

Неорганические материалы, 2020, T. 56, № 11, стр. 1175-1182
Синтез нитридов молибдена
В. М. Орлов 1, *, Р. Н. Осауленко 2, В. Я. Кузнецов 1
1 Институт химии и технологии редких элементов и минерального сырья им. И.В. Тананаева – обособленное подразделение ФИЦ “Кольский научный центр Российской академии наук”
184209 Мурманская обл., Апатиты, Академгородок, 26А, Россия
2 Петрозаводский государственный университет
185910 Республика Карелия, Петрозаводск,
пр. Ленина, 33, Россия
* E-mail: v.orlov@ksc.ru
Поступила в редакцию 30.12.2019
После доработки 02.07.2020
Принята к публикации 02.07.2020
Аннотация
Исследовано влияние прекурсора на условия синтеза и характеристики нитридов молибдена. Азотирование порошков Мо, МоO3 и MgМоO4 вели в токе аммиака при температуре 500–800°С. Использование в качестве прекурсора наноразмерного порошка молибдена позволяет снизить температуру и длительность синтеза. Установлена возможность аммонолиза непосредственно двойного оксида MgМоO4. С его использованием получен продукт с удельной поверхностью до 29 м2/г, что в 2–3 раза больше достигнутого при азотировании MoO3. Во всех случаях продукты синтеза представлены γ- и β-фазами Mo2N с кубической и тетрагональной решетками соответственно.
ВВЕДЕНИЕ
Развитие техники невозможно без использования новых функциональных материалов, например таких, как нитриды переходных металлов [1]. Одним из них является нитрид молибдена Mo2N. Он существует в двух модификациях: тетрагональной β и кубической гранецентрированной γ [2]. Mo2N рассматривается как перспективный катализатор различных процессов, в том числе синтеза аммиака, гидрообессеривания (HDS) топлив и гидроденитризации (HDN) нефти [3–5].
Существует несколько способов получения порошков нитридов молибдена. Авторы [6] исследовали возможность получения нитридов азотированием металлического порошка молибдена со средним размером частиц 2–7 мкм. Попытка получения нитридов в атмосфере азота при температуре 400–1000°С не дала положительных результатов. Азотированием в токе аммиака в течение 4 ч при температуре 700°С был получен нитрид Mo2N с содержанием азота 6.8%. Известно несколько способов синтеза Mo2N с использованием в качестве прекурсора пентахлорида молибдена [7–9]. В работе [7] γ-Mo2N получали взаимодействием MoCl5 с Ca3N2 при давлении 57 кбар. Добавление в шихту 3 молей NH4Cl позволило получить кристаллический нитрид при нормальном давлении. Нанокристаллический γ-Mo2N со средним размером частиц 30 нм получали взаимодействием металлического натрия с безводным MoCl5 и NH4Cl в автоклаве при температуре 550°С [8]. В работе [9] синтез наноразмерного MoNx осуществляли аммонолизом пентахлорида молибдена в обезвоженном хлороформе в течение 8 ч. Затем растворитель выпаривали при температуре 100°С в токе сухого аммиака. Полученный продукт подвергали термообработке при температуре 600–800°С в атмосфере аммиака в течение 10 ч. С ростом температуры термообработки от 600 до 800°С величина х изменилась с 1.321 до 0.769, а удельная поверхность уменьшилась с 49 до 16 м2/г.
