БИОФИЗИКА, 2020, том 65, № 5, с. 837-859
МОЛЕКУЛЯРНАЯ БИОФИЗИКА
УДК 577.32
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
© 2020 г. И.Ю. Петрушанко, В.А. Митькевич, А.А. Макаров
Инcтитут молекуляpной биологии им. В.А. Энгельгаpдта PАН, 119991, Моcква, ул. Вавилова, 32
E-mail: irina-pva@mail.ru
Поступила в редакцию 15.11.2019 г.
После доработки 17.06.2020 г.
Принята к публикации 19.06.2020 г.
Обзор посвящен молекулярным механизмам редокс-регуляции Na,K-АТФазы. Na,K-АТФаза со-
здает трансмембранный градиент ионов натрия и калия, необходимый для жизнедеятельности всех
клеток животных, а также является рецептором для кардиотонических стероидов, регулирующих
пролиферацию и апоптоз клеток. Функционирование Na,K-АТФазы зависит от окислительно-вос-
становительного (редокс) статуса клеток. Изначально было показано, что фермент ингибируется
при окислительном стрессе, однако теперь ясно, что редокс-регуляция Na,K-АТФазы является
сложным процессом и не объясняется одним лишь окислительным повреждением белка. Актив-
ность фермента максимальна при физиологической концентрации кислорода, снижаясь как при
гипоксии, так и при гипероксии, а также при падении или возрастании внутриклеточного глутати-
она. Таким образом, активность Na,K-АТФазы максимальна в определенном диапазоне редокс-
условий. В настоящее время стало очевидным, что нарушение активности Na,K-АТФазы при ряде
патологий, таких как гипоксия, ишемия, диабет, болезнь Альцгеймера, связано именно с измене-
нием редокс-статуса клеток. Рецепторная функция Na,K-АТФазы также зависит от редокс-статуса
клеток, что необходимо учитывать при рассмотрении действия кардиотонических стероидов на
клетки и ткани. В обзоре особое внимание уделено редокс-модификациям тиоловых групп различ-
ных субъединиц Na,K-АТФазы и рассмотрению процессов регуляции, в которые они вовлечены в
норме и патологии. Понимание молекулярных механизмов редокс-регуляции поможет предотвра-
тить нарушение функционирования Na,K-АТФазы при патологических условиях и снизить повре-
ждение клеток.
Ключевые слова: Na,K-ATФаза, редокс-регуляция, глутатионилирование, нитрозилирование, окисле-
ние, остатки цистеина, кардиотонические стероиды.
DOI: 10.31857/S0006302920050014
Na,K-АТФазы при ряде патологий связано имен-
Na,K-АТФаза - трансмембранный белок, ко-
но с изменением редокс-статуса клеток [7, 8].
торый за счет энергии гидролиза АТФ создает
Ранее неоднократно отмечалось, что актив-
трансмембранный градиент Na+ и K+. Данный
градиент используется для транспорта других
ность Na,K-АТФазы снижается при недостатке
ионов, аминокислот и глюкозы, поддержания
кислорода, и это происходит гораздо раньше, чем
истощение клеток по АТФ, которое может приве-
внутриклеточного pH и уровня Cа2+ [1-3]. Транс-
сти к ингибированию фермента [11-13]. Сниже-
портная и гидролитическая активности Na,K-
ние активности наблюдалось и при окислитель-
АТФазы чувствительны к редокс-статусу клеток
ном стрессе [4, 14, 15]. В дальнейшем было пока-
[4-8]. Они максимальны при физиологической
зано, что снижение активности Na,K-АТФазы
концентрации кислорода, снижаясь как при ги-
происходит не только при гипоксии, но и при ги-
поксии, так и гипероксии, а также при падении
пероксии [6], а избыток глутатиона, как и его не-
или возрастании внутриклеточного глутатиона
достаток, может также привести к снижению ак-
[5, 6]. Соотношение восстановленного (GSH) и
тивности фермента [5]. Таким образом, актив-
окисленного (GSSG) глутатиона определяет тио-
ность Na,K-АТФазы имеет максимум в
ловый редокс-статус клетки и изменяется при ги-
определенном диапазоне редокс-условий. Во-
поксии, окислительном стрессе и многих патоло-
прос о механизмах ее редокс-регуляции является
гиях
[9,
10]. Нарушение функционирования
крайне актуальным, потому что нарушение рабо-
ты этого фермента критично для жизнеспособно-
Сокращения: GSH - восстановленный глутатион, GSSG -
окисленный глутатион, КТС
- кардиотонические
сти клеток. В электровозбудимых тканях функци-
стероиды, АФК - активные формы кислорода.
онирование Na,K-АТФазы необходимо еще и для
837
838
ПЕТРУШАНКО и др.
нормального формирования потенциала дей-
тозольную петли, которые формируют три основ-
ствия. Именно нарушение функционирования
ных цитозольных домена: N-конец белка и малая
Na,K-АТФазы в ткани мозга и сердца при ише-
цитозольная петля формируют актуаторный до-
мии и гипоксии является одним из самых ранних
мен (А), большая петля формирует нуклеотид-
и критичных событий, приводящих к нарушению
связывающий домен (N) и фосфорилируемый до-
работы ткани и последующей гибели клеток [7, 8].
мен (P). β-Субъединица (55 кДа - гликозилиро-
При этом вопрос о редокс-регуляции Na,K-
ванная часть, 36-38 кДа - белковая часть) содер-
АТФазы не ограничивается только регуляцией
жит один трансмембранный домен. Большая
активности фермента. В последнее время появля-
часть β-субъединицы формирует внеклеточный
ется все больше работ, свидетельствующих об
домен. Она необходима для транспорта вновь
огромном значении Na,K-АТФазы как рецептора
синтезированной Na,K-АТФазы в мембрану, а
к кардиотоническим стероидам (КТС) [16-20].
также для правильной ориентации α-субъедини-
Эти вещества, которые ранее рассматривались
цы в цитоплазматической мембране и для окклю-
как экзогенные ядовитые соединения, специфи-
зии K+ [21]. В течение каталитического цикла
чески ингибирующие Na,K-АТФазу, затем были
Na,K-АТФаза проходит две основные конформа-
найдены в организме животных и человека, и в
ции: E1 и E2 [1, 7, 22]. Согласно циклу Альберса-
настоящее время рассматриваются как важные
Поста [1, 7, 22], цитозольный Na+ связывается с
гормоноподобные регуляторы. Na,K-АТФаза яв-
ферментом в Е1-конформации, обладающей вы-
ляется единственным известным рецептором
соким сродством к АТФ. Это приводит к гидро-
КТС. Связывание их c Na,K-АТФазой приводит
лизу АТФ, фосфорилированию фермента (E1P) и
к активации целого ряда сигнальных каскадов,
окклюзии трех ионов Na+. Конформационный
опосредующих пролиферацию и апоптоз клеток.
переход E1P-E2P приводит к снижению сродства
КТС активно использовались человечеством на
к Na+ и возрастанию сродства к K+. Происходит
протяжении сотен лет для лечения сердечной не-
высвобождение Na+ во внеклеточную среду и свя-
достаточности. Однако они очень токсичны и вы-
зывание двух ионов K+. Это приводит к дефосфо-
зывают ряд серьезных побочных эффектов. В на-
рилированию фермента и окклюзии ионов калия
стоящее время благодаря исследованиям рецеп-
(E2(2K+)). В этой конформации фермент связы-
торной функции Na,K-АТФазы причины многих
вает АТФ с низким сродством, что способствует
из этих эффектов стали ясны. Тем не менее такие
исследования еще только начинаются, и в этом
переходу E2-E1 и высвобождению K+ в цитозоль.
Это приводит к возрастанию сродства к АТФ. Та-
направлении больше вопросов, чем ответов. По-
ким образом, при низких концентрациях АТФ,
нимание того, как изменяется рецепторная функ-
еще достаточных для фосфорилирования, фер-
ция Na,K-АТФазы при окислительном стрессе,
гипоксии или иных изменениях редокс-статуса
мент будет ингибироваться, поскольку станет не-
очень важно для выявления особенностей регуля-
возможным связывание АТФ в сайте с низким
ции рецепторной функции фермента, а также для
сродством и уход K+ [1].
правильного использовании КТС в терапевтиче-
Данные по ограниченному трипсинолизу
ских целях.
Na,K-АТФазы
[23,
24], Fe-катализируемому
Влияние редокс-статуса на функционирова-
окислительному расщеплению [25], флуоресцен-
ние белка возможно как вследствие непосред-
ции фермента [26], а также тепловой денатурации
ственной модификации его остатков, так и опо-
[27, 28] в конформациях E1 и E2 указывают на
средованно, например, через редокс-чувстви-
различное относительное расположение доменов
тельные киназы. В данном обзоре фокус смещен
Na,K-АТФазы в этих состояниях. С появлением
на аспект редокс-регуляции Na,K-АТФазы путем
пространственной структуры Na,K-АТФазы в
непосредственной модификации остатков цисте-
E1P- [29] и E2P-конформациях [30] многие дан-
ина субъединиц Na,K-АТФазы.
ные о доменной организации фермента были
подтверждены, однако, к сожалению, структуры
фермента в E1-конформации до сих пор нет. Со-
СТРУКТУРА И ФУНКЦИЯ Na,K-АТФазы
гласно нынешним представлениям, E1-конфор-
Функциональный мономер Na,K-АТФазы со-
мация является более открытой в цитозоль, чем
стоит из каталитической α-субъединицы и регу-
E2- и E2P-конформации [23-30].
ляторной β-субъединицы
[1]. α-Субъединица
В настоящее время известны четыре изофор-
(100-113 кДа) имеет десять трансмембранных до-
мы α-субъединицы и три изоформы β-субъеди-
менов, содержит участки связывания АТФ, Na+,
ницы. В отличие от повсеместно распространен-
K+ и осуществляет гидролиз АТФ, а также транс-
ной α1-изоформы распределение других изоформ
порт ионов. Кроме того, на α-субъединице лока-
α-субъединицы тканеспецифично. α2-Изоформа
лизован участок связывания КТС. Как С-, так и
играет важную роль в регуляции сократимости
N-конец α-субъединицы находятся в цитозоле.
клеток и распространена в скелетных мышцах и
α-Субъединица содержит малую и большую ци-
сердце [31-34], а также в глиальных клетках [35].
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
839
α3-Изоформа распространена в нейронах, сет-
2
59.55
[GSH]
чатке глаза и встречается также в сердце [34-36].
Ehc = -240
−
log
Соотношение [GSH]/
2
[GSSG]
Созревание нейронной сети сопровождается уве-
[GSSG] является основным индикатором редокс-
личением уровня экспрессии α3-субъединицы
статуса клетки. Величина редокс-потенциала этой
[37], в то время как α2-субъединица важна для
пары коррелирует с биологическим статусом клет-
модуляции нейрональной активности в неона-
ки, составляя при пролиферации около -240 мВ,
тальный период, но во взрослом мозге встречает-
при дифференцировке -200 мВ и при апоптозе -
ся только в астроцитах [33]. α4 встречается только
170 мВ [10]. Как следует из приведенного выше
в тестикулах [32, 38]. β1- и β2-изоформы в различ-
выражения, редокс-потенциал пары
2GSH/
ных пропорциях встречаются в разных тканях,
GSSG зависит не только от соотношения окис-
тогда как β3-изоформа экспрессируется только в
ленной и восстановленной формы, но и от кон-
некоторых тканях, например в сетчатке [36]. Изо-
центрации GSH. Это обусловлено тем, что из двух
зимы фермента, содержащие одинаковые изо-
молекул GSH образуется одна молекула GSSG. В
формы β-субъединицы, но разные изоформы α-
то же время для пар НАД-Н/НАД+ и НАДФ-
субъединицы, отличаются по чувствительности к
Н/НАДФ+ редокс-потенциал зависит только от
КТС, причем показано, что важную роль в дан-
ной селективности играет остаток сахара КТС,
соотношения окисленной и восстановленной
агликоны не обладают селективностью в отноше-
форм. В клетках и тканях соотношение
нии изоформ α-субъединицы [39]. Изозимы, со-
НАДФ-Н/НАДФ+ около 100/1, тогда как НАД-
стоящие из разных α- и β-изоформ, имеют раз-
Н/НАД+ - от 1/10 до 1/1000 [41]. В то время как
личную чувствительность к субстратам, сердеч-
НАДФ-Н является основным источником элек-
ным гликозидам и окислительному стрессу
тронов для биосинтеза и участвует в реакции вос-
[7, 39].
становления глутатиона, НАД+ участвует в про-
цессах катаболизма и служит стоком электронов
[10].
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЙ
Изначально понятие редокc-статуса использо-
СТАТУС КЛЕТКИ
валось для описания соотношения восстановлен-
ной и окисленной пар молекул. Однако уже мно-
Окислительно-восстановительный (редокс)
статус клетки определяется соотношением вос-
го лет его используют в более широком смысле
становленных и окисленных форм молекул. В
для того, чтобы описать окислительно-восстано-
клетках животных много редокс-пар молекул, ко-
вительную среду клетки. Зачастую это понятие не
очень хорошо определено. В одной из ключевых
торые вовлечены в поддержание внутриклеточ-
работ по этой тематике [10] было высказано пред-
ного редокс-статуса, но самой распространенной
ложение характеризовать редокс-статус среды
является пара 2GSH/GSSG [9, 10]. Глутатион
(γ-глутамилцистеинилглицин) - основной низ-
как сумму редокс-потенциалов всех пар, каждый
комолекулярный тиол клеток животных. Он не
из которых умножен на концентрацию восста-
только определяет окислительно-восстанови-
новленного компонента пары. Хотя редокс-по-
тельный статус клетки, поддерживает SH-группы
тенциал пары НАДФ-Н/НАДФ+ ниже, чем ре-
белков в восстановленном состоянии, защищает
докс-потенциал пары 2GSH/GSSG, количество
клетку от активных форм кислорода (АФК), но и
восстановленного НАДФ-Н (0.1 мМ) [42] суще-
участвует в выводе ксенобиотиков. Окисленная
ственно ниже, чем GSH (5-10 мМ). Поэтому об-
форма глутатиона представляет собой гексапеп-
щий окислительно-восстановительный статус
тид, две молекулы глутатиона, соединенные SS-
клетки зависит в основном от пары 2GSH/GSSG,
мостиком. В норме содержание в клетке: GSH -
которую также называют главным редокс-буфе-
1-10 мM, GSSG - 50-500 мкM [10]. Следователь-
ром клетки [10].
но, в покоящихся клетках [GSH]/[GSSG] > 100.
При изменении редокс-статуса клетки ряд
Но в состоянии окислительного стресса это соот-
белков изменяет свое функционирование [9]. В
ношение может существенно снизиться:
1
<
большинстве случаев окислительные модифика-
< [GSH]/[GSSG] < 10 [10]. GSH синтезируется в
ции тиолов (SH) остатков цистеина обеспечива-
цитозоле в ходе двух ферментативных АТФ-зави-
ют редокс-зависимую регуляцию активности бел-
симых реакций, катализируемых глутамин-ци-
ков. Данные модификации можно разделить
стеин-лигазой и глутатион-синтетазой, и затем
на обратимые: cульфенирование (S-OH); глута-
транспортируется в различные компартменты
тионилирование (S-SG) - присоединение глута-
клетки [40]. Восстановление GSSG до GSH ката-
тиона к тиоловой группе белка с образованием
лизируется глутатион-редуктазой, которая в ка-
дисульфидного мостика; нитрозилирование
честве субстрата использует НАДФ-Н.
(S-NO) - присоединение NO и необратимые:
Редокс-потенциал, определяемый парой
сульфинирование (S-O2H) и сульфонирование
GSSG/GSH [10], при 25°C и pH 7.0, составляет
(S-O3H). В то время как обратимые модифика-
БИОФИЗИКА том 65
№ 5
2020
840
ПЕТРУШАНКО и др.
ции вовлечены в сигнальные функции и редокс-
гипоксии, так и в условиях гипероксии [6]. Таким
регуляцию функции белка, необратимые моди-
образом, было установлено, что активность
фикации приводят к потере контроля над регуля-
Na,K-АТФазы чувствительна к изменению ре-
цией функции белка и его последующей дегра-
докс-статуса клеток, реализующегося в случае
дации.
окислительного стресса, изменения уровня внут-
риклеточных тиолов и уровня оксигенации.
РЕДОКС-ЧУВСТВИТЕЛЬНОСТЬ
Na,K-АТФазы
МЕХАНИЗМЫ РЕДОКС-
Функционирование Na,K-АТФазы зависит от
ЧУВСТВИТЕЛЬНОСТИ Na,K-АТФазы
окислительно-восстановительного статуса кле-
ток. В первую очередь было обнаружено ингиби-
Основные механизмы редокс-чувствительно-
рование активности Na,K-АТФазы при развитии
сти Na,K-АТФазы можно разделить на две груп-
окислительного стресса, которое сопровождается
пы - прямая модификация тиоловых групп
снижением количества восстановленных SH-
остатков цистеина субъединиц белка и редокс-за-
групп α-субъединицы [4, 14, 20]. Затем было уста-
висимое фосфорилирование фермента [7, 8].
