

Нейрохимия, 2020, T. 37, № 2, стр. 173-182
Анализ роли хинонредуктазы 2 в механизме противопаркинсонического действия афобазола
И. А. Кадников 1, Д. Н. Воронков 2, М. В. Воронин 1, С. Б. Середенин 1
1 Федеральное государственное бюджетное научное учреждение
“Научно-исследовательский институт фармакологии им. В.В. Закусова”
Москва, Россия
2 Федеральное государственное бюджетное научное учреждение
“Научный центр неврологии”
Москва, Россия
Поступила в редакцию 21.06.2019
После доработки 15.07.2019
Принята к публикации 30.07.2019
Аннотация
На экспериментальной модели болезни Паркинсона изучено содержание дофамина в стриатуме, плотность TH+ нейронов в черной субстанции и двигательная активность мышей при введении препарата афобазол, взаимодействующего с Sigma-1 и МТ1 рецепторами, а также регуляторными сайтами МАО А и хинонредуктазы 2 (NQO2, MT3 рецептор), и его основного метаболита М-11, селективно взаимодействующего лишь с регуляторным сайтом NQO2. Препараты вводили внутрибрюшинно в течение 14 дней с началом курса через 30 мин после унилатерального интрастриатного введения 5 мкг 6-гидроксидофамина (6-OHDA). Афобазол и М-11 проявляли нейропротекторные свойства. Во всех экспериментах эффективная доза афобазола (2.5 мг/кг) оказалась значимо ниже в сравнении с М-11 (7.5 мг/кг). В использованной экспериментальной модели показан вклад NQO2 в противопаркинсоническое действие афобазола наряду с другими мишенями препарата.
Список сокращений
АФК – активные формы кислорода
ГВК – гомованилиновая кислота
ДА – дофамин
ДОФУК – дигидроксифенилуксуная кислота
М-11 – 2-[2-(3-оксоморфолин-4-ил)-этилтио]-5-этоксибензимидазола гидрохлорид
6-OHDA – 6-гидроксидофамин
DAPI – 4',6-диамидино-2-фенилиндол
DAQ – дофамин-хинон
DHBA – 3,4-дигидроксибензиламин
МТ3 рецептор – мелатониновый рецептор третьего типа
NQO2 – хинонредуктаза 2
Sigma1R – сигма-1 рецептор
SNc – компактная часть черной субстанции
TH – тирозингидроксилаза
ВВЕДЕНИЕ
Болезнь Паркинсона – распространенное нейродегенеративное заболевание, клинически проявляющееся брадикинезией, тремором в покое, мышечной ригидностью и постуральной неустойчивостью. Симптомы болезни Паркинсона обусловлены гибелью дофаминергических нейронов компактной части черной субстанции (Substantia nigra pars compacta, SNc) и их терминалей в стриатуме. К настоящему времени не существует эффективных способов медикаментозной терапии заболевания [1]. Поэтому чрезвычайно актуален поиск новых фармакологических мишеней, регуляция которых способна обеспечить защиту и восстановление функциональной активности нейронов нигростриатного пути.
Морфологические и нейрохимические особенности нейронов SNc обуславливают их восприимчивость к повреждающим влияниям как в наследственных, так и идиопатической формах болезни Паркинсона [2–5]. Основными механизмами повреждения нейронов хинонными производными дофамина (DAQ) считаются ковалентное связывание со свободным цистеином и тиольными группами белков [6], усиление продукции АФК в окислительно-восстановительных циклах [7], образование ДА аддуктов с ДНК [8, 9]. В нейронах SNc дофамин (ДА) способен автоокисляться с образованием DAQ и АФК [10]. Поэтому важная роль в процессе повреждения клетки DAQ принадлежит хинонредуктазе 2 (NQO2, EC 1.10.5.1). Этот флавопротеин экспрессируется в головном мозге и обладает способностью восстанавливать хинонные производные катехоламинов с образованием их нестабильных хинольных форм и усиливать образование АФК [11–13]. Фермент обладает регуляторным сайтом, тождественным мелатониновому рецептору третьего типа (МТ3) [14]. У пациентов с болезнью Паркинсона чаще встречается мутантный вариант гена NQO2 с высокой экспрессией фермента, что согласуется с усилением продукции АФК в условиях in vitro [15]. Одним из доноров электронов для NQO2 служит N‑метилдигидроникотинамид (N-methyldihydronicotinamide) [16]. У больных обнаружено повышение уровня фермента никотинамид N-метилтрансферазы, синтезирующего окисленную форму кофактора (N-methylnicotinamide) [17, 18]. Не исключено, что NQO2 может действовать и как флавиновый переключатель (flavin redox switch), который в зависимости от наличия в клетке субстратов фермента и окислительно-восстановительного потенциала FAD меняет конформацию, приобретает способность вступать во взаимодействие с различными низкомолекулярными веществами и влиять на активность белков [19–21]. Известно, что дофамин способствует активации белка p53 и развитию апоптоза нейронов [22], тогда как ингибиторы p53 оказывают защитное действие на дофаминергические нейроны [23, 24]. Эти соединения нормализуют двигательную активность при моделировании болезни Паркинсона [23]. Показано, что при угнетении митохондриального дыхания NQO2 способен к перемещению в ядро, где стабилизирует и активирует p53. Применение shRNA или ингибитора NQO2 препятствует стабилизации p53 [25]. Литературные данные указывают на нейропротекторную активность лигандов МТ3 рецептора, ингибирующих фермент [26, 27].
