

Журнал неорганической химии, 2023, T. 68, № 3, стр. 318-324
Гидротермально-микроволновой синтез ортофосфатов церия(IV)-аммония (NH4)2Ce(PO4)2 ⋅ H2O и NH4Ce2(PO4)3
И. В. Тронев a, b, Е. Д. Шейченко a, b, Л. С. Разворотнева a, b, Э. А. Труфанова a, b, П. В. Минакова a, b, Т. О. Козлова a, А. Е. Баранчиков a, В. К. Иванов a, b, *
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Национальный исследовательский университет “Высшая школа экономики”
101000 Москва, ул. Мясницкая, 20, Россия
* E-mail: van@igic.ras.ru
Поступила в редакцию 28.09.2022
После доработки 11.11.2022
Принята к публикации 17.11.2022
- EDN: JEWBVV
- DOI: 10.31857/S0044457X22601869
Аннотация
Проанализирована возможность получения кристаллических двойных ортофосфатов церия(IV) (NH4)2Ce(PO4)2 ⋅ H2O и NH4Ce2(PO4)3 в условиях гидротермального синтеза с одновременной микроволновой обработкой. Показано, что указанные ортофосфаты в однофазном состоянии могут быть получены в диапазоне температур 130–190°С при продолжительности синтеза ≥5 мин, при этом фазовый состав продуктов синтеза определяется мольным соотношением аммиака и ортофосфорной кислоты в реакционной смеси. Кратковременный (5 мин) низкотемпературный (130°С) гидротермальный синтез в условиях микроволнового воздействия приводит к получению (NH4)2Ce(PO4)2 ⋅ H2O и NH4Ce2(PO4)3 с размером частиц ~70 и ~200 нм соответственно. При более высоких температуре и продолжительности обработки (190°С и 24 ч) размер частиц указанных фаз увеличивается до ~200 и ~500 нм соответственно. Впервые определено значение оптической ширины запрещенной зоны для (NH4)2Ce(PO4)2 ⋅ H2O, составившее 2.8–3.1 эВ для непрямого и прямого переходов соответственно.
ВВЕДЕНИЕ
Известные на сегодняшний день кристаллические ортофосфаты четырехвалентного церия, за исключением Ce(PO4)(HPO4)0.5(H2O)0.5 [1, 2], характеризуются трехмерным структурным каркасом, в каналах которого могут находиться молекулы воды или положительно заряженные ионы [3]. Особенности кристаллической структуры ортофосфатов церия(IV) позволяют рассматривать их в качестве ионообменных материалов и сорбентов [4–6], при этом близость химических свойств церия(IV), тория(IV) и урана(IV), а также образование термодинамически стабильного монацита в результате отжига ортофосфатов церия(IV) делают данные соединения перспективными для иммобилизации радиоактивных элементов [7]. Оптические характеристики ортофосфатов церия(IV), в частности высокое поглощение в УФ-видимом диапазоне и биосовместимость, позволяют рассматривать их в качестве перспективных компонентов солнцезащитной косметики [8–11].
В качестве основного метода синтеза кристаллических ортофосфатов церия(IV) используют гидротермальную обработку [12–14]. Этот синтетический подход является достаточно времязатратным: продолжительность гидротермального синтеза ортофосфатов церия(IV), по имеющимся данным [15–17], составляет от нескольких часов до нескольких суток. В то же время для промышленного применения важным критерием является высокая скорость синтеза неорганических материалов. Эффективной альтернативой гидротермальной обработке является гидротермально-микроволновой метод, обеспечивающий быстрый и равномерный нагрев реакционных смесей, за счет чего достигается меньшая продолжительность синтеза [18–20].
Ранее нами был предложен способ селективного получения ортофосфатов церия(IV)-аммония (NH4)2Ce(PO4)2 ⋅ H2O и NH4Ce2(PO4)3 [13], включающий в себя гидротермальную обработку церийфосфатных растворов в смеси с водным раствором аммиака при 180°С в течение суток. Основной задачей настоящей работы стал анализ возможности получения ортофосфатов церия(IV)-аммония гидротермально-микроволновым методом с целью существенного сокращения температуры и продолжительности синтеза данного класса материалов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных веществ использовали Ce(NO3)3 ⋅ 6H2O (ч. д. а.), H3PO4 (85%-ный водный раствор, ч. д. а.), водный раствор аммиака (~25%, ч. д. а.), изопропанол (ос. ч.), дистиллированную воду.
Синтез ортофосфатов церия(IV)-аммония проводили в соответствии с ранее опубликованной методикой [13]. На первом этапе осаждением из раствора нитрата церия(III) водным раствором аммиака получали диоксид церия. Далее навеску CeO2 (0.1 г) растворяли в 5 мл ортофосфорной кислоты при 90°C и постоянном перемешивании. К полученному раствору добавляли 35 мл 0.5 или 3 М водного раствора аммиака и наблюдали образование гелеобразного осадка. Гидротермальную (ГТ) и гидротермально-микроволновую (ГТМВ) обработку реакционных смесей проводили в тефлоновых автоклавах объемом 100 мл (степень заполнения 40%) при температурах 190 и 130°C в течение 24 ч, 30 и 5 мин. Длительную (24 ч) гидротермальную обработку осуществляли, помещая автоклавы в сушильный шкаф Binder, предварительно разогретый до заданной температуры. Кратковременную (5 или 30 мин) обработку проводили с использованием гидротермально-микроволновой системы Milestone Ethos UP. После обработки полученные осадки очищали многократной промывкой водой с промежуточным центрифугированием до достижения проводимости маточного раствора 0.1–0.2 мСм и высушивали на воздухе при 50°C.
Данные порошковой рентгеновской дифракции получали на дифрактометре Bruker D8 Advance с использованием CuKα1,2-излучения в диапазоне углов 2θ 5°–80° с шагом 0.02° 2θ и временем накопления сигнала не менее 0.2 с на точку. Определение размеров областей когерентного рассеяния (ОКР) в ходе полнопрофильного анализа дифрактограмм осуществляли с помощью программного обеспечения TOPAS 4.2.
Морфологию и химический состав образцов анализировали с помощью растрового электронного микроскопа Carl Zeiss NVision 40, оснащенного детектором Oxford Instruments X-MAX, и растрового электронного микроскопа Tescan Amber, оснащенного детектором Oxford Instruments Ultim Max 100, в диапазоне ускоряющих напряжений 1–20 кВ.
Инфракрасные спектры пропускания образцов регистрировали на спектрометре Инфралюм ФТ08 в диапазоне частот 400–4000 см–1 с разрешением 4 см–1.
Спектры оптического поглощения регистрировали на спектрофотометре Ocean Optics QE65000 в интервале длин волн от 200 до 800 нм с использованием галоген-дейтериевой лампы Ocean Optics DH-2000 и интегрирующей сферы Ocean Optics ISP‑50‑8‑R в режиме диффузного отражения. Накопление спектров осуществляли с выдержкой 20 с и усреднением по пяти спектрам.
Для физико-химического анализа полученных материалов использовали оборудование ЦКП ФМИ ИОНХ РАН.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Согласно данным рентгенофазового анализа, дифрактограмма продукта гидротермальной обработки (24 ч, 190°С) реакционной смеси, полученной смешением церийфосфатного раствора с 3 М раствором NH4OH, характеризуется набором рефлексов, соответствующих однофазному (NH4)2Ce(PO4)2 ⋅ H2O (рис. 1а) [16]. Дифрактограмма продукта, полученного в аналогичных условиях с использованием 0.5 М водного раствора аммиака, соответствует однофазному NH4Ce2(PO4)3 (рис. 1б) [13]. Уточненные в результате полнопрофильного анализа полученных дифрактограмм параметры элементарных ячеек составили: а = 17.5057(82), b = 6.7856(27), с = = 8.0141(36) Å, β = 102.669(21)° для фазы NH4Ce2(PO4)3 и а = 6.8849(39), b = 6.8916(42), с = 17.7050(56) Å для фазы (NH4)2Ce(PO4)2 ⋅ H2O, синтезированных при 130°C в течение 5 мин, что удовлетворительно согласуется с данными работ [13] и [16] соответственно. Обе фазы характеризуются трехмерными каркасными структурами, в тоннелях которых находятся катионы аммония. Формирование однофазных ортофосфатов церия(IV)-аммония различного состава при использовании реакционных смесей с различным мольным соотношением ортофосфорной кислоты и аммиака соответствует ранее опубликованным сведениям [13] и схоже с экспериментальными данными, полученными при анализе продуктов гидротермального синтеза ортофосфатов тория [21]. Механизм формирования различных церийфосфатных фаз в близких условиях достоверно не установлен, однако можно предположить, что состав твердофазных продуктов определяется составом комплексов церия(IV) с фосфатными лигандами, образующихся при смешении церийфосфатных растворов и аммиака.
Рис. 1.
Дифрактограммы однофазных продуктов гидротермальной и гидротермально-микроволновой обработки церийфосфатных гелей, полученных осаждением водным раствором аммиака с концентрацией 3 М (а) при 190°С, 24 ч (1), 190°С, 30 мин (2), 190°С, 5 мин (3), 130°С, 5 мин (4); 0.5 М (б) при190°С, 24 ч (1), 190°С, 30 мин (2), 190°С, 5 мин (3), 130°С, 5 мин (4).

Снижение температуры синтеза до 130°C и продолжительности ГТМВ-обработки до 5 мин приводит к формированию твердофазных продуктов с тем же фазовым составом, что и при использовании обычного гидротермального синтеза (рис. 1). Оценка размеров ОКР показала, что уменьшение температуры и продолжительности синтеза ведет к уменьшению размера кристаллитов (табл. 1).
Таблица 1.
Размер областей когерентного рассеяния для продуктов гидротермальной и гидротермально-микроволновой обработки церийфосфатных гелей в смеси с водными растворами аммиака
| Вид обработки | Концентрация раствора аммиака, М | Температура, °С | Продолжительность | Фазовый состав | Размеры ОКР, нм |
|---|---|---|---|---|---|
| ГТ | 3 | 190 | 24 ч | (NH4)2Ce(PO4)2 ⋅ H2O | >100 |
| ГТМВ | 3 | 190 | 30 мин | 100 | |
| ГТМВ | 3 | 190 | 5 мин | 70 | |
| ГТМВ | 3 | 130 | 5 мин | 50 | |
| ГТ | 0.5 | 190 | 24 ч | NH4Ce2(PO4)3 | 80 |
| ГТМВ | 0.5 | 190 | 30 мин | 70 | |
| ГТМВ | 0.5 | 190 | 5 мин | 55 | |
| ГТМВ | 0.5 | 130 | 5 мин | 30 |
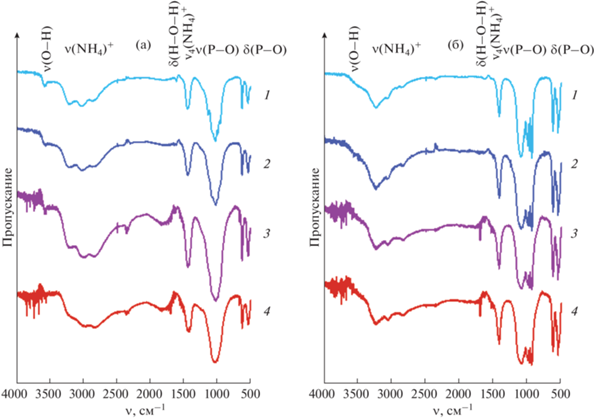
Данные ИК-спектроскопии (рис. 2) подтверждают результаты рентгенофазового анализа и согласуются с литературными данными для (NH4)2Ce(PO4)2 ⋅ H2O и NH4Ce2(PO4)3 [13], указывая на то, что изменение температуры и продолжительности гидротермальной обработки не влияет на состав получаемых соединений. В ИК-спектрах NH4Ce2(PO4)3 и (NH4)2Ce(PO4)2 ⋅ H2O присутствуют полосы поглощения с максимумами при 2800–3300 и 1430 см–1, относящиеся к валентным и деформационным колебаниям иона ${\text{NH}}_{4}^{ + }$ соответственно [22–25]. Полосы поглощения в области 1100–900 и 650–500 см–1 соответствуют валентным и деформационным колебаниям ортофосфат-аниона [26, 27]. ИК-спектр (NH4)2Ce(PO4)2 ⋅ H2O содержит полосы поглощения молекул воды с максимумами при 3550–3660 и 1600 см–1, отвечающие валентным колебаниям О–H и деформационным колебаниям H–O–H соответственно [28]. При этом широкие слабые полосы поглощения в этих же областях ИК-спектра NH4Ce2(PO4)3, по всей видимости, относятся к колебаниям молекул сорбированной воды.
Рис. 2.
ИК-спектры продуктов гидротермальной и гидротермально-микроволновой обработки церийфосфатных гелей, полученных осаждением водным раствором аммиака с концентрацией 3 М (а) при 190°С, 24 ч (1), 190°С, 30 мин (2), 190°С, 5 мин (3), 130°С, 5 мин (4); 0.5 М (б) при 190°С, 24 ч (1), 190°С, 30 мин (2), 190°С, 5 мин (3), 130°С, 5 мин (4).
Результаты локального рентгеноспектрального микроанализа всех полученных образцов дополнительно подтверждают приписанный им химический состав – (NH4)2Ce(PO4)2 ⋅ H2O и NH4Ce2(PO4)3. Так, соотношение Ce : P в продуктах ГТ- и ГТМВ-обработки реакционных смесей, полученных смешением церийфосфатного раствора с 3 и 0.5 М растворами NH4OH, составляет 1 : 2 и 1 : 1.5 соответственно.
Полученные данные указывают на то, что использование микроволнового нагрева при реализации гидротермального синтеза ортофосфатов церия(IV)-аммония позволяет получить твердофазные продукты за время ~5 мин, недостижимое при традиционной гидротермальной обработке. Как правило, высокие скорости фазообразования, наблюдаемые при гидротермально-микроволновом синтезе, связывают с быстрым и равномерным нагревом растворов и суспензий, что позволяет создать условия для формирования большого числа зародышей малорастворимых соединений [19, 29]. При этом зачастую в условиях ГТМВ-обработки размер частиц формирующихся фаз оказывается значительно меньше, чем при ГТ-синтезе в схожих условиях [30]. Гидротермально-микроволновой синтез показал свою эффективность при получении нанокристаллических оксидов, гидроксидов и халькогенидов металлов [19, 29], а также при синтезе ряда ортофосфатов, в первую очередь кристаллических и аморфных ортофосфатов кальция [31, 32], ортофосфатов алюминия [33], циркония [34] и церия [10].
Данные растровой электронной микроскопии подтверждают выявленную на основании результатов рентгенофазового анализа тенденцию к уменьшению размеров частиц с уменьшением температуры и продолжительности гидротермального синтеза. Как было указано выше, это связано с тем, что температура и продолжительность гидротермальной, равно как и гидротермально-микроволновой обработки, определяют особенности протекания процессов зародышеобразования и роста частиц. Более высокая скорость нагрева (20–30 град/мин), характерная для ГТМВ-обработки, способствует тому, что превалируют процессы зародышеобразования, а использование длительной обработки позволяет получать более крупные и сформированные частицы. Из рис. 3 видно, что фаза (NH4)2Ce(PO4)2 ⋅ ⋅ H2O представлена частицами, имеющими форму усеченных октаэдров, размер которых уменьшается от ~200 до ~70 нм с уменьшением температуры до 130°С и продолжительности синтеза до 5 мин. Фаза NH4Ce2(PO4)3, полученная при 190°С в течение 24 ч, представлена агрегатами, содержащими взаимно ориентированные вытянутые частицы размером ~500 нм, в то время как в продукте гидротермально-микроволновой обработки при 130°С присутствуют индивидуальные частицы длиной ~200 нм.
Рис. 3.
РЭМ-фотографии продуктов гидротермальной и гидротермально-микроволновой обработки церийфосфатных гелей, полученных осаждением водным раствором аммиака с концентрацией 3 М при 190°С, 24 ч (а), 190°С, 5 мин (б), 130°С, 5 мин (в); 0.5 М при190°С, 24 ч (г), 190°С, 5 мин (д), 130°С, 5 мин (е).

На рис. 4 представлены спектры поглощения в УФ-видимой области ортофосфатов церия(IV)-аммония NH4Ce2(PO4)3 и (NH4)2Ce(PO4)2 ⋅ H2O, полученных гидротермальной и гидротермально-микроволновой обработкой. Максимумы поглощения этих соединений находятся при ~375 нм, что соответствует поглощению в УФ-А-диапазоне. Видно, что спектры поглощения каждой из фаз практически идентичны, из этого можно сделать вывод о незначительном влиянии параметров синтеза и микроструктуры образцов на их УФ-защитные свойства. При этом в длинноволновой области поглощение NH4Ce2(PO4)3 выше по сравнению с поглощением (NH4)2Ce(PO4)2 ⋅ H2O, что обеспечивает наибольший фактор защиты от УФ-А-излучения.
Рис. 4.
Спектры поглощения продуктов гидротермальной и гидротермально-микроволновой обработки церийфосфатных гелей, полученных осаждением водным раствором аммиака с концентрацией 0.5 М при 190°С, 24 ч (1), 130°С, 5 мин (2); 3 М при 190°С, 24 ч (3), 130°С, 5 мин (4).

Из полученных спектров с использованием уравнения Кубелки–Мунка были определены значения оптической ширины запрещенной зоны (Eg) образцов:
где R – отражение. Поскольку достоверно неизвестно, какой тип переходов реализуется в исследуемых веществах, в случае непрямого перехода анализировали график зависимости (F · hν)1/2 от hν, а в случае прямого ‒ (F · hν)2 от hν [35].Рассчитанные значения Eg в случае NH4Ce2(PO4)3 составили 2.6 и 2.9 эВ для непрямого и прямого переходов соответственно, что согласуется с данными [11]. Аналогичные значения для (NH4)2Ce(PO4)2 ⋅ H2O составили 2.8 и 3.1 эВ. Отметим, что оценка оптической ширины запрещенной зоны для (NH4)2Ce(PO4)2 ⋅ H2O проведена впервые. Полученные результаты подтверждают перспективность использования ортофосфатов церия(IV) в качестве УФ-протекторных материалов. Возможность получения фаз NH4Ce2(PO4)3 и (NH4)2Ce(PO4)2 ⋅ H2O с разным размером частиц позволяет создавать материалы на их основе с контролируемыми физико-химическими характеристиками, в том числе материалы для солнцезащитной косметики.
ЗАКЛЮЧЕНИЕ
Предложен экспрессный метод получения ортофосфатов церия-аммония состава (NH4)2Ce(PO4)2 ⋅ H2O и NH4Ce2(PO4)3 с помощью гидротермально-микроволновой обработки. Установлено, что для формирования этих фаз достаточно обработки при 130°С в течение 5 мин, что может быть достигнуто при использовании микроволнового нагрева в ходе гидротермального синтеза. Изменение параметров синтеза влияет на микроструктуру конечных продуктов, в частности, приводит к уменьшению размеров частиц при уменьшении температуры и продолжительности гидротермальной обработки. Выявлено, что фаза NH4Ce2(PO4)3 характеризуется более высоким фактором защиты от УФ-А-излучения по сравнению с (NH4)2Ce(PO4)2 ⋅ H2O.
Список литературы
Nazaraly M., Wallez G., Chanéac C. et al. // Angew. Chem. Int. Ed. 2005. V. 44. P. 5691. https://doi.org/10.1002/anie.200501871
Nazaraly M., Wallez G., Chanéac C. et al. // J. Phys. Chem. Solids. 2006. V. 67. P. 1075. https://doi.org/10.1016/j.jpcs.2006.01.028
Козлова Т.О., Баранчиков А.Е., Иванов В.К. // Журн. неорган. химии. 2021. Т. 66. № 12. С. 1647. https://doi.org/10.31857/s0044457x21120102
Bevara S., Achary S.N., Patwe S.J. et al. // AIP Conf. Proc. 2016. V. 1731. P. 1. https://doi.org/10.1063/1.4948206
Nazaraly M., Quarton M., Wallez G. et al. // Solid State Sci. 2007. V. 9. P. 672. https://doi.org/10.1016/j.solidstatesciences.2007.04.021
Achary S.N., Bevara S., Tyagi A.K. // Coord. Chem. Rev. 2017. V. 340. № March. P. 266. https://doi.org/10.1016/j.ccr.2017.03.006
Romanchuk A.Y., Shekunova T.O., Larina A.I. et al. // Radiochemistry. 2019. V. 61. № 6. P. 719. https://doi.org/10.1134/S1066362219060134
Sato T., Li R., Sato C. et al. // Phosphorus Res. Bull. 2007. V. 21. P. 44. https://doi.org/10.3363/prb.21.44
Sato T., Yin S. // Phosphorus Res. Bull. 2010. V. 24. P. 43. https://doi.org/10.3363/prb.24.43
Sato T., Sato C., Yin S. // Phosphorus Res. Bull. 2008. V. 22. P. 17. https://doi.org/10.3363/prb.22.17
Kozlova T.O., Popov A.L., Kolesnik I.V. et al. // J. Mater. Chem. B. 2022. V. 10. № 11. P. 1775. https://doi.org/10.1039/d1tb02604f
Nazaraly M., Chanéac C., Ribot F. et al. // J. Phys. Chem. Solids. 2007. V. 68. P. 795. https://doi.org/10.1016/j.jpcs.2007.03.010
Shekunova T.O., Istomin S.Y., Mironov A.V. et al. // Eur. J. Inorg. Chem. 2019. V. 2019. № 27. P. 3242. https://doi.org/10.1002/ejic.201801182
Kozlova T.O., Mironov A.V., Istomin S.Y. et al. // Chem. A Eur. J. 2020. V. 26. № 53. P. 12188. https://doi.org/10.1002/chem.202002527
Lai Y., Chang Y., Wong T. et al. // Inorg. Chem. 2013. V. 52. № 23. P. 13639.
Salvado M.A., Pertierra P., Trobajo C. et al. // J. Am. Chem. Soc. 2007. V. 129. № 36. P. 10970.
Shekunova T.O., Baranchikov A.E., Ivanova O.S. et al. // J. Non-Cryst. Solids. 2016. V. 447. P. 183. https://doi.org/10.1016/j.jnoncrysol.2016.06.012
Zhu Y.J., Chen F. // Chem. Rev. 2014. V. 114. № 12. P. 6462. https://doi.org/10.1021/cr400366s
Meng L.Y., Wang B., Ma M.G. et al. // Mater. Today Chem. 2016. V. 1–2. P. 63. https://doi.org/10.1016/j.mtchem.2016.11.003
Moreira M.L., Mambrini G.P., Volanti D.P. et al. // Chem. Mater. 2008. V. 20. № 16. P. 5381. https://doi.org/10.1021/cm801638d
Salvadó M.A., Pertierra P., Bortun A.I. et al. // Inorg. Chem. 2008. V. 47. № 16. P. 7207. https://doi.org/10.1021/ic800818c
Petit S., Righi D., Madejová J. // Appl. Clay Sci. 2006. V. 34. № 1–4. P. 22. https://doi.org/10.1016/j.clay.2006.02.007
Petit S., Righi D., Madejová J. et al. // Clay Miner. 1999. V. 34. P. 543.
Kloprogge J.T., Broekmans M., Duong L.V. et al. // J. Mater. Sci. 2006. V. 41. № 11. P. 3535. https://doi.org/10.1007/s10853-005-5909-5
Xu Y., Feng S., Pang W. et al. // Chem. Commun. 1996. № 11. P. 1305. https://doi.org/10.1039/CC9960001305
Brandel V., Clavier N., Dacheux N. // J. Solid State Chem. 2005. V. 178. № 4. P. 1054. https://doi.org/10.1016/j.jssc.2005.01.005
Skogareva L.S., Shekunova T.O., Baranchikov A.E. et al. // Russ. J. Inorg. Chem. 2016. V. 61. № 10. P. 1219. https://doi.org/10.1134/S0036023616100181
Hadrich A., Lautie A., Mhiri T. et al. // Vib. Spectrosc. 2001. V. 26. P. 51.
Yang G., Park S.-J. // Materials (Basel). 2019. V. 12. № 7. P. 1177. https://doi.org/10.3390/ma12071177
Maksimov V.D., Meskin P.E., Churagulov B.R. // Inorg. Mater. 2007. V. 43. № 9. P. 988. https://doi.org/10.1134/S0020168507090142
Zhou H., Zhang M., Kong S. et al. // Mater. Lett. 2016. V. 180. P. 239. https://doi.org/10.1016/j.matlet.2016.05.165
Qi C., Zhu Y.-J., Sun T.-W. et al. // Chem. An Asian J. 2015. V. 10. № 11. P. 2503. https://doi.org/10.1002/asia.201500667
Sakintuna B., Yürüm Y. // J. Porous Mater. 2010. V. 17. № 6. P. 727. https://doi.org/10.1007/s10934-009-9344-x
Yu Y.-H., Chen Y.-P., Zeng M. et al. // Mater. Lett. 2016. V. 163. P. 158. https://doi.org/10.1016/j.matlet.2015.10.039
Kolesnik I.V., Aslandukov A.N., Arkhipin A.S. et al. // Crystals. 2019. V. 9. № 7. P. 332. https://doi.org/10.3390/cryst9070332
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии