БИОФИЗИКА, 2019, том 64, № 5, с. 917-932
БИОФИЗИКА КЛЕТКИ
УДК 577.3
МИОКАРДИАЛЬНЫЕ α2-АДРЕНОРЕЦЕПТОРЫ - ПОТЕНЦИАЛЬНЫЕ
МИШЕНИ ДЛЯ ЗАЩИТЫ ОТ ГИПЕРТРОФИИ И СЕРДЕЧНОЙ
НЕДОСТАТОЧНОСТИ
© 2019 г. О.Ю. Пименов*, М.Х. Галимова*, Э.В. Евдокимовский*, А.С. Аверин**,
О.В. Накипова**, С. Рейес***, А.Е. Алексеев* ***
*Институт теоретической и экспериментальной биофизики РАН,
142290, Пущино Московской области, ул. Институтская, 3
**Институт биофизики клетки РАН - обособленное подразделение ФИЦ ПНЦБИ РАН,
142290, Пущино Московской области, ул. Институтская, 3
***Department of Cardiovascular Medicine, Center for Regenerative Medicine, Stabile 5, Mayo Clinic, Rochester, MN, USA
E-mail: polegiteb@gmail.com, alekseev.alexey@mayo.edu
Поступила в редакцию 07.06.2019 г.
После доработки 07.06.2019 г.
Принята к публикации 08.07.2019 г.
Нарушение механизмов сердечной адаптации к катехоламиновой перегрузке приводит к сердечной
гипертрофии и недостаточности. Используемые подходы к лечению этого недуга во многом не оправ-
дывают терапевтических ожиданий, что требует поиска новых лечебных стратегий, направленных на
переход от лечения вторичных факторов сердечной недостаточности (нейрогормональная активация,
ренальная дисфункция и пр.) к улучшению сердечной структуры и функции. Мы обнаружили экс-
прессию изоформ α2-адренорецепторов не только во взрослых кардиомиоцитах, но и на всем протя-
жении развития эмбриональных сердец из стволовых клеток. В дополнение к известному ограниче-
нию активности симпатоадреналовой системы, в кардиомиоцитах α2-адренорецепторы обладают
собственным защитно-адаптационным потенциалом. Данный обзор представляет анализ α2-адрено-
рецепторной сигнализации в кардиомиоцитах, которая препятствует Са2+-перегрузке и ангиотензи-
нергической сердечной гипертрофии. Мы полагаем, что при десенситизации α2-адренорецепторов в
кардиомиоцитах в условиях развития сердечной недостаточности, ткань-специфичные клеточные
или генные терапии, направленные на восстановление/усиление α2-адренорецепторной сигнализа-
ции, могут противодействовать сердечной гипертрофии и недостаточности.
Ключевые слова: клеточная терапия, генная терапия, Са2+-гомеостаз, внутриклеточная сигнализация,
ангиотензин, G-белки.
DOI: 10.1134/S0006302919050120
α2-АДРЕНОРЕЦЕПТОРЫ В СТРЕССОВОЙ
пациентов, исследование ЭПОХА-ХСН) прини-
АДАПТАЦИИ ОРГАНИЗМА
мает масштаб национальной проблемы, на реше-
ние которой, по оценкам специалистов, требует-
По данным Российского кардиологического
ся более 120 млрд руб. ежегодно [1]. Несмотря на
общества распространенность хронической сер-
определенные успехи, имеющиеся терапевтиче-
дечной недостаточности в нашей стране (~17 млн
ские подходы к лечению этого заболевания тре-
буют дальнейшего совершенствования. Даже с
использованием современных терапевтических
Сокращения: AR - адренергические рецепторы (adrenocep-
tors), PLC - фосфолипаза С, PKC - протеинкиназа С, методов ежегодный показатель смертности от
PKA - протеинкиназа А, Akt/PKB - Akt-киназа/протеи-
хронической сердечной недостаточности состав-
новая киназа В, eNOS - эндотелиальная NO-синтаза,
ляет более 20% [1-3].
cGMP - циклический гуанозинмонофосфат, SERCA -
Ca2+-АТФаза саркоплазматического ретикулума, ICaL -
Установлено, что одним из факторов развития
входящий ток через потенциал-зависимые Са2+-каналы
L-типа, PKG - сGMP-зависимая протеинкиназа, АТ1R -
сердечной недостаточности является избыточная
ангиотензиновый рецептор первого типа, RGS - регуля- хроническая симпатическая (адренергическая)
тор G-белковой сигнализации (Regulator of G-protein Sig- стимуляция сердечной мышцы [2]. Так, напри-
naling), SH-крысы - спонтанно гипертензивные крысы
(Spontaneously Hypertensive Rats), GRK - киназа G-белок-
мер, хроническое нервно-психическое перена-
связанных рецепторов (G-protein coupled Receptor Kinase).
пряжение, гипертензия, перегрузка сердца объе-
917
918
ПИМЕНОВ и др.
мом и другие проявления реакции организма на
связанных α2-AR приводит к уменьшению уров-
стресс ведут к компенсаторной перестройке сер-
ня цAМФ и активности Са2+-каналов, а также от-
дечной мышцы (гипертрофической кардиомио-
крытию К+-каналов, подавляя механизм экзоци-
патии), адаптирующей сердце к повышенным
тоза [20,21]. Таким образом, α2-AR-сигнализация
энергетическим затратам [1,4-7]. При этом нару-
рассматривается как механизм отрицательной
шение адаптационных механизмов сердца или
обратной связи, подавляющий выброс катехола-
исчерпание их ресурсов приводит к развитию
минов из окончаний симпатических нейронов и
сердечной недостаточности [3,8].
надпочечника [22,23]. Согласно установленной
Реакция на стресс периферических органов
роли α2-AR, их фармакологические агонисты ис-
опосредуется симпатоадреналовой системой че-
пользуются как обезболивающие, седативные,
рез выброс катехоламинов (адреналин, норадре-
гипотензивные, гипотермические и прочие сред-
налин) из нервных окончаний и секрецию в кро-
ства, имеющие, к сожалению, множественные
воток из клеток надпочечника. Катехоламины
побочные действия [24,25] (см. таблицу).
действуют через адренергические рецепторы
α2-AR имеют четыре изоформы, обозначаемые
(AR), связанные с Gα/Gβγ-белковыми комплек-
α2A, α2B, α2C и кодируемые генами Adra2A,
сами. Изначально на основе клеточных ответов
Adra2B и Adra2C соответственно [26,27]; а также
адренорецепторы были дифференцированы на α-
два подтипа α2D, кодируемые генами Adra2Da и
и β-рецепторы [9,10]. Позднее, вследствие более
Adra2Db [28]. Все виды млекопитающих, включая
детального изучения фармакологических и
человека, имеют только первые три изоформы α2-
структурных особенностей, адренорецепторы
AR, в то время как остальные позвоночные (за ис-
были разделены на три типа, α1-AR, α2-AR и β-
ключением крокодилов) экспрессируют все гены,
AR, каждый из которых имеет три или более ре-
кодирующие эти рецепторы [29]. У млекопитаю-
цепторных подтипа [11]. В то время как актива-
щих присутствие α2A- и α2C-изоформ ярко выра-
ция α1-AR преимущественно вызывает диссоци-
жено в центральной нервной системе, в то время
ацию Gαq/Gβγ-комплекса, β-AR и α2-AR дей-
как все три изоформы широко представлены в пе-
ствуют через диссоциацию комплексов,
риферических тканях. Исследование животных с
состоящих из
«Gαs-стимулирующего» (Gs) и
генетическим прерыванием экспрессии Adra2A и
«Gαi-ингибирующего» (Gi) типов белков соответ-
Adra2C показало, что недостаток α2A-изоформы
ственно [5,12,13]. На сегодняшний день иденти-
приводит к общему увеличению уровня норадре-
фицировано по крайней мере 20 Gα-, 6 Gβ- и
налина в плазме крови, в то время как α2C-изо-
12 Gγ-подтипов G-белков. Такое многообразие
форма преимущественно контролирует секрецию
катехоламинов из хромаффинных клеток надпо-
может обеспечить около 1500 комбинаторных ва-
чечника по Са2+-зависимому механизму обратной
риантов клеточной сигнализации, что, в совокуп-
ности с множеством изоформ эффекторов, взаи-
связи, как в нейронных окончаниях [30,31]. Оцен-
ка потентности норадреналин-зависимого подав-
модействующих по отдельности или синергиче-
ски с Gα- и Gβγ-белками, определяет
ления нейротрансмиттерного выброса в этих ген-
но-модифицированных моделях животных, вы-
многообразие тканевых ответов на адренергиче-
явила примерно десятитикратно большее сродство
скую стимуляцию [14-16].
норадреналина к α2C-AR (Ki = 650 нM) по сравне-
В синапсах, где α1-AR и β-AR стимулируют
нию с α2А-AR (Ki = 5,8 мкM) [32]. Исследованный
нейрональную активность и выброс нейротранс-
функциональный вклад этих изоформ рецептора
миттеров, α2-AR имеют противоположное, по-
на различных частотах нейрональной стимуляции
давляющее действие. Действительно, присут-
показал, что α2C-AR подавляет выброс нейро-
ствующие в избыточном количестве в мозге Gq-
трансмиттеров при низком уровне активности
связанные α1-AR активируют фосфолипазу С
симпатических нейронов, в то время как α2А-AR -
(PLC), которая через гидролиз фосфатидилино-
при высоком [32,33].
зитола-4,5-бифосфата увеличивает внутрикле-
Так как устойчивая симпатическая стимуля-
точный уровень Са2+ и стимулирует изоформы
ция ассоциируется с гипертрофией сердца и раз-
протеинкиназы С (PKC) [17,18]. Активация β-AR
витием сердечной недостаточности, было пред-
через Gs стимулирует аденилилциклазу и продук-
положено, что нарушение функции α2-AR также
цию цAМФ, что ведет к активации протеинкина-
будет служить фактором риска развития сердеч-
зы А (PKA) [19]. В свою очередь, каталитическая
ных дисфункций. Действительно, повышенная
субъединица PKA фосфорилирует и таким обра-
по сравнению с контрольными животными
зом активирует потенциал-зависимые Са2+-ка-
смертность в экспериментальной группе мышей с
налы, увеличивая секрецию нейротрансмиттеров
генетическим «нокаутом» обеих изоформ α2А- и
[5]. И напротив, активация пресинаптических Gi-
α2С-AR была ассоциирована с увеличением
БИОФИЗИКА том 64
№ 5
2019
МИОКАРДИАЛЬНЫЕ α2-АДРЕНОРЕЦЕПТОРЫ
919
Побочные эффекты применения агонистов α2-AR
Агонисты
Клинические проявления
Побочные эффекты
Ссылки
Клонидин
Гипертония
После остановки приема препаратов - возвратная
[34, 35]
(гуанабенз,
гипертензия (ребаунд-эффект), тахикардия;
гуанфацин)
сопутствующие симптомы, такие как головная
[36, 37]
боль, беспокойство, тремор, потливость,
тошнота, рвота
Интра- или
При интратекальной инфузии - тяжелая
[38]
послеоперационня боль,
гипотензия, низкий индекс системного
хроническая боль
сосудистого сопротивления;
импотенция
[39, 40]
Беспокойство,
Сонливость, усталость, сухость во рту,
[41, 42]
дефицит внимания
брадикардия, раздражительность, боль в горле,
и гиперактивность
бессонница, запор, повышение температуры тела,
боль в ушах, тошнота, гипотензия, головная боль
Послеоперационная дрожь
Седативный эффект
[43]
Симптомы абстиненции
[44]
Значительная гипотензия
Тиназидин
Спастичность, мышечно-лицевая
Астения, сухость во рту, головокружение,
[45-49]
боль, мышечный спазм и судороги
сонливость, желудочно-кишечные расстройства
Дексмедетомидин
Спинальная анестезия, седация в
Гипотония, брадикардия, остановка функции
[50-52]
интенсивной терапии
синусного узла, тошнота, рвота, лихорадка,
гипоксия, тахикардия, анемия, тахифилаксия
уровня циркулирующих катехоламинов, отягчен-
нергическую стимуляцию. Действительно, ре-
ной гипертрофией левого желудочка сердца,
зультаты недавних исследований позволяют
фиброзом и сердечной недостаточностью [23].