В ФГБНУ “НИИ фармакологии им. В.В. Закусова” разработан анксиолитик афобазол (5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид) [28]. Установлены лигандные свойства препарата к МТ3 рецептору (Ki = 9.7E-7 M) [29] и его способность ингибировать NQO2 [30]. Другими мишенями афобазола являются шаперон Sigma1R (Ki = 5.9E-6 M), регуляторный сайт МАО‑А (Ki = 3.6E-6 M), мелатониновый рецептор МТ1 (Ki = 1.6E-5 M) [29]. В экспериментах in vitro и in vivo установлена нейропротекторная активность афобазола [31–33]. При моделировании болезни Паркинсона введением 6‑OHDA афобазол препятствовал снижению уровня ДА в стриатуме опытных мышей [34]. Однако при мультитаргетных взаимодействиях препарата вклад NQO2 в противопаркинсонический эффект остаётся неизвестным. В наших исследованиях найден удобный инструмент для анализа участия NQO2 в фармакодинамике афобазола. Оказалось, что основной метаболит афобазола соединение 2-[2-(3-оксоморфолин-4-ил)-этилтио]-5-этоксибензимидазола гидрохлорид (М-11) [35] среди молекулярных мишеней афобазола взаимодействует лишь с МТ3 рецептором (Ki = 3.9E-7 M) [29]. М-11 ингибирует фермент в сопоставимых с афобазолом концентрациях [36]. Таким образом, анализ различий в эффектах афобазола и М-11 позволит оценить вклад NQO2 в эффекты исходного соединения. Поэтому целью настоящей работы стало сравнительное изучение действия афобазола и его метаболита М-11 при моделировании болезни Паркинсона интрастриатным введением 6-OHDA.
МАТЕРИАЛЫ И МЕТОДЫ
Реактивы. В работе использованы следующие реактивы: 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид (афобазол) и его основной метаболит 2-[2-(3-оксоморфолин-4-ил)-этилтио]-5-этоксибензимидазола гидрохлорид (М-11) (ФГБНУ “НИИ фармакологии имени В.В. Закусова”, Россия), аскорбиновая кислота, NaCl, HClO4, сахароза, формальдегид, поликлональные антитела к тирозингидроксилазе Т8700, вторичные антитела, конъюгированные с флуорохромом CF488 SAB4600045, 4',6-диамидино-2-фенилиндол (DAPI), среда FluoroShield (Sigma-Aldrich, США), Тритон-Х100 (Amresco, США) хлоралгидрат (Serva, Германия), 3,4-дигидроксибензиламин (DHBA), дофамин (ДА), дигидроксифенилуксусная кислота (ДОФУК), гомованилиновая кислота (ГВК), KH2PO4, H3PO4, лимонная кислота, EDTA-Na2, октансульфоновая кислота, ацетонитрил (Fluka, США), среда Tissue Tek O.C.T (Fisher Scientific, США).
Животные. Эксперименты выполнены на мышах-самцах аутбредной линии ICR (CD-1) массой 25–30 г (n = 74), полученных из НПП Питомника лабораторных животных ФИБХ. Мышей содержали в условиях вивария (20–22°С, относительная влажность 30–70%, 12-ти часовой световой цикл) по 5–10 особей в пластиковых клетках со свободным доступом к пище и воде. Согласно решению комиссии ФГБНУ “НИИ фармакологии им. В.В. Закусова” по биоэтике все процедуры в исследовании соответствовали этическим принципам обращения с животными.
Моделирование болезни Паркинсона интрастриатным введением 6-OHDA. За 30 мин до операции животное анестезировали хлоралгидратом (400 мг/кг, внутрибрюшинно) и помещали в стереотаксические рамки (Stoelting Motorized Stereotaxis, Stoelting Co., Великобритания), где опытным животным однократно вводили 6‑OHDA в правый стриатум в координатах A = = 0.4; L = 1.8; V = –3.5, относительно брегмы [37]. Концентрация 6-OHDA составляла 5 мкг на 1 мкл раствора, содержащего 0.9% NaCl и 0.02% аскорбиновой кислоты. Опытным животным вводили 1 мкл раствора 6-OHDA со скоростью 0.5 мкл/мин гамильтоновским шприцом с иглой из нержавеющей стали (30 gauge), иглу извлекали через 2 мин после окончания инъекции. Ложнооперированным животным вводили 1 мкл контрольного раствора, содержащего 0.9% NaCl и 0.02% аскорбиновой кислоты, в тех же стереотаксических координатах.
Схема введения препаратов. Афобазол в дозе 2.5 мг/кг, М-11 в дозах 2.5 и 7.5 мг/кг и плацебо (вода для инъекций) вводили в/б ежедневно на протяжении 14 сут с началом курса через 30 мин после интрастриатной инъекции 6-OHDA или контрольного раствора. Растворы препаратов вводили из расчета 0.1 мл на 10 г массы животного. Животные были разделены на 5 экспериментальных групп: ложнооперированные, получавшие плацебо (n = 14); опытные, получавшие плацебо (n = 16); опытные, получавшие афобазол (n = 14); опытные, получавшие М-11 в дозе 2.5 мг/кг (n = = 15) и опытные, получавшие М-11 в дозе 7.5 мг/кг (n = 15).
Тест “вращающийся стержень”. Исследование двигательной активности мышей линии ICR (CD-1) в тесте “вращающийся стержень” проводили на экспериментальной установке Rota-rod/RS LE 8500 (диаметр стержня 3.2 см) (Panlab/Harvard Apparatus). Для адаптации мышей в установке и исключения из исследования малоподвижных животных в эксперимент включены две обучающие сессии. Первую обучающую сессию проводили на 12 сут после введения 6-OHDA. Каждое животное дважды помещалось в экспериментальную установку при скорости вращения стержня 4 об./мин с перерывом не менее 60 мин. Вторую обучающую сессию проводили на 13 сутки после введения 6-OHDA при скорости вращения стержня 10 об./мин. Манипуляции с животными были аналогичны первой обучающей сессии. Животные, находившиеся на вращающемся стержне менее минуты в одной из обучающих сессий, исключались из эксперимента [38].
На 14 сут после введения 6-OHDA исследовали способность мышей удерживаться в тесте при постоянной и увеличивающейся скоростях вращения стержня. Для этого экспериментальное животное помещали на вращающийся стержень с постоянной скоростью 20 об./мин и засекали время до падения животного со стержня на платформу. Во втором варианте теста экспериментальное животное помещали на стержень, вращающийся с начальной скоростью 4 об./мин и замеряли время до падения на платформу. Максимальное число оборотов (40 об./мин) [39] достигалось за 5 мин. Каждое животное проходило оба варианта теста по 3 раза с перерывом 30 мин между попытками. Замер времени прекращали после 120 с удерживания животного на стержне при фиксированной скорости вращения и 180 с при возрастающей. Для статистической обработки данных каждого варианта теста отбиралось максимальное время удерживания из трех попыток. Двигательная активность в тесте определена для 5 экспериментальных групп: ложнооперированные, получавшие плацебо (n = 7); опытные, получавшие плацебо (n = 9); опытные, получавшие афобазол (n = 9); опытные, получавшие М-11 в дозе 2.5 мг/кг (n = 10) и опытные, получавшие М-11 в дозе 7.5 мг/кг (n = 10).
Метод ВЭЖХ-ЭД. ДА и его метаболиты определяли в поврежденном и интактном стриатумах головного мозга мышей методом высокоэффективной жидкостной хроматографии с электрохимической детекцией (ВЭЖХ-ЭД). Через 14 сут после введения 6-OHDA мышей декапитировали и извлекали головной мозг. Левый и правый стриатумы выделяли при температуре тающего льда (0–4°С) на фильтровальной бумаге, смоченной 0.32 М раствором сахарозы. Каждый выделенный стриатум замораживали в жидком азоте (–196°С), взвешивали и хранили при температуре –80°C. Для определения содержания моноаминов и их метаболитов выделенный стриатум гомогенизировали в 0.1 M НClO4 с добавлением в качестве внутреннего стандарта 3,4-дигидоксибензиламина (DHBA) в концентрации 0.25 нмоль/мл в шаровом гомогенизаторе Tissue LyserLT (Quiagen, Германия) с частотой 45 ударов в минуту в течение 5 мин. Пробы центрифугировали при 10 000× g в течение 10 мин при 4°C. Надосадочную жидкость в объеме 20 мкл наносили на аналитическую колонку Kromasil C-18 4.6 × 150 (Dr.Maisch, Германия) с помощью автосемплера SIL-20 ACHT (Shimadzu, Япония). Моноамины и их метаболиты разделяли на колонке с использованием в качестве подвижной фазы 0.1 М цитратно-фосфатного буфера, содержащего 0.3 мМ октансульфоната натрия, 0.1 мМ ЭДТА и 8% ацетонитрила (рН 3.0). Определение моноаминов и их метаболитов осуществляли на хроматографической станции Shimadzu LC-20 Prominence (Shimadzu, Япония) с использованием электрохимической ячейки ESA 5011 (Е1 = –175; Е2 = +250) и электрохимического детектора Coulochem III (ESA, США). Скорость потока подвижной фазы составляла 1 мл/мин. Полученные результаты обрабатывали на ПК с использованием программно-аппаратного комплекса “Мультихром 1.5” (Ampersand, Россия). Анализ моноаминов в стриатумах проводили в 5 экспериментальных группах: ложнооперированные, получавшие плацебо (n = 14); опытные, получавшие плацебо (n = 16); опытные, получавшие афобазол (n = 14); опытные, получавшие М-11 в дозе 2.5 мг/кг (n = 15) и опытные, получавшие М-11 в дозе 7.5 мг/кг (n = 15).
Иммунгистохимический анализ. Через 14 сут после введения 6-OHDA мышей декапитировали и извлекали головной мозг. Мозг животных фиксировали в 4% формалине, приготовленном на фосфатном солевом буфере (PBS, pH 7.2–7.4) 24 ч. Образцы выдерживали в 30% растворе сахарозы, и заливали средой Tissue Tek O.C.T. после чего на замораживающем микротоме Sakura Tissue Tek Cryo3 готовили серию фронтальных срезов среднего мозга, толщиной 12 мкм в области черной субстанции. Срезы перед окрашиванием подвергали процедуре тепловой демаскировки антигена в микроволновой печи (0.1 М цитратный буфер, pH 6.0, 600 W, 5 мин), после остывания их выдерживали в PBS с 0.1% Тритон X-100 в течение часа. Для выявления дофаминовых нейронов черной субстанции срезы инкубировали 16 ч с кроличьими поликлональными антителами к тирозингидроксилазе (TH, Sigma, T8700), в разведении 1 : 500. Связывание иммуноглобулинов кролика визуализировали при помощи антител козы, конъюгированных с флуорохромом CF488 (Sigma) [40]. Ядра клеток подкрашивали с помощью DAPI. Срезы заключали в среду FluoroShield под покровные стекла и исследовали под флуоресцентным микроскопом Nikon Eclipse, c соответсвующим набором фильтров. Изображения получали при помощи цифровой камеры Nikon DS-Qi.
