БИОФИЗИКА, 2019, том 64, № 3, с. 446-456
МОЛЕКУЛЯРНАЯ БИОФИЗИКА
УДК 577.212.2
БАЗА ДАННЫХ ПОТЕНЦИАЛЬНЫХ СДВИГОВ РАМКИ СЧИТЫВАНИЯ
В КОДИРУЮЩИХ ПОСЛЕДОВАТЕЛЬНОСТЯХ ИЗ РАЗЛИЧНЫХ
ГЕНОМОВ ЭУКАРИОТ
© 2019 г. Ю.М. Суворова*, В.М. Пугачева*, Е.В. Коротков* **
*Федеральный исследовательский центр «Фундаментальные основы биотехнологии» РАН,
119071, Москва, Ленинский просп., 33/2
**Национальный исследовательский ядерный университет «МИФИ», 115409, Москва, Каширское шоссе, 31
E-mail: genekorotkov@gmail.com
Поступила в редакцию 06.02.2019 г.
После доработки 06.02.2019 г.
Принята к публикации 25.02.2019 г.
Разработан новый банк данных, содержащий потенциальные сдвиги рамки считывания (potential
reading frame shifts). Для поиска потенциальных сдвигов рамки считывания использован новый
математический метод, основанный на использовании генетического алгоритма и динамического
программирования. Банк данных содержит координаты потенциальных сдвигов рамки считывания
для кодирующих последовательностей 76 эукариотических геномов из геномного браузера Ensembl,
dex.cgi. Из всех проанализированных геномов примерно 23% кодирующих последовательностей
имеют сдвиг рамки считывания. Ошибки первого и второго рода составляют приблизительно 11% и
30%. Одновременно с банком данных создан Web-сервер для поиска потенциальных сдвигов рамки
использован для поиска потенциальных сдвигов рамки считывания во вновь определенных
кодирующих последовательностях.
Ключевые слова: сдвиг рамки считывания, динамическое программирование, позиционно-весовая
матрица, кодирующая последовательность.
DOI: 10.1134/S0006302919030049
сборке геномов [10]. Ошибки сдвига рамки счи-
Одной из известных мутаций является сдвиг
рамки считывания в гене, который может приво-
тывания в ридах очень часто наблюдаются для
дить к созданию мутантных белков [1]. С этой му-
пиросеквенирования и PacBio. Это приводит к
тацией могут быть связаны различные наслед-
тому, что после точки сдвига рамки считывания
ственные и онкологические заболевания [2-5].
возникает ошибочная аминокислотная последо-
Сдвиг рамки считывания в гене создают вставки и
вательность, что затрудняет аннотацию генов.
делеции оснований, не кратные трем основаниям
Существуют специальные программы для поиска
[6,7], и сдвиги границ экзонов и интронов [8].
и коррекции сдвигов рамки считывания для этих
Аминокислотная последовательность полностью
методов [11,12]. Поиск таких ошибок необходим
меняется после позиции сдвига рамки [9], что мо-
для более точной аннотации генов. Образование
жет привести к изменению или потере функции,
альтернативного белка возможно при програм-
либо не вызвать никаких изменений. Поиск сдви-
гов рамки считывания важен, потому что сдвиги
мируемом сдвиге рамки считывания. В этом слу-
рамки считывания могут вносить большой вклад
чае сама рибосома программируемо сдвигается на
в эволюцию белковых последовательностей. Со-
одно основание [13]. Такие сдвиги рамки считы-
временные базы данных содержат последователь-
вания лежат вне задач данного исследования. В
ности, в которых могут быть сдвиги рамки считы-
данной работе мы создали базу данных, где содер-
вания из-за неточностей при секвенировании и
жатся потенциальные сдвиги рамки считывания
(potential reading frame shifts), связанные с воз-
Сокращения: cds - кодирующая последовательность, HMM -
hidden Markov models.
можными вставками или делециями нуклеотидов
446
БАЗА ДАННЫХ ПОТЕНЦИАЛЬНЫХ СДВИГОВ РАМКИ СЧИТЫВАНИЯ
447
не кратные трем основаниям в кодирующих по-
триплетной периодичности возникает одновре-
следовательноcтях (cds).
менно со сдвигом рамки считывания. Для поиска
сдвига фазы триплетной периодичности исполь-
Для поиска сдвига рамки считывания суще-
зуются преобразование Фурье [28], динамическое
ствует два класса математических методов. Пер-
программирование [18], вэйвлет-преобразование
вый класс содержит методы, основанные на вы-
[29] и другие методы [30]. Метод преобразования
равнивании последовательностей. Для поиска
Фурье применим только для последовательно-
сдвига рамки считывания проводится поиск ами-
стей длиннее 750 оснований и с сильно выражен-
нокислотных последовательностей, гомологич-
ной триплетной периодичностью [28]. Примене-
ных аминокислотным последовательностям, по-
ние динамического программирования [18] также
лученным из изучаемой cds, но в других рамках
не может выявить сдвиги рамки считывания при
считывания. Если среди найденных последова-
слабо выраженной триплетной периодичности.
тельностей есть хотя бы одна подобная, то можно
говорить о существовании сдвига рамки считыва-
В данной работе мы применили новый метод
ния в исследуемой последовательности. Для по-
поиска потенциальных сдвигов рамки считыва-
иска подобий часто используется программа Blast
ния в cds из геномов эукариот. Применяемый
[7, 9, 14]. Однако если подобные последователь-
подход лишен недостатков HMM, связанных с
ности не обнаруживаются, то определить суще-
тем, что для HMM требуется использование вы-
ствование сдвига рамки считывания в cds невоз-
борки генов. Такая выборка может сильно усред-
можно. Также помешать сделать вывод о суще-
нить частоты k-слов, которые встречаются в от-
ствовании сдвига рамки считывания может
дельных генах, что в свою очередь может сильно
низкий уровень подобия. Для работы с слабо по-
усреднить корреляции между нуклеотидами по
добными последовательностями были разработа-
сравнению с корреляциями, которые наблюда-
ны специальные программы [15]. Обнаружение
ются в отдельных генах. HMM также настраива-
сдвигов рамки считывания методами второго
ется на корреляции нуклеотидов, которые харак-
класса происходит посредством анализа cds ab in-
терны для обучающей выборки. В то же время
itio [16-18]. На основе этих методов были разра-
корреляции нуклеотидов в анализируемом гене
ботаны программы FrameD [16, 19], FragGeneS-
могут быть совершенно другими, чем в обучаю-
can [20], программа HMM-frame [21]. Программа
щей выборке. Это будет приводить к тому, что
GeneTack часто применяется для поиска сдвигов
при помощи HMM не получится достоверно за-
рамки считывания [17, 22]. Этой программой мо-
регистрировать сдвиги фазы. Ранее нам удалось
гут быть найдены сдвиги рамки считывания, воз-
при помощи генетического алгоритма опреде-
никшие в результате мутаций или ошибок секве-
лить для каждого гена наилучшую матрицу три-
нирования. Она ищет сдвиги там, где предполага-
плетной периодичности [31]. В данной работе
ется эволюционное происхождение двух подряд
этот подход был расширен посредством исполь-
идущих генов от одного гена, т.е. сдвиг рамки
зования матрицы, учитывающей корреляцию со-
считывания разбивает один длинный ген на два
седних оснований. Это означает, что наилучшие
новых. В программе GeneTack используются
корреляции нуклеотидов могут быть определены
скрытые модели Маркова (hidden Markov models
для каждого изучаемого гена индивидуально.