новлено, что тканеспецифичные изоформы α2 и
Наибольшее количество остатков цистеина
α3 более чувствительны к окислению, чем кон-
ститутивная α1-изоформа [4, 20, 43-45]. При
содержит каталитическая α-субъединица. В зави-
этом присутствие миллимолярных концентраций
симости от изоформы их количество составляет
23 или 24 остатка, 15 из которых расположены в
АТФ защищало фермент от окисления гидрок-
цитозоле и доступны для модификаций. Работы
сильными радикалами [46], а также от ингибиро-
вания цистеин-модифицирующим реагентом
по точечному мутагенезу остатков цистеина с за-
[47]. Проведенное сравнение последовательно-
меной их на аланин или серин, а также полная за-
стей изоформ показало, что α1- и α2-изоформы
мена всех остатков цистеина не позволили вы-
явить ни одного остатка цистеина, критичного
имеют равное число остатков цистеина, а изо-
для функционирования Na,K-АТФазы [54]. Му-
форма α3 имеет один дополнительный цистеин,
таким образом, более высокая чувствительность к
тант α1-субъединицы Na,K-АТФазы овцы с пол-
окислению изоформ α2 и α3 по сравнению с изо-
ной заменой остатков цистеина демонстрирует
формой α1 не связана с большим количеством
снижение K0.5 для Na+, двукратное возрастание
тиоловых групп в тканеспецифичных изоформах
K0.5 для K+ и не влияет на Km для АТФ. Хотя заме-
[7].
ны незначительно влияют на активность, ста-
Необратимое окисление тиоловых групп дела-
бильность функционального мономомера α-β
ет невозможным их дальнейшие редокс-зависи-
снижается. При этом замена Сys242 у овцы (соот-
мые модификации, вследствие чего белок стано-
ветствующего Сys244 у крысы) на аланин делает
вится нечувствительным к изменению редокс-
клетки очень чувствительными к кардиотониче-
статуса клетки [48]. Na,K-ATФаза с необратимо
скому стероиду уабаину [54]. Замены цитозоль-
окисленными тиоловыми группами подвергается
ных остатков цистеина могут приводить к инги-
деградации [49]. Активность Na,K-АТФазы мак-
бированию активности Na,K-АТФазы максимум
симальна в определенном диапазоне внутрикле-
в два раза, при этом K0.5 для Na+ не меняется, а
точных редокс-условий. Как истощение нейро-
сродство к K+ и АТФ изменяется. Большинство
нальных клеток по глутатиону, так и их загрузка
остатков цистеина, замена которых влияет на ак-
глутатионом приводит к ингибированию транс-
тивность фермента, локализовано в большой ци-
портной активности Na,K-АТФазы [5].
тозольной петле M4-M5. Поскольку замены не
Одним из первых свидетельств о редокс-чув-
вызывают существенного изменения активности
ствительности Na,K-АТФазы является ее инги-
[54], а модификация остатков цистеина может су-
бирование при снижении уровня кислорода.
щественно менять функционирование фермента
Способность Na,K-АТФазы отвечать на сниже-
[8, 48], можно заключить, что цистеины важны не
ние уровня кислорода была показана на различ-
непосредственно для активности Na,K-АТФазы,
ных типах клеток, включая нейроны, кардиомио-
а для редокс-чувствительной регуляции ее актив-
циты, эритроциты, гепатоциты, гладкомышеч-
ности. Тиоловые группы остатков цистеина α-
ные клетки, эпителиальные клетки легкого и т.д.
субъединицы Na,K-АТФазы не образуют SS-мо-
[11-13, 50-53]. Однако позже было установлено,
стиков [55]. Данные по мутагенезу также свиде-
что не только гипоксия, но и гипероксия может
тельствуют о том, что SS-мостики α-субъедини-
снижать активность фермента. Так, на грануляр-
цы не нужны для фолдинга и активности белка.
ных клетках мозжечка крыс было показано, что
Тиоловые группы α-субъединицы находятся в
активность Na,K-АТФазы имеет максимум при
восстановленном состоянии или могут быть мо-
физиологическом парциальном давлении кисло-
дифицированы: окислены, нитрозилированы
рода pO2 (3-5 кПа) и снижается, как в условиях
или глутатионилированы [8, 48, 55].
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
841
β-Субъединица имеет семь остатков цистеина.
бен катализировать как реакцию глутатионили-
Из них шесть находятся на внеклеточной части
рования, так и деглутатионилирования в зависи-
белка и образуют три SS-мостика, а один (Cys46)
мости от уровня внутриклеточного НАДФ-Н.
является восстановленным и локализован вблизи
Глутатионилирование α-субъединицы и его вли-
трансмембранной части белка, погружаясь и вы-
яние на активность Na,K-АТФазы. α-Субъедини-
ходя из мембраны в ходе каталитического цикла,
ца Na,K-АТФазы имеет около 23 остатков цисте-
вследствие чего он может подвергаться модифи-
ина (23 в изоформах α1 и α2 и 24 - в α3-изоформе)
кациям, в частности глутатионилированию
[7]. Около 15 цистеинов локализованы в цитозоле
[56, 57].
и, таким образом, могут подвергаться редокс-за-
Третья субъединица (FXYD), являющаяся тка-
висимым модификациям. Было обнаружено, что
неспецифичной, содержит в зависимости от изо-
α-субъединица Na,K-АТФазы подвергается глу-
формы один или два остатка цистеина, один из
татионилированию [48, 59]. На очищенном белке
которых может подвергаться глутатионилирова-
из солевых желез утки и почек кролика было по-
нию [57].
казано, что инкубация Na,K-АТФазы с GSSG ин-
дуцирует увеличение глутатионилирования α1-
субъединицы и приводит к ингибированию фер-
РЕГУЛЯЦИЯ АКТИВНОСТИ,
мента вплоть до полной его инактивации [48].
ОБУСЛОВЛЕННАЯ
Уровень глутатионилирования β-субъединицы
ГЛУТАТИОНИЛИРОВАНИЕМ
при этом не меняется. Инкубация с GSSG приво-
СУБЪЕДИНИЦ Na,K-АТФазы
дит к дозозависимому снижению активности
фермента, развивающемуся во времени. Ингиби-
Полученные данные демонстрировали, что
рующее действие GSSG является бифазным:
окисление тиоловых групп Na,K-АТФазы не мо-
жет объяснить наблюдаемое значительное инги-
быстрое взаимодействие фермента с GSSG при-
бирование фермента при гипоксии, поскольку
водит к снижению активности на 80%, затем мед-
даже полностью окисленный белок вследствие
ленное взаимодействие приводит к полной инак-
тивации белка. Для очищенного фермента из со-
инкубации с высокими концентрациями переки-
левых желез утки и кролика величина IC50
си водорода обладает активностью [4, 14, 15]. Тот
факт, что индуцированное изменение уровня
cоставляет около 60 мкМ [48]. Обработка сарко-
GSH в клетке существенно изменяет активность
леммальной фракции гомогената сердца крысы
фермента [5], а при гипоксии редокс-статус кле-
GSSG также приводила к росту глутатионилиро-
ток и соотношение GSH/GSSG меняется [6], поз-
вания α-субъединицы и ингибированию Na,K-
волил предположить, что глутатион может напря-
АТФазы на 80% [48, 59]. При этом преинкубация
мую модифицировать Na,K-АТФазу. Субъедини-
сарколеммальной фракции со 100%-м кислоро-
цы Na,K-АТФазы содержат тиоловые группы, а
дом в течение 30 мин или преинкубация очищен-
это значит, что при изменении редокс-статуса, в
ного белка в течение нескольких часов с 20% кис-
частности при окислительном стрессе, возможно
лорода приводила к исчезновению чувствитель-
S-глутатионилирование - образование смешан-
ности Na,K-АТФазы к действию GSSG, не влияя
ных дисульфидов между цистеинами белка и глу-
существенным образом на активность фермента
татионом. Возможные пути глутатионилирова-
[48]. Вероятно, это связано с окислением тиоло-
ния белков подробно описаны в работах [9, 58]. В
вых групп белка и утрате их способности взаимо-
частности, глутатионилирование возможно при
действовать с GSSG. Важно отметить, что инги-
взаимодействии тиоловой группы белка с GSSG
биторный эффект GSSG на сарколеммальную
или при взаимодействии частично окисленной
фракцию сердца значительно снижался при акти-
тиоловой группы с GSH [58]:
вации NO-синтаз [59]. Это может быть связано со
снижением глутатионилирования тиоловых
P-S- + GSSG → P-SSG + GS-,
групп вследствие нитрозилирования. Инкубация
P-SOH + GSH → P-SSG + H2O.
очищенного фермента с восстановителем, напри-
Соответственно, развитие окислительного
мер, дитиотреитолом снижает глутатионилирова-
стресса, сопровождающееся ростом GSSG и
ние α-субъединицы и восстанавливает актив-
окислением белков, будет приводить к индукции
ность фермента. Также деглутатионилирование и
глутатионилирования [9, 58]. Эта модификация
восстановление активности фермента достигает-
защищает тиоловые группы белков от необрати-
ся при инкубации с глутаредоксином и НАДФ-H
мого окисления, а в некоторых случаях приводит
[48]. Индукция глутатионилирования α-субъеди-
к изменению функционирования белка. При
ницы Na,K-АТФазы была продемонстрирована
нормализации редокс-условий в клетке равнове-
не только на очищенном белке из почек и соле-
сие смещается в сторону реакции деглутатиони-
вых желез, но и на ряде клеточных линий, ткани
лирования, протекающей с участием специаль-
сердца, мышечной ткани в разных видах живот-
ных ферментов, таких как глутаредоксин [58].
ных: утке, кролике, свинье, мыши, крысе, слепы-
Необходимо отметить, что глутаредоксин спосо-
ше и человеке [48, 55, 59-67]. Это свидетельствует
БИОФИЗИКА том 65
№ 5
2020
842
ПЕТРУШАНКО и др.
о том, что механизм глутатионилирования α-
вых нуклеотидов (АТФ, АДФ, АМФ) ингибиро-
субъединицы является общим для различных ор-
вание Na,K-АТФазы GSSG нарушается [48, 64].
ганов и тканей в различных организмах.
Наиболее эффективным защитным действием
В мышечных тканях кроме α1-субъединицы
обладал АТФ, поскольку меньшие его концентра-
ции требовались для нивелирования эффекта ин-
экспрессируется α2-субъединица, которая, бу-
дучи солокализована с Na/Ca-обменником, игра-
гибирования GSSG [64]. Это может объясняться
ет важную роль в регуляции мышечного сокраще-
тем, что связывание АТФ приводит к изменению
ния [32, 68]. На ткани сердца и скелетных мыш-
конформации белка, в частности, к сближению
цах было показано, что глутатионилированию
фосфорилируемого и нуклеотидсвязывающего
доменов, переводя фермент в E1-закрытое состо-
подвергается как α1-, так и α2-изоформа Na,K-
АТФазы [48, 62-65]. При этом чувствительность
яние, что делает регуляторные цистеины недо-
α2-изоформы к глутатионилированию выше, чем
ступными для взаимодействия с GSSG [71].
чувствительность α1-изоформы [48]. На препара-
Было высказано предположение, что основ-
те из сердца крысы было показано, что концен-
ным регуляторным остатком является Cys452,
трация GSSG, ингибирующая α2β-изофермент
находящийся вблизи участка связывания АТФ
на 50%, в шесть раз ниже, чем концентрация, ин-
[48]. Согласно данным молекулярного моделиро-
гибирующая α1β-изофермент (IC50 cоставляет
вания, внесение отрицательного заряда вслед-
около 44 и 265 мкМ соответственно) [48]. Это хо-
ствие глутатионилирования может нарушать свя-
рошо коррелирует c большей чувствительностью
зывание АТФ. Это также соответствовало дан-
α2-изоформы к окислению
[69]. Хотя число
ным масс-спектрометрии, согласно которым при
остатков цистеина в этих изоформах одинаково,
инкубации с GSSG увеличивается уровень глута-
α2-изоформа отличается от α1 дополнительным
тионилирования остатков Cys454, 456, 459, лока-
цистеином в актуаторном домене (Cys 236) и от-
лизованных в нуклеотидсвязывающем домене
сутствием цистеина в нуклеотидсвязывающем
вблизи сайта связывания АТФ и Сys244, локали-
домене (Cys 458). Ранее было высказано предпо-
зованного в малой цитозольной петле, формиру-
ложение [7], что большая чувствительность к
ющей актуаторный домен [48]. Однако экспери-
окислению фермента с α2-изоформой по сравне-
менты по точечному мутагенезу показали, что
нию с α1-изоформой может быть следствием
наиболее критичным для ингибирования актив-
сдвига равновесия Е2-Е1 в сторону более откры-
ности при глутатионилировании являются вовсе
той в цитозоль конформации E1 [23]. Еще одной
не Cys454, 456, 459, а остаток Cys244 [60]. При за-
возможной причиной является разное окружение
мене этого остатка на аланин Na,K-АТФаза теря-
остатков цистеина в этих изоформах. Анализ двух
ет способность ингибироваться при инкубации с
ближайших аминокислот вблизи остатков цисте-
GSSG. В настоящее время причина снижения ак-
ина в последовательности α1- и α2-изоформ по-
тивности при глутатионилировании Cys244 не
казал, что для остатков Сys 206 и 579 в α2-изофор-
совсем ясна. Остаток Cys244 может приближать-
ме по сравнению с α1-изоформой происходит
ся к АТФ-связывающему сайту, когда Na,K-
замена незаряженных аминокислот на положи-
АТФаза находится в E2-конформации. В кон-
тельно заряженные, что может приводить к сдви-
формации E2 АТФ связывается с белком с низ-
гу pK тиоловых групп цистеинов и, следователь-
ким сродством и способствует уходу K+ [7]. Веро-
но, к их большей доступности для редокс-моди-
ятно, глутатионилирование Cys244 нарушает
фикаций [8].
данный процесс.
Возрастание уровня глутатионилирования
Эффективность глутатионилирования α-субъ-
α-субъединицы приводит к значительному сни-
единицы зависит от конформации фермента, что
жению активности фермента, вплоть до его пол-
связано с изменением доступности остатков ци-
ного ингибирования [48]. Такое значительное из-
стеина [72]. Максимальное увеличение уровня
менение активности белков при глутатионилиро-
глутатионилирования α-субъединицы при инку-
вании может наблюдаться в случае, если
бации с GSSG наблюдается в случае, когда фер-
глутатионилирующийся остаток цистеина нахо-
мент находится в E1-конформации. В E2-кон-
дится вблизи активного центра, как в случае с
формации эффективность глутатионилирования
альдозоредуктазой [9, 70]. Было установлено, что
снижается. Самая низкая эффективность глута-
причиной ингибирования Na,K-АТФазы являет-
тионилирования наблюдается в E2P- и E2P-уаба-
ся нарушение связывания адениновых нуклеоти-
ин-конформациях. Как было сказано выше, при
дов [48]. Вследствие этого преинкубация Na,K-
изменении конформации меняется взаимное
АТФазы с GSSG нарушает дозозависимую акти-
расположение цитозольных доменов. Так, со-
вацию фермента АТФ. С другой стороны, преин-
гласно данным ограниченного трипсинолиза, в
кубация Na,K-АТФазы с возрастающими кон-
E1 конформации большая и малая цитозольные
центрациями АТФ приводит к дозозависимому
петли находятся отдельно друг от друга, в то вре-
снижению ингибирующего действия GSSG [48].
мя как в E2-конформации они приближены друг
В присутствии высоких концентраций аденино-
к другу [23]. Таким образом, можно было предпо-
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
843
ложить, что в Е1-конформации большая часть
стично устраняла эти модификации, что приводи-
остатков цистеина доступна для модификации.
ло к незначительному увеличению активности фер-
Проведенный анализ существующих структур
мента. В работе были использованы три химиче-
Na,K-АТФазы показал, что действительно в E1P-
ских восстановителя с различными стандартными
конформации количество доступных растворите-
редокс-потенциалами: трис(2-карбоксиэтил)фос-
лю остатков цистеина максимально, в то время
фин, дитионит натрия (Na2S2O4) и боргидрид на-
как в E2P- и E2P-уабаин-конформациях оно
трия (NaBH4), которые имеют окислительно-вос-
существенно ниже [72]. Таким образом, эффек-
становительные потенциалы -290 мВ, -416 мВ и
тивность глутатионилирования коррелирует с ко-
-1024 мВ соответственно. Максимальную эф-
личеством остатков цистеина, доступных рас-
фективность в снятии глутатионилирования,
творителю [72]. Обнаруженная зависимость эф-
нитрозилирования и снижении уровня окисле-
фективности глутатионилирования от кон-
ния продемонстрировал самый сильный восста-
формации фермента может пролить свет на роль
новитель - боргидрид натрия. Только под дей-
лигандов и белков-партнеров во влиянии на ре-
ствием этого восстановителя наблюдалось досто-
докс-модификацию тиоловых групп Na,K-
верное возрастание активности белка, однако оно
АТФазы и регуляцию его активности. В частно-
составляло менее 20%. В то же время фермента-
сти, связывание уабаина будет снижать эффек-
тивная система глутаредоксин/глутатионредук-
тивность глутатионилирования фермента. Кроме
таза деглутатионилировала нативную Na,K-
того, смещение равновесия E2-E1 может быть
АТФазу в меньшей степени, чем химические вос-
причиной большей чувствительности к глутатио-
становители (на 20%), но, в отличие от них, при-
нилированию тканеспецифичной α2-изоформы
водила к значительному увеличению активности
по сравнению с α1-изоформой.