В программе ImageJ (NIH), подсчитывали плотность TH-позитивных (TH+) нейронов с ядрами, попадающими в плоскость среза, при увеличении объектива ×40. Подсчет проводили на 3–6 срезах с каждого животного, взятых на разных уровнях черной субстанции (в ростральном, медиальном, каудальном отделах). На каждом срезе на ипси- и контралатеральной сторонах относительно введения 6-OHDA клетки подсчитывали в 5–7 полях зрения (площадью 0.044 мм2). Полученные данные усредняли для каждого животного. Изменения плотности нейронов в черной субстанции на стороне повреждения выражали в процентах, относительно противоположного полушария. Содержания TH определяли в группах ложнооперированных животных (n = 6) и опытных животных, получавших плацебо (n = 7), афобазол (n = 9) опытные, получавшие М-11 в дозе 2.5 мг/кг (n = 6) и опытные, получавшие М-11 в дозе 7.5 мг/кг (n = 8).
Статистическая обработка экспериментальных данных. Соответствие полученных данных нормальному распределению проверяли критериями Д’Агостино–Пирсона и Шапиро–Уилка. Так как распределение данных содержания ДА, его метаболитов и времени удерживания мышей на вращающемся стержне в значительной части выборок отличалось от нормального, то для дальнейшей обработки использовали методы непараметрической статистики. Оценку статистической значимости различий проводили с применением знаково-рангового критерия Вилкоксона и непараметрического аналога дисперсионного анализа по Краскелу–Уоллису с обработкой методом множественных сравнений по Данну (Kruskal–Wallis test, Dunn’s post hoc). Оценку статистической значимости различий плотности TH+ нейронов проводили с применением однофакторного дисперсионного анализа с обработкой методом множественных сравнений по Holm–Sidak. Табличные данные представлены в виде медиан и нижнего и верхнего квартилей (Mdn (q25–75)). Данные на гистограммах представлены в виде медиан (минимум–максимум) (Mdn (min–max) с указанием среднего значения. Статистическая обработка и визуализация полученных данных осуществлялись с помощью программного пакета GraphPad Prism 8.0.1 (GraphPad Software, San Diego California USA (www.graphpad.com).
РЕЗУЛЬТАТЫ
На 14-е сут после инъекции 6-OHDA в правый стриатум мышей ICR, получавших плацебо, зарегистрировано более чем двукратное снижение уровня ДА по сравнению с их интактным стриатумом и поврежденным стриатумом ложнооперированных мышей (рис. 1). Внутрибрюшинное ведение афобазола в дозе 2.5 мг/кг на протяжении 14-ти дней с началом курса через 30 мин после интрастриатного введения 6-OHDA статистически значимо повышало уровень ДА в поврежденном стриатуме опытных мышей и восстанавливало его содержание до значений, зафиксированных у ложнооперированных животных (рис. 1). Хроническое введение М-11 в дозе 2.5 мг/кг не изменяло содержания ДА в поврежденном стриатуме опытных животных (рис. 1). Однако увеличение дозы М-11 до 7.5 мг/кг вызывало увеличение уровня ДА в поврежденном стриатуме, аналогично действию афобазола. Афобазол и М-11 в обеих дозах не влияли на уровень ДА в интактном стриатуме опытных мышей в сравнении с контрольными значениями (рис. 1).
Рис. 1.
Действие афобазола и М-11 на содержание дофамина в стриатуме мышей при моделировании
болезни Паркинсона. Примечания. Данные представлены в виде Mdn (min–max). “+” – среднее
значение. Л.О. – ложнооперированные животные. 6-OHDA – опытные животные.  – интактный стриатум,
– интактный стриатум,  – поврежденный стриатум. Kruskal–Wallis test, Dunn’s post hoc: ** p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом ложнооперированных
животных, получавших плацебо. ## p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных, получавших плацебо. mp < 0.05, mmp < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных получавших М-11 в дозе 2.5 мг/кг. Wilcoxon test: ^ p < < 0.05, ^^ p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом в
отдельной экспериментальной группе.
– поврежденный стриатум. Kruskal–Wallis test, Dunn’s post hoc: ** p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом ложнооперированных
животных, получавших плацебо. ## p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных, получавших плацебо. mp < 0.05, mmp < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных получавших М-11 в дозе 2.5 мг/кг. Wilcoxon test: ^ p < < 0.05, ^^ p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом в
отдельной экспериментальной группе.

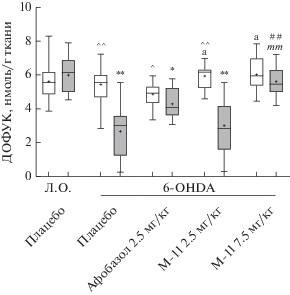
Снижение уровня ДОФУК в поврежденном стриатуме опытных животных соответствует уменьшению содержания ДА в данной экспериментальной группе (рис. 2). Несмотря на восстанавливающее влияние афобазола на ДА в группе опытных животных, содержание ДОФУК в повреждённом стриатуме сохранилось на более низком уровне в сравнении с ложнооперированными животными (рис. 2). Количество ДОФУК в интактном стриатуме животных, получавших афобазол, также оказалось значимо ниже по сравнению с интактным стриатумом животных, получавших М-11 в обеих дозах (рис. 2). Соединение М-11 в дозе 2.5 мг/кг не оказывало влияния на уровень ДОФУК в поврежденном 6-OHDA стриатуме. В дозе 7.5 мг/кг метаболит М-11 вызывал повышение уровня ДОФУК до контрольных значений (рис. 2).