Несмотря на такое многообразие подходов, основным способом синтеза нитридов молибдена из-за относительной простоты его реализации можно считать аммонолиз МоО3 [10–13]. Авторы [10, 11] использовали температурно-программированную реакцию, т.е. повышение температуры с заданной скоростью. Нагрев МоО3 в токе аммиака от комнатной температуры до 623 К продолжался 0.5 ч. От 623 до 732 К скорость нагрева составляла 40 или 100 К/ч и далее до 973 К – 100 или 200 К/ч. Выдержка при конечной температуре длилась 1 ч. Полученный продукт дополнительно обрабатывали в токе водорода при температуре 673 К в течение 3 ч. Он представлял собой порошок γ-Mo2N в некоторых случаях с небольшой примесью Мо или МоО2. В зависимости от условий аммонолиза удельная поверхность изменялась от 4 до 116 м2/г. Нитриды молибдена γ-Mo2N и β-Mo2N0.78 с использованием в качестве прекурсора МоО3 получали в работе [12]. Синтез γ-Mo2N осуществляли в токе NН3 при скорости нагрева 5.6°С/мин до 357°С, 0.5°С/мин в интервале 357– 447°С и затем 2.1°С/мин до 785°С. Выдержка при конечной температуре составляла 5 ч. Удельная поверхность полученного γ-Mo2N 85 м2/г. β-Mo2N0.78 синтезировали обработкой МоО3 в токе смеси H2 : N2 в соотношении 3 : 1 при температуре 700°С и выдержке 2 ч. Удельная поверхность нитрида 9 м2/г. Нитрид Mo3N2 с мезопористой структурой и удельной поверхностью 45 м2/г в виде нанопроволок был получен аммонолизом нанопроволок МоО3 в трубчатой печи при температуре 700°С с выдержкой в течение 3 ч [13]. По мнению авторов, мезопористая структура нитрида, обусловлена перегруппировкой структуры оксида металла в нитрид, что приводит к образованию пор в материале.
Использование нанопорошков металла в качестве прекурсора для синтеза нитридов в токе аммиака позволяет значительно снизить температуру и сократить длительность процесса [14, 15]. Поэтому представляло интерес исследовать условия азотирования нанопорошков молибдена, получаемых магниетермическим восстановлением двойных оксидов молибдена [16], а также возможность получения нитридов с увеличенной удельной поверхностью аммонолизом непосредственно двойного оксида молибдена.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных материалов для азотирования использовали порошок молибдена с удельной поверхностью 4.8 м2/г (расчетный размер частиц 130 нм), полученный магниетермическим восстановлением молибдата кальция; молибдат MgMoO4, синтезированный методом спекания [17]; а также МоО3 квалификации “х. ч.” в качестве прекурсора сравнения. Удельная поверхность МоО3 и MgMoO4 составляла 0.4 и 1.5 м2/г соответственно. Реактор (рис. 1) представлял собой кварцевую ампулу диаметром 40 и длиной 700 мм. Кварцевые лодочки с материалом помещали в зоне печи с равномерным нагревом. Температуру измеряли с помощью хромель-алюмелевой термопары, горячий спай которой располагался над лодочкой с материалом. Температуру и скорость ее подъема поддерживали с помощью регулятора температуры Термодат-13К2. Подробно методика эксперимента описана ранее [14].
Рис. 1.
Схема экспериментальной установки для азотирования: 1 – баллон с аммиаком, 2 – манометр, 3 – вентиль тонкой регулировки, 4 – ротаметр, 5 – кварцевый реактор, 6 – лодочка с материалом, 7 – печь сопротивления, 8 – трубка подачи аммиака, 9 – экраны, 10 – термопара, 11 – термодат-13К2, 12 – колба, 13 – водяной затвор.
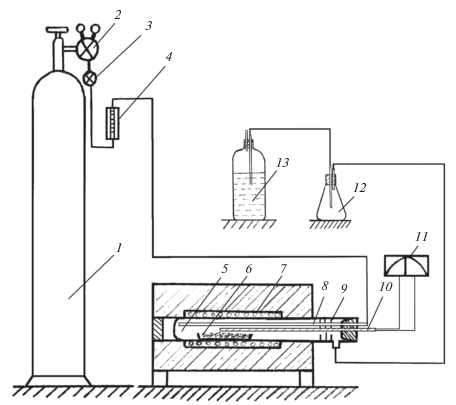
Процесс вели со скоростью подъема температуры 8 К/мин до 500–800°С. Длительность выдержки при максимальной температуре 1–5 ч. Скорость пропускания аммиака 150–200 мл/мин. После окончания выдержки реторту извлекали из печи, продукты реакции охлаждали в токе аммиака. Длительность охлаждения до температуры 100°С составляла 15 мин.
