

Высокомолекулярные соединения (серия А), 2020, T. 62, № 2, стр. 112-120
РЕЛАКСАЦИОННОЕ ПОВЕДЕНИЕ ТЕРМОПЛАСТИЧНОГО ПОЛИИМИДА Р-ОДФО В АМОРФНОМ СОСТОЯНИИ
А. М. Камалов a, *, М. Э. Борисова a, А. Л. Диденко b, Н. А. Никонорова b, В. М. Светличный b, В. Е. Смирнова b, Р. А. Кастро c, В. Е. Юдин b
a Санкт-Петербургский политехнический университет Петра Великого
195251 Санкт-Петербург, Политехническая ул., 29, Россия
b Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр., 31, Россия
c Российский государственный педагогический университет имени А.И. Герцена
191186 Санкт-Петербург, наб. реки Мойки, 48, Россия
* E-mail: spb.kamalov@gmail.com
Поступила в редакцию 11.04.2019
После доработки 06.05.2019
Принята к публикации 13.07.2019
Аннотация
Методами термостимулированных токов деполяризации и диэлектрической спектроскопии исследована молекулярная подвижность пленок поли((4,4'-бис-(4-аминофенокси)бифенила) имидо-1,3-бис-(3,3'-4,4'-дикарбоксифенокси)бензола). Обнаружено существование двух областей релаксации дипольной поляризации (β и α), обусловленных локальной подвижностью в стеклообразном состоянии и кооперативной сегментальной подвижностью в высокоэластическом состоянии соответственно. Температурные зависимости времени релаксации для β-процесса подчиняются уравнению Аррениуса, а для α-процесса – уравнению Фогеля–Таммана–Гессе. Появление третьего пика βI связано с релаксацией объемного гомозаряда. Идентифицированы молекулярные механизмы всех наблюдаемых процессов.
ВВЕДЕНИЕ
В настоящее время хорошо разработанные методики синтеза позволяют получать полиимиды самой разнообразной химической структуры и с большой вариацией физических свойств [1, 2]. Особый интерес представляют термопластичные ароматические полиимиды, используемые в микроэлектронике, электротехнике, перспективные в качестве газоразделительных мембран, электретов, связующих для получения композиционных материалов [3–6]. Разработанный в Институте высокомолекулярных соединений РАН термопластичный частично-кристаллический полиимид Р-ОДФО на основе 1,3-бис-(3,3'-4,4'-дикарбоксифенокси)бензола (диангидрид Р) и 4,4'-бис-(4-аминофенокси)бифенила (диамин ОДФО) имеет отличительную способность к рекристаллизации с воспроизводимой степенью кристалличности. Полиимид Р-ОДФО может служить основой для совершенно новых материалов в качестве матрицы или молекулы, поэтому важно изучить молекулярное движение Р-ОДФО в широком спектре частот воздействия электрического поля.
Свойства полиимидов и полимерных композитов на их основе хорошо исследованы методами динамического механического анализа, дифференциально сканирующей калориметрии. Определены их механические, теплофизические характеристики и температурный интервал их применения [4–6]. Большой интерес представляет изучение ПИ методом диэлектрической спектроскопии и термостимулированных токов деполяризации (ТСД), которые могут предоставить дополнительную информацию о структуре и молекулярной подвижности полимерных цепей на всех уровнях их молекулярной организации. Времена релаксации и энергии активации релаксационных процессов зависят от молекулярных взаимодействий и определяются химической структурой и природой полимера [7, 8]. С помощью методов диэлектрической спектроскопии и ТСД в ПИ различной химической структуры обнаружены три области дипольной релаксации: γ-, β- и α-процессы [9–12]. Дополнительная информация о релаксационных явлениях в ПИ может быть получена путем изучения их объемно-зарядовой поляризации [13, 14].
Цель настоящей работы – установление особенностей процессов диэлектрической релаксации и переноса заряда в термопластичном полиимиде Р-ОДФО на основе диангидрида Р и диамина ОДФО методами диэлектрической и термоактивационной спектроскопии.
МЕТОДИКА ЭКСПЕРИМЕНТА
В работе исследованы релаксационные процессы в аморфных пленках термопластичного полиимида Р-ОДФО [15]

Для измерений использовали пленки толщиной ~50–100 мкм, полученные из 20%-ного раствора полиамидокислоты молекулярной массы Mw ~ 30 × 103, высушенные при 60°С в течение 1 суток [16]. Термообработку пленок ПАК для перевода их в ПИ проводили на стекле в режиме ступенчатого прогревания при 100, 200 и 300°С с выдержкой образцов в термостате при каждой температуре в течение одного 1 ч. Перед измерениями пленки дополнительно прогревали до 250°С. Температуру переходов определяли на динамическом калориметре “DSC 204 Phoenix” фирмы “NETZSCH” в инертной среде при скорости подъема температуры 5 град/мин [16, 17].
Термостимулированные токи деполяризации для образца Р-ОДФО измеряли в короткозамкнутой и в открытой цепи. При измерении в открытой цепи между диэлектриком и измерительным электродом помещали изолирующую прокладку из пленки ПТФЭ толщиной 20 мкм. Спектры ТСД получали с помощью электрометра В7-30. Поляризацию пленок с алюминиевыми фольговыми электродами толщиной 10 мкм проводили в электрическом поле Е = 107 В/м в течение 80 мин при температуре 200°C и последующим охлаждением со скоростью 10 град/мин до комнатной температуры. Затем при нагревании образца со скоростью 2 град/мин измеряли токи деполяризации в отсутствие внешнего электрического поля.
Диэлектрические спектры получали на широкополосном диэлектрическом спектрометре “Concept-21” (“Novocontrol Technologies GmbH”) с автоматическим частотным анализатором высокого разрешения ALPHA-ANB. Пленки прессовали между алюминиевыми электродами (диаметр верхнего электрода 20 мм) при температуре на ∼30°С выше температуры стеклования. Температурно-частотные зависимости диэлектрической проницаемости ε/, фактора диэлектрических потерь ε//, угла диэлектрических потерь tgδ получали в интервале частот 10–1–(3 × 106) Гц и температур 20–350°С.
ТЕРМОАКТИВАЦИОННАЯ СПЕКТРОСКОПИЯ АМОРФНЫХ ПЛЕНОК Р-ОДФО
Поляризация пленки Р-ОДФО в сильном электрическом поле приводила к накоплению как гомо- так и гетерозаряда (рис. 1). Гомозаряд образовался в пленке за счет внедрения носителей заряда из электрода. Механизм возникновения заряда в пленке связан либо с инжекцией носителей из электрода (E ~ 107 В/м), либо с поляризацией Максвелла–Вагнера за счет повышенной проводимости приповерхностных слоев пленки. Образование гетерозаряда может быть обусловлено различными видами поляризации – дипольно-ориентационной и приэлектродной.
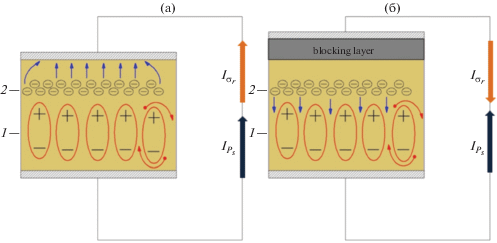
Рис. 1.
Схематическое описание релаксации заряда в режиме короткозамкнутой (а) и открытой (б) цепи. 1 – гетерозаряд Ps, 2 – гомозаряд σr.
Спектры токов ТСД полиимида Р-ОДФО, полученные при измерении в различных контактных режимах (в короткозамкнутой и открытой цепи), приведены на рис. 2. Измерения в режиме короткозамкнутой и открытой цепи позволяют уточнить механизмы релаксационных процессов [19].
Рис. 2.
Зависимости токов ТСД пленки Р-ОДФО, измеренные в режимах короткозамкнутой (1) и открытой (2) цепи.
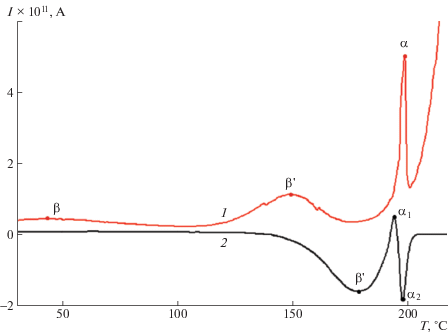
На спектре ТСД, измеренном в режиме короткозамкнутой цепи (рис. 2, кривая 1), имеется три максимума, которые обозначены как β-, β1- и α-процессы. Сопоставление с данными работ [9–12] по диэлектрической релаксации в полиимидах позволяют отождествить пики β и α на спектре ТСД с локальной подвижностью диаминной и диангидридной частей макромолекулы и с ее сегментальной подвижностью. Кроме того, на спектре тока ТСД наблюдается β1-максимум (рис. 2). Направление тока ТСД в короткозамкнутой цепи соответствует направлению тока разрядки образца, что обусловлено разрушением поляризации (гетерозаряд) и движением носителей заряда (гомозаряд) через приповерхностный слой, обладающей повышенной проводимостью (рис. 1а).
Для уточнения природы максимума β1 токи ТСД измерены в режиме как открытой, так и закороченной цепи (рис. 2, кривые 2 и 1 соответственно). В режиме короткого замыкания носители перемещаются через приповерхностный слой диэлектрика, а в режиме открытой цепи – через толщу диэлектрика. Направления движения носителей объемного заряда, следовательно, тока ${{I}_{{{{\sigma }_{r}}}}}$ во внешней цепи определяются контактными условиями. При измерении в открытой цепи направления тока ТСД во внешней цепи совпадает с током зарядки. Из сравнения токов ТСД, определенных в различных контактных условиях, видно, что имеет место инверсия направления тока ТСД и смещение максимума β1 в область высоких температур (рис. 2) при переходе от режима закороченной к режиму открытой цепи. Это позволяет связать максимум β1 с релаксацией гомозаряда, когда носители движутся через толщу полимера [20, 21] (рис. 1б). Смещение максимума β1 в область высоких температур свидетельствует о пониженной проводимости толщи ПИ по сравнению с приповерхностной слоем полимера.
Пик α обусловлен двумя механизмами релаксации заряда: разрушением дипольно-сегментальной поляризации (пик α1) и релаксацией объемного заряда (пик α2), что следует из частичной инверсии α максимума (рис. 2. кривая 2). Температура максимумов, измеренная в режиме короткозамкнутой цепи, характеризующая релаксационные переходы в полиимиде для β-, βI- и α-пиков, составляет 44, 149 и 198°С соответственно.
Анализ спектров токов ТСД проводили на основе представлений о суперпозиции дискретных элементарных дебаевских максимумов $\sum\nolimits_{1,2,3}^n {{{J}_{k}}} $, описываемых кинетикой первого порядка [22]. В данном случае для каждого максимума величина плотности тока ТСД JТСД может быть представлена выражением
(1)