Рис. 2.
Действие афобазола и М-11 на содержание ДОФУК в стриатуме мышей при моделировании
болезни Паркинсона. Примечания. Данные представлены в виде Mdn (min–max). “+” – среднее
значение. Л.О. – ложнооперированные животные. 6-OHDA – опытные животные.  – интактный стриатум,
– интактный стриатум,  – поврежденный стриатум. Kruskal–Wallis test, Dunn’s post hoc: * p < 0.05, ** p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом ложнооперированных
животных, получавших плацебо. ##p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных, получавших плацебо. mmp < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных получавших М-11 в дозе 2.5 мг/кг. a p < 0.05 – статистически значимые различия по сравнению с интактным стриатумом опытных
животных получавших афобазол в дозе 2.5 мг/кг. Wilcoxon test: ^ p < 0.05, ^^ p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом в
отдельной экспериментальной группе.
– поврежденный стриатум. Kruskal–Wallis test, Dunn’s post hoc: * p < 0.05, ** p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом ложнооперированных
животных, получавших плацебо. ##p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных, получавших плацебо. mmp < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных получавших М-11 в дозе 2.5 мг/кг. a p < 0.05 – статистически значимые различия по сравнению с интактным стриатумом опытных
животных получавших афобазол в дозе 2.5 мг/кг. Wilcoxon test: ^ p < 0.05, ^^ p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом в
отдельной экспериментальной группе.
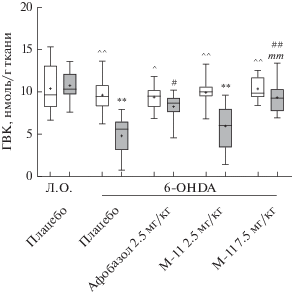
Уровень ГВК в поврежденном стриатуме опытных животных, получавших плацебо, понижался по сравнению с контралатеральным стриатумом и поврежденным стриатумом ложнооперированных животных (рис. 3). Соединение М-11 в дозе 2.5 мг/кг не влияло на уровень ГВК (рис. 3). Афобазол в дозе 2.5 мг/кг и М-11 в дозе 7.5 мг/кг статистически значимо повышали содержание ГВК в поврежденном стриатуме по сравнению с группой плацебо, которое, однако, не достигало уровня контралатерального стриатума (рис. 3). В отличие от М-11 в дозе 7.5 мг/кг действие афобазола и М-11 в дозе 2.5 мг/кг статистически значимо не различались.
Рис. 3.
Действие афобазола и М-11 на содержание ГВК в стриатуме мышей при моделировании болезни
Паркинсона. Примечания. Данные представлены в виде Mdn (min–max). “+” – среднее значение.
Л.О. – ложнооперированные животные. 6-OHDA – опытные животные.  – интактный стриатум,
– интактный стриатум,  – поврежденный стриатум. Kruskal–Wallis test, Dunn’s post hoc: ** p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом ложнооперированных
животных, получавших плацебо. # p < 0.05, ## p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных, получавших плацебо. mm p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных получавших М-11 в дозе 2.5 мг/кг. Wilcoxon test: ^ p < 0.05, ^^ p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом в
отдельной экспериментальной группе.
– поврежденный стриатум. Kruskal–Wallis test, Dunn’s post hoc: ** p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом ложнооперированных
животных, получавших плацебо. # p < 0.05, ## p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных, получавших плацебо. mm p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных
животных получавших М-11 в дозе 2.5 мг/кг. Wilcoxon test: ^ p < 0.05, ^^ p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом в
отдельной экспериментальной группе.
Через 14 сут после введения 6-OHDA наряду с падением уровня ДА в стриатуме зафиксировано уменьшение плотности ТН+ нейронов в ипсилатеральной SNc (рис. 4) и почти двукратное снижение их количества относительно контралатеральной стороны (рис. 5). Афобазол в дозе 2.5 мг/кг статистически значимо увеличивал плотность и относительное количество TH+ нейронов в ипсилатеральной SNc (рис. 4, 5). Соединение М-11 в дозе 2.5 мг/кг не влияло на количество ТН+ нейронов в SNc опытных мышей. При увеличении дозы М-11 до 7.5 мг/кг метаболит статистически значимо увеличивал количество ТН+ нейронов в ипсилатеральной SNc (рис. 4, 5).
Рис. 4.
Влияние афобазола и М-11 на число ТН+ нейронов в ипсилатеральной черной субстанции при моделировании болезни Паркинсона. Примечания. Л.О. – ложнооперированные животные. 6-OHDA – опытные животные. Снимки сделаны при увеличении объектива ×10.

Рис. 5.
Влияние афобазола и М-11 на относительное количество TH+ нейронов в ипсилатеральной черной субстанции при моделировании болезни Паркинсона. Примечания. Данные представлены в виде Mdn (min–max). “+” – среднее значение. Л.О. – ложнооперированные животные. 6-OHDA – опытные животные. Kruskal–Wallis test, Dunn’s post hoc: ** p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом ложнооперированных животных, получавших плацебо. # p < 0.05, ## p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных животных, получавших плацебо. mm p < 0.01 – статистически значимые различия по сравнению с поврежденным стриатумом опытных животных получавших М-11 в дозе 2.5 мг/кг.

