БИОФИЗИКА, 2020, том 65, № 3, с. 421-438
МОЛЕКУЛЯРНАЯ БИОФИЗИКА
УДК 577.3
СВОБОДНО-РАДИКАЛЬНАЯ ПРИРОДА МОЛЕКУЛ МОНООКСИДА
АЗОТА КАК ФАКТОР, ОПРЕДЕЛЯЮЩИЙ ИХ ПРЕВРАЩЕНИЕ В ЖИВЫХ
ОРГАНИЗМАХ В ИОНЫ НИТРОЗОНИЯ
© 2020 г. А.Ф. Ванин
Федеральный исследовательский центр химической физики им. Н.Н. Семёнова РАН, 119334, Москва, ул. Косыгина, 4
Институт регенеративной медицины Первого Московского государственного медицинского университета
имени И.М. Сеченова МЗ РФ, 119991, Москва, ул. Трубецкая, 8/2
E-mail: vanin@polymer.chph.ras.ru
Поступила в редакцию 13.03.2020 г.
После доработки 13.03.2020 г.
Принята к публикации 16.03.2020 г.
Получены новые результаты, подтверждающие наши предыдущие данные о способности биядер-
ной формы биологически активных динитрозильных комплексов железа с тиолсодержащими ли-
гандами (глутатионом или N-ацетил-L-цистеином) выступать в качестве доноров катионов нитро-
зония, обеспечивающих образование S-нитрозотиолов при распаде этих комплексов в кислой среде
как в аэробных, так и анаэробных условиях. Появление катионов нитрозония в составе биядерных
динитрозильных комплексов железа определяется реакцией диспропорционирования свободно-
радикальных молекул монооксида азота при их связывании в ходе синтеза этих комплексов с кати-
онами Fe2+ (по две на один катион). При окислении тиолсодержащих лигандов в динитрозильных
комплексах железа или их блокаде тиолспецифическими реагентами выделяющиеся при распаде
этих комплексов катионы нитрозония при нейтральных значениях рН подвергаются гидролизу,
превращаясь в анионы нитрита. Аналогичное превращение имеет место при распаде при нейтраль-
ных значениях рН моноядерной формы динитрозильных комплексов железа с лигандами нетиоло-
вой природы. Установлено, что образование S-нитрозотилов при распаде биядерных динитрозиль-
ных комплексов железа с тиолсодержащими лигандами при кислотных значениях рН может блоки-
роваться при двух-трехкратном избытке свободных тиолов (не включенных в состав комплексов) по
отношению к содержанию самих комплексов. Эта блокада обусловлена восстановлением катионов
нитрозония тиолами при участии ионов железа как катализаторов до нейтральных молекул NO, вы-
свобождающихся из биядерных динитрозильных комплексов железа. Таким образом обе формы ди-
нитрозильных комплексов, возникающие в живых организмах, могут функционировать в них не
только как доноры одного из универсальных регуляторов метаболических процессов монооксида
азота, но и как доноры катионов нитрозония, инициирующих процесс S-нитрозирования низкомо-
лекулярных и высокомолекулярных (белковых) тиолов.
Ключевые слова: динитрозильные комплексы железа, оксид азота, S-нитрозотиолы, электронный
парамагнитный резонанс.
DOI: 10.31857/S0006302920030011
публикациях, - оксид азота - NO), функциони-
В настоящее время установлено, что во всех
рующее в живых организмах в качестве одного из
представителях живого мира - животных и чело-
универсальных регуляторов разнообразных мета-
веке, растениях и бактериях - ферментативным
болических процессов [1]. Его биологическое
путем непрерывно вырабатывается простейшее
действие не опосредовано клеточными рецепто-
химическое соединение - монооксид азота (или,
рами: оксид азота легко проникает через клеточ-
как сейчас его принято называть в биологических
ные мембран и, связываясь с гемовой группой
Сокращения: М-ДНКЖ - моноядерные динитрозильные
разнообразных гемсодержащих ферментов, на-
комплексы железа, ЭПР - электронный парамагнитный
пример, с гуанилатциклазы, активирует (или ин-
резонанс, Б-ДНКЖ - биядерные динитрозильные ком- гибирует) эти ферменты.
плексы железа, ДНКЖ-GSH - динитрозильные комплек-
Наряду с биологической активностью NO не
сы железа с глутатионом, GS-NO - S-нитрозоглутатион,
NAC - N-ацетил-L-цистеин, ДНКЖ-NAC - динитро- менее важным представляется биологическое
зильные комплексы железа с N-ацетил-L-цистеином, действие ионизованной формы этого агента - ка-
ЭДТА - этилендиаминтетраацетат, МНКЖ - мононитро-
зильные комплексы железа.
тиона нитрозония (NO+), связывание которого с
421
422
ВАНИН
тиоловыми группами белков приводит к образо-
а другой
- в анион нитроксила (NO-) (по
ванию соответствующих S-нитрозотиолов (RS-
реакции 1):
NO), резко изменяющих реактивность этих бел-
ков [2-4].
2NO NO+ + NO-.
(1)
Как было показано нами в работе [8], суще-
В связи с активной ролью катиона NO+ в био-
ствование такой реакции в газообразном NO сле-
логических процессах встает вопрос, каким обра-
дует из факта превращения этого газа в NO2 и
зом в живых организмах может достигаться иони-
N2O в соответствии с реакцией 2, характерной
зация (одноэлектронное окисление) молекул NO
для этого газа при давлении несколько десятков
до NO+? Большинство исследователей связывает
атмосфер (11, 12):
это с окислением NO кислородом до NO2. В обра-
зующемся аддукте - триоксиде азота (NO-NO2) -
3NO = NO2 + N2O.
(2)
взаимное одноэлектронное окисление-восста-
Как показано в работе [8], уравнение, характе-
новление свободно-радикальных молекул NO и
ризующее реакцию 1, превращается в уравнение,
NO2 (реакция их диспропорционирования) при-
характеризующее реакцию 2, после умножения
водит к трансформации триоксида азота в аддукт
обеих его частей на 2 и добавления к ним двух мо-
NO+-NO2-. В отсутствие тиолов ион нитрозония
лекул воды, обеспечивающих гидролиз ионов
в этом аддукте должен гидролизоваться, превра-
NO+ и NO- с образованием соответственно мо-
щаясь при связывании с ионами гидроксила и со-
лекул азотистой кислоты (HNO2) и молекул нит-
хранении нейтральных значений рН (в буферном
роксила (HNO). Последующее диспропорциони-
растворе), в анион нитрита [2-7]. В присутствии
рование этих молекул в соответствии с реакция-
тиолов, характеризующихся существенно более
ми 3 и 4
высоким, чем анион гидроксила, сродством к
2HNO2 ⇐ H2O + NO + NO2,
(3)
ионам нитрозония, последние, связываясь с тио-
лами, образуют S-нитрозотиолы. В кислой среде
2НNO ⇐ H2O + N2O
(4)
оба компонента аддукта NO+-NO2- превращают-
приводит соответственно к их превращению в ди-
ся в отсутствие тиолов в молекулы азотистой кис-
оксид, монооксид (оксид) и закись азота и тем са-
лоты (НNO2), тогда как в присутствии тиолов - в
мым (после удаления из обеих частей модифици-
молекулы S-нитрозотиолов [5, 6].
рованного уравнения одной молекулы воды и од-
ной молекулы NO) к превращению этого
Такого рода превращения, продемонстриро-
уравнения в уравнение 2. Таким образом, при вы-
ванные автором в работе [8] для чисто химиче-
соких давлениях оксид азота может вести себя как
ских систем, как оказалось, не имеют отношения
кислотный ангидрид, обеспечивающий образо-
к процессам образования S-нитрозотиолов в жи-
вание азотистой кислоты.
вых организмах. Проведенные на клеточных
культурах и микроорганизмах исследования по-
Такое поведение молекул NO не обнаружи-
казали, что генерация в них NO действительно
вается при давлении в одну атмосферу и ниже,
приводит к появлению в этих клетках как низко-
т.е. при концентрации этого агента, продуци-
молекулярных, так белковых S-нитрозотиолов.
руемого в живых организмах. Очевидно, в этом
Однако этот процесс равноэффективно идет как в
случае концентрация аддуктов (NO+-NO-) не-
присутствии, так и в отсутствие кислорода [9, 10].
достаточна для образования в реакции гидро-
Это означает, что образование S-нитрозотиолов,
лиза заметного количества азотистой кислоты,
т.е. необходимое для этого окисление NO до
так что молекулы NO не проявляют себя в этих
NO+, может происходить в живых организмах и в
условиях в качестве кислотного ангидрида. Тем
анаэробных условиях - в отсутствие кислорода.
не менее, как было предположено автором в
работах [8, 13], диспропорционирование моле-
Возможно ли это? Да, если принять во внима-
кул NO может идти и в этих условиях при нали-
ние свободнорадикальную природу оксида
чии в растворе ионов Fe2+, характеризующихся
азота - агента, выбранного Природой в качестве
высоким сродством к молекулам NO [14]. Свя-
одного из универсальных регуляторов метаболи-
зывание двух молекул NO с ионом Fe2+ с обра-
ческих процессов у всех представителей живого
зованием железодинитрозильного [Fe(NO)2]-
мира. Дело в том, что для свободных радикалов
характерна реакция диспропорционирования,
фрагмента - так называемого железодинитро-
т.е. реакция взаимного одноэлектронного окис-
зильного [Fe(NO)2]-мотива, с включением в
ления-восстановления, реализующаяся при взаи-
его молекулярные орбитали π-орбиталей NO и
модействии двух молекул свободных радикалов.
d-орбиталей железа, обеспечивает перенос не-
Именно эта реакция может обеспечивать превра-
спаренного электрона в этом мотиве с одной
щение одной молекулы NO в катион нитрозония,
молекулы NO на другую, т.е. их реакцию дис-
БИОФИЗИКА том 65
№ 3
2020
СВОБОДНО-РАДИКАЛЬНАЯ ПРИРОДА МОЛЕКУЛ МОНООКСИДА АЗОТА
423
пропорционирования - реакцию 1. Последую-
гандного окружения железа с последующим
щее протонирование появляющегося в этом
диспропорционированием молекул нитрокси-
мотиве иона нитроксила приводит к выходу
ла (HNO) и появлением вместо них в соответ-
этого иона, как это показано на схеме 1, из ли-
ствии с уравнением 4 закиси азота и воды:
+1/2 (N O
+
H
O)
HNO
2
2
H+
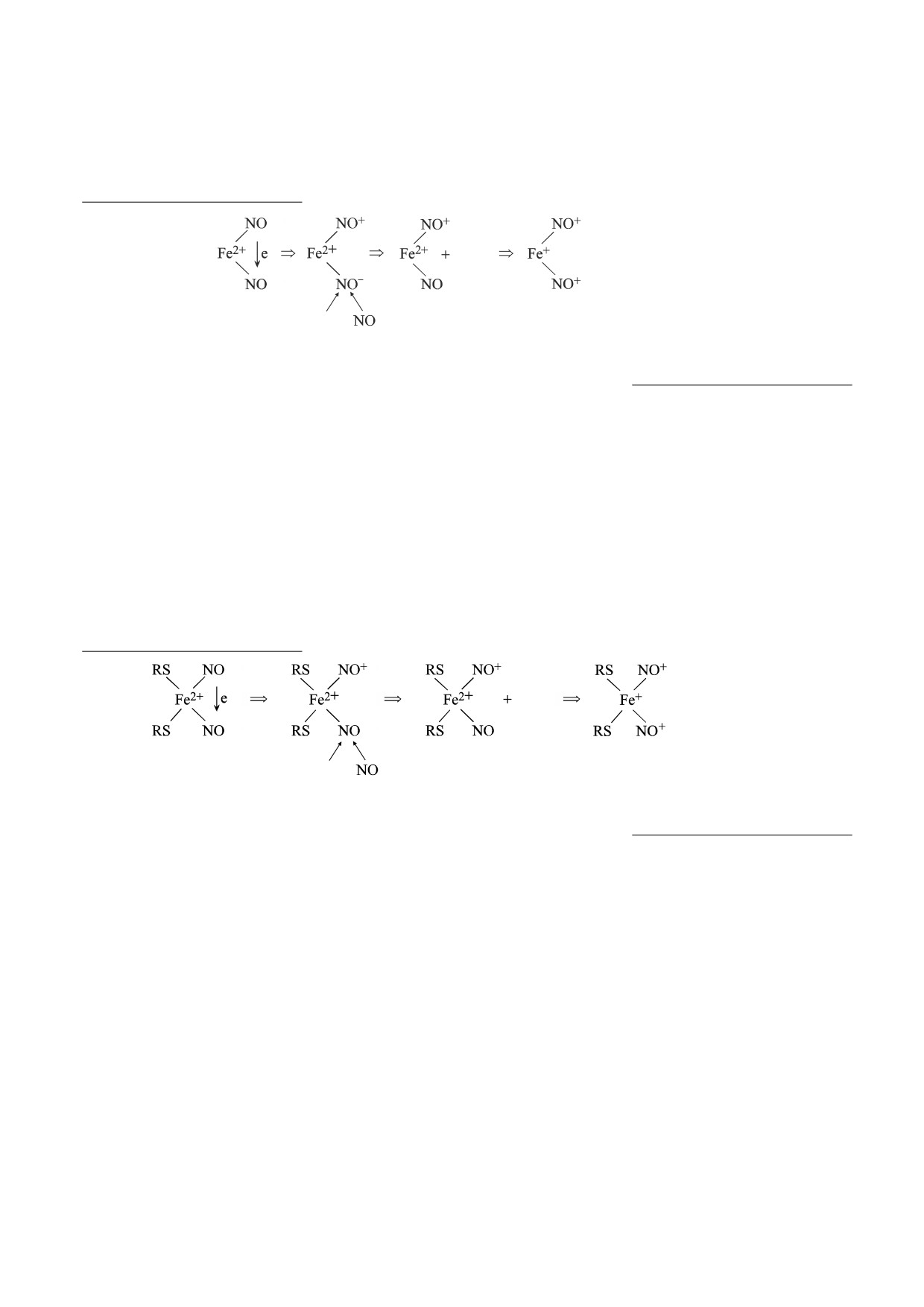
Схема 1. Предполагаемый механизм образования [Fe(NO)2]-мотива в реакции молекул NO
c ионом Fe2+ [8, 13, 15-19].