фермента (до 30%). Таким образом, фермента-
Для того чтобы установить, как влияет глута-
тивная восстановительная система специфиче-
тионилирование на конформацию фермента, был
ски влияет на глутатионилирование регулятор-
проведен анализ трипсинолиза α-субъединицы
ных остатков цистеина α1-субъединицы Na,K-
для ряда конформаций Na,K-АТРазы, глутатио-
АТФазы. На белке из почек кролика, обработан-
нилированной в различной степени [74]. Деглута-
ном восстановителями дититотретитолом, мер-
тионилирование α-субъединицы приводит к уве-
каптоэтанолом и трис(2-карбоксиэтил)фосфи-
личению скорости трипсинолиза для фермента в
ном, также было отмечено снижение исходного
E1- и E2-конформациях. Дополнительное увели-
глутатионилирования белка и некоторое повы-
чение уровня глутатионилирования фермента в
шение активности [66]. Во всех случаях полное
конформации E2P приводит к увеличению содер-
снятие глутатионилирования с нативного белка
жания более высокомолекулярных фрагментов
оказалось невозможным [66, 67].
по сравнению с нативным и деглутатионилиро-
Кроме регуляторного глутатионилирования α-
ванным препаратами. Другими словами, глутати-
субъединицы, которое влияет на активность фер-
онилирование уменьшает количество доступных
мента и может быть снято восстановителями, бы-
сайтов для протеолиза по сравнению с неглутати-
ло обнаружено также базальное глутатионилиро-
онилированным белком. Это свидетельствует о
вание. Это глутатионилирование не может быть
том, что глутатионилирование влияет на конфор-
удалено при обработке нативного белка восста-
мацию фермента и его чувствительность к трип-
новителями. Однако обработка восстановителя-
синолизу [74]. Таким образом, можно предпола-
ми денатурированного белка приводит к деглута-
гать, что глутатионилирование α-субъединицы
тионилированию [55]. Было высказано предпо-
вследствие влияния на конформацию Na,K-
ложение, что связывание глутатиона с рядом
АТФазы может изменять ее взаимодействие с
остатков цистеина происходит в процессе синте-
белками-партнерами.
за белка, и в нативном белке данные остатки ци-
Поскольку Na,K-АТФаза является редокс-
стеина уже недоступны для модификации. Дей-
чувствительным ферментом, при исследовании
ствительно, на клетках фибробластов мыши ли-
ее свойств важно представлять, в каком состоя-
нии SC1 было показано, что длительная гипоксия
нии находятся тиоловые группы белка. В одной
в течение нескольких суток (72 ч) приводит к воз-
из работ были охарактеризованы модификации
растанию уровня базального глутатионилирова-
остатков цистеина α1-субъединицы очищенного
ния Na,K-АТФазы, в то время как при острой ги-
препарата Na,K-АТФазы из солевых желез утки
поксии происходит увеличение только регуля-
[67]. Было установлено, что α1-субъединица
торного глутатионилирования [55]. Тщательный
Na,K-АТФазы содержит глутатионилированные,
анализ структур Na,K-АТФазы (PBD коды: 3B8E,
нитрозилированные и окисленные до SOH и
3KDP, 3WGU, 3WGV, 4HYNT) показал, что вбли-
SO2H остатки цистеина. Данные модификации
зи некоторых остатков цистеина (Cys206-Cys244;
были зарегистрированы как с помощью Вестерн-
Cys454-Cys458-Cys459; Cys700-Сys369; Cys601)
блоттинга, так и с помощью масс-спектрометрии.
имеются полости с неразрешенной электронной
Инкубация с химическими восстановителями ча-
плотностью [55]. Они не могут быть объяснены
БИОФИЗИКА том 65
№ 5
2020
844
ПЕТРУШАНКО и др.
присутствием воды и по размеру соответствуют
глутатионилирования β-субъединицы под дей-
молекуле глутатиона. Глутатионилированные
ствием ONOO- наблюдалось также на очищен-
остатки белков часто регистрируют с помощью
ном белке из почек свиньи [56]. Инкубация с ди-
масс-спектроскопии, однако детекция глутатио-
тиотреитолом приводила к существенному сни-
нилированных белков в кристаллических струк-
жению уровня глутатионилирования. Интересно
турах очень редка. Впервые подход по обнаруже-
отметить, что в присутствии ингибитора NO-син-
нию глутатионилированных остатков цистеина с
таз (L-NAME) общий уровень глутатионилирова-
помощью анализа неразрешенной электронной
ния был ниже и не возрастал под действием ан-
плотности вблизи остатков цистеина в структуре
гиотензина
[56]. Позже глутатионилирование
белка (митохондриального АВС транспортера)
β-субъединицы было показано не только для сер-
был описан в работе [74]. Анализ структур α-субъ-
дечной мышцы, но и для скелетных мышц и глад-
единицы Na,K-ATФазы показал, что в полости с
кой мускулатуры [62, 63, 75]. В отличие от глута-
неразрешенной плотностью направлены тиоло-
тионилирования α-субъединицы, индукция глу-
вые группы двух остатков цистеинов, при этом
татионилирования Cys46 приводит лишь к
глутатион может быть связан только с одним из
частичному ингибированию фермента, его ак-
них [55]. Дисульфидные связи в α-субъединице
тивность снижается на 20% [56].
не обнаружены [55]. Анализ первичной последо-
Предполагают, что эффект ингибирования
вательности вблизи цистеиновых групп белков
Na,K-АТФазы связан с нарушением взаимодей-
показал, что, действительно, только в ближайшей
ствия между α- и β-субъединицами [56], посколь-
окрестности одного из цистеинов имеются поло-
ку в результате глутатионилирования β-субъеди-
жительно заряженные аминокислотные остатки,
ницы снижается коиммунопреципитация этих
смещающие pK тиоловой группы, что увеличива-
субъединиц. К сожалению, в ряде случаев реги-
ет способность данного цистеина глутатионили-
страция глутатионилирования β-субъединицы не
роваться. В паре Cys204-Cys242 это Cys204, в па-
сопровождалась оценкой глутатионилирования
ре Cys367-Cys698 - Cys698; в паре Cys452-
α-субъединицы, поэтому вычленить вклад глута-
Cys456 - Cys456. Было высказано предположе-
тионилирования каждой из субъединиц сложно
ние, что базальное глутатионилирование, проис-
[75]. Глутатионилирование β-субъединицы за-
ходящее в момент синтеза белка, может лежать в
медляет скорость перехода E1-E2P и скорость
основе так называемой редокс-памяти, обуслав-
перехода E2-E1-E2P [56].
ливая изменение свойств белка, синтезированно-
Необходимо отметить, что Cys46 находится
го в измененных редокс-условиях. Вероятно, ба-
вблизи мембраны и заглублен в нее в случае
зальное глутатионилирование Na,K-АТФазы
2K+-E2P-конформации
[76]. На очищенном
является котрансляционной модификацией, из-
белке из почек свиньи было показано, что дан-
меняющей свойства белка для адаптации к дли-
ный остаток доступен для глутатионилирования
тельному изменению редокс-условий [55]. Дан-
в E1ATP- и E1Na(3)-конформации [77]. Так, для
ный аспект редокс-регуляции белков еще пред-
фермента в E1ATP-конформации уровень глута-
стоит изучить.
тионилированной β-субъединицы почти в де-
Глутатионилирование β-субъединицы и его вли-
сять раз выше, чем в E2-конформации, а для E1-
яние на активность Na,K-АТФазы. У β-субъедини-
Na(3)-конформации в два с половиной раза вы-
цы есть только один восстановленный остаток
ше, чем в E2-конформации. При этом разницы
цистеина, который может подвергаться редокс-
между глутатионилированием в E2- и E2K-кон-
модификациям, в частности, глутатионилирова-
формациях не наблюдалось. Кроме того, инку-
нию - Cys46. В отличие от α-субъединицы инку-
бация белка с ONOO- приводила к существен-
бация фермента с GSSG не приводит к измене-
ному возрастанию глутатионилирования в E1-
нию уровня глутатионилирования β-субъедини-
конформации (более чем в два раза), но не меня-
цы, что было показано на очищенных препаратах
ла глутатионилирования белка, находящегося в
белка из солевых желез утки [48]. Впервые глута-
E2-конформации [77]. Наблюдаемые эффекты
тионилирование β1-субъединицы было обнару-
связаны с тем, что Сys46 в E2-конформации
жено в ткани сердца кролика [56]. Увеличение
практически недоступен. При этом ингибирова-
уровня глутатионилирования β1-субъединицы
ние активности при инкубации белка с ONOO-
наблюдалось под действием пероксинитрита
и GSH также наблюдалось гораздо более суще-
(ONOO-), перекиси водорода, параквата и акти-
ственное, если белок находился в E1Na(3)-, а не
вации HAДФ-H-оксидазы ангиотензином II [56].
в E2-конформации. Данный эффект наблюдал-
Преинкубация с супероксиддисмутазой приводи-
ся как на очищенном белке из почек свиньи, так
ла к снижению глутатионилирования индуциро-
и на кардиомиоцитах свиньи, однако характер
ванного паракватом или активацией HAДФ-H-
ингибирования различался вследствие того, что
оксидазы. Таким образом, образование суперок-
в кардиомиоцитах кроме β1-субъединицы при-
сид-анион-радикала вовлечено в индукцию глу-
сутствует нечувствительная к глутатионилиро-
татионилирования β-субъединицы. Увеличение
ванию β2-субъединица [77]. Преинкубация кар-
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
845
диомиоцитов с уабаином, который фиксирует
навливает реакционную способность данного
фермент в E2P-конформации, предотвращает
остатка цистеина. И, наоборот, замена положи-
глутатионилирование индуцированное ONOO-
тельно заряженного остатка на глицин в FXYD1
[77]. Таким образом, присутствие уабаина сни-
лишает его описанной функции. Эти данные сви-
жает уровень глутатионилирования β-субъеди-
детельствуют в пользу гипотезы о том, что поло-
ницы при окислительном стрессе. В то время
жительно заряженные аминокислотные остатки в
как глутатионилирование β1-субъединицы в
ближайшем окружении остатка цистеина, сдви-
конформации E2 минимально, коиммунопре-
гая pK тиоловой группы, повышают эффектив-
ципитация β1- и α-субъединицы, напротив, вы-
ность глутатионилирования [80]. Уровень глута-
ше в E2-конформации. Глутаредоксин, снижая
тионилирования FXYD возрастает при инкуба-
глутатионилирование β-субъединицы, повыша-
ции с ONOO-. Инкубация с дитиотреиолом
ет коиммунопреципитацию α- и β-субъединиц.
снимает глутатионилирование. Активатор адени-
Полученные данные свидетельствуют о том, что
латциклазы форсколин, индуцирующий увеличе-
глутатионилирование β-субъединицы нарушает
ние глутатионилирования β1-субъединицы [81],
ее взаимодействие с α-субъединицей [77]. С дру-
приводит также к возрастанию глутатионилиро-
гой стороны, доступность Cys46 может возрас-
вания FXYD1 [57]. Супероксиддисмутаза предот-
тать при нарушении взаимодействия между α- и
вращает индукцию глутатионилирования, вы-
β- субъединицами [78].
званную форсколином, что еще раз подтверждает
важность супероксид-анион-радикала в процессе
Трансмембранные домены изоформ β-субъ-
индукции глутатионилирования.
единицы β2 и β3 имеют не очень высокую гомо-
логию с β1 (от 57 до 61%) [79] и, вероятно, вслед-
Инкубация кардиомиоцитов с ангиотензин-
ствие этого изоформы β2 и β3 не способны глута-
ном II приводит к возрастанию уровня глутатио-
нилирования FXYD1. Также увеличение глутати-
тионилироваться. Предполагают, что Cys46 в
данных изоформах недоступен для модификаций
онилирования было обнаружено в зоне инфарк-
[56].
та. Было установлено, что глутатионилирование
Глутатионилирование FXYD-субъединицы и его
и ингибирование β1 может быть обращено добав-
влияние на активность Na,K-АТФазы. Хотя функ-
лением FXYD3. Однако мутантная безцистеино-
вая форма FXYD не оказывает подобного дей-
циональный мономер Na,K-АТФазы состоит из
ствия. В сердцах FXYD-/-мышей уровень глута-
α- и β-субъединиц, третья субъединица из семей-
ства FXYD-белков может связываться с фермен-
тионилирования β-субъединицы был повышен.
том и влиять на его активность. Семейство FXYD
Таким образом, предполагают, что FXYD играет
содержит около семи небольших трансмембран-
важную роль в деглутатионилировании β1-субъ-
единицы, восстанавливая ее активность, и,
ных белков, которые могут модулировать актив-
следовательно, вовлечена в динамическую регу-
ность Na,K-АТФазы. Эти белки в зависимости от
изоформы содержат от одного до двух цистеинов,
ляцию активности Na,K-АТФазы.
которые могут быть доступны для глутатионили-
Установлено, что глутатионилирование субъ-
рования. Впервые глутатионилирование было
единицы FXYD влияет на ее взаимодействие с α-
показано для субъединицы FXYD1, экспрессиру-
и β-субъединицами, которое было оценено c по-
ющейся в сердце и известной как фосфолемман
мощью коиммунопреципитации. Возрастание
[57]. Глутатионилирование FXYD1 облегчает де-
уровня глутатионилирования FXYD вследствие
глутатионилирование β1-субъединицы и таким
инкубации с ангиотензином или в зоне инфарк-
образом обращает окислительное ингибирование
та приводит к снижению взаимодействия FXYD
Na,K-АТФазы. Субъединица FXYD содержит два
с α-субъединицей, тогда как взаимодействие
остатка цистеина (Сys40 и Сys42 у FXYD1 челове-
FXYD с β-субъединицей возрастает [75]. До на-
ка). С помощью точечного мутагенеза было пока-
стоящего времени не ясен механизм деглутатио-
зано, что FXYD1 подвергается глутатионилиро-
нилирования β-субъединицы в присутствии
ванию по остатку Cys42 [57]. Подверженность
FXYD. Это может происходить как вследствие
данного остатка глутатионилированию обуслов-
реакции тиол-дисульфидного обмена между
лена тем, что остаток Сys42, в отличие от Сys40,
этими субъединицами, так и вследствие того,
имеет в своем окружении положительно заря-
что глутатионилированная FXYD-субъединица,
женные аминокислоты [57]. Сys42 в FXYD1 и
которая сильнее взаимодействует с β-субъеди-
FXYD7 фланкирован остатками аргинина и лизи-
ницей, предотвращает ее глутатионилирование.
на. Изоформа FXYD2, в которой Сys42 фланки-
Анализ структур показывает, что способные глу-
рован не двумя, а одной положительно заряжен-
татионилироваться остатки цистеина β-субъеди-
ной аминокислотой, обладает меньшей способ-
ницы и FXYD расположены далеко, что делает не-
ностью глутатионилироваться и вследствие этого
возможным прямой тиол-дисульфидный обмен
не способна снижать глутатионилирование β1-
[75]. Необходимо отметить, что в отношении фос-
субъединицы. Введение второго положительно
фолеммана (FXYD1), присоединение которого ре-
заряженного остатка рядом с цистеином восста-
гулирует активность Na,K-АТФазы в сердце, су-
БИОФИЗИКА том 65
№ 5
2020
846
ПЕТРУШАНКО и др.
ществуют противоречивые данные о роли влия-
культуре клеток фибробластов мыши линии SC1
ния его модификаций, фосфорилирования,
[61]. При гипоксии происходит снижение про-
пальмитоилирования (присоединение пальми-
дукции NO, развитие окислительного стресса и
тиновой кислоты) и глутатионилирования в
рост GSSG [48, 59]. Так, было показано, что в тка-
условиях окислительного стресса на активность
ни сердца при гипоксии происходит смещение
Na,K-АТФазы [82], которые в настоящее время
баланса GSH/GSSG в сторону GSSG [48], и ре-
пока не решены.
