

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 2, стр. 139-152
Синтез серебряных композитов ультрадисперсного политетрафторэтилена и его фракций в среде сверхкритического диоксида углерода и исследование их структуры
Э. Е. Саид-Галиев a, *, С. С. Абрамчук a, А. Р. Хохлов a, А. В. Наумкин a, В. Н. Таланова a, З. Н. Нысенко b, Э. В. Штыкова c, В. В. Волков c, K. A. Abd-Elsalam d
a Институт элементоорганических соединений имени А.Н. Несмеянова Российской академии наук
119991 Москва, ул. Вавилова, 28, Россия
b Институт органической химии им. Н.Д. Зелинского Российской академии наук
119991 Москва, Ленинский пр., 47, Россия
c Институт кристаллографии им. А.В. Шубникова Российской академии наук
Федеральное государственное учреждение
Федеральный научно-исследовательский центр “Кристаллография и фотоника” Российской академии наук
119333 Москва, Ленинский пр., 59, Россия
d Agricultural Research Center Plant Pathology Research Institute, NanoBiotechnology Lab.
12619 Giza, Egypt
* E-mail: ernest@ineos.ac.ru
Поступила в редакцию 13.08.2019
После доработки 25.10.2019
Принята к публикации 06.11.2019
Аннотация
Ультрадисперсный политетрафторэтилен был разделен на растворимые и нерастворимые фракции в сверхкритическом диоксиде углерода. Исходный полимер и его растворимые и нерастворимые фракции импрегнированы (1,5-циклооктадиен)-1,1,1,5,5,5-гексафторацетилацетонатом серебра (I) в растворе сверхкритического диоксида углерода при термодинамических условиях 65°С, 8 МПа в течение 6.6 ч с последующим восстановлением металла в атмосфере водорода при 65°С, 1.2 МПа, 6 ч. Содержание металла по данным рентгенофлуоресцентного анализа достигает 2.4% по массе. Ag-полимерные композиты исследованы методами рентгенофлуоресцентного анализа, просвечивающей электронной микроскопии, малоуглового рентгеновского рассеяния и рентгеновской фотоэлектронной спектроскопии. Показано, что серебро в виде наночастиц с размером 1.0–7.5 нм локализуется в основном в растворимой низкомолекулярной фракции.
ВВЕДЕНИЕ
Ультрадисперсный политетрафторэтилен торговой марки “Forum” [1], получаемый термическим переделом отходов тефлона [2–4], привлекает в последние годы внимание исследователей, так как требует поиска областей применения. Ранее было предложено использовать его в качестве добавки к моторным маслам для создания тонких антифрикционных слоев в двигателях автомобилей [5], оболочек парафиновых капсул [6], в качестве супергидрофобизирующих покрытий [7, 8], в частности для текстильных материалов [9–12]. Для расширения областей применения он нуждается в функционализации. Одно из направлений функционализации – создание на его основе металлополимерных композитов. Особенно интересны свойства нанокомпозитов, поскольку наночастицы благодаря размерному эффекту приобретают новые физические свойства по сравнению с объемным состоянием или частицами микронных размеров [13].
Ранее мы сообщали о создании нанометаллокомпозитов на основе различных полимерных [14–18], карбоновых [19–21] и неорганических [22] материалов с помощью флюидной технологии в среде сверхкритического СО2 и в сочетании с металло-паровым синтезом.
Настоящая работа посвящена функционализации ультрадисперсного ПТФЭ с помощью флюидной технологии наночастицами серебра для придания ему дополнительно к супергидрофобизирующим антимикробных, каталитических и других свойств. Это может быть использовано при разработке многофункциональных материалов, в частности в текстильной промышленности, получении гетерогенных катализаторов низкотемпературного окисления газов и др.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Образец порошкообразного ультрадисперсного ПТФЭ получен из Института химии Дальневосточного отделения Российской академии наук. Ранее химический состав и морфология ультрадисперсного ПТФЭ были изучены в работах [23–25]. Экспериментальный образец, по данным ИК-спектроскопии, содержит заметное количество трифторметильных боковых групп со строением ~CF2–CF(CF3)–CF2~ и терминальных групп –СF=CF2 [26]. Спектр комбинационного рассеяния опытного образца, измеренный ранее [26] при комнатной температуре, был плохого качества, с сильным фоном и низким отношением сигнал/шум, что не позволило зарегистрировать слабые линии в области 400–700 см–1. Однако частоты наблюдающихся интенсивных линий отличаются от частот спектра стандартного тефлона лишь на несколько обратных сантиметров [27]. Спектр содержит линии 289ср и 385ср – деформационные колебания CF в группе F–C–F, 732ос – сложное колебание с участием валентных колебаний CF, 750осл – деформационные + валентные колебания CF, 1216сл – колебания связей CF, 1300сл – валентные колебания СС и 1380ср – смешанные колебания связей CF и СС [28].
Из этого следует, что в основном данный образец ультрадисперсного ПТФЭ, как и классический “Фторопласт-4”, имеющий химическое строение главной цепи (CF2–CF2)n, находится в конформации спирали 157 (повторяющееся звено спирали состоит из 15 групп CF2, образующих семь витков) [26]. Исследуемый образец ультрадисперсного ПТФЭ – кристаллический материал, имеющий воспроизводимый при повторном нагревании фазовый переход при –33°С [25, 29], отнесенный к изменению конформации, а именно, к изменению направления закручивания спирали [25]. Он разлагается в интервале температур 60–550°С в две ступени с разной скоростью [29], проявляет сублимационные свойства и теряет массу в сверхкритическом СО2 (23.3 мас. % при 125°С и 30 МПа) [26].
(1,5-циклооктадиен)-1,1,1,5,5,5-гексафторацетилацетонат серебра (I),

99%, CODAg[hfАcАc] (“Aldrich”) и СО2 с чистотой 99.997%, использовали без дополнительной очистки. Водород, применяемый в качестве восстановителя металла комплекса, – квалификации ч., этанол – квалификации х.ч.
Работу вели на стандартной установке для получения жидкого СО2, схема которой, включая реактор объемом 25 см3, приведена в работе [17].
Методика фракционирования ультрадисперсного ПТФЭ в сверхкритическом СО2
Навеску высушенного порошка взвешивали на весах с разрешением 10–5 г, помещали в контейнер – короткую стеклянную трубку высотой 25 мм с впаянным фильтром Шота № 3, закрытую сверху колпачком из фильтровальной бумаги. Трубку закрепляли в середине реактора. На дне реактора размещали мешатель в тефлоновой оболочке. После загрузки порошкообразного ультрадисперсного ПТФЭ реактор погружали в баню с теплоносителем – силиконовой жидкостью № 5, установленную на магнитной мешалке типа “RET basic C” (IKA, Германия) с обогревом и датчиком температуры фирмы “IKATRON ETS-D5”, включенным в электронную схему мешалки (точность поддержания температуры ±0.5°С). По достижении рабочей температуры создавали заданное давление с помощью цилиндрического ручного пресса фирмы “High Pressure Equipment” (СШA) и включали перемешивание. Время опыта составляло 7 ч. После завершения фракционирования реактор охлаждали до комнатной температуры, проводили декомпрессию и вскрывали. Извлекали контейнер, взвешивали, откачивали в вакуумном шкафу при комнатной температуре в течение 30 мин для удаления остаточных СО2 и воды и затем снова взвешивали. В процессе работы полимер в отдельных опытах разделяли на фракции.
Методика синтеза металлокомпозита
Были получены металлополимерные композиты на основе исходного ультрадисперсного ПТФЭ и образцов а, в, г, д (табл. 1). Для проведения импрегнации на дно реактора (в отдельных экспериментах) загружали навески исходного ультрадисперсного ПТФЭ 0.3 г (300 × 10–5 моль/л) и комплекс CODAg[hfАcАc] 0.071 г (16.7 × 10–5 моль/л) из расчета 1 г-атом Ag на 18 звеньев полимера. Импрегнацию вели при термодинамических условиях 65°С, 8 МПа в течение 6.6 ч. Аппаратурное оформление такое же, как и при фракционировании, за исключением реактора, рабочий объем которого составлял 10 см3.
По достижении заданной температуры, как и при фракционировании, подавали жидкий СО2, который в реакторе переходил в сверхкритическое состояние. Комплекс растворялся при работающей мешалке в сверхкритическом СО2 и диффундировал в матрицу вследствие возникновения диффузионного и дрейфового потоков из-за наличия градиента концентрации [30]. По завершении импрегнации реактор охлаждали, проводили декомпрессию. Затем следовал этап восстановления металлокомплекса до металла. Реактор повторно нагревали до 65°С, заполняли водородом до давления 1.2 МПа и выдерживали при работающей мешалке в течение 6 ч. Затем охлаждали, вскрывали и полученный продукт, имеющий светло-коричневый цвет, промывали двукратно этанолом на плотном стеклянном фильтре для удаления не восстановленного комплекса. Далее продукт сушили на воздухе при 65°С в течение 2 ч и в вакууме при 20°С в течение 3 ч.
Аналогичным образом проводили импрегнацию выделенных фракций: в реактор загружали 0.084 г (84 × 10–5 моль/л) порошкообразной фракции ультрадисперсного ПТФЭ, не растворимой при 65°С и 13 МПа, или фракций, растворимой и не растворимой при 125°С и 30 МПа и 0.02 г (5 × × 10–5 моль/л) кристаллического порошкообразного (1,5-циклооктадиен)-1,1,1,5,5,5-гексафторацетилацетоната серебра (I) и магнитный мешатель в тефлоновой оболочке. Реактор герметизировали, продували током СО2, помещали в баню с силиконовой жидкостью № 5 в качестве теплоносителя, установленную на магнитной мешалке с обогревом “RCT basic” IKA WERKE и нагревали до температуры 65°С, которую регулировавали датчиком температуры фирмы “IKATRON ETS-D5” с точностью ±0.5°С. С помощью ручного цилиндрического пресса создавали давление 10–12 МПа и выдерживали в этих условиях при перемешивании в течение 7 ч. Затем реактор охлаждали и проводили декомпрессию. Восстановление металла вели в атмосфере Н2 при 1.2 МПа и 65°С в течение 7 ч. Затем реактор охлаждали, вскрывали и носитель с иммобилизованными наночастицами металла промывали на фильтре Шотта № 3 этанолом для освобождения от невосстановленного комплекса и продуктов его деструкции. Окончательно композит сушили в вакуумном шкафу при 60°С в течение 2 ч.
Методы анализа
Рентгенофлуоресцентный анализ. Концентрацию серебра в порошкообразном композите определяли на рентгенофлуоресцентном анализаторе “VRA 30” (Германия) по линии AgKα-спектра рентгеновской флуоресценции. Для возбуждения рентгеновской флуоресценции использовали рентгеновскую трубку с Mo-анодом в режиме 50 кВ × 20µA. Измеряли высоту пика над фоном от образца пленки и стандарта. Образцы в виде дисков диаметром 1 см, предварительно взвешенные на весах ВЛР-20, закрепляли на липкой ленте в держателе образцов спектрометра 10/16 Al. Стандартный образец для калибровки спектрометра готовили прессованием таблетки из мелкодисперсного полистирола с навеской AgNO3, содержащей 28% Ag, так, что в слое стандарта массой 1.23 мг высота пика AgKα создавалась серебром массой 1.23 × 0.28 = 0.34 мг. Сравнивая высоту пиков от образца и стандарта, находили массу серебра в образце и соответственно концентрацию серебра.
Малоугловое рентгеновское рассеяние. Измерения проводили методом традиционного малоуглового рентгеновского рассеяния на усовершенствованном лабораторном дифрактометре АМУР-К (разработка Специального конструкторского бюро Института кристаллографии РАН, Москва) на фиксированной длине волны излучения λ = = 0.1542 нм с применением геометрии Кратки [31]. Модификация прибора и использование детектора с улучшенными параметрами позволили использовать более широкую область значений волновых векторов в интервале 0.12 < s < 6.0 нм–1 (s = 4π sinθ/λ, 2θ – угол рассеяния). Полученные экспериментальные данные были нормированы на интенсивность падающего пучка, после чего в них вводилась поправка на коллимационные искажения в соответствии со стандартной процедурой [32].
Первичную обработку полученных экспериментальных данных малоуглового рассеяния проводили с помощью программы PRIMUS [33].
Для анализа распределения по размерам в ультрадисперсном ПТФЭ использовали диалоговую программу GNOM [34]. В этом случае объемные функции распределения по размерам DV(R) находили, решая интегральное уравнение вида
Анализ брэгговских пиков на кривых малоуглового рассеяния выполняли с помощью программы PEAK [33]. Размер кристаллитов L определялся из полуширины максимума интенсивности первого пика, соответствующего углу рассеяния 2θ:
Степень разупорядоченности в полимерной системе Δ/d находили по формуле
где d = 2π/s – период структуры, межплоскостное расстояние, βs – полная ширина на полувысоте максимума интенсивности рассеяния (в радианах), наблюдаемого на угле рассеяния 2θ, соответствующего волновому вектору s, а Δ – среднеквадратичное отклонение от расстояния между ближайшими регулярно упакованными структурными мотивами [35].Рентгеновская фотоэлектронная спектроскопия. Рентгеновские фотоэлектронные спектры получали на спектрометре “Axis Ultra DLD” (“Kratos”) с использованием монохроматического AlKα-излучения при мощности рентгеновской пушки 150 Вт. Обзорные спектры и спектры высокого разрешения регистрировали при энергии пропускания 160 и 40 эВ соответственно. Обзорный спектр записывался с шагом 1 эВ, спектры высокого разрешения – с шагом 0.1 эВ. Размер анализируемой области составлял около 300 × 700 мкм2. Образцы закрепляли на держателе с помощью двусторонней адгезионной ленты и исследовали при комнатной температуре и остаточном давлении в камере спектрометра, не превышающем 10–8 мм рт. ст. Проведенная предварительно калибровка шкалы энергии спектрометра отвечала следующим значениям пиков стандартных образцов (очищенных ионным распылением поверхностей металлов): Au 4f7/2 – 83.96 эВ, Cu 2p3/2 – 932.62 эВ, Ag 3d5/2 – 368.21 эВ. Для устранения эффекта зарядки образцов спектры снимали с использованием нейтрализатора. Калибровку спектров осуществляли по низкоэнергетическому состоянию, выделенному в спектре F 1s, которому была приписана энергия 689.67 эВ [36]. Вычитание фона неупругих потерь энергии электронов проводили по методу Ширли с учетом ряда особенностей [37]. Концентрацию элементов рассчитывали с учетом функции пропускания спектрометра и коэффициентов элементной чувствительности фирмы, входящих в программное обеспечение спектрометра.
Просвечивающая электронная микроскопия. Анализ выполняли на электронном микроскопе “Carl Zeiss Leo 912 AB OMEGA” при ускоряющем напряжении 100 кВ.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Ранее было показано, что ультрадисперсный ПТФЭ при экспозиции в сверхкритическом СО2 уже при давлении 13 МПа и 25°С теряет до 9% массы. Эти потери растут с увеличением давления и температуры и при 125°С и 30 МПа достигают ~23% по массе [26].
Ранние исследования показали, что в реакторе удается собрать лишь до 30–50% массы потерянной фракции. Наиболее вероятное объяснение этого факта – частичный пиролиз наиболее низкомолекулярной фракции при нагревании в среде сверхкритического СО2 с образованием газообразных и легколетучих продуктов и удаление этих продуктов в процессе декомпрессии. Ранее пиролиз ультрадисперсного ПТФЭ был подробно изучен в работах [23–25, 38–50] и было показано, что в интервале 50–100°С происходит сублимация фторуглеводородов С3–С14. Фторпарафины хорошо растворяются в сверхкритическом СО2 [51]. Следует при этом заметить, что именно фторпарафины составляют наиболее низкомолекулярную фракцию ультрадисперсного ПТФЭ [48].
Вопрос о механизме процесса, в результате которого происходит потеря массы навеской ультрадисперсного ПТФЭ в сверхкритическом СО2, не является предметом настоящей работы. Вместе с тем вопрос о растворимости ультрадисперсного ПТФЭ важен для практики, поскольку данный полимер планировали в дальнейшем использовать для супергидрофобизации текстильных волокон [9–12]. С экономической точки зрения при большом объеме производства весьма невыгодно было бы использовать исходный полимер целиком. Целесообразнее было бы отделять и работать только с растворимой в сверхкритическом СО2 фракцией (15–20% массы полимера). Однако эксперимент показал, что собрать эту фракцию в практически разумных количествах не удается, такой путь фактически является экономически нецелесообразным. Свойства этой фракции, полученной в сверхкритическом СО2, не позволяют отделять и использовать ее для супергидрофобизации и поэтому остается выбирать только исходный полимер целиком или обратиться к сублимации при повышенных температурах, хотя данный процесс является весьма медленным [4].
Импрегнированные материалы были исследованы методами рентгенофлуоресцентного анализа, просвечивающей электронной микроскопии, малоуглового рентгеновского рассеяния и рентгеновской фотоэлектронной спектроскопии (РФЭС). Содержание наночастиц Ag в ультрадисперсном ПТФЭ после восстановления металла составляет, по данным рентгенофлуоресцентного анализа, 2.3 ± 0.5% по массе. Результаты исследования ультрадисперсного ПТФЭ, импрегнированного комплексом серебра, методом просвечивающей электронной микроскопии (ПЭМ) приведены на рис. 1.
Рис. 1.
Микрофотография крупного кристалла серебра (а) и дебаеграмма ультрадисперсного ПТФЭ после импрегнации его комплексом CODAg[hfАcАc] в сверхкритическом СО2 (65°С, 8 МПа, 8 ч) и последующем восстановлении металла водородом (1.2 МПа, 65°С, 6 ч) (б).

При ПЭМ-анализе исходного импрегнированного полимера в нем обнаруживаются крупные монокристаллы. Анализ монокристаллов методом электронной дифракции показал, что они обладают гранецентрированной кубической решеткой, совпадающей с решеткой монокристаллов серебра. Структуру полученного композита анализировали также методом малоуглового рентгеновского рассеяния (рис. 2).
Рис. 2.
Экспериментальные кривые малоуглового рассеяния исходного ультрадисперсного ПТФЭ (1) и ультрадисперсного ПТФЭ, импрегнированного в сверхкритическом СО2 комплексом CODAg[hfАcАc] (65°C, 8 МПа, 8 ч) и восстановленного водородом (65°С, 1.2 МПа, 6 ч) (2) (а); разностная кривая малоуглового рассеяния от сформированных в образце 1 наночастиц серебра (б); объемные функции распределения по размерам рассеивающих неоднородностей в матрице ультрадисперсного ПТФЭ (1) и распределение по размерам наночастиц серебра в этой матрице (2) (в).

Разностная кривая малоуглового рассеяния была получена вычитанием кривой малоуглового рентгеновского рассеяния от исходного ультрадисперсного ПТФЭ из кривой рассеяния от ультрадисперсного ПТФЭ с наночастицами серебра для получения рассеяния только от наночастиц металла. Как видно, объемные распределения по размерам рассеивающих неоднородностей в исходном образце ультрадисперсного ПТФЭ и сформированных в нем наночастиц серебра имеет бимодальный характер: присутствуют две фракции – мелких и крупных образований. Однако исходный образец характеризуется значительно более широким распределением по размерам. Наночастицы серебра после восстановления имеют размер порядка 3 нм в узкой фракции мелких частиц и не более 15 нм для более крупных образований, в то время как размер неоднородностей в исходной матрице может достигать 60 нм.
Поскольку в работе [48] было показано, что пиролиз и сублимация ультрадисперсного ПТФЭ начинается выше 50°С, а по данным ТГА при 70°С (что допустимо, так как материал не стандартный и разные партии могут различаться по составу и морфологии), мы провели импрегнацию и восстановление металла при 50°С. Полученный композит после тщательной экстракции невосстановленного комплекса в этаноле и сушки в вакууме был проанализирован методами рентгенофлуоресцентного анализа, ПЭМ и малоуглового рентгеновского рассеяния (рис. 3, 4). По данным рентгенофлуоресцентного анализа, содержание серебра составило 1.8%. Причиной снижения содержания серебра явилась низкая температура, при которой резко понижается скорость импрегнации. На микрофотографии (рис. 3) видны мелкие сферические частицы металла размером 6–30 нм, но в то же время наблюдаются и крупные кристаллы размером более 100 нм.
Рис. 3.
Микрофотография исходного полимера, импрегнированного раствором CODAg[hfАcАc] в сверхкритическом CО2 с последуюшим восстановлением металла в водороде (1.2 МПа, 50°С).

Рис. 4.
Экспериментальные кривые малоуглового рассеяния исходного образца УПТФЭ (1) и образца с наночастицами серебра (импрегнация и восстановление при 50°С) (2) (а), а также объемные функции распределения по размерам в этих образцах (б).

На рис. 4а видно, что кривые малоуглового рентгеновского рассеяния исходного образца и содержащего наночастицы серебра очень слабо различаются. Это может объясняться небольшим содержанием наночастиц серебра в образце 2. Оба образца полидисперсны и не упорядочены – не наблюдаются брэгговские пики на кривых рассеяния. Возможное кристаллическое состояние наночастиц серебра можно обнаружить только при более высоких значениях волновых векторов (≫10 нм–1), что недоступно для исследования методом малоуглового рассеяния.
Однако небольшая разность интенсивностей рассеяния образцов с наночастицами серебра и без них позволяет вычислить и сравнить объемные функции распределения по размерам в этих образцах (рис. 4б).
Объемные функции распределения по размерам рассеивающих неоднородностей в образце ультрадисперсного ПТФЭ и распределение по размерам в образце с наночастицами серебра также отличаются незначительно. Обе функции DV(R) имеют бимодальное распределение, характерное для образцов данного типа, где присутствует сравнительно узкая фракция с небольшими размерами рассеивающих неоднородностей порядка 2–3 нм и широкая фракция с размерами до 40 нм. Основное различие состоит в более высокой амплитуде функции DV(R) фракции малых размеров для образца, содержащего наночастицы серебра. Это объясняется более высокой электронной плотностью металла по сравнению с электронной плотностью полимера, поскольку амплитуда функции DV(R) пропорциональна электронной плотности. Однако увеличение амплитуды объемной функции распределения по размерам DV(R) не наблюдается для фракции более крупных неоднородностей. Отсюда можно сделать вывод, что в отсутствие пиролиза и при малых концентрациях серебра формирование наночастиц серебра происходит почти исключительно в микропорах ультрадисперсного ПТФЭ.
Фракции 2, не растворимые при 65°С, 13 МПа и 75°С, 125°С и 30 МПа были также исследованы методом ПЭМ и малоуглового рентгеновского рассеяния. При анализе малоуглового рассеяния этих образцов было установлено, что их кривые отличаются незначительно. Наибольшее отличие наблюдали для образца 1, в то время как кривые для образцов 2 и 3 практически совпадают. Для образца 1 можно было получить разностную кривую малоуглового рассеяния и оценить распределение по размерам наночастиц серебра в матрице (рис. 5).
Рис. 5.
Экспериментальные кривые малоуглового рассеяния (а) и рассчитанные с помощью программы GNOM объемные распределения по размерам рассеивающих неоднородностей (б): 1 – исходный ультрадисперсный ПТФЭ; 2 – фракция, не растворимая при 65°С и 13 МПа СО2; 3 – фракция, не растворимая при 125°С, 13 МПа СО2; 4 – образец 2 с импрегнированным комплексом CODAg[hfАcАc] и после восстановления металла.

Видно, что экспериментально полученные кривые малоуглового рентгеновского рассеяния для всех образцов сходны между собой, имеют близкие профили и амплитуды, которые отличаются друг от друга только средней частью. Последнее свидетельствует о некоторых структурных различиях в образцах. Однако рассеяние образцов 2 и 3 (две центральные кривые рассеяния на основном рисунке) практически совпадает и, следовательно, можно предположить, что структуры этих образцов практически идентичны.
В то же время на вставке видно, что амплитуда рассеяния на графике функции ${{D}_{v}}(R)$ для образцов 2 и 3 различается, что может объясняться присутствием более плотных неоднородностей в матрице после обработки при более высокой температуре. Отделение фракции 65°С, 13 МПа не изменяет градиент плотности на границе полимерное тело–пора (кривые 1 и 2), а при росте температуры до 125°С этот градиент становится весьма значительным (кривая 3).
Не растворимые при 75 и 125°C, 30 МПа фракции ультрадисперсного ПТФЭ, импрегнированные комплексом серебра и после восстановления металла, также анализировали методом малоуглового рентгеновского рассеяния.
Для ультрадисперсного ПТФЭ с инкорпорированными наночастицами серебра кривые малоуглового рентгеновского рассеяния имеют некоторое различие только для фракции, не растворимой в условиях 65°C, 13 МПа (в образце еще сохраняется большая часть низкомолекулярной фракции). Для образцов, не растворимых при 75 и 125°C, 30 МПа с инкорпорированным серебром (в которых низкомолекулярная фракция почти полностью отсутствует) (образцы 2 и 4) кривые малоуглового рентгеновского рассеяния практически совпадают с кривыми рассеяния исходных образцов 1 и 3 или даже идут несколько ниже. Это означает, что интенсивность малоуглового рассеяния, которая пропорциональна электронной плотности рассеивающих объектов, для образцов 2 и 4, содержащих металл, оказывается не выше, а парадоксальным образом практически равна интенсивности исходного материала. Поскольку темный цвет образцов с инкорпорированными наночастицами серебра свидетельствует о присутствии в образцах металла, объяснить данное явление можно тем, что практически весь металл оказался на поверхности матриц и не проник в глубину полимера, т.е. наночастицы серебра образовались не внутри матрицы, а в поглощающем поверхностном слое металла.
По-видимому, этот факт указывает на то, что в режиме экстракции при 65°С и 13 МПа низкомолекулярная менее плотная фракция, содержащая поры, способная импрегнироваться комплексом металла, еще не полностью удалена. В то же время при 75 и 125°С и 30 МПа эта фракция почти полностью экстрагирована, и нерастворимые фракции имеют структуру, близкую к Фторопласту-4, плотному кристаллическому полимеру, почти не набухающему в сверхкритическом СО2.
Далее такими же методами была исследована фракция 1, растворимая при 125°C и 30 МПа (рис. 6). Из рис. 6 следует, что наночастицы локализуются в основном в растворимой низкомолекулярной фракции, и их размеры лежат в нанометровой области в интервале 1.0–7.5 нм. Максимум распределения приходится на область 3.5–4.0 нм.
Рис. 6.
Микрофотография фракции 1 ультрадисперсного ПТФЭ, растворимой при 125°C и 30 МПа, импрегнированной комплексом серебра с последующим восстановлением металла (а) и гистограмма распределения наночастиц серебра по размеру (б).

Исследования растворимой фракции методом малоуглового рентгеновского рассеяния дали следующие результаты: значительно более выразительные кривые были получены для образцов первой фракции ультрадисперсного ПТФЭ (рис. 7).
Рис. 7.
Экспериментальные кривые малоуглового рассеяния первой фракции образцов ультрадисперсного ПТФЭ: 1 – первая фракция 125°C; 30 МПа, 2 – первая фракция с частицами Ag.

Наличие ярко выраженных брэгговских пиков на кривых малоуглового рассеяния от образцов первой фракции ультрадисперсного ПТФЭ свидетельствует об образовании упорядоченной, квазикристалличской структуры как в исходном образце, так и в образце с импрегнированными наночастицами серебра, что совпадает с результатами работы [29]. Образование наночастиц металла вызывает заметные изменения квазикристаллической структуры растворимой фракции ультрадисперсного ПТФЭ: меняется положение брэгговских пиков на кривой рассеяния и амплитуда их максимумов.
Основные характеристики пиков приведены в табл. 2.
Таблица 2.
Структурные характеристики образцов первой фракции ультрадисперсного ПТФЭ
| Образец | s1, нм–1 | $\bar {d}$1, нм | s2, нм–1 | $\bar {d}$2, нм | s3, нм–1 | $\bar {d}$3, нм | s4, нм–1 | $\bar {d}$4, нм | L, нм | Δ/$\bar {d}$ |
|---|---|---|---|---|---|---|---|---|---|---|
| Ультрадисперсный ПТФЭ | 1.5 | 4.1 | 2.3 | 2.7 | 3.0 | 2.1 | 4.5 | 1.4 | 42 | 0.10 |
| Ультрадисперсный ПТФЭ-Ag | 1.4 | 4.3 | 2.0 | 3.1 | 2.9 | 2.1 | 4.0 | 1.6 | 59 | 0.09 |
Поскольку кривые малоуглового рассеяния в сильной степени “зашумлены”, определение точного положения всех четырех максимумов и соответствующих межплоскостных расстояний не представляется возможным. Однако данные табл. 2 позволяют предположить, что положения брэгговских пиков соответствуют возможному наложению двух ламелярных упаковок в каждом из образцов: на обеих кривых малоуглового рентгеновского рассеяния имеются ламели s1–s3 и s2–s4 с эквидистантным расположением брэгговских пиков. Характеристический размер этих ламелей d1 увеличивается после инкорпорации наночастиц серебра с 4.1 нм до 4.3 нм.
Следовательно, можно говорить об образовании металлических наночастиц внутри матрицы – интенсивность рассеяния возрастает после инкорпорации наночастиц серебра (рис. 7, кривая 2). Однако провести вычитание рассеяния матрицей из кривой рассеяния образца, содержащего металл, в данном случае нельзя, поскольку структура матрицы изменилась. Более того, определение по размерам в подобных структурах также является некорректным из-за присутствия брэгговских пиков на кривых рассеяния. Можно только предположить, что наночастицы располагаются между ламелями и их средний размер не превышает 4 нм. Однако возможно образование длинных кластеров, состоящих из таких наночастиц разного размера, так как форма кривых малоуглового рассеяния в области самых малых угловых векторов свидетельствует о высокой степени полидисперсности и наличии крупных частиц с линейными размерами более 100 нм (что подтверждается данными ПЭМ).
Для изучения химического состояния металла в композите образцы исследовали методом РФЭС. В обзорных РФЭС-спектрах наряду с пиками, характерными для исследуемых образцов, наблюдаются пики, соответствующие примесным атомам кислорода.
Результаты количественного анализа, полученные из обзорных спектров на основе коэффициентов элементной чувствительности приведены в табл. 3.
Таблица 3.
Элементный состав образцов, определенный из обзорных спектров
| Образец | C, ат. % | O, ат. % | Ag, ат. % | F, ат. % | F/C |
|---|---|---|---|---|---|
| Ультрадисперсный ПТФЭ (С38F61.8О0.2) | 38.03 | 0.15 | – | 61.82 | 1.63 |
| Ультрадисперсный ПТФЭ-Ag-композит (С32.6F66.8Ag0.2O0.4) | 32.64 | 0.35 | 0.16 | 66.85 | 2.05 |
Спектры С 1s и F 1s образцов ультрадисперсного ПТФЭ и ультрадисперсного ПТФЭ-Ag представлены на рис. 8. Спектры заметно различаются по форме, что указывает на разную относительную концентрацию химических групп. После осаждения серебра наблюдается уширение спектров, свидетельствующее об увеличении неоднородности поверхности и уменьшении относительной концентрации групп CF2.
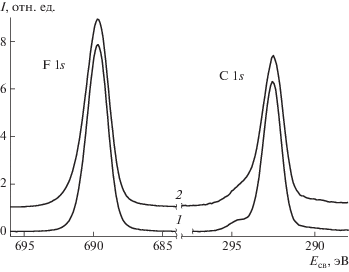
На рис. 9 показано разложение спектров F 1s и C 1s на компоненты, а в табл. 4 – численные характеристики состояний (энергия связи Есв, ширина пиков W и относительная интенсивность Iотн). Спектры C 1s описаны тремя состояниями с энергией связи около 294.5, 292.5 и 290.5 эВ, которые могут быть приписаны группам CF2, CF3 и CFxOy. Спектры F 1s представлены в виде совокупности основного состояния с энергией связи 689.7, соответствующей группе CF2, и, согласно разложению спектра C 1s на компоненты, – группе CFxOy с энергией связи 691.2 и 691.5 эВ для образцов ультрадисперсного ПТФЭ и ультрадисперсного ПТФЭ-Ag соответственно. Различие в величинах энергий связи групп CFxOy обусловлено разной концентрацией кислорода. Энергии связи пика F 1s групп CF2 и CF3 достаточно близки [36], поэтому в спектре F 1s не выделено соответствующее состояние.
Рис. 9.
Разложение спектров С 1s и F 1s образцов ультрадисперсного ПТФЭ (1) и ультрадисперсного ПТФЭ-Ag (2) на компоненты.

Таблица 4.
Энергии связи (Есв), ширины пиков (W) и относительные интенсивности химических состояний (Iотн), выделенных в спектрах F 1s и С 1s
| Спектр | Группа | Есв | W | Iотн |
|---|---|---|---|---|
| F 1s | CF2 | 689.7/689.7 | 1.47/1.63 | 0.95/0.90 |
| CFx | 691.2/691.5 | 1.47/1.65 | 0.05/0.10 | |
| С 1s | CFx | 290.5/290.4 | 1.60/1.60 | 0.04/0.06 |
| CF2 | 292.5/292.5 | 1.06/1.20 | 0.84/0.75 | |
| CF3 | 294.2/294.0 | 1.70/1.70 | 0.12/0.19 |
Спектр Ag 3d образца ультрадисперсного ПТФЭ-Ag с энергией связи пиков Ag 3d5/2 и 3d3/2, равной 369.23 и 375.23 эВ, что почти на 1 эВ превышает энергию связи состояния Ag0 для массивного образца, представлен на рис. 10 [52]. Такой сдвиг может быть приписан размерному эффекту. Однако поскольку, согласно результатам количественного анализа и разложению спектров C 1s на компоненты, происходит одновременное увеличение отношения F/C c 1.63 (ультрадисперсный ПТФЭ) до 2.05 (ультрадисперсный ПТФЭ-Ag) и концентрации кислорода соответственно с 0.15 до 0.35 (табл. 3), относительной концентрации группы CF3 с 0.12 до 0.19 (табл. 4), его можно приписать образованию комплекса с участием группы CF3 и кислорода, например такого, как AgOC(O)CF3. Энергия связи пика Ag 3d5/2 для AgOC(O)CF3 равна 368.8 эВ (значение, наиболее близкое к экспериментальному, – 369.23) [53]. Это предположение подкрепляется обнаружением в спектрах C 1s и F 1s состояния CFxOy, наряду с состоянием CF3. Такое соединение могло бы получиться при термоокислении за счет внутреннего кислорода или кислорода, содержащегося в структуре ультрадисперсного ПТФЭ, с разрывом координационных связей Ag → O и Ag → COD в исходном комплексе и образованием соединения Ag–O–C(O)–CF3.

ЗАКЛЮЧЕНИЕ
Для исследования возможности и целесообразности иммобилизации наночастиц серебра в матрице ультрадисперсного ПТФЭ с помощью сверхкритического СО2 и изучения их распределения в ней полимер был расфракционирован в сверхкритическом СО2 по температуре и давлению. Выделены фракции, не растворимые при 75 и 125°С, 30 МПа и 65 и 125°С, 13 МПа, а также фракция, растворимая при 125°С, 30 МПа. Исходный полимер и его фракции были импрегнированы металлоорганическим комплексом CODAg [hfАcАc] в растворе сверхкритического СО2 с последующим восстановлением металла в атмосфере водорода. Содержание металла в полимере, его химическое состояние и структура полученных композитов исследованы методами рентгенофлуоресцентного анализа, ПЭМ, малоуглового рентгеновского рассеяния и РФЭС. Растворимая фракция при выделении испытывает пиролитический распад, а ее оставшаяся часть в зависимости от температуры и давления, составляет 10–50% от потери массы полимером, причем потеря массы обусловлена как растворением, так, вероятно, и сепарацией. Импрегнирование исходного ультрадисперсного ПТФЭ и его фракций комплексом серебра позволяет получить нанокомпозиты с содержанием серебра 1.8–2.3%. Распределение по размерам наночастиц Ag в ультрадисперсном ПТФЭ, по данным малоуглового рентгеновского рассеяния, является бимодальным. Размеры частиц в исходном ПЭТФ лежат в интервале 1–15 нм, для растворимой фракции, по данным ПЭМ, в интервале 1.0–7.5 нм с максимумом при 4 нм. Имеются частицы с размерами более 100 нм. Наночастицы металла в основном локализуются в растворимой фракции. В нерастворимых фракциях из-за их высокой плотности и кристаллической структуры (по которым они фактически близки к стандартному ПТФЭ) диффузия вглубь полимера не происходит, и частицы в основном локализуются на поверхности. Наличие ярко выраженных брэгговских пиков на кривых малоуглового рассеяния от образцов растворимой фракции ультрадисперсного ПТФЭ свидетельствует об образовании упорядоченной, квазикристаллической структуры (что совпадает с данными работы [29]), которая наблюдается и в образце с импрегнированными наночастицами серебра (рис. 7, кривая 2). Образование наночастиц металла вызывает некоторые изменения квазикристаллической структуры ультрадисперсного ПТФЭ. Заметный рост амплитуды рассеяния после импрегнации серебра свидетельствует о высокой степени восстановления металла. По данным табл. 2 можно сделать предположение, что положения брэгговских пиков соответствует возможному наложению двух ламелярных упаковок в каждом из образцов: на обеих кривых малоуглового рентгеновского рассеяния имеются ламели s1–s3 и s2–s4 с эквидистантным расположением брэгговских пиков. При этом характеристический размер ламелей d1 увеличивается после иммобилизации наночастиц серебра с 4.1 до 4.3 нм. Можно предположить, что наночастицы располагаются между ламелями и их средний размер не превышает 4 нм. Однако возможно образование длинных кластеров, состоящих из таких наночастиц разного размера – форма кривых малоуглового рассеяния свидетельствует о высокой степени полидисперсности и наличии крупных частиц с линейными размерами более 100 нм (что подтверждается результатами анализа методом ПЭМ).
Из полученных данных также следует, что рациональное решение об использовании для целей супергидрофобизации текстильных и других материалов с помощью флюидной технологии только растворимой в сверхкритическом СО2 фракции ультрадисперсного ПТФЭ на самом деле является экономически нецелесообразным из-за ее низкого выхода, вследствие пиролиза. В связи с этим для придания гидрофобизируемым материалам дополнительно еще и бактерицидных свойств импрегнировать серебром имеет смысл только исходный УПТФЭ. Но наночастицы серебра, локализованные в основном в растворимой фракции, будут переходить в раствор и сорбироваться при декомпрессии реактора на гидрофобизируемых материалах, придавая им дополнительно бактерицидные свойства.
Работа выполнена при поддержке Президиума РАН (проект П13-2019-1) и в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН в части измерений и анализа данных рентгеновского рассеяния, а также при поддержке Министерства науки и высшего образования Российской Федерации с использованием научного оборудования Центра исследования строения молекул ИНЭОС РАН.
Список литературы
Товарный знак Forum № 140122, 25. 04. 1997.
Цветников А.К. Пат. 1763210 РФ. 1993.
Пaт. 2035308 PФ. 1995.
Цветников А.К., Бузник В.М., Матвиенко Л.А. Пат. 2100376 РФ. 1995.
www.74rif.ru/tribo-tomflon.html.
Галлямов М.О., Бузник В.М., Цветников А.К., Винокур Р.А., Никитин Л.Н., Саид-Галиев Э.Е., Лебедева О.В., Хохлов А.Р., Schaumburg K. // Докл. РАН. 2003. Т. 392. № 1. С. 77.
Галлямов М.О., Бузник В.М., Цветников А.К., Винокур Р.А., Никитин Л.Н., Саид-Галиев Э.Е., Хохлов А.Р., Schaumburg K. // Хим. физика. 2004. Т. 23. № 6. С. 76.
Галлямов М.О., Никитин Л.Н., Николаев А.Ю., Образцов А.Н., Бузник В.М., Хохлов. A.Р. // Коллоид. журн. 2007. Т. 69. № 4. С. 448.
Киселев М.Г., Кумеева Т.Ю., Завадский А.В., Пуховский Ю.П. // Журн. Росс. хим. об-ва им. Д.И. Менделеева. 2002. Т. 46. № 1 (Ч. 1). С. 116.
Пророкова Н.П., Кумеева Т.Ю., Завадский А.Е., Никитин Л.Н. // Хим. волокна. 2009. № 1. С. 26.
Пророкова Н.П., Кумеева Т.Ю., Хореев А.В., Бузник В.М., Никитин Л.Н. // Хим. волокна. 2010. № 2. С. 31.
Пророкова Н.П., Бузник В.М., Кирюхин Д.П., Никитин Л.Н. // Хим. технология. 2010. Т. 11. № 4. С. 213.
Помогайло А.Д., Розенберг А.С., Уфлянд И.Е. Наночастицы металлов в полимерах. М.: Химия, 2000.
Саид-Галиев Э.Е., Гамзазаде А.И., Краснов А.П., Галлямов М.О., Николаев А.Ю., Хохлов А.Р., Штыкова Э.В. // Матер. конф. “Современная химическая физика. ХХ симпозиум”. Туапсе, 2008. С. 26.
Said-Galiev E.E., Stakhanov A.I., Blagodatskikh I.V., Kobitskaya E.M., Khokhlov A.R., Naumkin A.V., Volkov I.O., Volkov V.V., Shtykova E.V., Dembo K.A., Pisarev S.A. // Polymer Science B. 2010. V. 52. № 3–4. P. 165.
Shtykova E.V., Dembo K.A., Volkova V.V., Said-Galiev E.E., Stakhanov A.I., Khokhlov A.R. // Nanotechnologies in Russia. 2009. V. 4. № 9–10. P. 700.
Said-Galiev E.E., Gamzazade A.I, Grigor’ev T.E., Khokhlov A.R., Bakuleva N.P., Lyutova I.G., Shtykova E.V., Dembo K.A., Volkov V.V. // Nanotechnologies in Russia. 2011. V. 6. № 5–6. P. 341.