Вместо иона нитроксила в окружение железа
будучи сильными π-донорами электронной плот-
включается третья молекула NO, в результате же-
ности, могут обеспечить перенос этой плотности
лезо в железодинитрозильном мотиве приобрета-
на железо и нитрозильные лиганды. Происходя-
ет d7-электронную конфигурацию с соответству-
щее в результате этого переноса снижение поло-
ющей резонансной структурой железодинитро-
жительного заряда на этих лигандах ослабляет
зильного мотива - [Fe+(NO+)2].
взаимодействие последних с ионами гидроксила,
предотвращая тем самым гидролиз нитрозильных
Очевидно, что наличие в этом мотиве легко
лигандов, в результате в соответствии со схемой 2
гидролизуемых NO+-лигандов делает указанную
возникают стабильные парамагнитные моно-
резонансную структуру неустойчивой. Эту не-
ядерные динитрозильные комплексы железа (М-
устойчивость можно устранить введением в коор-
ДНКЖ) с тиолсодержащими лигандами (форму-
динационную сферу железодинитрозильного мо-
тива тиолсодержащих (RS-) лигандов, которые,
ла [(RS-)2 Fe+(NO+)2]:
-
-
-
-
-
HNO
+1/2 (N O
+
H
O)
2
2
–
-
-
-
-
H+
Схема 2. Предполагаемая схема образования моноядерных ДНКЖ с тиолсодержащими лигандами в реакции
двухвалентного железа, тиолов и газообразного NO [8, 13, 15-19].
Именно эти комплексы впервые были обнару-
живых организмах, М-ДНКЖ с тиолсодержащи-
жены нашей группой сначала в дрожжевых клет-
ми лигандами были обнаружены во многих био-
ках (1964-1965 гг.) [20, 21], а затем в тканях жи-
системах, способных продуцировать оксид азота
вотных и птиц (1967 г.) [22] по характерному для
[25-36].
этих комплексов сигналу электронного парамаг-
Результаты многолетних исследований приро-
нитного резонанса (ЭПР) со средним значением
ды и биологической активности этих комплексов
g-фактора, равным 2.03 (сигнала 2.03), приведен-
позволили нам в последние годы предположить,
ному на рис. 1. В 1964 г. слабый пик при g = 2.03
что ДНКЖ с тиолсодержащими лигандами (и не
был зарегистрирован Д. Мэллардом и М. Кентом
только М-ДНКЖ, но и обнаруженная позже в
в химически индуцированной гепатоме крыс [23],
биосистемах биядерная форма этих комплексов -
а в 1965 г. такой же сигнал (точнее, только его
часть - пик при g⊥ = 2.035) был зарегистрирован
Б-ДНКЖ, формула [(RS-)2Fe2+(NO+)4]) могут
Б. Коммонером с сотрудниками в печени крыс
функционировать в живых организмах в качестве
при инициировании у них гепатоканцрогенеза
«рабочей формы» оксида азота - они обеспечива-
[24]. В 1990-е годы, когда оксид азота был при-
ют стабилизацию и доставку NO, а также его ка-
знан в качестве одного из эндогенных универ-
тионной формы - ионов нитрозония - к мише-
сальных регуляторов метаболических процессов в
ням их биологического действия [18, 19].
БИОФИЗИКА том 65
№ 3
2020
424
ВАНИН
(а)
(б)
(д)
g =
2.03
g = 2.004
g = 2.003
g =
2.03
9
g = 2.004
g = 2.0
g =
2.03
1
(в)
(г)
g = 2.037
2.0
Сутки
7
Грудная
мышца
2
голубя
14
Печень
21
кролика
3
35
4
49
g=2.005
g=2.035
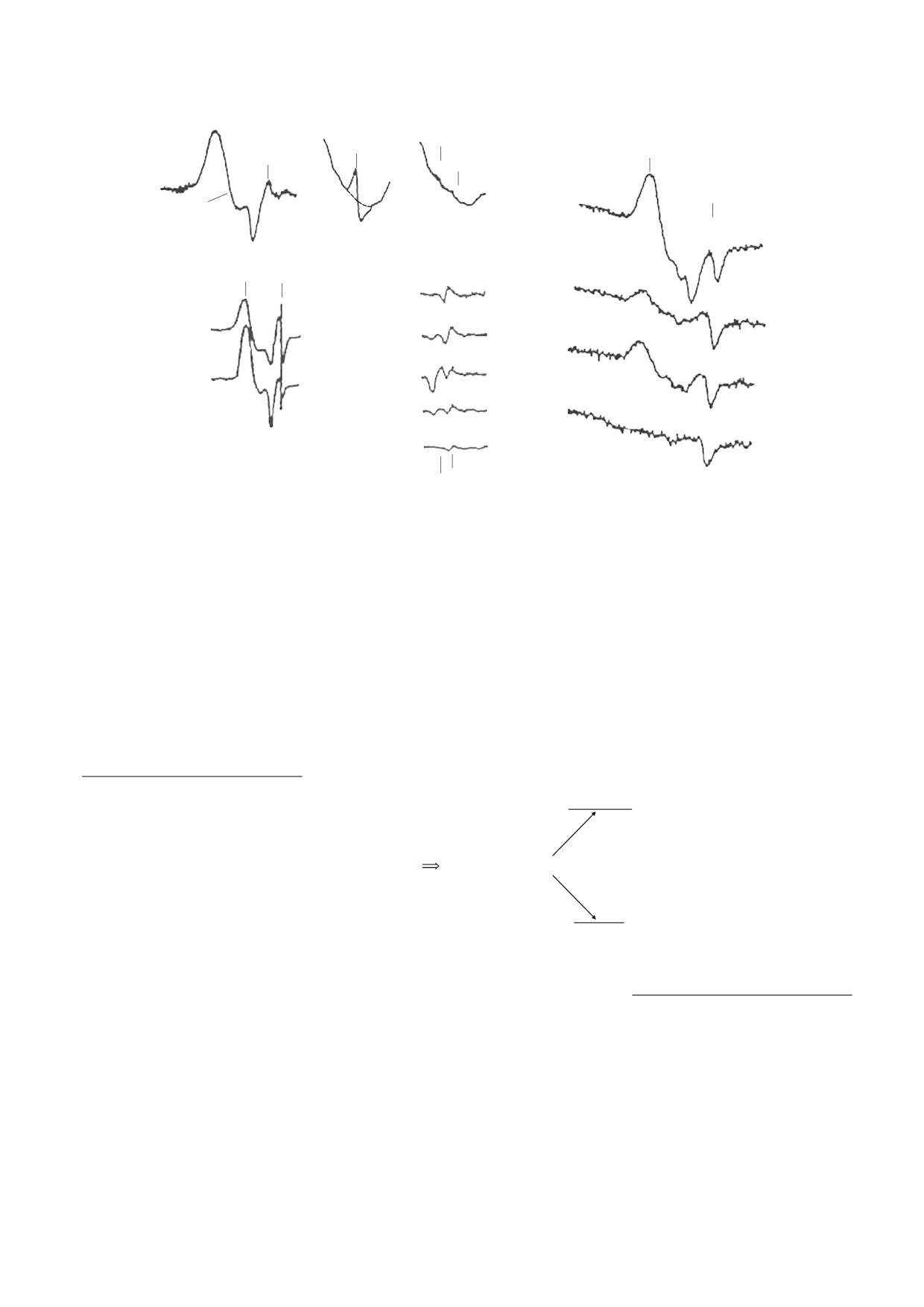
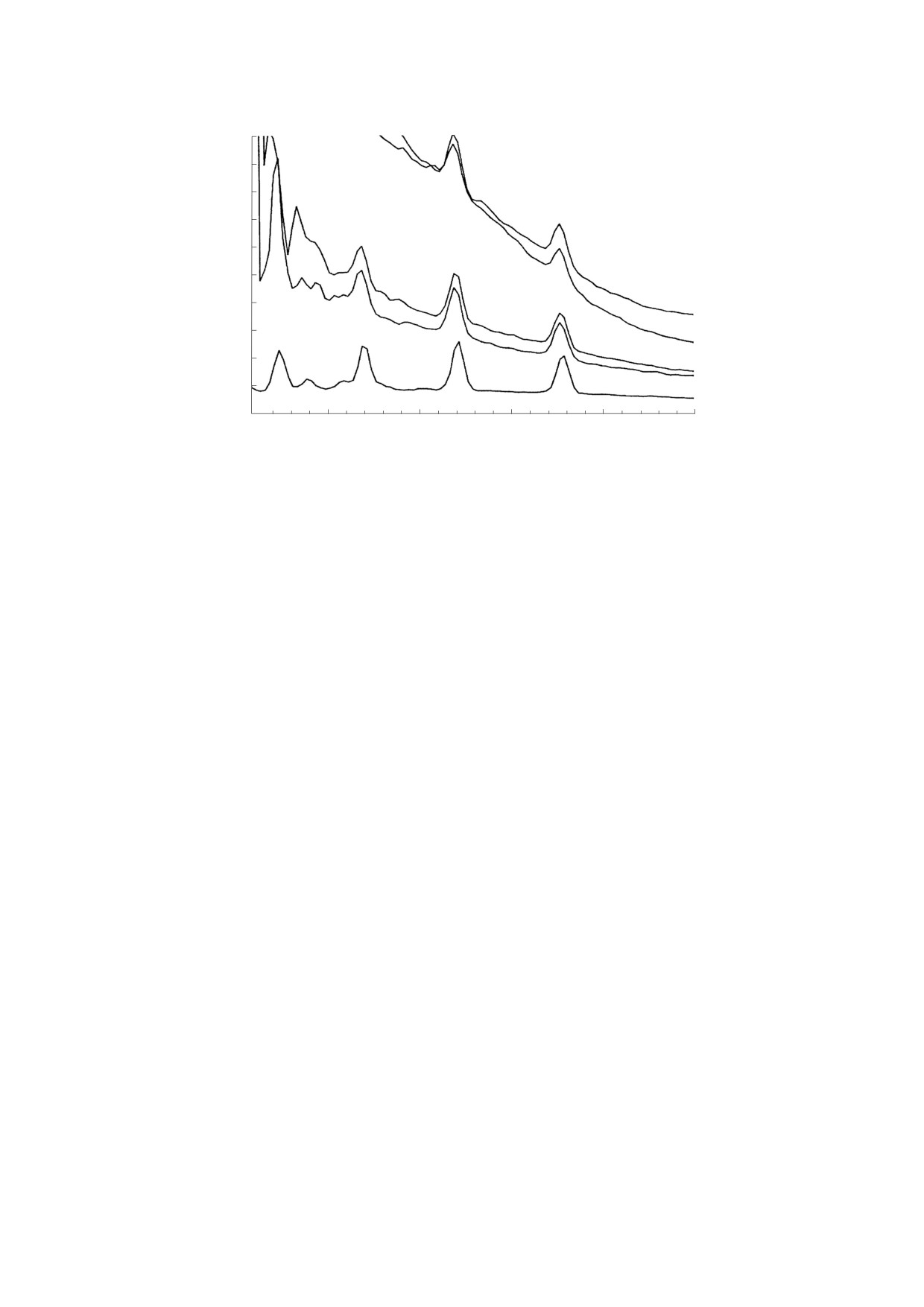
Рис. 1. Сигнал с g = 2.03, впервые зарегистрированный в 1960-е годы: (а) - в дрожжевых клетках [20, 21]; (б) - в
химически индуцированной гепатоме крыс [23]; (в) - в тканях голубей и кроликов [22]; (г) - в печени крыс на 1-
45 сутки после введения в их диету гепатоканцерогенов [24]. (д) - Сигнал с g = 2.03, зарегистрированный в 1990-е годы
в активированных макрофагах мышей в присутствии cубстрата NO-синтаз L-аргинина (1), L-аргинина и ингибитора
NO-синтаз N-метил-L-аргинина (2), в отсутствие L-arg и NMMA (3) или в присутствии только NMMA (4) [25].
Цель настоящей публикации состояла в выяс-
вать образование S-нитрозотиолов, а при блокаде
нении поставленного ранее в работах автора [8,
тиолов соответствующими реагентами или их
13] вопроса о способности ДНКЖ с тиолсодержа-
окислении - образование нитрита. При этом в
щими лигандами выступать в качестве доноров не
соответствии со схемой 3 уровень S-нитрозотио-
только нейтральных молекул NO, но и ионов нит-
лов или нитритов должен быть быть равен коли-
розония. Другими словами, могут ли эти ком-
плексы сами по себе, как это показано на схеме 3,
честву высвобождающихся при этом нейтраль-
при их распаде в присутствии тиолов иницииро-
ных молекул NO.
H +NO+-
2
OH-
–
2+
+
2
(RS ) Fe+(NO+)
2
Fe
+ NO + NO
RS-
RS-NO
Схема 3. Предполагаемая схема распада М-ДНКЖ с тиолсодержащими лигандами, приводящего при наличии
или отсутствии (блокаде) тиолов в растворе соответственно к накоплению S-нитрозотиолов или нитрита [8, 13].
