker L. // Micropor. Mesopor. Mater. 2004. V. 74. P. 171.
Goa Y., Wu P., Tatsumi T. // J. Phys. Chem. B. 2004. V. 108. P. 4242.
Fang X.Q., Wang Q., Zheng A.M., Liu Y.M., Wang Y.N., Deng X.J., Wu H.H., Deng F., He M.Y., Wu P. // Catal. Sci. Technol. 2012. V. 2. P. 2433.
Xu L., Ding J.H., Yang Y.L., Wu P. // J. Catal. 2014. V. 309. P. 1.
Yang Y.L., Ding J.H., Wang B.S., Wu J., Zhao C., Gao G.H., Wu P. // J. Catal. 2014. V. 320. P. 160.
Lu X.Q., Wu H.H., Jiang J.G., He M.Y., Wu P. // J. Catal. 2016. V. 342. P. 173.
Roth W.J., Nachtigall P., Morris R.E., Čejka J. // Chem. Rev. 2014. V. 114. P. 4807.
Opanasenko M.V., Roth W.J., Čejka J. // Catal. Sci. Technol. 2016. V. 6. P. 2467.
Yang X.-Y., Chen L.-H., Li Y., Rooke J.C., Sanchez C., Su B.-L. // Chem. Soc. Rev. 2017. V. 46. P. 481.
Zhang K., Xu L.-L., Jiang J.-G., Calin N., Lam K.-F., Zhang S.-J., Wu H.-H., Wu G.-D., Albela B., Bonneviot L. // J. Am. Chem. Soc. 2013. V. 135. P. 2427.
Möller K., Bein T. // Chem. Mater. 2017. V. 29. P. 371.
Morey M.S., Bryan J.D., Schwarz S., Stucky G.D. // Chem. Mater. 2000. V. 12. P. 3435.
Coelho J.V., Oliveira L.C.A., Moura F.C.C., de Souza P.P., Silva C.A., Batista K.B., da Silva M.J. // Appl. Catal. A Gen. 2012. V. 419–420. P. 215.
Kleitz F., Kim T.-W., Ryoo R. // Langmuir. 2006. V. 22. P. 440.
Ramanathan A., Subramaniam B. // Molecules. 2018. V. 23. P. 263.
Ramanathan A., Maheswari R., Barich D.H., Subramaniam B. // Micropor. Mesopor. Mater. 2014. V. 190. P. 240.
Yan W., Ramanathan A., Ghanta M., Subramaniam B. // Catal. Sci. Technol. 2014. V. 4. P. 4433.
Kilos B., Aouine M., Nowak I., Ziolek M., Volta. J. C. // J. Catal. 2004. V. 224. P. 314.
Ramanathan A., Subramaniam B., Badloe D., Hanefeld U., Maheswari R. // J. Porous Mater. 2012. V. 19. P. 961.
Oyama S.T. In: Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis. Amsterdam, The Netherlands: Elsevier, 2008. P. 3.
Yan W., Ramanathan A., Patel P.D., Maiti S.K., Laird B.B., Thompson W.H., Subramaniam B. // J. Catal. 2016. V. 336. P. 75.
Telalovic S., Ramanathan A., Mul G., Hanefeld U. // J. Mater. Chem. 2010. V. 20. P. 642.
Maiti S.K., Ramanathan A., Thompson W.H., Subramaniam B. // Ind. Eng. Chem. Res. 2017. V. 56. P. 1999.
Ramanathan A., Zhu H., Maheswari R., Subramaniam B. // Micropor. Mesopor. Mater. 2018. V. 261. P. 158.
Дополнительные материалы отсутствуют.
Инструменты
Нефтехимия