СОДЕРЖАНИЕ
ВВЕДЕНИЕ.......................................................229
РОЛЬ ПЕПТИДОВ В ОРГАНИЗМЕ................231
ПРИМЕНЕНИЕ ПЕПТИДОВ В БИОФАРМАЦЕВТИКЕ................................231
СОВРЕМЕННЫЕ ПОДХОДЫ К ПОЛУЧЕНИЮ СИНТЕТИЧЕСКИХ ПЕПТИДНЫХ ПРЕПАРАТОВ........................232
ОСНОВНЫЕ НАПРАВЛЕНИЯ РАЗВИТИЯ НОВОГО ПОКОЛЕНИЯ ПЕПТИДНЫХ ПРЕПАРАТОВ........................234
СТРАТЕГИИ ПОВЫШЕНИЯ ПРОНИ- ЦАЕМОСТИ И ПРОТЕОЛИТИЧЕСКОЙ СТАБИЛЬНОСТИ ПЕПТИДОВ .....................234
СИНТЕЗ ЦИКЛИЧЕСКИХ ПЕПТИДОВ И ПЕПТИДОМИМЕТИКОВ...........................236
ЗАКЛЮЧЕНИЕ................................................239
СПИСОК ЛИТЕРАТУРЫ................................239
ВВЕДЕНИЕ
Известно, что вещества, состоящие из аминокислот, называют белками – это важнейшие компоненты жизнедеятельности всего живого на Земле. Аминокислоты соединены между собой в цепочки химической амидной связью, называемой пептидной.
Исследования химической структуры и биологических свойств веществ пептидно-белковой природы стали возможны в начале XX века, когда ученым-химикам, в первую очередь Э. Фишеру, удалось разработать методы избирательного синтеза пептидов, состоящих из нескольких аминокислот [1].
В настоящее время в мировой фармацевтической индустрии зарегистрировано >5000 фармацевтических субстанций на основе природных и синтетических органических соединений и только ~80 препаратов из них пептидной природы (65 зарубежных и 14 российских).
Для удобства работы принято условно классифицировать пептиды по размерам пептидной цепи. Все молекулы, состоящие из 50 и более аминокислотных остатков (а.о), называют белками, а менее длинные последовательности – пептидами. В свою очередь, пептиды тоже подразделяют на полипептиды (20–50 а.о.), олигопептиды (10–20 а.о.) и короткие пептиды (мини-пептиды, 2–10 а.о.). Все эти разделения довольно условны, и с развитием методов получения белков и пептидов различия, в первую очередь технологического свойства, постепенно стираются.
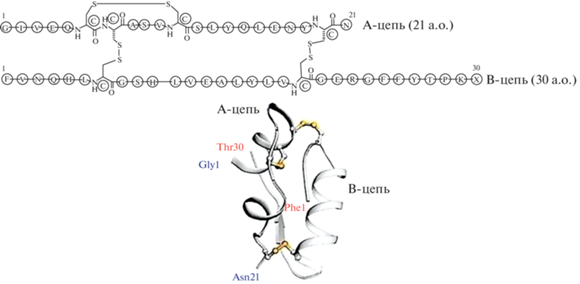
В качестве примера на рис. 1 приведены аминокислотная последовательность и трехмерная структура исторически одного из наиболее важных соединений – инсулина (пограничного, по определению, между полипептидами и белками) [2].
РОЛЬ ПЕПТИДОВ В ОРГАНИЗМЕ
Пептиды широко распространены в природе. Пептидный пул присутствует во всех клеточных организмах и представляет собой уникальный класс фармакологических соединений, находящийся между малыми органическими молекулами (small molecules), используемыми в фармацевтике в качестве лекарственных препаратов, и белками, хотя биохимически и терапевтически они отличны от обеих групп [3, 4].
Пептиды – универсальные регуляторы многочисленных процессов, протекающих в живых организмах. Пептиды представляют собой фрагменты белков, образующиеся при их частичном гидролизе. При нарушении пептидэргической регуляторной функции в работе клеток происходит сбой, влекущий за собой расстройство жизнедеятельности и функционирования поврежденного органа и всего организма. Пептиды также регулируют эмоциональные реакции, сон, сексуальное поведение, агрессию, проявляют анальгетические и многие другие свойства [5]. Пептиды отвечают и за стимуляцию или подавление иммунитета [6]. Некоторые пептиды также способствуют выведению из организма радионуклидов и солей тяжелых металлов [7]. Использование пептидов в качестве терапевтических средств эволюционировало в течение времени и продолжает развиваться вместе с появлением новых подходов как к их синтезу, так и к современным методам лечения.
Пептиды – основной пример того, как природа создает из одного гена, кодирующего соответствующий белок, множество регулируемых функциональных пептидов, действующих в организме в нужных местах и в нужное время. Известно, что белки на начальной стадии процессинга гидролизуются сложной системой из более чем 500 протеаз, действующих на внутриклеточные сайты, откуда, после секреции, пептиды транспортируются во внеклеточную среду для выполнения своих функций с последующим регулируемым гидролизом до аминокислот [8].
