

Нейрохимия, 2022, T. 39, № 3, стр. 228-242
Особенности продукции и трансплацентарного переноса тиреоидных гормонов в системе “мать-плод” при гипергомоцистеинемии
А. Д. Щербицкая 1, 2, А. А. Коваленко 1, Ю. П. Милютина 2, Д. С. Васильев 1
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
2 Научно-исследовательский институт акушерства,
гинекологии и репродуктологии им. Д.О. Отта
Санкт-Петербург, Россия
Поступила в редакцию 16.03.2022
После доработки 25.03.2022
Принята к публикации 26.03.2022
- EDN: TKZBQS
- DOI: 10.31857/S1027813322030104
Аннотация
Накопление в организме матери аминокислоты гомоцистеина – гипергомоцистеинемия – отмечается при несоблюдении сбалансированного питания во время беременности, недостатке некоторых витаминов и генетических дефектах ферментов метионинового цикла. При этом повышается риск развития микротромбозов в плаценте, а у потомства наблюдается отставание развития ткани головного мозга, гибель нейронов и глиоз, приводящие к нарушению когнитивных функций. Проведено исследование тиреоид-зависимой системы регуляции развития мозга плода при хронической пренатальной гипергомоцистеинемии, вызванной дозированной метиониновой нагрузкой у самок крыс. Анализировали содержание тироксина и трийодтиронина в крови беременных самок, содержание их переносчика транстиретина, экспрессии рецепторов (TR-α, TR-β), трансмембранных переносчиков (Oatp1c1, Mct8) и дейодиназ (Dio2, Dio3) в ткани плаценты и мозга плода. Несмотря на отсутствие заметных нарушений снабжения плода тиреоидными гормонами со стороны матери, было обнаружено негативное влияние гипергомоцистеинемии на уровень экспрессии Dio3, Oatp1c1 и Mct8 в нервной ткани плода на Е14, а также снижение экспрессии TR-α и повышение Dio3 в плаценте в конце беременности. Подобный эффект высокого уровня гомоцистеина на функциональное состояние плаценты и на чувствительность развивающегося мозга у потомства к трофическому действию тиреоидных гормонов может являться причиной отставания в процессе развития нервной ткани плода.
ВВЕДЕНИЕ
Отставание развития головного мозга является неспецифической реакцией плода на любое стрессовое воздействие во внутриутробном периоде [1]. В частности, отставание развития структуры и функций головного мозга было отмечено при наиболее частых видах воздействий на систему “мать-плод”: гипоксического [2, 3] и токсического [4, 5]. Вместе с тем механизмы отставания в развитии недостаточно изучены. Есть основания полагать, что одна из причин задержки развития плода может быть связана с нарушением трофической и/или гормональной регуляции роста плода со стороны организма матери [6].
Хорошо известно, что тиреоидные гормоны, тироксин (Т4) и трийодтиронин (Т3), играют важную роль в регуляции общего развития и формирования головного мозга в раннем онтогенезе [7, 8]. В частности, известно об их влиянии на процессы миграции нейробластов [9], дифференцировки нейронов [10] и глиальных элементов [11, 12]. Помимо прямого влияния тиреоидных гормонов на экспрессию целого ряда генов, необходимых для нормального формирования нервной ткани, возможно опосредованное влияние через модуляцию продукции нейротрофических факторов [13–15]. В период эмбриогенеза тиреоидные гормоны попадают в плод через плаценту, и их недостаточное поступление из организма матери может приводить к серьeзным патологиям развития плода, в первую очередь его головного мозга [16]. Известно, что, начиная с 3-го дня после имплантации эмбриона крысы и до созревания его собственной системы синтеза тиреоидных гормонов, стимуляция развития головного мозга этими гормонами полностью зависит от снабжения со стороны организма матери. На поздних стадиях эмбриогенеза роль собственной продукции тиреоидных гормонов эмбрионом возрастает, однако зависимость от материнского Т4 остаeтся до самого рождения [17]. Установлено, что даже в случае достаточного уровня собственного синтеза гормонов плодом, гипотиреоз матери может вызвать нарушение нормального формирования ЦНС [18]. Вместе с тем, состояние системы собственного синтеза гормонов плодом на поздней стадии эмбриогенеза крайне важно для обеспечения нормальной экспрессии нейрональных генов в клетках кортикальных отделов мозга [19].
Врожденный гипотиреоз достаточно часто встречается в клинической практике и бывает вызван либо сниженной продукцией тиреоидных гормонов, либо нарушением их действия на уровне клетки. Так, под влиянием тиреотоксических препаратов, принимаемых во время беременности (большие дозы йода, соли лития, бромиды, Мерказолил, некоторые транквилизаторы), может развиться патология щитовидной железы. Другими распространенными факторами еe развития являются аутоиммунный тиреоидит у беременных, влияние внутриутробных инфекций, а также токсических и химических веществ [20, 21]. Влияние внешних факторов среды и стрессоров признаeтся не менее важным, однако изучено значительно слабее, чем действие медико-биологических факторов. В 15% случаев причиной врожденного гипотиреоза является недостаточный синтез тиреоидных гормонов или тканевых рецепторов к ним [22]. Встречается врожденный гипотиреоз вследствие генетически обусловленных дефектов (выработка аномального тиреотропного гормона и тиреолиберина), либо внутриутробной аномалии гипоталамо-гипофизарной системы. Возможна пожизненная инвалидизация больного, если диагноз “врожденный гипотиреоз” устанавливался слишком поздно. Распространeнными симптомами врожденного гипотиреоза у детей являются вялость, утомляемость и задержка физиологического развития. Сходное отставание физиологического развития (вес, сроки открытия глаз и отделения наружного уха от кожи головы), формирования двигательных реакций и нарушения когнитивных функций наблюдались и в раннем онтогенезе крысят, перенесших пренатальную гипоксию [3] и гипергомоцистеинемию (ГГЦ) [4, 5], что позволяет рассматривать дефицит тиреоидных гормонов как одну из потенциальных причин патологии развития мозга потомства при действии неблагоприятного фактора на систему “мать-плод” в период беременности.
Пренатальная ГГЦ связана с накоплением в организме матери аминокислоты гомоцистеина (ГЦ). Она отмечается при несоблюдении сбалансированного питания во время беременности, недостатке некоторых витаминов [23, 24] и генетических дефектах ферментов метионинового цикла [25, 26]. У потомства наблюдается отставание развития мозга, гибель нейронов и глиоз в кортикальных отделах [27, 28]. При избытке ГЦ повышается риск развития тромбозов, в том числе в плаценте. Изменение микроциркуляции приводит к целому ряду акушерских осложнений, включая развитие гипоксии плода и хронической фетоплацентарной недостаточности, и, как следствие, к рождению детей с низкой массой тела и развитию целого ряда нарушений в период раннего онтогенеза [29]. Известно, что гипоксическое воздействие, как в перинатальный [30], так и постнатальный период [31] влияет на регуляцию тиреоид-опосредованного сигналинга. Развитие гипоксии плода, либо оксидативного стресса вследствие действия ГЦ [32] позволяет предполагать возможность нарушений тиреоид-опосредованных механизмов регуляции развития головного мозга при пренатальной ГГЦ. Данные о влиянии ГЦ на функции тиреоидной системы скудны и отрывочны [33]. В литературе, посвящённой модели пренатальной ГГЦ на грызунах, практически отсутствуют данные о регуляции выработки тиреоидных гормонов и их трансплацентарного переноса в системе “мать-плод”. Действие ГЦ на молекулярные механизмы, регулирующие онтогенетическое развитие мозга плода изучено слабо. Необходимость экспериментальной проверки гипотезы о возможности развития раннего гипотиреоза под влиянием пренатальной ГГЦ определила тематику данного исследования тиреоид-зависимой системы регуляции развития мозга у крыс при хронической ГГЦ в период беременности. В рамках настоящего исследования впервые рассматривались динамика содержания тиреоидных гормонов в организме матери и плода, уровень их основного переносчика транстиретина (ТТР) в плаценте, экспрессия рецепторов (TR-α, TR-β), трансмембранных переносчиков (Oatp1c1, Mct8) и дейодиназ (Dio2, Dio3). Нарушение регуляции каждого из этих элементов способно вызывать изменения всей системы тиреоид-зависимой модуляции развития ЦНС. Высокая социальная значимость и распространённость заболеваний ЦНС, связанных с дефицитом тиреоидных гормонов определяет необходимость проведения такого исследования.
МЕТОДЫ ИССЛЕДОВАНИЯ
Животные. Работа проводилась на беременных самках крыс линии Вистар (5–6 мес.), которые были разделены на 2 группы (по 15–19 животных). Первая группа состояла из животных, находившихся на стандартном рационе, и их плодов, взятых на 14-й и 20-й день пренатального развития (Е14 и Е20 соответственно). Вторую группу составили самки крыс, получавшие метиониновую нагрузку на фоне стандартного рациона, и их плоды на те же дни развития.
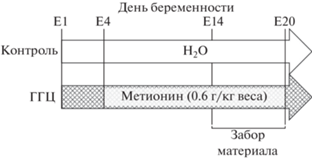
При проведении работ использовали разработанный ранее метод моделирования ГГЦ, основанный на дозированной метиониновой нагрузке, создаваемой путем принудительного перорального введения экспериментальным животным водного раствора L-метионина (0.6 г на кг веса животного), ежедневно, начиная с четвертого дня после оплодотворения и до родоразрешения [34, 35]. Самкам крыс контрольной группы в эти же сроки дополнительно перорально вводили воду (рис. 1). На Е14 и Е20 у самок крыс забирали кровь, у плодов – кровь, цельный мозг, плаценту и амниотическую жидкость. На различные сроки беременности в сыворотке крови самок и их плодов на Е20, а также мозге плодов и амниотической жидкости были определены уровни тиреоидных гормонов. В плаценте проводился анализ содержания белка ТТР. В мозге плода и частях плаценты исследовали уровень экспрессии генов рецепторов к тиреоидным гормонам TR-α и TR-β, переносчиков Oatp1c1, Mct8, дейодиназ Dio2 и Dio3.
Рис. 1.
Схема моделирования гипергомоцистеинемии (ГГЦ) матери. Е1 – 1-й день беременности, Е5 – 5-й день беременности, Е14 – 14-й день беременности, Е20 – 20-й день беременности.
Тиреолибериновый тест. Для исследования влияния хронической ГГЦ крыс в период беременности на уровень гормонов щитовидной железы в сыворотке крови в условиях стимуляции тиреолиберином тиреоидной оси моделирование состояния ГГЦ у беременных самок проводили как описано выше. Через 1 ч после введения метионина или воды животным из каждой группы интраназально вводили тиреолиберин в дозе 300 мкг/кг в 20 мкл физиологического раствора [36] на Е5, Е14 и Е20 (рис. 2). Через 3 ч после введения метионина или воды, когда уровень ГЦ в сыворотке крови достигает пиковых значений [35], и через 2 ч после введения тиреолиберина у самок забирали кровь из десны [37]. Для контроля фоновых значений уровней тиреоидных гормонов у самок также забирали кровь до введения тиреолиберина. Объем выборок составлял от 6 до 8 особей.
Рис. 2.
Схема эксперимента с тиреолиберином. Е5 – 5-й день беременности, Е14 – 14-й день беременности, Е20 – 20-й день беременности.

Иммуноферментный анализ. Сравнительное исследование уровней общего трийодтиронина (Т3общ), свободной фракции трийодтиронина (Т3св), общего тироксина (Т4общ) и свободной фракции тироксина (Т4св) в сыворотке крови, амниотической жидкости и мозге плода проводили методом иммуноферментного анализа с использованием коммерческих наборов реагентов (Вектор-Бест, Россия). В качестве проб мозга плода брали супернатант, полученный путем центрифугирования (16 000 g, 20 мин, +4°С) гомогената нервной ткани, приготовленного в 0.01 М PBS.
ПЦР в реальном времени. Для анализа экспрессии генов в мозге плода и плаценте использовали метод полимеразной цепной реакции в реальном времени (ртПЦР). Объeм выборок составлял от 7 до 10 особей. Плаценту предварительно отмывали от крови в 0.01 М PBS и делили на материнскую (МЧП) и плодную (ПЧП). Выделение тотальной РНК производилось с использованием реагента ExtractRNA (Евроген, Россия) согласно протоколу производителя с модификациями. Концентрацию РНК измеряли с помощью спектрофотометра NanoDrop 2000 (Thermo Fisher Scientific, США). Обратную транскрипцию образцов проводили с использованием oligo dT праймера и обратной транскриптазы MMLV RT (Promega, Madison, WI, USA) по протоколу производителя. Для количественной ПЦР в реальном времени готовили реакционную смесь, содержащую 0.8 мкл образца кДНК, 0.75 ед. TaqM-полимеразы (Алкор-Био, Санкт-Петербург, Россия), 200 нМ специфических прямых и обратных праймеров, 100 нМ (200 нМ для Actb) зонды TaqMan, 3.5 мМ MgCl2 и 250 мкМ dATP/dTTP/dCTP/dGTP в 10 мкл общего объема 1× TaqM-реакционного буфера. Для детекции накопления ПЦР-продукта использовали специфические флуоресцентные зонды по типу линейно-разрушаемых проб (технология TaqMan). В эксперименте использовали флуорофоры FAM, ROX и HEX, гасители флуоресценции BHQ-1 и BHQ-2. Последовательности праймеров и зондов представлены в табл. 1. Амплификацию проводили на приборе CFX96 Real-Time System (Bio-Rad, США). Все пробы анализировали двукратно. Регистрацию флуоресценции производили на окончании этапа отжига праймеров. Полученные результаты анализировали в программе REALTIMEPCR методом пороговой линии. Для определения относительного количества мРНК в исследуемых образцах, полученные значения Ct для генов интереса нормировали по среднему геометрическому значений Ct двух генов “домашнего хозяйства” фосфоглицераткиназы (Pgk1) и 14-3-3z (Ywhaz) с использованием 2–ΔΔCt метода [38].
Таблица 1.