- HMM) и алгоритм Витерби. Для работы про-
В данной работе мы провели финальное вы-
граммы GeneTack требуется обучающая выборка
равнивание изучаемой последовательности отно-
для настройки программы, что существенно
сительно найденной матрицы. В ходе этого вы-
ограничивает ее возможности. В обучающей вы-
равнивания мы определяли места потенциальных
борке статистические свойства каждой cds усред-
сдвигов рамки считывания. В cds из генома
няются, что увеличивает число ошибок первого
A. thaliana было обнаружено 9930 cds, имеющих
рода и значительно уменьшает мощность метода.
потенциальные сдвиги рамки считывания (при
Программа GeneMarkS [23] используется для обу-
уровне ошибок первого и второго рода в 11% и
чения. GС-содержание генов также оказывает
30%). Это составляет примерно 21% от всех cds из
сильное влияние на поиск сдвигов рамки считы-
этого организма. Аналогичные результаты были
вания.
получены для 76 геномов, накопленных в базе
Триплетная периодичность cds часто исполь-
данных по адресу: ftp://ftp.ensembl.org/pub/re-
зуется в методах по поиску сдвигов рамки считы-
lease-86/embl/. Все полученные результаты со-
вания. Почти все cds содержат триплетную пери-
браны в базу данных потенциальных сдвигов рам-
одичность, которая возникает из-за неравномер-
ки считывания, которая находится по адресу:
ного использования кодонов различными
организмами [24, 25]. Многие программы по по-
cgi. Одновременно с банком данных был создан
иску кодирующих районов используют это свой-
сервер для поиска потенциальных сдвигов рамки
ство [26, 27]. Также известно [18], что сдвиг фазы
считывания в любых cds, который находится по
БИОФИЗИКА том 64
№ 3
2019
448
СУВОРОВА и др.
Таблица
1. Матрица триплетной периодичности
ствует сочетанию 12, k = 2 соответствует сочета-
Т(3,4), полученная для последовательности S=atcg-
нию 23, k = 3 соответствует сочетанию 31 в после-
tagctgacagtcga длиной 18 нуклеотидов
довательности A. Таким образом, номер столбца k
матрицы M совпадает со значением a(i - 1). Но-
1
2
3
мер строки матрицы M для каждой пары основа-
a
2
1
2
ний рассчитаем как n = s(i - 1) + (s(i) - 1)×4. Мат-
рица М заполняется как m(a(i - 1), n)) = m(a(i - 1),
t
0
2
2
n) + 1 для i от 1 до N. В результате мы получаем
матрицу размерности 3×16. В данной работе мы
c
1
1
2
использовали перекодировку нуклеотидов в чис-
g
3
2
0
ла как: 1 = a, 2 = t, 3 = c, 4 = g. Матрица M(k,n)
учитывает как неоднородность использования
нуклеотидов в матрице T, так и корреляцию со-
седних оснований в последовательности S. Вве-
сервере можно ввести любую cds и получить ко-
дем следующие обозначения. Пусть S0 - последо-
ординаты возможных сдвигов рамки считывания.
вательность, которая была в cds до сдвига рамки
считывания. Последовательность S образовалась
из последовательности S0 при сдвиге рамки счи-
МАТЕМАТИЧЕСКИЕ МЕТОДЫ И АЛГОРИТМЫ
тывания посредством делеции одного основания
в центре последовательности. Для последователь-
Общее описание метода. В данной работе мы
ности S0 можно рассчитать матрицу M0(k,n), а для
рассматривали триплетную периодичность как
последовательности S - матрицу M(k,n). Если бы
статистическое свойство, позволяющее обнару-
матрица M0(k,n) была известна, то относительно
жить сдвиги рамки в анализируемых генах. Три-
несложно найти местоположение вставки или де-
плетная периодичность задается матрицей Т(3,4),
леции в последовательности S. Это можно сде-
где признаками столбцов являются позиции ос-
лать, если построить глобальное выравнивание
нований в кодонах (1, 2 или 3), а признаками
при помощи динамического программирования
строк являются нуклеотиды [32]. Матрица Т(3,4)
для последовательности S относительно матрицы
хорошо учитывает отличие частот оснований в
W0(k,n). Матрица W0(k,n) есть позиционно-весо-
каждой позиции кодона от частот оснований во
всей нуклеотидной последовательности. Рас-
вая матрица, которая создается с использованием
смотрим последовательность S = atcgtagctgacagtc-
частотной матрицы M0(k,n) по формуле (1) (см. ни-
ga длиной N = 18. Для заполнения матрицы созда-
же). По этой же формуле можно также рассчитать
ем последовательность A, элементы которой a(i)
W(k,n) для частотной матрицы M(k,n).
= imod3 + + 1 для всех i от 1 до N. Здесь mod обо-
Такое выравнивание можно сделать так, как
значает остаток от деления числа i на 3. Тогда по-
это было выполнено в работе [33]. В результате
следовательность A = 123123123123123123. Матри-
мы рассчитываем максимальное значение функ-
ца триплетной периодичности Т(3,4) заполняется
ции сходства mF0 последовательности S относи-
как t(s(i),a(i)) = t(s(i),a(i)) + 1 для всех i от 1 до N.
тельно матрицы W0(k,n). Однако нет сохранен-
Результат показан в табл. 1. Сумма всех элементов
матрицы равна N. Однако матрица T не учитывает
ной матрицы M0(k,n), мы не можем построить
корреляцию между соседними основаниями.
W0(k,n), и местоположение вставки или делеции
Пусть последовательность S имеет четыре три-
оснований в последовательности S является не-
плета: ATA, TAT, CGC, GCG. Пусть также они бу-
известным. Это также значит, что мы не можем
дут расположены с равной вероятностью и в про-
рассчитать функцию сходства mF0. Задача в таком
извольном порядке. В этом случае частоты осно-
случае и состоит в том, чтобы найти такую матри-
ваний в каждом столбце матрицы T будут равны
цу W2(k,n), которая была бы наиболее близка к
частотам оснований во всей последовательности
матрице W0(k,n). Для нахождения матрицы
S. Это означает, что найти корреляцию между по-
следовательностями оснований ДНК при помо-
W2(k,n) будем использовать генетический алго-
щи матрицы T невозможно. Однако это можно
ритм, который был разработан нами в работе [31].
сделать, если использовать матрицу M(k,n). У
Начало генетического алгоритма состоит в том,
этой матрицы признаками столбцов являются па-
что мы генерируем определенное множество WR
ры оснований, которые встречаются в позициях
случайных позиционно-весовых матриц размер-
i - 1 и i последовательности S, i пробегает от 1 до
ностью 3 на 16. Эти матрицы являются «организ-
N. В последовательности A в этом случае встреча-
мами» для генетического алгоритма, а функция
ются сочетания 12, 23 и 31. Пронумеруем столбцы
сходства выполняет роль целевой функции. Затем
матрицы M следующим образом: k = 1 соответ-
мы применяем генетический алгоритм с целью
БИОФИЗИКА том 64
№ 3
2019
БАЗА ДАННЫХ ПОТЕНЦИАЛЬНЫХ СДВИГОВ РАМКИ СЧИТЫВАНИЯ
449
создания такой матрицы W2(k,n), которая будет
максимизировать функцию сходства mF.
Пусть матрица W2(k,n) имеет максимальное
значение функции сходства mF1 при проведении
глобального выравнивания последовательности
S. При помощи генетического алгоритма мы
ищем такую матрицу W2(k,n), которая имеет
функцию сходства mF1, как угодно близкую к зна-
чению mF0. Это означает, что разница (mF0 - mF1)
стремится к нулю при стремлении числа итера-
ций генетического алгоритма к бесконечности.
Глобальное выравнивание также позволяет опре-
делить координаты потенциальных сдвигов рам-
ки считывания в последовательности S. Блок-
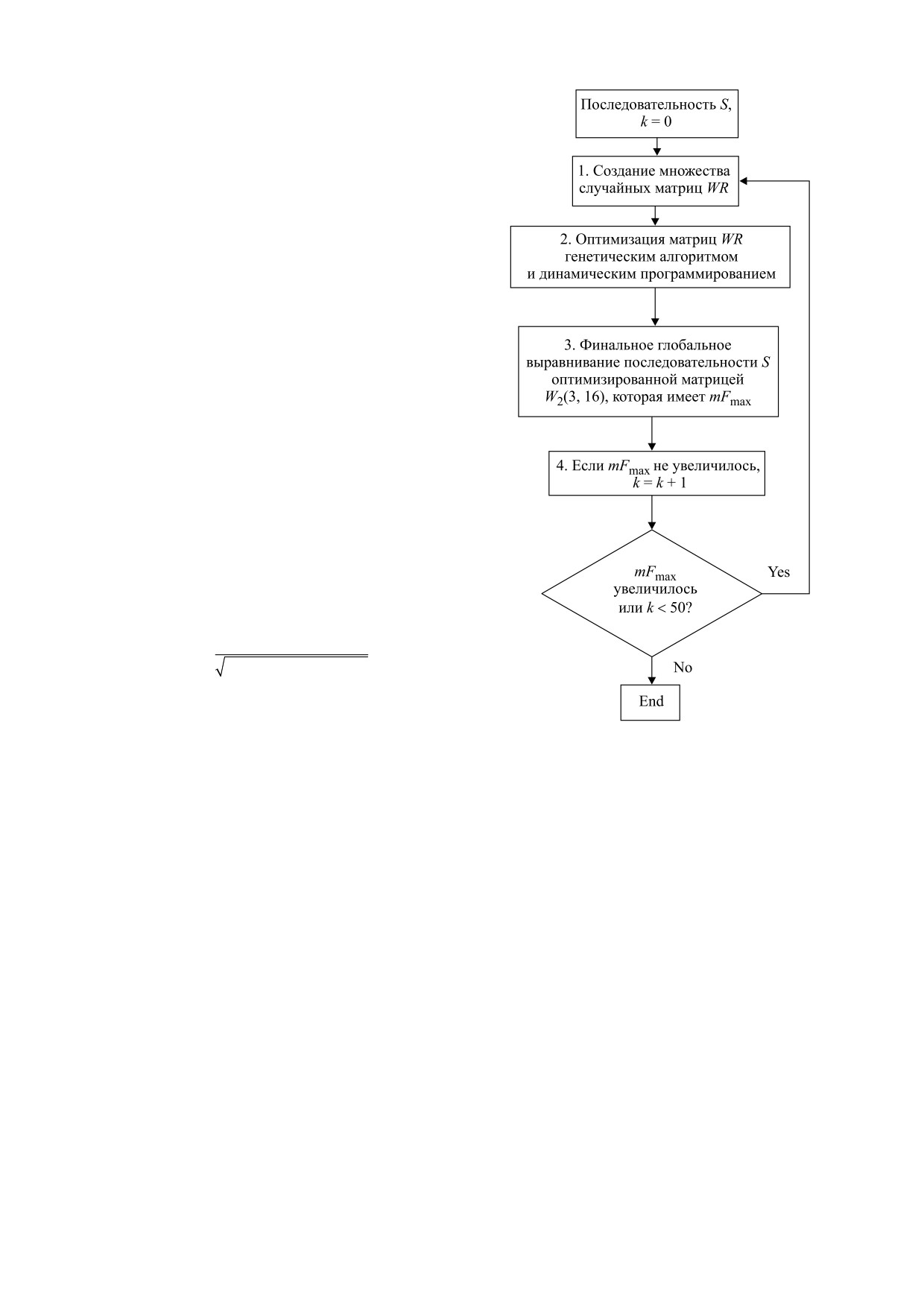
схема алгоритма показана на рис. 1. Рассмотрим
более детально по пунктам этот алгоритм.
Расчет позиционно-весовых матриц и создание
множества случайных матриц. Пусть последова-
тельность S содержит cds. Для создания случай-
ной матрицы W(3,16) из множества случайных
матриц WR мы случайно перемешивали нуклео-
тиды в последовательности S и создавали после-
довательность SR. Алгоритм случайного переме-
шивания был подробно описан ранее [31]. Затем
при помощи последовательности SR мы заполня-
ли матрицу M(3,16), как это было описано в пунк-
те «Общее описание метода». Затем мы рассчиты-
вали матрицу WR(3,16) по формуле:
M
(
k,n
)
−
(N
−1)
2
p n
WR
(
k,n
)
=
(N
-1)
p n)(1
−
p n))
(1)
2
2
Здесь p2(n) = p(l)p(m), где p(l) и p(m) - вероят-
ность встретить нуклеотид типа l или m в последо-
вательности SR (l ∈ {a,t,c,g}, m ∈ {a,t,c,g}); p(n) =
q(n)/N, q(n) - количество нуклеотидов типа l в по-
следовательности SR; N - длина последователь-
Рис. 1. Блок-схема алгоритма, применяемого для
ности SR.
процедуры оптимизации случайных матриц M(3,16)
При таком расчете мы учитываем как неодно-
из множества MR с целью поиска матрицы с
родности в частотах нуклеотидов в каждой пози-
наибольшим mFmax.
ции кодона в последовательности SR, так и кор-
реляцию соседних оснований. Это связано с тем,
что рассчитывается ожидаемое число каждой из
матрицы Wt(3,16). Суть трансформации состоит в
16 пар соседних оснований для каждой позиции k
том, чтобы сумма квадратов элементов матрицы
матрицы W(3,16). Последовательность S случай-
была равна R2. При трансформации поддержива-
но перемешивалась 500 раз и всего было создано
500 матриц для множество WR.
лось постоянство константы Kd , что позволяет
Оптимизация матрицы W2(3,16) генетическим
сохранять среднее значение элементов матрицы
алгоритмом и динамическим программированием.
Wt в расчете на один нуклеотид последовательно-
Функция сходства F в динамическом программи-
сти S. Метод трансформации матрицы W2 в мат-
ровании выступает как целевая функция для ге-
рицу Wt, выбор констант R2 и Kd и генетический
нетического алгоритма. Пусть мы имеем ген или
алгоритм подробно был рассмотрен нами в работе
cds в виде последовательности S. Длина последо-
вательности равна N. Последовательность S вы-
[31]. Для этого использовали следующую итера-
равнивалась относительно трансформированной
тивную процедуру:
БИОФИЗИКА том 64
№ 3
2019
450
СУВОРОВА и др.
Для них мы брали d = 10000, что исключает созда-
ние таких вставок или же делеций.
Одновременно с делециями в последователь-
ности S могут происходить делеции одного или
двух столбцов матрицы Wt(a(i),n). Введение таких
столбцов соответствует переходам F(i-3,j-2) → F(i-2,
j-1) → F(i-1,j-1) → F(i,j) и F(i-4,j-2) → F(i-3,j-1) → F(i-2,
j-1) → F(i-1,j-1) → F(i,j). Для этих переходов мы не
учитывали корреляции между соседними основа-
ниями. Вместо матрицы Wt(a(i),n) для этих двух
Рис. 2. Иллюстрация для выбора индекса k при деле-
переходов мы использовали матрицу Et(x,n):
ции двух оснований в последовательности S. В этом
случае k = s(j-3). (Это третий вариант выбора k в пунк-
те «Оптимизация матрицы W2(3,16) генетическим ал-
t
t
E x,n)
=
0,25
∑
W x2
i
+
(n
−
1)
×
4)
(3)
горитмом и динамическим программированием»).
i=1,4
Здесь уже n меняется от 1 до 4 и при использо-
вании этой матрицы n = s(j). Это означает, что
t
⎧
F(i
−
1,
j
−
1)
+
W ai),n)
⎫
(2)
при делециях столбцов корреляция соседних ос-
⎪
⎪
F i, j)
=
max
⎨
F i
j
−
1)
−
d
⎬
нований ДНК не учитывается. Это вполне допу-
⎪
⎪
стимо в том случае, когда число делеций или же
F
(i
−
1, j)
−
d
⎩
⎭
вставок небольшое.
Нулевые строка и столбец матрицы F заполня-
где i меняется от 1 до (N + 2), а j меняется от 1 до
лись отрицательными числами: F(0,j) и F(i,0) рав-
N. Здесь n = s(k) + (s(j) - 1)×4, а значение a(i) бы-
ны 0 для i от 1 до N+2, а j от 1 до N, а значения
ло определено в пункте «Общее описание мето-
F(0,0), F(1,0), ..., F(2,0) также равны 0. Матрица
да». Введение параметра n связано с тем, что в
использовалась также для перехода из нулевого
матрице W и Wt учитываются пары оснований.
столбца в первый столбец матрицы F и из нулевой
Это означает, что мы должны определить преды-
строки в первую строку матрицы F. Выбор цены
дущее основание, которое мы уже включили в
за вставку или делецию d (d = 25,0) был сделан
выравнивание. Поэтому индекс k рассчитывался
так, как мы это делали ранее [31]. Матрица обрат-
по уже созданным переходам в матрице F в зави-
ных переходов заполнялась одновременно с мат-
симости от того, какое предыдущее основание из
рицей F, как это обычно делается при поиске гло-
последовательности S было включено в выравни-
бального выравнивания. Потом мы строили вы-
вание. В зависимости от индекса k мы брали зна-
равнивание последовательности S относительно
чение n. Есть три варианта для индекса k.
t
матрицы
по матрице обратных переходов и
W
1. Если предыдущее основание из последова-
определяли максимальное значение функции
тельности S (которое мы уже взяли в выравнива-
сходства mF в одной из трех ячейках матрицы
ние) есть s(j-1), то мы берем k = j-1 и n = s(j-1) +
F(N,N), F(N+1,N), F(N+2,N).
(s(j)-1)×4. Это соответствует переходам по ячей-
кам матрицы F(i-2,j-2) → F(i-1,j-1) → F(i,j).
Целевая функция для генетического алгорит-
ма была выбрана как mF. «Организм» в генетиче-
2. Если предыдущее основание в выравнива-
ском алгоритме представляет собой матрицу
нии есть s(j-2), то мы берем k = j-2 и n = s(j-2) +
W(3,16). Генетический алгоритм был рассмотрен
(s(j) - 1)×4. Это соответствует переходам из ячей-
ранее в работе [31], там можно найти и подробно-
ки F(i-2,j-3) → → F(i-1,j-2) → F(i-1,j-1) → F(i,j).
сти его применения. Множество случайных мат-
Эти переходы соответствует пропуску основания
риц WR (пункт «Расчет позиционно-весовых мат-
s(j-1).
риц и создание множества случайных матриц
3. Если предыдущее основание есть s(j-3), то
WR») было сформировано, оно содержало
мы берем k = j-3 и n = s(j-3) + (s(j) - 1)×4. Это со-
500 матриц, каждая размерностью 3 на 16. Это
ответствует переходам из ячейки F(i-2,j-4) → F(i-1,
множество участвовало в работе генетического
j-3) → F(i-1,j-2) → F(i-1,j-1) → F(i,j).
алгоритма. В результате работы генетического ал-
горитма определялась одна матрица из множе-
На рис. 2 схематично показаны эти переходы.
ства WR, которая имеет максимальное значение
Они соответствуют пропуску основания s(j-1) и
mF. Назовем его mFmax. Окончание генетического
s(j-2) в выравнивании. Так как мы изучаем три-
плетную матрицу, то делеции больше чем два ос-
алгоритма происходило после того, как значение
нования в последовательности S блокировались.
mFmax переставало увеличиваться на протяжении
БИОФИЗИКА том 64
№ 3
2019
БАЗА ДАННЫХ ПОТЕНЦИАЛЬНЫХ СДВИГОВ РАМКИ СЧИТЫВАНИЯ
451
Рис. 3. Схема разделения mFmax на V1, V2 и V3 (см. пункт «Расчет количественной меры для поиска сдвига фазы
триплетной периодичности»).
50 итераций. В среднем до этого момента требует-
создали множество последовательностей SR. По-
следовательности генов имеют различную длину
ся около 9 · 103 итераций.
и различную триплетную периодичность [18], по-
В итоге в конце работы генетического алгорит-
этому для каждого гена необходим свой расчет V0.
ма мы имеем значение mFmax и соответствующую
Во множестве SR содержится 103 последователь-
матрицу W(3,16). Назовем ее Wmax(3,16). Кроме
ностей S, кодоны в которых были случайно пере-
матрицы мы имеем построенное выравнивание
мешаны. Таким перемешиванием уничтожаются
последовательности S относительно столбцов
все возможные сдвиги фазы триплетной перио-
матрицы Wmax(3,16). Именно это выравнивание
дичности. Мы выбирали значение V0 для каждого
позволяет нам определить координаты потенци-
гена, при котором было не более 20 последова-
альных сдвигов рамки считывания в последова-
тельностей во множестве SR, для которых V > V0.
тельности S.
Только такие гены имеют статистически значи-
мые вставки или делеции. Сдвиги фазы триплет-
Расчет количественной меры для поиска сдвига
ной периодичности отсутствовали в генах, если
фазы триплетной периодичности. Значение mFmax,
V2 + V3 = 0.
которое характеризует триплетную периодич-
ность в последовательности S, само по себе мало
информативно. Интерес представляет статисти-
РЕЗУЛЬТАТЫ И ДИСКУССИЯ
ческая значимость найденных сдвигов фазы три-
Геномы, которые были изучены. Всего было
плетной периодичности. Вставки или делеции
изучено 76 эукариотических геномов из базы дан-
символов в выравнивании последовательности S
ных Еnsembl (последовательности cds для каждо-
относительно матрицы Wmax(3,16) указывают на
го генома были взяты из ftp://ftp.ensembl.