взглянуть по-новому на функциональную роль и
Однако ткань-специфическое возобновление
терапевтический потенциал α2-AR в клетках
миокарда.
экспрессии этих изоформ рецептора в симпати-
ческих нейронах приводило лишь к частичному
восстановлению вентрикулярного ответа на внут-
НАРУШЕНИЯ ФУНКЦИИ α2-AR
ривенное введение агониста α2-AR [53]. Вместе с
И РАЗВИТИЕ СЕРДЕЧНЫХ ПАТОЛОГИЙ
тем было показано, что предварительное конди-
Известны несколько генных модификаций в
ционирование изолированных сердец с исполь-
изоформах α2-AR человека, способных вносить
зованием агониста α2-AR дексмедетомидина сти-
вклад в развитие сердечных дисфункций. В ос-
мулировало фосфорилирование внеклеточной
новном такие модификации приводят к замене
сигнал-регулируемой киназы
1/2, Akt-киназы
или потере аминокислотных остатков в третьей
(протеиновой киназы В) и эндотелиальной NO-
внутриклеточной петле (рис. 1) и вызывают нару-
синтазы (eNOS) в миоцитах левого желудочка
шения взаимодействия рецепторов с G-белками
сердца и улучшало восстановление сердечной
[56-59]. Например, обнаруженная потеря трех
функции после периода ишемии-реперфузии
глютаматов в кислотном участке третьей внутри-
[54,55]. Эти наблюдения указывают на заметную
клеточной петли α2В-AR рассматривается как ге-
роль α2-AR в сердечной мышце, где они могут ло-
нетический фактор риска острого коронарного
кально контролировать сердечный ответ на адре-
синдрома, но, однако, не гипертензии [59]. Этот
БИОФИЗИКА том 64
№ 5
2019
920
ПИМЕНОВ и др.
β1-AR (β1Arg389), приводящей к примерно двух-
кратному увеличению активности этих рецепто-
ров по сравнению с контрольным типом рецепто-
ра (β1Gly389) [62], также рассматривались как
группа риска, склонная к развитию сердечной не-
достаточности. Однако, вопреки ожиданию,
β1Arg389-пациенты тоже не были склонны к раз-
витию сердечной недостаточности [61]. И лишь
относительно узкая подгруппа, имеющая одно-
временно мутацию α2СDel322-325, увеличиваю-
щую уровень норадреналина, и мутацию
β1Arg389, увеличивающую чувствительность кар-
диомиоцитов к норадреналину, имела ярко выра-
женную склонность к развитию сердечной дис-
функции [61]. Возможно, отсутствие выраженно-
го фенотипического заболевания у пациентов с
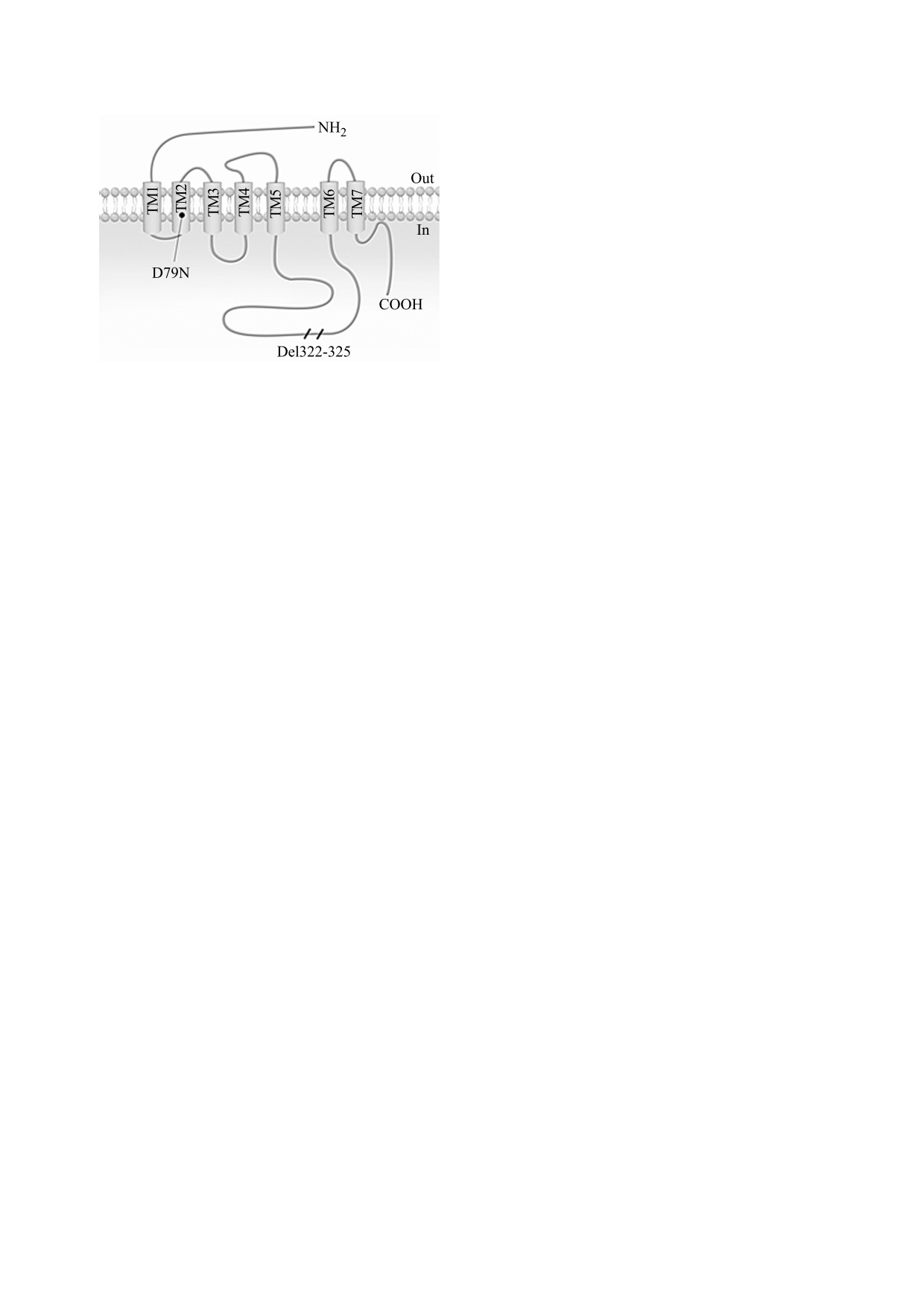
Рис. 1. Структурная схема α2-AR, показывающая рас-
мутацией α2СDel322-325 является следствием
пределение трех внеклеточных и трех внутриклеточ-
сохраняющегося контроля уровня катехоламинов
ных петель, соединяющих семь трансмембранных
другой, не мутированной, изоформой α2-AR.
альфа-спиралей (ТМ1 - ТМ7). Считается, что внут-
Нельзя исключить и вероятность того, что другие
риклеточный участок С-концевой последовательно-
сти заякорен с плазматической мембраной посред-
изоформы α2-AR в мембране вентрикулярных
ством S-пальмитирования цистеинового остатка.
миоцитов могут компенсировать дисфункцию
Мутация аспартата в позиции 79 второго трансмем-
α2СDel322-325-AR при стимуляции сердца по-
бранного участка последовательности α2А-AR
вышенным уровнем катехоламинов [63].
(D79N) приводит к потере способности рецептора
активировать К+-каналы, но не влияет на его способ-
ность подавлять активность потенциал-зависимых
Са2+-каналов, а также понижать уровень цAМФ. Вы-
ЭКСПРЕССИЯ α2-AR
деленный участок в третьей внутриклеточной петле,
И α2-АДРЕНОРЕЦЕПТОРНАЯ
соединяющей ТМ5 и ТМ6, соответствует полимор-
физму, приводящему к потере четырех аминокислот
СИГНАЛИЗАЦИЯ
(G-A-G-P) в структуре α2С-AR (Del322-325) и на-
В КАРДИОМИОЦИТАХ
рушению взаимодействия рецептора с G-белковым
комплексом.
В то время как активация β-AR и α1-AR явля-
ется хорошо изученным механизмом увеличения
миокардиальной сократимости [5,64], α2-AR тра-
же генетический полиморфизм у пациентов сред-
диционно рассматривались как рецепторы, не
них лет был ассоциирован с увеличением рисков
обладающие никаким воздействием на сердеч-
острого инфаркта миокарда и внезапной сердеч-
ную мышцу или имеющие очень ограниченное
ной смерти у лиц моложе 55 лет. Предполагается,
воздействие. Несколько ранее проведенных ис-
что пациенты с мутированным рецептором
следований не обнаружили прямого инотропного
склонны к проявлениям спазма коронарных ар-
эффекта α2-AR ни на сердце в целом, ни на изо-
терий вблизи участка стеноза, что, утяжеляя фор-
лированную папиллярную мышцу [65,66]. Спо-
му коронарного синдрома, увеличивает смерт-
собность агонистов (клонидин, дексмедетоми-
ность [60].
дин) оказывать антигипертензивный и брадикар-
диальный эффекты через активацию α2-AR в
Поскольку устойчивая симпатическая стиму-
коронарных артериях и иннервирующих сердеч-
ляция связана с развитием сердечной недостаточ-
ные ткани нейронах, была подтверждена ткань-
ности, особый интерес вызвала группа с генети-
специфическим подавлением экспрессии изо-
ческим полиморфизмом α2С-AR (α2СDel322-
форм рецептора или мутацией D79N, нарушаю-
325), нарушающим способность этих рецепторов
щей функцию рецептора (рис. 1) [22,25]. При
подавлять выброс норадреналина [23,58] (рис. 1).