$\begin{gathered} {{J}_{{{\text{ТСД}}}}} = {{J}_{{{\text{макс}}}}}{\text{exp}}\left[ {\frac{W}{k}\left( {\frac{1}{{{{T}_{{{\text{макс}}}}}}} - \frac{1}{T}} \right)} \right] \times \\ \times \,{\text{exp}}\left\langle { - \frac{W}{{kT_{{{\text{макс}}}}^{2}}}\mathop \smallint \limits_{{{T}_{{{\text{макс}}}}}}^T {\text{exp}}\left[ {\frac{W}{k}\left( {\frac{1}{{{{T}_{{{\text{макс}}}}}}} - \frac{1}{{T{\kern 1pt} '}}} \right)} \right]dT{\kern 1pt} '} \right\rangle \\ \end{gathered} $Здесь плотность тока в максимуме Jмакс выражается как
(2)
$\begin{gathered} {{J}_{{{\text{макс}}}}} = \frac{{{{P}_{0}}\beta W}}{{kT_{{{\text{макс}}}}^{2}}} \times \\ \, \times {\text{exp}}\left\{ { - \frac{W}{{kT_{{{\text{макс}}}}^{2}}}\mathop \smallint \limits_{{{T}_{0}}}^{{{T}_{{{\text{макс}}}}}} {\text{exp}}\left[ {\frac{W}{k}\left( {\frac{1}{{{{T}_{{{\text{макс}}}}}}} - \frac{1}{{T{\kern 1pt} '}}} \right)} \right]dT{\kern 1pt} '} \right\}, \\ \end{gathered} $Время релаксации τ описывается выражением
(3)
$\tau = {{\tau }_{{{\text{макс}}}}}{\text{exp}}\left( {\frac{W}{{kT}} - \frac{W}{{k{{T}_{{{\text{макс}}}}}}}} \right)$Зависимость вектора поляризованности от времени
(4)
$P\left( t \right) = {{P}_{0}}{\text{exp}}\left( { - \mathop \smallint \limits_0^t \frac{{dt}}{\tau }} \right).$В основе расчета энергии активации лежат характерные значения плотности тока в максимуме Jмакс и температуры максимума Tмакс, которые определяются по экспериментальным спектрам токов ТСД; при этом значение энергии активации W варьируется. Результат математического моделирования представлен в табл. 1.
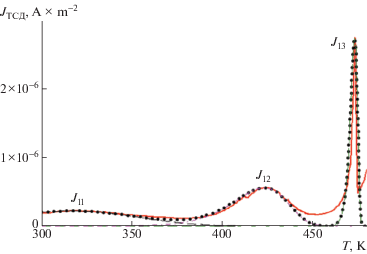
Экспериментально измеренные (сплошная линия) и рассчитанные (точки) спектры ТСД для аморфной пленки представлены на рис. 3.
Рис. 3.
Экспериментальная (сплошная линия) и расчетная (точки) зависимости плотности тока от температуры.
Из максимумов спектров токов ТСД были рассчитаны температурные зависимости времен релаксации. Зависимости lgτ = f(1000/T), полученные по формуле (5), приведены на рис. 4.
(5)
$\tau \left( T \right) = \frac{{Q\left( T \right)}}{{J\left( T \right)}} = \frac{{\frac{1}{r}\int\limits_T^\infty {J\left( T \right)dT} }}{{J\left( T \right)}},$
Наклон прямых соответствует энергиям активации, которые оценены методом математического моделирования.
ДИЭЛЕКТРИЧЕСКОЕ ПОВЕДЕНИЕ Р-ОДФО
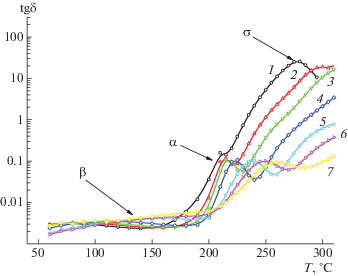
Температурные зависимости тангенса дельта tgδ = f(T) для Р-ОДФО при различных частотах представлены на рис. 5. На зависимости tgδмакс видны два релаксационных максимума β (в стеклообразном состоянии) и α (в высокоэластическом состоянии). Эти области связаны с процессами релаксации дипольно-релаксационной поляризации, что подтверждается смещением максимумов с ростом частоты в сторону высоких температур. При температурах, превышающих α- процесс, наблюдали резкий рост диэлектрических потерь и область tgδмакс, обусловленную проводимостью диэлектрика.
Рис. 5.
Температурные зависимости tgδ для Р-ОДФО при частоте 0.1 (1), 1 (2), 10 (3), 102 (4), 103 (5), 104 (6) и 106 Гц (7).
Анализ диэлектрических спектров проводили, описывая комплексную диэлектрическую проницаемость ε* эмпирическим уравнением Гаврилиака–Негами [23]:
(6)
$\varepsilon {\text{*}}(\omega ) - {{\varepsilon }_{\infty }} = \sum\limits_{k = 1}^n {\frac{{\Delta {{\varepsilon }_{k}}}}{{{{{\{ 1 + {{{(i\omega {{\tau }_{{{\text{Г}}{{{\text{Н}}}_{k}}}}})}}^{{{{\alpha }_{k}}}}}\} }}^{{{{\beta }_{k}}}}}}}} $Здесь ε* = ε'(ω) – iε''(ω), ω = 2πf, Δε = εs – ε∞ – инкремент диэлектрической проницаемости (εs = ε' при ω → 0, ε∞ = ε' при ω → ∞); τГН – характеристическое время релаксации Гаврилиака–Негами; αГН и βГН – расчетные параметры, отвечающие расширению и асимметрии распределения времен релаксации соответственно. При температурах выше Тс в формуле (6) учитывали вклад, обусловленный проводимостью $\frac{{{{\sigma }_{{{\text{dc}}}}}a}}{{{{\varepsilon }_{v}}{{\omega }^{s}}}}$, где σdc – удельная проводимость на постоянном токе, a – постоянная, имеющая размерность [a] = [Гц]s– 1, s < 1, ${{\varepsilon }_{v}}$ – диэлектрическая проницаемость вакуума [24]; τмакс определяли из частотных зависимостей ε'' = φ(f) по частотному положению $\varepsilon _{{{\text{макс}}}}^{{''}}$ по формуле (2) [24]:
(7)
${{\tau }_{{{\text{макс}}}}} = {{\tau }_{{{\text{ГН}}}}}{{\left[ {\frac{{\sin \left( {\frac{{\pi ({{\alpha }_{{{\text{ГН}}}}}){{\beta }_{{{\text{ГН}}}}}}}{{2({{\beta }_{{{\text{ГН}}}}} + 1)}}} \right)}}{{\sin \left( {\frac{{\pi ({{\alpha }_{{{\text{ГН}}}}})}}{{2({{\beta }_{{{\text{ГН}}}}} + 1)}}} \right)}}} \right]}^{{1/({{\alpha }_{{{\text{ГН}}}}})}}}$Диэлектрические спектры (частотные зависимости ε'' = φ(f)) для Р-ОДФО в области β-процесса надежно описаны одним процессом Гаврилиака–Негами (рис. 6).
Рис. 6.
Частотные зависимости ε'' в области β-процесса для Р-ОДФО при 60 (1), 80 (2), 100 (3), 120 (4), 140 (5), 160 (6) и 180°С (7). Точки – эксперимент, сплошные линии – расчет по формуле Гаврилиака–Негами.
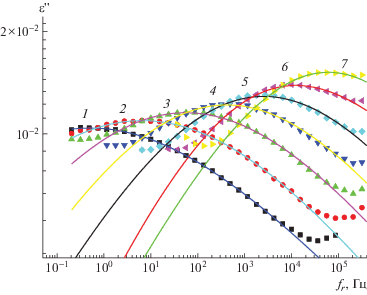
В области α-процесса диэлектрические спектры описаны в интервале температур 220–340°С как сумма вкладов за счет α-процесса и проводимости, а в интервале температур 260–320°С как сумма трех компонентов: α-процесса, проводимости и поляризации Максвелла–Вагнера–Силларса (рис. 7).
Рис. 7.
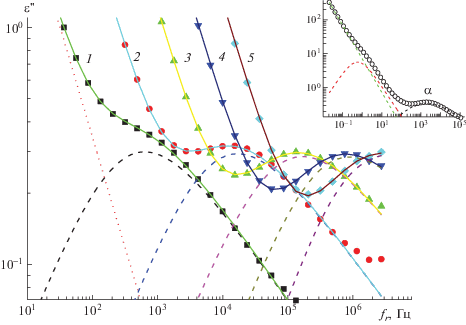
Частотные зависимости ε'' в области α-процесса для Р-ОДФО при 240 (1), 260 (2), 280 (3), 300 (4) и 320°С (5). Точки – экспериментальные точки, сплошные линии – описание по формуле Гаврилиака–Негами, штриховые линии – вклад за счет α-процесса; пунктирная линия – вклад за счет проводимости при 240°С. На вставке – разделение диэлектрического спектра при 260°С на α-процесс и поляризацию Максвелла–Вагнера–Силларса.
Значения τмакс, рассчитанные по формуле Гаврилиака–Негами для Р-ОДФО в области β-, α-процессов и поляризации Максвелла–Вагнера–Силларса, приведены на рис. 8.
Рис. 8.
Зависимости –lgτмакс от обратной температуры для Р-ОДФО для β- (1), α- (2) процессов и поляризации Максвелла–Вагнера–Силларса (3). Точки – вычисления τмакс по формуле Гаврилиака–Негами. Звездочкой отмечены значения fe, вычисленные из спектров ТСД для β-, β1- и α-процессов.
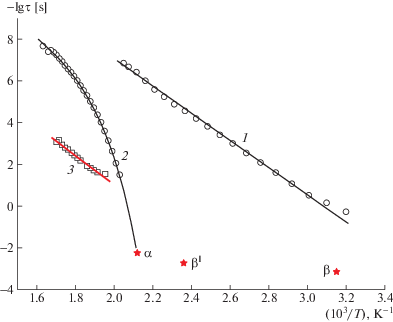
Для β-процесса зависимость –lgτмакс от обратной температуры линейная и описывается уравнением Аррениуса
в котором τ0 = τмакс при Т → ∞, Wa – энергия активации, R – универсальная газовая постоянная. Параметры уравнения (3): –lgτ0 = 19.4 с и Wа = = 1.26 эВ. Линейная зависимость –lgτмакс = φ(1/T) типична для локальных форм молекулярной подвижности, описываемых моделью Дебая, когда процесс переориентации диполей определяется только внутримолекулярными взаимодействиями; при этом значения энергии активации не зависит от температуры.Для α-процесса зависимость –lgτмакс = φ(1/T) криволинейна и хорошо описывается эмпирическим уравнением Фогеля–Таммана–Гессе [26, 27]:
где τ0, В и Т0 – расчетные параметры, не зависящие от температуры (τ0 – предэкспоненциальный множитель, В – активационный параметр, T0 – так называемая температура Фогеля). Для Р-ОДФО параметры уравнения (9) τ0, В и Т0 составляют 12.0 с, 1917 и 414 К соответственно.Методы диэлектрической спектроскопии и токов термостимулированной деполяризации являются “внутренне” связанными, поскольку процессы релаксации дипольной поляризации кинетических единиц, несущих полярную группу, дают области максимума на зависимостях диэлектрических потерь ε'' = φ(f) и на температурных зависимостях тока деполяризации I = φ(T), а результаты обоих методов могут быть сопоставлены при эквивалентной частоте fe [28–30].
Когда температурная зависимость времени релаксации подчиняется уравнению Аррениуса, fe определяется как [28, 29]:
(r – скорость нагревания, Tмакс – температура максимума пика на спектре тока деполяризации). Согласно расчету, для β-процесса fe = 1.22е–4, т.е. –lgτмакс = –3.9, для β1-процесса fe = 3.45е–4, т.е. –lgτмакс= –3.46.Для кооперативных форм молекулярной подвижности и, в частности, для α-процесса, эквивалентная частота определяется как [30]:
Обозначения те же, что и в формулах (9) и (10). Для α-процесса расчет по формуле (11) дает значение fe = 1.96e–3, т.е. –lgτмакс = –2.7.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
При исследовании полиимидов, различающихся большой вариацией структуры, диэлектрическим методом обнаружены по крайней мере три релаксационных процесса: α, β и γ, для которых были предложены молекулярные механизмы [9–12]. Полагали, что γ-процесс обусловлен локальной подвижностью фенильных колец с примыкающими к ним простыми эфирными группами в диаминной части макромолекулы, а также подвижностью связанных молекул воды [11, 12]. Для γ-процесса температура tgδмакс не зависела от строения ПИ и составляла примерно –100°С (при 1 Гц). В настоящей работе условия эксперимента были за пределами проявления γ-процесса.
Относительно молекулярного механизма β-процесса высказывали предположения, что он связан с подвижностью в диангидридной части макромолекулы [11], вращением пара-фениленовых звеньев в диаминной части [9] или коррелированными локальными движениями в обеих частях макромолекулы [9, 11, 12]. Для β-процесса температура tgδмакс зависит от строения, термической предыстории и морфологии образца и лежит в интервале 50–150°С (при 1 Гц). Исходя из химической структуры Р-ОДФО, можно полагать, что β-процесс отражает локальные движения фенильных колец в диаминной и диангидридной частях макромолекулы с примыкающими к ним эфирными и амидными полярными группами. Иными словами, β-процесс является наложением нескольких мод молекулярной подвижности с близкими временами релаксации.
Что касается α-процесса, его можно однозначно связать с переходом через температуру стеклования. На это прежде всего указывает нелинейность температурных зависимостей –lgτмакс = φ(1/T) и большая кажущаяся энергия активации, что характерно для кооперативных форм молекулярной подвижности, когда энергия активации изменяется с температурой. К кооперативной форме молекулярной подвижности относится в первую очередь сегментальная подвижность, связанная с переходом в высокоэластическое состояние. Молекулярным источником α-процесса является крупномасштабная сегментальная подвижность хребта макромолекулы. Температурно-частотные координаты α-процесса (зависимости –lgτмакс = = φ(1/T)) отделяют область стеклообразного состояния полимера (справа) от высокоэластического состояния (слева). Температура стеклования исследованных образцов, определенная традиционной для диэлектрического метода процедурой – экстраполяцией зависимости –lgτмакс = φ(1/T), описанной уравнением Фогеля–Таммана–Гессе, к lgτмакс = 0 (τмакс = 1 с) оказалась равна 210°С.
Значения fe для β-, β1- и α-процесса для ТСД спектров Р-ОДФО приведены на рис. 8. Видно, что для α-процесса, наблюдаемого диэлектрическим методом, есть хорошая корреляция с данными ТСД. Это позволяет утверждать, α-пик на кривой ТСД обусловлен сегментальной подвижностью. Для β-процесса различие между температурами пиков на ТСД и диэлектрической кривой составляет ~35°С. При этом, β1-процесс, связанный с релаксацией объемного гомозаряда, не имеет аналога на диэлектрических зависимостях. Можно полагать, что объемный гомозаряд, накапливаемый при длительной поляризации образца в сильном электрическом поле при температуре, близкой к Тс, существенно изменяет спектр токов ТСД и непосредственно влияет на β-процесс, что приводит к отличию энергий активаций и времен релаксаций.
ЗАКЛЮЧЕНИЕ
Методом термоактивационной и диэлектрической спектроскопии исследована молекулярная подвижность пленок термопластичного ароматического полиимида Р-ОДФО. Диэлектрические и ТСД-спектры свидетельствовали о наличии β- и α-процессов в стеклообразном и высокоэластическом состоянии соответственно. Сопоставление значений времен релаксации и эквивалентных частот, определенных для β- и α-процессов, показывает, что они обусловлены локальной подвижностью в диаминной и диангидридной частях макромолекулы и сегментальной подвижностью соответственно.
Кроме того, в спектрах токов ТСД имеет место третий релаксационный β1-процесс, который вызван релаксацией объемного гомозаряда. Влияние объемного гомозаряда приводит к появлению широкого пика на спектре ТСД, который накладывается на β-процесс и смещает его температуру относительно β-релаксации, наблюдаемой на диэлектрических спектрах.
Спектры ТСД и диэлектрической спектроскопии аморфного Р-ОДФО дают информацию о диэлектрических характеристиках в широком интервале температур и частот электрического поля, что необходимо при расчете и эксплуатации электроизоляционных конструкций и устройств. Накопленный объемный заряд в полиимиде Р-ОДФО определяет электрическую прочность и ресурс работы изоляции [31].
Список литературы
Bryant R.G. Polyimides. New York: Wiley, 2002.
Sroog C.E. // Encyclopedia of Polymer Science and Technolog. New York: Wiley, 1969. V. 11. P. 247.