Нуклеотидные последовательности праймеров и зондов
| Ген | Прямой праймер (5' → 3') Обратный праймер (5' → 3') Зонд (5' → 3') |
Температура отжига праймеров, °C |
|---|---|---|
| Dio2 NM_031720.5 |
CGTCATCCTCAAGTGTCCCC TGGTACGCGCACATTACCTT HEX-ACGTGCGACAGTGAAGCGGA-BHQ2 |
62 |
| Dio3 NM_017210.4 |
GCCCGTTGGTGCTCAATTTT GGTGGGCTTCCTCGATGTAG ROX-ACCTGACCACCGTTCATGGCGCGGA-BHQ2 |
60 |
| TR-α NM_031134.2 |
TGAGCACTACGTCAACCACC CTCTGCACTTCTCTCTCCTTCA FAM-TCCGCACTTCTGGCCCAAGC-BHQ1 |
62 |
| TR-β NM_012672.3 |
CCTTAGTCTGCTGGAGGACG AGCTCTGGCATTCCCTTATTCA ROX-2CGCGTGGTGGTACCAAGTTCCA-BHQ2 |
62 |
| Oatp1c1 NM_053441.1 |
CATGTGTGGGGACAATGGGA CCCATCATGCCTGACCAGTT FAM-TGCGTCGGCTTGTCTTGCTGGCTGT-BHQ1 |
60 |
| Mct8 NM_147216.2 |
TACCGTATCTGGGCCTTTGG AGCACCCAGGTTTCCTTGAT HEX-CGCTGCTGCTGCCCTTGGTTACTTCGT-BHQ2 |
60 |
| Ywhaz NM_013011 |
GATGAAGCCATTGCTGAACTTG GTCTCCTTGGGTATCCGATGTC ROX-TGAAGAGTCGTACAAAGACAGCACGC-BHQ2 |
60 |
| Pgk1 NM_053291 |
ATGCAAAGACTGGCCAAGCTAC AGCCACAGCCTCAGCATATTTC HEX-TGCTGGCTGGATGGGCTTGGA-BHQ2 |
60 |
Вестерн-блот анализ. Для определения уровня ТТР методом Вестерн-блот ткани плаценты, полученные на Е14 и Е20, предварительно отмывали от крови в 0.001М PBS. Гомогенаты ткани готовили в лизирующем буфере, содержащем 50 мM Tris-HCl (pH 7.4), 150 мM NaCl, 0.1% Triton X-100, 0.01% SigmaFast protease inhibitor cocktail (Sigma, S8830-20TAB) и далее центрифугировали (16000 g, 20 мин, +4°С). Содержание белка в супернатанте определяли по методу Бредфорда. Электрофорез белков проводили в 15%-м полиакриламидном геле, загружая по 70 мкг белка на лунку [39]. После электрофореза белки переносили на PVDF мембрану, которую блокировали в 0.1%-м Tween20 на фосфатном буфере, содержащем 5% сухого молока, и инкубировали с первичными моноклональными антителами LS-B2607-50 (Life Bioscience) в разведении 1 : 1000. В качестве белка сравнения использовали β-актин, выявляемый первичными антителами (А5060, Sigma, 1 : 1000). Далее мембраны инкубировали с HRP-конъюгированными вторичными антителами против IgG мыши (Abcam, разведение 1 : 4000) или кролика (Abcam, разведение 1 : 5000). Визуализацию фракций белков осуществляли при помощи BioRad Chemidoc Touch (BioRad, USA) с использованием набора реагентов Optiblot ECL Ultra Detect Kit (1.2pg-2ng) (Abcam, ab133409). Денситометрический анализ проводили в программе ImageLab (BioRad, USA), при этом определяли величину отношения оптической плотности полос ТТР (16 кДа) к оптической плотности β-актина (45 кДа). Сопоставление контрольной (n = 9) и ГГЦ (n = 9) групп проводили по полученным отношениям.
Статистическая обработка данных производилась с использованием программы “Statisticа 10”. Результаты обрабатывались с использованием H‑критерия Крускала-Уоллиса и U-критерия Манна-Уитни. Данные были проверены на однородность дисперсий с помощью критерия Левена, нормальность распределения оценивалась критерием Шапиро-Уилка. Для анализа зависимых переменных применяли Т-критерий Вилкоксона. Для оценки тесноты связей между показателями применялся коэффициент корреляции Кендалла. Данные в тексте и на рисунках представлены в виде Me [25%, 75%] (Ме – медиана, 25% и 75% – 1-й и 3-й квартили). Критический уровень значимости при проверке статистических гипотез принимался равным 0.05. В данном исследовании использовался двойной слепой метод. Исследователи, осуществлявшие измерения и анализ данных, не знали, к какой группе принадлежали исследуемые животные, поскольку не участвовали в назначении экспериментальных групп. Расчет оптимального размера выборок проводился на основе заранее проведенного пилотного эксперимента с использованием программы (http://www.openepi.com/ SampleSize/SSPropor.htm).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Тиреолибериновый тест используется в мировой практике как функциональный тест на состояние тиреоидной системы. При этом введение тиреолиберина (тиреотропин-рилизинг-гормон), являющегося одним из представителей класса рилизинг гормонов гипоталамуса, вызывает усиление секреции передней долей гипофиза тиреотропного гормона, который в свою очередь оказывает стимулирующее влияние на выработку гормонов щитовидной железы (тироксина и трийодтиронина). В нашем эксперименте уровень Т3св и Т4св, а также Т3общ и Т4общ закономерно повышался (Wilcoxon test, p ≤ 0.05) у самок крыс после введения тиреолиберина на Е5, Е14 и Е20 (рис. 3). При этом стимулирующий эффект тиреолиберина на содержание тиреоидных гормонов у беременных самок с ГГЦ не отличался от такового у контрольной группы. Однако стоит отметить тот факт, что величина подъема Т3общ на Е14 у крыс с хронической ГГЦ был значимо меньше (Mann-Whitney U Test, p ≤ 0.01), чем у самок контрольной группы, но к Е20 возвращался к нормальным значениям.
Рис. 3.
Уровни общего и свободного трийодтиронина (Т3общ и Т3св), общего и свободного тироксина (Т4общ и Т4св) до и через 2 ч после введения тиреолиберина в контрольной группе (n = 6–7) самок крыс и в группе с гипергомоцистеинемией (ГГЦ, n = 8) на различные сроки беременности. Е5 – 5-й день беременности, Е14 – 14-й день беременности, Е20 – 20-й день беременности. Данные представлены в виде Me [25%, 75%] (Ме – медиана, 25% и 75% – 1-й и 3-й квартили). * Отличие значений до и после введения тиреолиберина (Wilcoxon test, p ≤ 0.05).

Внутригрупповое исследование показало, что концентрация Т3св после введения тиреолиберина снижается (Kruskal-Wallis test, p ≤ 0.05) с течением беременности в контрольной группе крыс, а также у самок с хронической ГГЦ (Kruskal-Wallis test, p ≤ 0.05, p ≤ 0.01). Сходные изменения были обнаружены при анализе содержания Т4св у самок крыс, которым в течение беременности вводили раствор метионина, после стимуляции тиреолиберином (Kruskal-Wallis test, p ≤ 0.05). Нами было показано снижение от Е5 до Е20 уровня Т4общ у контрольных крыс при взятии крови до введения тиреолиберина (Kruskal-Wallis test, p ≤ 0.05), при этом анализ не выявил достоверных отличий в концентрации гормона у данных крыс после стимуляции тиреоидной оси. Уменьшение концентрации Т4общ в течение беременности было установлено в сыворотке крови самок крыс из группы с ГГЦ до и после введения им тиреолиберина (Kruskal-Wallis test, p ≤ 0.05, p ≤ 0.01, p ≤ 0.001).
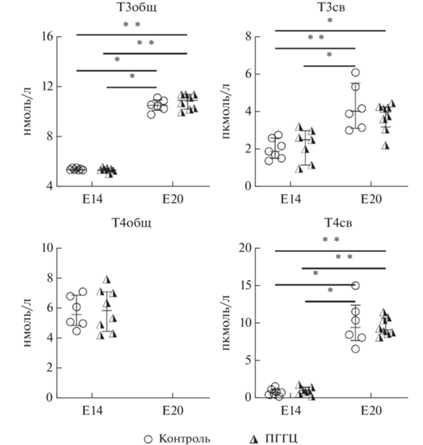
В рамках настоящего исследования было впервые проведено сравнительное исследование уровней T3общ, Т3св, Т4общ и Т4св в сыворотке крови крыс на различные сроки беременности (рис. 4) и у их плодов на Е20 (рис. 5). Установлено, что ГГЦ не приводила к изменению содержания Т3общ и Т3св, а также уровня Т4св в сыворотке крови самок крыс ни на Е14, ни на Е20. При этом, в данном эксперименте было отмечено снижение (Mann-Whitney U Test, p ≤ 0.05) концентрации Т3общ с течением беременности у контрольных самок, тогда как в группе с ГГЦ такой динамики не наблюдалось. Сходные отличия также были обнаружены при исследовании уровня Т4общ у контрольной группы самок (Mann-Whitney U Test, p ≤ 0.05). Показано также, что хроническая ГГЦ привела к повышению уровня Т4общ в сыворотке крови самок крыс к Е20 (Mann-Whitney U Test, p ≤ 0.05). Исследование уровня гормонов щитовидной железы в сыворотке крови плодов на Е20 значимых (Mann-Whitney U Test) изменений не выявило (рис. 5).
Рис. 4.
Содержание гормонов щитовидной железы в сыворотке крови самок крыс контрольной группы (n = 5–6) и в группе с гипергомоцистеинемией (ГГЦ, n = 6–8) на различные сроки беременности. Е14 – 14-й день беременности, Е20 – 20-й день беременности. Данные представлены в виде Me [25%, 75%] (Ме – медиана, 25% и 75% – 1-й и 3-й квартили). * Отличие контрольных значений от группы ГГЦ (Mann-Whitney U Test, p ≤ 0.05); # отличие содержания гормона с течением беременности (Mann-Whitney U Test, p ≤ 0.05).

Рис. 5.
Содержание тиреоидных гормонов в сыворотке крови плодов крыс контрольной группы (n = 6–7) и в группе с пренатальной гипергомоцистеинемией (ПГГЦ, n = 7–9) на 20-й день внутриутробного развития (Е20). Данные представлены в виде Me [25%, 75%] (Ме – медиана, 25% и 75% – 1-й и 3-й квартили).

Проведенный анализ взаимосвязей с помощью Тау-критерия Кендалла выявил отрицательную связь между уровнем Т3общ в сыворотке крови матери и плода из контрольной группы (τ = = –0.8 при р ≤ 0.05), тогда как в группе с ГГЦ расчет ранговых коэффициентов корреляции Кендалла позволил установить наличие положительной ассоциации между концентрацией Т3общ у матери и ее плода (τ = 0.619 при р ≤ 0.05).
Исследование уровней тиреоидных гормонов было также проведено в мозге (рис. 6) и амниотических жидкостях (рис. 7) эмбрионов и плодов крыс на Е14 и на Е20. Показано, что хроническая ГГЦ матери во время беременности не приводила к изменению содержания тиреоидных гормонов в данных тканях (Kruskal-Wallis test), что согласуется с результатами, полученными при анализе этих маркеров в сыворотке крови плодов. Несмотря на то, что в нашем эксперименте не было обнаружено различий в уровнях Т3 и Т4 в амниотических жидкостях контрольных плодов и развивавшихся в условиях ГГЦ матери, были получены данные о динамике изменения концентрации данных гормонов в процессе внутриутробного развития (Kruskal-Wallis test, p < 0.05). Так, нами было показано, что уровни Т3общ и Т3св, а также Т4св возрастают в амниотической жидкости в процессе развития плода, в то время как концентрация Т4общ на Е20 снижается и становится ниже порога детектирования, что может быть связано с пределом чувствительности тест-системы.
Рис. 6.
Содержание тиреоидных гормонов в мозге плодов крыс на 14-й и 20-й день внутриутробного развития (Е14 и Е20 соответственно) в контрольной группе (n = 6–8) и при пренатальной гипергомоцистеинемии (ПГГЦ, n = 5–8). Данные представлены в виде Me [25%, 75%] (Ме – медиана, 25% и 75% – 1-й и 3-й квартили).
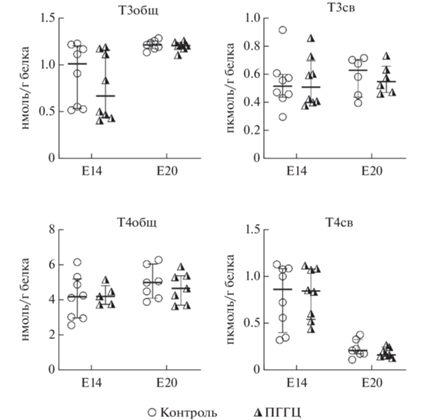
Рис. 7.
Содержание гормонов щитовидной железы в амниотической жидкости плодов крыс на 14-й и 20-й день внутриутробного развития (Е14 и Е20, соответственно) в контрольной группе (n = 6–7) и при пренатальной гипергомоцистеинемии (ПГГЦ, n = 7–8). Данные представлены в виде Me [25%, 75%] (Ме – медиана, 25% и 75% – 1-й и 3-й квартили). * Отличие значений между группами (Kruskal-Wallis test, p ≤ 0.05); ** отличие значений между группами (Kruskal-Wallis test, p ≤ 0.01).
Результаты анализа уровня экспрессии генов интереса в мозге плодов крыс, ПЧП и МЧП на Е14 и Е20 представлены в табл. 2. В ходе исследования было выявлено, что у плодов крыс преобладает экспрессия TR-α. В мозге продукция мРНК TR-α оставалась неизменной на Е14 и на Е20. Однако в обеих частях плаценты на Е20 экспрессия этого гена была значительно снижена при ГГЦ (Mann-Whitney U Test, p ≤ 0.01). В то же время экспрессия TR-β не менялась в мозге плода и плаценте. Было выявлено, что в мозге плода на Е20 и ПЧП ген Dio2 практически не экспрессируется, а в МЧП его экспрессия при ГГЦ остается на уровне контроля. В мозге плодов, развивающихся в условиях ГГЦ матери, нами показано снижение экспрессии гена дейодиназы Dio3 (Mann-Whitney U-test, p ≤ 0.05), осуществляющей инактивацию тиреоидных гормонов, а также гена Oatp1c1 (Mann-Whitney U-test, p ≤ 0.05) и Mct8 (Mann-Whitney U-test, p ≤ 0.05). Напротив, в МЧП нами было отмечено повышение экспрессии Dio3 как на Е14, так и на Е20.
Таблица 2.
Уровень экспрессии генов интереса
| Ген | Мозг плода | ПЧП | МЧП | ||||
|---|---|---|---|---|---|---|---|
| контроль | ПГГЦ | контроль | ПГГЦ | контроль | ПГГЦ | ||
| TR-α | Е14 | 1.00 [0.81, 1.12] |
0.81 [0.76, 0.86] |
0.97 [0.83, 1.03] |
0.96 [0.72, 1.10] |
3.79 [1.18, 5.08] | 4.30 [3.26, 7.22] |
| Е20 | 1.13 [0.84, 1.16] | 1.10 [1.06, 1.16] | 0.84 [0.80, 1.23] | 0.55 [0.52, 0.58]** | 0.94 [0.84, 1.00] | 0.65 [0.58, 0.74]** | |
| TR-β | Е14 | 1.06 [0.80, 1.44] |
1.09 [0.71, 1.43] |
1.28 [0.67, 1.59] |
0.90 [0.25, 1.49] | 3.35 [2.51, 4.30] | 4.42 [1.78, 10.08] |
| Е20 | 1.25 [0.59, 2.13] | 1.04 [0.16, 2.09] | 1.03 [0.74, 1.13] | 1.16 [0.68, 1.27] | 1.03 [0.94, 1.09] | 1.10 [0.74, 1.63] | |
| Dio3 | Е14 | 1.04 [0.87, 1.15] |
0.69 [0.35, 0.71]* |
0.86 [0.69, 1.82] | 1.49 [0.82, 2.11] | 0.93 [0.78, 1.39] | 1.82 [1.81, 1.96]* |
| Е20 | 0.89 [0.89, 0.93] | 1.03 [0.89, 1.39] | 0.67 [0.62, 0.71] | 0.71 [0.59, 1.13] | 0.77 [0.73, 1.43] | 1.96 [1.26, 2.46]* | |
| Dio2 | Е14 | 2.66 [0.44, 6.08] |
3.60 [2.84, 5.37] | ||||
| Е20 | 0.96 [0.60, 1.46] | 0.92 [0.60, 1.71] | |||||
| Oatp1c1 | Е14 | 0.97 [0.84, 1.10] |
0.53 [0.48, 0.72]* |
||||
| Е20 | 0.93 [0.86, 0.96] | 1.00 [0.96, 1.11] | |||||
| Mct8 | Е14 | 0.95 [0.93, 1.11] |
0.60 [0.42, 0.84]* |
||||
| Е20 | 0.99 [0.94, 1.06] | 0.95 [0.90, 1.05] | |||||
Уровень экспрессии генов TR-α, TR-β, Dio2, Dio3, Oatp1c1 и Mct8 в мозге плода крыс, плодной (ПЧП) и материнской части (МЧП) плаценты на 14-й и 20-й день внутриутробного развития (Е14 и Е20, соответственно) в контрольной группе (n = 5–8) и при пренатальной гипергомоцистеинемии (ПГГЦ, n = 6–8). Данные представлены в виде Me [25%, 75%] (Ме – медиана, 25% и 75% – 1-й и 3-й квартили). * Отличие значений между группами (Mann-Whitney U Test, p ≤ 0.05); ** отличие значений между группами (Mann–Whitney U Test, p ≤ 0.01).