В тесте “вращающийся стержень” при постоянной скорости вращения медиана времени удерживания опытных животных, получавших плацебо, уменьшилась по сравнению с ложнооперированными животными (табл. 1). Хроническое введение М-11 в дозе 2.5 мг/кг не изменяло время удерживания опытных животных (табл. 1). Афобазол в дозе 2.5 мг/кг увеличивал время удерживания на стержне при фиксированной скорости вращения в сравнении с опытными животными, получавшими плацебо и М-11 в дозе 2.5 мг/кг. Увеличение дозы М-11 до 7.5 мг/кг приводило к увеличению времени удерживания опытных животных на стержне, аналогично действию афобазола. Схожие зависимости были зарегистрированы в тесте с возрастающей скоростью вращения стержня (табл. 1). Медиана времени удерживания на стержне опытных животных, получавших плацебо, в этих условиях снизилась в 1.85 раза по сравнению с ложнооперированными животными (табл. 1). Соединение М-11 в дозе 2.5 мг/кг не влияло на токсическое действие 6-OHDA. Афобазол или М-11 в дозе 7.5 мг/кг нивелировали действие 6-OHDA, приводя время удерживания на стержне этих животных к контрольным значениям. (табл. 1).
Таблица 1.
Влияние 14-дневного введения афобазола и М-11 на время удерживания мышей на стержне при фиксированной и возрастающей скоростях вращения
| Экспериментальные группы | Время удерживания, с | ||
|---|---|---|---|
| 20 об/мин | 4–40 (об/мин) | ||
| Л.О. | Плацебо (n = 7) |
120 (116–120) |
180 (170–180) |
| 6-OHDA | Плацебо (n = 9) |
17 (13–20.5) **p = 0.0003 |
97 (71–123.5) **p = 0.008 |
| Афобазол 2.5 мг/кг (n = 9) |
92 (72.5–118.5) #p = 0.011 mmp = 0.006 |
178 (154–180) #p = 0.02 mmp = 0.007 |
|
| М-11 2.5 мг/кг (n = 10) |
15 (13.5–22) **p = 0.0001 |
72 (54.5–104) **p = 0.0003 |
|
| М-11 7.5 мг/кг (n = 10) |
88.5 (34–118.5) #p = 0.034 mp = 0.019 |
175.5 (156.8–180) #p = 0.029 mmp = 0.001 |
|
Данные представлены в виде Mdn (q25–75). n – количество животных в группе. Л.О. – ложнооперированные животные. 6‑OHDA – опытные животные. Kruskal–Wallis test, Dunn’s post hoc: ** p < 0.001 – статистически значимые различия по сравнению с группой ложнооперированных животных, получавших плацебо. # p < 0.05 – статистически значимые различия по сравнению с группой опытных животных, получавших плацебо. m p < 0.05, mm p < 0.01 – статистически значимые различия по сравнению с группой опытных животных, получавших М-11 в дозе 2.5 мг/кг.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Содержание ДА и его метаболитов в стриатуме контрольных и опытных животных соответствуют ранее опубликованным данным [34]. Наряду со снижением уровня ДА в стриатуме опытных животных выявлено уменьшение числа ТН+ нейронов SNc, что соответствует ретроградному токсическому действию 6-OHDA [37, 38]. На увеличенной выборке животных повторно показана способность афобазола восстанавливать уровень ДА в стриатуме опытных мышей [34]. Совместно с повышением содержания ДА в стриатуме введение афобазола приводило к увеличению количества TH+ нейронов SNc, что свидетельствует о нейропротекторной активности препарата [38, 41–43]. Нейрохимические изменения, выявленные при курсовом применении афобазола, сопровождались нормализацией двигательной активности опытных животных в тесте “вращающийся стержень”, что подтверждает наши предыдущие данные [44] и указывает на возможность предотвращения двигательных нарушений при профилактическом введении препарата. Судя по литературным данным, все известные белковые мишени афобазола могут опосредовать антипаркинсоническое действие препарата [45–49]. В качестве удобного инструмента для изучения вклада NQO2 в механизмы действия афобазола нами предложено сравнение эффектов препарата с действием его метаболита М‑11. Ранее на модели менадионовой генотоксичности в суспензии клеток костного мозга мышей афобазол и его метаболит М-11, селективно взаимодействующий с МТ3 рецептором, проявляли цитопротекторное свойства в разных концентрациях [50, 51]. Эти результаты согласуются с настоящим исследованием in vivo, где афобазол был эффективен в дозе 2.5 мг/кг, а М-11 лишь при увеличении дозы до 7.5 мг/кг вызывал восстановление уровня ДА стриатума, увеличение количества ТН+ нейронов ипсилатеральной SNc и нормализацию двигательной активности опытных животных. Полученные данные демонстрируют роль NQO2 в развитии нейропротекторного действия афобазола и соответствуют ранее опубликованным работам. Так, внесение ингибитора NQO2 в культуры клеток нейробластомы или нейронов, инкубируемых в присутствии косубстрата и адренохрома, снижало продукцию АФК [52]. Ингибиторы фермента препятствовали снижению активности митохондрий и повышали выживаемость гиппокампальных нейронов при действии менадиона или депривации ростовой среды [26]. На модели культуры астроцитов показано негативное влияние параквата на процесс аутофагии, которое устранялось ингибированием NQO2 [47]. Нокаутные по гену NQO2 мыши демонстрировали более высокую обучаемость в тесте вращающийся стержень, что выражалось в увеличении времени удерживания [26].