Проведенные ранее на 0.5 мМ растворах Б-
гандов кислородом воздуха, либо связыванием
ДНКЖ с глутатионом (Б-ДНКЖ-GSH) экспери-
этих групп с ртутьсодержащим агентом - пара-
менты, казалось бы, полностью подтвердили это
хлормеркурийбензоатом - в последних двух опы-
предположение [8, 13]. Распад комплексов в этих
тах при нейтральных значениях рН раствора.
работах был инициирован либо протонировани-
Предполагалось, что в обоих случаях Б-ДНКЖ
ем тиолсодержащих лигандов, достигавшимся
сначала распадались на моноядерные формы, ко-
прогреванием растворов Б-ДНКЖ в сильно кис-
торые затем начинали распадаться на составляю-
лой среде, окислением тиоловых групп этих ли-
щие их компоненты, как это показано на схеме 3.
БИОФИЗИКА том 65
№ 3
2020
СВОБОДНО-РАДИКАЛЬНАЯ ПРИРОДА МОЛЕКУЛ МОНООКСИДА АЗОТА
425
При этом половина высвобождающихся в кис-
лизоваться при участии только ионов Fe2+ или
лой среде нитрозильных лигандов (как в аэроб-
эти ионы выступали в качестве промежуточных
ных, так и в анаэробных условиях) обнаружива-
участников - катализаторов реакции восстанов-
лась в форме NO+ в составе S-нитрозоглутатиона
ления катионов нитрозония глутатионом как
(GS-NO), тогда как при распаде ДНКЖ в раство-
представителем тиолсодержащих соединений -
ре при нейтральных значениях рН также только
этот вопрос в наших предыдущих работах [8, 13]
половина нитрозильных лигандов превращалась
не изучался.
в анионы нитрита (очевидно, в результате гидро-
В настоящей работе сделана попытка преодо-
лиза высвобождающихся из ДНКЖ ионов нитро-
леть этот недостаток и тем самым окончательно
зония).
ответить на вопрос, действительно ли ДНКЖ с
Что касается другой половины нитрозильных
тиолсодержащими лигандами могут функциони-
лигандов, то в соответствии со схемой 3 она долж-
ровать в живых организмах не только в качестве
на была высвобождаться из Б-ДНКЖ в форме га-
доноров газообразных (нейтральных) молекул
зообразных (летучих) молекул NO. Однако оцен-
NO, но и катионов нитрозония.
ка уровня этого газа, высвобождавшегося в тече-
ние часа в анаэробных условиях из подкисленных
12 мМ растворов Б-ДНКЖ с глутатионом при их
МАТЕРИАЛЫ И МЕТОДЫ
прогреве, показала, что оба нитрозильных лиган-
да, входивших в состав железо-динитрозильного
Материалы. В экспериментах были использо-
мотива, переходили в форму нейтральных моле-
ваны ферросульфат (Fluka, Швейцария), восста-
кул NO. Ни GS-NO, ни нитрит в растворах распа-
новленный глутатион, N-ацетил-L-цистеин
дающихся ДНКЖ в этих опытах не обнаружи-
(NAC), неокупроин и нитрит натрия (все произ-
вались.
водства компании Sigma, США). Газообразный
Для объяснения полученного несоответствия
NO был получен в реакции ферросульфата и нит-
со схемой 3 было предположено, что GS-NO, ко-
рита натрия в 0.1 M HCl с последующей очисткой
торый мог возникать в этих условиях, распадался
NO от примеси NO2 методом низкотемператур-
при каталитическом действии на него примесной
ной сублимации в вакуумированном сосуде [17].
меди, уровень которой мог повышаться, напри-
мер, в результате использования при синтезе Б-
Синтез Б-ДНКЖ и М-ДНКЖ с глутатионом
ДНКЖ-GSH в сравнительно высокой (12 мМ)
или N-ацетил-L-цистеином. При синтезе
концентрации ферросульфата (до 24 мМ) [8, 13].
ДНКЖ с глутатионом (ДНКЖ-GSH) или N-
Возможен был и другой путь превращения всех
ацетил-L-цистеином (ДНКЖ-NAC) как пред-
нитрозильных лигандов при распаде Б-ДНКЖ-
ставителями тиолсодержащих лигандов ис-
GSH в нейтральные молекулы NO. Он мог реали-
пользовалась способность S-нитрозотиолов
зоваться в результате одноэлектронного восста-
продуцировать ДНКЖ с тиолсодержащими
новления катионов нитрозония ионами Fe2+, вы-
(RS-) лигандами в реакции двух молекул RS-
свобождающимися в соответствии со схемой 3 из
NO c ионом двухвалентного железа и двумя мо-
Б-ДНКЖ. Мог ли этот процесс эффективно реа-
лекулами тиолов (схема 4):
–
-
–
•
-
-
+ RS
-
RS +•SR
-
–
-
–
-
+ RS-
Схема 4. Предполагаемый механизм образования М-ДНКЖ с тиолсодержащими лигандами в реакции между
Fe2+-ионами, тиолами (RS-) и S-нитрозотиолами (RS-NO+) [37].
В соответствии со схемой 3 при связывании с
следующего обратимого превращения М-ДНКЖ
Fe2+ ионом двух разных резонансных структур
в биядерную Б-ДНКЖ-форму, происходящего в
соответствии со схемой
5, то, поскольку
RS-NO/NO•+-RS• и NO+-RS- перенос электро-
тиолы включаются в эти комплексы в ионизован-
на от NO• в первой из них на ион нитрозония в
ной RS--форме, из-за низкого содержания по-
другой структуре может сразу же из-за распада S-
следней при нейтральных значениях рН подавля-
нитрозотиольных лигандов приводить к форми-
ющая часть М-ДНКЖ, в частности М-ДНКЖ-
рованию
железодинитрозильного
[(RS-)2
GSH, в этих условиях должна димеризоваться в
Fe+(NO+)2]-мотива М-ДНКЖ. Что касается по-Б-ДНКЖ:
БИОФИЗИКА том 65
№ 3
2020
426
ВАНИН
D
2.5
g = 2.04
g = 2.014
1
×15
2.0
1.5
2
×1
1
2
3
323
328
333
1.0
Длина волны, нм
0.5
0.0
200
300
400
500
600
700
Длина волны, нм
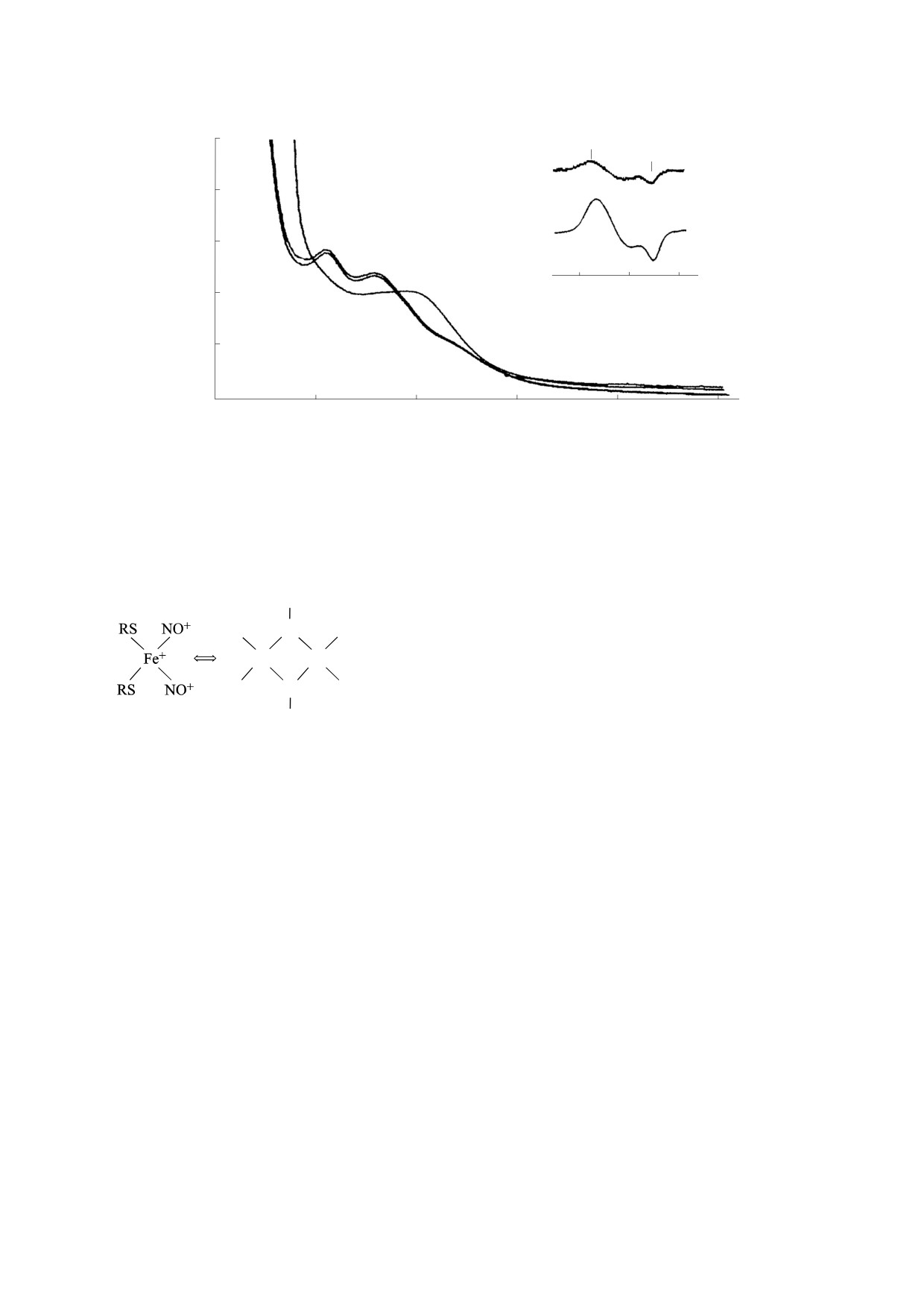
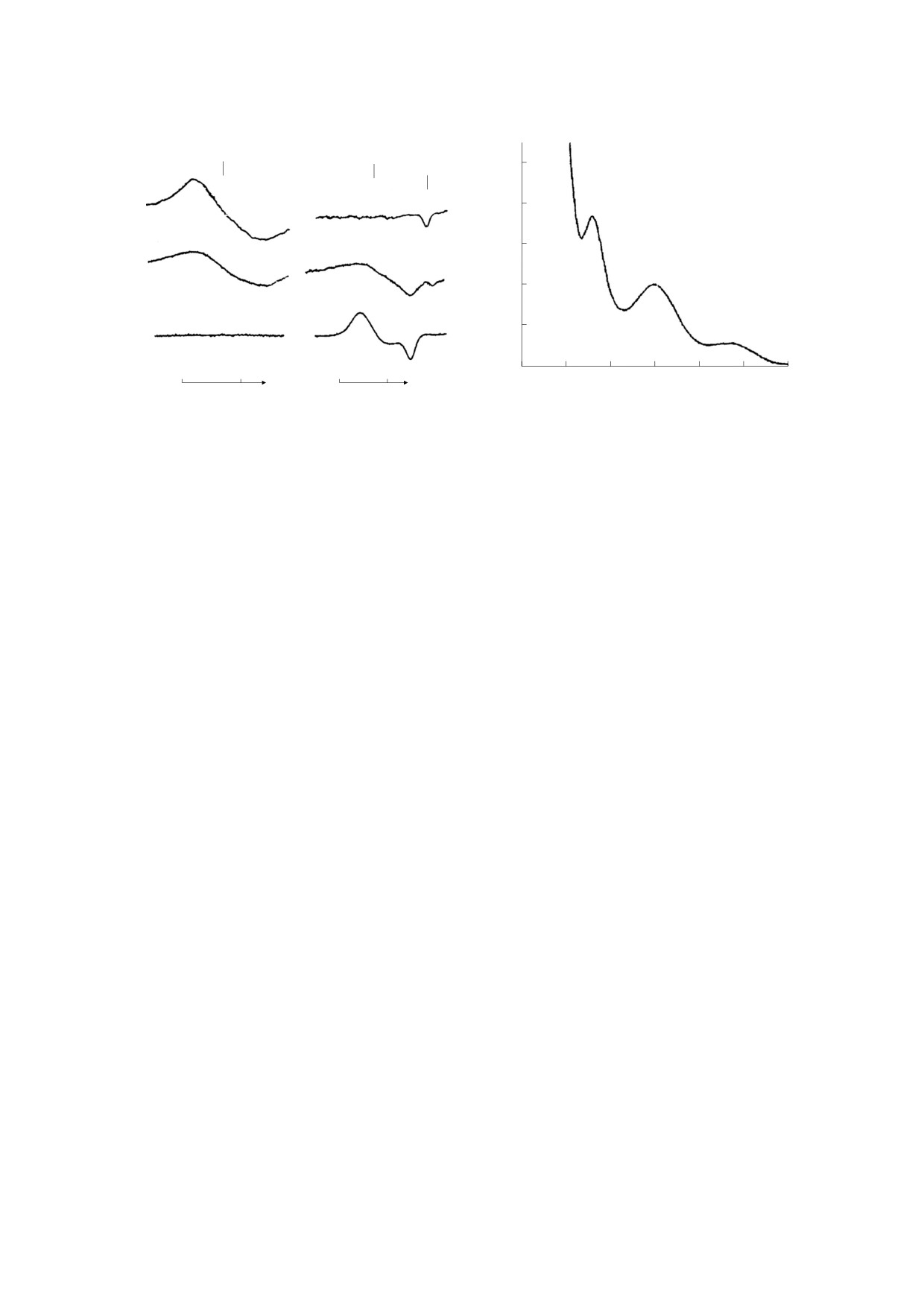
Рис. 2. Спектры оптического поглощения Б-ДНКЖ-GSH, зарегистрированные в водном растворе при рН 7.4
и 1.0 (кривые 1 и 2 соответственно). Кривая 3 - спектр поглощения М-ДНКЖ-GSH, зарегистрированный в растворе
Б-ДНКЖ-GSH в присутствии трехкратного избытка глутатиона и рН 11.0. На врезке - сигналы с g = 2.03 (кривые 1
и 2), зарегистрированные при 77 К в растворах Б-ДНКЖ-GSH, характеризующихся спектрами оптического
поглощения, представленными соответственно кривыми 1 и 3. Справа у кривых на врезке указано усиление
радиоспектрометра в отн. ед. Точно такие же спектры оптического поглощения и сигнал с g = 2.03 характерны
соответственно для Б-ДНКЖ-NAC и М-ДНКЖ-NAC.