Perena J.M. // Angew. Makromol. Chem. 1982. V. 106. P. 61.
Yudin V.E., Svetlichnyi V.M. // Polymer Science C. 2016. V. 58. № 1. P. 16.
Юдин В.Е., Светличный В.М. // Рос. хим. журн. 2009. Т. 53. № 4. С. 75.
Yudin V.E., Svetlichnyi V.M., Shumakov A.N., Schechter R., Harel H., Marom G. // Composites. A. 2008. V. 39. P. 85.
Hedvig P. Dielectric Spectroscopy of Polymers. Budapest: Akademiai Kiado, 1977.
Riande E., Diaz-Calleja R. Electrical properties of Polymers. New York: Marcel Dekker, Inc. 2004. P. 638.
Sun Z., Dong L., Zhuang Y., Cao L., Ding M., Feng Z. // Polymer. 1992. V. 33. P. 4728.
Cheng S.Z.D., Chalmes T.M., Gu Y., Yoon Y., Harris F.W., Cheng J., Fone M., Koenig J.L. // Macromole. Chem. Phys. 1995. V. 196. P. 1439.
Chisca S., Musteata V.E., Sava I., Bruma M. // Eur. Polym. J. 2011. V. 47. P. 1176.
Jacobs J.D., Arlen M.J., Wang D.H., Ounaies Z., Berny R., Tan L., Garrett P.H., Vaia R.A. // Polymer. 2010. V. 51. P. 3139.
Gaur M.S., Tiwari R.K. // J. Plastic Film Sheeting. 2009. V. 25. P. 271.
Lu S.X., Cebe P., Capel M. // J. Appl. Polym. Sci. 1995. V. 57. P. 1359.
Svetlichnyi V.M., Kudryavtsev V.V. // Polymer Science B. 2003. V. 45. № 6. P. 140.
Smirnova V.E., Gofman I.V., Ivan’kova E.M., Didenko A.L., Krestinin A.V., Zvereva G.I., Svetlichnyi V.M., Yudin V.E. // Polymer Science A. 2013. V. 55. № 4. P. 268.
Yudin V.E., Svetlichnyi V.M., Gubanova G.N., Grigor’ev A.I., Sukhanova T.E., Gofman I.V., Didenko A.L., Popova E.N., Fedorova G.N., Kudryavtsev V.V. // Polymer Science A. 2002. V. 44. № 2. P. 148.
Borisova M.E., Kamalov A.M., Smirnova V.E., Yudin V.E. // Polymer Science A. 2018. V. 60. № 3. P. 278.
J. van Turnhout // Polym. J. 1971. V. 2. № 2. P. 173.
Khare P.K., Keller J.M., Datt S.C. // Bull. Mater. Sci. 1999. V. 22. P. 109.
Tiwari A., Gupta A.K., Bajpai R., Keller J.M. // Indian J. Phys. 2011. V. 85. P. 1581.
Борисова М.Э., Койков С.Н. Физика диэлектриков. Л.: ЛГУ, 1979.
Havriliak S., Negami S. // Polymer. 1967. V. 8. P. 161.
Vallerien S.U., Kremer F., Boeffel C. // Liq. Cryst. 1989. V. 4. P. 79.
Diaz-Calleja R. // Macromolecules. 2000. V. 33. P. 8924.
Vogel D.H. // Phys. Z. 1921. V. 22. P. 645.
Donth E. The Glass Transition: Relaxation Dynamics in Liquids and Disopdered Materials. Berlin: Springer, 2001. P. 418.
Mano J.F., Mura-Ramos J.J. // Thermochim. Acta. 1998. V. 323. P. 65.
Mura-Ramos J.J., Mano J.F. // Thermochim. Acta. 1996. V. 285. P. 347.
Boersma A., Turnhout J. van, Wubbenhorst M. // Macromolecules. 1998. V. 31. № 21. P. 7453.
Fu M., Dissado L. A., Chen G., Fothergill J. // IEEE Trans. Dielectrics Electrical Insulation. 2008. V. 15. № 3. P. 851.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)