В нашем исследовании выявлены количественные различия в нейропротекторном действии афобазола и М-11. Эффективная доза афобазола оказалась в три раза меньше в сравнении с М-11. Схожие зависимости действия афобазола и М-11 от концентраций были установлены ранее в экспериментах in vitro [50, 51], где с использованием селективных лигандов доказан вклад Sigma1R в цитопротекторное действие афобазола [50]. В настоящей работе оба препарата восстанавливали уровень ДА стриатума опытных животных, однако афобазол значимо не влиял на содержание ДОФУК, что можно связать с его ингибирующим влиянием на МАО-А [53].
ЗАКЛЮЧЕНИЕ
Таким образом, полученные данные позволяют сделать заключение о вкладе NQO2 в механизмы нейропротекторного действия афобазола в использованной экспериментальной модели болезни Паркинсона. Установленные результаты и ранее выполненные исследования показали, что в фармакодинамике афобазола шаперон Sigma1R и NQO2 аддитивно формируют нейропротекторный эффект.
Список литературы
Poewe W., Seppi K., Tanner C.M., Halliday G.M., Brundin P., Volkmann J., Schrag A.E., Lang A.E. // Nat. Rev. Dis. Primers. 2017. V. 3. P. 17013.
Pissadaki E.K., Bolam J.P. // Front. Comput. Neurosci. 2013. V. 7. P. 13.
Harris J.J., Jolivet R., Attwell D. // Neuron. 2012. V. 75. № 5. P. 762–777.
Bolam J.P., Pissadaki E.K. // Mov. Disord. 2012. V. 27. № 12. P. 1478–1483.
Guzman J.N., Sanchez-Padilla J., Wokosin D., Kondapalli J., Ilijic E., Schumacker P.T., Surmeier D.J. // Nature. 2010. V. 468. № 7324. P. 696–700.
Sulzer D., Zecca L. // Neurotox. Res. 2000. V. 1. № 3. P. 181–195.
Bolton J.L., Trush M.A., Penning T.M., Dryhurst G., Monks T.J. // Chem. Res. Toxicol., 2000. V. 13. № 3. P. 135–160.
Zahid M., Saeed M., Yang L., Beseler C., Rogan E., Cavalieri E.L. // IUBMB Life. 2011. V. 63. № 12. P. 1087–1093.
Gurusamy P., Muthukumar K., Rajesh S., Muneeswaran G., Perumal S., Karunakaran C. // J. Struct. Biol. 2012. V. 180. № 1. P. 125–131.
Herrera A., Munoz P., Steinbusch H.W.M., Segura-Aguilar J. // ACS Chem. Neurosci. 2017. V. 8. № 4. P. 702–711.
Fu Y., Buryanovskyy L., Zhang Z. // J. Biol. Chem. 2008. V. 283. № 35. P. 23829–23835.
Cassagnes L.E., Perio P., Ferry G., Moulharat N., Antoine M., Gayon R., Boutin J.A., Nepveu F., Reybier K. // Free Radic. Biol. Med. 2015. V. 89. P. 126–134.
Reybier K., Perio P., Ferry G., Bouajila J., Delagrange P., Boutin J.A., Nepveu F. // Free Radic. Res. 2011. V. 45. № 10. P. 1184–1195.
Boutin J.A., Ferry G. // J. Pharmacol. Exp. Ther. 2019. V. 368. № 1. P. 59–65.
Wang W., Le W.D., Pan T., Stringer J.L., Jaiswal A.K. // J. Gerontol. A. Biol. Sci. Med. Sci. 2008. V. 63. № 2. P. 127–134.
Calamini B., Santarsiero B.D., Boutin J.A., Mesecar A.D. // Biochem. J. 2008. V. 413. № 1. P. 81–91.
Parsons R.B., Smith S.W., Waring R.H., Williams A.C., Ramsden D.B. // Neurosci. Lett. 2003. V. 342. № 1–2. P. 13–16.
Aoyama K., Matsubara K., Kondo M., Murakawa Y., Suno M., Yamashita K., Yamaguchi S., Kobayashi S. // Neurosci. Lett. 2001. V. 298. № 1. P. 78–80.
Leung K.K., Shilton B.H. // Biochemistry. 2015. V. 54. № 51. P. 7438–7448.
Leung K.K., Shilton B.H. // J. Biol. Chem. 2013. V. 288. № 16. P. 11242–11251.
Senda T., Senda M., Kimura S., Ishida T. // Antioxid. Redox Signal. 2009. V. 11. № 7. P. 1741–1766.
Daily D., Barzilai A., Offen D., Kamsler A., Melamed E., Ziv I. // Cell. Mol. Neurobiol. 1999. V. 19. № 2. P. 261–276.
Duan W., Zhu X., Ladenheim B., Yu Q.S., Guo Z., Oyler J., Cutler R.G., Cadet J.L., Greig N.H., Mattson M.P. // Ann. Neurol. 2002. V. 52. № 5. P. 597–606.
Nair V.D. // Apoptosis. 2006. V. 11. № 6. P. 955–966.
Khutornenko A.A., Roudko V.V., Chernyak B.V., Vartapetian A.B., Chumakov P.M., Evstafieva A.G. // Proc. Natl. Acad. Sci. USA. 2010. V. 107. № 29. P. 12828–12833.
Benoit C.E., Bastianetto S., Brouillette J., Tse Y., Boutin J.A., Delagrange P., Wong T., Sarret P., Quirion R. // J. Neurosci. 2010. V. 30. № 38. P. 12690–12700.