Продукты, полученные с использованием в качестве прекурсора MgMoO4, для удаления оставшегося MgO выщелачивали 2 раза по 1.5 ч 10%-ным раствором HCl. Порошок нитрида промывали дистиллированной водой до нейтральной реакции раствора и сушили при температуре 70°С в течение 8 ч.
Фазовый состав продуктов реакции определяли на дифрактометре ДРФ-2 с CuKα-излучением и графитовым монохроматором. Идентификацию фаз осуществляли по базе дифрактометрических данных PDF-2 ICPDS-ICCD 2002. Для уточнения параметров кристаллической структуры использовали автоматизированный рентгеновский дифрактометр ДРОН-4, метод Ритвельда и базу данных структур неорганических кристаллов ICSD, Карлсруе, 2017. На приборе Micromeritics TriStar II 3020 измеряли удельную поверхность адсорбционным статическим методом БЭТ и параметры пористости методом BJH. Содержание азота и кислорода определяли методом газо-адсорбционной хроматографии в сочетании с импульсным нагревом на анализаторе К-671.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Взаимодействие порошка молибдена и его оксидных соединений с аммиаком может протекать по следующим реакциям:
(1)
${\text{8Мо}} + {\text{4N}}{{{\text{H}}}_{{\text{3}}}} = {\text{4М}}{{{\text{о}}}_{{\text{2}}}}{\text{N}} + {\text{6H}}_{{\text{2}}}^{ \uparrow },$(2)
${\text{2Мо}} + {\text{2N}}{{{\text{H}}}_{{\text{3}}}} = {\text{2МоN}} + {\text{3H}}_{{\text{2}}}^{ \uparrow },$(3)
${\text{4Мо}}{{{\text{О}}}_{{\text{3}}}} + {\text{8N}}{{{\text{H}}}_{{\text{3}}}} = {\text{2М}}{{{\text{о}}}_{{\text{2}}}}{\text{N}} + {\text{12}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{3N}}_{{\text{2}}}^{ \uparrow },$(4)
$\begin{gathered} {\text{4MgMo}}{{{\text{O}}}_{{\text{4}}}} + {\text{8N}}{{{\text{H}}}_{{\text{3}}}} = {\text{2M}}{{{\text{o}}}_{{\text{2}}}}{\text{N}} + \\ + \,\,{\text{4MgO}} + {\text{12}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{3N}}_{{\text{2}}}^{ \uparrow }. \\ \end{gathered} $Азотирование порошка молибдена. Содержание азота в продуктах азотирования порошка молибдена и их удельная поверхность в зависимости от температуры выдержки приведены на рис. 2. Уже после часовой выдержки при температуре 500°С содержание азота в порошке составило 3.9% и достигло максимального значения при повышении температуры синтеза до 600°С (рис. 2а). Удельная поверхность нитридов, синтезированных в интервале 500–700°С, на уровне исходного порошка молибдена и несколько снижается после повышения температуры до 800°С (рис. 2б). На рис. 3 представлены рентгенограммы исходного порошка молибдена и полученных при разных температурах продуктов. Уже после азотирования в течение 1 ч при температуре 500°С появляются слабые и широкие пики новых фаз: тетрагональной β-Mo2N и кубической гранецентрированной γ-Mo2N (рентгенограмма 2 на рис. 3). С увеличением длительности выдержки и температуры интенсивность пиков фаз нитридов растет, а интенсивность максимумов фазы Mo уменьшается. После азотирования в течение 3 ч при температуре 800°С максимумы, соответствующие Mo, отсутствуют (рентгенограмма 5 на рис. 3).