докс-потенциал пары GSH/GSSG сдвигается в
Итак, все три изоформы Na,K-АТФазы могут
более окисленную область [59]. Это приводит к
подвергаться глутатионилированию. При этом
снижению нитрозилирования и росту глутатио-
нилирования α-субъединицы Na,K-ATФазы [48,
максимальное изменение активности, вплоть до
полного ингибирования, наблюдается в случае α-
59]. Возрастание уровня глутатионилирования и
субъединицы за счет нарушения связывания
ингибирование фермента наблюдаются как при
АТФ. Глутатионилирование β-субъединицы при-
гипоксии на изолированном сердце [48, 59], так и
водит к ингибированию фермента на 20% вслед-
при инкубации животного в гипоксийной камере
[59]. Также S-глутатионилирование α1-субъеди-
ствие нарушения взаимодействия α- и β-субъеди-
ниц. Как для α-, так и для β-субъединицы макси-
ницы Na,K-ATФазы наблюдалось в клетках фиб-
мальная эффективность глутатионилирования
робластов мыши линии SC1 при их инкубации в
наблюдается для белка в E1-конформации, сни-
течение трех с половиной часов в условиях гипо-
жаясь в E2- и E2P-конформациях. Связывание
ксии (0.2% pO2 и 0.05% pO2) [55, 61].
уабаина с ферментом, фиксирующее Na,K-
Поскольку снижение уровня АТФ увеличивает
АТФазу в E2P-конформации, приводит к резкому
эффективность глутатионилирования [64], то в
снижению способности к глутатионилированию
этом случае процесс глутатионилирования α-субъ-
α- и β-субъединиц. Необходимо отметить, что в
единицы должен протекать существенно быстрее.
случае α-субъединицы глутатионилированию
Было высказано предположение, что глутатиони-
подвержены не только повсеместно распростра-
лирование α-субъединицы Na,K-АТФазы, приво-
ненная α1-, но и тканеспецифичная α2-изофор-
дящее к ее ингибированию, является одним из эта-
ма, тогда как в случае только одна из трех изо-
пов адаптации, позволяющим предотвратить исто-
форм - β1, а в случае с FXYD только некоторые из
щение клеток по АТФ [8, 48] в первые минуты
изоформ, в частности FXYD1 и FXYD7.
гипоксии. Доля АТФ, которая расходуется на под-
держание работы Na,K-ATФазы, довольно суще-
РОЛЬ ГЛУТАТИОНИЛИРОВАНИЯ
ственна - от 20% внутриклеточных запасов в мы-
И НИТРОЗИЛИРОВАНИЯ СУБЪЕДИНИЦ
шечной ткани до 80% в головном мозге [84]. Дей-
Na,K-АТФазы В РЕГУЛЯЦИИ АКТИВНОСТИ
ствительно, замена в α-субъединице регуляторных
ФЕРМЕНТА ПРИ РАЗЛИЧНЫХ
остатков цистеина на аланин предотвращает инги-
ПАТОЛОГИЯХ
бирование фермента окисленным глутатионом и
приводит к снижению жизнеспособности клеток
Изменение уровня глутатионилирования
HEK293, экспрессирующих данный белок, в усло-
Na,K-АТФазы может являться причиной измене-
виях гипоксии (1% O2, 24 ч) [60]. Как было сказано
ния активности фермента при патологиях, сопро-
выше, наиболее критичным остатком, глутатиони-
вождающихся нарушением редокс-статуса.
лирование которого приводит к существенному
Роль глутатионилирования и нитрозилирования
снижению активности, является Cys244 [60]. Кро-
α-субъединицы в регуляции активности Na,K-
ме того, было установлено, что инкубация клеток
АТФазы при гипоксии и ишемии. Ингибирование
SC1 с веществами, индуцирующими глутатиони-
Na,K-АТФазы является одним из самых ранних и
лирование Na,K-АТФазы, приводит к лучшей вы-
критичных ответов клеток на гипоксию [6, 8, 48,
живаемости клеток в условиях гипоксии и препят-
59, 83]. Как было описано ранее, оно наблюдается
ствует существенному снижению АТФ [61].
в самых разных тканях. Ингибирование Na,K-
АТФазы приводит к быстрой диссипации гради-
Схема редокс-регуляции активности Na,K-
ента Na+ и K+ и гибели клетки. Однако, с другой
АТФазы вследствие модификации регуляторных
стороны, ингибирование Na,K-АТФазы начина-
остатков цистеина α-субъединицы приведена на
ется существенно раньше, чем развивается значи-
рис. 1. Фермент находится в динамическом рав-
тельный окислительный стресс, и не может быть
новесии между различными состояниями. Его
объяснено просто необратимым окислением
максимальная активность наблюдается в опреде-
остатков цистеина фермента [8]. Обнаружено,
ленных редокс-условиях. В полностью активном
что ключевым фактором, приводящим к ингиби-
ферменте регуляторные остатки цистеина не мо-
рованию фермента при гипоксии, является глута-
дифицированы. Окислительный стресс, в том
тионилирование каталитической α-субъединицы
числе вызванный гипоксией, приводит к окисле-
фермента [48]. Этот эффект был обнаружен в го-
нию SH-групп регуляторных цистеинов до SOH и
могенате сердца крысы [48, 59] и затем показан в
последующему глутатионилированию вследствие
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
847
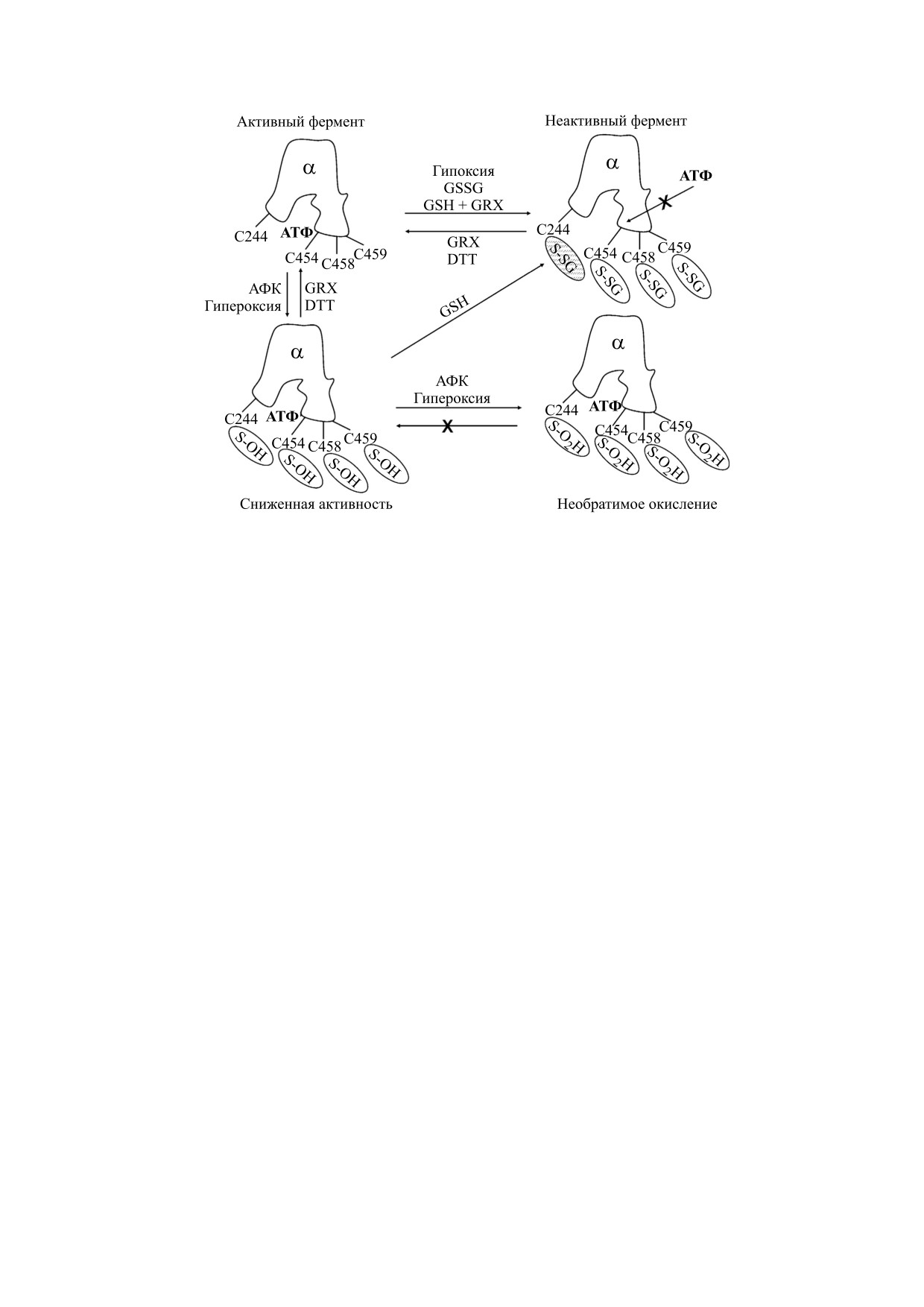
Рис. 1. Редокс-регуляция активности Na,K-АТФазы вследствие модификации остатков цистеина α-субъединицы. На
схеме отмечены регуляторные остатки цистеина α-субъединицы. В полностью активном ферменте они не модифици-
рованы. Окислительный стресс, в том числе вызванный гипоксией, приводит к окислению SH-групп регуляторных
остатков цистеина до SOH и последующему глутатионилированию вследствие их взаимодействия с GSH или к глута-
тионилированию SH-групп вследствие реакции тиол-дисульфидного обмена с GSSG, уровень которого возрастает.
Истощение по АТФ усиливает эффективность глутатионилирования. Главную роль в ингибировании фермента игра-
ет глутатионилирование остатка Cys244 (выделено штриховкой). В результате нарушается связывание АТФ. Ингиби-
рование вследствие глутатионилирования регуляторных цистеинов обратимо при восстановлении нормальных ре-
докс-условий, реакция деглутатионилирования может катализироваться, в частности, глутаредоксином (GRX). В слу-
чае сильного окислительного стресса тиоловые группы не успевают глутатионилироваться и возможно их
необратимое окисление до SO2H и SO3H. Фермент с необратимо окисленными группами подвергается деградации.
их взаимодействия с GSH или к глутатионилиро-
к снижению повреждения фибробластов мыши
ванию SH-групп вследствие реакции тиол-ди-
линии SC1 при гипоксии [61]. На кардиомиоци-
сульфидного обмена с GSSG, уровень которого
тах из сердца крысы было показано, что данные
возрастает. Истощение по АТФ усиливает эффек-
вещества, в особенности этилглутатион и нитро-
тивность глутатионилирования. Глутатионили-
зоглутатион, способны снижать повреждающее
рование защищает тиоловые группы белка от не-
действие гипоксии и существенно увеличивать
обратимого окисления. При этом глутатионили-
время нормальной сократимости клеток [65].
рование регуляторного остатка Cys244 приводит
Наибольшим защитным эффектом обладал нит-
к нарушению связывания АТФ и инактивации
розоглутатион, который не только увеличивал
фермента. Однако эта инактивация обратима при
время сократимости клеток, сохраняя форму
восстановлении нормальных редокс-условий. В
Ca2+-пика близкой к нормальной, но и более чем
частности, реакция деглутатионилирования мо-
на треть ускорял восстановление нормальной со-
жет катализироваться глутаредоксином. В том
кратительной функции сердца [65]. Тканеспеци-
случае, если окислительный стресс сильный и
фичная α2-субъединица более важна для регуля-
тиоловые группы не успевают глутатионилиро-
ции уровня кальция и сократимости кардиомио-
ваться, возможно их необратимое окисление до
цитов, чем конститутивная α1-субъединица,
SO2H и SO3H. Фермент с необратимо окисленны-
которая осуществляет контроль транспорта Na+ и
ми группами подвергается деградации (рис. 1).
K+ [68, 85, 86]. Оказалось, что инкубация кардио-
Было обнаружено, что инкубация клеток с ве-
миоцитов с нитрозоглутатионом приводит к уве-
ществами, индуцирующими глутатионилирова-
личению уровня глутатионилирования α2-субъ-
ние α-субъединицы Na,K-АТФазы, - проникаю-
единицы, не изменяя глутатионилирование α1
щим аналогом глутатиона этилглутатионом, нит-
[65]. Это соответствует полученным ранее дан-
розоглутатионом, N-ацетилцистеином приводят
ным о том, что α2-изоформа сердца крысы более
БИОФИЗИКА том 65
№ 5
2020
848
ПЕТРУШАНКО и др.
чувствительна к глутатионилированию, чем α1
ляет активность Na,K-АТФазы. Регуляторное
[48]. Предполагают, что специфические ингиби-
глутатионилирование - механизм адаптации кле-
торы α2 способны индуцировать положительный
ток к окислительному стрессу и гипоксии.
ионотропный эффект, не вызывая перегрузку
У ряда животных, способных переживать ги-
кардиомиоцитов по Са2+ и аритмию [85]. По-ви-
поксию, процесс диссипации градиента Na+ и K+
димому, в определенных концентрациях нитро-
сильно замедлен за счет того, что при ингибиро-
зоглутатион может работать как селективный ин-
вании Na,K-АТФазы реализуется закрытие ион-
гибитор α2 в кардиомиоцитах [65]. Однако пока
ных каналов, так называемый «канальный арест»
непонятно, опосредовано ли защитное действие
[87]. При этом деятельность животного прекра-
нитрозоглутатиона при гипоксии только глутати-
щается [88-91]. Отмечалось, что способность к
онилированием α2-субъединицы или в это вовле-
выживанию в условиях гипоксии сохраняется да-
чены и другие механизмы.
же в срезах ткани мозга гибернирующих живот-
Регуляция активности Na,K-АТФазы при ги-
ных, а уабаин ее повышает [92]. Кроме того, есть
поксии тесно связана с изменением редокс-ста-
устойчивые к гипоксии виды, такие как черепаха,
туса, а также уровнем глутатионилирования и
а также ведущие подземный образ жизни живот-
нитрозилирования белка. Важно отметить, что
ные: голый землекоп (Heterocephalus glaber) и сле-
гипоксия изолированного сердца крысы приво-
пыш (Spalax mole rate), которые способны быть
дит не только к повышению уровня глутатиони-
активными при гипоксии. В условиях понижен-
лирования α-субъединицы [48, 59], но и сниже-
ного кислорода у этих животных, в отличие от
нию уровня ее нитрозилирования [59]. Подробно
крыс, не наблюдается ингибирования Na,K-
это было исследовано на модели изолированного
АТФазы. Было показано, что при критичном pO2
сердца крысы. При гипоксии редокс-потенциал,
слепыши (Spalax judaei и Spalax galili) по крайней
обусловленный парой GSH/GSSG, сдвигается в
мере в течение 20 мин способны поддерживать
более окисленную область. Так, в гомогенате
редокс-статус клеток достаточно восстановлен-
сердца крысы он изменяется от -266 мВ (при 20%
ным для того, чтобы Na,K-АТФаза не ингибиро-
pO2) до -256 мВ (при 5% pO2). При этом произ-
валась [59]. Интересно отметить, что у слепышей
водство NO в условиях гипоксии снижается [59].
локализация остатков цистеина такая же, как и у
Это связано с тем, что NO-синтазы обладают низ-
крысы, и они тоже способны подвергаться глута-
ким сродством к кислороду [7]. В результате уро-
тионилированию. Так, при инкубации с GSSG
вень нитрозилирования α-субъединицы Na,K-
гомогената сердечной ткани слепышей происхо-
АТФазы снижается, а уровень глутатионилирова-
дит индукция глутатионилирования Na,K-АТФа-
ния возрастает. В присутствии ингибитора NO-
зы и ингибирование фермента [59]. Таким об-
синтаз (L-NAME) уровень NO снижается, и сдвиг
разом, у этого вида гипоксийно устойчивых жи-
редокс-потенциала в окисленную область проис-
вотных присутствуют регуляторные остатки
ходит даже в условиях нормоксии. Уровень NO в
цистеина, которые, по-видимому, достаточно
нормоксии и гипоксии в присутствии этого инги-
консервативны.