Janda E., Parafati M., Aprigliano S., Carresi C., Visalli V., Sacco I., Ventrice D., Mega T., Vadala N., Rinaldi S., Musolino V., Palma E., Gratteri S., Rotiroti D.,Mollace V. // Br. J. Pharmacol. 2013. V. 168. № 1. P. 46–59.
Seredenin S.B., Voronina T.A., Neznamov G.G., Blednov Iu A., Badyshtov B.A., Viglinskaia I.V., Kozlovskaia M.M., Kolotilinskaia N.V., Iarkova M.A., Savel’ev V.L., Garibova T.L., Val’dman E.A. // Vestn. Ross. Akad. Med. Nauk. 1998. V. @. № 11. P. 3–9.
Seredenin S.B., Voronin M.V. // Eksp. Klin. Farmakol. 2009. V. 72. № 1. P. 3–11.
Kadnikov I.A., Voronin M.V., Seredenin S.B. // Pharm. Chem. J. 2014. V. 47. № 10. P. 514–516.
Zenina T.A., Gavrish I.V., Melkumyan D.S., Seredenina T.S., Seredenin S.B. // Bull. Exp. Biol. Med. 2005. V. 140. № 2. P. 194–196.
Galaeva I.P., Garibova T.L., Voronina T.A., Seredenin S.B. // Bull. Exp. Biol. Med. 2005. V. 140. № 5. P. 535–537.
Kraineva V.A., Seredenin S.B. // Bull. Exp. Biol. Med. 2010. V. 149. № 2. P. 204–207.
Voronin M.V., Kadnikov I.A., Seredenin S.B. // Neurochem. J. 2019. V. 13. № 1. P. 49–56.
Seredenin S.B., Viglinskaya A.O., Mozhaeva T.Y., Kolyvanov G.B., Litvin A.A., Avdyunina N.I., Savel’ev V.L., Zherdev V.P. // Eksp. Klin. Farmakol. 2008. V. 71. № 2. P. 50–52.
Kadnikov I.A., Voronin M.V., Seredenin S.B. // Farmakokinetika i farmakodinamika. 2018. № 3. P. 27–30.
Alvarez-Fischer D., Henze C., Strenzke C., Westrich J., Ferger B., Hoglinger G.U., Oertel W.H.,Hartmann A. // Exp. Neurol. 2008. V. 210. № 1. P. 182–193.
Goes A.T.R., Jesse C.R., Antunes M.S., Lobo Ladd F.V., Lobo Ladd A.A.B., Luchese C., Paroul N., Boeira S.P. // Chem. Biol. Interact. 2018. V. 279. P. 111–120.
Brooks S.P., Dunnett S.B. // Nat. Rev. Neurosci. 2009. V. 10. № 7. P. 519–529.
Grigoriev I.P., Vasilenko M.S., Sukhorukova E.G.,Korzhevskii D.E. // Morfologiia. 2010. V. 138. № 6. P. 60–63.
Sadan O., Bahat-Stromza M., Barhum Y., Levy Y.S., Pisnevsky A., Peretz H., Ilan A.B., Bulvik S., Shemesh N., Krepel D., Cohen Y., Melamed E., Offen D. // Stem Cells Dev. 2009. V. 18. № 8. P. 1179–1190.
Aguiar L.M., Nobre H.V., Jr., Macedo D.S., Oliveira A.A., Freitas R.M., Vasconcelos S.M., Cunha G.M., Sousa F.C., Viana G.S. // Pharmacol. Biochem. Behav. 2006. V. 84. № 3. P. 415–419.
Chan H.H., Kumar S., Zhuo L. // Eur. J. Pharmacol. 2013. V. 715. № 1–3. P. 405–413.
Kadnikov I.A., Voronin M.V., Seredenin S.B. // Farmakokinetika i farmakodinamika, 2018. V. № 3. P. 3–8.
Francardo V., Geva M., Bez F., Denis Q., Steiner L., Hayden M.R., Cenci M.A. // Neurotherapeutics. 2019. V. 16. № 2. P. 465–479.
Francardo V., Bez F., Wieloch T., Nissbrandt H., Ruscher K.,Cenci M.A. // Brain. 2014. V. 137. № Pt 7. P. 1998–2014.
Janda E., Lascala A., Carresi C., Parafati M., Aprigliano S., Russo V., Savoia C., Ziviani E., Musolino V., Morani F., Isidoro C., Mollace V. // Autophagy. 2015. V. 11. № 7. P. 1063–1080.
Riederer P., Laux G. // Exp. Neurobiol., 2011. V. 20. № 1. P. 1–17.
Mack J.M., Schamne M.G., Sampaio T.B., Pertile R.A., Fernandes P.A., Markus R.P., Prediger R.D. // Oxid. Med. Cell. Longev. 2016. V. 2016. P. 3472032.
Voronin M.V., Kadnikov I.A. // Pharmacol. Res. Perspect. 2016. V. 4. № 6. P. e00273.
Kadnikov I.A., Voronin M.V., Seredenin S.B. // Bull. Exp. Biol. Med. 2015. V. 159. № 1. P. 44–47.
Cassagnes L.E., Chhour M., Perio P., Sudor J., Gayon R., Ferry G., Boutin J.A., Nepveu F., Reybier K. // Free Radic. Biol. Med. 2018. V. 120. P. 56–61.
Voronin M.V., Aksenova L.N., Buneena O.A., Medvedev A.E. // Bull. Exp. Biol. Med. 2009. V. 148. № 1. P. 23–25.
Дополнительные материалы отсутствуют.