координаты сдвиги фазы. Значение mFmax было
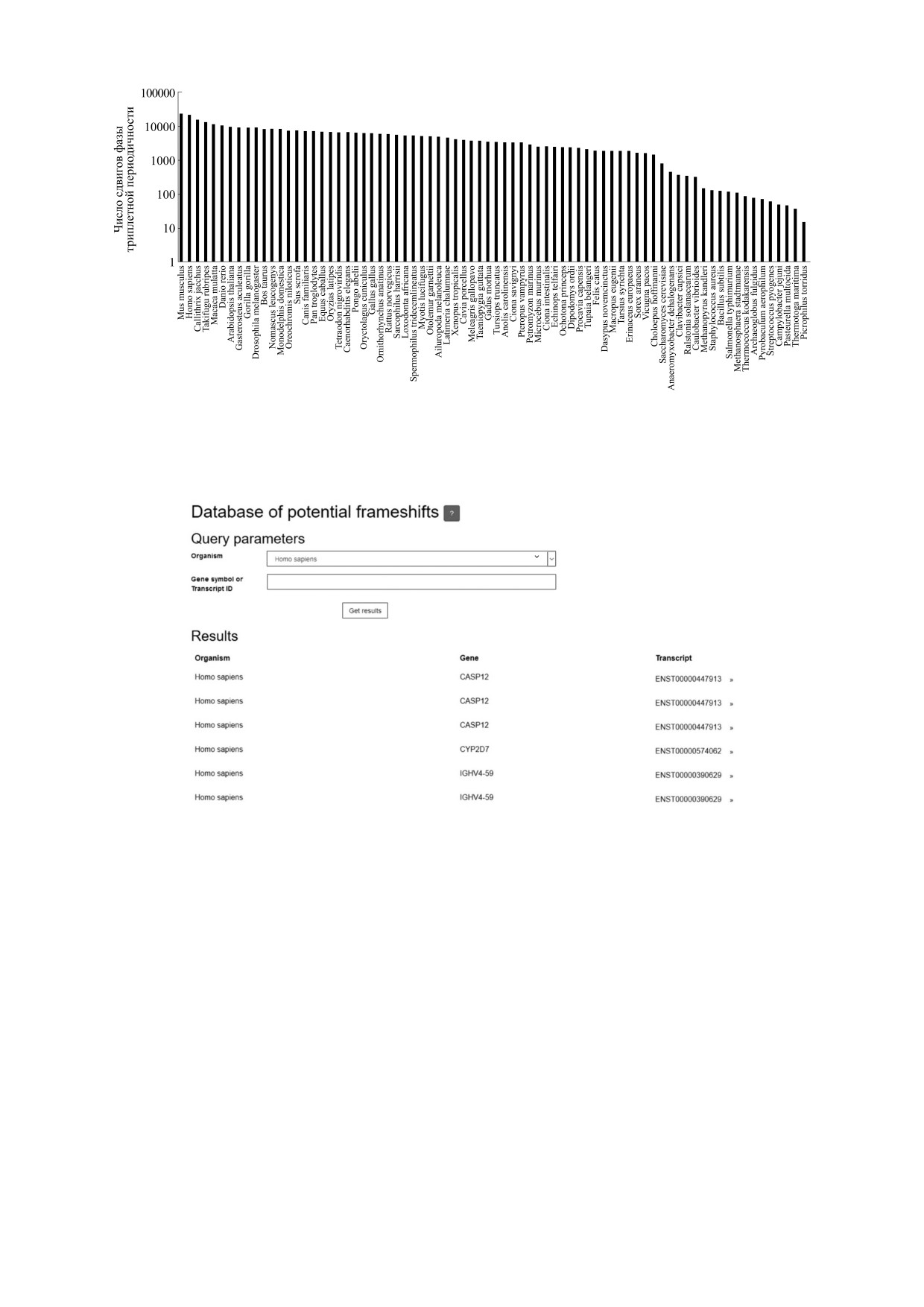
org/pub/release-86/embl/). Из рис. 4 видно, что
разделено на три части для расчета количествен-
примерно 23% cds в среднем из каждого эукарио-
ной меры сдвига фазы триплетной периодично-
тического организма содержат сдвиги рамки счи-
сти. Участки последовательности S, где позиции
тывания. Созданный банк данных находится по
столбцов матрицы и рамка считывания в после-
довательности S совпадают между собой, принад-
index.cgi. Первая страница базы данных показана
лежат первой части. На рис. 3 этот район обозна-
на рис. 5. Из этого рисунка видно, что пользова-
чен как V1. Районы последовательности S, где
телю предоставляется возможность выбрать ге-
имеются совпадения 1 → 2, 2 → 3, 3 → 1 принадле-
ном, для которого он хочет найти cds с потенци-
жат второй части (рис. 3, V2). Совпадения вида
альным сдвигом рамки считывания. Уже в
1 → 3, 2 → 1, 3 → 2 принадлежат к третьей части (рис.
выбранном организме предоставляется возмож-
3, V3). Сумма V1 + V2 + V3-kd равна mFmax, где k -
ность выбрать cds либо общим списком, либо
число вставок или делеций, d - цена за вставку
произвести поиск нужной cds по символу гена,
или делецию из формулы 2. Матрица Wmax(3,16)
либо же по идентификатору транскрипта. Если
транскрипт выбран, то пользователю предостав-
не имеет связи с рамкой считывания. Поэтому
ляется следующая информация:
1) матрица
циклические перестановки матрицы Wmax(3,16)
Wmax(3,16); 2) координата найденных сдвигов фа-
производились для того, чтобы V1 ≥ V2 и V1 ≥ V3.
зы; 3) выравнивание cds относительно матрицы
Мы выбрали сумму V2 + V3 как количественную
меру характеризующую статистическую значи-
Wmax(3,16).
мость сдвигов фазы триплетной периодичности.
Рассмотрим пример этой информации для тран-
Для каждой анализируемой cds было рассчитано
скрипта ENST00000583951 из генома Dasypus novem-
значение V = V2 + V3. Пороговое значение для V =
cinctus (девятипоясный броненосец). Это тран-
V0 было рассчитано. Если V < V0, то сдвиг рамки
скрипт гена (AKAP10), который кодирует A-kinase
считывания в cds отсутствует. Свое пороговое V0
anchoring protein 10 [34]. В этом транскрипте была
выбиралось для каждой cds. Для расчета V0 мы
совершена вставка нуклеотида с в 71-й позиции. В
БИОФИЗИКА том 64
№ 3
2019
452
СУВОРОВА и др.
Рис. 4. По оси абсцисс показаны геномы эукариотических организмов, которые были проанализированы и которые
содержатся в базе данных. По оси ординат показано число сдвигов фазы триплетной периодичности, которое было
найдено в cds в каждом эукариотическом организме.
Рис. 5. Первая страница базы данных данных потенциальных сдвигов рамки считывания в кодирующих последова-
результате этого произошел сдвиг фазы триплетной
W2(3,16) генетическим алгоритмом и динамическим
периодичности после
71-й позиции. Матрица
программированием»). Из табл. 2 видно, что веса
пар могут существенно отличаться. Например, вес
Wmax(l,n) (l = 1,2,3, n=1,2,...,16) показана в табл. 2.
пары TG для i = 1 (т.е. в первой фазе) составляет 11,6.
Для удобства восприятия она была переведена в
Это означает, что вес пары TG в том случае, если T
трехмерную матрицу W3max(i,j,k). Индекс i в этой мат-
будет в третьей фазе, а G будет в первой фазе (см.
рице соответствует индексу l матрицы Wmax(l,n). Ин-
пункт «Общее описание метода»), будет равен 11,6.
декс j матрицы получается как j = n - 4×int((n-1)/4),
Присутствуют в матрице также и большие отрица-
где n = 1,2,...,16. Индекс k рассчитывается как:
тельные значения. Например для i = 2 пара TA имеет
k=4×int((n-1)/4). Индекс j и k показывают пару ос-
вес, равный -6,7. Это означает, что вес пары TA бу-
нований c номером n = j + (k-1)×4 (см. пункты «Об-
дет равен -6,7 в том случае, если T будет в первой
щее описание метода» и «Оптимизация матрицы
фазе, а A будет во второй фазе. Таким образом, мы
БИОФИЗИКА том 64
№ 3
2019
БАЗА ДАННЫХ ПОТЕНЦИАЛЬНЫХ СДВИГОВ РАМКИ СЧИТЫВАНИЯ
453
Таблица 2. Матрица Wmax(3,16), полученная для поис-
считывания. Видно, что наибольшее число cds
ка возможных сдвигов рамки считывания последова-
содержат один или два потенциальных сдвига
тельности транскрипта ENST00000583951
рамки считывания. Количество cds, которое со-
держит более пяти сдвигов рамки считывания,
A
T
C
G
является незначительным.
1
A
-3,4
0,7
-2,4
2,4
Оценка числа ошибок первого и второго рода.
Для определения количества ошибок первого и
1
T
второго рода мы взяли все кодирующие последо-
-3,8
-2,3
-0,6
11,6
вательности из генома A. thaliana, число которых
составляет 48322 cds. Затем из этих последова-
1
C
-2,0
0,5
1,3
-4,5
тельностей множество последовательностей RN
1
G
было создано путем случайного перемешивания
-2,2
-1,9
-0,2
-0,9
кодонов. В этом множестве заведомо не будет
сдвигов фазы триплетной периодичности при со-
2
A
-1,7
-0,2
-3,5
-1,1
хранении статистических свойств исходной ко-
дирующей последовательности. Множество RN
2
T
-6,7
-0,2
6,3
-3,0
позволяет оценить число ошибок первого рода
(true positive). Мы применили разработанный на-
2
C
0,1
-2,9
2,5
-3,5
ми подход к анализу множества RN и оказалось,
что в этих последовательностях можно найти
2
G
5,7
-1,0
0,9
0,0
1098 последовательностей со сдвигом фазы три-
плетной периодичности, где V2 + V3 было больше
3
A
1,8
-0,2
-1,4
1,5
порогового уровня. В них содержится 1549 сдви-
гов рамки считывания. Если учесть, что в геноме
3
T
-4,8
-1,3
0,6
-0,1
A. thaliana нашим методом было найдено
9930 cds, то уровень ошибок первого рода состав-
3
C
-3,0
13,0
-1,2
-3,5
ляет примерно 15%. Мы проделали такое изуче-
ние для всех проанализированных геномов и мак-
3
G
-3,1
-2,9
-1,2
-0,9
симальное значение ошибок первого рода не пре-
вышало 23%.
Интересно также определить число ошибок
учитываем не только отличие частот по позициям
второго рода и мощность метода. Для этого мы со-
периода от средних частот оснований в последова-
здавали множество последовательностей RD из ко-
тельности S, но также учитываем корреляцию со-
дирующих последовательностей из генома
седних оснований. Полученное с помощью матри-
A. thaliana с длиной более 500 нуклеотидов. Кодо-
цы W3max(i,j,k) выравнивание cds для транскрипта
ны в этих последовательностях были случайно пе-
ремешаны. Затем в случайное положение каждой
ENST00000583951 показано в табл. 3.
последовательности не ближе 100 нуклеотидов от
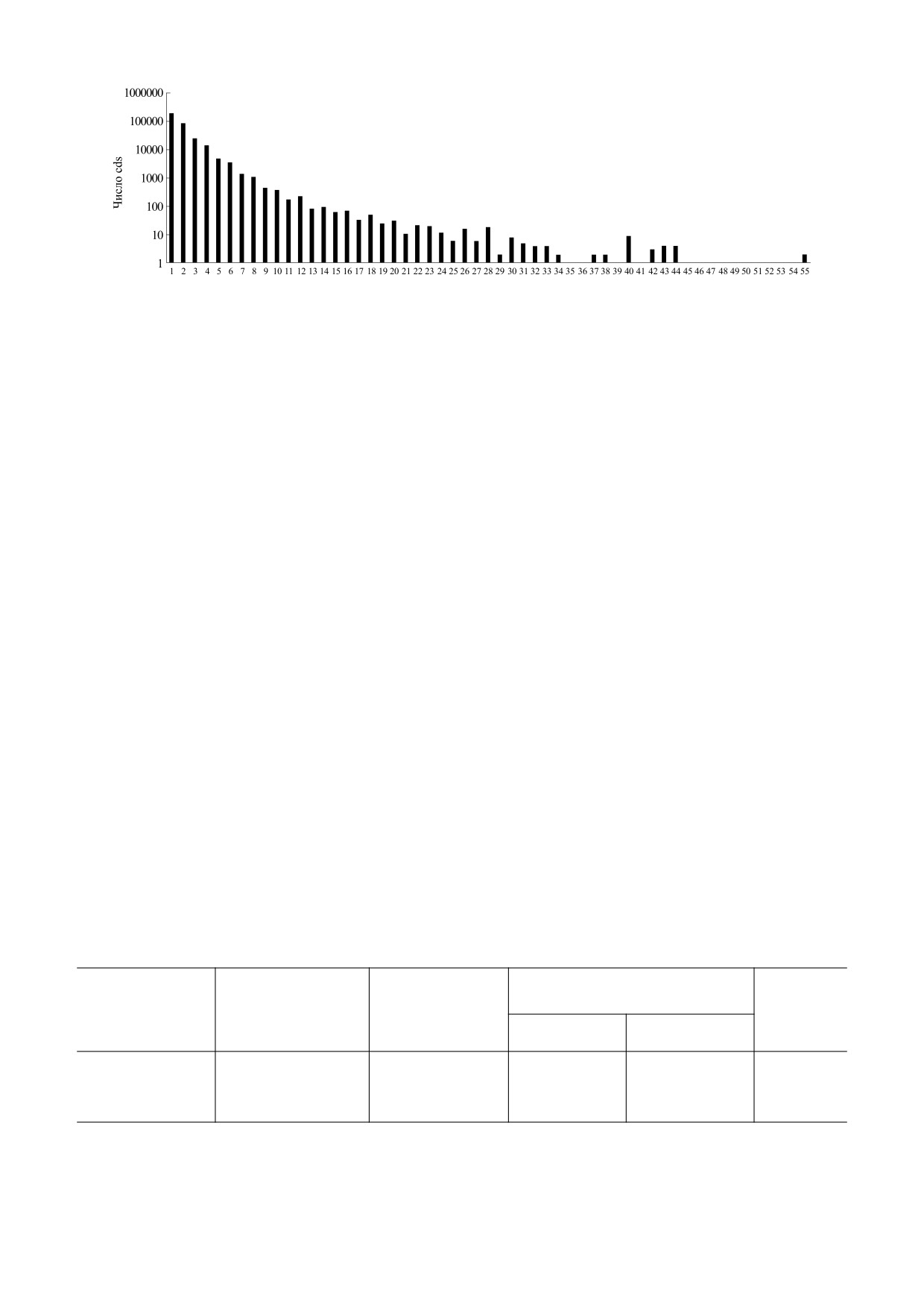
На рис. 6 показано распределение числа cds по
начала и конца каждой последовательности вноси-
суммарному числу потенциальных сдвигов рамки
лась случайная делеция одного основания. Эти по-
Таблица 3. Выравнивание cds для транскрипта ENST00000583951 относительно матрицы W3max(i,j,k). Вставка
одного нуклеотида в позиции 71 отмечена звездочкой
0001 CTGGCTCTGTTGGCCCTCCTGATGAGTCTCACCCAGGGAGTTCTGACAGCTCTGCGTCTC
0001 231231231231231231231231231231231231231231231231231231231231
0061 AGGAAGATGACATTTGGAAGAGTCAGTGACTTGGGGCAATTCATCCGAGAATCTGAGCCT
0061 2312312312*3123123123123123123123123123123123123123123123123
0121 GAACCTGATGTAAGGAAATCAAAAGGATCCATGTTCTCACAAGCTATGAAGAAATGGGTG
0120 123123123123123123123123123123123123123123123123123123123123
0181 CAAGGAAATACTGATGAGGCCCAGGAAGAGCTAGCTTGGAAGATTGCTAAAATG
0180 123123123123123123123123123123123123123123123123123123
БИОФИЗИКА том 64
№ 3
2019
454
СУВОРОВА и др.
Рис. 6. Распределение числа cds, содержащихся в базе данных потенциальных сдвигов рамки считывания, по числу
сдвигов триплетной периодичности.
следовательности были проанализированы с по-
тельностей в геноме) и Genetack (для поиска по-
мощью разработанного алгоритма, и в итоге полу-
тенциальных сдвигов рамки считывания) [35].
чены результаты, представленные в табл. 4. Из
Программа Genetack ищет случаи потенциальных
этой таблицы видно, что всего удалось выявить
сдвигов рамки, которые привели к разбиению од-
28357 последовательностей во множестве RD, для
ной гипотетической кодирующей последователь-
которых (V2 + V3) ≥ V0 и положение находится в
ности на две независимых (в современных базах
интервале
±50 от искусственной делеции из
данных они обычно представлены двумя отдель-
40621 последовательностей со сдвигами. Это пока-
ными генами). Результатом работы программы
зывает, что уровень ошибок второго рода составля-
GeneTack-GM являются предсказанные коорди-
ет 30%, т.е. мощность метода составляет 70%. Так-
наты гена (обычно гипотетического гена) и коор-
же метод достаточно точно предсказывает место-
дината сдвига внутри этого гена.
положение сдвигов, так как только у
1128
Всего в геноме A. thaliana программой Gene-
последовательностей были найдены сдвиги вне
Tack-GM [22] было найдено в mRNA 2067 потен-
района ±50 от точки делеции. Также следует отме-
циальных сдвигов рамки считывания, тогда как
тить, что метод не создает значительного количе-
нам удалось обнаружить 14951 (табл. 5). Следует
ства случайных сдвигов, так как на 29485 последо-
отметить, что мы в данной работе анализировали
вательностей со статистически значимыми сдвига-
только cds, тогда как в GeneTack database содер-
ми (28357 + 1128) приходится 29888 сдвигов, т.е.
жатся данные для mRNA, которые также имеют
403 сдвига обусловлены чисто случайными факто-
некодирующие последовательности. Поэтому мы
рами.
разделили найденные 2067 сдвигов рамки считы-
Сравнительный анализ полученных результатов
вания на три группы. К первой группе отнесли
с более ранними публикациями. Интересно срав-
потенциальные сдвиги рамки считывания, кото-
нить полученные нами результаты с полученны-
рые находятся только внутри cds не ближе 50 nt от
ми ранее результатами по поиску сдвигов рамки
начала и конца кодирующего участка. Во второй
считывания в cds из эукариотических геномов. В
группе сдвиг содержится на расстоянии не боль-
первую очередь это относится к геномам, резуль-
шем 50 nt от концов cds, а в третьей группе он
таты изучения которых, накопленные в GeneTack
приходится на некодирующие районы mRNA. К
database [22], были получены с использованием
первой группе относится 485, ко второй группе
программы GeneTack-GM. Данная программа
относится 710, а в третью группу входит 872 сдви-
представляет собой объединение программ Gene-
га рамки считывания, найденные программой
Mark (для выделения кодирующих последова-
GeneTack-GM. Если учесть, что нам удалось вы-
Таблица 4. Поиск сдвигов фазы триплетной периодичности во множестве RD
Число
Число последовательностей
Общее число
Название
последователь-
с V2 + V3 ≥ V0
Общее число
последовательностей
организма
ностей
сдвигов
в множестве RD
внутри ±50
вне ±50
с V2 + V3 ≠ 0
A. thaliana
40612
31203
28357
1128
29888
Anaeromyxobacter
3460
3458
2975
68
3668
dehalogenans
БИОФИЗИКА том 64
№ 3
2019
БАЗА ДАННЫХ ПОТЕНЦИАЛЬНЫХ СДВИГОВ РАМКИ СЧИТЫВАНИЯ
455
Таблица 5. Число cds содержащих потенциальные сдвиги рамки считывания в шести исследованных эукариоти-
ческих геномах
Число потенциальных
Число потенциальных
Число cds c потенциальными
Название организма
framesfift mutations
framesfift mutations
framesfift mutations
из работы [22]
A. thaliana
14954
9930
2067
C. elegans
10411
5941
611
D. melanogaster
31873
8833
2616
H. sapiens
20795
13285
7395
R. norvegicus
9811
5768
703
X. tropicalis
6518
4228
529
явить 14951 потенциальный сдвиг рамки считы-
Сдвиги рамки считывания могут принимать уча-
вания из генома A. thaliana, то разработанный на-
стие в образовании новых генов. Изучение эволю-
ми алгоритм примерно в семь раз эффективнее,
ции генов привлекает внимание исследователей
чем программа GeneTack-GM. Если сравнивать
много лет и исследования в этой области значи-
результаты, относящиеся к первой группе, то та-
тельно ускорились после определения последова-
кое различие будет еще больше, так как более 70%
тельностей многих прокариотических и эукарио-
обнаруженных нами сдвигов относится к первой
тических генов. В настоящее время считается, что
группе. Естественно, что такое сравнение кор-
новые гены происходят путем дупликации суще-
ректно проводить только с первой группой. По-
ствующих генов [36]. Поэтому большое количе-
хожие результаты также были получены для неко-
ство генов объединены в семейства на основе по-
торых других эукариотических геномов (табл. 5),
добия их аминокислотных или нуклеотидных по-
где мы обнаруживаем в несколько раз больше по-
следовательностей
[37]. Также известно, что
тенциальных сдвигов рамки считывания.