битора не отличается [59]. В результате в присут-
ствии ингибитора NO-синтаз уровень нитрози-
Роль глутатионилирования α-субъединицы в ре-
лирования α-субъединицы снижается в условиях
гуляции активности Na,K-АТФазы при болезни
нормоксии и в условиях гипоксии уже не меняет-
Альцгеймера. К изменению уровня глутатионили-
ся, при этом уровень глутатионилирования в нор-
рования α-субъединицы Na,K-АТФазы могут
моксии возрастает, а в гипоксии нет [59]. На гра-
приводить патологии, сопровождающиеся изме-
нулярных клетках мозжечка крыс было показано,
нением редокс-статуса. Так, например, на ранних
что активность Na,K-АТФазы максимальна при
стадиях развития болезни Альцгеймера происхо-
физиологическом содержании кислорода (3-5%
дит нарушение редокс-статуса нейрональных
pO2), снижаясь при гипоксии и гипероксии. При
клеток, в частности, снижение содержания GSH
этом L-NAME приводит к исчезновению макси-
[93-95]. При этом активность Na,K-АТФазы в
мума активности Na,K-АТФазы при физиологи-
этих клетках снижается [96-99]. Восстановление
ческих концентрациях кислорода [6], а активация
уровня глутатиона даже рассматривают в качестве
NO-синтаз существенно снижает ингибирова-
возможной стратегии терапии при болезни Альц-
ние, индуцируемое GSSG [59]. Очевидно, кисло-
геймера [95]. Было установлено, что патогенный
род-индуцированная регуляция Na,K-АТФазы
пептид бета-амилоид (Аβ) способен непосред-
опосредована переключением между глутатиони-
ственно взаимодействовать с Na,K-АТФазой и
лированием и нитрозилированием регуляторных
ингибировать ее активность в течение 30-60 мин
тиоловых групп, локализованных на каталитиче-
инкубации [99, 100]. Это быстрое ингибирование
ской α-субъединице фермента [59]. Таким об-
не связано с его проникновением в клетку [100].
разом, нитрозилирование и глутатионилирова-
Однако при более длительных временах инкуба-
ние - две конкурирующие редокс-модификации
ции необходимо также учитывать возможное ин-
α-субъединицы, и изменение их уровня опреде-
гибирование Na,K-АТФазы в результате измене-
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
849
ния редокс-статуса клеток под действием Аβ-
типах волокон, при этом для β1-субъединицы в
пептида. Было показано, что инкубация клеток
окислительных волокнах достоверного возраста-
нейробластомы человека SH-SY-5Y в течение 24 ч
ния не наблюдается, но оно наблюдается для β2-
с Аβ-пептидом приводит к существенному сни-
субъединицы в гликолитических волокнах [62].
жению уровня восстановленного глутатиона, из-
Необходимо отметить, что ранее считалось, что
менению соотношения GSH/GSSG и индукции
β2-субъединица глутатионилироваться неспо-
глутатионилирования α-субъединицы Na,K-
собна [77]. Хотя и в окислительных, и в гликоли-
АТФазы [101]. Таким образом, увеличение уров-
тических волокнах GSSG вследствие глутатиони-
ня глутатионилирования Na,K-АТФазы является
лирования Na,K-АТФазы приводит к ингибиро-
одной из причин снижения активности фермен-
ванию фермента, наибольшее ингибирование
та, наблюдаемого на ранних этапах развития бо-
наблюдается в окислительных волокнах. Таким
лезни Альцгеймера [101]. В связи с этим стано-
образом, предполагают, что глутатионилирова-
вится ясна причина неудачной попытки восста-
ние играет роль в регуляции работы Na,K-АТФа-
новить активность Na,K-АТФазы в клетках
зы в скелетных мышцах. В частности, глутатио-
мозга, подверженных действию Аβ-пептида, глу-
нилирование, индуцированное окислительным
татионом и его синтетическими аналогами [98],
стрессом, особенно важно при длительной на-
наоборот, под действием этих веществ актив-
грузке. Было показано, что при физической на-
ность Na,K-АТФазы еще более снижалась. По-
грузке и β2-адренергической стимуляции в мыш-
видимому, это связано с тем, что происходило
цах человека возрастает уровень глутатионилиро-
еще большее увеличение глутатионилирования
вания β1-субъединицы, тогда как уровень
Na,K-АТФазы. В пользу этого свидетельствует
глутатионилирования α-субъединицы практиче-
тот факт, что проникающий аналог глутатиона
ски не меняется [63]. Таким образом, глутатиони-
(этилглутатион) приводит к ингибированию
лирование способствует комплексной регуляции
Na,K-АТФазы в нейрональных клетках [5], и при
Na,K-АТФазной функции в скелетных мышцах
инкубации клеток с этилглутатионом и нитро-
человека. В частности, глутатионилирование
зоглутатионом глутатионилирование α-субъеди-
Na,K-АТФазы может объяснить снижение мак-
ницы Na,K-АТФазы возрастает [61]. Таким обра-
симальной активности Na,K-АТФазы после фи-
зом, необходимо тщательно подбирать соедине-
зической нагрузки, которое может быть вовлече-
ния и их рабочий диапазон концентраций для
но в развитие мышечной усталости [63].
восстановления нормального редокс-статуса кле-
Роль глутатионилирования β-субъединицы в регу-
ток при болезни Альцгеймера. Просто загрузка
ляции активности Na,K-АТФазы в норме и патоло-
клеток глутатионом в условиях окислительного
гии. В настоящее время описано несколько физио-
стресса может лишь увеличить уровень глутатио-
логических факторов, приводящих к изменению
нилирования белков. Можно предложить ис-
глутатионилирования β-субъединицы. Как было
пользовать для восстановления редокс-статуса
сказано выше, увеличение глутатионилирования
клеток более сильные восстановители, такие как
β-субъединицы приводит к снижению активности
НАДФ-H [101]. В последнее время появляется все
Na,K-АТФазы, которое, однако, менее выражено,
больше свидетельств того, что изменение уровня
чем при глутатионилировании α-субъединицы
нитрозилирования и глутатионилирования бел-
и составляет около 20% [56]. Условия, в которых
ков является важным аспектом патогенеза болез-
наблюдают возрастание глутатионилирования α- и
ни Альцгеймера [102]. Можно надеяться, что
β-субъединиц, различаются [48, 56, 59, 63]. Увели-
вскоре появятся способы для нормализации ре-
чение уровня глутатионилирования β-субъедини-
докс-статуса клеток и восстановления нормаль-
цы наблюдали при стимуляции HAДФ-H-оксида-
ного функционирования белков при патологии.
зы, которая может быть вызвана фосфорилирова-
нием субъединицы HAДФ-H-оксидазы про-
Роль глутатионилирования Na,K-ATФазы в раз-
теинкиназой C вследствие активации β1/β2-адре-
ных типах мышц. Глутатионилирование α-субъ-
норецепторов или обработки миокарда ангиотен-
единицы Na,K-АТФазы наблюдается не только в
зином II. Более того, увеличение глутатионилиро-
сердечной мышце, но и в других типах мышц. В
вания β-субъединицы наблюдалось в зоне инфарк-
окислительных мышечных волокнах скелетных
та сердца овцы [56]. Похожий эффект достигается
мышц уровень глутатионилировнаия α-субъеди-
экспонированием кардиомиоциоцитов с актива-
ницы почти в два раза выше, чем в гликолитиче-
тором аденилатциклазы форсколином и последу-
ских мышечных волокнах [62]. При этом отмече-
ющей активацией протеинкиназой C [81] или пря-
но, что базальный уровень глутатионилирования
мой обработкой кардиомитоцитов пероксинитри-
β1-субъединицы в обоих типах мышц выше, чем
α-субъединицы. Дитиотреитол снижает глутатио-
том (ONOO-) [56]. Обработка гладкомышечных
нилирование и приводит к возрастанию активно-
клеток сосудов нитрозоглутатионом также приво-
сти фермента. Инкубация с GSSG приводит к
дит к возрастанию глутатионилирования Сниже-
возрастанию глутатионилирования α-субъедини-
ние уровня супероксид-анион-радикала (O2•-) с
цы как в окислительных, так и в гликолитических
помощью супероксиддисмутазы предотвращает
БИОФИЗИКА том 65
№ 5
2020
850
ПЕТРУШАНКО и др.
глутатионилирование β-субъединицы. Также
дефосфорилированию p47phox и ее диссоциации от
предотвратить глутатионилирование позволяет
мембранных субъединиц. В результате предотвра-
стимуляция гуанилатциклазы, поскольку это ме-
щается глутатионилирование β1-субъединицы и
шает фосфорилированию и активации HAДФ-H-
вызванное этим ингибирование Na,K-АТФазы
оксидазы [103]. Механизм индукции глутатиони-
[107]. Таким образом, сверхэкспрессия β3-адрено-
лирования β-субъединицы при обработке клеток
рецептора, наблюдаемая в сердце крыс с диабетом
ангиотензином был изучен достаточно подробно.
[108], возможно, играет важную компенсаторную
Ангиотензин II активирует адренорецептор, что
роль и позволяет снизить уровень окислительного
приводит к активации протеинкиназы C и фосфо-
стресса и вызванное этим ингибирование Na,K-
рилированию p47phox-субъединицы HAДФ-H-ок-
АТФазы.
сидазы. В результате происходит транслокация
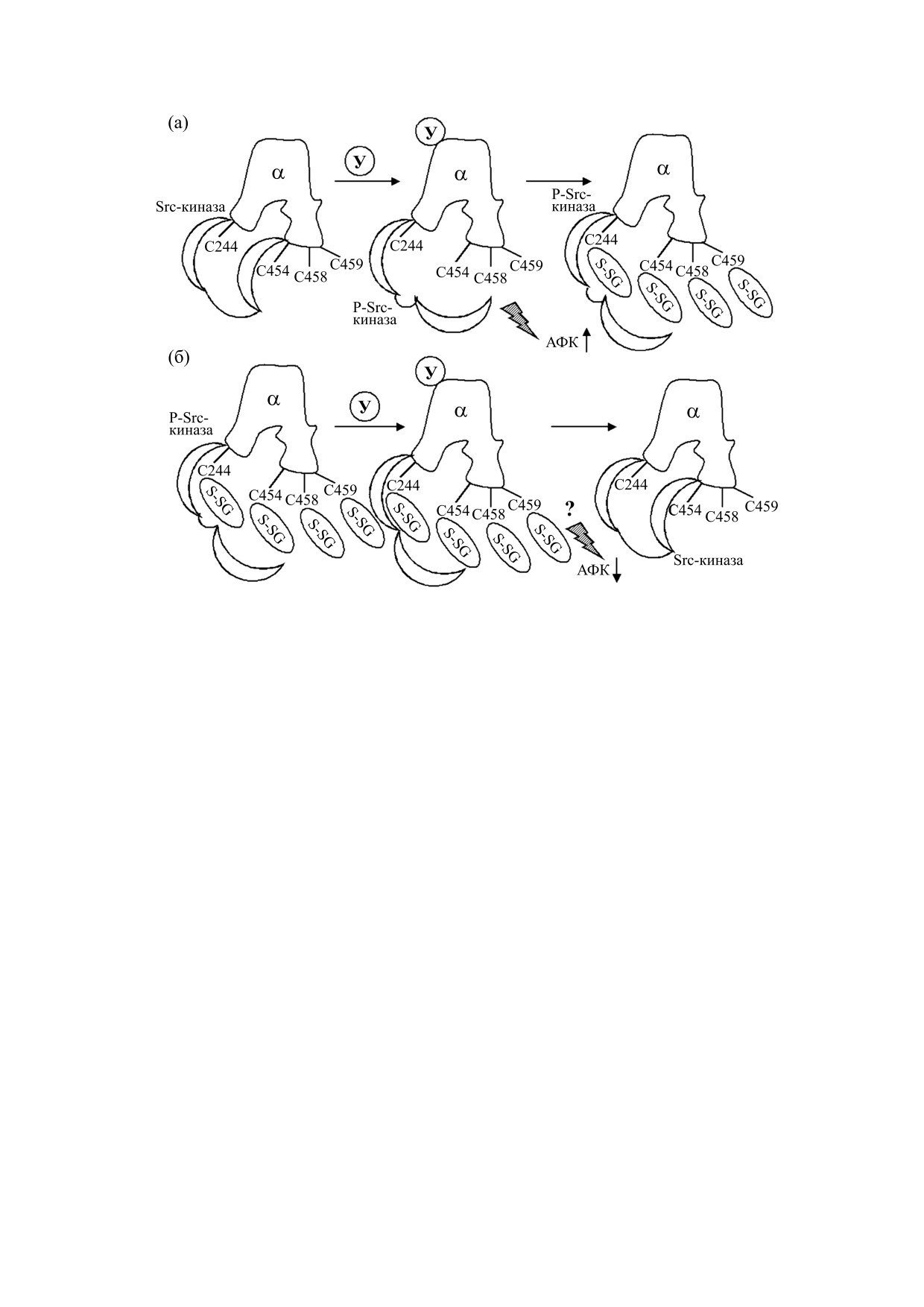
Подводя итог вышесказанному, можно пред-
p47phox, приводящая к активации HAДФ-H-окси-
ложить следующую схему регуляции глутатио-
дазы и образованию O2•-. Взаимодействие O2•- с
нилирования β-субъединицы и субъединицы се-
NO приводит к образованию пероксинитрита и
мейства FXYD (рис. 2). Физиологические сти-
индуцирует глутатионилирование [81]. Стимуля-
мулы, приводящие к активации НАДФ-Н-
ция гуанилатциклазы индуцирует осуществляемое
оксидазы (ангеотинезин II, активация β1/β2-ад-
фосфатазой PP2A-зависимое дефосфорилирова-
ренорецепторов, гипергликемия), приводят к
ние p47phox, препятствует активации HAДФ-H ок-
росту супероксид-анион-радикала, образова-
сидазы и нарушает глутатионилирование β1-субъ-
нию пероксинитрита и индукции глутатионили-
единицы [103]. Этот сдвиг в сторону деглутатиони-
рования β-субъединицы. В результате наруша-
лирования может приводить к активации Na,K-
ется взаимодействие α- и β-субъединиц и сни-
АТФазы. Также было показано, что ангиотензин
жается транспортная активность фермента. По-
ингибирует Na,K-АТФазу в гладкомышечных
видимому, образование ONOO- важно для глу-
клетках легочной артерии путем индукции глута-
татионилирования β-субъединицы, поскольку в
тионилирования β-субъединицы [104].
присутствии супероксиддисмутазы или ингиби-
тора NO-синтаз, а также ловушки ONOO- глута-
В отличие от активации β1/β2-адренорецепто-
тионилирование снижается. Глутатионилирова-
ров активация β3-адренорецептора, напротив,
ние возрастает также при непосредственной
снижает S-глутатионилирование β1-субъедини-
цы [104] и приводит к активации Na,K-АТФазы
обработке клеток ONOO-, H2O2 или нитрозоглу-
[105]. В мышах с делецией β3-адренорецептора
татионом. В присутствии FXYD1 под действием
отмечается повышенный уровень окислительно-
вышеописанных стимулов происходит индук-
го стресса и возрастание глутатионилирования
ция ее глутатионилирования, что затрудняет
β1-субъединицы. Ингибирование NO-синтазы
глутатионилирование β-субъединицы и предот-
снимает этот эффект [105]. Необходимо отме-
вращает ингибирование фермента. Обработка
тить, что сверхэкспрессия β3-адренорецептора
восстановителями или восстановление нор-
наблюдается при нарушении функции миокарда
мальных редокс-условий приводят к деглутатио-
[106]. Можно предполагать, что она является
нилированию данных субъединиц. Эта реакция
компенсаторным механизмом. Об этом же свиде-
катализируется глутаредоксином (рис. 2).
тельствуют данные, полученные при гипергли-
Таким образом, глутатионилирование всех
кемии.
трех субъединиц вовлечено в редокс-регуляцию
Гипергликемия, индуцированная в организме
активности Na,K-АТФазы. В зависимости от
кролика блокадой инсулинового рецептора, при-
условий и физиологических стимулов каждое из
водит к активации HAДФ-H-оксидазы [107]. При
них играет свою роль, что обеспечивает регуля-
активации HAДФ-H-оксидазы происходит глута-
цию активности Na,K-АТФазы и адаптационный
тионилирование эндотелиальных NO-синтаз, в ре-
ответ клеток.
зультате чего в большей степени генерируется
O•-, чем NO [107]. Возрастание O•- приводит к
РЕДОКС-РЕГУЛЯЦИЯ РЕЦЕПТОРНОЙ
увеличению уровня глутатионилирования β1-субъ-
ФУНКЦИИ Na,K-АТФазы
единицы и ингибированию Na,K-АТФазы в кар-
диомиоцитах [107]. При этом активация β3-адре-
Na,K-АТФаза является единственным извест-
норецептора с помощью обработки агонистом
ным рецептором к эндогенным КТС [16-20], ко-
снижает уровень окислительного стресса, вызван-
торые были впервые выделены из растений более
ного гипергликемией. Это происходит в результате
200 лет назад и издавна применялись для лечения
восстановления работы эндотелиальных NO-син-
хронической сердечной недостаточности. Не-
таз, возможно, за счет его связывания с белком
смотря на достаточно низкий терапевтический
теплового шока Hsp90, и синтезируемый NO сни-
индекс вследствие близости терапевтических и
жает активацию HAДФ-H оксидазы через канони-
токсичных доз, некоторые КТС, например дигок-
ческий гуанилатциклазный путь, приводящий к
син, до сих пор активно используются в терапии
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
851
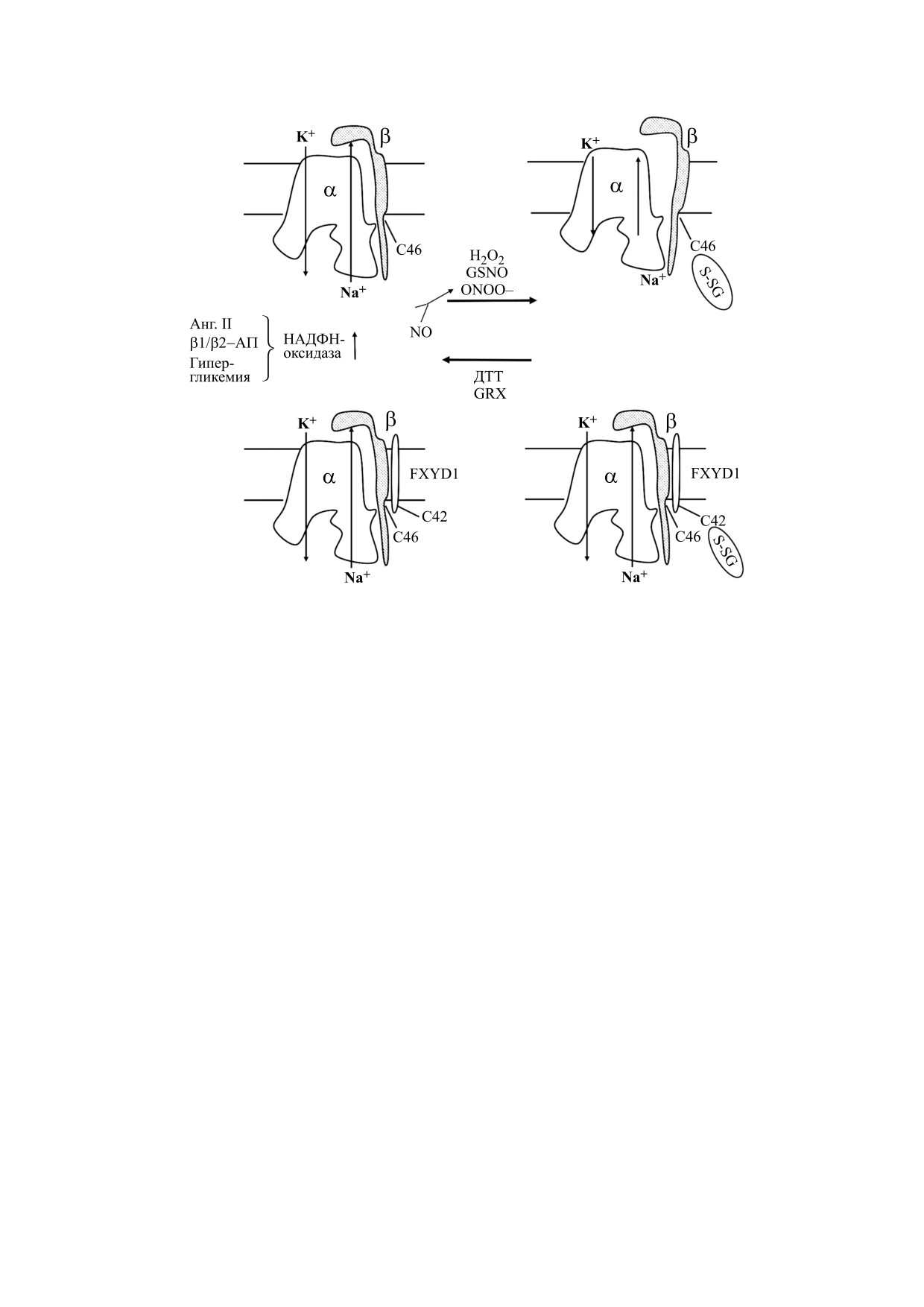
Рис. 2. Редокс-регуляция активности Na,K-АТФазы вследствие модификации остатков цистеина β-субъединицы и
FXYD1. Физиологические стимулы, приводящие к активации НАДФ-Н-оксидазы (ангеотинезин II (Анг. II), активация
β1/β2-адренорецепторов (β1/β2-АР), гипергликемия) приводят к росту супероксид-анион-радикала (О2·-), образова-
нию пероксинитрита (ONOO-) и индукции глутатионилирования β-субъединицы. В результате нарушается взаимодей-
ствие α- и β-субъединиц и снижается транспортная активность фермента. Обработка клеток ONOO-, H2O2 или нитро-
зоглутатионом (GSNO) также приводит к индукции глутатионилирования. В присутствии третьей субъединицы FXYD1
под действием данных стимулов в первую очередь происходит индукция ее глутатионилирования, что затрудняет глута-
тионилирование β-субъединицы и предотвращает ингибирование фермента. Обработка восстановителями, такими как
дитиотреитол (ДТТ), или восстановление нормальных редокс-условий при катализе глутаредоксином (GRX), приводят
к деглутатионилированию данных субъединиц. .
[109]. Связывание КТС c внеклеточной частью α-
идного ядра находится пятичленное лактоновое
субъединицы приводит к фиксации фермента в
кольцо, а у буфадиенолидов - шестичленное лак-
E2P-конформации и останавливает его каталити-
тоновое кольцо. Ряд КТС содержит остатки саха-
ческий цикл. Лечебное действие КТС основано
ра. Карденолиды впервые были выделены из рас-
на том, что ингибирование Na,K-АТФазы сердца
тений, а буфадиенолиды - из амфибий. В орга-
приводит к возрастанию уровня натрия, что вы-
низме человека обнаружены представители обоих
зывает активацию Na+/Ca2+-обменника, возрас-
классов. Первым в плазме крови человека был об-
танию уровня внутриклеточного Ca2+ и в конеч-
наружен карденолид, эдогенный уабаин [116], а
ном итоге к увеличению силы сердечных сокра-
затем в плазме и моче был выявлен буфадиено-
щений [109]. В настоящее время понятно, что
лид, маринобуфагенин [115, 117, 118]. КТС обна-
КТС не только ингибируют Na,K-АТФазу, но и
руживаются в крови в наномолярных и субнано-
вызывают активацию целого ряда сигнальных
молярных концентрациях. Оба эндогенных КТС
каскадов [17, 110].
являются вазоконстрикторами [119]. Увеличение
уровня маринобуфагенина наблюдается при
Изначально КТС были найдены в растениях и
ишемии миокарда [115] и почечной ишемии [120].
на коже некоторых видов жаб, а затем обнаруже-
ны в крови позвоночных [111-115]. Все КТС со-
КТС вовлечены в развитие некоторых заболева-
держат в своем составе стероидное (циклопен-
ний, в том числе гипертонии [121]. Присутствие
танпергидрофенантреновое) ядро. КТС делятся
различных КТС в организме предполагает, что их
на два класса - карденолиды и буфадиенолиды. У
действие на рецепторную функцию Na,K-АТФазы
карденолидов в семнадцатом положении стеро-
отличается. Действительно, различные КТС мо-
БИОФИЗИКА том 65
№ 5
2020
852
ПЕТРУШАНКО и др.
гут обуславливать разные биологические ответы
генерацию АФК и в других типах клеток: нейро-
[122]. Так, несмотря на то, что маринобуфагенин
нальных [142], раковых [143], поджелудочной же-
и уабаин практически одинаково ингибируют
лезы [144], почек [145, 146]. В присутствии анти-
Na,K-АТФазу почечных эпителиальных клеток,
оксиданта N-ацетилцистеина, снижающего уро-
концентрации этих ингибиторов, приводящие к
вень АФК, сигналлинг нарушается [145]. В то же
гибели 50% клеток, отличаются на два порядка
время устойчивое увеличение АФК в клетках
[123]. Эксперименты с очищенным препаратом
миокарда на уровне, сопоставимом с уабаином,
Na,K-АТФазы показали, что связывание уабаина
также способно активировать каскады, вовлечен-
и маринобуфагенина индуцирует разные кон-
ные в сигналлинг Na,K-АТФазы, в частности,
формационные изменения в молекуле белка, что
приводит к росту Ca2+, активации Erk1/2 и гипер-
может быть причиной связывания различных
трофических маркерных генов [147]. Так, добав-
белков партнеров и активации разных сигналь-
ление к культуре кардиомиоцитов глюкозоокси-
ных каскадов [124].
дазы, приводящее к возрастанию уровня H2O2,
КТС выполняют роль гормонов и продуциру-
активирует сигнальные каскады через Na,K-
ются средним мозгом и надпочечниками при ги-
АТФазу [138].
поксии и в ответ на определенные стимулы, такие
Как Src-киназа, так и кавеолин (структурный
как ангиотензин II, ацетилхолин, вазопрессин,
белок кавеол), вовлеченные в рецепторную функ-
катехоламины [125]. КТС играют важную роль в
цию Na,K-АТФазы, являются редокс-чувстви-
патофизиологии эссенциальной гипертонии,
тельными и необходимы для формирования ре-
преэклампсии, конечной стадии почечной недо-
докс-чувствительной сигнальной платформы
статочности, застойной сердечной недостаточно-
[148-154]. Ряд данных свидетельствует о том, что
сти и сахарного диабета [126]. Функция эндоген-
базальный уровень АФК необходим для стимуля-
ных КТС до конца не установлена, однако
ции каскадов, опосредованных связыванием уа-
очевидно, что их действие опосредовано связыва-
баина с Na/K-ATФазой и активации Src-киназы
нием с Na,K-АТФазой, которое приводит к акти-
[145, 146]. Показано, что в Src-киназе есть ре-
вации сигнальных каскадов, регулирующих ги-
докс-чувствительные остатки цистеина, окисле-
бель и пролиферацию клеток [20, 127]. Одним из
ние которых до SOH приводит к ее активации при
основных и наиболее исследованных компонен-
возрастании АФК, например, при НАДФ-Н-опо-
тов этих каскадов является Src-киназа. Это тиро-
средованном сигналлинге [153]. Таким образом,
зиновая киназа, участвующая в регуляции целого
применение антиоксидантов требует тщательно-
ряда клеточных процессов, в том числе роста, вы-
го рассмотрения с учетом того, что поддержание
живаемости и дифференцировки клеток [128,
редокс-статуса в физиологическом диапазоне
129]. Активность Src-киназы может регулиро-
важно для эффективного АФК-сигналлинга
ваться активирующим и ингибирующим фосфо-
[155].
рилированием по остаткам Tyr416 и Tyr527 соот-
Поскольку уабаин индуцирует рост АФК, воз-
ветственно. Фосфорилирование по остатку
никает вопрос, не может ли сам по себе рост АФК
Tyr527 приводит к инактивации киназы, в то вре-
приводить к индукции сигнального каскада, ин-
мя как при дефосфорилировании Tyr527 она ста-
дуцированного Na,K-АТФазой. Добавление пе-
новится активной, а фосфорилирование по
рекиси водорода или глюкозооксидазы стимули-
Tyr416 приводит к еще большему возрастанию ее
рует Na,K-АТФазный сигналлинг в клетках по-
активности
[130-132]. Часть молекул Na,K-
чек [145-147] и кардиомитоцитах [138, 145, 147].
АТФазы образует комплекс с Src-киназой. Свя-
Обработка антиоксидантами (N-ацетилцистеи-
зывание с Na,K-АТФазой уабаина приводит к
ном или витамином E) нарушает индукцию
высвобождению киназного домена Src-киназы из
Na,K-АТФаза/Src-сигналлинга уабаином и глю-
комплекса, что приводит к активации Src-киназы
козооксидазой [145, 146]. При этом ингибирова-
[133] и возрастанию уровня ее активирующего
ние c-Src и Erk1/2 отменяет эффекты АФК-инду-
фосфорилирования [128]. Действие уабаина на
цированного синтеза белка, на который не влияет
фосфорилирование Src-киназы не зависит от
хелатирование внутриклеточного Ca2+ [138]. Не-
концентраций внутриклеточных ионов, поэтому
обходимо отметить, что ингибирование с-Src-ки-
предполагают, что ингибирование уабаином
назы антиоксидантами может блокировать уаба-
Na,K-АТФазы, наблюдающееся при высоких
ин-стимулированную генерацию АФК, но не
концентрациях уабаина, и активация Src-зависи-
влияет на индуцированное уабаином увеличение
мого сигналлинга реализуются по двум независи-
мо регулируемым механизмам [134, 135-137].
Ca2+ [156]. Таким образом, можно сделать вывод,
что возрастание Ca2+, вызванное уабаином, не яв-
На изолированных кардиомиоцитах крысы
ляется необходимым для роста АФК [157].
было показано, что активация сигнальных каска-
дов через Na,K-АТФазу уабаином приводит к
Необходимо отметить, что низкие концентра-
увеличению генерации АФК [138-141]. Также уа-
ции уабаина приводят к клатрин-зависимому эн-
баин стимулирует Na,K-АТФазный сигналлинг и
доцитозу Na,K-АТФазы, ассоциированной с Src-
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
853
киназой, что может приводить к снижению сиг-
ина, которые вызывают различные биологиче-
нальной функции и активности Na,K-АТФазы со
ские ответы, приводит к разным изменениям
временем [158]. Есть основания предполагать, что
конформации фермента [172]. В настоящее время
изменение уровня Na,K-АТФазы может зависеть
все большее развитие получает представление о
от изоформного состава фермента. Так, уабаин
Na,K-АТФазе как о «стыковочной станции» для
приводил к увеличению мРНК α1- и β1-изоформ
различных белков. Известны более десяти бел-
и снижал при этом уровень мРНК α2- и β2-изо-
ков, которые могут связываться с ней и взаимо-
форм [159]. Длительное повышение уровня КТС
действие с некоторыми из них регулируется КТС
приводит к развитию гипертрофии и фиброза в
[165]. Очевидно, что изменение конформации
тканях сердца и почек [160, 161].
Na,K-АТФазы играет в регуляции связывания
белков-партнеров важную роль. Так, например,
В настоящее время существует следующая мо-
некоторые белки (PI3-киназа, кавеолин, анки-
дель связывания Na,K-АТФазы с Src-киназой:
рин, проапоптотический белок Bcl-2) имеют пе-
SH2-домен Src-киназы связан с актуаторным до-
рекрывающиеся сайты связывания в актуаторном
меном Na,K-АТФазы, а ее киназный домен - с
домене Na,K-АТФазы и, следовательно, не могут
нуклеотидсвязывающим доменом Na,K-АТФазы
связываться одновременно [164, 173-175]. Можно
[162-164]. Взаимодействие Src-киназы с актуа-
предположить, что КТС, стабилизирующие
торным доменом более сильное и сохраняется все
Na,K-АТФазу в разных конформациях, создают
время, а вот взаимодействие с нуклеотидсвязыва-
преимущества для связывания с ней разных бел-
ющим доменом нарушается при связывании уа-
ков-партнеров.
баина, что приводит к высвобождению киназного
Несмотря на широкое применение экзоген-
домена и активации киназы. КТС-индуцирован-
ных КТС и активное исследование роли эндоген-
ная активация Src-киназы в дальнейшем может
ных КТС, долгое время оставалось неясным, как
приводить к активации митоген-активируемых
гипоксия и иные изменения редокс-статуса клет-
киназ, индуцировать каскад через фосфолипазу C
ки влияют на рецепторную функцию Na,K-
или влиять на сигнальный путь, опосредованный
АТФазы. Как уже было отмечено выше, некото-
PI3-киназой [165]. Для реализации рецепторной
рые виды млекопитающих отвечают на гипоксию
функции через связывание с Src-киназой важно,
увеличением уровня КТС в крови. Высказыва-
чтобы в составе Na,K-АТФазы была α1-изоформа
лись предположения, что уабаин может защи-
[164, 166], поскольку именно у этой изоформы
щать сердце от ишемии-реперфузии за счет акти-
есть участок взаимодействия с Src [167]. Однако
вации Src-киназы [176]. На клетках фибробластов
интересно отметить, что фермент, имеющий в со-
мыши линии SC1 [177] и на клетках эмбриональ-
ставе тканеспецифичную α3-изоформу каталити-
ной почки человека [60] было обнаружено, что в
ческой субъединицы, реагирует на стимуляцию
условиях гипоксии действие уабаина на клетки
низкими дозами уабаина с активацией Erk1/2-ки-
изменяется. В условиях острой гипоксии инкуба-
назы, но не Src-киназы [166, 168]. Таким образом,
ция клеток с уабаином не приводит к активации
возможен клеточный сигналлинг, независимый
Src-киназы, которое наблюдается в клетках, на-
от активации Src-киназы.
ходящихся при 20% кислорода [177]. Напротив, ее
Необходимо отметить также, что кроме моде-
активирующее фосфорилирование снижается по
ли прямого взаимодействия Src-киназы с Na,K-
сравнению с необработанными уабаином клетка-
АТФазой существуют и другие, менее исследо-
ми, находящимися в гипоксии [60, 177]. При этом
ванные модели, например модель, объясняющая
в условиях острой гипоксии уровень активирую-
активацию Src-киназы под действием уабаина ее
щего фосфорилирования Src-киназы повышен
временным взаимодействием с комплексом α1-
по сравнению с контролем [60, 177]. Вероятно,
Na,K-АТФаза-кавеолин-1 [169], или модель, в
это связано с активацией Src-киназы под дей-
которой активация c-Src-киназы регулируется
ствием повышенного уровня АФК вследствие
изменением соотношения АТФ/АДФ и низкими
модификации остатков цистеина [153], посколь-
концентрациями Na+ и K+ (обе эти модели были
ку, как уже говорилось выше, уровень АФК в
протестированы лишь в бесклеточной системе)
условиях гипоксии возрастает [8, 177]. Добавле-
[170, 171]. Было высказано предположение, что
ние уабаина, которое в обычных условиях вызы-
поскольку все модели объединяет то, что в ре-
вает рост АФК, в условиях гипоксии приводит к
зультате стабилизируется E2P-конформация бел-
снижению индуцированного гипоксией роста
ка, а переход E2P-E1P замедляется (в присут-
АФК [177]. Возможно, именно вследствие этого
ствии КТС и/или из-за энергетического статуса
при добавлении уабаина активация Src-киназы
клетки), то именно это замедление регулирует
снижается [60]. Важно отметить, что высокие до-
сигнальную функцию Na,K-АТФазы [157]. Веду-
зы уабаина, которые при 20% pO2 через 24 ч инку-
щая роль изменения конформации Na,K-АТФазы
бации приводят к снижению жизнеспособности
в реализации сигналлинга представляется наибо-
клеток, в условиях гипоксии (0.05% pO2) не ока-
лее вероятной. В пользу этого свидетельствует тот
зывают цитотоксического действия или даже по-
факт, что связывание маринобуфагенина и уаба-
вышают жизнеспособность клеток [177]. Таким
БИОФИЗИКА том 65
№ 5
2020
854
ПЕТРУШАНКО и др.
Рис. 3. Схематичное представление механизма редокс-регуляции рецепторной функции Na,K-АТФазы вследствие
модификации остатков цистеина α-субъединицы. (а) - В условиях нормоксии Src-киназа связана с актуаторным и
нуклеотидсвязывающим доменом Na,K-АТФазы. Связывание уабаина (У) приводит к диссоциации киназного
домена Src-киназы от нуклеотидсвязывающего домена Na,K-АТФазы и активации Src-киназы. Вследствие этого
начинается рост АФК, что приводит к глутатионилированию регуляторных остатков цистеина Na,K-АТФазы в
нуклеотидсвязывающем домене и присоединение к нему киназного домена Src-киназы становится невозможным.
(б) - Гипоксия и последующий рост АФК приводят к глутатионилированию регуляторных остатков цистеина и
диссоциации Src-киназы от нуклеотидсвязывающего домена даже в отсутствие уабаина, что приводит к ее активации.
Связывание уабаина c Na,K-АТФазой в условиях гипоксии приводит к снижению АФК, что индуцирует
деглутатионилирование Na,K-АТФазы и ингибирование Src-киназы вследствие восстановления связывания ее
киназного домена с нуклеотидсвязывающим доменом Na,K-АТФазы.
образом, в условиях гипоксии рецепторная функ-
создана трехмерная модель комплекса «Na,K-
ция Na,K-АТФазы существенно меняется, что
АТФаза:Src-киназа» на основе α1β1-изозима
необходимо учитывать при терапевтическом ис-
Na,K-АТФаза из почек свиньи (PDB 3wgu) и че-
пользовании КТС. С другой стороны, в присут-
ловеческой Src-киназы (PDB 2src). Докинг Src-
ствии уабаина клетки становятся менее чувстви-
киназы и нуклеотидсвязывающего домена Na,K-
тельны к гипоксии. Следовательно, одной из фи-
АТФазы показал, что остатки цистеина Cys454-
зиологических функций КТС действительно
458-459 находятся вблизи интерфейса взаимодей-
может быть их протекторное действие при ише-
ствия этих белков. Более того, по данным моде-
мии и гипоксии. Вопрос о механизме изменения
лирования S-глутатионилирование остатков
рецепторной функции при гипоксии пока остает-
Cys458 и Cys459 нарушает связывание Src-киназы
ся открытым.
с нуклеотидсвязывающим доменом Na,K-АТФа-
зы [60]. Таким образом, одной из причин измене-
Эксперименты по точечному мутагенезу пока-
ния рецепторной функции Na,K-ATФазы при ги-
зали, что замена остатка Cys244 на аланин, кото-
поксии является нарушение взаимодействия глу-
рая приводит к утрате регуляторного цистеина и
татионилированной Na,K-АТФазы с Src-
предотвращает ингибирование фермента GSSG, не
киназой.
влияет на рецепторную функцию Na,K-АТФазы.
Однако замена трех остатков цистеина Cys454-
Можно предположить следующий механизм
458-459 на аланин нарушает активацию Src-кина-
редокс-чувствительности рецепторной функции
зы в ответ на уабаин или гипоксию, делая ее не-
Na,K-АТФазы (рис. 3). В условиях нормоксии
чувствительной к данным стимулам [60]. Была
связывание уабаина приводит к диссоциации ки-
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
855
назного домена Src-киназы от нуклеотидсвязы-
ляции транспортной и рецепторной функции
вающего домена Na,K-АТФазы и активации Src-
Na,K-АТФазы дадут ответы на вопросы, постав-
киназы [162-164]. Вследствие этого начинается
ленные в данном обзоре.
рост АФК [157, 177], что приводит к глутатиони-
лированию регуляторных остатков цистеина
Na,K-АТФазы в нуклеотидсвязывающем домене,
ФИНАНСИРОВАНИЕ РАБОТЫ
и присоединение к нему киназного домена Src-
Работа выполнена при финансовой поддержке
киназы становится невозможным [60]. Гипоксия
Российского научного фонда (проект № 19-14-
и последующий рост АФК приводят к глутатио-
00374).
нилированию регуляторных остатков цистеина и
вызывают диссоциацию Src-киназы от нуклеоти-
дсвязывающего домена даже в отсутствие уабаи-
КОНФЛИКТ ИНТЕРЕСОВ
на, что приводит к ее активации [60, 177]. Связы-
Авторы заявляют об отсутствии конфликта
вание уабаина c Na,K-АТФазой в условиях гипо-
интересов.
ксии приводит к снижению АФК [177], что, по-
видимому, индуцирует деглутатионилирование
СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ
Na,K-АТФазы и последующее ингибирование
Src-киназы [60, 177] вследствие восстановления
Настоящая работа не содержит описания ис-
связывания ее киназного домена с нуклеотидсвя-
следований с использованием людей и животных
зывающим доменом Na,K-АТФазы (рис. 3).
в качестве объектов.
Таким образом, есть несколько причин ре-
СПИСОК ЛИТЕРАТУРЫ
докс-чувствительности рецепторной функции
1. J. H. Kaplan, Annu. Rev. Biochem. 71, 511 (2002).
Na,K-АТФазы: во-первых, это редокс-зависимые
2. S. N. Orlov, A. A. Platonova, P. Hamet, and R. Gry-
модификации остатков цистеина самой Na,K-
gorczyk, Am. J. Physiol. Cell. Physiol. 305 (4), C361
АТФазы, которые могут влиять на связывание с
(2013).
Src-киназой и взаимодействие с КТС; во-вторых,
3. M. J. Shattock, M. Ottolia, D. M. Bers, et al., J. Physi-
изменение активности Src-киназы и иных участ-
ol. (Lond). 593, 1361 (2015).
ников сигнального каскада и, в-третьих, петля
4. D. Dobrota, M. Matejovicova, E.G. Kurella, and
обратной связи, заключающаяся в том, что свя-
A. A. Boldyrev, Cell. Mol. Neurobiol. 19 (1),
141
зывание КТС с Na,K-АТФазой само влияет на ре-
(1999).
докс-статус клеток [60, 157].
5. I. Petrushanko, N. Bogdanov, E. Bulygina, et al., Am.
J. Physiol. Regul. Integr. Comp. Physiol. 290, R916
(2006).
ЗАКЛЮЧЕНИЕ
6. I. Y. Petrushanko, N. B. Bogdanov, N. Lapina, et al., J.
Подводя итог, можно заключить, что редокс-
Gen. Physiol. 130, 389 (2007).
зависимые модификации тиоловых групп Na,K-
7. A. Bogdanova, I. Petrushanko, A. Boldyrev, and
АТФазы - глутатионилирование и нитрозилиро-
M. Gassmann, Curr. Enzyme Inhibit. 2, 37 (2006).
вание - играют важную роль в регуляции транс-
8. A. Bogdanova, I. Y. Petrushanko, P. Hernansanz-Agu-
портной и рецепторной функции фермента в
stín, and A. Martínez-Ruiz, Front. Physiol. 7, 314
норме и патологии. В зависимости от условий и
(2016).
физиологических стимулов определяющим для
9. J. J. Mieyal, M. M. Gallogly, S. Qanungo, et al., Anti-
регуляции активности Na,K-АТФазы и адаптаци-
oxid. Redox Signal. 10, 1941 (2008).
онного ответа клеток может являться изменение
10. F. Q. Schafer and G. R. Buettner, Free Radic. Biol.
уровня глутатионилирования/нитрозилирования
Med. 30 (11), 1191 (2001).
каталитической (α) или регуляторных субъеди-
11. S. Suzuki, M. Noda, M. Sugita, et al., J. Appl. Physiol.
ниц фермента (β, FXYD). Однако пока нет полно-
87 (3), 962 (1999).
го понимания согласованной редокс-регуляции
12. R. Carini, M. G. De Cesaris, R. Splendore, et al.,
всех трех субъединиц Na,K-АТФазы. Необходи-
Hepatology 31 (1), 166 (2000).
мо создание единой картины регуляции различ-
13. A. Y. Bogdanova, O. O. Ogunshola, C. Bauer, and M.
ных субъединиц фермента при изменении ре-
J. Gassmann, Membr. Biol. 195 (1), 33 (2003).
докс-условий, обеспечивающей изменение его
функционирования как целого. Только в этом
14. A. Boldyrev and E. Kurella, Biochem. Biophys. Res.
случае будет возможно предотвращать и коррек-
Commun. 222 (2), 483 (1996).
тировать редокс-зависимые нарушения функци-
15. E. G. Kurella, O. V. Tyulina, and A. A. Boldyrev, Cell.
онирования фермента, жизненно необходимого
Mol. Neurobiol. 19 (1), 133 (1999).
для каждой клетки нашего организма. Мы наде-
16. A. Y. Bagrov, J. I. Shapiro, and O. V. Fedorova, Phar-
емся, что дальнейшие исследования редокс-регу-
macol. Rev. 61, 9 (2009).
БИОФИЗИКА том 65
№ 5
2020
856
ПЕТРУШАНКО и др.
17. A. O. Del Toro, L. Jimenez, L. Hinojosa, et al., Car-
46. K. Y. Xu, J. L. Zweier, and L.C. Becker, Ann. N. Y.
diol. Res. Pract. 2019, 8646787 (2019).
Acad. Sci. 834, 680 (1997).
18. M. Haas, H. Wang, J. Tian, and Z. Xie, J. Biol. Chem.
47. C. Gatto, S. J. Thornewell, J. P. Holden, and J. H. Ka-
277, 18694 (2002).
plan, J. Biol. Chem. 274 (35), 24995 (1999).
19. M. Simonini, P. Casanova, L. Citterio, et al., Int. J.
48. I. Yu. Petrushanko, S. Yakushev, V. A. Mitkevich, et al.,
Mol. Sci. 19 (7), 1948 (2018).
J. Biol. Chem. 287, 32195 (2012).
20. J. X. Xie, X. Li, and Z. Xie. IUBMB Life 65 (12), 991
49. F. Thevenod and J. M. Friedmann, FASEB J. 13, 1751
(2013).
(1999).
21. D. C. Chow and J. G. Forte, J. Exp. Biol. 198, 1 (1995).
50. A. Ziegelhoffer, K. Kjeldsen, H. Bundgaard, et al.,
22. G. Scheiner-Bobis, Eur. J. Biochem. 269, 2424 (2002).
Gen. Physiol. Biophys. 19 (1), 9 (2000).
23. L. Segall, Z. Z. Javaid, S. L. Carl, et al., J. Biol. Chem.
51. M. Erecinska and I. A. Silver, Prog. Neurobiol. 43(1),
278, 9027 (2003).
37 (1994).
24. P. L. Jorgensen, Ann. N. Y. Acad. Sci. 986, 22 (2003).
52. M. Inoue, N. Fujishiro, and I. Imanaga J. Physiol. 519,
25. C. Lupfert, E. Grell, V. Pintschovius, et al., Biophys. J.
385 (1999).
81, 2069 (2001).
53. T. Clausen, Physiol. Rev. 83 (4), 1269 (2003).
26. S. J. Karlish, J. Bioenerg. Biomembr. 12, 111 (1980).
54. H. G. Shi, L. Mikhaylova, A. E. Zichittella, and
27. И. Ю. Петрушанко, В. А. Митькевич, В. А. Борзова
J. M. Arguello, Biochim. Biophys. Acta 1464,
177
и др., Биофизика 54 (6), 1019 (2009).
(2000).
28. A. V. Grinberg, N. M. Gevondyan, N. V. Grinberg, and
55. V. A. Mitkevich, I. Y. Petrushanko, Y. M. Poluektov,
V. Y. Grinberg, Eur. J. Biochem. 268 (19), 5027 (2001).
et al., Oxid. Med. Cell Longev. 9092328 (2016).
29. R. Kanai, H. Ogawa, B. Vilsen, et al., Nature 502, 201
56. G. A. Figtree, C. C. Liu, S. Bibert, et al., Circ. Res. 105
(2013).
(2), 185 (2009).
30. J. P. Morth, B. P. Pedersen, M. S. Toustrup-Jensen,
et al., Nature 450, 1043 (2007).
57. S. Bibert, C. C. Liu, G. A. Figtree, et al., J. Biol. Chem.
286, 18562 (2011).
31. T. Clausen, Physiol. Rev. 83, 1269 (2003).
58. J. J. Mieyal, M. M. Gallogly, S. Qanungo, et al., Anti-
32. J. Lingrel, A. Moseley, I. Dostanic, et al., Ann. N. Y.
oxid. Redox Signal. 10 (11), 1941 (2008).
Acad. Sci., 986, 354 (2003).
59. S. Yakushev, M. Band, M. C. Tissot VanPatot, et al.,
33. A. E. Moseley, S. P. Lieske, R. K. Wetzel, et al., J. Biol.
Chem. 278, 5317 (2003).
Am. J. Physiol. Heart Circ. Physiol. 303, H1332 (2012).
34. P. Ostadal, A. B. Elmoselhi, I. Zdobnicka, et al., Bio-
60. I.Y. Petrushanko, V. A. Mitkevich, V. A. Lakunina,
chem. Biophys. Res. Commun. 306, 457 (2003).
et al., Redox Biol. 13, 310 (2017).
35. L. Peng, P. Martin-Vasallo, and K. J. Sweadner, J.
61. И. Ю. Петрушанко, О. В. Симоненко, К. М. Бур-
Neurosci. 17, 3488 (1997).
нышева и др., Молекуляр. биология 49, 175 (2015).
36. R. K. Wetzel, E. Arystarkhova, and K. J. Sweadner, J.
62. C. Juel, PLoS One 9 (10), e110514 (2014).
Neurosci. 19, 9878 (1999).
63. C. Juel, M. Hostrup, and J. Bangsbo, Physiol. Rep. 3
37. Sundaram S.M., D. Safina, A. Ehrkamp, et al., Neuro-
(8), e12515 (2015).
chem. Int. 128, 163 (2019).
64. С. Мэн, И. Ю. Петрушанко, Е. А. Климанова и др.,
38. G. Blanco, Ann. N. Y. Acad. Sci. 986, 536 (2003).
Биохимия 79 (2), 209 (2014).
39. A. Katz, Y. Lifshitz, E. Bab-Dinitz, et al., J. Biol.
65. Y. M. Poluektov, I. Y. Petrushanko, N. A. Undrovinas,
Chem. 285 (25), 19582 (2010).
et al., Sci. Rep. 9 (1), 4872 (2019).
40. A. Scirè, L. Cianfruglia, C. Minnelli, et al., Biofactors
66. Е. А. Дергоусова, И. Ю. Петрушанко, Е. А. Клима-
45 (2), 152 (2019).
нова и др., Молекуляр. биология 52 (2), 289 (2018).
41. J. G. Reich and E. E. Sel’kov, Energy metabolism of the
67. E. A. Dergousova, I. Y. Petrushanko, E. A. Klimanova,
cell: a theoretical treatise (Acad. Press, N. Y., 1981).
et al., Biomolecules 7 (1), 18 (2017).
42. H. Sies, In Metabolic Compartmentation (Acad. Press,
68. S. Despa, J. B. Lingrel, and D. M. Bers, Cardiovasc.
Lond., 1982), p. 205.
Res. 95, 480 (2012).
43. А. Болдырев, Е. Булыгина, О. Герасимова и др.,
Биохимия 69 (4), 530 (2004).
69. Z. Xie, M. Jack-Hays, Y. Wang, et al., Biochem. Bio-
phys. Res. Commun. 207, 155 (1995).
44. A. A. Boldyrev, In: Molecular and Therapeutical Aspects
of RedOx Biochemistry, Ed. by T. Bahorun and A. Gu-
70. A. Chandra, S. Srivastava, J. M. Petrash, et al., Bio-
rib-Fakim (OICA, 2003), p. 59.
chemistry 36 (50), 15801 (1997).
45. Z. Xie, M. Jack-Hays, Y. Wang, et al., Biochem. Bio-
71. I. Y. Petrushanko, V. A. Mitkevich, A. A. Anashkina,
phys. Res. Commun. 207, 155 (1995).
et al., Sci. Rep. 4, 5165 (2014).
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
857
72. Y. M. Poluektov, E. A. Dergousova, O. D. Lopina,
98. C. Kairane, R. Mahlapuu, K. Ehrlich, et al., Curr. Alz-
et al., Biochem. Biophys. Res. Commun. 510 (1), 86
heimer Res. 11, 79 (2014).
(2019).
99. C. A. Dickey, M. N. Gordon, D. M. Wilcock, et al.,
73. Е. А. Дегоусова, Ю. М. Полуэктов, Е. А. Климано-
BMC Neurosci. 2, 1 (2005).
ва и др., Биохимия 83 (8), 1220 (2018).
100. I. Y. Petrushanko, V. A. Mitkevich, A. A. Anashkina,
74. V. Srinivasan, A. J. Pierik, and R. Lill, Science 343
et al., Sci. Rep. 6, 27738 (2016).
(6175), 1137 (2014).
101. В. А. Лакунина, И. Ю. Петрушанко, К. М. Бурны-
75. C. C. Liu, K. Galougahi, R. M. Weisbrod, et al., Free
шева и др., Докл. РАН 473 (3), 374 (2017).
Radic. Biol. Med. 65, 563 (2013).
102. R. R. Dyer, K. I. Ford, and R. A. S. Robinson, Meth-
76. H. Ogawa, T. Shinoda, F. Cornelius, and C. Toyoshi-
ods Enzymol. 626, 499 (2019).
ma, Proc. Natl. Acad. Sci. USA 106, 13742 (2009).
103. K. Chia, C. C. Liu, E. J. Hamilton, et al., Am. J.
77. C. C. Liu, A. Garcia, Y. A. Mahmmoud, et al., J. Biol.
Physiol. Cell Physiol. 309, 239 (2015).
Chem. 287, 12353 (2012).
104. S. M. Rahaman, K. Dey, T. Chakraborti, and
78. A. Garcia, N. D. Eljack, M. A. Sani, et al., Biochim.
S. Chakraborti, Ind. J. Biochem. Biophys. 52 (2), 119
Biophys. Acta 1848, 2430 (2015).
(2015).
79. S. P. Barwe, S. Kim, S. A. Rajasekaran, et al., J. Mol.
105. H. Bundgaard, C. C. Liu, A. Garcia, et al., Circula-
Biol. 365, 706 (2007).
tion. 122, 2699 (2010).
80. I. Dalle-Donne, R. Rossi, G. Colombo, et al., Trends
106. S. Moniotte, L. Kobzik, O. Feron, et al., Circulation
Biochem. Sci. 34, 85 (2009).
103, 1649 (2001).
81. C. N. White, C. C. Liu, A. Garcia, et al., J. Biol. Chem.
107. K. Galougahi, C. C. Liu, A. Garci, et al., Am. J. Phy-
285, 13712 (2010).
siol. Cell. Physiol. 309 (5), 286 (2015).
82. D. Pavlovic, W. Fuller, and M. J. Shattock, J. Mol. Cell.
108. V. M. Altan, E. Arioglu, S. Guner, and A. T. Ozcelikay,
Cardiol. 61, 83 (2013).
Heart Fail. Rev. 12, 58 (2007).
83. W. Fuller, V. Parmar, P. Eaton, et al., Cardiovasc. Res.
109. F. M. Botelho, F. Pierezan, B. Soto-Blanco, and
57, 1044 (2003).
M. M. Melo, Toxicon. 158, 63 (2019).
84. R. G. Boutilier, J. Exp. Biol. 204 (18), 3171 (2001).
110. A. A. Shiyan, L. V. Kapilevich, A. V. Lopachev, et al.,
85. M. Habeck, et al., J. Biol. Chem. 291, 23159 (2016).
PLoS One 14 (9), e0222767 (2019).
86. H.-J. Apell, Rev. Physiol. Biochem. Pharmacol. 150, 1
111. J. M. Hamlyn, M. P. Blaustein, S. Bova, et al., Proc.
(2003).
Natl. Acad. Sci. USA 88, 6259-6263 (1991).
87. L. T. Buck and M. E. Pamenter, Comp. Biochem.
112. R. S. Kieval, V. P. J. Butler, F. Derguini, et al., Am.
Physiol. Part B: Biochem. Mol. Biol. 224, 61 (2018).
Coll. Cardiol. 11, 637 (1988).
88. L. T. Buck and P. W. Hochachka, Am. J. Physiol. 265,
113. D. Lichtstein, I. Gati, and H. J. Ovadia, Cardiovasc.
R1020 (1993).
Pharmacol. 22, S102 (1993).
89. P. W. Hochachka, L. T. Buck, C. J. Doll, and
114. A. Hollman, Br. Heart J. 54 (3), 258 (1985).
S. C. Land, Proc. Natl. Acad. Sci. USA 93, 9493
115. A. Y. Bagrov, O. V. Fedorova, R. I. Dmitrieva, et al.,
(1996).
Hypertension 31, 1097 (1998).
90. P. Hylland, S. Milton, M. Pek, et al., Neurosci. Lett.
116. J. H. Ludens, M. A. Clark, D. W. Du Charme, et al.,
235, 89 (1997).
Hypertension 17, 923 (1991).
91. M. P. Wilkie, M. E. Pamenter, S. Alkabie, et al., Comp.
117. O. V. Fedorova, P. A. Doris, and A. Y. Bagrov, Clin.
Biochem. Physiol. C. Toxicol. Pharmacol. 148, 355
Exp. Hypertens. 20, 581-591 (1998).
(2008).
92. A. P. Ross, S. L. Christian, H. W. Zhao, and K. L. Drew,
118. H. C. Gonick, Y. Ding, N. D. Vaziri, et al., Clin. Exp.
J. Cereb. Blood Flow Metab. 26, 1148 (2006).
Hypertens. 20, 617 (1998).
93. W. M. Johnson, A. L. Wilson-Delfosse, and
119. A. Y. Bagrov and O. V. Fedorova, J. Hypertens. 16,
J. J. Mieyal, Nutrients 4, 1399 (2012).
1953 (1998).
94. S. Akterin, R. F. Cowburn, A. Miranda-Vizuete, et al.,
120. J. Tian, S. Haller, S. Periyasamy, et al., Hypertension
Cell Death and Differentiation 13, 1454 (2006).
56, 914 (2010).
95. C. B. Pocernich and D. A. Butterfield, Biochim. Bio-
121. W. Schoner and G. Scheiner-Bobis, Am. J. Cardiova-
phys. Acta. 1822 (5), 625 (2012).
sc. Drugs 7, 173 (2007).
96. L. N. Zhang, Y. J. Sun, S. Pan, et al., Fundam. Clin.
122. M. Dvela, H. Rosen, T. Feldmann, et al., Pathophysi-
Pharmacol. 27, 96 (2013).
ology 14, 159 (2007).
97. V. M. Vitvitsky, S. K. Garg, R. F. Keep, et al., Biochim.
123. O. A. Akimova, A. Y. Bagrov, O. D. Lopina, et al., J.
Biophys. Acta 822, 1671 (2012).
Biol. Chem. 280, 832 (2005).
БИОФИЗИКА том 65
№ 5
2020
858
ПЕТРУШАНКО и др.
124. E. A. Klimanova, I. Y. Petrushanko, V. A. Mitkevich,
150. P. N. Seshiah, D. S. Weber, P. Rocic, et al., Circ. Res.
et al., FEBS Lett. 589, 2668 (2015).
91, 406 (2002).
125. C. De Angelis and G. T. Haupert, Am. J. Physiol. 274,
151. R. M. Touyz, G. Yao, and E. L. Schirin, Arterioscler.
F182 (1998).
Thromb. Vasc. Biol. 23, 981 (2003).
126. A. Y. Bagrov, J. I. Shapiro, and O. V. Fedorova, Phar-
152. K. J. Bubb, A. B. Birgisdottir, O. Tang, et al., Free
macol. Rev. 61, 9 (2009).
Radic. Biol. Med. 109, 61 (2017).
127. R. S. El-Mallakh, K. S. Brar, and R. R. Yeruva, Insects
153. D. E. Heppner, C. M. Dustin, C. Liao, et al., Nat.
Commun. 9 (1), 4522 (2018).
10 (4), 102 (2019).
154. E. Giannoni and P. Chiarugi, Antioxid. Redox Signal.
128. M. Haas, H. Wang, J. Tian, and Z. Xie, J. Biol. Chem.
20 (13), 2011 (2014).
277, 18694 (2002).
155. A. C. Montezano, R. M. Touyz, Antioxid. Redox Sig-
129. G. A. Knock, V. A. Snetkov, Y. Shaifta, et al., Cardio-
nal. 20 (1), 164 (2014).
vasc. Res. 80, 453 (2008).
156. J. Tian, X. Gong, and Z. Xie, Am. J. Physiol. Heart
130. A. MacAuley and J. A. Cooper, Mol. Cell. Biol. 9,
Circ. Physiol. 281 (5), H1899 (2001).
2648 (1989).
157. R. D. Pratt, C. R. Brickman, C. L. Cottrill, et al., Int.
131. W. Xu, S. C. Harrison, and M. J. Eck, Nature 385, 595
J. Mol. Sci. 19, 2600 (2018).
(1997).
158. J. Liu, M. Liang, L. Liu, et al., Kidney Int. 67, 1844
132. S. W. Cowan-Jacob, G. Fendrich, P. W. Manley, et al.,
(2005).
Structure 13, 861 (2005).
159. R. Hosoi, T. Matsuda, S. Asano, et al., J. Neurochem.
133. J. Tian, T. Cai, Z. Yuan, et al., Mol. Biol. Cell. 17, 317
69 (5), 2189 (1997).
(2006).
160. D. J. Kennedy, S. Vetteth, S. M. Periyasamy, et al.,
134. J. Liu, J. Tian, M. Haas, et al., J. Biol. Chem. 275,
Hypertension 47, 488 (2006).
27838 (2000).
161. F. K. Khalaf, P. Dube, A. Mohamed, et al., Int. J. Mol.
135. M. Haas, A. Askari, and Z. Xie, J. Biol. Chem. 275,
Sci. 19(9), E2576 (2018).
27832 (2000).
162. M. Banerjee, Q. Duan, Z. Xie, PLoS One 10, e0142119
136. A. Aydemir-Koksoy, J. Abramowitz, and J. C. Allen, J.
(2015).
Biol. Chem. 276, 46605 (2001).
163. Z. Li, T. Cai, J. Tian, et al., J. Biol. Chem. 284, 21066
(2009).
137. M. Haas, H. Wang, J. Tian, Z. Xie, J. Biol. Chem. 277,
164. Z. Li and Z. Xie, Pflug. Arch. 457, 635 (2009).
18694 (2002).
165. L. Reinhard, H. Tidow, M. J. Clausen, and P. Nissen,
138. L. Liu, J. Li, J. Liu, et al., Free Radic. Biol. Med. 41,
Cell. Mol. Life Sci. 70, 205 (2013).
1548 (2006).
166. L. V. Karpova, E. R. Bulygina, and A. A. Boldyrev,
139. J. Liu, J. Tian, M. Haas, et al., J. Biol. Chem. 275,
Cell Biochem. Funct. 28 (2), 135 (2010).
27838 (2000).
167. H. Yu, X. Cui, J. Zhang, et al., Am. J. Physiol. Cell
140. J. Tian, J. Liu, K. D. Garlid, et al., Mol. Cell Biochem.
Physiol. 314, C202 (2018).
242, 181 (2003).
168. N. Madan, Y. Xu, Q. Duan, et al., Am. J. Physiol. Cell
141. Z. Xie, P. Kometiani, J. Liu, et al., J. Biol. Chem. 274,
Physiol. 00199, 02016 (2016).
19323 (1999).
169. E. Yosef, A. Katz, Y. Peleg, et al., J. Biol. Chem. 291,
142. A. Boldyrev, E. Bulygina, M. Yuneva, and W. Schoner,
11736 (2016).
Ann. N. Y. Acad. Sci. 986, 519 (2003).
170. K. M. Weigand, H. G. Swarts, N. U. Fedosova, et al.,
143. Y. T. Huang, S. C. Chueh, C. M. Teng, J. H. Guh,
Biochim. Biophys. Acta 1818, 1269 (2012).
Biochem. Pharmacol. 67, 727 (2004).
171. M. E. Gable, S. L. Abdallah, S. M. Najjar, et al., Bio-
chem. Biophys. Res. Commun. 446, 1151 (2014).
144. M. Kajikawa, S. Fujimoto, Y. Tsuura, et al., Diabetes
51, 2522 (2002).
172. E. A. Klimanova, I. Yu. Petrushanko, V. A. Mitkevich,
et al., FEBS Lett. 589, 2668 (2015).
145. Y. Yan, A. P. Shapiro, S. Haller, et al., J. Biol. Chem.
173. P. K. Lauf, T. Alqahtani, K. Flues, et al., Am. J.
288, 34249 (2013).
Physiol. Cell Physiol. 308, C51 (2015).
146. Y. Yan, A. P. Shapiro, B. R. Mopidevi, et al., J. Am.
174. P. K. Lauf, J. Heiny, J. Meller, et al., Cell. Physiol.
Heart Assoc. 5, e003675 (2016).
Biochem. 3, 257 (2013).
147. Y. Wang, Q. Ye, C. Liu, et al., Free Radic. Biol. Med.
175. G. A. Morrill, A. B. Kostellow, and A. Askari, Steroids
71, 415 (2014).
77, 1160 (2012).
148. A. Y. Zhang, F. Yi, G. Zhang, et al., Hypertension 47,
176. P. Pasdois, C. L. Quinlan, A. Rissa, et al., Am. J. Phy-
74 (2006).
siol. Heart Circ. Physiol. 292, H1470 (2007).
149. W. Han, H. Li, V. A. M. Villar, et al., Hypertension 51,
177. В. А. Лакунина, К. М. Бурнышева, В. А. Митьке-
481 (2008).
вич и др., Молекуляр. биология 51, 172 (2017).
БИОФИЗИКА том 65
№ 5
2020
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕДОКС-РЕГУЛЯЦИИ Na,K-АТФазы
859
Molecular Mechanisms of Redox Regulation of the Na,K-ATPase
I.Yu. Petrushanko, V.A. Mitkevich, and A.A. Makarov
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences,
ul. Vavilova 32, Moscow, 119991 Russia
This review is dedicated to the molecular mechanisms of redox regulation of the Na,K-ATPase. The Na,K-
ATPase creates a transmembrane gradient for the ions of sodium and potassium, which is necessary for the
vital activity of all animal cells, and is also a receptor for cardiotonic steroids that regulate cell proliferation
and apoptosis. The functioning of the Na,K-ATPase depends on redox status in the cells. Initially, it was
shown that the enzyme is inhibited during oxidative stress, but it is now clear that redox regulation of Na,K-
ATPase activity is a complex process and cannot be explained only by oxidative damage to the protein. The
activity of the enzyme is maximal under physiological oxygen concentration and decreased by both hypoxia
and hyperoxia, as well as due to decreased or increased intracellular glutathione concentration. Thus, the
Na,K-ATPase reaches its maximal activity within a specific range of redox conditions. Today, it is obvious
that a disturbance of the Na,K-ATPase activity in a number of pathologies such as hypoxia, ischemia, diabe-
tes, Alzheimer's disease is associated with a change in redox status in the cells. Receptor function of the Na,K-
ATPase also depends on redox status in the cells and should be taken into account while studying the effects
of cardiotonic steroids on cells and tissues. In this review special focus is placed on redox modifications of
thiol groups of various Na,K-ATPase subunits and on study of the regulatory processes in which these sub-
units are involved in normal and pathological conditions. Gaining insight into the molecular mechanisms of
redox regulation can help us to understand what is needed to prevent the impairment of Na,K-ATPase func-
tion in pathological conditions thereby reducing cell damage.
Keywords: Na,K-ATPase, redox regulation, glutathionylation, nitrosylation, oxidation, cysteine residues, cardio-
tonic steroids
БИОФИЗИКА том 65
№ 5
2020