R
раствора был повышен до нейтральных значений
(до рН 7.3-7.5) добавлением нескольких капель
-
+
-
+
NO
S
NO
100 мМ раствора NaOH, в результате чего розовая
2
Fe+
Fe+
+ 2RS-
окраска раствора, характерная для GS-NO, сме-
нялась в течение пяти-шести часов на темно-ко-
-
+
-
+
NO
S
NO
ричневую, характерную для образующегося Б-
ДНКЖ-GSH.
R
Как следует из схемы 4, при равных концен-
Схема 5. Обратимое взаимопревращение М-ДНКЖ и
Б-ДНКЖ с тиолсодержащими лигандами [17].
трациях Fe2+ и GS-NO (10 мМ) последний полно-
В соответствии с ранее разработанным нашей
стью включался в Fe2+-динитрозильный мотив
группой протоколом синтеза Б-ДНКЖ на основе
Б-ДНКЖ, что существенно снижало риск загряз-
схемы 4 [38], синтез Б-ДНКЖ-GSH был осу-
нения раствора нитритом, не включившимся в Б-
ществлен следующим образом. К 10 мл 15 мМ
ДНКЖ («свободным нитритом»). Что касается
HEPES-буфера при рН 7.4 последовательно до-
Fe2+-ионов, то только 50% из них (5 из 10 мМ)
бавлялись 20 или 15 мМ глутатиона, 10 мМ фер-
включались при нейтральных значениях рН в об-
росульфата и 10 мМ нитрита натрия. Понижение
разующиеся Б-ДНКЖ. Другая половина осажда-
рН раствора HEPES после добавления в него глу-
лась в форме железогидроокисных комплексов, и
татиона (до рН 3.0-3.5) обеспечивало полное рас-
ее можно было полностью удалить из раствора его
творение добавляемого ферросульфата, а
пропусканием через бумажный фильтр. Получен-
также полное превращение нитрита в GS-NO,
ные таким образом растворы Б-ДНКЖ-GSH за-
образующийся в реакции протонированного нит-
мораживали в жидком азоте и хранили в холо-
рита с глутатионом. Об эффективности образова-
дильнике при -18°С с последующим разморажи-
ния GS-NO можно было судить по кинетике на-
ванием перед использованием в экспериментах.
растания полосы оптического поглощения
этого соединения на длине волны 334 нм, а также
Что касается глутатиона, содержание которого
по более слабой полосе на 546 нм с коэффициен-
при синтезе Б-ДНКЖ в полтора или два раза пре-
тами экстинкции, равными соответственно
вышает содержание железа, соответствующая его
0.94 M-1см-1 и 0.017 M-1см-1 [38].
часть - 10 или 5 мМ - не включалась в Б-ДНКЖ,
По окончании образования GS-NO, происхо-
т.е. сохранялась в свободном состоянии. Количе-
дившего в течение одного-полутора часов, рН ство тиола, включившегося в эти комплексы, бы-
БИОФИЗИКА том 65
№ 3
2020
СВОБОДНО-РАДИКАЛЬНАЯ ПРИРОДА МОЛЕКУЛ МОНООКСИДА АЗОТА
427
(а)
(б)
(в)
D
1.5
0.7
2
1
-0.1
190
245
300
Длина волны, нм
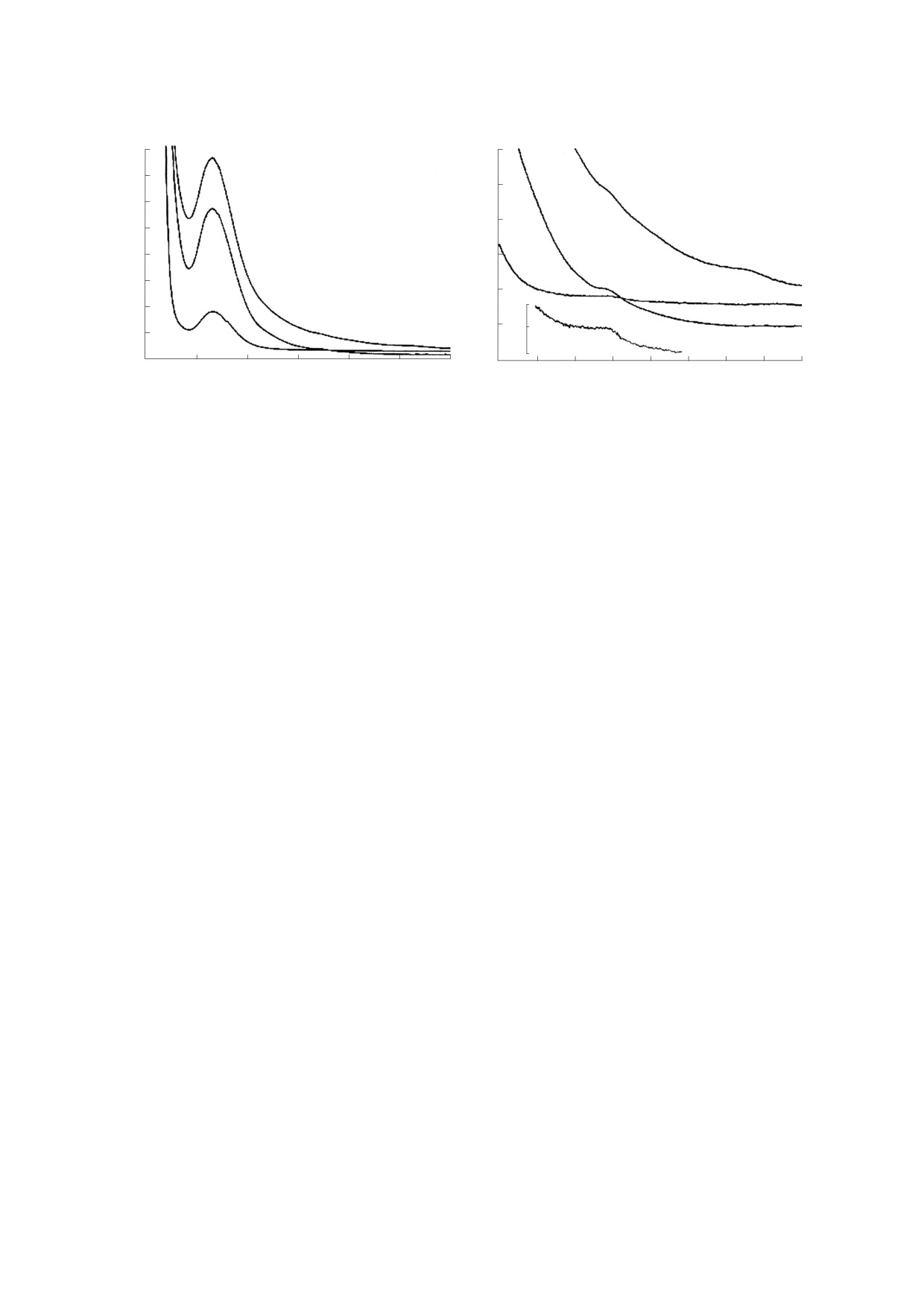
Рис. 3. Схемы модифицированных аппаратов Тунберга, использовавшихся для получения ДНКЖ с лигандами
нетиоловой природы (а) или для измерения оптического поглощения NO и NO2 в газовой фазе (б); (в) - спектр
оптического поглощения газообразного NO (четыре эквидистантных узких полосы, кривая 1) и суммарное
оптическое поглощение газовой смеси NO + NO2 (кривая 2).
ло равно в соответствии со схемой 4 количеству
спектр поглощения раствора трансформировался
включенного в них железа.
и становился характерным для спектра М-ДН-
Концентрация полученных Б-ДНКЖ-GSH
КЖ-GSH с полосой на 390 нм с ε = 4700 М-1см-1
[17].
была определена оптическим методом по интен-
сивности характерных для этих комплексов по-
Аналогичным образом были синтезированы
лос поглощения на 310 и 360 нм в спектре погло-
Б-ДНКЖ-NАС. Различие состояло лишь в том,
щения, приведенном на рис. 2, с коэффициента-
что синтез S-нитрозо-N-ацетил_L-цистеина (S-
ми экстинкции ε, равными соответственно 4600 и
NAC), необходимого для образования этих ком-
3700 М-1см-1 (в пересчете на один атом железа)
плексов, заканчивался через 20-25 мин после до-
[17, 38]. Концентрация комплексов, полученных
бавления в соответствующий раствор нитрита, а
в этих условиях, составляла 4.5-5.0 мМ. Таким
синтез Б-ДНКЖ-NАС, начинавшийся после
образом, при использовании 20 или 15 мМ глута-
подщелачивания раствора S-NAC и ферросуль-
тиона при синтезе Б-ДНКЖ-GSH отношение со-
фата, заканчивался через три-четыре часа. Соот-
держания свободного глутатиона к содержанию
ношение NAC и железа, использованных при
комплексов составляло соответственно 2 : 1 или
синтезе Б-ДНКЖ-NAC, составляло 3 : 1 (30 и
1 : 1.
10 мМ) или 1.5 : 1 (15 и 10 мМ). Концентрация
нитрита натрия составляла 10 мМ. Отношение
Что касается моноядерной формы ДНКЖ-
содержания свободного NAC к содержанию ком-
GSH, о ее наличии в растворе можно было судить
плексов, синтезированных при 30 и 15 мМ NAC и
по сигналу 2.03, приведенному на врезке на
10 мМ железа, составляло соответственно 4 : 1 и
рис. 2, со следующими значениями g-фактора:
1 : 1.
g⊥ = 2.04, g = 2.014, gср = 2.03. Интенсивность
этого сигнала при нейтральных значениях рН со-
Синтез ДНКЖ с лигандами нетиоловой приро-
ответствовала включению в М-ДНКЖ не более
ды. Синтез М-ДНКЖ с лигандами нетиоловой
5% от общего содержания железа, включавшегося
природы - водой, фосфатом и этилендиаминтет-
суммарно в обе формы ДНКЖ. При повышении
раацетатом (ЭДТА) - проводили в модифициро-
до 10-11 рН раствора, в который при синтезе
ванном аппарате Тунберга, схема которого пред-
ДНКЖ вводили 20-30 мМ глутатиона, интенсив-
ставлена на рис. 3а, в соответствии с описанным
ность сигнала 2.03 повышалась в 20-30 раз
ранее подходом [17]. Процедура синтеза состояла
(рис. 2, врезка), так что все железо в растворе об-
в следующем. В верхнюю часть аппарата помеща-
наруживалось в составе М-ДНКЖ. При этом
ли 0.5 мл дистиллированной воды, а в нижнюю
БИОФИЗИКА том 65
№ 3
2020
428
ВАНИН
часть - 4.5 мл 30 мМ HEPES-буфера при рН 7.3-
тализаторов этого процесса. Естественно, что для
7.5 без каких-либо добавок или содержавшего
проверки этой возможности необходимо было
100 мМ фосфата или ЭДТА. После пяти-десяти-
оценить эффективность образования GS-NO при
минутного выдерживания этой системы в вакуу-
прогреве подкисленных растворов Б-ДНКЖ-
ме, необходимого для удаления воздуха из воды, в
GSH в анаэробных условиях в зависимости от
верхнюю часть аппарата (в дистиллят) вводили
уровня свободного (не включенного в ДНКЖ)
ферросульфат железа, снова откачивали воздух из
глутатиона в растворе. Ожидалось, что содержа-
аппарата и затем вводили в него газообразный
ние GS-NO должно было повышаться по мере
NO под давлением 100-150 мм рт. ст. на установ-
снижения этого уровня до минимального (нуле-
ке, описанной в работе [39]. При последующем
вого) значения. Такая ситуация могла, очевидно,
пяти-десятиминутном встряхивании аппарата
реализоваться, если при синтезе Б-ДНКЖ глута-
раствор ферросульфата в верхней его части при-
тион и железо вводили бы в равном количестве.
обретал зеленую окраску, обусловленную образо-
Однако при таком соотношении глутатиона и
ферросульфата оказалось невозможным синтези-
ванием монотирозильного комплекса Fe2+; далее
ровать Б-ДНКЖ-GSH в концентрации, равной
этот раствор смешивался с раствором, помещен-
ным в нижней части аппарата. После десятими-
концентрации этих комплексов при использова-
нии при их синтезе более высокого соотношения
нутного встряхивания аппарат откачивали от NO,
количества глутатиона и ферросульфата, напри-
а полученный раствор замораживали в жидком
мер при соотношениях 2 : 1 или 3 : 1. Такое равен-
азоте для последующего изучения оптическими и
ство удалось достигнуть только при соотношении
ЭПР-методами.
глутатиона и ферросульфата при синтезе Б-ДН-
Регистрация спектра оптического поглощения
КЖ-GSH, равном 1.5 : 1.0, например при концен-
газообразного NO. Для регистрации спектра опти-
трациях глутатиона и ферросульфата, равных 15 и
ческого поглощения газообразного NO, выделяв-
10 мМ, и концентрации нитрита, равной 10 мМ.
шегося из Б-ДНКЖ-GSH при его распаде, ис-
Судя по спектру оптического поглощения раство-
пользовали модифицированный аппарат Тунбер-
ра полученных таким образом комплексов
га (схема представлена на рис. 3б), содержавший
(рис. 4, кривая 2), их концентрация была близка к
припаянную к нему кювету цилиндрической
концентрации Б-ДНКЖ-GSH, синтезированных
формы с кварцевыми торцевыми стенками. Ап-
при соотношении глутатиона и ферросульфата,
парат помещали в спектрофотометр таким обра-
равном 2 : 1, (рис. 4, кривая 1). В обоих случаях
зом, чтобы световой луч в нем проходил вдоль оси
растворы перед их записью на спектрофотометре
кюветы. Характерный спектр регистрируемого
разбавляли в десять раз.
при этом оптического поглощения газообразного
NO и примесного газообразного NO2 показаны
При последующем прогреве при 80°С в анаэ-
на рис. 3в. Спектр поглощения газообразного NO
робных условиях этих двух неразбавленных под-
характеризовался четырьмя эквидистантными
кисленных растворов Б-ДНКЖ-GSH наблюда-
лось полное исчезновение исходных спектров по-
узкими полосами поглощения в области 190-
глощения и появление спектра поглощения,
200 нм, тогда как газообразный NO2 давал в этой
характерного для GS-NO (рис. 4, кривые 3-5), с
области широкую бесструктурную полосу погло-
интенсивной полосой на 334 нм и слабой полосой
щения [38, 40]. Вакуумная сублимация смеси
на 543 нм, о которых было сказано в методиче-
NO + NO2 приводила в результате удаления NO2
ской части. При этом, судя по интенсивности
к полному исчезновению бесструктурного опти-
этих полос, концентрация GS-NO, образующих-
ческого поглощения этого газа.
ся в растворах Б-ДНКЖ-GSH с низким содержа-
Для оценки количества газообразного NO, вы-
нием глутатиона, соответствовала (в полном со-
свобождавшегося из Б-ДНКЖ-GSH, использо-
ответствии со схемой 3) включению в GS-NO по-
вали стандартный образец газообразного NO при
ловины нитрозильных лигандов, входивших в
давлении, определявшемся ртутным манометром
состав Б-ДНКЖ (рис. 4, кривые 4 и 5), что суще-
на вакуумной установке, описанной нами в рабо-
ственно превышало уровень GS-NO, возникших
те [39].
в растворах растворах Б-ДНКЖ-GSH с более вы-
соким содержанием глутатиона (рис. 4, кривая 3).
Очевидно, именно последнее, в соответствии с
РЕЗУЛЬТАТЫ
высказанным выше предположением, обеспечи-
S-Нитрозирующее действие Б-ДНКЖ-GSH и
вало превращение подавляющей катионов нит-
Б-ДНКЖ-NAC на включенные в них тиолсодержа-
розония не в GS-NO, а в NO так, как это могло
щие лиганды. Как указывалось во введении, обра-
иметь в наших ранних опытах [8, 13], о которых
зование GS-NO при распаде Б-ДНКЖ-GSH мог-
говорилось выше. При этом следует отметить не-
ло ослабляться в результате одноэлектронного
большой сдвиг (на несколько нм) центра погло-
восстановления катионов нитрозония молекула-
щения основной полосы GS-NO на рис. 4 с 334
ми глутатиона при участии ионов железа как ка-
нм в коротковолновую область. Этот сдвиг был
БИОФИЗИКА том 65
№ 3
2020
СВОБОДНО-РАДИКАЛЬНАЯ ПРИРОДА МОЛЕКУЛ МОНООКСИДА АЗОТА
429
D
D
0.3
3.0
4
4
3
0.2
5
2.0
1
1
0.1
2
2
3
400
500
600
1.0
Длина волны, нм
5
0.0
300
400
500
600
Длина волны, нм
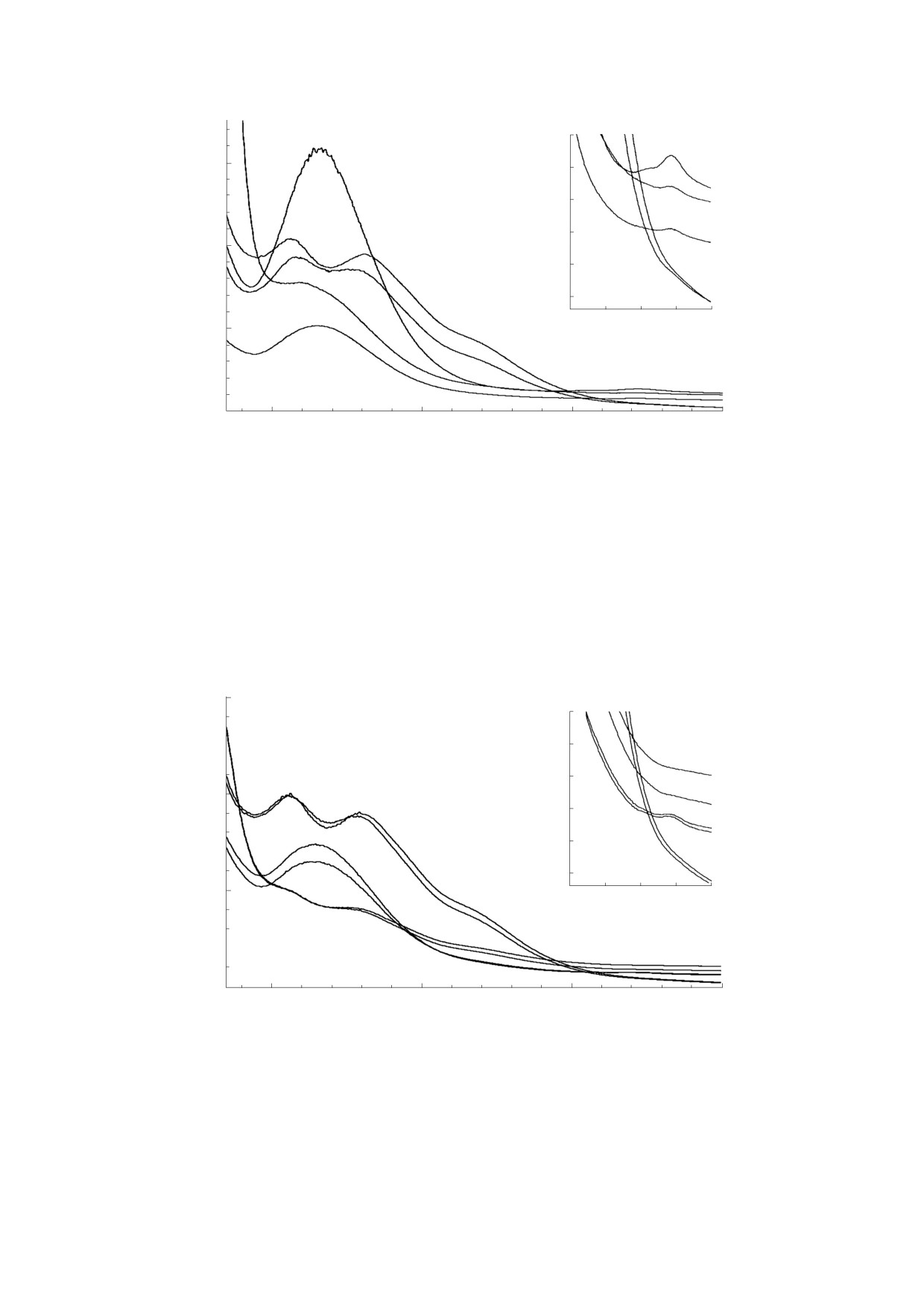
Рис. 4. Спектры поглощения растворов Б-ДНКЖ-GSH, синтезированных при соотношениях концентраций
глутатиона и ферросульфата (нитрита), равных 2 : 1 (20 мМ и 10 мМ, кривая 1) или 1,5 : 1 (15 мМ и 10 мМ, кривая 2).
Растворы разбавлены в десять раз. Кривые 3 и 4 - характерные для GS-NO спектры поглощения растворов тех же
комплексов, подвергнутых нагреванию при 80°С в анаэробных условиях в течение восьми-девяти минут без
разбавления растворов. Спектры характеризуются слегка сдвинутой в коротковолновую область полосой поглощения
на 334 нм и слабой полосой на 543 нм. Кривая 5 - спектр поглощения раствора, характеризующегося кривой 4, после
разбавления этого раствора в четыре раза. На врезке приведены спектры поглощения всех этих растворов,
зарегистрированные при более высокой чувствительности спектрометра в области полосы поглощения GS-NO на
543 нм.
D
3.0
D
0.3
3
0.2
4
2.0
5
2
6
1
2
0.1
5
1
6
1.0
400
500
600
Длина волны, нм
3,4
0.0
300
400
500
600
Длина волны, нм
Рис. 5. Спектры поглощения растворов Б-ДНКЖ-NAC, синтезированных при соотношениях концентраций NAC и
ферросульфата, равных 3 : 1 (30 мМ и 10 мМ, кривая 1) или 1,5 : 1 (15 мМ и 10 мМ, кривая 2). Растворы разбавлены в
десять раз. Кривые 3, 4 и 5, 6 - соответственно спектры поглощения растворов тех же комплексов, разбавленных в два
раза и подвергнутых нагреванию при 800С в анаэробных условиях в течение семи-девяти или двух-трех минут.
Спектры 5 и 6 с полосами поглощения на 334 и 543 нм обусловлены образующимся S-нитрозо-N-ацетил-L-
цистеином. На врезке приведены спектры поглощения всех этих растворов, зарегистрированные при более высокой
чувствительности спектрометра в области полосы поглощения S-нитрозо-N-ацетил-L-цистеина на 543 нм.
БИОФИЗИКА том 65
№ 3
2020
430
ВАНИН
обусловлен оптическим поглощением ионов
ионов меди - неокупроина [41], проведенные в
настоящей работе, показали, что связывание
Fe3+, сохранявшихся в растворе после синтеза
примесной меди этим хелатором не влияло на по-
комплексов.
явление соответствующих S-нитрозотиолов в
Такой же результат был получен в аналогич-
растворах Б-ДНКЖ при их распаде.
ных опытах на растворах Б-ДНКЖ-NAC, синте-
зированных при соотношениях концентраций
Таким образом, есть основание полагать, что
NAC : ферросульфат : нитрит натрия, равных со-
обнаруженное ранее в работах [8, 13] высвобож-
ответственно 30 : 10 : 10 мМ и 15 : 10 : 10 мМ
дение нитрозильных лигандов из Б-ДНКЖ-GSH
(рис. 5). В этих опытах прогрев раствора Б-ДН-
в подкисленных растворах, прогревавшихся
КЖ-NAC-комплексов, синтезированных при
при 80°С в анаэробных условиях, только в
трехкратном избытке глутатиона по сравнению с
форме нейтральных молекул NO было обусловле-
концентрацией ферросульфата, приводил к вос-
но восстановительным действием глутатиона на
становлению до NO всех катионов нитрозония,
высвобождающиеся из этих комплексов катионы
высвобождавшихся из Б-ДНКЖ-NAC при про-
нитрозония. Этот эффект мог сниматься в ре-
греве их подкисленных растворов в анаэробных
зультате окисления избытка глутатиона в раство-
условиях. Об этом свидетельствует отсутствие да-
рах Б-ДНКЖ-GSH кислородом при выдержива-
же следовых количеств S-нитрозо-N-ацетил-L-
ние этих растворов, что, по-видимому, имело ме-
цистеина в спектрах поглощения раствора, пред-
сто в наших экспериментах, описанных в работах
ставленных кривыми 3 и 4.
[8, 13].
Что касается количества S-нитрозо-N-ацетил-
В связи с этим не исключено, что упоминав-
L-цистеина, образовавшегося при распаде Б-
шееся выше образование S-нитрозотиолов
ДНКЖ-NAC, синтезированных при соотноше-
при прогреве подкисленных 0.5 мМ растворов
нии NAC : ферросульфат (нитрит), равным 1.5 : 1,
Б-ДНКЖ при 80°С на воздухе вне зависимости от
оно составляло не более 30% от общего количе-
соотношения тиолов и железа в растворах этих
ства нитрозильных лигандов, входивших в состав
комплексов могло быть обусловлено быстрым
Б-ДНКЖ-NAC. Не исключено, что образование
окислением избытка тиолов кислородом воздуха
S-нитрозо-N-ацетил-L-цистеина при распаде
в ходе повышения температуры растворов от ком-
этих комплексов в анаэробных условиях частич-
натной температуры до 80С.
но подавлялось воcстановительным действием
Образование нитрита при распаде Б-ДНКЖ-
NAC на катионы нитрозония даже при сравни-
GSH в анаэробных условиях под действием ферри-
тельно малом содержании этого тиолсодержаще-
цианида калия. В соответствии с предложенным
го соединения. Как следует из вышеизложенного,
на схеме 3 с механизмом распада ДНКЖ с тиолсо-
такой эффект не обнаруживался при распаде ана-
держащими лигандами, например, в результате
логичных комплексов Б-ДНКЖ-GSH, сопро-
блокады тиоловых групп соответствующими реа-
вождашемся включением в GS-NO половины
гентами или их окисления, вместо S-нитрозотио-
нитрозильных лигандов в этих комплексах. Не
лов в растворах Б-ДНКЖ должны были при ней-
исключено, что это различие было обусловлено
тральных значениях рН накапливаться анионы
более высокой восстановительной активностью
нитрита, причем в количестве, равном в моляр-
NAC по сравнению с аналогичной характеристи-
ном выражении количеству высвобождающихся
кой глутатиона.