Said-Galiyev E., Nikitin L., Shtykova E., Volkov V., Dembo C., Lebedeva O., Khokhlov A. Abstrs. “10 European Meeting on Supercritical Fluids”. Colmar (France), 2005. P. 3.
Said-Galiyev E.E., Nikolaev A.Yu., Levin E.E., Lavrentyeva E.K., Gallyamov M.O., Polyakov S.N., Tsirlina G.A., Petrii O.A., Khokhlov A.R. // J. Solid State Electrochem. 2011. V. 15. P. 623.
Григорьев Т.Е., Саид-Галиев Э.Е., Николаев А.Ю., Кондратенко М.С., Эльманович И.В., Галлямо М.О., Хохлов А.Р. // Росс. нанотехнологии. 2011. Т. 6. № 3. С. 14.
Said-Galiyev E., Gallyamov M., Nikolaev A., Abramchuk S., Tsirlina G., Khokhlov A. // Mater. 7 Int Scientific and Practical conference “Supercritical Fluids: fundamentals, technologies, innovations. Kazan, 2007. P. 34.
Said-Galiev E.E., Vasil’kov A.Yu., Khokhlov A.R., Shtykova E.V., Dembo K.A., Erkey C. // Russ. J. Phys. Chem. A. 2012. V. 86. № 10. P. 1597.
Игнатьева Л.Н., Бузник В.М. // Рос. хим. журн. 2008. Т. 52. № 3. С. 139.
Бузник В.М., Курявый В.Г. // Рос. хим. журн. 2008. Т. 52. № 3. С. 131.
Bousnik V.M., Kirik S.D., Solovyov L.A., Tsvetnikov A.K. Powder Difraction. 2004. V. 19. № 2. P. 1.
Said-Galiev E.E., Nikitin L.N., Nikolaev A.Yu., Vopilov Yu.E., Garbuzova I.A., Khokhlov A.R., Buznik V.M. // Polymer Science A. 2015. V. 57. № 3. P. 271.
Пейнтер П., Коулмен М., Кениг Дж. Теория колебательной спектроскопии (приложение к полимерным материалам). М.: Мир,
Kurbakova A.P., Leites L.A., German L.S., Kurykin M.A. // J. Fluorine Chem. 1996. V. 77. P. 169.
Said-Galiev E.E., Buzin M.I., Korlykov A.A., Goloveshkin A.S., Buznik V.M., Khokhlov A.R. // Polymer Science A. 2016. V. 58. № 1. P. 42.
Соболь Э.Н., Баграташвили В.Н., Соболь А.Э., Хоудл С.М. // Докл. АН СССР. 1997. V. 356. № 6. С. 777.
Могилевский Л.Ю., Дембо А.Т., Свергун Д.И., Фейгин Л.А. // Кристаллография. 1984. Т. 29. № 3. С. 587.
FeiginL.A., Svergun D.I. Structure Analysis by Small-Angle X-ray and Neutron Scattering: New York: Plenum
Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. // J. Appl. Crystallogr. 2003. V. 36. P. 1277.
Svergun D.I. // J. Appl. Cryst. 1992. V. 25. P. 495.
Vainshtein B.K. Diffraction of X-rays by chain molecules. Amsterdam; London; New York: Elsevier, 1966.
Beamson G., Briggs D. High resolution XPS of Organic Polymers. Chichester: Wiley, 1992.
Herrera-Gomez A., Bravo-Sanche M., Ceballos-Sanchez O., Vazquez-Lepe M.O. // Surf. Interface Anal. 2014. V. 46. № 10–11. P. 897.
Ignatieva L.N., Buznik V.M. // Rus. Chem. J. 2008. V. 52. P. 66.
Ignatieva L.N., Kuryavyi V.G., Tsvetnikov A.K. // KONA. 2008. V. 28. P. 98.
Buznik V.M., Ignat’eva L.N., Kuryavyi V.G., Merkulov E.B., Savchenko N.N., Ustinov A.Yu., Slobodyuk A.B., Nikitin L.N. // Polymer Science A. 2010. V. 52. № 12. P. 1334.
Ignatieva L.N., Gorbenko O.M. Kuryavy V.G., Sukhoverkhov S.V., Slobodyu A.B., Bouznik V.M., Macromolecules. 2011. V. 1. P. 5–11.
Ignatieva L.N., Buznik M. // Rus. J. Phys. Chem. V. 79. P. 1631.
Ignatieva L.N., Buznik V.M. // J. Fluorine Chem. 2011. V. 132. P. 724.
Lebedev Y.A., Ignatieva L.N. // Russ. J. Crystallogr. 2010. V. 55. P. 651.
Ignatieva L.N., Buznik M. // Fluorine Chem. 2012. V. 132. P. 17.
Buznik V.M., Vopilov Yu.E., Yurkov G.Yu., Ignat’ieva L.N., Toropov A.N., Smirnov M.A. // Polymer Science A. 2015. V. 57. № 4. P. 415.
Ignatieva L.N., Buznik V.M. // J. Structural Chemistry. 2016. V. 57. № 5. P. 892.
Ignatieva L.N., Gorbenko O.M., Kuryvyi V.G., Savchenko N.N., Pavlov A.D., Mashtalyar D.V., Bouznik V.M. // J. Fluorine Chem. 2013. V. 156. P. 246.
Игнатьева Л.Н., Бузник В.М. Матер. X Всерос. конф. “Химия фтора”. Томск, 2015. С. 67.
Цветников А.И. // Матер. X Всерос. конф. “Химия фтора”. Томск, 2015. С. 170.
Будков Ю.А., Киселев М.Г. // Тез. докл. IX науч.-практич. конф. международным участием “Сверхкритические флюиды (СКФ): фундаментальные основы, технологии, инновации”. Сочи, 2017. OR-6.
Naumkin A.V., Kraut-Vass A., Gaarenstroom S.W., Powell C.J. NIST X-ray Photoelectron Spectroscopy Database, Version 4.1 Gaithersburg: National Institute of Standards and Technology, 2012. http://srdata.nist.gov/xps/.
Wagner C.D. //Disc. Faraday Soc. 1975. V. 60. P. 291.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)