Поэтому ожидалось, что эти пациенты должны
этом обнаруженная экспрессия поли(A)+РНК
быть подвержены высокому риску развития сер-
α2A-, α2B- и α2C-изоформ в сердечных мышцах
дечной недостаточности. Однако проведенные
крыс была ничтожной по сравнению с экспресси-
клинические исследования не подтвердили этот
ей этих изоформ в нейронах, почках, печени или
прогноз, обнаружив достаточно слабую корреля-
сосудах (аорте) [67]. Однако более поздние иссле-
цию между полиморфизмом α2СDel322-325 и
дования подтвердили белковую экспрессию всех
развитием болезни [61]. С другой стороны, паци-
изоформ α2-AR в ткани целого сердца, и только
енты с повышенной реакцией миокардиальной
α2A- и α2C-изоформ в вентрикулярных миоцитах
мышцы на катехоламины вследствие мутации в
грызунов и человека [54,68]. В наших исследова-
БИОФИЗИКА том 64
№ 5
2019
МИОКАРДИАЛЬНЫЕ α2-АДРЕНОРЕЦЕПТОРЫ
921
Рис. 2. (а) - Фрагмент образов ДНК-микрочипов, демонстрирующий профиль экспрессии генов, специфичных для
сердечной ткани (обозначения справа) на уровне эмбриональной стволовой клетки (R1), на разных стадиях
эмбрионального развития (дни Е7,5 - Е18,5), через три дня после рождения (D3) и через три месяца взросления (А или
ADULT) в левом (LV) и правом (RV) желудочках сердца. (б) - Профили экспрессии генов изоформ α2-АR (Adra2a,
Adra2b и Adra2c) на разных стадиях кардиогененеза, полученные при помощи ДНК-микрочипов.
ниях экспрессия α2A-, α2B- и α2C-изоформ об-
ния должна поддерживать запасы Са2+ в сарко-
наружилась не только во взрослых кардиомиоци-
плазматическом ретикулуме. Действительно,
тах [69], но и на всем протяжении эмбрионально-
установлено, что подавление eNOS уменьшает
го развития сердца, а также в кардиомиоцитах,
сердечный выброс, развиваемое максимальное
дифференцированных из эмбриональных ство-
давление и скорость его развития в левом желу-
ловых клеток [70,71] (рис. 2). Это показывает, что
дочке [80,81], а следовательно, eNOS-зависимое
α2-AR могут быть вовлечены в механизмы кар-
стимулирование SERCA через активацию α2-AR
диогенеза, а следовательно, в будущем могут ис-
в кардиомиоцитах должно обеспечивать высокую
пользоваться как мишени для манипуляции в ре-
эффективность сокращения миокарда, улучшая
генерационной клеточной терапии сердца.
сердечную функцию.
Используя L-аргинин, субстрат для клеточно-
Более того, активация этого же сигнального
го NO-синтеза, присутствие которого было обя-
пути от α2-AR через PI3K-Akt/PKB-NO-cG-
зательным на всем протяжении выделения кар-
MP-PKG приводила к подавлению входящего
диомиоцитов и последующих эксперименталь-
ных процедур, мы обнаружили, что NO и
Са2+-тока через потенциал-зависимые Са2+ ка-
циклический гуанозинмонофосфат (cGMP) яв-
налы L-типа (ICaL) [63] (рис. 2). α2-AR-зависимое
ляются центральными внутриклеточными мес-
уменьшение ICaL может включать несколько ве-
сенджерами α2-AR-сигнализации в вентрикуляр-
роятных механизмов: 1) непосредственно зависи-
ных миоцитах [63,69]. Проведенный ингибитор-
мое от сGMP-зависимой протеинкиназы (PKG)
ный анализ показал, что агонист α2-AR гуанабенз
фосфорилирование канального белка, снижаю-
стимулировал продукцию NO, активируя изо-
щее вероятность его открывания [82,83]; 2) cG-
форму eNOS, но не нейрональную NO-синтазу,
MP-PKG-зависимая стимуляция протеиновых
через PI3K-Akt/PKB-сигнальный путь
[69]
фосфатаз PP1 и/или PP2A [63,84], которая ведет к
(рис. 3). Известно, что активация Akt/PKB и
дефосфорилированию и снижению активности
eNOS вызывает фосфорилирование и S-нитроли-
Са2+-каналов [85,86]; 3) α2-AR-Gi-белок-зави-
зирование фосфоламбана [72-75] и Ca2+-АТФазы
симая стимуляция фосфодиэстераз, ведущая к
саркоплазматического ретикулума (SERCA)
падению уровня сAMP [87-89] и, наконец, 4) по-
[76,77] (рис. 2), активируя закачку Са2+ в ретику-
давляющий ICaL эффект субъединицы Gβγ
лум, что в наших экспериментах сопровождалось
подавлением спонтанных Са2+-осцилляций в
[16,90]. В то время как дефосфорилирование, ка-
тализируемое PP1 и PP2A, действительно проти-
изолированных кардиомиоцитах [69,78,79]. Реа-
лизация этого сигнального механизма в условиях
водействует PKA-зависимой активации ICaL, воз-
реального сопряжения возбуждения и сокраще-
можный эффект противодействия этих се-
БИОФИЗИКА том 64
№ 5
2019
922
ПИМЕНОВ и др.
Рис. 3. Схема взаимодействия основных сигнальных путей регуляции внутриклеточного Са2+ в кардиомиоцитах от
α2-AR, связанных с Gi белковым комплексом, и рецепторами, связанными с Gs- и Gq-белками, на основе проведен-
ного ингибиторного анализа. В целях упрощения проиллюстрированы лишь выборочные сигнальные пути регуляции
внутриклеточного Са2+. Обозначения: Gi, Gq, Gs - G-белки; PI3K - фосфатидилинозитол-3-киназа; PDK - фосфа-
тидилинозитол-зависимая киназа; Akt(PKB) - Akt-киназа (киназа B); eNOS - эндотелиальная NO-синтаза; SERCA -
саркоплазматическая Са2+-ATФаза; RyR - рианодиновый рецептор; PLB - фосфоламбан; sCG - растворимая гуани-
латциклаза; PKG - сGMP-зависимая протеинкиназа; PP1 и PP2A - фосфатазы 1 и 2A; RGS - регулятор G-белковой
сигнализации, при фосфорилировании PKG может противодействовать гипертрофическим последствиям активации
связанных с Gq рецепторов; PKA - протеиновая киназа A, которая в ответ на активацию β-адреноцептора и увеличе-
ние уровня цAМФ фосфорилирует Ca2+-каналы L-типа, активируя вход в клетку внешнего Са2+, и индуцирует вы-
брос Са2+ из саркоплазматического ретикулума; PC-PLC - фосфатидилхолин-специфическая фосфолипаза C;
DAG - диацилглицерол; PKC - протеиновая киназа С, PKC-зависимое фосфорилирование фосфатазного ингибито-
ра I1 увеличивает активность PP1; IP3 - инозитол трифосфат, который через взаимодействие с соответствующим ре-
цептором (IP3R) вызывает выброс Ca2+ из саркоплазматического ретикулума. Внутриклеточный Са2+ определяет со-
кращение сердечной мышцы. Пунктирная стрелка обозначает предполагаемую непосредственную cGMP-зависимую
активацию PP1 и PP2A через PKG, уменьшающие число фосфорилированных Ca2+-каналов. Значки «плюс» и «ми-
нус» обозначают активацию и блокирование Са2+-каналов в ответ на действие протеинкиназ. Сигнальные пути от ад-
ренергических α1- и β-, а также ангиотензиновых АТ1-рецепторов приводят к увеличению уровня свободного Са2+,
способствуя гипертрофической перестройке сердца. Активация α2-AR инициирует кардиопротекторные сигнальные
пути, способные противодействовать Са2+-перегрузке, вызванной активацией рецепторов, связанных с Gs- и Gq-бел-
ками.
рин/треониновых фосфатаз PKG-зависимому
виях симпатической стимуляции норадренали-
ном, агонистом как β-, так и α-адреноцепторов,
фосфорилированию Са2+ каналов остается неиз-
блокатор α2-AR йохимбин значительно увеличи-
вестным. Тем не менее есть основания полагать,
вал ICaL при концентрациях норадреналина, пре-
что в вентрикулярных миоцитах при стимуляции
катехоламинами α2-адренорецепторная сигнали-
вышающих 10 нM [63]. Согласно установленному
зация сдвигает киназно-фосфатазный баланс
сигнальному механизму, блокирование α2-AR
йохимбином, в присутствии норадреналина, уве-
клетки, что ведет к утилизации свободного Са2+,
препятствуя развитию гипертрофии и сердечной
личивало внутриклеточные Ca2+ выбросы, вы-
недостаточности [91-93]. Действительно, в усло-
званные стимуляцией одиночного кардиомиоци-
БИОФИЗИКА том 64
№ 5
2019
МИОКАРДИАЛЬНЫЕ α2-АДРЕНОРЕЦЕПТОРЫ
923
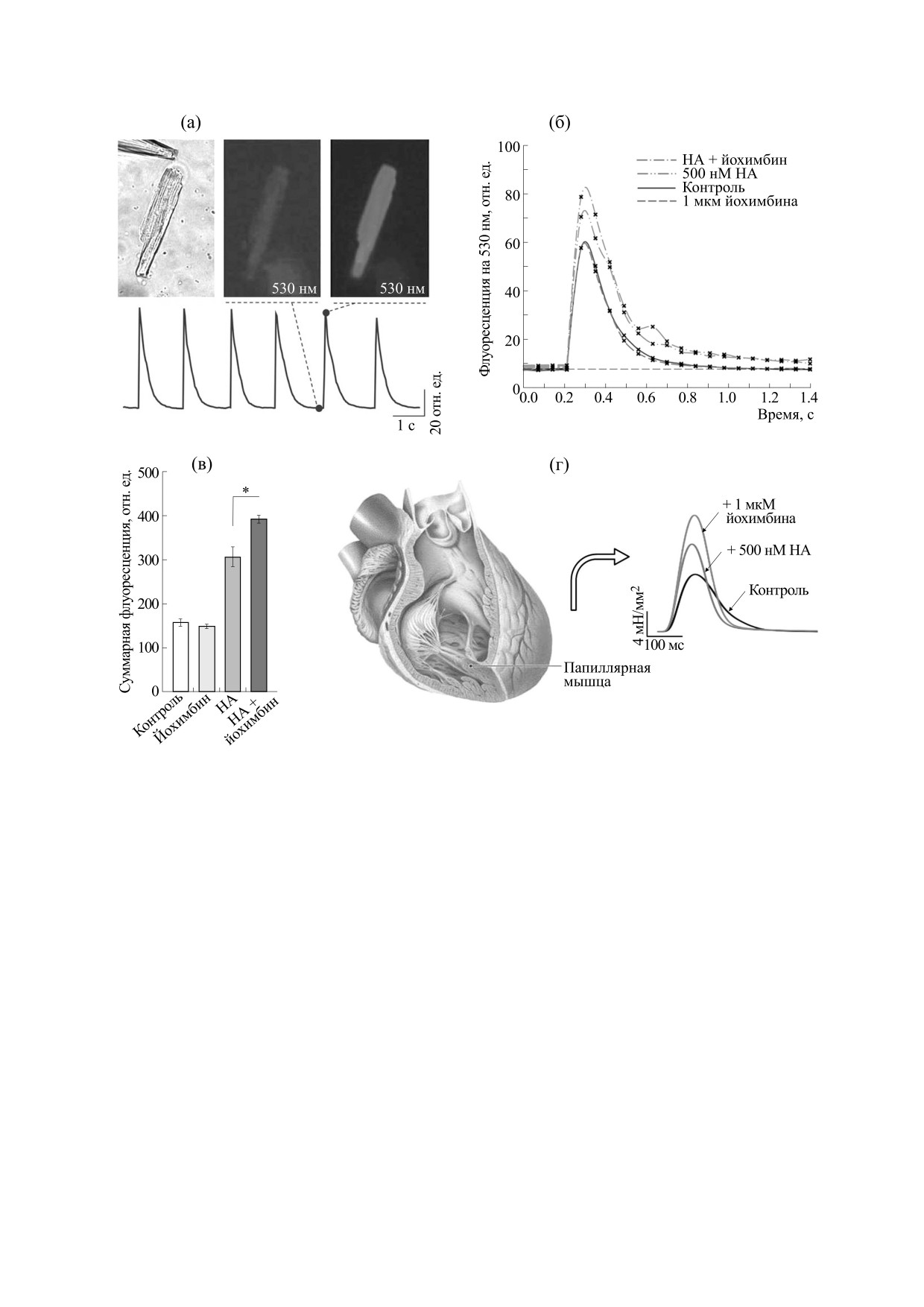
Рис. 4. Активация α2-AR уменьшает внутриклеточную Са2+-нагрузку. (а) - Одиночный кардиомиоцит, нагруженный
Са2+-красителем Fluo-4AM, в проходящем свете и флюоресцирующий на длине волны 530 нм в ответ на стимуляцию
с частотой 1 Гц через приближенный стеклянный электрод. (б) - Сравнительные одиночные Са2+-переходы,
измеренные последовательно в контроле, в присутствии антагониста α2-AR йохимбина и в присутствии
норадреналина, который вызывал значительное увеличение внутриклеточного Са2+. Блокирование йохимбином
активированных норадреналином α2-AR вызывало дополнительную внутриклеточную Са2+-нагрузку. (в) -
Статистика, соответствующая панели (б). (г) - Записи изометрического сокращения изолированной папиллярной
мышцы при стимуляции с частотой 1 Гц. Согласно увеличению внутриклеточного Са2+ в присутствии норадреналина
и йохимбина, блокирование α2-AR усиливало норадреналин-зависимую сократительную нагрузку папиллярной
мышцы (адаптировано из работы [63]).
та, что приводило к положительному инотропно-
ПЕРЕСЕЧЕНИЕ α2-АДРЕНОРЕЦЕПТОРНЫХ
му эффекту, увеличивающему сократительную
СИГНАЛЬНЫХ ПУТЕЙ
нагрузку миокарда [63] (рис. 4). Это указывает на
С ПРОГИПЕРТРОФИЧЕСКИМИ
то, что в дополнение к известной способности
СТИМУЛАМИ В ВЕНТРИКУЛЯРНЫХ
нейрональных α2-AR подавлять активность сим-
МИОЦИТАХ
патических нейронов, α2-AR на поверхности
Устойчивая стимуляция α1- и β-AR катехола-
кардиомиоцитов, в противовес адренергической
минами вызывает увеличение внутриклеточного
стимуляции α1- и β-AR, препятствует клеточной
уровня Са2+, что определяет формирование ком-
Са2+-перегрузке, оптимизируя расходы энерге-
тических ресурсов во время сердечного ответа на
плекса Са2+/кальмодулин и активацию кальци-
стресс (рис. 5).
нурин- и РКС-зависимых транскрипционных
БИОФИЗИКА том 64
№ 5
2019
924
ПИМЕНОВ и др.
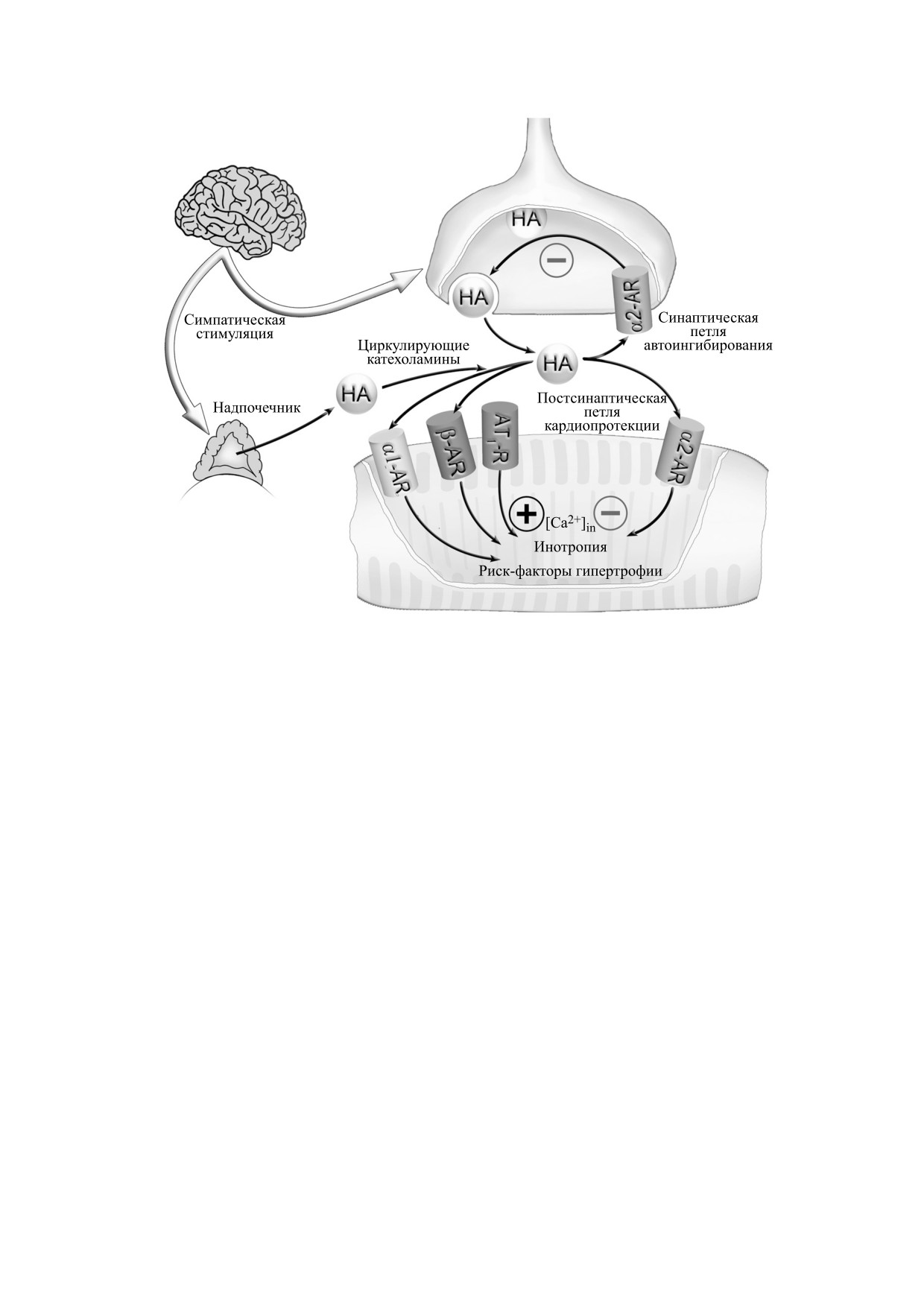
Рис. 5. α2-AR по механизму обратной связи контролирует симпатоадреналовую стимуляцию. В синапсах симпатиче-
ских нейронов α2-AR в ответ на активацию норадреналином (НА) инициирует автоингибиторную петлю, подавляю-
щую выброс норадреналина. Сарколеммальные α2-AR в кардиомиоцитах при активации норадреналином иницииру-
ют клеточную кардиопротекторную реакцию, которая ослабляет мобилизацию внутриклеточного Са2+ ([Са2+]in),
миокардиальную сократительную нагрузку (инотропия) и экспрессию риск-факторов гипертрофии, вызванные акти-
вацией других адренорергических и ангиотензиновых рецепторов.
факторов [94-96]. Нарушение контроля внутри-
жими G11-белками, включают локализованную в
клеточного уровня Са2+ увеличивают экспрес-
плазматической мембране PLC [18,103], которая
сию и активность этих ферментов, что приводит к
стимулирует Са2+-зависимые (PKCα, PKCβ) и
патологическим перестройкам сердечной ткани
Са2+-независимые (PKCδ, PKCε) изоформы PKC
[97,98]. Однако ряд недавних исследований пока-
и вызывает выброс Са2+ из саркоплазматического
зал, что долговременная активация Gq-связан-
ретикулума [97,104-106]. В изолированных кар-
ных α1-AR может по крайней мере частично ни-
диомиоцитах активация фосфатидилхолин-спе-
велировать чрезмерную стимуляцию β-AR в усло-
цифической PLC через стимуляцию некой изо-
виях сердечной недостаточности [99]. С другой
формы PKC подавляет продукцию NO, блокируя
стороны, активация других Gq-связанных рецеп-
eNOS, и уменьшает активность SERCA [69], т.е.
торов, таких как ангиотензиновые рецепторы
вызывает эффект, которому может противодей-
первого типа (АТ1R), которые являются основ-
ствовать сигнализация от α2-AR (рис. 3). Соглас-
ными мишенями ренин-ангиотензиновой систе-
но доступным нам данным, непосредственная
мы в сердечной мышце, помимо аритмического и
экспериментальная проверка противодействия
апоптотического эффектов, также инициализи-
сигнальных путей от α2-AR с АТ1R или α1-AR в
рует клеточную программу гипертрофической
кардиомиоцитах не проводилась. Однако в на-
перестройки миокарда [100-102]. На сегодняш-
ших исследованиях на изолированных кардио-
ний день сложился консенсус, что гипертрофиче-
миоцитах активация α2-AR подавляла спонтан-
ские стимулы через активацию рецепторов, свя-
ные Са2+-осцилляции, вызванные утечкой Са2+
занных с Gq-белками или с функционально схо-
из саркоплазматического ретикулума, в то время
БИОФИЗИКА том 64
№ 5
2019
МИОКАРДИАЛЬНЫЕ α2-АДРЕНОРЕЦЕПТОРЫ
925
как активация сигнального пути PC-PLC-
четырехкратному увеличению уровня ангиотен-
DAG-PKC, инициированного имидазолиновым
зина II в сердце. Показано, что у этих животных
рецептором, вызывала обратный эффект генера-
добавка в питьевую воду в течение четырех недель
дозы гуанабенза, не влияющей на кровяное дав-
ции Са2+-волн [69]. При этом следует отметить,
ление и частоту сердечных сокращений, вызыва-
что возникновение Са2+-волн и PKC-зависимое
ло достоверное уменьшение толщины задней
увеличение уровня внутриклеточного Са2+ также
стенки левого желудочка и тенденцию к сниже-
наблюдались в присутствии агониста α1-AR фе-
нию вентрикулярной массы
[122]. Таким
нилэфрина или агониста АТ1R ангиотензина II
образом, данное исследование впервые проде-
[107,108]. Более того, сообщалось, что Gq-сигна-
монстрировало способность периферических α2-
лизация может непосредственно взаимодейство-
AR противодействовать развитию гипертрофии
вать с PI3K и Akt [109,110], которые являются эф-
сердечной мышцы в условиях повышения уровня
ангиотензина II в сердце.
фекторами сигнального каскада α2-AR в кардио-
миоцитах [63,69] (рис. 3).