из Б-ДНКЖ нейтральных молекул NO. Такого
При прогреве подкисленных 0.5 мМ растворов
рода опыты с окислением тиоловых групп в Б-
всех этих комплексов при 80°С на воздухе не об-
ДНКЖ-GSH на воздухе показали, что действи-
наруживалось какого-либо различия в их поведе-
тельно нитрит, появляющийся в растворе, в соот-
нии. Все они, судя по исчезновению окраски,
ветствии со схемой 3 включал в себя ровно поло-
распадались в течение 0.5-1.0 минуты с образова-
вину нитрозильных лигандов, входивших в состав
нием соответствующих S-нитрозотиолов в кон-
Б-ДНКЖ [8, 13]. Однако в экспериментах с бло-
центрации, соответствующей включению в них
кадой тиоловых групп в этих комплексах пара-
половины нитрозильных лигандов (или равной
хлормеркурийбензоатом в анаэробных условиях
концентрации комплексов, рассчитанной на
нитрит не обнаруживался, а оптическим методом
один атом железа), как это было описано ранее в
регистрировался только газообразный NO в ко-
работах [8, 13].
личестве, соответствующем включению в NO 75-
Что касается возможного влияния примеси
80% нитрозильных лигандов Б-ДНКЖ [8, 13].
меди как агента, разрушающего S-нитрозотиолы
Было предположено, что нитрит в этих опытах
и тем самым возможно блокировавшего появле-
возникал, однако подкисление растворов Б-ДН-
ние GS-NO при распаде Б-ДНКЖ-GSH в анаэ-
КЖ в ходе их распада приводило к его превраще-
робных условиях, о чем было предположено в ра-
нию в азотистую кислоту, реакция диспропрцио-
ботах [8, 13], эксперименты с добавлением в раз-
нирования которой приводила к накоплению в
ных концентрациях сильного хелатора
газовой фазе дополнительного количества NO
БИОФИЗИКА том 65
№ 3
2020
СВОБОДНО-РАДИКАЛЬНАЯ ПРИРОДА МОЛЕКУЛ МОНООКСИДА АЗОТА
431
D
1.0
0.8
5
0.6
4
0.4
3
0.2
2
1
0.0
200
210
220
230
240
Длина волны, нм
Рис. 6. Изменение оптического спектра NO в газовой фазе, зарегистрированного в модифицированном аппарате
Тунберга, схема которого приведена на рис. 3б, в ходе распада 16 мл 8 мМ раствора Б-ДНКЖ-GSH в 200 мМ HEPES-
буфере при рН 7.4, вызванного добавлением к раствору феррицианида калия (FeCN) в конечной концентрации 80 мМ
с последующим подкислением раствора. Кривая 1 - спектр поглощения стандартного образца газообразного NO
(125 микромолей в 100 мл свободного объема аппарата Тунберга. Кривые 2 и 3 - четыре эквидистантные полосы
поглощения NO, зарегистрированные через 15 и 20 мин после добавления FeCN к раствору Б-ДНКЖ-GSH при
рН 7.4, нагретому до 60°С, в откачанном аппарате Тунберга. Кривые 4 и 5 - первые две (из четырех) эквидистантных
полос поглощения NO, зарегистрированные в повторно откачанном аппарате Тунберга через 15 и 20 мин после
подкисления раствора серной кислотой. Дрейф нулевой линии в спектрах обусловлен загрязнением торцевых стенок
цилиндрической кюветы аппарата Тунберга частицами берлинской лазури, образующейся в реакции FeCN c
двухвалентным железом, высвобождающимся из Б-ДНКЖ.
(наряду с выделяющимся из распадающихся Б-
твора газообразного NO, уровень которого в ап-
ДНКЖ).
парате оценивали по его спектру поглощения, ре-
гистрируемому в цилиндрической кювете, присо-
В связи с этим необходимо было показать, что
единенной к аппарату. Через 20 мин этот уровень
распад Б-ДНКЖ с тиолсодержащими лигандами
достигал плато, соответствующего высвобожде-
в анаэробных условиях, вызванный удалением
нию в форме NO половины нитрозильных лиган-
этих лигандов из координационной сферы желе-
дов, что было равно количеству Б-ДНКЖ-GSH
за, действительно при нейтральных значениях рН
в пересчете на один атом железа в этих комплек-
приводит к накоплению в растворе анионов нит-
сах - 128 микромолей в 100 мл свободного объема
рита, образующихся в результате гидролиза кати-
аппарата Тунберга, равного свободному объему
онов нитрозония, высвобождающихся из Б-ДН-
этого аппарата после введения в него 16 мл рас-
КЖ. Такого рода результат был получен в настоя-
твора Б-ДНКЖ-GSH.
щей работе при изучении распада Б-ДНКЖ-
GSH, вызванного в анаэробных условиях окисле-
После этого газообразный NO, высвободив-
нием тиоловой группы в глутатионе феррициани-
шийся из Б-ДНКЖ, откачивали вакуумным насо-
дом калия (FeCN) (рис. 6). 16 мл 8мМ раствора Б-
сом, затем в верхнюю часть аппарата после его от-
ДНКЖ-GSH в 200 мМ HEPES-буфере при рН 7.4
крытия на воздухе вводили несколько капель
помещали в модифицированный аппарат Тун-
концентрированной серной кислоты, которая
берга (рис. 3б) с последующей откачкой воздуха
смешивалась с оставшимся в нижней части аппа-
из этого аппарата. Предполагалось, что высокая
рата раствором после откачки воздуха из аппара-
концентрация буфера в этом растворе должна бы-
та. Предполагалось, что нитрит, который образо-
ла обеспечить сохранение нейтрального значения
вался в результате гидролиза катионов нитрозо-
рН, которое могло снижаться в результате гидро-
ния, высвободившегося из Б-ДНКЖ в
лиза катионов нитрозония, высвобождаюшихся
количестве, равном (в соответствии со схемой 3)
из Б-ДНКЖ.
половине нитрозильных лигандов в исходных Б-
Сразу же после добавления FeCN (80 мМ) к
ДНКЖ-GSH, должен был после превращения
раствору Б-ДНКЖ-GSH при 60°С в анаэробных
нитрита в азотистую кислоту перейти в форме NO
условиях начиналось бурное выделение из рас-
в газовую фазу и обеспечить появление соответ-
БИОФИЗИКА том 65
№ 3
2020
432
ВАНИН
D
0.5
0.4
0.3
0.2
0.1
0.0
190
200
210
220
230
240
Длина волны, нм
Рис. 7. Верхняя кривая - спектр поглощения газообразного NO, высвободившегося из 15 мл 10 мМ раствора нитрита
в течение 1 ч при 60°С после его подкисления до рН 2.0. Нижняя кривая - спектр поглощения стандартного образца
газообразного NO (75 микромолей NO в 100 мл свободного объема аппарата Тунберга).
ствующих полос поглощения. Действительно,
Затем из 37.5 микромолей азотистой кислоты
удалось зарегистрировать две первые длинновол-
снова образуются по ~18 микромолей NO и NO2,
новые полосы, интенсивность которых через
и далее процесс повторяется по описанному вы-
20 мин после подкисления раствора достигала
ше пути, так что в его конце в газовой фазе долж-
плато. Зарегистрировать остальные две коротко-
но накапливаться ~100 микромолей NO, или ~
волновые полосы поглощения NO было невоз-
70% от исходного количества нитрита.
можно из-за сдвига нулевой линии вследствие за-
Таким образом результаты и этих эксперимен-
грязнения торцевых стенок цилиндрической кю-
тов находятся в полном соответствии с механиз-
веты аппарата Тунберга попадавшими на них
мом распада Б-ДНКЖ с тиолсодержащими ли-
частицами берлинской лазури, образующейся в
гандами, приведенным на схеме 3. Они показыва-
реакции FeCN c высвобождавшимся из Б-ДНКЖ
ют, что действительно в ходе этого процесса из
двухвалентным железом.
этих комплексов могут высвобождаться катионы
Количество NO, перешедшего из подкислен-
нитрозония в количестве, равном половине нит-
ного нитрита в газовую фазу, оказалось равным
розильных лигандов в этих комплексах или их
~90 микромолей на 100 мл свободного объема ап-
концентрации в пересчете на один атом железа в
парата Тунберга, что соответствовало превраще-
этих комплексах. Сохранение этих катионов в
нию в NO 70% нитрита, предположительно обра-
условиях устранения тиолсодержащих лигандов
зовавшегося при распаде Б-ДНКЖ-GSH, вы-
(в результате окисления) показывает, что ионы
званном FeCN. Эксперименты, поставленные с
Fe2+ сами по себе без участия тиолов не способны
целью проверки эффективности превращения
были восстанавливать катионы нитрозония.
нитрита в NO в кислой среде, результаты которых
приводятся на рис. 7, показали, что действитель-
Двухвалентное железо катализирует в анаэроб-
но примерно 70% нитрита превращаются в этих
ных условиях превращение молекул NO в анионы
условиях в NO. Из 15 мл 10 мМ раствора нитрита
нитрита. Вывод, сделанный в предыдущем разде-
натрия, т.е. из 150 микромолей нитрита, в анаэ-
ле, подтверждается результатами исследований
робных условиях при рН 2.0 высвобождалось
распада М-ДНКЖ с лигандами нетиоловой при-
примерно
100 микромолей NO
- ~70% от
роды со спином S = 1/2, существование которых
150 микромолей.
было продемонстрировано еще в 1960-е годы
уже в первом ЭПР-исследовании низкоспиновых
Эту величину легко получить из следующих
М-ДНКЖ с различными анионными лигандами
расчетов. Протонизация 150 микромолей нитрита
[42]. Последующие исследования, проведенные в
приводит к его превращению в 150 микромолей
1980-е годы нашей группой, показали, что наряду
азотистой кислоты, которая в результате реакции
с указанными низкоспиновыми М-ДНКЖ, ха-
диспропорционирования (реакция 3, представ-
рактеризующимися сравнительно узкими сигна-
ленная во введении) дает по 75 микромолей NO и
лами ЭПР со значениями g-фактора в диапазоне
NO2. Далее пары молекул NO2 в ходе гидролиза
2.05-2.014 (gср = 2.03), в тех же растворах, исходно
превращаются в равном количестве (37.5 микро-
молей) в молекулы азотистой и азотной кислот.
содержащих ионы Fe2+ и лиганды нетиоловой
БИОФИЗИКА том 65
№ 3
2020
СВОБОДНО-РАДИКАЛЬНАЯ ПРИРОДА МОЛЕКУЛ МОНООКСИДА АЗОТА
433
(а)
(б)
D
g = 4.0
g =
2.03
g = 2.0
1.25
1
1.00
0.75
2
0.50
0.25
3
0.00
200
500
800
11 мТл В
4.5 мТл
В
Длина волны, нм
Рис. 8. (а) - Спектры ЭПР нитрозильных комплексов железа с ЭДТА (спектр 1), водой (спектр 2) и NAC (спектр 3),
зарегистрированные при 77 К; (б) - спектры оптического поглощения, характерные для нитрозильных комплексов
железа с ЭДТА или водой.
природы, такие как фосфат, цитрат или аскорбат,
встряхивания аппарата, когда раствор железа в
при их обработке газообразным NO возникают
дистиллированной воде приобретал зеленую
также высокоспиновые (с S = 3/2) мононитро-
окраску, смешивали его с раствором в нижней ча-
зильные комплексы железа (МНКЖ), характери-
сти аппарата и после последующего пятиминут-
зующиеся сравнительно широким сигналом ЭПР
ного встряхивания в присутствии NO аппарат от-
со значениями g-фактора в диапазоне 4.0-2.0
качивали с последующим быстрым заморажива-
[43-45]. Такая же смесь низкоспиновых и высо-
нием в жидком азоте полученного раствора в
коспиновых нитрозильных комплексов железа
ампуле для ЭПР-исследований диаметром 4 мм.
(соответственно ДНКЖ и МНКЖ) возникала
В полном соответствии с результатами нашей
при обработке оксидом азота чисто водных рас-
предыдущей работы
[43] в растворах Fe2+ c
творов Fe2+, так что в этих комплексах в качестве
HEPES-буфером были зарегистрированы сигна-
лигандов выступали молекулы воды [43-45]
лы ЭПР высокоспиновых МНКЖ и низкоспино-
В настоящей работе были синтезированы нит-
вых ДНКЖ c молекулами воды в качестве лиган-
розильные комплексы железа с ЭДТА, водой и
дов (рис. 8а, спектр 2), тогда как растворах Fe2+ c
(для сравнения) с NAC, спектры ЭПР которых
ЭДТА или с NAC - сигналы ЭПР соответственно
представлены на рис. 8а. На рис. 8б приведены
только высокоспиновых МНКЖ с ЭДТА, харак-
спектры оптического поглощения, характерные
теризующиеся значениями g⊥ = 4.0 и g|| = 2.0), или
для МНКЖ с лигандами нетиоловой природы, в
низкоспиновых М-ДНКЖ с NAC, характеризую-
нашем случае - для МНКЖ с ЭДТА или молеку-
щиеся значениями g⊥ =2.04, g||.=2.014 (gср = 2.03)
лами воды.
(рис. 8a, спектры 1 и 3).