После завершения работ по расшифровке генома человека, определивших новое направление в науке, названное геномикой, возникло множество новых разнообразных комплексных “омикс-технологий”. Одним из важнейших комплексных направлений, следующим после геномики, на котором ученые сосредоточили свое внимание, стала протеомика, еще более комплексная программа исследования структуры и функции белков в организме. Геномная и протеомная эра стимулировали работы, в результате которых исследователи смогли идентифицировать молекулярные характеристики и структуры рецепторов для многих важных эндогенных пептидов [9, 10].
На базе новой парадигмы стало понятно, что следующим логичным этапом следует ожидать рождение нового раздела биоорганической химии – пептидомики, направленного на исследования процессинга белков и детальное изучение пептидов как продуктов белкового гидролиза. Пептидомика, выросшая из протеомики, обеспеченная современными технологиями, в настоящее время, помимо синтетического направления, занимается всесторонним качественным и количественным анализом пептидов в биологических образцах [11, 12]. Основные задачи пептидомики – всестороннее картирование протеолитических фрагментов белков, идентификация возможных пептидных биомаркеров различных заболеваний, определение пептидных гормонов и других сигнальных молекул. Комплексные биологические системы, обычно исследуемые в пептидомике, требуют систематической экстракции различных типов пептидов для достижения успешного анализа. Методами пептидомики были обнаружены и выделены из различных тканей, клеток и органелл животных новые пептидные молекулы и установлены их последовательности с помощью современной методологии секвенирования [13].
Постоянно растущий интерес вызывают биологически активные пептиды, действующие на центральную нервную систему. В центре внимания находятся нейропептиды, в особенности нейромедиаторы, модуляторы нервной активности, эндогенные пептиды опиоидного действия и многие другие [14]. Один из примеров – важный класс эндогенных нейроактивных мессенджеров – нейропептиды [15], они участвуют в регуляции большинства физиологических процессов у животных. Пептидные гормоны первоначально синтезируются на рибосоме в виде препрогормона. Этот предшественник с высокой молекулярной массой содержит N-концевой сигнальный пептид, направляющий белок в эндоплазматический ретикулум, где сигнальный пептид отщепляется сигнальной пептидазой. Прогормон обычно не обладает значительной биологической активностью. Большинство прогормонов и про-нейропептидов содержат в своих последовательностях несколько пептидов меньшего размера с различными биологическими активностями. Эти прогормоны обычно подвергаются посттрансляционной обработке – образованию дисульфидных связей или химическим модификациям, таким как N-гликозилирование, O-гликозилирование, ацетилирование или амидирование. Прогормоны подвергаются дальнейшему специфическому протеолитическому расщеплению, а затем сортируются в секреторные гранулы. Многие гормоны (вазопрессин, пролактин, окситоцин, АКТГ, брадикинин и др.) относятся к пептидам [16].
ПРИМЕНЕНИЕ ПЕПТИДОВ В БИОФАРМАЦЕВТИКЕ
Современная пептидомика и направление пептидной биофармацевтики не отставали от научных инноваций, исследуя новые соединения и молекулярные мишени, используя новые стратегии химии для расширения молекулярного разнообразия, а также путем разработки улучшенных фармацевтических свойств пептидов и пептидомиметиков. Поиск и выделение новых биологически активных пептидов из различных источников, определение спектра их активностей расширяют возможности использования пептидных соединений. Современные технологии позволяют выделять и исследовать в качестве кандидатов потенциальных терапевтических средств пептиды из животных, морских организмов и растений, а также антимикробные пептиды из амфибий и других микроорганизмов [17].
Пептиды представляют уникальную терапевтическую нишу и занимают заметное место в развивающейся биофармацевтике. Они перспективны как инструменты для решения проблем, связанных с усовершенствованием специфики действия лекарств, а также для поиска новых соединений и их аналогов, селективно связывающихся на молекулярном уровне с белковыми мишенями [18]. Изучение биологической активности пептидов в различных моделях и исследование их влияния на физико-химические процессы в организме показало несомненную перспективность дальнейшего использования этих соединений в медицинских целях.
Новым этапом развития медицинской науки и практики стала разработка персонализированной медицины. Потребовался больший акцент на эффективность, безопасность и специфичность действия препаратов и методов диагностики. Основываясь на последних достижениях в области пептидомики, стало возможным определять с помощью пептидов новые биомаркеры на ранних стадиях заболевания [19, 20].
Принимая во внимание положительные терапевтические свойства пептидов, многие разработчики лекарств увеличили количество своих потенциальных разработок за счет новых пептидов и пептидомиметиков. Поэтому потенциальный рынок пептидных препаратов стал более интенсивно развиваться, когда, понимая его перспективность, на него обратили внимание разработчики лекарственных средств из глобального фармацевтического сектора [21].
Большинство пептидных препаратов (~85%) получают химическим синтезом, что в значительной степени связано с развитием современных методов пептидного синтеза, и лишь для 15% пептидных лекарств используют рекомбинантные методы. Технологии химического синтеза предоставляют большие возможности для модификаций молекул, в том числе с использованием неприродных аминокислот, внедрением псевдопептидных связей и других модификаций, недоступных имеющимся рекомбинантным методам [22].
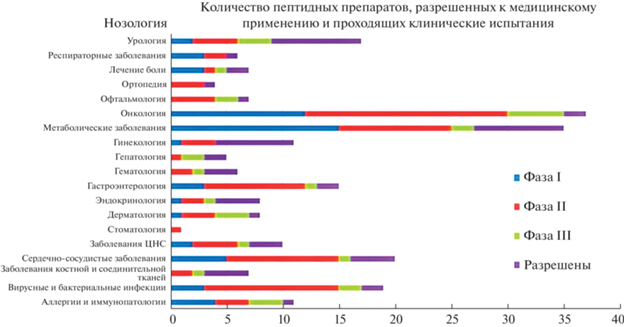
Пептиды исследуют по широкому спектру имеющихся и перспективных показаний, представляющих большой интерес для клинического применения. В настоящее время стратегия применения пептидных препаратов сместилась от гормональной терапии и диагностики рака в сторону лечения более широкого спектра заболеваний. Продолжает расти интерес к применению пептидов при таких заболеваниях, как диабет, остеопороз, сердечно-сосудистые заболевания, анемия, синдром раздраженного кишечника, болезнь Кушинга, рассеянный склероз и др. (рис. 2) [23].
СОВРЕМЕННЫЕ ПОДХОДЫ К ПОЛУЧЕНИЮ СИНТЕТИЧЕСКИХ ПЕПТИДНЫХ ПРЕПАРАТОВ
Несмотря на то что рынок пептидно-белковых лекарственных препаратов занимает небольшую часть (2%) мирового рынка лекарств, количество разрешенных пептидно-белковых препаратов увеличивается интенсивнее, чем остальной рынок традиционных лекарств [23]. Производство пептидов и белков сложное и более дорогостоящее [24] по сравнению с малыми молекулами, которые относительно легче синтезировать, поэтому такие препараты обычно более дешевые, чем пептидно-белковые.
Пептиды, как правило, имеют низкую проницаемость через клеточные мембраны, ограниченную стабильность и низкую пероральную биодоступность, поэтому преимущественный путь их введения в организм – это подкожные (SC), внутримышечные (IM) или внутривенные (IV) инъекции. В настоящее время увеличивается количество разработок пептидных препаратов, применяемых интраназально в виде капель и/или спреев [25].
Сравнение положительных и отрицательных свойств пептидов и химических молекул приведено в табл. 1.
Таблица 1.
Сравнение положительных и отрицательных свойств пептидов и химических молекул
| Химические молекулы | Пептиды | ||
|---|---|---|---|
| 80% рынка | 2% рынка | ||
| + | Низкая себестоимость синтеза | – | Высокая себестоимость синтеза |
| + | Высокая мембранная проницаемость | – | Низкая мембранная проницаемость |
| + | Высокая стабильность in vivo | – | Низкая стабильность in vivo |
| + | Высокая биодоступность per os | – | Низкая биодоступность per os |
| – | Низкая гидролитическая стабильность | ||
| – | Ограниченность способов введения в организм | ||
| – | Высокая вероятность перекрестного взаимодействия с другими мишенями (рецепторами) | + | Низкая вероятность перекрестного взаимодействия с другими мишенями (рецепторами) |
| + | Высокая селективность и аффинность к рецепторам | ||
| + | Легкость идентификации структуры | ||
| + | Множественность биомишеней in vivo | ||
| + | Низкая токсичность и иммуногенность | ||
| + | Меньшая конкуренция среди дженериков |
На начальных этапах развития пептидной фармацевтики малочисленность зарегистрированных пептидных лекарственных препаратов по сравнению с малыми химическими молекулами была обусловлена несколькими объективными факторами. К ним относятся короткий период полувыведения многих пептидных молекул, которые были первыми синтетическими пептидными лекарственными средствами, из-за присутствия в организме многочисленных пептидаз и экскреторных механизмов, гидролизующих природные пептиды. Еще одним препятствием, замедлявшим внедрение пептидов в качестве лекарственных средств, выступает низкая пероральная биодоступность [26–28]. Поскольку пероральная доставка часто рассматривается как предпочтительная для пациентов, наличие на ранних этапах только парентеральных форм сделала пептидные лекарственные препараты менее привлекательным вариантом в случае необходимости их хронического амбулаторного приема. В ряде случаев пептиды становятся успешными препаратами, поскольку проявляют множественное (плейотропное) действие на различные мишени в организме [29].
Новые стратегии химического синтеза и адресной доставки препаратов позволяют изменять фармакокинетические, физическо-химические свойства и специфичность молекулы. Для этого проводят модификации аминокислот или пептидной цепи, включение неприродных аминокислот, конъюгацию пептидов с носителями для увеличения периода полувыведения или улучшения растворимости [30, 31].
Отметим, что синтез пептидов – это одно из самых сложных направлений синтетической органической химии, поскольку получение простейшего пептида, состоящего из двух аминокислот, требует до четырех стадий синтеза. Химия пептидов, как раздел органической химии, постепенно развивалась в мире наряду с совершенствованием общих методов органического синтеза и очистки.
В “допептидомную эру” большой прорыв в направлении развития пептидной химии произошел в середине 1950-х гг. Ученым удалось выделить в индивидуальном виде ряд пептидных гормонов и белков и установить их химические структуры.
Впервые, в начале 1950-х гг., под руководством В. дю Винье после установления структуры гормона окситоцина был осуществлен его полный химический синтез [32]. Эта работа увенчалась Нобелевской премией по химии в 1955 г. “за выделение, структурную идентификацию и общий синтез циклического пептида окситоцина” [33]. Успехи команды В. дю Винье заложили фундамент пептидной фармацевтики. Усилия исследователей во многих странах были направлены на выделение биологически активных пептидов из различных организмов, установление их строения и воспроизведение синтетическим путем.
Несмотря на все сложности синтеза, отсутствие индустрии обеспечения специализированными реактивами и эффективной системы очистки получаемых пептидных молекул, в мире стали разрабатывать не только лабораторные, но и технологические методы для создания промышленного производства лекарственных препаратов на основе пептидов.
Индивидуальные синтетические пептидные лекарственные препараты появились на мировом фармацевтическом рынке в начале 1960-х гг. Большие усилия были приложены для получения первых синтетических гормонов: окситоцина, вазопрессина, производных лютенизирующего гормона. Впоследствии в ряде лабораторий начались “состязания” по первому синтезу белка. Группами американских (Р. Меррифилд) и западногерманских (Г. Цан) химиков практически одновременно был завершен полный синтез инсулина – первый химический синтез белка [34].
Фактически регулярное внедрение в практику синтетических пептидных лекарственных препаратов началось в начале 1970-х гг. Зарубежные компании выводили на фармацевтический рынок пептидные гормоны, антибиотики, противоопухолевые препараты.
ОСНОВНЫЕ НАПРАВЛЕНИЯ РАЗВИТИЯ НОВОГО ПОКОЛЕНИЯ ПЕПТИДНЫХ ПРЕПАРАТОВ
Механизм действия биологически активных веществ обычно заключается в их взаимодействии со специфическими рецепторами. Каждый рецептор обладает характерной пространственной структурой участка, взаимодействующего с биологически активным веществом, и их структуры должны соответствовать друг другу, учитывая не только последовательность аминокислот, но и стереоспецифичность по отношению к активному центру рецептора. У большинства лекарственных препаратов существует прямая взаимосвязь между пространственной структурой, стереоспецифичностью и фармакологической активностью.
Многие синтетические ксенобиотики (непептидные лекарственные препараты) существуют в виде смеси двух или большего числа пространственных изомеров, отличающихся своими биологическими свойствами. Такие рацемические препараты могут вызывать серьезные побочные эффекты (известный пример – препарат Талидомид [35]). Выявление фармакокинетических и фармакодинамических особенностей отдельных изомеров открывает перспективы для совершенствования уже известных лекарственных средств. Лишь 15% синтетических химических препаратов, находящихся на европейских рынках, производится в виде отдельных стереоизомеров, остальные 85% представляют собой смеси изомеров. В то же время пептидные препараты получают, как правило, в виде индивидуальных изомеров и выбирать из них те, которые обладают наиболее выраженными эффектами и/или наименьшей токсичностью [36, 37].
Поскольку природные пептиды легко гидролизуются протеазами, которые распространены по всему организму, то протеолиз – основной путь элиминации большинства пептидов. Поэтому важно определять основные структурные свойства при взаимодействии между пептидом и его мишенью, чтобы проводить успешные модификации с целью повышения стабильности и сохранения биологической активности исследуемого соединения в качестве кандидата на создание лекарственного препарата. Природные немодифицированные пептиды имеют короткий период полураспада [4] и низкую проницаемость через клеточные мембраны, поэтому их активность ограничена, как правило, внеклеточными мишенями [23, 38]. Но даже при таких ограничениях, благодаря уникальным свойствам, пептиды остаются перспективными кандидатами для разработки новых лекарственных средств. Преимущества природных пептидов перед ксенобиотиками – высокие константы связывания с белковыми мишенями, легкая идентификация субстрата связывания, высокая целевая специфичность, широкий спектр клинического применения, низкая токсичность и иммуногенность и многие другие [38]. Низкая пероральная биодоступность (<1%) [40, 41] немодифицированных пептидов в основном обусловлена коротким периодом полураспада (минуты), вызванным протеолизом в крови, почках или печени, а также быстрым почечным клиренсом при первичном прохождении желудочно-кишечного тракта и печени [23, 40, 42]. Несмотря на сложность разработки перорально стабильных пептидов и пептидомиметиков, во многих лабораториях интенсивно разрабатываются такие препараты [43, 44]. Подходы к повышению стабильности пептидов постоянно совершенствуются, создавая новые варианты структурных модификаций. Одним из очевидных решений стабилизации гидролитической лабильности лекарственных препаратов, содержащих природные пептиды, выступает синтез модифицированных аналогов природных пептидов, ранее зарегистрированных как парентеральные лекарственные средства. Модификации аналогов основаны на введении замен в различные части исходной молекулы для стабилизации, а иногда и изменения ее структуры, спектра и даже направления действия, с перспективой получения нового поколения препаратов с улучшенными фармацевтическими свойствами.
СТРАТЕГИИ ПОВЫШЕНИЯ ПРОНИЦАЕМОСТИ И ПРОТЕОЛИТИЧЕСКОЙ СТАБИЛЬНОСТИ ПЕПТИДОВ
Замена природных аминокислот – одна из стратегий, используемых для предотвращения гидролиза; для этого проводят модификации по сайтам, подвергающимся гидролизу, с последующим замещением исходной аминокислоты [10]. В качестве заместителей могут быть D-аминокислоты, β-аминокислоты, дегидроаминокислоты и другие неприродные производные аминокислот, а также различные производные олефинов. Такие модификации способствуют улучшению стабильности и увеличению периода полувыведения препаратов из плазмы [41, 43, 45, 46].
Описаны примеры, когда устойчивые к деградации протеазами пролин и гидроксипролин вводили в места расщепления, заменяя легко подвергающиеся гидролизу аминокислоты, что улучшало стабильность препарата в организме [46, 47]. Для этих же целей также использовали N-метилирование или введение N-метил-аминокислот [48, 49].
Одновременное включение D-аминокислот и N-метилирование амидных связей может значительно повысить метаболическую стабильность, создавая дополнительные стерические препятствия. Известно много структурных модификаций, включающих N-алкилирование, для повышения биологической активности и метаболической стабильности пептидов [50–55].
Множество протеолитических ферментов в крови, плазме, печени или почках – экзопептидазы, аминопептидазы и карбоксипептидазы – гидролизуют пептидные последовательности с N- и С-концов. Поэтому N-ацилирование и C-амидирование часто также повышают устойчивость модифицированных пептидов к протеолизу [50–55].
Общепринятым вариантом для повышения жесткости, образования внутримолекулярных водородных связей и уменьшения межмолекулярного гидратирования выступает циклизация линейных молекул [50–55].
Деградация экзопептидазами может быть подавлена путем введения N-концевых D-аминокислот, восстановления С-концевого карбоксила в соответствующий спирт [50–55].
Получает распространение метод химических “сшивок” боковых групп аминокислот в пептидной цепи посредством “сшивки” боковых остатков аминокислот в цепи углеводородными “вставками” либо образованием лактамных мостиков для стабилизации спиральности, повышения стабильности и внутриклеточной проницаемости пептидов, так называемый метод “stapled peptides” [50–55].
Еще один современный подход для повышения стабильности и создания пролонгированной формы нового соединения – конъюгация пептидов с макромолекулами. Для этих целей применяют различные полимеры, например, полиэтиленгликоль (ПЭГ) [50–55]. ПЭГилирование пептидов позволяет эффективно снижать потенциальную иммуногенность, сохранять биологическую активность и замедлять их ферментативный гидролиз [50–55]. Для стабилизации пептидов и защиты от протеолиза используют некоторые жирные кислоты, с которыми конъюгируют пептиды, или инкапсулируют пептид в липосомы, нано/микрочастицы или мицеллы с большей молекулярной массой. Метод конъюгации приводит к увеличению периода полураспада и повышению биодоступности пептидных препаратов. В результате конъюгации пептидов с липидами получают липопептидные производные, сочетающие биологические свойства пептидов и липидов и обладающие улучшенной эффективностью и селективностью. Липидизация пептидов приводит к образованию амфифильных пептидных конъюгатов с повышенной биодоступностью и увеличению их транспорта через клеточные мембраны [50–55].
В последнее время получила распространение новая концепция создания полноразмерных энантиомерных D-пептидов, замещающих все соответствующие L-аминокислоты. Такие пептиды (D-пептиды) увеличивают период полураспада целевого продукта и значительно улучшают его стабильность [55, 56].
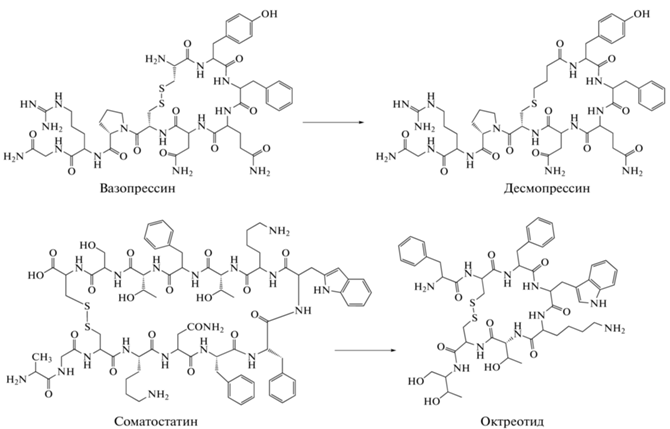
Один из первых классических примеров успешных модификаций природных пептидных молекул – гормон вазопрессин, содержащий L‑Arg и имеющий период полураспада у человека 10–35 мин [57]. Аналог вазопрессина, в котором L-Arg заменен на D-Arg, получил название десмопрессин, и его период полураспада составляет ~4 ч [58]. Похожий пример – октреотид – это аналог соматостатина, применяемого для лечения желудочно-кишечных опухолей. Этот пептид имеет укороченную последовательность соматостатина (8 вместо 14 а.о.) и с заменами L-аминокислот D-аминокислотами (рис. 3) [59, 60].
СИНТЕЗ ЦИКЛИЧЕСКИХ ПЕПТИДОВ И ПЕПТИДОМИМЕТИКОВ
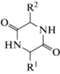
Минимальные циклические структуры пептидных соединений – 2,5-дикетопиперазины (2,5-ДКП), представляющие собой циклодипептиды, полученные конденсацией двух α-аминокислот (рис. 4).
На основе ДКП можно генерировать большое число различных структур для поиска новых лидерных соединений [61]. Производные 2,5-ДКП часто встречаются (как в виде простых незамещенных 2,5-ДКП-структур, так и более сложных молекулярных конструкций) в разнообразных натуральных продуктах, грибах, бактериях, во многих растениях и у млекопитающих. Множество антибиотиков – производные дикетопиперазинов [61]. В качестве примера можно привести лекарственные препараты от простых циклических дипептидов, таких как производное димера циклосерина или каиромицина B [62], до сложных сопряженных полиядерных систем, таких как бицикломицин [63, 64].
Сама природа создала молекулы, удобные для модификаций, поскольку 2,5-ДКП состоит из центрального фрагмента (центроида или scaffold), в который можно вводить различные заместители (рис. 5). 2,5-ДКП представляют собой шестичленные гетероциклические соединения с ограниченным конформационным набором положений. 2,5-ДКП устойчивы к протеолизу и весьма привлекательны для структурно-функциональных исследований при поиске новых потенциальных лекарственных препаратов. Эти конформационно ограниченные хиральные центроиды имеют шесть положений, доступных для структурной модификации различными функциональными группами с определенной стереохимией. Структура 2,5-ДКП позволяет проводить модификации не только по всем шести позициям молекулы, но и получать стереохимические изомеры по всем четырем положениям оптических центров. 2,5-ДКП имеют жесткий каркас, способный имитировать предпочтительную конформацию, ограничивая подвижность аминокислот, встроенных в его структуру. 2,5-ДКП, состоящие из трифункциональных аминокислот, содержат различные функциональные группы, которые дополнительно могут быть использованы как для “нащупывания” положений мишени (с которыми эта молекула вступает во взаимодействие), так и служить линкерами для присоединения различных фармакофоров (рис. 6) [64].
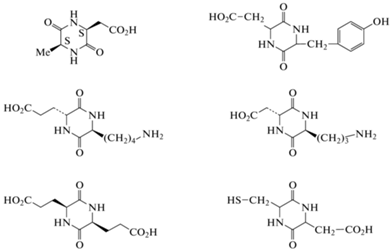
Рис. 6.
Общая формула платформы для синтеза библиотек пептидомиметиков на основе замещенных 2,5-ДКП, где A, D – биологически активные фармакофоры или фрагменты пептидных соединений; L1 и L2 – биогидролизуемые линкеры; m, n – количество CH2-групп (от 0 дo 4); R5 – возможные производные фармакофоров, присоединенные по атомам азота. *S или R – оптическая ориентация по положениям 3 и 6 атомов углерода [63].
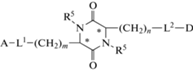
Функциональные группы (фармакофоры) должны легко подвергаться метаболическим превращениям, например, при присоединении к центроиду сложноэфирными связями, чтобы легко гидролизоваться в организме. 2,5-ДКП относительно легко синтезировать и присоединять к ним большое разнообразие заместителей, которыми служат различные аминокислоты, используемые в качестве строительных блоков. Набор заместителей позволяет варьировать многие физико-химические характеристики молекулы, такие как строение, размер, форму, липофильность, дипольный момент, электростатический заряд, функциональные группы. Все это позволяет in silico моделировать структуры для направленного дизайна библиотек производных 2,5-ДКП [65].
Понимание влияния стерических особенностей на физиологическую активность молекулы позволяет с помощью стереоспецифичных методов синтеза получать лекарственные препараты, обладающие наибольшей эффективностью и/или наименьшей токсичностью. При поиске новых лидерных соединений следует избегать присутствия в центроиде или заместителях группировок, способных придавать получаемым соединениям токсические свойства. Одна из причин различающейся физиологической активности стереоизомеров лекарственных препаратов – разница в их проникновении в организм. Это может быть связано с особенностями строения 2,5-ДКП, свойствами биологических мембран, которые сами построены из оптически активного, асимметрического материала, а также с наличием в мембранах транспортных систем, осуществляющих перенос метаболитов через мембраны [66].
Получил распространение подход с использованием производных 2,5-ДКП [67] как в виде коротких аналогов пептидов, так и в виде “вставок” в различные положения цепи, с варьированием местоположения 2,5-ДКП в N-, C-конце молекулы или внутри пептидной цепи для повышения гидролитической стабильности и возможности перорального применения получаемой молекулы [68].
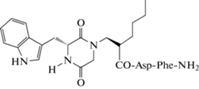
Примером таких модификаций могут служить исследования аналогов гормона холецистокинина (CCK). Производные ССК4 были синтезированы путем замены последовательности N-концевого дипептида Trp-Met на дикетопиперазиновое кольцо, полученное циклизацией аминокислот Gly и Trp, к которым далее по атому азота был присоединен трипептидный фрагмент Nle-Asp-Phe-NH2 (рис. 7), показавший положительные результаты связывания и селективности с ССК-рецепторами [67]. Проведенные эксперименты показали, что активность проявлял только аналог с R-конфигурацией углерода, находящегося у Сα-атома индольного кольца, тогда как соединения с S-конфигурацией не имели сродства к CCK-рецепторам (рис. 8).
В процессе изучения биологической активности изомеров дипептида Glu-Trp были получены новые сведения о влиянии химической и оптической структуры на биологические свойства этих изомеров. Показано, что L-Glu-L-Trp-OH, L‑Glu-(L-Trp-OH)-OH, а также их структурные и стереоизомеры D-Glu-D-Trp-OH и D-Glu-(D-Trp-OH)-ОН проявляют реципрокное действие на клетки тимуса (тимоциты) in vitro: L-L-пептиды обладают иммуностимулирующими свойствами, а D-D-пептиды – иммунодепрессивными [69]. Позже мы показали, что L-Glu-L-Trp-ONa (Тимоген®) и его структурный и оптический D‑изомер D-Glu-(D-Trp-OH)-ONa (Тимодепрессин®) оказывают сходное действие на гемопоэз: на гемопоэтических клетках-предшественниках in vitro и in vivo продемонстрировано, что L-L-пептиды обладают гемостимулирующими свойствами, а D-D-пептиды – гемосупрессорными [6].
Современные тенденции разработки пептидных лекарственных средств нового поколения демонстрируют растущий спрос на неинвазивные, предпочтительно пероральные, лекарственные формы. В лаборатории биофармацевтики ИБХ РАН разработан оригинальный метод создания пептидомиметиков на основе разветвленных 2,5-ДКП. Соответственно, была создана новая химическая платформа на основе разветвленных производных 2,5-ДКП для получения протеазорезистентных неинвазивных биологически активных пептидомиметиков [6, 64, 69]. Следуя этому подходу, были синтезированы серии перорально стабильных аналогов Тимогена®, эффективно стимулирующих восстановление нарушенного кроветворения и пролиферацию интактных клеток костного мозга [69], и перорально активных иммуно- и гемосупрессорных аналогов Тимодепрессина® [6].
Таким образом, производные биоактивных 2,5-ДКП привлекают все больший интерес исследователей, занимающихся поиском и разработкой новых лекарственных препаратов.
Возможности использовать для модификации все шесть положений гетероцикла и построение центроида из трифункциональных аминокислот позволили провести синтез ряда сложных структур, содержащих этот центроид. В результате сотрудничества различных специалистов удалось получить несколько новых лекарственных препаратов на основе 2,5-ДКП [65].
Методология химии пептидов постепенно развивалась в мире наряду с совершенствованием подходов к синтезу органических соединений. Большой прорыв в этом направлении произошел после первого успешного синтеза пептида окситоцина [32].
Как упоминалось выше, со времени первого синтеза окситоцина начался процесс создания мировой пептидной фармацевтики. Вскоре и в СССР стали развиваться работы в этом перспективном направлении. В конце 1960-х годов в Москве, Ленинграде, Киеве были созданы лаборатории химии природных соединений, занимавшиеся исследованиями пептидов. Несмотря на все сложности синтеза, отсутствие специализированных реактивов и эффективной приборной базы для очистки получаемых пептидных молекул, в СССР стали разрабатывать не только лабораторные, но и технологические методы для создания лекарственных препаратов на основе пептидов. В лабораториях СССР также проводили синтез и исследования пептидов, структуры которых были установлены к тому времени – окситоцина, вазопрессина, АКТГ, соматостатина и др. [70, 71].
Зарубежные фармацевтические компании выводили на рынок пептидные гормоны, антибиотики, противоопухолевые препараты. В СССР не отставали от темпов исследований зарубежных коллег, также проводились работы по синтезу различных пептидов, депсипептидов, ионофоров и других пептидных соединений и созданию препаратов на их основе. Пионерами в этом направлении были ученые из Москвы (ИХПС АН СССР, ныне Институт биоорганической химии им. М.М. Шемякина и Ю.А. Овчинникова РАН), МГУ им. М.В. Ломоносова, Институт молекулярной генетики АН СССР, Институт фармакологии МЗ СССР), Ленинграда (ЛГУ, Институт высокомолекулярных соединений АН СССР и Институт особо чистых биопрепаратов – ВНИИ ОЧБ) и Новосибирска (НПО “Вектор”).
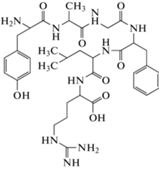
Первыми зарегистрированными в СССР в 1988 и 1989 гг. оригинальными отечественными пептидными лекарственными препаратами были Даларгин [72] (рис. 8) и Тимоген [73]. Даларгин стал первым в мире лекарством, созданным на основе синтетического нейропептида – аналога лейцин-энкефалина. Тимоген – природный иммунокорректор – пептид, первоначально выделенный из экстракта тимуса и впоследствии полученный синтетическим путем [73].
Была разработана промышленная технология получения пептидных лекарственных препаратов и налажено их производство на первом в СССР опытно-промышленном производстве биопрепаратов при Всесоюзном кардиологическом центре МЗ СССР. В это же время проводились работы по синтезу и созданию лекарственных препаратов в ряде других лабораторий и институтов страны. Были созданы препараты Семакс, Дельтаран, Ликопид, Иммунофан. В более позднее время на российский рынок вышли другие оригинальные препараты: Тимодепрессин, Селанк и Ноопепт [74–79].
В СССР и России за период до 2015 г. созданы и внедрены в производство ~30 оригинальных синтетических лекарственных препаратов, из них 14 препаратов представляют собой пептиды (табл. 2). Если сравнить достижения тех лет отечественных разработчиков с зарубежными, то получится, что из ~5000 химических препаратов ~15 отечественных (~0.1%), а из 80 пептидных препаратов – 14 отечественных, что составляло почти 20% процентов мировых разработок.
Таблица 2.
Отечественные оригинальные пептидные препараты
| № | Препарат | Активное вещество | Показания к применению |
|---|---|---|---|
| 1 | Даларгин | Tyr-D-Ala-Gly-Phe-Leu-Arg | Противоязвенное средство |
| 2 | Тимоген | Glu-Trp | Иммуномодулятор |
| 3 | Семакс | Met-Glu-His-Phe-Gly-Pro | Инсульт и ишемия мозга |
| 4 | Ликопид | GLcNAc(β1–4)Mur NAc | Иммуномодулятор |
| 5 | Имунофан | Arg-α-Asp-Lys-Val-Tyr-Arg | Иммуномодулятор |
| 6 | Тимодепрессин | γ-D-Glu-D-Trp | Иммуносупрессор |
| 7 | Гепон | Thr-Glu-Lys-Lys-Arg-Arg-Glu-Thr- Val-Glu-Arg-Glu-Lys-Glu | Противовирусное средство |
| 8 | Седатин | Arg-Tyr-D-Ala-Phe-Gly | Стресс-протектор (разрешен в ветеринарии) |
| 9 | Бестим (аналог Тимогена) | γ-D-Glu-L-Trp | Иммуномодулятор |
| 10 | Ноопепт | Этиловый эфир N-фенилацетил-
L-пролилглицина 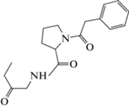 |
Ноотропное средство |
| 11 | Дельтаран | Trp-Ala-Gly-Gly-Asp-Ala-Ser-Gly-Glu | Стресс-протектор |
| 12 | Стемокин | Ile-Glu-Trp | Стимулятор гемопоэза, иммуно-модулятор |
| 13 | Селанк | Thr-Lys-Pro-Arg-Pro-Gly | Анксиолитик (транквилизатор) |
| 14 | Аллокин-альфа | His-Gly-Val-Ser-Gly-His-Gly- Gln-His-Gly-Val-His-Gly | Иммуномодулятор |
ЗАКЛЮЧЕНИЕ
Основные этапы развития химии пептидов и понимание важной роли этих соединений в процессах жизнедеятельности практически всех живых организмов создали предпосылки для интенсивной разработки методов анализа, синтеза и практического применения пептидов в медицине, ветеринарии и сельском хозяйстве. Последовательное внедрение пептидных лекарственных препаратов в медицинскую и ветеринарную практику стимулировало создание современной научной, технологической и клинической базы для расширения исследований “от идеи до аптеки”, и постепенно было сформировано пептидное направление биофармацевтики. Учитывая значительные успехи советских и российских пептидных исследований и разработок, есть надежда, что образовавшееся отставание от международных пептидных лабораторий будет преодолено, и отечественная биофармацевтика возобновит свое успешное развитие.