процессы склейки генов (gene fusion) [38], перета-
совка экзонов (exon shuffling) [39], альтернатив-
Такое различие в результатах может быть свя-
ный сплайсинг (alternative splicing) [40] и горизон-
зано с тем, что метод HMM настраивается по
тальный перенос генов (lateral gene transfer) [41]
множеству выбранных mRNA [22] и используют-
являются основными механизмами по диверген-
ся статистические свойства усредненные по мно-
ции генов и создании разнообразия генов в гено-
жеству последовательностей и эти свойства фик-
ме и соответствующих им белков. Однако такими
сированы. Это усреднение как раз и может приво-
механизмами трудно быстро создать принципи-
дить к тому, что многочисленные сдвиги фазы
ально новую последовательность, а сдвиг рамки
будут пропускаться. В разработанном нами алго-
считывания может это сделать быстро. В связи с
ритме такого усреднения не происходит, метод
этим было предположено ранее, что мутации ти-
настраивается на триплетную периодичность, ко-
па сдвиг рамки считывания могут играть суще-
торая существует в каждом гене или cds. Это озна-
ственную роль в создании новых генов [7,9]. В
чает что разработанный нами математические
метод персонально для каждой анализируемой
этих работах высказано предположение, что если
cds или гена находит оптимальную корреляцион-
белок по тем или иным причинам выведен из-под
ную матрицу с учетом возможности вставок или
давления отбора, то сдвиг рамки считывания мо-
делеций нуклеотидов. Финальное выравнивание
жет закрепиться в гене. Именно это явление мы,
изучаемой последовательности против получен-
вероятно, наблюдаем в данной работе, так как бо-
ной оптимизационной матрицы дает выравнива-
лее 20% cds из различных геномов эукариот со-
ние последовательности и координаты потенци-
держат потенциальные сдвиги рамки считыва-
альных сдвигов рамки считывания. Важно еще
ния. Остается только вопрос: как после сдвига
отметить, что для поиска потенциальных сдвигов
рамки считывания образуется белковая последо-
рамки считывания нашим методом требуеются
вательность, имеющая какой-либо биологиче-
только последовательности гена или cds и ника-
ский смысл? Можно предполагать, что генетиче-
кая другая информация больше не нужна. В этом
ский код хорошо адаптирован к таким изменени-
состоит основное улучшение метода по сравне-
ям, и он позволяет в результате сдвига рамки
нию с использованием метода HMM для опреде-
считывания получать биологически осмыслен-
ления сдвигов рамки считывания.
ные последовательности.
БИОФИЗИКА том 64
№ 3
2019
456
СУВОРОВА и др.
СПИСОК ЛИТЕРАТУРЫ
22. 22. I. Antonov, P. Baranov, and M. Borodovsky, Nucl.
Acids Res. 41, D152 (2013).
1. J. D. Watson, et al., Molecular biology of the gene
23. R. K. Azad and M. Borodovsky, Brief. Bioinform. 5
(Pearson Education, Inc. 2013).
(2),118 (2004).
2. Y. Ogura, et al., Nature 211, 603 (2001).
24. G. Gutiérrez, J. L. Oliver, and A. Marín, J. Theor. Biol.
3. M. C. Iannuzzi, et al., Am. J. Hum. Genet. 48 (2), 227
167 (4), 413 (1994).
(1991).
25. V. R. Chechetkin and A. Yu. Turygin, J. Theor. Biol.
4. W. K. Chung, C. Kitner, and B. J. Maron, Cardiol.
175 (4), 477 (1995).
Young 21, 345 (2011).
26. J. Gao, et al., J. Biomed. Biotechnol. 2005 (2), 139
5. X. Xu, et al., Gene 519, 343 (2013).
(2005).
6. J. L. Wood and J. Chen, in DNA Repair, Genetic Insta-
27. C. Yin and S. S.-T. Yau, J. Theor. Biol. 247 (4), 687
bility, and Cancer (World Scientific Publishing Co. Pte.
(2007).
Ltd., 2007), pp. 1-22.
28. H. Masoom, et al., in Proc. 2006 IEEE Symp. on Com-
7. K. Okamura, et al., Genomics 88 (6), 690 (2006).
put. Intelligence in Bioinformatics and Comput. Biology
8. S. M. Berget, J. Biol. Chem., 270 (6), 2411 (1995).
(CIBCB’06)
(2006).
DOI:
10.1109/CIB-
9. J. Raes and Y. Van De Peer, Trends Genet. 21 (8), 428
CB.2006.330971.
(2005).
29. L. Wang and L. D. Stein, BMC Bioinformatics 11 (1),
10. S. L. Sheetlin, et al., Bioinformatics 30 (24), 3575
550 (2010).
(2014).
30. M. A. Korotkova, N. A. Kudryashov, and E. V. Korot-
11. Y. Zhang and Y. Sun, BMC Bioinformatics 12, 198
kov, Genomics. Proteomics. Bioinformatics 9 (4), 158
(2011).
(2011).
12. N. Du and Y. Sun, Bioinformatics 32 (17), i529 (2016).
31. V. Pugacheva, A. Korotkov, and E. Korotkov, Stat.
Appl. Genet. Mol. Biol. 15 (5), 381 (2016).
13. R. Ketteler, Frontiers in Genetics 19 (3), 242 (2012).
32. F. E. Frenkel and E. V. Korotkov, Gene 421 (1-2), 52
14. A. A. Mironov, P. S. Novichkov, and M. S. Gelfand,
(2008).
Bioinformatics 17 (1), 13 ( 2001).
33. A. A. Laskin, Mol. Biol. (Moscow) 37 (4), 663 (2003).
15. M. Gîrdea, L. Noé, and G. Kucherov, Algorithms Mol.
Biol. 5 (1), 6 (2010).
34. L. J. Huang, Proc. Natl. Acad. Sci. USA 94 (21), 11184
(1997).
16. T. Schiex, et al., Nucl. Acids Res. 31 (13), 3638 (2003).
35. I. Antonov, et al., Nucl. Acids Res. 41 (13), 6514 (2013).
17. I. Antonov and M. Borodovsky, J. Bioinform. Comput.
Biol. 8 (3), 535 (2010).
36. S. Ohno, Evolution by Gene Duplication (Springer Berlin
Heidelberg, 1970).
18. F. Frenkel and E. V. Korotkov, DNA Research 16 (2),
105 (2009).
37. E. V. Koonin, Rev. Genet. 39 (1), 309 (2005).
19. J. Gouzy, S. Carrere, and T. Schiex, Bioinformatics 25
38. T. M. Thomson, et al., Genome Res. 10 (11), 1743
(5), 670 (2009).
(2000).
20. M. Rho, H. Tang, and Y. Ye, Nucl. Acids Res. 38 (20),
39. W. Gilbert, Nature 271 (5645), 501 (1978).
e191 (2010).
40. M. Hiller, et al., Genome Biol. 6 (7), R58 (2005).
21.
21. Y. Zhang and Y. Sun, BMC Bioinformatics 12 (1),
41. H. Ochman, Curr. Opin. Genet. Dev. 11 (6), 616
198 (2011).
(2001).
A Database of Potential Reading Frame Shifts in Coding Sequences
from Different Eukaryotic
Yu.M. Suvorova*, V.M. Pugacheva*, and E.V. Korotkov* **
*Institute of Bioengineering, Research Center of Biotechnology of the Russian Academy of Sciences,
Leninsky prosp. 33/2, Moscow, 119071 Russia
**National Research Nuclear University (Moscow Engineering Physics Institute),
Kashirskoye Shosse 31, Moscow, 115409 Russia
In this paper we offer a new databank containing potential reading frame shifts in coding sequences. A new
mathematical method based on the use of a genetic algorithm and dynamic programming was used to search
for potential shifts in the reading frame. The data bank includes coordinates of potential reading frame shifts
for coding sequences of 76 eukaryotic genomes from Ensembl genomic browser, version 86. The database is
mately 23% of coding sequences have a reading frame shift. Errors of the first and second kind are about 11%
and 30%. Simultaneously with the data bank, a Web-server has been created to search for potential shifts in
for potential shifts of the reading frame in newly defined coding sequences.
Keywords: reading frame shift, dynamic programming, position-weight matrix, coding sequence
БИОФИЗИКА том 64
№ 3
2019