В данной работе нами было впервые проведено исследование содержания ТТР в плаценте при нормальном протекании беременности и в условиях повышенного уровня ГЦ в организме матери (рис. 8). Проведенный анализ не выявил различий в уровне ТТР в ткани плаценты между контрольной и экспериментальной (ГГЦ) группами ни на Е14 (Mann-Whitney test U-test), ни на Е20 (Mann-Whitney test U-test).
Рис. 8.
Содержание транстиретина (TTР) в плаценте на 14-й (E14) и 20-й (E20) дни беременности у крыс при нормальном протекании беременности (контроль, n = 9) и при пренатальной гипергомоцистеинемии (ПГГЦ, n = 9). Данные об отношении оптической плотности ТТР к β-актину представлены в виде Me [25%, 75%] (Ме – медиана, 25% и 75% – 1-й и 3-й квартили). На верхней картинке показан репрезентативный вестерн блот белка ТТР (16 кДа) и β-актина (45 кДа).

ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Известно лишь небольшое число работ, посвященных изучению возможного влияния ГГЦ во время беременности на состояние тиреоидной системы матери и плода как у человека, так и на животных моделях. Зачастую в научной литературе рассматривается вопрос об эффектах гипо- или гипертиреоза на обмен ГЦ в организме. Так, некоторые клинические исследования показывают, что у пациентов с гипотиреозом концентрация Т3св и Т4св негативно коррелирует в уровнем ГЦ [40, 41] ввиду пониженной активности фермента 5,10-метилтетрагидрофолатредуктазы (MTHFR), что, вероятно, связано с влиянием гормонов щитовидной железы на доступность ФМН и ФАД [42]. У пациентов с тиреотоксикозом сообщается о положительной связи уровня T4св и ГЦ [43]. Повышение концентрации Т4св вместе с ГЦ также наблюдалось у беременных женщин и новорожденных детей [33], что по данным авторов было опосредовано эффектами тиреоидных гормонов на уровни фолиевой кислоты и витамина В12. Исследования на животных показывают, что гипертиреоз приводит к более высоким концентрациям ГЦ [44, 45], и предполагается влияние гормонов щитовидной железы на активность метионинсинтазы [40], которая зависит от доступности витамина B12. Вместе с этим в исследованиях, проведенных в горных регионах Китая, было обнаружено, что одновременное отклонение от нормального уровня тиреоидных гормонов и ГЦ в сыворотке крови матери увеличивает в 3 раза риск развития дефектов нервной трубки плода [46].
В связи с тем, что свидетельств возможного влияния повышенного содержания ГЦ в организме на регуляцию выработки тиреоидных гормонов в литературе недостаточно, нами было проведено исследование уровня Т3 и Т4 в сыворотке крови самок крыс на различные сроки беременности и в тканях их плодов. Результаты настоящего исследования подтверждают и несколько расширяют полученные другими авторами данные, которые показывают, что у крыс происходят важные изменения уровней гормонов щитовидной железы на последних стадиях физиологической беременности. Так, было установлено, что концентрация Т4 и Т3 в плазме крыс снижается от Е17 к Е22 и ближе к родоразрешению составляет примерно 30% от уровня, отмечаемого у небеременных самок [47]. Мы также обнаружили снижение содержания Т3общ и Т4общ у контрольных крыс с течением беременности (от Е14 к Е20) в то время, как у самок с повышенным уровнем ГЦ такой динамики не наблюдалось. Полученные в ходе тиреолиберинового теста данные дают основание полагать, что хроническое введение метионина не оказывает негативного эффекта на секрецию тиреотропного гормона или на его рецептор в щитовидной железе самок крыс. Вместе с этим, обнаруженное на Е20 повышение уровня Т4общ в сыворотке крови самок крыс с хронической ГГЦ может негативно влиять на функционирование не только материнского организма перед родоразрешением, но и на систему “мать-плацента-плод” в целом.
По данным литературы, в тканях плода крыс обнаруживаются небольшие количества Т4 и Т3 с 11-го дня эмбрионального развития (E11) [48]. Показано, что уровень Т4св в сыворотке плода может зависеть от концентрации Т4-связывающих белков самого плода, а также от уровня общего и свободного Т4 матери, преодолевших плацентарный барьер. При этом, исследователями было установлено, что Т4-связывающая способность белков в крови плода определяется онтогенетически, не зависит от состояния щитовидной железы матери и намного превышает то количество Т4, которое поступает к плоду [16]. Таким образом, доступность Т4св для эмбриональных тканей в конечном итоге зависит от уровня тиреоидных гормонов в крови матери. Начало же активного функционирования щитовидной железы у плода совпадает с моментом полного созревания портальных сосудов гипофиза, которое наблюдается на Е17.5–E18 у крыс (при начале беременности на Е0) [16]. Далее доля тиреоидных гормонов плода в его тканях увеличивается, поэтому к моменту рождения около 17.5% фетального пула Т4 [49] и 47% фетального пула Т3 имеют материнское происхождение. Поэтому закономерно, что в нашем исследовании не было выявлено значимых изменений в концентрации тиреоидных гормонов в сыворотке крови плодов на Е20.
Известно, что амниотическая жидкость представляет собой сложную среду, содержащую не только метаболиты плода, но питательные вещества и факторы роста, способствующие развитию плода и поступающие за счет быстрой двунаправленной диффузии между плодом и амниотической жидкостью через еще не ороговевшую кожу плода [50]. Более того, исследования на животных показали, что у плода присутствуют адаптивные и/или защитные механизмы от последствий дефицита йода, такие как поглощение йода за счет повышенной экспрессии симпортера Na-I (NIS) [51]. Таким образом, делается вывод о том, что плод может поглощать йод из амниотической жидкости для синтеза своих собственных тиреоидных гормонов [52]. Несмотря на это, работы, посвященные анализу тиреоидных гормонов в амниотической жидкости, единичны и данные в них противоречивы. Так, в одном исследовании было показано, что оценка уровня гормонов щитовидной железы в амниотической жидкости, особенно Т3, может помочь в диагностике дисфункции щитовидной железы плода [53]. При этом, в более поздней работе указывается, что анализ концентрации тиреоидных гормонов и ТТГ в амниотической жидкости не дают надежного прогноза состояния щитовидной железы плода или новорожденного [54]. На животных же моделях исследования содержания данных гормонов проводятся еще реже.
Вместе с этим, исследования, посвященные изучению уровня Т3 и Т4 в развивающемся мозге эмбрионов и плодов крыс, как в норме, так и при патологии развития, малочисленны, а полученные в них данные ограничены, в основном, периодом, совпадающим с началом функционирования щитовидной железы плода (Е17.5–E18), или следующими за ним днями (до E21) [55, 56]. У мышей T4 в мозге обнаруживается по крайней мере с E16 [57], то есть непосредственно перед началом работы щитовидной железы плода, которое у мышей отмечается примерно с E16.5 [58], при этом T3 детектируется с E18 [59]. Несмотря на то, что ГГЦ матери не оказывала влияния на уровень Т3 и Т4 ни в амниотической жидкости, ни в мозге плода, нами были получены новые данные об изменении содержания исследуемых гормонов в процессе роста и развития эмбрионов крыс, начиная с Е14.