Особый интерес может представлять RGS -
НЕДОСТАТОЧНОСТЬ
регулятор G-белковой сигнализации (англ. Regu-
α2-АДРЕНОРЕЦЕПТОРНОЙ
lator of G-protein Signaling), обнаруженный в клет-
СИГНАЛИЗАЦИИ В МОДЕЛИ
ках гладкой мускулатуры [111]. Предполагается,
СЕРДЕЧНОЙ ГИПЕРТРОФИИ
что RGS, как потенциальный эффектор сигналь-
Спонтанно гипертензивные крысы (SH-кры-
ной цепочки от α2-AR, может препятствовать
сы, англ. Spontaneously Hypertensive Rats) представ-
прогипертрофической активации Gq-связанных
ляют собой хорошо известную модель животных
рецепторов [112,113] (рис. 3). Известно, что акти-
с генетически обусловленной гипертонией, кото-
вация cGMP-зависимой PKG вызывает фосфо-
рые, подобно человеку, склонны с возрастом к
рилирование RGS, который вследствие этого уве-
развитию гипертрофии и сердечной недостаточ-
личивает активность GTPазы субъединицы Gα,
ности [123]. В то время как перестройки клеточ-
инактивируя G-белок, и прерывает проведение
ного матрикса и микротубул, наряду с дерегуля-
сигнала от ассоциированного рецептора [114].
цией путей апоптоза, контроля внутриклеточного
В ряде аспектов сигнальные пути, активиро-
Са2+, β-адренергической стимуляции и митохон-
ванные α2-AR, схожи с сигнальными путями от
дриальной функции, являются характерными
β3-AR, которые в миокардиальных клетках тоже
особенностями болезненного фенотипа SH-
ассоциированы с Gi-белком [115]. Предполагает-
крыс, механизмы, ответственные за нарушение
ся, что активация β3-AR также способствует под-
тканевой функций сердца и перехода от гипер-
держанию сердечной функции через NO/cGMP
трофии к сердечной недостаточности, остаются
сигнальный механизм [116,117]. Таким образом,
лишь частично изученными [123-125]. Важно от-
особым свойством активации α2-AR в кардио-
метить, что развитие гипертрофии и дисфункции
миоцитах является их противодействие Gq-
левого желудочка сердца у SH-крыс, по-видимо-
му, не определяется исключительно гипертони-
PLC-PKC-зависимой сигнализации, что подчер-
ей, а скорее связано с другими факторами [124].
кивает перспективность использования кардио-
Проведенные исследования показали, что даже
протекторных особенностей α2-AR в условиях
превращение SH-крыс в нормотензивных живот-
симпатоадреналовой и ангиотензинергической
ных при помощи периферийной симпатэктомии
перегрузки сердца. Поскольку внутриклеточные
не останавливает развитие гипертрофии и сер-
сигнальные каскады, опосредованные разными
дечной недостаточности [126,127]. Основным ме-
изоформами PKC, в целом сходятся на продук-
ханизмом, вносящим решающий вклад в пере-
ции таких маркеров гипертрофии, как TGFβ, p38,
стройку сердца у SH-крыс является повышенная
NFκB и др. [101,118-120], есть основания пола-
симпатическая активность как на системном
гать, что активация α2-AR в кардиомиоцитах мо-
уровне, так и в сердечной ткани [124]. Повышен-
жет влиять на экспрессию этих маркеров, препят-
ная по сравнению с сердцами контрольных крыс
ствуя развитию гипертрофии и сердечной недо-
симпатическая активность наблюдается в левых
статочности (рис. 5). В связи с этим являются
желудочках сердец SH-крыс в возрасте от 4 до 50
показательными недавние исследования, прове-
недель, что, в свою очередь, усиливает гиперто-
денные на гипертрофической модели трансген-
нию после того, как сердечная и васкулярная ги-
ных крыс [TGR(hAGT)L1623]. Эти животные
пертрофии развились полностью [128]. Оценка
экспрессируют человеческую изоформу ангио-
изменений систолического и диастолического
тензиногена, устойчивого к расщеплению крыси-
ным ренином [121], но подверженного действию
внутриклеточного Са2+ по сравнению с кон-
сердечной химазы, что приводит к локальному
трольными животными выявила в целом наруше-
БИОФИЗИКА том 64
№ 5
2019
926
ПИМЕНОВ и др.
ние регуляции внутриклеточного Са2+ у SH-крыс
миоцитах, было предложено как средство, пре-
пятствующее перестройке сердечной ткани и раз-
[124].
витию сердечной недостаточности [131,138,139].
Наши исследования показали, что активация
Подавление клеточного ответа на активацию
α2-AR агонистами в кардиомиоцитах SH-крыс
α2-AR в сердцах SH-крыс может быть также при-
оказалась существенно ослабленной по сравне-
чиной нарушения функции эффекторов в соот-
нию с кардиомиоцитами нормотензивных жи-
ветствующей сигнальной цепочке. Действитель-
вотных [63]. Причиной такой дерегуляции мог бы
но, уровень белковой экспрессии еNOS был об-
быть генетический полиморфизм α2-AR или
наружен уменьшенным у молодых SH-крыс,
связанных эффекторов, однако подобные со-
однако в возрасте одного года оказался уже в два
ставляющие онтогенеза SH-крыс остаются неиз-
раза выше по сравнению с контрольными живот-
вестными [129,130]. Удивительно, что подавление
ными [140]. Такой профиль экспрессии NOS на-
эффективности α2-адренорецепторной сигнали-
вряд ли может объяснить обнаруженную в кар-
зации сопровождается многократным увеличени-
диомиоцитах SH-крыс неспособность α2-AR по-
ем уровня экспрессии мРНК каждой из изоформ
вышать скорость продукции NO и эффективно
α2-AR [63]. Вероятно, подавление сигнализации
подавлять ICaL, тем более, что уровень cGMP в
через α2-AR, обнаруженное у SH-крыс, является
этих животных остается неизменным [140]. Шун-
следствием увеличения экспрессии β-аррестина,
тирование сигнального звена eNOS-sGC с ис-
характерной для механически перегруженных
пользованием донора NO или проницаемого ана-
сердец [131,132]. β-Аррестин, действуя в коорди-
лога cGMP показали, что в кардиомиоцитах SH-
нации со своим кофактором GRK - киназой G-
крыс NO- и cGMP-зависимые пути сигнализа-
белок-связанных рецепторов, разобщает рецеп-
ции, по-видимому, нарушены [63]. Вероятно, в
тор и соответствующий G-белок, что приводит в
этих условиях, ослабление функционального от-
результате к функциональной десенситизации и
вета на активацию α2-AR у SH-крыс вносит вклад
интернализации рецептора [131,133,134]. В дей-
в гипертрофическую перестройку сердца, в то
ствительности на сегодняшний день мало что из-
время как увеличенная экспрессия изоформ α2-
вестно о механизмах десенситизации/интернали-
AR является следствием адаптационной клеточ-
зации α2-AR в кардиомиоцитах. Однако в других
ной программы, реализуемой в попытке ком-
тканях показано, что десенситизация некоторых
пенсировать симпатоадреналовую перегрузку
изоформ α2-AR к их агонистам осуществляется
сердца. Представляется важным определить, яв-
посредством GRK-зависимого фосфорилирова-
ляются ли обнаруженные дефекты α2-AR-сигна-
ния рецепторов [135,136]. Сравнение аминокис-
лизации особенностью SH-крыс или они харак-
лотной последовательности третьей внутрикле-
терны для других моделей сердечной недостаточ-
точной петли в различных изоформах α2-AR -
ности. Аналогичный профиль дерегуляции α2-
части структуры, вовлеченной во взаимодействие
адренорецепторной сигнализации в других моде-
рецептор-аррестин - при фосфорилировании
лях животных с вызванной гипертрофией будет
сериновых и треониновых остатков выявило низ-
означать, что коррекция соответствующих сиг-
кую степень гомологии, что предполагает нали-
нальных путей могла бы быть основой для разра-
чие различных механизмов десенситизации изо-
ботки новых терапевтических принципов проти-
форм этого рецептора [137]. В связи с этим только
водействия развитию миокардиальной гипертро-
α2А- и α2В-изоформы являются субстратами для
фии и декомпенсации в направлении сердечной
фосфорилирования GRK и связывания с β-арре-
недостаточности.
стином, ведущего к десенситизации этих типов
рецептора в условиях кратковременной актива-
ции адреналином [137,138]. В условиях долговре-
α2-АДРЕНОРЕЦЕПТОРЫ В ТЕРАПИИ
менной активации степень десенситизации изо-
СЕРДЕЧНОЙ ГИПЕРТРОФИИ
форм α2-AR может быть выражена следующим
И НЕДОСТАТОЧНОСТИ
соотношением: α2A = α2B > α2C. В то время как
в условиях долговременной активации десенси-
Несмотря на значительный прогресс, достиг-
тизация α2А- и α2В-изоформ обуславливается
нутый в последние годы, в лечении сердечной не-
как интернализацией, так и падением уровня бел-
достаточности с использованием блокаторов
ковой экспрессии, десенситизация α2С-изофор-
β-AR, агонистов альдостерона, ангиотензин-пре-
мы является исключительно результатом падения
вращающего фермента, ресинхронизирующей
уровня G-белка [135,137]. Поскольку GRK-зави-
терапии и трансплантации сердца, данное забо-
симая десенситизация α2-AR вносит вклад в па-
левание по-прежнему является причиной высо-
тофизиологию сердца, подавление действия β-
кой смертности [1-3,141], что требует дальнейше-
аррестина напрямую или опосредованно, напри-
го усовершенствования имеющихся терапевтиче-
мер, через регуляцию экспрессии GRK в кардио-
ских подходов. Например, блокаторы β-AR и
БИОФИЗИКА том 64
№ 5
2019
МИОКАРДИАЛЬНЫЕ α2-АДРЕНОРЕЦЕПТОРЫ
927
ренин-ангиотензиновой системы, являясь наи-
SERCA [69] и к NO-независимому стимулирова-
более широко используемыми лекарствами для
нию фосфатазной PP1-активности [120]. Показа-
лечения сердечной недостаточности, не оправды-
но, что генетическое прерывание экспрессии ге-
вают в полной мере терапевтических ожиданий
на, кодирующего PKCα, обращало развитие кар-
[142,143]. В частности, блокаторы β-AR не спо-
диомиопатии, связанной со сверхэкспрессией
собны полностью подавить симпатическую пере-
PP1 [120]. Этот результат полностью согласуется с
грузку сердца вазоактивными симпатическими
установленным фактом развития сердечной дис-
котрансмиттерами, такими как допамин или ней-
функции вследствие увеличения общей фосфа-
ропептид Y [144-146].
тазной активности, а также увеличения белковой
экспрессии и активности PKC [155-158]. В свою
В этой связи использование агонистов α2-AR
очередь, PKC-зависимое снижение активности
как агентов, снижающих активность симпатоад-
SERCA приводит не только к уменьшению уров-
реналовой системы в целом и, следовательно,
ня Са2+ в саркоплазматическом ретикулуме, но, в
уровень соответствующих нейротрансмиттеров,
конечном итоге, и к падению общего уровня Са2+
может показаться оправданным подходом для ле-
вследствие его откачки из клетки мембранными
чения сердечной недостаточности. Однако не-
смотря на наличие ожидаемого эффекта клони-
Са2+-АТФазами [120]. Вызванный таким образом
дина у контрольных пациентов, у больных сер-
отрицательный инотропный эффект у пациентов
дечной недостаточностью этот агонист вызывал
исследования MOXCON со средней и тяжелой
лишь незначительное снижение уровня норадре-
формами хронической сердечной недостаточно-
налина по причине десенситизации и падения
сти (сердечный выброс <35%) мог иметь самые
количества α2-AR [147,148]. В качестве альтерна-
губительные последствия.