Синтез обоих типов нитрозильных комплек-
Следует отметить, что использование раствора
сов железа - высокоспиновых МНКЖ и низкос-
Fe2+ в дистиллированной воде было обязатель-
пиновых ДНКЖ - проводили следующим обра-
ным для синтеза указанных комплексов, по-
зом. В нижнюю часть аппарата Тунберга, приве-
скольку только в дистиллированной воде при
денного на рис. 3а, вводили в 4 мл воды ЭДТА
рН 5.0-5.5 можно было полностью растворить
(100 мМ)
+ HEPES-буфер (30 мМ), только
соль двухвалентного железа. После контакта с NO
HEPES-буфер (30 мМ) или 50 мМ NAC + HEPES-
буфер (30 мМ) - все при нейтральных значениях
ионы Fe2+, связавшиеся с NO, могли включаться
рН, а в верхнюю часть аппарата - 1 мл дистилли-
в водный раствор в нижней части аппарата, со-
рованной воды при рН 5.0-5.5. После откачки
державший при нейтральных значениях рН
воздуха в верхнюю часть аппарата вводили сухой
ЭДТА, HEPES или NAC с образованием соответ-
порошок FeSO4⋅H2O, так что его концентрация
ствующих МНКЖ с ЭДТА или молекулами воды
и М-ДНКЖ с водой или NAC.
при последующем смешивании растворов в ниж-
ней и верхней частях аппарата составляла 20 мМ.
В отличие от ДНКЖ с тиолсодержащими ли-
После вторичной откачки воздуха в аппарат вво-
гандами, как МНКЖ, так и М-ДНКЖ с молеку-
дили газообразный NO и после пятиминутного
лами воды в качестве лигандов сохранялись в вод-
БИОФИЗИКА том 65
№ 3
2020
434
ВАНИН
(а)
(б)
D
D
2.0
0.3
2
1.5
3
0.2
1.0
2
0.1
1
0.5
0.10
3
1
1а
0.08
0.0
0.0
200
400
600
800
400
500
600
700
800
Длина волны, нм
Длина волны, нм
Рис. 9. Образование GS-NO как индикатора превращения оксида азота (NO) в анионы нитрита, появлявшегося при
распаде М-ДНКЖ с молекулами воды. М-ДНКЖ были синтезированы путем обработки водных 30 мМ растворов
HEPES газообразным NO в присутствии 20 мМ ионов Fe2+ (кривые 2 и 3). Кривая 1 получена при такой же обработке
растворов HEPES, но без добавления к ним ионов Fe2+. Приводятся зарегистрированная в указанных растворах
полоса оптического поглощения GS-NO на 334 нм (а) и зарегистрированная при большей чувствительности
спектрометра полоса на 543 нм (б). На врезке (кривая 1а) - увеличенный фрагмент кривой 1.
ных растворах при нейтральных значениях рН
сов железа с ЭДТА. Эти комплексы существуют
только в присутствии газообразного NO. При от-
только в форме высокоспиновых МНКЖ, не об-
качке последнего через 10-20 мин эти комплексы
разуя (как это следует из рис. 8) их динитрозиль-
полностью распадались, при этом высвобождав-
ных аналогов
шиеся из них ионы Fe2+ выпадали из раствора в
Образование GS-NO в водных растворах, об-
форме нерастворимых в воде гидроокисных ком-
плексов, резко повышавших мутность раствора.
работанных газообразным NO, наблюдалось и в
Последнее устранялось подкислением получив-
отсутствие экзогенных ионов Fe2+ в этих раство-
шихся суспензий до рН 1-2. Если после этого в
рах (рис. 9, кривая 1). Этот процесс был обуслов-
полученные таким образом растворы сразу после
лен наличием в газообразном NO примеси NO2.
их подкисления вводили тиолы, например глута-
Его уровень существенно снижался при очистке
тион в концентрации 50 мМ, в спектре оптиче-
NO от примеси NO2 методом низкотемператур-
ского поглощения растворов появлялись полосы
на 334 и 543 нм, характерные для GS-NO (рис. 9).
ной сублимации в вакуумированном сосуде.
Характерно, что при этом по мере растворения
Эксперименты, проведенные на растворах
гидроокисных осадков железа из-за ослабления
нитрозильных комплексов железа с фосфатом,
интенсивности светорассеяния растворов базо-
представленных, как и ДНКЖ с молекулами во-
вая линия снижалась практически до нуля (рис. 9,
кривые 2 и 3).
ды, высокоспиновыми МНКЖ и низкоспиновы-
ми М-ДНКЖ, показали, что распад последних
Появление полос поглощения GS-NO, оче-
также (в соответствии со схемой 3) сопровождает-
видно, было обусловлено S-нитрозированием
ся высвобождением нитрита, образующим GS-
глутатиона молекулами азотистой кислоты, обра-
NO при подкислении растворов нитрита с после-
зующейся при подкислении нитрита, который в
соответствии со схемой 3 должен был возникать
дующим добавлением к ним глутатиона (данные
при распаде М-ДНКЖ с молекулами воды, как
не приводятся).
лигандами нетиоловой природы. Что касается
Таким образом, включение молекул NO в
возможности S-нитрозирования глутатиона вы-
М-ДНКЖ с лигандами нетиоловой природы в ка-
сокоспиновыми МНКЖ с молекулами воды, од-
на из резонансных структур которых описывается
честве нитрозильных лигандов может обеспечи-
вать превращение половины из них в катионы
как Fe+NO+ и могла бы выступать в качестве до-
нитрозония. Высвобождаясь из комплексов,
нора катиона нитрозония, такая возможность для
указанных комплексов не реализуется. В пользу
эти катионы превращаются в результате гидроли-
этого свидетельствует факт отсутствия S-нитро-
за при нейтральных значениях рН в анионы нит-
зирующей активности у нитрозильных комплек-
рита.
БИОФИЗИКА том 65
№ 3
2020
СВОБОДНО-РАДИКАЛЬНАЯ ПРИРОДА МОЛЕКУЛ МОНООКСИДА АЗОТА
435
ОБСУЖДЕНИЕ
воспроизводстве этих агентов существование
указанной системы как самоподдерживающейся,
Способность ДНКЖ с тиолсодержащими ли-
гандами высвобождать при распаде нейтральные
саморегулирующейся системы реакций 1 и 2, ре-
молекулы NO и катионы нитрозония делает воз-
акций химического равновесия, устанавливаю-
можной их регенерацию по двум путям, приводи-
щихся между М-ДНКЖ и Б-ДНКЖ, а также меж-
мым на схеме 6 - в реакции Fe2+, газообразного
ду ними и составляющими их компонентами.
NO и тиолов (реакции 1) и реакции Fe2+, S-нит-
Так, в зависимости от соотношения уровней NO
розотиолов (RS-NO) и тиолов (реакции 2). Эти
и RS-NO в этой системе может попеременно до-
реакции, в соответствии с их механизмами, ука-
минировать либо реакция 1, либо реакция 2. По
занными на схемах 2 и 4, необратимы из-за пре-
мере расходования NO в реакции 1 и накопления
вращения NO и тиолов соответственно в закись
RS-NO cинтез М-ДНКЖ начинает протекать по
азота и дисульфиды. Это превращение можно
реакции 2 и, наоборот, по мере расходования RS-
рассматривать в качестве «сгорания» основных
«топливных компонентов системы», показанной
NO и накопления NO снова начинает доминиро-
на схеме 6 - NO и тиолов, обеспечивающих при
вать реакция 1.
-
2+
2RS + Fe
+ 3 NO
N O2
1
1
+ RS-
+ RS-
–
–
2+
–
-
[(RS ) Fe+(NO+) ]
[(RS ) Fe+(NO+)
Fe
+ NO + RS + NO++ RS
2
2
4
2
2
- RS-
- RS-
RS-NO
2
RS-SR
2
–
2+
2RS + Fe
+ 2RS
-NO
Схема 6. Саморегулирующаяся система реакций химического равновесия, устанавливающихся между М-
и Б-ДНКЖ, а также между ними и составляющими их компонентами.
Таким образом, для этой системы не исключа-
тральных значениях рН в нитрит-анионы - ко-
ется колебательный режим ее существования и
нечный продукт распада рассматриваемой систе-
более того появление автоволн, о чем свидетель-
мы, что и наблюдалось в эксперименте (рис. 6).
ствуют результаты наших исследований, опубли-
При нейтральных значениях рН этот процесс был
кованных в работах [19, 46].
необратимым, поскольку при этих рН нитрит сам
по себе не мог инициировать образования
Самоподдерживающийся, саморегулирую-
ДНКЖ. В этом случае рассматриваемую систему
щийся режим существования системы, приведен-
можно было реконструировать только при добав-
ной на схеме 6, вполне возможен в живых орга-
лении в нее тиолов с последующим их превраще-
низмах, в которых может поддерживаться на ста-
ционарном уровне концентрация «топливных»
нием в S-нитрозотиолы в реакции с нитритом в
кислой среде. Последующее повышение рН до
компонентов указанной системы -NO и тиолов.
В отсутствие такого воспроизводства эта система
нейтральных значений в присутствии ионов Fe2+
перестает функционировать, но может снова на-
должно было (по реакции 2 на схеме 6) обеспе-
чать работать при возобновлении ее снабжения
чить образование М-ДНКЖ и Б-ДНКЖ.
NO или тиолами, причем этот процесс может раз-
Как показали наши предыдущие исследования
виваться по следующим сценариям. При блокаде
поставки NO указанная система должна сразу же
[8, 13] и исследования, изложенные в настоящей
останавливаться, и ее последующая реконструк-
работе, распад химической системы, приведен-
ция, а именно возобновление в ней соответству-
ной на схеме 6, можно было инициировать прото-
ющих реакций, может начаться сразу после нача-
нированием тиолсодержащих лигандов в Б-ДН-
ла снабжения системы газообразным NO. При за-
КЖ, обеспечивающим разрыв связи между иона-
медлении снабжения системы тиолами,
ми железа и атомами тиоловой серы в этих
вызванном, например, их окислением или блока-
комплексах. Этот процесс усиливался при подо-
дой тиоловыми реагентами, катионы нитрозония
греве их кислотных растворов. Высвобождающи-
должны вместо реакции S-нитрозирования
еся при этом протонированные тиолы могли свя-
включаться в реакцию гидролиза, приводящую к
зываться с катионами нитрозония и образовы-
превращению катионов нитрозония при ней-
вать с ними S-нитрозотиолы, что наблюдалось
БИОФИЗИКА том 65
№ 3
2020
436
ВАНИН
как в прошлых исследованиях [8, 13], так и в на-
разовании указанных комплексов. В результате
стоящей работе (рис. 5 и 6). Вместе с тем, как по-
электронная конфигурация железа в этой группе
казал эксперимент, протонированные тиолы бы-
переходит из d7- в d8-конфигурацию, а при после-
ли не способны связываться с ионами Fe2+ и тем
дующем связывании с железом второй молекулы
самым обеспечивать при участии S-нитрозотио-
NO и тиолсодержащей молекулы, приводяшем к
лов образование в кислой среде соответствующих
окончательному образованию М-ДНКЖ, желе-
ДНКЖ. Это и приводило к обнаруживаемому на-
зодинитрозильная группа в этих комплексах на-
коплению в этой среде S-нитрозотиолов в коли-
чинает характеризоваться d9-электронной кон-
честве, равном половине нитрозильных лиган-
фигурацией железа или, как представлено в рабо-
дов, имевшихся в исходных Б-ДНКЖ.
те
[54], структурой
{Fe(NO)2}9. В качестве
Предложенный механизм образования
экспериментального факта, подтверждающего
М-ДНКЖ и Б-ДНКЖ с тиолсодержащими ли-
вышеизложенное предположение о механизме
гандами основывается на предположении о воз-
образования парамагнитных М-ДНКЖ с тиолсо-
можности реакции диспропорционирования мо-
держащими лигандами, авторы работы [53] ука-
лекул NO при их связывании с ионами Fe2+ (схе-
зывают на обнаруженное ими появление в рас-
мы 1 и 2). Такая возможность обсуждается и
творе синтезируемых комплексов тиильного ра-
другими авторами [47-49], но в отличие от меха-
дикала (RS·), образующегося в результате
низма, описанного в данной работе, они объяс-
одноэлектронного окисления тиола железомоно-
няют, исходя этого предположения, образование
нитрозильной (Fe-NO) группой.
при распаде ДНКЖ только диоксида и закиси
Вывод о локализации на d-орбиталях железо-
азота (NO2 и N2O) как результата гидролиза обра-
динитрозильной группы в М-ДНКЖ и Б-ДНКЖ
зующихся при диспропорционировании двух мо-
с тиолсодержащими лигандами девяти электро-
лекул NO - катиона нитрозония и аниона нит-
нов находится в полном соответствии с пред-
роксила. Что касается направления наших иссле-
ставлениями многих исследователей этих ком-
дований, они касаются изучения возможности
плексов, основанными на результатах рентгено-
образования в ходе этих процессов S-нитрозо-
структурных кристаллографических исследова-
тиолов, т.е реакции, способной успешно конку-
ний М-ДНКЖ и Б-ДНКЖ с тиолсодержащими
рировать с реакцией гидролиза катионов нитро-
лигандами [55-60]. Поскольку, согласно этим ис-
зония. В этом отношении основной результат на-
следованиям, железодинитрозильная группа,
стоящей работы состоит в том, что образование
включая атомы тиоловой серы, характеризуется в
S-нитрозотиолов при распаде М-ДНКЖ и Б-ДН-
кристаллическом состоянии тетраэдрической
КЖ с тиолсодержащими лигандами может суще-
пространственной структурой, то парамагнетизм
ственно затрудняться из-за восстановления кати-
моноядерной формы ДНКЖ с S = ½ может обес-
онов нитрозония тиолами при посредстве ионов
печиваться локализацией на верхних t2g (dxz, dyz и
железа как катализаторов этого процесса.
dxy) орбиталях пяти электронов (девять электро-
Следует отметить, что предположение о роли
нов суммарно на всех d-орбиталях). При этом
реакции диспропорционирования молекул NO в
предполагается, что при растворении ДНКЖ ука-
процессе образования ДНКЖ восходит (в соот-
занная тетраэдрическая пространственная струк-
ветствии с информацией, приводимой в работах
тура этих комплексов и соответствующая ей элек-
[50, 51]) еще к пионерским работам Джозефа
тронная структура полностью сохраняется, что
Пристли - великого английского ученого, от-
полностью согласуется с выводами авторов рабо-
крывшего в XYIII веке не только азот, но и моно-
ты [53] о d9-электронной конфигурации железа в
оксид азота, а также реакцию последнего с желе-
ДНКЖ, синтезируемых в растворе.
зом, приводящей как к образованию железонит-
розильных комплексов, так и к реализации
Таким образом в настоящее время существуют
реакции диспропорционирования молекул NO
два взаимоисключающих представления об элек-
до закиси и диоксида азота.
тронной структуре ДНКЖ с тиолсодержащими
В последнее время был предложен другой ме-
лигандами, характеризующейся либо d9-, либо
ханизм образования М и Б-ДНКЖ с тиол-содер-
d7-электронной конфигурацией железа в железо-
жащими лигандами в водных растворах в ходе ре-
динитрозильных фрагментах этих комплексов.
акции ионов Fe2+, тиолов и газообразного NO
Вопрос о справедливости какого-либо из этих
[52, 53]. Авторы этих работ полагают, что пара-
представлений остается открытым. Единствен-
магнетизм возникающих при этом М-ДНКЖ на
ное, что позволяет, по мнению автора настоящей
первой стадии их образования обеспечивается
работы, отдать предпочтение d7-конфигурации -
одноэлектронным восстановлением мононитро-
это то, что в ее рамках легко обьяснить на основе
зильной (Fe2+-NO ↔ Fe+-NO+) группы одним из
схемы 3 способность М-ДНКЖ и Б-ДНКЖ вы-
тиолсодержащих соединений, участвующих в об-
ступать в качестве доноров не только нейтраль-
БИОФИЗИКА том 65
№ 3
2020
СВОБОДНО-РАДИКАЛЬНАЯ ПРИРОДА МОЛЕКУЛ МОНООКСИДА АЗОТА
437
ных, газовых молекул NO, но и катионов нитро-
11. T. P. Melia, J. Inorg. Nucl. Chem. 27, 95 (1965).
зония, превращающихся либо в S-нитрозотиолы,
12. S. F. Agnew, B. I. Spansou, L. H. Jones, et al., J. Phys.
либо в анионы нитрита.
Chem. 89, 1678 (1985).
13. A. F. Vanin, Cell. Biochem. Biophys. 77, 279 (2019).
БЛАГОДАРНОСТИ
14. Q. Liu, K. Yu, P. Yi, et al., Environment. Sci. Pollution
Автор выражает благодарность В.Д. Микояну
Res. 28, 19540 (2019).
за регистрацию спектров ЭПР (рис. 8), Р.Р. Боро-
15. A. F. Vanin, Nitric Oxide Biol. Chem. 21, 1 (2009).
дулину - за регистрацию спектров оптического
16. A. F. Vanin and D. Sh. Burbaev, Biophys. J. 14, 818836
поглощения (рис. 6 и 7) и Н.А. Ткачеву - за ком-
(2011).
пьютерную обработку рис. 4, 5 и 9.
17. A. F. Vanin, A. P. Poltorakov, V. D. Mikoyan, et al., Ni-
tric Oxide Biol. Chem. 23, 136 (2010).
ФИНАНСИРОВАНИЕ РАБОТЫ
18. A. F. Vanin, Nitric Oxide Biol. Chem. 54, 15 (2018).
Работа выполнена в рамках Государственного
19. A. F. Vanin, Dinitrosyl Iron Complexes as a “Working
задания Российского Федерального Агенства для
Form” of Nitric Oxide in Living Organisms, (Cambridge
научных организаций
(00008202014-0001б,
Scholars Publishing, Cambridge, UK, 2019).
№ АААА-А17-117040610310-6,
0082-2014-0008 и
№ АААА-А17-1170403100008-5, спонсирована
20. Р. М. Налбандян, А. Ф. Ванин и Л. А. Блюмен-
Российским академическим проектом «5-100», а
фельд, в сб. Тезисы конференции «Свободноради-
также финансирована в рамках гранта Россий-
кальные процессы в биологических системах» (1964),
ского фонда фундаментальных исследований
с. 18.
№ 18-04-00059а.
21. А. Ф. Ванин и Р. М. Налбандян, Биофизика 10, 167
(1965).
КОНФЛИКТ ИНТЕРЕСОВ
22. А. Ф. Ванин, Л. А. Блюменфельд и А. Г. Четвери-
Автор заявляет об отсутствии конфликта инте-
ков, Биофизика 12, 829 (1967).
ресов.
23. J. R. Mallard and M. Kent, Nature 204, 1192 (1964).
24. A. J. Vithaythil, B. Ternberg, and B. Commoner, Na-
СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ
ture 207, 1246 (1965).
Настоящая работа не содержит описания ка-
25. J. R. Lancaster and J. B. Hibbs, Proc. Natl. Acad. Sci.
ких-либо исследований с использованием людей
USA 87, 1223 (1990).
и животных в качестве объектов.
26. C. Pellat, Y. Henry, and J.-C. Drapier, Biochim. Bio-
phys. Res. Comm. 166, 119 (1990).
СПИСОК ЛИТЕРАТУРЫ
27. J.-C. Drapier, C. Pellat, and Y. Henry, J. Biol. Chem.
266, 10162 (1991).
1. L. J. Ignarro, Nitric Oxide Biology and Pharmacology
(Acad. Press, Zurich, Switzerland, 2000).
28. M. Lepoivre, J.-M. Flavan, and Y. Henry, J. Biol.
2. N. Hogg, Free Rad. Biol. Med. 28, 1478 (2000).
Chem. 267, 22294 (1992).
3. B. M. Gaston, J. Carver, A. Doctor, et al., Mol. Inter-
29. A. F. Vanin, P. I. Mordvintcev, S. Hauschildt, et al.,
vention 3, 253 (2003).
Biochem. Biophys. Acta 1177, 37 (1993).
4. D. Seth, D. T. Hess, A. Hausladen, et al., Mol. Cell 89,
30. J. Stadler, H. A. Bergonia, M. DiSilvio, et al., Arch.
451 (2018).
Biochim. Biophys. 302, 4 (1993).
5. D. A. Wink, R. W. Nims, J. F. Darbyshir, et al., Chem.
31. A. K. Nussler, D. A. Geller, M. A. Sweetland, et al.,
Res. Toxicol. 7, 525 (1994).
Biochim. Biophys. Res. Comm. 194, 826 (1993).
6. V. G. Khatitonov, A. R. Sandquist, and V. S. Sharma, J.
32. Y.-L. Geng, A.-D. Petersson, A. Wennmalm, et al.,
Biol. Chem. 270, 28158 (1995).
Exp. Cell Res. 214, 418 (1994).
7. N. S. Bryan, T. Rassaf, R. E. Maloney, et al., Proc.
33. N. R. Bastian, C.-Y. Yim, J. B. Hibbs, et al., J. Biol.
Natl. Acad. Sci. USA 101, 438 (2004).
Chem. 269, 5127 (1994).
8. A. F. Vanin, Austin J. Analyt. Pharmac. Chem. 5 (4),
34. K. Odoi, T. Akaike, H. Horie, et al., Cancer 77, 1598
id1109 (2018).
(1996).
9. C.A. Bosworth, J.C. Toledo, J.W. Zmiewski et al.,
35. B. Muller, A.Kleschyov, and J.-C. Stoclet, Br. J. Phar-
Proc. Natl. Acad. Sci. USA 106, 4671 (2009).
macol. 119, 1281 (1996).
10. M. V. Foster, L. Liu, M. Zeng, et al., Biochem. 48, 792
36. R. N. Watts, C. Hawkins, P. Ponka, et al., Proc. Natl.
(2009).
Acad. Sci. USA 103, 7670 (2006).
БИОФИЗИКА том 65
№ 3
2020
438
ВАНИН
37. A. F. Vanin, I. V. Malenkova, and V. A. Serezhenkov,
49. C. K. Brozek, J. T. Miller, S. A. Stoian, et al., J. Am.
Nitric Oxide Biol. Chem. 1, 1991 (1997).
Chem. Soc. 137, 7495 (2015).
38. R. R. Borodulin, L.N. Kubrina, V. D. Mikoyan, et al.,
50. E. W. Ainscough and A. M. Brodie, J. Chem. Educ. 72,
Nitric Oxide Biol. Chem. 29, 4 (2013).
686 (1995).
39. A. F. Vanin, R. R. Borodulin, and V. D. Mikoyan, Ni-
51. D. Michael and P. Mingos, Struct. Bond. 153, 1 (2014).
tric Oxide Biol. Chem. 66, 1 (2017).
52. J. C. M. Pereira, A. V. Iretskii, R.-M. Han, et al. J. Am.
40. J. Lique and D. R. Grosley, J. Chem. Phys. 111, 7405
Chem. Soc. 137, 328 (2015).
(1999).
41. A. P. Dicks, P. Herves Beloso, D. Lyn, and H. Wil-
53. D. R. Truzzi, O. Augusto, P. C. Ford, Chem. Comm.
liams, J. Chem. Soc. Perkin Trans. 2, 1429 (1997).
55, 9156 (2019).
42. C. C. MacDonald, W. D. Philips, and H. F. Mower, J.
54. J. H. Enemark and R. D. Feltham, Coordination
Am. Chem. Soc. 87, 3319 (1965).
Chem. Rev. 13, 339 (1974).
43. A. F. Vanin and D. I. Aliev, Studia Biophysica 93, 63
55. T. C. Harrop, Z. J. Tonzetich, E. Reisner, et al., J. Am.
(1983).
Chem. Soc. 130, 15602 (2008).
44. A. Wanat, T. Schneppensieper, G. Stochel, et al., In-
56. A. F. Shestakov, Yu. M. Shul`ga, N. S. Emel`yanova,
org. Chem. 41, 4 (2002).
et al., Rus. Chem. Bull. 56, 1289 (2009).
45. A. In-lam, M. Wolf, C. Wilfer, et al., Chem. Eur. 25,
57. R. Polukkody and M. Y. Darensbourg, Acc. Chem.
1304 (2019).
Res. 48, 2049 (2015).
46. A. F. Vanin, V. D. Mikoyan, N. M. Rubtsov, et al., Ni-
58. J. Fitzpatrick and E. Kim, Acc. Chem. Res. 48, 2453
tric Oxide Biol. Chem. 23, 175 (2010).
(2015).
47. P. C. Ford and I. M. Lorcovic, Chem. Rev. 102, 993
59. T.-T. Lu, Y.-M. Wang, C.-H. Hung, et al., Inorg.
(2002).
Chem. 57, 12425 (2018).
48. S. Stojanovic, D. Stanic, M. Nicolic, et al., Nitric Ox-
60. S.-L. Cho, C.-J. Liao, and T.-T. Lu, J. Biol. Inorg.
ide Biol. Chem. 11, 256 (2004).
Chem. 24, 495 (2019).
Free Radical Nature of Nitric Oxide Molecules as a Determinant
of Their Transformation into Nitrosonium Cations in Living Systems
A.F. Vanin
Semenov Federal Research Center of Chemical Physics, Russian Academy of Sciences, ul. Kosygina 4, Moscow, 119334 Russia
Institute of Regenerative Medicine, Sechenov First Moscow State Medical University of the Ministry of Health of Russian
Federation, Trubetskaya ul. 8/2, Moscow, 119991 Russia
The paper presents novel results that confirm our previous conclusion that the binuclear form of biologically
active dinitrosyl iron complexes (B-DNIC) with thiol-containing ligands (glutathione or N-acetyl-L-cyste-
ine) may act as a donor of nitrosonium cations which are responsible for S-nitrosothiol formation during B-
DNIC decomposition in acid solutions under aerobic/anaerobic conditions. The presence of nitrosonium
cations within the composition of B-DNIC is determined by a dispropoportionation reaction between free
radical NO molecules while binding to Fe2+ cations (two molecules for one ion) during B-DNIC synthesis.
When thiol-containing ligands are oxidized in DNIC or inhibited by thiol-specific reagents released during
decomposition of these DNIC at neutral pH values, nitrosonium cations undergo hydrolysis followed by
transformation into nitrite anions. Similar transformation occurs when mononuclear DNIC (M-DNIC)
with non-thiol ligands are decomposed at neutral pH values. It was found that S-nitrosothiol formation in
the decomposition process of B-DNIC with thiol-containing ligands at acid pH values can be prevented in
the presence of free thiol molecules (not present within the composition of B-DNIC) in two-threefold excess
with regard to the content of B-DNIC. This inhibition is due to reduction of nitrosonium cations to NO mol-
ecules induced by free thiol molecules with participation of iron ions as catalytic agents. Therefore, both
forms of DNIC (M- and B-DNIC) which form in living systems can act not only as the donors of nitric oxide
recognized now as one of universal regulators of metabolic processes but also as the donors of nitrosonium
cations that initiate the S-nitrosation process of low- and high-molecular (protein-bound) thiols.
Keywords: dinitrosyl iron complexes, NO, S-nitrosothiols, EPR
БИОФИЗИКА том 65
№ 3
2020