Известно, что белок ТТР является основным переносчиком Т3 и Т4 в жидких средах организма. Как было показано ранее, повышение содержания ГЦ, наблюдаемое при белково-энергетической недостаточности у людей, сопровождается снижением уровня ТТР в плазме крови [60–62]. Однако, проведенное в данной работе сопоставление содержания ТТР в плаценте на Е14 и Е20 при нормальном протекании беременности и в условиях повышенного уровня ГЦ в сыворотке крови матери показало отсутствие статистически значимого снижения содержания ТТР в плаценте крыс при ГГЦ, что можно объяснить как межвидовыми различиями объектов исследования, так и различиями в моделировании повышения содержания ГЦ в организме.
Хотя полученные нами данные не подтверждают первоначальную гипотезу о возможном изменении уровня выработки тиреоидных гормонов в организме беременных самок крыс с ГГЦ и нарушении их транспорта к плоду, следует отметить, что трофическое действие Т3 и Т4 на развитие головного мозга плода в условиях пренатальной ГГЦ определяется не только выработкой данных гормонов в организме матери и их трансплацентарным переносом в развивающийся плод. Не менее важным фактором в данном случае является тиреоид-опосредованный сигналинг в тканях развивающегося мозга, при оценке уровня которого необходимо учитывать активность нейрональных и глиальных дейодиназ [31], внутриклеточных транспортеров (например, Mct8, [63]), экспрессию рецепторных молекул [22], что значительно усложняет задачу исследователей. Каждый из этих элементов способен вызывать нарушения всей системы тиреоид-зависимой модуляции развития ЦНС.
По литературным данным, у крыс активность Dio2 обнаруживается в мозге плода уже на Е17 с ее возрастанием вплоть до дня, предшествующего рождению [64]. У мышей экспрессия мРНК Dio2 обнаруживается в головном мозге, по крайней мере, с E15 [59], свидетельствуя, что материнские тиреоидные гормоны необходимы развивающемуся мозгу плода еще до созревания его собственной щитовидной железы. В нашем исследовании уже на Е14 в мозге эмбрионов крыс была обнаружена экспрессия мРНК Dio3, осуществляющей инактивацию тиреоидных гормонов, и транспортеров Oatp1c1 и Mct8. Что касается переносчиков тиреоидных гормонов, то на данный момент нет исследований, описывающих пространственно-временной характер их экспрессии во время эмбрионального развития ни у крыс, ни у мышей. В мозге взрослой крысы до 80% связанного с ядeрными рецепторами Т3 образуется из Т4 [65], поэтому основным источником Т3 в головном мозге является реакция локального превращения Т4 в Т3 под действием Dio2. Чтобы попасть в мозг, циркулирующие Т4 и Т3 должны преодолеть барьеры головного мозга через специфические переносчики тиреоидных гормонов. В исследованиях, посвященных анализу расположения транспортеров у грызунов, было показано, что T3 и T4 проникают через ГЭБ посредством Mct8 во внеклеточную жидкость, где они напрямую достигают нервных клеток вблизи кровеносных сосудов [66, 67]; и что T4, но не T3, проходит через ГЭБ с помощью Oatp1c1 непосредственно в астроциты, контактирующие с кровеносными сосудами [68], и уже в астроцитах Т4 за счет Dio2 превращается в T3, который затем может транспортироваться к нейронам [69, 70]. Мозг плода же во время развития почти полностью зависит от T3, локально генерируемого Dio2. На беременных самках с гипотиреозом было показано, что T4, но не Т3, в физиологической концентрации может преодолевать барьеры как плаценты, так и головного мозга плода, нормализуя концентрацию T3 в мозге плода за счет дейодирования T4 и увеличивая экспрессию нейрональных генов [63, 71, 72]. Причина, по которой мозг плода нечувствителен к циркулирующему Т3, неизвестна и не может быть объяснена отсутствием транспортера Mct8, поскольку он экспрессируется в мозге во время внутриутробного развития плода [63]. Из вышесказанного следует исключительная важность данных о динамике экспрессии переносчиков тиреоидных гормонов и дейодиназ, при этом на модели пренатальной ГГЦ такие данные были получены впервые именно в рамках настоящей работы. Известно, что в ткани мозга Dio2 экспрессируется преимущественно в глиальных клетках. И поскольку процесс глиогенеза в рассматриваемый нами период внутриутробного развития еще не завершен, возможно, поэтому уровень экспрессии данного фермента оказывается недостаточным для его детекции. При этом, обнаруженное на Е14 в мозге плодов, развивающихся в условиях ГГЦ матери, снижение экспрессии Dio3, а также Oatp1c1 и Mct8, в контексте отсутствия изменения уровней Т3 и Т4 в сыворотке крови матери и мозге плода, с одной стороны, может быть компенсаторной реакцией организма, а с другой – негативно сказываться на развитии мозга плода, в частности, за счет регуляции экспрессии Т3-зависимых генов. Так, было показано, что высокий уровень ГЦ у матери может влиять на экспрессию генов, связанных как с сетью гормонов щитовидной железы матери, так и с регуляцией заращения нервной трубки посредством гомоцистеинилирования гистонов [46, 73]. Кроме того, у эмбрионов мышей при нарушении заращения нервной трубки отмечено повышение уровня ингибиторных модификаций гистонов в области промотора Dio3, при этом в тканях их матерей данных изменений обнаружено не было [74].
В то время как плод начинает вырабатывать собственные гормоны щитовидной железы только во второй половине беременности, транспорт и регуляция активности тиреоидных гормонов материнского происхождения плацентой играют важную роль в развитии и функционировании как самой плаценты, так и влияют на развитие плода на протяжении всей беременности. С самого начала гестации Т3, действуя через свои рецепторы, стимулирует выработку эстрадиола, ХГЧ и плацентарного лактогена, каждый из которых, в свою очередь, участвует в успешной адаптации матери к беременности, а также в развитии трофобласта и сосудистой сети плаценты. В плаценте человека TR-α и TR-β локализуются в ядрах клеток синцитиотрофобласта и цитотрофобласта [75]. В нашем же исследовании у крыс экспрессия данных рецепторов обнаруживалась как в МЧП, так и в ПЧП, что соответствует зонам спонгиотрофобласта и лабиринта плаценты крысы. Полученные данные о снижении уровня мРНК TR-α в МЧП и ПЧП на Е20 у крыс с повышенным уровнем ГЦ согласуются с результатами исследований влияния других токсических воздействий на мать во время беременности, например, потребление алкоголя [76].
В тканях матери и плода обнаружены три йодтиронин-дейодиназы (Dio1, Dio2, Dio3), но только Dio2 и Dio3 присутствуют и активны в плаценте [77]. Плацентарные дейодиназы регулируют доступность тиреоидных гормонов для клеток плаценты и крови плода. Предполагается, что, в частности, Dio3 играет ключевую роль в защите плода от чрезмерных концентраций гормонов щитовидной железы матери путем превращения Т4 и Т3 в их неактивные формы [78, 79]. При ограниченном потреблении питательных веществ в плацентах овец, у которых была зафиксирована задержка внутриутробного роста плода, в конце беременности отмечено повышение экспрессии мРНК Dio3 [80]. Стоит отметить, что ранее при моделировании пренатальной ГГЦ мы также наблюдали снижение массы тела и мозга плода на Е20 [34], а в данной работе нами было отмечено увеличение экспрессии мРНК Dio3 в МЧП. Это, наряду со снижением экспрессии мРНК TR-α в обеих частях плаценты, весьма вероятно, можно рассматривать как компенсаторную реакцию плаценты в ответ на более высокие концентрации Т4 в материнской крови при ГГЦ.
ЗАКЛЮЧЕНИЕ
Полученные результаты свидетельствуют о том, что несмотря на отсутствие заметных нарушений снабжения плода тиреоидными гормонами со стороны организма матери, в середине беременности (на Е14) наблюдается негативное влияние ГГЦ на уровень экспрессии Dio3, Oatp1c1 и Mct8 в нервной ткани плода, регулирующих обмен тиреоидных гормонов. Подобный эффект высокого уровня ГЦ на чувствительность развивающегося мозга к трофическому действию тиреоидных гормонов может являться причиной отставания в процессе развития нервной ткани плода. Вместе с тем, ГЦ может влиять на рост плода опосредованно, то есть нарушая функциональное состояние плаценты, осуществляющей транспорт и инактивацию избыточного количества тиреоидных гормонов материнского происхождения.
Список литературы
Rice D., Barone S. // Environmental Health Perspectives. 2000. V. 108. № suppl. 3. P. 511-533.
Nyakas C., Buwald B., Luiten P.G.M. // Progress in Neurobiology. 1996. V. 49. № 1. P. 1–51.
Dubrovskaya N.M., Zhuravin I.A. // Neuroscience and Behavioral Physiology. 2009. V. 40. № 2. P. 231–238.
Arutjunyan A., Kozina L., Stvolinskiy S., Bulygina Y., Mashkina A., Khavinson V. // Int. J. Clin. Exp. Med. 2012. V. 5. № 2. P. 179–185.
Gerasimova E., Yakovleva O., Burkhanova G., Khaertdinov N., Sitdikova G., Ziyatdinova G. // BioNanoScience. 2017. V. 7. № 1. P. 155–158.
Fowden A.L., Forhead A.J., Coan P.M., Burton G.J. // Journal of Neuroendocrinology. 2008. V. 20. № 4. P. 439–450.
Alemu A., Terefe B., Abebe M., Biadgo B. // Int. J. Reprod. Biomed. 2016. V. 14. № 11. P. 677–686.
Andersen S., Bruun N.H., Pedersen K.M., Laurberg P. // Thyroid. 2003. V. 13. № 11. P. 1069–1078.
Martínez-Galán J.R., Pedraza P., Santacana M., Escobar del Ray F., Morreale de Escobar G., Ruiz-Marcos A. // Journal of Clinical Investigation. 1997. V. 99. № 11. P. 2701–2709.
Enrique Silva J., Peter R. // Endocrinology. 1990. V. 126. № 2. P. 1276–1282.
Manzano J., Bernal J., Morte B. // International Journal of Developmental Neuroscience. 2007. V. 25. № 3. P. 171–179.
Lima F.R.S., Gervais A., Colin C., Izembart M., Neto V.M., Mallat M. // The Journal of Neuroscience. 2001. V. 21. № 6. P. 2028–2038.
Bernal J. // Nature Clinical Practice Endocrinology & Metabolism. 2007. V. 3. № 3. P. 249–259.
Wu Y., Koenig R.J. // Trends in Endocrinology & Metabolism. 2000. V. 11. № 6. P. 207–211.
Gould E., Butcher L.L. // The Journal of Neuroscience. 1989. V. 9. № 9. P. 3347–3358.
Morreale de Escobar G., Obregon M.J., Escobar del Rey F. // European Journal of Endocrinology. 2004. P. U25–U37.
Vulsma T., Gons M.H., de Vijlder J.J. // N. Engl. J. Med. 1989. V. 321. № 1. P. 13–16.
Haddow J.E., Palomaki G.E., Allan W.C., Williams J.R., Knight G.J., Gagnon J., O’Heir C.E., Mitchell M.L., Hermos R.J., Waisbren S.E., Faix J.D., Klein R.Z. // N. Engl. J. Med. 1999. V. 341. № 8. P. 549–555.
Morte B., Ceballos A., Diez D., Grijota-Martínez C., Dumitrescu A.M., Di Cosmo C., Galton V.A., Refetoff S., Bernal J. // Endocrinology. 2010. V. 151. № 5. P. 2381–2387.
Williams F.L.R., Simpson J., Delahunty C., Ogston S.A., Bongers-Schokking J.J., Murphy N., van Toor H., Wu S.-Y., Visser T.J., Hume R. // The Journal of Clinical Endocrinology & Metabolism. 2004. V. 89. № 11. P. 5314–5320.
Ares S., Quero J., Diez J., de Escobar G.M. // Journal of Pediatric Endocrinology and Metabolism. 2011. V. 24. № 11-12. P.
Hashimoto K., Curty F.H., Borges P.P., Lee C.E., Abel E.D., Elmquist J.K., Cohen R.N., Wondisford F.E. // Proceedings of the National Academy of Sciences. 2001. V. 98. № 7. P. 3998–4003.
Baydas G., Koz S.T., Tuzcu M., Nedzvetsky V.S., Etem E. // Int. J. Dev. Neurosci. 2007. V. 25. № 3. P. 133–139.
Blaise S.A., Nedelec E., Schroeder H., Alberto J.M., Bossenmeyer-Pourie C., Gueant J.L., Daval J.L. // Am. J. Pathol. 2007. V. 170. № 2. P. 667–679.
Zhang C., Cai Y., Adachi M.T., Oshiro S., Aso T., Kaufman R.J., Kitajima S. // Journal of Biological Chemistry. 2001. V. 276. № 38. P. 35867–35874.
Namekata K., Enokido Y., Ishii I., Nagai Y., Harada T., Kimura H. // Journal of Biological Chemistry. 2004. V. 279. № 51. P. 52961–52969.
Shcherbitskaia A.D., Vasilev D.S., Milyutina Y.P., Tumanova N.L., Mikhel A.V., Zalozniaia I.V., Arutjunyan A.V. // Cells. 2021. V. 10. № 6. P. 1536.
Shcherbitskaia A.D., Vasilev D.S., Milyutina Y.P., Tumanova N.L., Zalozniaia I.V., Kerkeshko G.O., Arutjunyan A.V. // Neurotoxicity Research. 2020. V. 38. № 2. P. 408–420.
Ars C.L., Nijs I.M., Marroun H.E., Muetzel R., Schmidt M., Steenweg-de Graaff J., van der Lugt A., Jaddoe V.W., Hofman A., Steegers E.A., Verhulst F.C., Tiemeier H., White T. // British Journal of Nutrition. 2016. V. 122. № s1. P. S1–S9.
Pereira D.N., Procianoy R.S. // Acta Paediatr. 2003. V. 92. № 3. P. 339–345.
Freitas B.C.G., Gereben B., Castillo M., Kalló I., Zeöld A., Egri P., Liposits Z., Zavacki A.M., Maciel R.M.B., Jo S., Singru P., Sanchez E., Lechan R.M., Bianco A.C. // Journal of Clinical Investigation. 2010. V. 120. № 6. P. 2206–2217.
Koz S.T., Gouwy N.T., Demir N., Nedzvetsky V.S., Etem E., Baydas G. // Int. J. Dev. Neurosci. 2010. V. 28. № 4. P. 325–329.
Barjaktarovic M., Steegers E.A.P., Jaddoe V.W.V., de Rijke Y.B., Visser T.J., Korevaar T.I.M., Peeters R.P. // The Journal of Clinical Endocrinology & Metabolism. 2017. V. 102. № 12. P. 4548–4556.
Arutjunyan A.V., Milyutina Y.P., Shcherbitskaia A.D., Kerkeshko G.O., Zalozniaia I.V., Mikhel A.V. // Biochemistry (Moscow). 2020. V. 85. № 2. P. 248–259.
Arutyunyan A.V., Milyutina Y.P., Zaloznyaya I.V., Pustygina A.V., Kozina L.S., Korenevskii A.V. // Neurochemical Journal. 2012. V. 6. № 1. P. 71–76.
Derkach K.V., Shpakova E.A., Titov A.K., Shpakov A.O. // International Journal of Peptide Research and Therapeutics. 2015. V. 21. № 3. P. 249–260.
Зильфян В.Н., Кумкумаджян В.А. // Журн. экспер. и клин. медицины. 1970. V. 10. № 4. P. 12–14.
Livak K.J., Schmittgen T.D. // Methods. 2001. V. 25. № 4. P. 402–408.
Schägger H. // Nature Protocols. 2006. V. 1. № 1. P. 16–22.
Orzechowska-Pawilojc A., Sworczak K., Lewczuk A., Babinska A. // Endocrine Journal. 2007. V. 54. № 3. P. 471–476.
Diekman M.J.M., Van Der Put N.M., Blom H.J., Tijssen J.G.P., Wiersinga W.M. // Clinical Endocrinology. 2001. V. 54. № 2. P. 197–204.
Orzechowska-Pawilojc A., Lewczuk A., Sworczak K. // Endokrynol Pol. 2005. V. 56. № 2. P. 194–202.
Colleran K.M., Ratliff D.M., Burge M.R. // Endocrine Practice. 2003. V. 9. № 4. P. 290–295.
Jacobs R.L., Stead L.M., Brosnan M.E., Brosnan J.T. // Canadian Journal of Physiology and Pharmacology. 2000. V. 78. № 7. P. 565–570.
Ozkan Y., Donder E., Guney H., Baydas G. // Neuro Endocrinol Lett. 2005. V. 26. № 5. P. 536–540.
Gu Y.-H., Zhang Q., Guo J., Wang F., Bao Y., Qiu Z., Zheng P., Ushijima M., Matsuura M., Xie X., Zhang T. // Journal of Trace Elements in Medicine and Biology. 2021. V. 68.
Calvo R., Obregon M.J., De Ona C.R., Ferreiro B., Del Rey F.E., De Escobar G.M. // Endocrinology. 1990. V. 127. № 1. P. 10–16.
Obregon M.J., Mallol J., Pastor R., Escobar G.M.D., Rey F.E.D. // Endocrinology. 1984. V. 114. № 1. P. 305–307.
De Escobar G.M., Calvo R., Obregon M.J., Del Rey F.E. // Endocrinology. 1990. V. 126. № 5. P. 2765–2767.
Underwood M.A., Gilbert W.M., Sherman M.P. // Journal of Perinatology. 2005. V. 25. № 5. P. 341–348.
Obregon M.-J., de Escobar G.M., de Mena R.M., Calvo R.M., Lavado-Autric R. // Endocrinology. 2013. V. 154. № 1. P. 529–536.
Velasco I., Sánchez-Gila M., Manzanares S., Taylor P., García-Fuentes E. // Journal of Clinical Medicine. 2020. V. 9. № 1. P. 177.
Chopra I.J., Crandall B.F. // New England Journal of Medicine. 1975. V. 293. № 15. P. 740–743.
Hollingsworth D.R., Alexander N.M. // The Journal of Clinical Endocrinology & Metabolism. 1983. V. 57. № 2. P. 349–355.
De Oña C.R., Obregón M.J., Del Rey F.E., De Escobar G.M. // Pediatric Research. 1988. V. 24. № 5. P. 588–594.
Escobar G.M.D., Pastor R., Obregon M.J., Rey F.E.D. // Endocrinology. 1985. V. 117. № 5. P. 1890–1900.
Dong H., You S.-H., Williams A., Wade M.G., Yauk C.L., Thomas Zoeller R. // Cerebral Cortex. 2015. V. 25. № 7. P. 1735–1745.
Fernández L.P., López-Márquez A., Santisteban P. // Nature Reviews Endocrinology. 2014. V. 11. № 1. P. 29–42.
Bárez-López S., Obregon M.J., Bernal J., Guadaño-Ferraz A. // Cerebral Cortex. 2018. V. 28. № 5. P. 1783–1793.
Ingenbleek Y., Kimura H. // Nutrition Reviews. 2013. V. 71. № 7. P. 413–432.
McCully K.S. // Ann Clin Lab Sci. 2011. V. 41. № 4. P. 301–314.
McCully K.S. // Comprehensive Physiology. 2015, P. 471–505.
Morte B., Bernal J., Morreale de Escobar G., Díez D., Grijota-Martínez C. // Endocrinology. 2011. V. 152. № 4. P. 1713–1721.
De Oña C.R., De Escobar G.M., Calvo R., Del Rey F.E., Obregon M.J. // Endocrinology. 1991. V. 128. № 1. P. 422–432.
Crantz F.R., Silva J.E., Larsen P.R. // Endocrinology. 1982. V. 110. № 2. P. 367–375.
Dumitrescu A.M., Liao X.-H., Weiss R.E., Millen K., Refetoff S. // Endocrinology. 2006. V. 147. № 9. P. 4036–4043.
Trajkovic M., Visser T.J., Mittag J., Horn S., Lukas J., Darras V.M., Raivich G., Bauer K., Heuer H. // Journal of Clinical Investigation. 2007. V. 117. № 3. P. 627–635.
Mayerl S., Müller J., Bauer R., Richert S., Kassmann C.M., Darras V.M., Buder K., Boelen A., Visser T.J., Heuer H. // Journal of Clinical Investigation. 2014. V. 124. № 5. P. 1987–1999.
Bárez-López S., Guadaño-Ferraz A. // Frontiers in Cellular Neuroscience. 2017. V. 11. № P.
Morte B., Bernal J. // Frontiers in Endocrinology. 2014. V. 5. P. 82.
Calvo R., Obregón M.J., Ruiz de Oña C., Escobar del Rey F., Morreale de Escobar G. // Journal of Clinical Investigation. 1990. V. 86. № 3. P. 889–899.
De Escobar G.M., Jesus Obregon M., De Oña C.R., Del Rey F.E. // Endocrinology. 1988. V. 122. № 4. P. 1521–1531.
Zhang Q., Bai B., Mei X., Wan C., Cao H., Dan L., Wang S., Zhang M., Wang Z., Wu J., Wang H., Huo J., Ding G., Zhao J., Xie Q., Wang L., Qiu Z., Zhao S., Zhang T. // Nature Communications. 2018. V. 9. № 1.
Li H., Bai B., Zhang Q., Bao Y., Guo J., Chen S., Miao C., Liu X., Zhang T. // Gene. 2015. V. 573. № 2. P. 254–260.
Knabl J., de Maiziere L., Hüttenbrenner R., Hutter S., Jückstock J., Mahner S., Kainer F., Desoye G., Jeschke U. // International Journal of Molecular Sciences. 2020. V. 21. № 11. P. 4056.
Shukla P.K., Sittig L.J., Ullmann T.M., Redei E.E. // Alcoholism: Clinical and Experimental Research. 2011. V. 35. № 3. P. 559–565.
Kanellopoulos-Langevin C., Sun Y.-N., Liu Y.-J., Zhang L., Ye Y., Lin L.-X., Li Y.-M., Yan Y.-Q., Chen Z.-P. // PLoS One. 2014. V. 9. № 4. P. e96047.
Patel J., Landers K., Li H., Mortimer R.H., Richard K. // Trends in Endocrinology & Metabolism. 2011. V. 22. № 5. P. 164–170.
Mortimer R.H., Galligan J.P., Cannell G.R., Addison R.S., Roberts M.S. // The Journal of Clinical Endocrinology & Metabolism. 1996. V. 81. № 6. P. 2247–2249.
Steinhauser C.B., Askelson K., Hobbs K.C., Bazer F.W., Satterfield M.C. // Domestic Animal Endocrinology. 2021. V. 77. № P. 106632.
Дополнительные материалы отсутствуют.