тивного подхода для снижения общего уровня ка-
Вместо подавления симпатоадреналовой си-
техоламинов было предложено использовать ак-
стемы через активацию центральных α2-AR, ко-
тивацию имидазолиновых рецепторов селектив-
торая имеет значительное число побочных эф-
ными агонистами, такими как моксонидин или
фектов (см. таблицу), представляется целесооб-
рилменидин, способными также снижать актив-
разным использовать ткань-специфическую
ность симпатической системы [149,150]. Тем не
активацию α2-AR в кардиомиоцитах. Такой под-
менее находящиеся уже на третьей фазе клиниче-
ход мог бы не только усилить кардиопротектор-
ские испытания моксонидина
(«MOXonidine
ный механизм устойчивости сердца к стрессу, но
CONgestive Heart Failure (MOXCON)») выявили
и повысить эффективность блокирования β-AR и
более высокую смертность в группе пациентов с
ренин-ангиотензиновой системы, используемых
сердечной недостаточностью, принимающих
в настоящее время для лечения сердечной недо-
препарат, что потребовало срочно приостановить
статочности. С этой точки зрения будущие ген-
испытания [151,152]. Было отмечено несколько
ные и клеточные терапии для коррекции или уси-
факторов, предположительно приведших к про-
ления α2-адренорецепторной сигнализации мо-
валу этого клинического испытания: 1) пониже-
гут рассматриваться в качестве альтернативных
ние симпатического тона могло иметь отрица-
или вспомогательных терапевтических подходов
тельный эффект для пациентов с пониженным
к лечению сердечной недостаточности (рис. 6).
базовым уровнем норэпинефрина; 2) усиленная
Показано, что доставка здоровых или модифици-
титрация препарата, превышающая в пять-десять
раз рекомендуемые антигипертензивные дозы,
рованных клеток в больное сердце наряду с мето-
дами генного перепрограммирования клеток сер-
могла приводить к полному подавлению симпа-
тической активности в сердце; 3) негативный эф-
дечной ткани может быть безопасной и иметь
определенную терапевтическую эффективность
фект могло иметь увеличение миокардиального
метаболизма [153,154]. Очевидно, что ошибки в
[159-162]. Однако разработка подобных иннова-
ционных подходов к использованию сарколем-
разработке и имплементации этого клинического
мальных α2-AR в вентрикулярных миоцитах
испытания связаны с отсутствием достаточного
требует решения по меньшей мере следующих
понимания действия симпатолитических соеди-
практических вопросов: 1) обеспечивают ли име-
нений центрального действия, которые могут во-
ющиеся изоформы α2-AR кардиопротекторное
влекать неисследованные альтернативные цен-
действие через общие или независимые друг от
тры связывания, регуляции, сигнальные пути, а
друга сигнальные механизмы?; 2) какие изофор-
также мобилизовать механизмы, ставшие более
выраженными в условиях гипертензии и сердеч-
мы α2-AR ответственны за контроль Са2+-гомео-
ной недостаточности. Важно отметить, что акти-
стаза и миокардиальную сократимость?; 3) как
вация имидазолиновых рецепторов в мембране
активация различных изоформ α2-AR сказывает-
кардиомиоцитов увеличивает активность PKC,
ся на экспрессии внутриклеточных маркеров ги-
что приводит к eNOS-зависимому подавлению
пертрофии и сердечной недостаточности?; 4) ка-
БИОФИЗИКА том 64
№ 5
2019
928
ПИМЕНОВ и др.
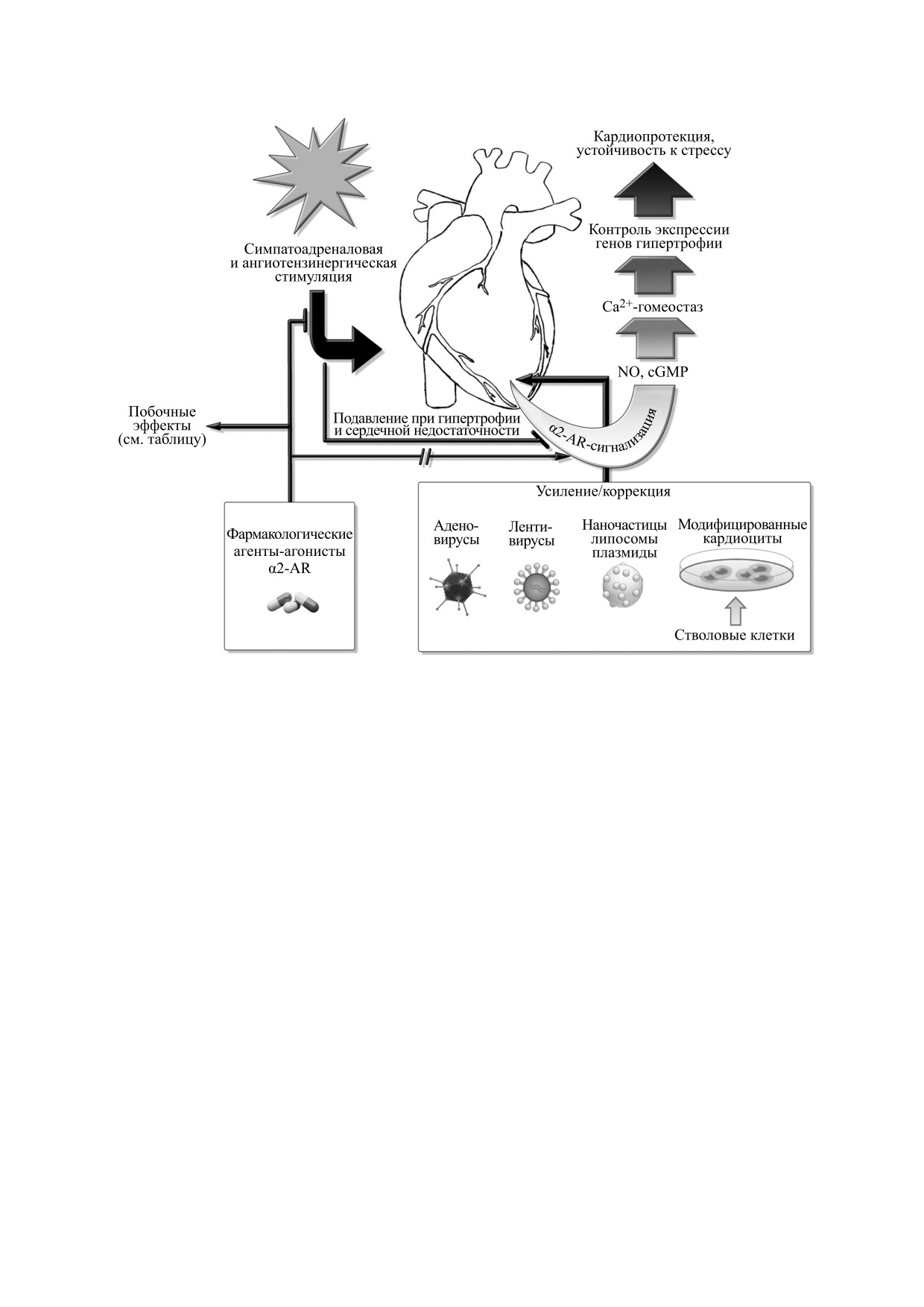
Рис. 6. В условиях десенситизации и дерегуляции сигнальных путей, связанных с α2-AR при развитии пост-
адаптационной гипертрофии и сердечной недостаточности, тканеспецифические усиление или коррекция α2-AR-
сигнализации в кардиомиоцитах с использованием генной или клеточной терапии могут служить альтернативным
подходом к обеспечению устойчивости сердечной функции к стрессовой гиперстимуляции.
кие механизмы ответственны за подавление α2-
ходы требуют дальнейшего совершенствования.
адренорецепторной сигнализации в условиях
Установлено, что одним из ключевых факторов
симпатоадреналовой перегрузки?
развития сердечной недостаточности является из-
быточная хроническая симпатическая (адренерги-
Таким образом, ткань-специфичная актива-
ческая) и ангиотензинергическая стимуляция сер-
ция α2-AR в кардиомиоцитах с использованием
дечной мышцы. В вентрикулярных кардиомиоци-
клеточной или генной терапии может оказаться
тах изоформы α2-AR (α2А, α2B и α2C), в
перспективным подходом к противодействию ги-
дополнение к известному контролю активности
пертрофической перестройке сердечной мышцы
симпатических нейронов, обладают кардиопро-
и лечению сердечной недостаточности. Кроме
текторными свойствами, ограничивающими мио-
того, усиление или коррекция сигнализации че-
кардиальный ответ на гиперадренергическую сти-
рез α2-AR в кардиомиоцитах может обеспечить
муляцию. Установленные внутриклеточные сиг-
стресс-устойчивый сердечный фенотип, избегая
нальные механизмы позволяют предположить, что
центрального подавления общей адаптационной
активация α2-AR может противодействовать про-
реакции организма, необходимой для выживания
гипертрофической активации адренергических и
в опасных для жизни условиях.
ангиотензиновых рецепторов. Следовательно,
ткань-специфическая активация α2-адренорецеп-
торов или усиление/коррекция их функции в кар-
ЗАКЛЮЧЕНИЕ
диомиоцитах могут быть использованы для разра-
Несмотря на успехи в лечении сердечной недо-
ботки комплементарных имеющимся или альтер-
статочности, существующие терапевтические под- нативных методов борьбы с развитием сердечной
БИОФИЗИКА том 64
№ 5
2019
МИОКАРДИАЛЬНЫЕ α2-АДРЕНОРЕЦЕПТОРЫ
929
недостаточности. Такой подход соответствует мне-
23.
M. Brede, F. Wiesmann, R. Jahns, et al., Circulation,
нию, что усовершенствование терапии сердечной
106 (19), 2491 (2002).
недостаточности требует смены парадигмы борьбы
24.
J. C. Hunter, D. J. Fontana, L. R. Hedley, et al., Br. J.
с вторичными последствиями болезни (такими как
Pharmacol. 122 (7), 1339 (1997).
нейрогормональная активация, нарушение гемо-
25.
L. B. MacMillan, L. Hein, M. S. Smith, et al., Science
динамики, аритмия, ренальная дисфункция и пр.)
273 (5276), 801 (1996).
на непосредственное воздействие на сердце с це-
26.
D. B. Bylund, H. S. Blaxall, L. J. Iversen, et al., Mol.
лью улучшения его структуры и функции [163].
Pharmacol. 42 (1), 1 (1992).
27.
J. W. Lomasney, W. Lorenz, L. F. Allen, et al., Proc.
Natl. Acad. Sci. USA 87 (13), 5094 (1990).
ФИНАНСИРОВАНИЕ РАБОТЫ
28.
J. O. Ruuskanen, H. Xhaard, A. Marjamäki, et al., Mol.
Работа выполнена при финансовой поддержке
Biol. Evol. 21 (1), 14 (2004).
Российского научного фонда (грант № 18-15-
29.
H. A. Céspedes, K. Zavala, M. W. Vandewege, et al.,
00198).
Gen. Comp. Endocrinol. 250, 85 (2017).
30.
M. Brede, G. Nagy, M. Philipp, et al., Mol. Endocri-
СПИСОК ЛИТЕРАТУРЫ
nol. 17 (8), 1640 (2003).
1. В. Ю. Мареев, Ф. Т. Агеев, Г. П. Арутюнов и др.,
31.
E. Moura, J. Afonso, L. Hein, et al., Br. J. Pharmacol.
Сердечная недостаточность 7, 379 (2013).
149 (8), 1049 (2006).
2. J. J. McMurray and M. A. Pfeffer, Lancet 365 (9474),
32.
L. Hein, J. D. Altman, and B. K. Kobilka, Nature 402
1877 (2005).
(6758), 181 (1999).
3. S. Neubauer, N. Engl. J. Med. 356 (11), 1140 (2007).
33.
M. Philipp, M. Brede, and L. Hein, Am. J. Physiol. 283
4. G. Eisenhofer, P. Friberg, B. Rundqvist, et al., Circula-
(2), R287 (2002).
tion 93 (9), 1667 (1996).
34.
B. C. Campbell and J. L. Reid., Int. J. Clin. Pharmacol.
5. N. Dzimiri, Pharmacol. Rev. 51 (3), 465 (1999).
Res. 5 (4), 215 (1985).
6. H. Ashrafian, C. Redwood, E. Blair, et al., Trends
35.
В. М. Делягин, У. Левано, Б. М. Блохин и др.
Genet. 19 (5), 263 (2003).
Практич. медицина 5 (44), 42 (2010).
7. I. Kehat and J. D. Molkentin, Circulation 122 (25),
36.
G. N. Karachalios, A. Charalabopoulos, V. Papalim-
2727 (2010).
neou, et al., Int. J. Clin. Pract. 59 (5), 562 (2005).
8. M. H. Drazner, Circulation 123 (3), 327 (2011).
37.
Е. В. Крюков, Н. П. Потехин, А. Н. Фурсов и др.,
9. R. P. Ahlquist, J. Auton. Pharmacol. 1 (1), 101 (1980).
Клин. медицина 94 (1), 52 (2016).
10. D. B. Bylund, Am. J. Physiol. 293 (6), E1479 (2007).
38.
F. Puskas, E. M. Camporesi, C. E. O’Leary, et al.,
11. D. B. Bylund, D. C. Eikenberg, J. P. Hieble, et al.,
Anesth. Analg. 97 (5), 1251 (2003).
Pharmacol. Rev. 46 (2), 121 (1994).
39.
G. Koman, A. Alfieri, J. Rachinger, et al., Pain Physi-
12. T. Gudermann, F. Kalkbrenner, and G. Schultz, Ann.
cian 15 (4), E523 (2012).
Rev. Pharmacol. Toxicol. 36 (1), 429 (1996).
40.
А. Д. Радченко, Артериальная гипертензия 3 (23),
13. M. I. Simon, M. P. Strathmann, and N. Gautam, Sci-
69 (2012).
ence 252 (5007), 802 (1991).
41.
D. F. Connor, K. E. Fletcher, and J. M. Swanson, J.
14. T. Gudermann, F. Kalkbrenner, E. Dippel, et al., Adv.
Am. Acad. Child Adolesc. Psychiatry 38 (12), 1551
Second Messenger Phosphoprotein Res.
31,
253
(1999).
(1997).
42.
A. Sharma and J. Couture, Ann. Pharmacother. 48 (2),
15. S. Offermanns and G. Schultz, Naunyn Schmiedebergs
209 (2014).
Arch. Pharmacol. 350 (4), 329 (1994).
43.
M. Panneer, P. Murugaiyan, and S. V. Rao., Anesth.
16. D. E. Clapham and E. J. Neer, Ann. Rev. Pharmacol.
Essays Res. 11 (1), 151 (2017).
Toxicol. 37 (1), 167 (1997).
44. M. S. Gold, A. C. Pottash, D. R. Sweeney, et al., JAMA
17. S. G. Birnbaum, Science 306 (5697), 882 (2004).
243 (4), 343 (1980).
18. K. B. Hubbard and J. R. Hepler, Cell. Signal. 18 (2),
45.
H. R. Henney III and M. Chez, Pediatric Drugs 11 (6),
135 (2006).
397 (2009).
19. R. Gilsbach and L. Hein, Handb Exp Pharmacol.
46.
P. W. Nance, W. A. Sheremata, S. G. Lynch, et al.,
(184), 261 (2008).
Arch. Neurol. 54 (6), 731 (1997).
20. L.-G. Wu and P. Saggau, Trends Neurosci. 20 (5), 204
47.
A. J. Wagstaff and H. M. Bryson, Drugs 53 (3), 435
(1997).
(1997).
21. R. J. Miller, Ann. Rev. Pharmacol. Toxicol. 38 (1), 201
48.
T. T. Батышева, Н. Ф. Попова, С. В. Петров и др.,
(1998).
Журн. неврологии и психиатрии им. С. С.
22. J. D. Altman, A. U. Trendelenburg, L. MacMillan, et
Корсакова 5, 57 (2010).
al., Mol. Pharmacol. 56 (1), 154 (1999).
49.
А. Б. Данилов, Рос. мед. журн. 20 (31), 1543 (2012).
БИОФИЗИКА том 64
№ 5
2019
930
ПИМЕНОВ и др.
50. J. A. Giovannitti, S. M. Thoms, and J. J. Crawford,
76. T. Adachi, R. M. Weisbrod, D. R. Pimentel, et al., Nat.
Anesth. Prog. 62 (1), 31 (2015).
Med. 10 (11), 1200 (2004).
51. Pediatric Advisory Committee Meet. on Precedex La-
77. X. Tong, A. Evangelista, and R. A. Cohen, Curr. Opin.
Pharmacol. 10 (2), 133 (2010).
tees/CommitteesMeetingMaterials/PediatricAdviso-
78. J. Hüser, D. M. Bers, and L. A. Blatter, Am. J. Physiol.
ryCommittee/ucm494275.htm
274 (5, Pt 2), H1800 (1998).
52. А. А. Еременко и Е. В. Чернова, Анестезиология и
79. Y. Bai, P. P. Jones, J. Guo, et al., Circ. Res. 113 (5), 517
реанимация 5, 4 (2013).
(2013).
53. R. Gilsbach, J. Schneider, A. Lother, et al., Cardiovasc.
80. B. Casadei and C. E. Sears, Prog. Biophys. Mol. Biol.
Res. 86 (3), 432 (2010).
82 (1-3), 67 (2003).
54. M. Ibacache, G. Sanchez, Z. Pedrozo, et al., Biochim.
81. B. D. Prendergast, V. F. Sagach, and A. M. Shah, Cir-
Biophys. Acta 1822 (4), 537 (2012).
culation 96 (4), 1320 (1997).
55. Y. Yoshikawa, N. Hirata, R. Kawaguchi, et al., Anesth.
82. L. H. Jiang, D. J. Gawler, N. Hodson, et al., J. Biol.
Analg. 126 (2), 443 (2018).
Chem. 275 (9), 6135 (2000).
56. P. Heinonen, M. Koulu, U. Pesonen, et al., J. Clin. En-
83. F. Schröder, G. Klein, B. Fiedler, et al., Cardiovasc.
docrinol. Metab. 84 (7), 2429 (1999).
Res. 60 (2), 268 (2003).
57. K. M. Small, S. L. Forbes, K. M. Brown, et al., J. Biol.
84. Z. Xu, S. Lee, and J. Han, Int. J. Biochem. Cell Biol.
Chem. 275 (49), 38518 (2000).
45 (8), 1577 (2013).
58. K. M. Small, S. L. Forbes, F. F. Rahman, et al., J. Biol.
85. W. H. duBell, M. S. Gigena, S. Guatimosim, et al., Am.
Chem. 275 (30), 23059 (2000).
J. Physiol. 282 (1), H38 (2002).
59. A. Snapir, P. Heinonen, T.-P. Tuomainen, et al., J. Am.
86. J. Hescheler, M. Kameyama, W. Trautwein, et al., Eur.
Coll. Cardiol. 37 (6), 1516 (2001).
J. Biochem. 165 (2), 261 (1987).
60. A. Snapir, J. Mikkelsson, M. Perola, et al., J. Am. Coll.
87. K. D. Keef, J. R. Hume, and J. Zhong, Am. J. Physiol.
Cardiol. 41 (2), 190 (2003).
281 (6), C1743 (2001).
61. K. M. Small, L. E. Wagoner, A. M. Levin, et al., N. En-
88. C.-J. Dong, Y. Guo, Y. Ye, et al., J. Neurosci. 34 (28),
gl. J. Med. 347 (15), 1135 (2002).
9432 (2014).
62. D. A. Mason, J. D. Moore, S. A. Green, et al., J. Biol.
89. A. M. Isidori, M. Cornacchione, F. Barbagallo, et al.,
Chem. 274 (18), 12670 (1999).
Cardiovasc. Res. 106 (3), 408 (2015).
63. Y. M. Kokoz, E. V. Evdokimovskii, A. V. Maltsev, et al.,
90. G. J. Stephens and S. Mochida, J. Physiol. 563 (3), 765
J. Mol. Cell. Cardiol. 100, 9 (2016).
(2005).
64. B.-E. Myagmar, J. M. Flynn, P. M. Cowley, et al., Circ.
91. D. M. Bers, D. A. Eisner, and H. H. Valdivia, Circ.
Res. 120 (7), 1103 (2017).
Res. 93 (6), 487 (2003).
65. I. D. Dukes and E. M. Vaughan Williams, J. Physiol.
92. M. Vassalle and C.-I. Lin, J. Biomed. Sci. 11 (5), 542
355, 523 (1984).
(2004).
66. M. Hongo, S. Fujisawa, T. Adachi, et al., J. Pharmacol.
93. X. H. T. Wehrens and A. R. Marks, Nat. Rev. Drug Dis-
Sci. 131 (2), 118 (2016).
cov. 3 (7), 565 (2004).
67. W. Lorenz, J. W. Lomasney, S. Collins, et al., Mol.
94. A. Terzic, M. Pucéat, G. Vassort, et al., Pharmacol.
Pharmacol. 38 (5), 599 (1990).
Rev. 45 (2), 147 (1993).
68. D. E. Berkowitz, D. T. Price, E. A. Bello, et al., Anes-
95. T. Zhang, S. Miyamoto, and J. H. Brown, Recent Prog.
thes. 81 (5), 1235 (1994).
Horm. Res. 59, 141 (2004).
69. A. V. Maltsev, Y. M. Kokoz, E. V. Evdokimovskii, et al.,
96. L. S. Maier, Front. Biosci. (Landmark Ed.) 14, 486
J. Mol. Cell. Cardiol. 68, 66 (2014).
(2009).
70. A. Martinez-Fernandez, X. Li, K. A. Hartjes, et al.,
97. J. O. Mudd and D. A. Kass, Nature 451 (7181), 919
Circ. Cardiovasc. Genet. 6 (5), 462 (2013).
(2008).
98. Y. K. Tham, B. C. Bernardo, J. Y. Y. Ooi, et al., Arch.
71. A. Martinez-Fernandez, T. J. Nelson, S. Reyes, et al.,
Toxicol. 89 (9), 1401 (2015).