Рис. 2.
Зависимости содержания азота (а) и удельной поверхности (б) от температуры азотирования порошка молибдена (выдержка 1 ч).
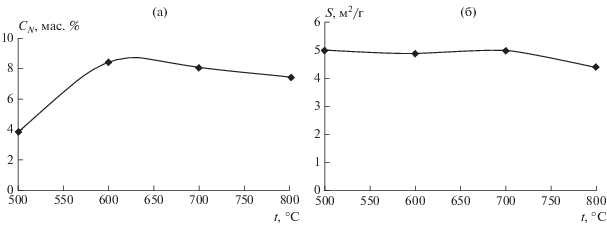
Рис. 3.
Рентгенограммы порошка Мо (1) и продуктов, полученных его азотированием при 500°С, 1 ч (2); 500°С, 3 ч (3); 800°С, 1 ч (4); 800°С, 3 ч (5).
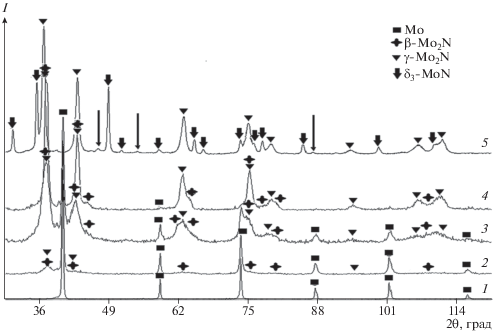
Данные о структуре и фазовом составе продуктов азотирования Mo в зависимости от условий синтеза приведены в табл. 1. Отсутствие фазы β‑Mo2N в продукте, полученном после выдержки в течение 2 ч при температуре 800°С, объясняется тем, что содержание азота в нем составило 9%. Второй фазой в этом случае является гексагональный нитрид δ3-MoN. Его структура характеризуется упорядоченными дефектами в подрешетке азота. Атомы азота расположены в каждой второй тригональной призматической пустоте, построенной из слоев атомов металла. Наличие такого рода дефектов подтверждается присутствием на рентгенограмме 5 (рис. 3) сверхструктурных отражений этой фазы, показанных стрелками [18].
Таблица 1.
Фазовый состав и параметры структуры продуктов азотирования порошка молибдена
| Фазы | № ICSD | Пр. гр. | a, Å | c, Å | Содержание, мас. % | ||
|---|---|---|---|---|---|---|---|
| ICSD | эксп. | ICSD | эксп. | ||||
| Исходный Mo | |||||||
| Mo | 52 267 | $Im\bar {3}m$ | 3.14737 | 3.146 | – | 100 | |
| Азотирование 500°С, 1 ч | |||||||
| Mo | 52 267 | $Im\bar {3}m$ | 3.14737 | 3.1460(6) | – | 71 | |
| β-Mo2N | 30 593 | ${{I{{4}_{1}}} \mathord{\left/ {\vphantom {{I{{4}_{1}}} {amd}}} \right. \kern-0em} {amd}}$ | 4.2 | 4.2318(2) | 8.0 | 7.9451(2) | 16 |
| γ-Mo2N | 172 802 | $Fm\bar {3}m$ | 4.16158 | 4.1714(5) | – | 13 | |
| Азотирование 500°С, 5 ч | |||||||
| Mo | 52 267 | $Im\bar {3}m$ | 3.14737 | 3.1424(8) | – | 58 | |
| β-Mo2N | 30 593 | ${{I{{4}_{1}}} \mathord{\left/ {\vphantom {{I{{4}_{1}}} {amd}}} \right. \kern-0em} {amd}}$ | 4.2 | 4.2261(8) | 8.0 | 7.9798(1) | 21 |
| γ-Mo2N | 172 802 | $Fm\bar {3}m$ | 4.16158 | 4.1887(9) | – | 21 | |
| Азотирование 800°С, 1 ч | |||||||
| Mo | 52 267 | $Im\bar {3}m$ | 3.14737 | 3.1468(9) | – | 3 | |
| β-Mo2N | 30 593 | ${{I{{4}_{1}}} \mathord{\left/ {\vphantom {{I{{4}_{1}}} {amd}}} \right. \kern-0em} {amd}}$ | 4.2 | 4.2126(1) | 8.0 | 8.0443(4) | 29 |
| γ-Mo2N | 172 802 | $Fm\bar {3}m$ | 4.16158 | 4.1828(5) | – | 68 | |
| Азотирование 800°С, 2 ч | |||||||
| γ-Mo2N | 172 802 | $Fm\bar {3}m$ | 4.16158 | 4.1710(2) | – | 70 | |
| δ3-MoN | 99 452 | $P{{6}_{3}}mc$ | 5.73659 | 5.7424(3) | 5.6188(4) | 5.6205(7) | 30 |
Азотирование оксидных соединений. Содержание азота (СN), кислорода (СО), поверхность (S) и фазовый состав продуктов, полученных азотированием МоО3 и MgMoO4 в зависимости от условий процесса, приведены в табл. 2, а их рентгенограммы на рис. 4 и 5. Состав продуктов, полученных азотированием MoO3 при температуре 600°С, свидетельствует о том, что процесс протекает в 2 стадии. Вначале происходит восстановление MoO3 водородом, выделяющимся при разложении аммиака, до MoO2, а затем замещение в последнем кислорода азотом. Подробные данные о структуре и фазовом составе продуктов, полученных аммонолизом MoO3, приведены в табл. 3.
Таблица 2.
Состав продуктов, полученных азотированием МоО3 и MgMoO4
| Прекурсор | t, °С | τ, ч | СN, % | СO, % | S, м2/г | Фазы |
|---|---|---|---|---|---|---|
| MoO3 | 600 | 1 | – | – | – | MoO2 |
| 3 | 2.5 | >12 | 2.6 | MoO2, Mo2N | ||
| 700 | 1 | 7.2 | – | 6.6 | Mo2N, MoN | |
| 3 | 6.9 | – | – | Mo2N, MoN | ||
| 800 | 2 | 6.5 | 2.1 | 11 | Mo2N, Mo | |
| MgMoO4 | 500 | 2 | – | – | – | MgMoO4* |
| 600 | 1 | – | – | – | MgMoO4*, MgO, Mo2N | |
| 600 | 2 | 4.2 | 5.2 | – | Mo2N | |
| 700 | 2 | 7.7 | 8.1 | 14.3 | Mo2N | |
| 800 | 1 | 8.1 | 3.4 | 22.3 | Mo2N | |
| 800 | 2 | 5.6 | 5.1 | 29 | Mo2N |
Рис. 4.
Рентгенограммы продуктов, полученных азотированием МоО3 при 600°С, 3 ч (1); 700°С, 1 ч (2); 800°С, 3 ч (3).
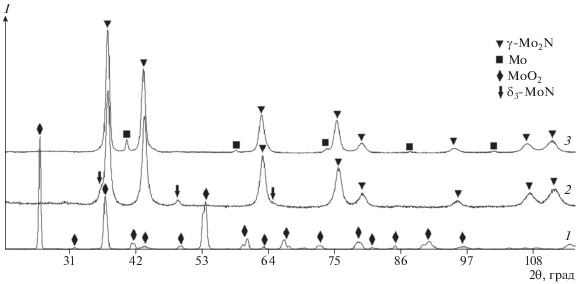
Рис. 5.
Рентгенограммы продуктов, полученных азотированием MgМоО4: а – температура 600°С, выдержка 1 ч (1), 2 ч (2); б – температура 800°С, выдержка 1 ч (1), 2 ч (2).