Circ. Cardiovasc. Genet. 7 (5), 667 (2014).
99. T. D. O’Connell, B. C. Jensen, A. J. Baker, et al., Phar-
72. A. G. Brittsan and E. G. Kranias, J. Mol. Cell. Cardiol.
macol. Rev. 66 (1), 308 (2014).
32 (12), 2131 (2000).
100.L. A. Capote, R. Mendez Perez, and A. Lymperopou-
73. D. Catalucci, M. V. G. Latronico, M. Ceci, et al., J. Bi-
los, Eur. J. Pharmacol. 763, 143 (2015).
ol. Chem. 284 (41), 28180 (2009).
101. K.-D. Schlüter and S. Wenzel, Pharmacol. Ther. 119
74. F. Garofalo, M. L. Parisella, D. Amelio, et al., Proc.
(3), 311 (2008).
Biol. Sci. 276 (1675), 4043 (2009).
102.T. Kashihara, T. Nakada, K. Kojima, et al., J. Physiol.
75. E. Filice, T. Angelone, E. M. De Francesco, et al., Cell.
595 (13), 4207 (2017).
Physiol. Biochem. 28 (1), 41 (2011).
103.M. J. Berridge, Physiol. Rev. 96 (4), 1261 (2016).
БИОФИЗИКА том 64
№ 5
2019
МИОКАРДИАЛЬНЫЕ α2-АДРЕНОРЕЦЕПТОРЫ
931
104.V. Goutsouliak and S. W. Rabkin, Gen. Pharmacol. 30
133.S. S. Ferguson, Pharmacol. Rev. 53 (1), 1 (2001).
(3), 367 (1998).
134.E. Reiter and R. J. Lefkowitz, Trends Endocrinol. Me-
105.Y. E. G. Eskildsen-Helmond, K. Bezstarosti, and D.
tab. 17 (4), 159 (2006).
H. W. Dekkers, J. Mol. Cell. Cardiol. 29, 2545 (1997).
135.S. B. Liggett, J. Ostrowski, L. C. Chesnut, et al. J. Biol.
106.G. W. Dorn and T. Force, J. Clin. Invest. 115 (3), 527
Chem. 267 (7), 4740 (1992).
(2005).
136.P. M. C. Lembo, M. H. Ghahremani, and P. R. Albert,
107.G. Bkaily, N. El-Bizri, M. Nader, et al., Peptides 26
Mol. Endocrinol. 13 (1), 138 (1999).
(8), 1418 (2005).
137. M. G. Eason and S. B. Liggett, J. Biol. Chem. 267 (35),
108.Z. Zeng, H. Zhang, N. Lin, et al., J. Pharmacol. Sci.
25473 (1992).
126 (1), 37 (2014).
138.Q. Wang, J. Feng, P. B. Allen, et al., Science 304
109.L. M. Ballou, H.-Y. Lin, G. Fan, et al., J. Biol. Chem.
(5679), 1940 (2004).
278 (26), 23472 (2003).
139.A. Lymperopoulos, in Progress in Molecular Biology
110. A. L. Howes, J. F. Arthur, T. Zhang, et al., J. Biol.
and Translational Science, Ed. by D. B. Teplow (Aca-
Chem. 278 (41), 40343 (2003).
demic Press, 2018), pp. 27-57.
111. M. Tang, G. Wang, P. Lu, et al., Nat. Med. 9 (12), 1506
140.A. Piech, C. Dessy, X. Havaux, et al., Cardiovasc. Res.
(2003).
57 (2), 456 (2003).
112. M. Klaiber, M. Kruse, K. Völker, et al., Basic Res. Car-
141. C. W. Yancy, M. Jessup, B. Bozkurt, et al., J. Am. Coll.
diol. 105 (5), 583 (2010).
Cardiol. 68 (13), 1476 (2016).
113. I. Shimizu and T. Minamino, J. Mol. Cell. Cardiol. 97,
142.B. Kveiborg, A. Major-Petersen, B. Christiansen, et
245 (2016).
al., Vasc. Health Risk Manag. 3 (1), 31 (2007).
114. G. Xie and P. P. Palmer, J. Mol. Biol. 366 (2), 349
143.E. Braunwald, Lancet 385 (9970), 812 (2015).
(2007).
144.A. S. Maisel, N. A. Scott, H. J. Motulsky, et al., Am. J.
115. 115. C. Gauthier, V. Leblais, L. Kobzik, et al., J. Clin.
Med. 86 (1), 43 (1989).
Invest. 102 (7), 1377 (1998).
145.X.-J. Du, Cardiovasc. Res. 50 (3), 443 (2001).
116. J.-L. Balligand, Cardiovasc. Res. 111 (2), 128 (2016).
146.R. Mittal, L. H. Debs, A. P. Patel, et al., J. Cell. Physi-
117. A. Cannavo and W. J. Koch, J. Cardiovasc. Pharmacol.
ol. 232 (9), 2359 (2017).
69 (2), 71 (2017).
147. C. C. Lang, C. M. Stein, R. A. Nelson, et al., Hyper-
tension 30 (3, Pt 1), 392 (1997).
118. J. Wang, X. Liu, A. S. Arneja, et al., Can. J. Cardiol. 15
(6), 683 (1999).
148.A. Aggarwal, M. D. Esler, F. Socratous, et al., J. Am.
Coll. Cardiol. 37 (5), 1246 (2001).
119. S. Ruf, M. Piper and K.-D. Schlüter, Pflugers Arch.
443 (3), 483 (2002).
149.G. A. Head, S. L. Burke, and C. K. Chan, Clin. Exp.
120.J. C. Braz, K. Gregory, A. Pathak, et al., Nat. Med. 10
Hypertens. 19 (5-6), 591 (1997).
(3), 248 (2004).
150.M. Hausberg, F. Tokmak, H. Pavenstädt, et al., J. Hy-
pertens. 28 (9), 1920 (2010).
121. C. M. Ferrario, J. VonCannon, Y. Jiao, et al., Am. J.
Physiol. 310 (8), H995 (2016).
151. A. J. S. Coats. Int. J. Cardiol. 71 (2), 109 (1999).
122.S. Reyes, J. Varagic, J. VonCannon, et al., Circulation
152.J. N. Cohn, M. A. Pfeffer, J. Rouleau, et al., Eur. J.
138 (Suppl. 1), A16308 (2018).
Heart Fail. 5 (5), 659 (2003).
123.O. H. L. Bing, C. H. Conrad, M. O. Boluyt, et al.,
153.S. Mukaddam-Daher, Can. J. Cardiol. 28 (5), 590
Heart Fail. Rev. 7 (1), 71 (2002).
(2012).
124.B. M. Palmer, Z. Chen, R. R. Lachapelle, et al., Am. J.
154.R. Mobini, M. Fu, P.-A. Jansson, et al., Clin. Sci. 110
Physiol. 290 (1), H463 (2006).
(3), 329 (2006).
125.Y. Tang, C. Mi, J. Liu, et al., Cardiovasc. Pathol. 23
155.N. Bowling, R. A. Walsh, G. Song, et al., Circulation
(2), 101 (2014).
99 (3), 384 (1999).
126.A. F. Cutilletta, L. Erinoff, A. Heller, et al., Circ. Res.
156.A. N. Carr, A. G. Schmidt, Y. Suzuki, et al., Mol. Cell.
40 (4), 428 (1977).
Biol. 22 (12), 4124 (2002).
127. A. F. Cutilletta, M. Benjamin, W. S. Culpepper, et al.,
157. J. Neumann, Basic Res. Cardiol. 97 (Suppl. 1), I91
J. Mol. Cell. Cardiol. 10 (8), 689 (1978).
(2002).
128.M. A. Adams, A. Bobik, and P. I. Korner, Hyperten-
158.A. L. Bayer, M. C. Heidkamp, N. Patel, et al., Mol.
sion 14 (2), 191 (1989).
Cell. Biochem. 242 (1-2), 145 (2003).
129.L. Sun, M. Chun, S. McArdle, et al., Life Sci. 53 (4),
159.A. Behfar, R. Crespo-Diaz, A. Terzic, et al., Nat. Rev.
PL45 (1993).
Cardiol. 11 (4), 232 (2014).
160.A. Terzic and A. Behfar, Trends Cardiovasc. Med. 26
130.S. Kobayashi, S. Umemura, N. Hirawa, et al., J. Hy-
(5), 395 (2016).
pertens. 12 (3), 235 (1994).
131. A. Lymperopoulos and A. Bathgate, in Progress in Mo-
161. H. Tani, T. Sadahiro, M. Ieda, et al., Int. J. Mol. Sci.
lecular Biology and Translational Science, Ed. by L. M.
19 (9), 2629 (2018).
Luttrell (Elsevier, Oxford, London, Amsterdam,
162.R. D. Singh, M. Hillestad, C. Livia, et al., Tissue Eng.
Waltham, San Diego, 2013), pp. 297-334.
Part A 25 (1-2), 145 (2019).
132.J. You, J. Wu, Q. Zhang, et al., Am. J. Physiol. 314 (3),
163.M. Gheorghiade, C. J. Larson, S. J. Shah, et al., Heart
H552 (2017).
Fail. 9 (5), e002727 (2016).
БИОФИЗИКА том 64
№ 5
2019
932
ПИМЕНОВ и др.
Myocardial α2-Adrenoceptors as Therapeutic Targets to Prevent Cardiac Hypertrophy
and Heart Failure
O.Y. Pimenov*, M.H. Galimova*, E.V. Evdokimovskii*, A.S. Averin**, O.V. Nakipova**,
S. Reyes***, and A.E. Alekseev* ***
*Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences,
Institutskaya ul. 3, Pushchino, Moscow Region, 142290 Russia
**Institute of Cell Biophysics, Russian Academy of Sciences, Institutskaya ul. 3, Pushchino, Moscow Region, 142290 Russia
***Department of Cardiovascular Medicine, Center for Regenerative Medicine, Stabile 5, Mayo Clinic, Rochester, MN, USA
Aberrancies in mechanisms of cardiac adaptation to catecholamine overflow can result in maladaptive cardiac
hypertrophy and heart failure. Currently available heart failure therapies fall short of expectations, warranting
their refining via the paradigm shift from treatment of heart failure secondary factors (neurohormonal activa-
tion, renal dysfunction etc.) to direct heart targeting with the goal of improving cardiac structure and function.
We have identified expression of α2-adrenoceptors isoforms not only in adult cardiomyocytes, but also through-
out development of the heart from embryonic stem cells. In addition to known suppression of the sympathoad-
renal system activity, α2-adrenoceptors play protective and adaptive roles in cardiomyocytes. This review pres-
ents the analysis of α2-adrenoceptor signaling in cardiomyocytes that counteracts intracellular Ca2+ overload
and angiotensin-induced cardiac hypertrophy. We suppose that under heart failure linked desensitization of
these receptors, cardiac- specific cell- or gene-based therapies aimed at repairing/amplification of α2-adreno-
ceptor signaling could offer prospects for the prevention of cardiac hypertrophy and heart failure.
Keywords: cell-based therapy, gene therapy, Ca2+ homeostasis, intracellular signaling, angiotensin, G-proteins
БИОФИЗИКА том 64
№ 5
2019