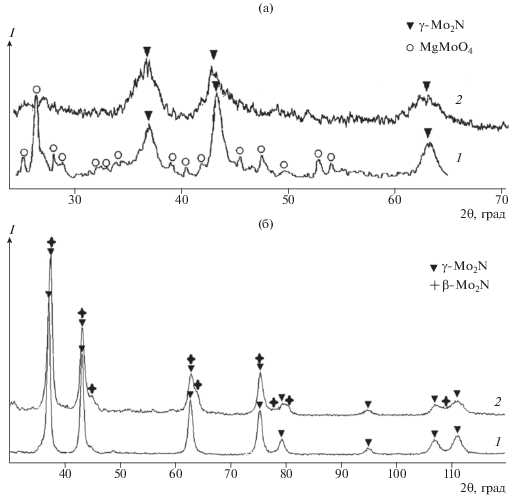
Таблица 3.
Фазовый состав и параметры структуры продуктов азотирования МоО3 и MgMoO4
| Фазы | № ICSD | Пр. гр. | a, Å | c, Å | Содержание, мас. % | ||
|---|---|---|---|---|---|---|---|
| ICSD | эксп. | ICSD | эксп. | ||||
| Азотирование МоО3, 700°С, 1 ч | |||||||
| γ-Mo2N | 172 802 | $Fm\bar {3}m$ | 4.16158 | 4.16669 | – | 91 | |
| δ3-Mo2N | 99 452 | $P{{6}_{3}}mc$ | 5.73659 | 5.73652 | 5.6188(4) | 5.60233 | 9 |
| Азотирование МоО3, 800°С, 2 ч | |||||||
| Mo | 52 267 | $Im\bar {3}m$ | 3.14737 | 3.14873 | – | 3 | |
| γ-Mo2N | 172 802 | $Fm\bar {3}m$ | 4.16158 | 4.17926 | – | 97 | |
| Азотирование MgMoO4, 800°С, 1 ч | |||||||
| γ-Mo2N | 172 802 | $Fm\bar {3}m$ | 4.16158 | 4.1768(5) | – | 100 | |
| Азотирование MgMoO4, 800°С, 3 ч | |||||||
| β-Mo2N | 30 593 | ${{I{{4}_{1}}} \mathord{\left/ {\vphantom {{I{{4}_{1}}} {amd}}} \right. \kern-0em} {amd}}$ | 4.2 | 4.2006(3) | 8.0 | 8.0739(2) | 35 |
| γ-Mo2N | 172 802 | $Fm\bar {3}m$ | 4.16158 | 4.1725(6) | – | 65 | |
Азотирование MgMoO4 при температуре 500°С в течение 2 ч не привело к изменению фазового состава исходного вещества (табл. 2). Однако уже после часовой выдержки при температуре 600°С в продуктах реакции наблюдается фаза Mo2N (рентгенограмма 1 на рис. 5а). После увеличения длительности выдержки до 2 ч весь молибден, входящий в состав соединения, образует нитрид γ-Mo2N (рентгенограмма 2 на рис. 5а). Рентгенограммы продуктов, полученных при более высокой температуре, приведены на рис. 5б. Уточненный с использованием метода Ритвельда фазовый состав и параметры решетки продуктов, полученных азотированием MgMoO4, приведен в табл. 3.
Анализ характеристик продуктов, полученных азотированием всех трех прекурсоров, показывает, что практически все они представлены смесью фаз β- и γ-Mo2N. Высокотемпературная фаза γ-Mo2N присутствует даже в образцах, полученных азотированием порошка молибдена при температуре 500°С. Это соответствует данным работы [19]. Авторы [19] установили, что увеличение содержания азота в нитриде с 28.7 до 34.5 ат. % (5.54–7.13 мас. %) приводит к уменьшению температуры фазового перехода β-Mo2N ↔ γ-Mo2N с 800 до 400°С. Присутствие высокотемпературной фазы γ-Mo2N можно объяснить двумя факторами: медленной кинетикой превращения γ-фазы в β-фазу и уменьшением порядка в подрешетке азота с увеличением содержания азота. При содержании 34.5 ат. % N тетрагональная решетка нитрида преобразуется в кубическую [19].
Обращает на себя внимание высокое содержание кислорода, особенно в продуктах азотирования молибдата. Однако присутствие оксидных фаз не обнаружено. Вероятно, значительна часть анализируемого кислорода сорбирована поверхностью порошка нитрида. Это подтверждается тем, что параметры решетки нитридов близки к эталонным. Сорбцией азота поверхностью, вероятно, объясняется и то, что при сверхстехиометрическом по отношению к нитриду Mo2N содержании азота в ряде образцов не наблюдается присутствия фазы MoN.
Пористая структура полученных продуктов существенно зависит от используемого прекурсора. Типичные кривые сорбции приведены на рис. 6. У продуктов, полученных азотированием Мо и МоО3 (рис. 6б, 6в), они по своему виду близки к типу II по классификации IUPAC, характерному для макропористых веществ [20]. Вид петли гистерезиса близок к типу D, присущему веществам с клиновидными порами. Совершенно другая пористая структура у продуктов, полученных аммонолизом MgМоО4. Кривые сорбции также соответствуют типу II, однако гистерезис практически отсутствует (рис. 6г). Объем пор составляет 0.1–0.18 см3/г. Это более чем в 5 раз превышает объем пор в продуктах, полученных азотированием Мо и МоО3. Такое изменение – следствие дополнительных макропор, образовавшихся в результате выщелачивания оксида магния, входящего в состав молибдата.

ЗАКЛЮЧЕНИЕ
Исследован состав продуктов, полученных азотированием металлического порошка и триоксида молибдена, а также молибдата MgМоО4 в токе аммиака в интервале 500–800°С. Использование в качестве прекурсора наноразмерного порошка молибдена с удельной поверхностью 4.8 м2/г (расчетный размер частиц 130 нм) позволило синтезировать продукт с содержанием азота на уровне 8% уже после часовой выдержки при температуре 600°С. Полученный результат показывает преимущество использованного метода, поскольку при азотировании порошка молибдена с размерами частиц 2–7 мкм для этого требовалась выдержка 4 ч при температуре 700°С [6].
Установлено, что аммонолиз оксида МоО3 протекает в 2 стадии. Вначале он восстанавливается до МоО2, а затем уже следует замещение кислорода азотом с образованием нитрида. Продукт с содержанием азота 7% получен после часовой выдержки при температуре 700°С. Кривые сорбции нитридов, полученных азотированием Мо и МоО3, по своему виду близки к типу II по классификации IUPAC, характерному для макропористых веществ.
Впервые показана возможность получения нитридов молибдена азотированием в токе аммиака двойного оксида MgМоО4. Его использование в качестве прекурсора вместо МоО3 позволило в 2–3 раза увеличить удельную поверхность полученных продуктов. Присутствие в частицах азотированного продукта дополнительных прослоек оксида магния привело к ранее не наблюдавшемуся изменению пористой структуры нитрида после их выщелачивания. Кривые сорбции также соответствуют типу II, однако гистерезис практически отсутствует.
Фазовый состав полученных при 700 и 800°С продуктов представлен в основном сочетанием низкотемпературного тетрагонального нитрида β-Mo2N и высокотемпературной кубической гранецентрированной фазы γ-Mo2N. Соотношение между ними зависит от температуры синтеза и содержания азота в нитриде.
Список литературы
Тот Л. Карбиды и нитриды переходных металлов: пер. с англ.; под ред. Гельда П.В. М.: Мир, 1974. 296 с.
Самсонов Г.В., Кулик О.П., Полищук В.С. Получение и методы анализа нитридов. Киев: Наукова думка, 1978. 320 с.
Volpe L., Boudart M. Ammonia Synthesis on Molybdenum Nltride // J. Phys. Chem. 1986. V. 90. № 20. P. 4874–4877.
Abe H., Cheung T.K., Bell A.T. The Activity of Transition Metal Nitrides for Hydrotreating Quinoline and Thiophene // Catal. Lett. 1993. V. 21. № 1–2. P. 11–18.
Oyama S.T. Preparation and Catalytic Properties of Transition Metal Carbides and Nitrides // Catal. Today. 1992. V. 15. № 2. P. 179–200.
Верхоглядова Т.С., Дубовик Г.Д., Самсонов Г.В. Азотирование порошков переходных металлов // Порошковая металлургия. 1961. № 4. С. 9–20.
O’Loughlin J.L., Wallace C.H., Knox M.S., Kaner R.B. Rapid Solid-State Synthesis of Tantalum, Chromium, and Molybdenum Nitrides // Inorg. Chem. 2001. V. 40. № 10. P. 2240–2245.
Maa J., Duc Y. A Convenient Thermal Reduction–Nitridation Route to Nanocrystalline Molybdenum Nitride (Mo2N) // J. Alloys Compd. 2008. V. 463. № 1–2. P. 196–199.
Choi D., Kumta P.N. Synthesis and Characterization of Nanostructured Niobium and Molybdenum Nitrides by a Two-Step Transition Metal Halide Approach // J. Am. Ceram. Soc. 2011. V. 94. № 8. P. 2371–2378.
Choi J.-G., Brenner J.R., Colling C.W. et al. Synthesis and Characterization of Molybdenum Nitride Hydrodenitrogenation Catalysts // Catal. Today. 1992. V. 15. № 2. P. 201–222.
Choi J.-G., Curl R.L., Thompson L.T. Molybdenum Nitride Catalysts. I. Influence of the Synthesis Factors on Structural Properties // J. Catal. 1994. V. 146. № 1. P. 218–227.
Mckaya D., Hargreavesa J.S.J., Ricob J.L. et al. The Influence of Phase and Morphology of Molybdenum Nitrides on Ammonia Synthesis Activity and Reduction Characteristics // J. Solid State Chem. 2008. V. 181. № 2. P. 325–333.
Lee K.-H., Lee Y.-W., Ko A.-R. et al. Single-Crystalline Mesoporous Molybdenum Nitride Nanowires with Improved Electrochemical Properties // J. Am. Ceram. Soc. 2013. V. 96. № 1. P. 37–39.
Орлов В.М., Кузнецов В.Я., Осауленко Р.Н. Аммонолиз магниетермических порошков тантала // Журн. неорган. химии. 2017. Т. 62. № 1. С. 38–43. https://doi.org/10.1134/S0036023617010132
Орлов В.М., Осауленко Р.Н. Свойства нитридов, полученных аммонолизом магниетермических порошков ниобия // Неорган. материалы. 2018. Т. 54. № 7. С. 675–680. https://doi.org/10.7868/S0002337X18070047
Колосов В.Н., Мирошниченко М.Н., Орлов В.М. Влияние химического состава прекурсоров и условий их восстановления на свойства магниетермических порошков молибдена // Неорган. материалы. 2017. Т. 53. № 10. С. 1081–1086. https://doi.org/10.7868/S0002337X17100098
Мирошниченко М.Н., Колосов В.Н., Макарова Т.И., Орлов В.М. Синтез молибдатов и вольфраматов кальция и магния // Изв. СПбГТИ(ТУ). 2017. № 38. С. 44–47.
Ganin A.Yu., Kienle L., Vajenine G.V. Synthesis and Characterisation of Hexagonal Molybdenum Nitrides // Solid State Chem. 2006. V. 179. № 8. P. 2339–2348.
Ettmayer P. Das System Molibdän-Stickstoff // Monatsh. Chem. 1970. B. 101. P. 127–140.
Sing K.S.W., Everett D.H., Haul R.A.W. et al. Reporting Physisorption Data for Gas/Solid Systems // Pure Appl. Chem. 1985. V. 57. № 4. P. 603–619.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы