БИОХИМИЯ, 2019, том 84, вып. 11, с. 1743 - 1758
УДК 577.152.13
РОЛЬ МИТОХОНДРИАЛЬНОГО КОМПЛЕКСА I
В ПОВРЕЖДЕНИИ ТКАНИ ГОЛОВНОГО МОЗГА
ПОСЛЕ ИШЕМИИ/РЕПЕРФУЗИИ
Обзор
© 2019
А. Галкин*
Division of Neonatology, Department of Pediatrics, Columbia University William Black Building,
10032 New York, NY USA; E mail: ag4003@cumc.columbia.edu
Поступила в редакцию 07.06.2019
После доработки 08.07.2019
Принята к публикации 09.07.2019
Ишемический инсульт и неонатальная гипоксически ишемическая энцефалопатия являются одними из
основных причин инвалидности у взрослых людей и новорожденных. Потребности клеток мозга в энергии
обеспечиваются за счет механизма окислительного фосфорилирования, протекающего в митохондриях.
Синдром ишемии/реперфузии характеризуется нарушением процесса образования АТP в митохондриях,
что приводит к гибели клеток мозга из за недостатка энергии. Митохондриальный комплекс I - фермента
тивный комплекс дыхательной цепи, характеризующийся наибольшей чувствительностью к ишемии/ре
перфузии. Механизмы ингибирования этого комплекса до сих пор не достаточно изучены. В настоящем об
зоре рассмотрены данные литературы, касающиеся нарушения структуры и функций митохондрий во вре
мя ишемии/реперфузии, а также предложены два различных механизма повреждения комплекса I, осно
ванные на результатах последних исследований в лаборатории автора. Одним из механизмов является обра
тимая диссоциация естественного кофактора фермента флавинмононуклеотида в условиях ишемии. Другой
механизм предполагает модификацию ключевого остатка цистеина комплекса I после перехода фермента
из активного конформационного состояния в деактивированное (А/Д переход). В представленном обзоре
описано потенциальное влияние этих двух процессов на развитие нарушений функционирования митохон
дрий при ишемии/реперфузии и кратко обсуждаются возможные нейропротекторные стратегии, направ
ленные на снижение степени повреждения клеток при ишемии/реперфузии.
КЛЮЧЕВЫЕ СЛОВА: инсульт, повреждение в результате ишемии/реперфузии, митохондрии, комплекс I,
флавин, тиоловые группы, нитрозилирование.
DOI: 10.1134/S0320972519110150
Повреждение головного мозга в результате
личением среднего возраста населения можно
ишемии/реперфузии происходит в патологи
ожидать, что в ближайшем будущем частота ин
ческих условиях, таких как ишемический ин
сультов будет только возрастать. Другим клини
сульт, остановка сердца, травматическое пов
ческим проявлением ишемии/реперфузии моз
реждение мозга или перинатальные гипокси
га являются перинатальные гипоксически ише
чески ишемические травмы. Инсульт стоит на
мические энцефалопатии, при которых смерт
пятом месте среди самых распространенных
ность новорожденных в 2010 г. во всем мире сос
причин смертности во всем мире. В течение
тавила 1,15 млн человек [1]. В целом, стоимость
жизни каждый шестой человек переносит ин
лечения и продления жизни выживших больных
сульт различной степени тяжести. В связи с уве
оценивается в триллионы долларов [2]. Несмот
ря на широкое распространение и разрушитель
ные последствия ишемии мозга, до сих пор ос
Принятые сокращения: А и Д формы - активная и
новными способами лечения инсульта остаются
деактивированная формы комплекса I соответственно;
А/Д переход - переход комплекса I из активного в деакти
механические или фармакологические способы
вированное состояние; АФК - активные формы кислоро
восстановления кровоснабжения или реперфу
да; ЭПФ - электронпереносящий флавопротеин β окис
зии. Также до сих пор нет полного понимания
ления жирных кислот; MCAo - middle cerebral artery occlu
молекулярных механизмов критических по
sion, окклюзия средней церебральной артерии; RET -
вреждений ткани мозга при синдроме ише
reverse electron transfer, обратный перенос электронов; Q -
убихинон.
мии/реперфузии. В настоящее время активные
* Автор является выпускником кафедры биохимии биоло
научные исследования направлены на разработ
гического факультета МГУ им. М.В. Ломоносова.
ку способов терапевтического вмешательства,
1743
1744
ГАЛКИН
снижающего степень начального повреждения
которые переносят электроны от своих субстра
тканей во время ишемии/реперфузии. В пред
тов к убихинону (Q): глицерол 3 фосфатдегид
ставленном обзоре проанализированы недавние
рогеназа (GPDH), расположенная на внешней
достижения в изучении роли митохондриально
стороне [7], и дегидрогеназа электронперенося
го комплекса I во время острой первичной и
щего флавопротеина (ЭПФ:Q оксидоредуктаза),
вторичной энергетической недостаточности
локализованная на внутренней стороне мембра
мозга при ишемии/реперфузии. В настоящей
ны митохондрий и участвующая в процессе
работе рассматриваются изменения в митохон
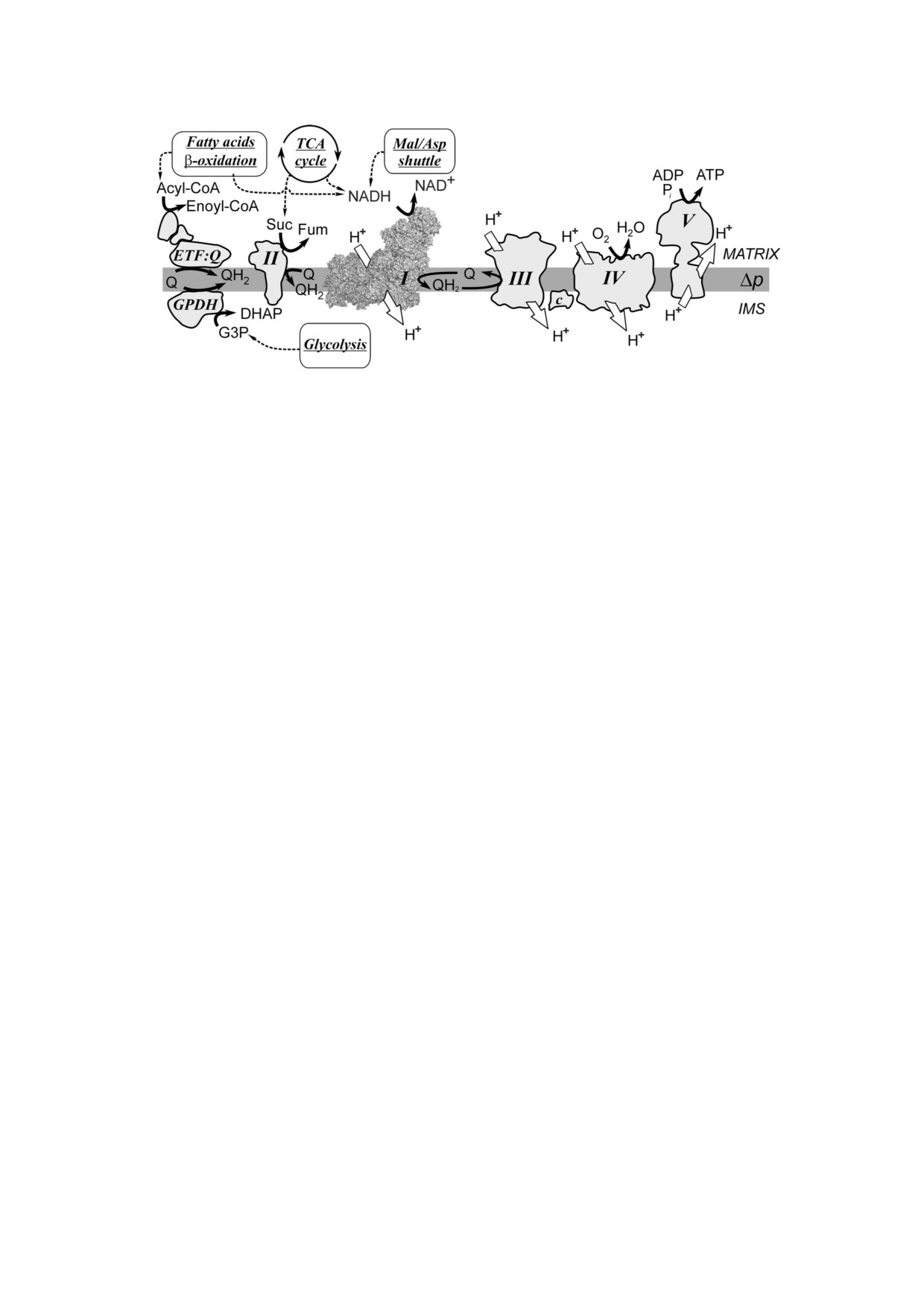
окисления жирных кислот [8] (рис. 1).
дриях, происходящие только в первые часы пов
Комплекс I или протон транслоцирующая
реждения тканей во время острого энергодефи
NADH дегидрогеназа (NADH:убихинон окси
цита; сведения о долговременных аспектах вли
доредуктаза) - это единственный фермент, от
яния ишемии/реперфузии на митохондриаль
вечающий в матриксе митохондрий за окисле
ный метаболизм читатель может найти в ранее
ние NADH и регенерацию NAD+, необходимого
опубликованных обзорах [3-6].
для стабильного протекания катаболических
процессов (цикл трикарбоновых кислот, β
окисление жирных кислот и процесс гликоли
ДЫХАТЕЛЬНАЯ ЦЕПЬ МИТОХОНДРИЙ
за). При переносе 2 х электронов от молекулы
И МИТОХОНДРИАЛЬНЫЙ КОМПЛЕКС I
NADH на убихинон комплекс I переносит 4
протона через мембрану, таким образом, обла
Работа нашего мозга главным образом зави
дая наибольшим «коэффициентом полезного
сит от функционирования митохондрий, выра
действия» по сравнению с остальными фермен
батывающих энергию в результате совместной
тами дыхательной цепи [9] (рис. 1).
работы ферментов метаболизма, дыхательной
Митохондриальный комплекс I представля
цепи и АТР синтазы (рис. 1). Углеводы, липиды
ет собой мембраносвязанный L образный фер
и аминокислоты разлагаются до более простых
мент, состоящий из гидрофобного мембранного
универсальных промежуточных продуктов, ко
и относительно гидрофильного периферийного
торые могут окисляться с высвобождением
домена, выступающего в митохондриальный
энергии. Окислительный процесс начинается с
матрикс. У млекопитающих этот фермент со
переноса электронов от субстратов через систе
стоит из 45 субъединиц, и функции многих из
му переносчиков, образующих дыхательную
них до сих пор не установлены. Из 14 консерва
цепь, расположенную во внутренней мембране
тивных субъединиц комплекса, составляющих
митохондрий. Ферментные комплексы дыха
каталитическую единицу бактериального фер
тельной цепи в несколько стадий переносят
мента, 7 гомологичных мембранных полипепти
электроны на конечный акцептор электронов -
дов эукариот кодируются митохондриальной
молекуляный кислород (рис. 1). Комплексы I,
ДНК (ND субъединицы) [10]. Ферментативный
III и IV дыхательной цепи используют энергию,
комплекс можно подразделить на три функцио
высвобождающуюся в ходе окислительно вос
нальных модуля: периферический N модуль, с
становительных реакций, для векторного пере
которым связывается молекула NADH; цент
носа протонов через мембрану и таким образом
ральный Q модуль, где происходит восстанов
создают протондвижущую силу (Δp) (рис. 1).
ление убихинона; и расположенный в мембране
Обратное движение протонов из межмембран
P модуль, где происходит транслокация прото
ного пространства в митохондриальный мат
нов через мембрану [11]. Все редокс центры
рикс приводит к синтезу АТР из АDР и неорга
фермента локализованы в модулях N и Q. Комп
нического фосфата комплексом V. В ходе опи
лекс I содержит одну молекулу прочно, но не
санного выше процесса обеспечиваются пот
ковалентно связанного флавинмононуклеотида
ребности клеток мозга в энергии. Основная
(FMN) и восемь железо серных кластеров. Фла
часть энергии, запасенная в виде АТР, необхо
вин непосредственно принимает электроны,
дима для осуществления ионного транспорта во
поступающие от NADH матрикса, и далее пере
время распространения потенциала действия по
дает их на цепочку последовательно располо
нервному волокну.
женных железо серных кластеров, обеспечива
В отличие от схемы, изложенной в большин
ющих перенос электронов в примембранной об
стве учебников биохимии, в которой электроны
ласти на молекулу убихинона. На конечном эта
поставляются в дыхательную цепь только на
пе восстановления убихинона, скорее всего,
уровне комплексов I и II, в этом процессе участ
происходит образование промежуточного про
вуют и другие ферменты. В головном мозге на
дукта - семихинона, участвующего в энергети
внутренней митохондриальной мембране рас
ческом сопряжении [12, 13]. Высвобождение
положены две дополнительные дегидрогеназы,
энергии на последнем этапе переноса электро
БИОХИМИЯ том 84 вып. 11 2019
КОМПЛЕКС I ПРИ ИШЕМИИ/РЕПЕРФУЗИИ МОЗГА
1745
Рис. 1. Схема процесса окислительного фосфорилирования во внутренней мембране митохондрий. Несколько типов де
гидрогеназ транспортируют электроны от субстратов на убихинон (Q) в мембране. ЭПФ:убихинон оксидоредуктаза
(ETF:Q) переносит электроны, высвободившиеся на стадии дегидрирования различных ацил КоА в процессе окисления
жирных кислот в матриксе. На внешней стороне внутренней мембраны глицерол 3 фосфатдегидрогеназа (GPDH) окис
ляет образующийся в процессе гликолиза глицерол 3 фосфат (G3P) с образованием дигидроксиацетонфосфата (DHAP).
Комплекс II или сукцинатдегидрогеназа цикла трикарбоновых кислот окисляет янтарную кислоту (Suc) до фумаровой
(Fum). Комплекс I (NADH:убихинон оксидоредуктаза) или NADH дегидрогеназа представлен в виде структурной моде
ли (PDB 6G2J, атомы показаны в виде сфер). Комплекс I окисляет NADH, образующийся в процессе окисления жирных
кислот, цикла трикарбоновых кислот и малат аспартатного челночного механизма (Mal/Asp shuttle). Комплексы I, III и
IV дыхательной цепи митохондрий транслоцируют протоны (H+) через мембрану (белые стрелки) создавая протондвижу
щую силу (Δp), использующуюся комплексом V для синтеза ATP
нов приводит к конформационным изменени
восстанавливать NAD+ в реакции обратного пе
ям, передающимся на P модуль фермента, где
реноса электронов (RET, reverse electron trans
происходит трансмембранный перенос прото
fer). Во время обратного переноса электроны
нов ND субъединицами [14].
переходят от высокопотенциальных редокс
В физиологических условиях комплекс I
центров фермента на центры с более отрица
окисляет NADH и переносит электроны на мо
тельным окислительно восстановительным по
лекулу убихинона. В этот процесс вовлечены
тенциалом по направлению к FMN. Эта энерго
почти все редокс центры фермента. NADH:уби
потребляющая реакция требует наличия высо
хинон редуктазная реакция чувствительна к
кого соотношения QH2/Q и протондвижущей
действию хиноноподобных ингибиторов фер
силы на мембране. Эти условия выполняются
мента, таких как ротенон и пиерицидин. В то же
при окислении сукцината, глицерол 3 фосфата
время комплекс I способен катализировать и
или жирных кислот. Соответствующие дегидро
другие реакции [15]. NADH может быть окислен
геназы образуют QH2, а протондвижущая сила
так называемыми искусственными акцепторами
возникает благодаря совместной работе комп
электронов (феррицианид, гексааминоруте
лексов III и IV. Впервые поддерживаемая сукци
ний), которые вступают в реакцию с ферментом
натом реакция обратного переноса электронов в
на уровне FMN или первых железо серных
митохондриях была показана in vitro более 60 лет
кластеров. Эта реакция протекает в N модуле,
назад [16, 17]. Недавно этот процесс снова при
на нее не действуют специфические гидрофоб
влек внимание в связи с исследованиями in vivo,
ные ингибиторы, и она не зависит от генных мо
где было установлено значительное повышение
дификаций субъединиц, входящих в состав Q
уровня сукцината, приводящее к условиям, спо
или P модуля. Комплекс I может также восста
собствующим протеканию обратного переноса
навливать молекулярный кислород и образовы
электронов в некоторых патологических ситуа
вать активные формы кислорода (АФК) в виде
циях, например при ишемии в различных тка
супероксид аниона (О.-) или пероксида водоро
нях [18-22]. Обратный перенос электронов так
да (H2O2). В физиологических условиях комп
же обеспечивает наиболее высокую скорость
лекс I, вероятно, является основным произво
продукции АФК комплексом I в интактных ми
дителем АФК в митохондриальной дыхательной
тохондриях или субмитохондриальных частицах
цепи.
[23-29]. Следует отметить, что во время окисле
Особо следует отметить, что прямая реакция
ния сукцината интактными митохондриями,
окисления NADH убихиноном обратима.
после восстановления всего пула NAD+ в мат
Комплекс I может окислять мембранный QH2 и
риксе, реакция переходит в стационарные усло
15 БИОХИМИЯ том 84 вып. 11 2019
1746
ГАЛКИН
вия, где все редокс центры комплекса I восста
(т.е. при наличии обоих субстратов: убихинона и
новлены и уровень обратного переноса электро
NADH) Д форма комплекса I превращается в А
нов определяется скоростью одноэлектронного
форму в течение нескольких минут, хотя кинети
восстановления кислорода комплексом I (обра
ка активации in situ определена не до конца. Фи
зование супероксид аниона). Скорее всего, в
зиологические эффекторы, такие как свободные
этих условиях восстановленный или полувос
жирные кислоты [58] и ионы кальция [56], могут
становленный флавин комплекса I напрямую
значительно увеличивать время перехода из Д в
реагирует с кислородом с образованием АФК
А форму, в то время как ионы натрия немного
[24, 30-34].
увеличивают скорость активации [59].
Комплекс I дыхательной цепи митохондрий
С использованием метода ковалентных
млекопитающих может существовать в двух
кросс сшивок и флюоресцентных меток мы оп
формах - активной (A ) и деактивированной
ределили, что структурные изменения при А/Д
(Д ), отличающихся друг от друга структурной
переходе происходят рядом с убихинон связы
организацией и кинетическими свойствами, что
вающим участком вблизи границы раздела мо
было впервые показано в лаборатории А.Д. Ви
дулей Q и P [40, 44, 60]. Установлено, что крити
ноградова [35-38]. Эти две различные каталити
чески важный тиоловый остаток (Cys39) гибкой
ческие формы фермента были описаны в препа
гидрофильной петли мембранной субъединицы
ратах митохондрий in vitro [39-45] и в различных
ND3 доступен для модификации только в Д , но
тканях ex vivo [46-52], что указывает на роль
не в А форме [37, 40]. Согласно результатам
A/Д перехода в физиологической регуляции ак
дифференциального мечения, деактивация
тивности фермента. В предыдущих обзорах бы
приводит к повышению доступности и других
ли подробно рассмотрены различия в кинетике
субъединиц, таких как мембранно связанная
и структуре между A и Д формами фермента, а
ND1 и NADPH связывающая субъединица
также были приведены диагностические тесты,
NDUFA9 [44, 60]. Удивительно, но мы не обна
позволяющие различать эти формы [53-55], и
ружили повышения доступности какой либо из
поэтому свойства A/Д перехода будут рассмот
субъединиц в A форме [55]. Таким образом, ве
рены только кратко.
роятно, что Д форма соответствует «расслаб
Комплекс I в условиях in situ или in vitro пред
ленной», в то время как A форма соответствует
ставляет собой равновесную смесь A и Д форм
«напряженной» конформации фермента. В
[53, 55]. При физиологических температурах при
дальнейшем это предположение получило подт
отсутствии субстратов равновесие сдвигается
верждение после анализа данных криоэлек
в сторону каталитически неактивной (деактиви
тронной микроскопии фермента, выделенного
рованной) Д формы. В присутствии обоих
из митохондрий сердца быка [61] и дрожжей
субстратов (NADH и убихинона) Д форма спо
[62]. Две различные конформационные формы
собна медленно переходить в каталитически ак
комплекса I были также описаны для фермента,
тивную A форму путем изменения конформа
выделенного из сердечной мышцы мыши [63].
ции во время одного или нескольких медленных
Было показано, что значительные изменения
(1-4 мин-1) каталитических полуоборотов фер
также происходят в трансмембранных спиралях
мента [53, 55]. Активация может осуществляться
двух ND субъединиц, кодируемых митохондри
только в прямой NADH:убихинон редуктазной
альной ДНК. Во время деактивации фермента
реакции, но не во время RET, так как в Д форме
несколько критических аминокислотных остат
перенос электронов от убихинола на терминаль
ков поворачиваются внутри липидного бислоя
ный железо серный кластер заблокирован [31,
примерно на 90° вокруг оси α спирали. Струк
56]. Деактивация фермента in situ происходит
туры обеих каталитических форм доступны в
при ишемии тканей или метаболической гипок
PDB формате (PDB 6G2J и 6G72) [63].
сии, когда перенос электронов на уровне комп
Несмотря на то что комплекс I был объектом
лекса IV остановлен, что приводит к прекраще
интенсивных исследований более 50 лет, его
нию дыхания, почти полному восстановлению
структура была расшифрована относительно не
мембранного пула убихинона (повышению со
давно [64-66], а механизм регуляции активности
отношения QH2/Q) [46-51]. При отсутствии
фермента in situ до сих пор не выяснен. Наруше
субстратного убихинона комплекс I перестает
ние функционирования этого фермента является
функционировать, что при физиологических
одной из причин возникновения наследственных
температурах вызывает А/Д переход. Интерес
митохондриальных паталогий [67], процессов
но, что после остановки кровообращения деак
старения [68] и нейродегенеративных заболева
тивация комплекса I происходит быстрее в го
ний [69]. Комплекс I также является основным
ловном мозге, чем в сердечной ткани [57]. При
источником и одновременно мишенью для
реоксигенации и понижении отношения QH2/Q
действия АФК в патологических состояниях [55].
БИОХИМИЯ том 84 вып. 11 2019
КОМПЛЕКС I ПРИ ИШЕМИИ/РЕПЕРФУЗИИ МОЗГА
1747
ФИЗИОЛОГИЧЕСКИЕ ИЗМЕНЕНИЯ,
внеклеточной жидкостью. При этом внутрикле
ПРОИСХОДЯЩИЕ ВО ВРЕМЯ
точная концентрация ионов натрия в 7 раз
ИШЕМИИ/РЕПЕРФУЗИИ МОЗГА
меньше, а концентрация ионов кальция на пять
порядков ниже их концентрации вне клеток.
Прежде чем детально обсудить влияние ише
Отсутствие АТР приводит к остановке энергоза
мии/реперфузии на митохондриальный комп
висимых ионных насосов и нарушению ионно
лекс I, следует кратко рассмотреть изменения
го баланса. В итоге происходит падение потен
процесса энергетического обмена в клетках
циала на плазматической мембране и резкое
мозга, происходящие после остановки цереб
увеличение концентрации ионов натрия внутри
рального кровообращения. Количество глюко
клеток. В результате активности Na+/Ca2+ об
зы и альтернативных субстратов, таких как гли
менника плазматической мембраны происходит
коген, лактат и жирные кислоты, значительно
значительное повышение концентрации ионов
превышает доступное количество кислорода,
кальция как в цитоплазме, так и внутри митохо
необходимое для их окисления [5]. В любой
ндрий, и развитие отека мозга [74, 75]. В то же
конкретный момент времени количество кисло
время в ходе анаэробного гликолиза происходит
рода, связанного с гемоглобином и в растворен
переработка остаточных углеводов с образова
ном виде, оценивается как ~250 мкмоль на один
нием лактата и закисление цитоплазмы клеток.
мозг [70]. При средней скорости потребления
Степень закисления зависит от начальной кон
кислорода во время бодрствования около
центрации глюкозы и гликогена в клетках мозга
1,88 ммоль/мин/мозг [71] этого количества до
до возникновения ишемии и в дальнейшем вли
статочно для поддержания метаболизма в тече
яет на степень нарушения ионного баланса кле
ние ~10 c, что примерно соответствует времени
ток мозга. Кроме того, нейроны также исключи
потери сознания после полной остановки моз
тельно чувствительны к так называемой глута
гового кровообращения. У мышей кислород
матной эксайтотоксичности. Повышение кон
мозга истощается полностью в течение всего
центрации ионов кальция приводит к высво
нескольких секунд после остановки сердца [72].
бождению из синаптических везикул нейротра
Более 75% кислорода, поступающего в мозг,
нсмиттера глутамата. В результате ишемическо
потребляется нейронами и астроцитами [5], и
го нарушения поглощения глутамата астроцита
поэтому эти клетки наиболее чувствительны к
ми, происходит активация глутаматных ионот
недостатку кислорода. Дефицит кислорода ока
ропных рецепторов, повышающая энергозави
зывает сильное влияние на метаболические про
симую электрическую активность нейронов.
цессы, происходящие в митохондриях, которые
Это приводит к еще большему нарушению ион
обеспечивают клетки АТР в результате окисле
ного баланса и дальнейшему снижению энерге
ния субстратов, образующихся в процессе гли
тического потенциала клетки, усиливающим
колиза, цикла трикарбоновых кислот и окисле
процесс повреждения нейронов [5].
ния жирных кислот (рис. 1). У мышей и крыс
Постоянная ишемия приводит в конечном
падение уровня АТР происходит в течение 2-5 с
счете к гибели мозговой ткани в результате по
после прекращения мозгового кровообращения
вреждения нейронов и олигодендроцитов в об
[3, 73]. Остановка работы дыхательной цепи ми
ласти, в которую перекрытая артерия поставляла
тохондрий и образования АТР в результате ише
кислород и питательные вещества [76]. Необхо
мии определяется как первичная энергетичес
димым условием для выживания клеток являет
кая недостаточность. При отсутствии кислорода
ся восстановление снабжения кислородом или
останавливается процесс переноса электронов в
реоксигенация, поэтому кровоток должен быть
комплексе IV. Это приводит к полному восста
восстановлен в течение 15-30 мин (реперфузия).
новлению всех подвижных переносчиков элект
Ткань, подвергшуюся воздействию ишемии,
ронов (NAD(P)H и убихинон), а также практи
можно формально подразделить на «ядро», в ко
чески всех редокс центров дыхательной цепи.
тором наблюдается резкое снижение содержа
Ввиду избытка метаболических субстратов про
ния кислорода, и окружающую его «пенумбру»,
исходит быстрое накопление интермедиатов ка
в которой остаточный кровоток обеспечивает
таболизма (сукцинат, свободные жирные кисло
минимальную доставку кислорода. Клетки «яд
ты, ацил КоА и глицерол). В свою очередь,
ра» и «пенумбры» отличаются друг от друга по
энергетический кризис, вызванный резким сни
динамике падения уровня АТР, степени увеличе
жением скорости образования АТР, оказывает
ния содержания лактата, уровню закисления и
отрицательное влияние на ионный гомеостаз.
концентрации ионов кальция в цитоплазме. Во
Так, в физиологических условиях механизм
время реперфузии «ядро» и «пенумбра» также
ионного транспорта поддерживает внутри клет
различаются по динамике восстановления уров
ки 30× градиент ионов калия по сравнению с
ня кислорода и скорости его потребления, что
БИОХИМИЯ том 84 вып. 11 2019
15*
1748
ГАЛКИН
отражает различия в степени повреждения кле
компрессионной ишемии мозга кроликов было
ток и состоянии микроциркуляторного русла.
показано существенное снижение АDP стиму
Необратимо поврежденные клетки в области
лируемого дыхания на малате и глутамате через
«ядра» в конечном итоге умирают, и через не
20-40 мин после увеличения внутричерепного
сколько недель эта область превращается в очаг
давления [83, 84]. Аналогичные результаты были
поражения, состоящий из некротизирующей
получены при изучении двусторонней ишемии
ткани. Клетки «пенумбры», в которых поддер
мозга у монгольских песчанок. В этом случае бы
живается неполный уровень энергетического
ло показано, что после 60 мин ишемии митохон
метаболизма, могут выжить в зависимости от
дриальное дыхание падало на 50% [85]. Уже ис
длительности ишемии и интенсивности реокси
ходя из ранних работ, можно сделать вывод, что
генации [77]. Реперфузия приводит к возобнов
ишемия мозга ингибирует митохондриальное
лению функционирования дыхательной цепи
дыхание только в случае использования субстра
митохондрий и восстановлению уровня АТР, от
тов NAD+ зависимых дегидрогеназ, генерирую
вечающего потребностям клеток в энергии. Ран
щих NADН для комплекса I.
ние стадии реперфузии также ассоциируются с
Патофизиология первичной энергетической
краткосрочным увеличением уровня АФК, кото
недостаточности во время ишемии, продемон
рые, вероятно, образуются несколькими фер
стрированная еще в ранних работах, усложняет
ментативными системами, включая митохон
ся тем, что при возобновлении кровоснабжения
дрии. Это приводит к развитию окислительного
во время реперфузии или реоксигенации тя
стресса и дальнейшему повреждению ткани. В
жесть исходного повреждения ткани затем усу
нескольких моделях ишемии/реперфузии мозга
губляется. Реперфузия, необходимая для выжи
in vivo было показано, что уровень АТР и интен
вания ткани, в то же время вызывает множест
сивность окислительного фосфорилирования
венные непрямые или вторичные последствия,
могут снова снижаться спустя несколько часов
влияющие на жизнеспособность клеток в пора
после реперфузии. Это явление принято назы
женной области. Результаты, полученные с ис
вать вторичной энергетической недостаточ
пользованием модели глобальной ишемии моз
ностью, и природа его до сих пор не ясна [3-5].
га у крыс, свидетельствуют о почти полном вос
становлении дыхания с малатом и глутаматом
или сукцинатом через 30 мин после возобновле
ВЛИЯНИЕ ИШЕМИИ/РЕПЕРФУЗИИ
ния кровотока, несмотря на то что в результате
ТКАНИ МОЗГА НА СОСТОЯНИЕ
ишемии дыхание на всех субстратах было значи
МИТОХОНДРИАЛЬНОГО КОМПЛЕКСА I
тельно снижено [86, 87]. В этих работах, выпол
ненных с использованием моделей как полной,
Ишемия/реперфузия мозга и митоходриальная
так и неполной ишемии, дыхание с малатом и
дыхательная цепь. Еще в первых работах, выпол
глутаматом было нарушено в большей степени,
ненных с использованием электронной микро
чем в случае дыхания, поддерживаемого сукци
скопии и гистохимического анализа, были выяв
натом. Однако при неполной ишемии через
лены набухание митохондрий и разрушение их
30 мин реперфузии наблюдали дальнейшее сни
внутренней мембраны после долговременной
жение интенсивности дыхания. Это свидетель
ишемии [78, 79]. При окрашивании срезов пост
ствует о том, что тяжесть повреждения ткани
ишемических тканей тетразолиевыми солями
определяют глубина ишемии и интенсивность
снижение активности NADН зависимых дегид
реперфузии [86, 87]. Возможный сценарий раз
рогеназ происходило раньше, чем нарушение
вития поражения ткани в результате ише
сукцинат зависимой активности [78, 80]. Сейчас
мии/реперфузии становится более сложным в
эти результаты можно интерпретировать как
связи с феноменом так называемой вторичной
прямое доказательство наибольшей чувстви
недостаточности энергии. Так, кратковремен
тельности комплекса I к действию ишемии по
ная локальная ишемия у крыс приводила к двух
сравнению с другими компонентами дыхатель
кратному падению ADP стимулируемого дыха
ной цепи. Впервые ингибирование дыхания ми
ния с использованием малата и пирувата, кото
тохондрий после кислородной депривации было
рое сопровождалось полным восстановлением
продемонстрировано на модели глобальной
активности через 1 ч после реперфузии и посте
ишемии обескровленных крыс [81, 82]. Уже по
пенным вторичным снижением интенсивности
сле двухминутной ишемии скорость дыхания
дыхания в течение 3-24 ч [88]. Этот эффект за
митохондрий при использовании в качестве
висел от участка ткани мозга, который был взят
субстрата глутамата составляла только 25% от
для проведения анализа. Анализ дыхания пре
уровня потребления кислорода образцами, по
парата интактных митохондрий в сходной моде
лученными из контрольных животных. В модели
ли [89] подтвердил изменения интенсивности
БИОХИМИЯ том 84 вып. 11 2019
КОМПЛЕКС I ПРИ ИШЕМИИ/РЕПЕРФУЗИИ МОЗГА
1749
дыхания после ишемии/реперфузии, когда сна
дриальной глицерол 3 фосфатдегидрогеназы,
чала происходило снижение комплекс I зависи
которая переносит электроны на пул хинонов и
мого дыхания после ишемии, реоксигенация
далее на комплексы III-IV [92]. Пара сукцинат
вызывала быстрое восстановление дыхания, ко
и глутамат обеспечивает дыхание через комп
торое затем снова снижалось.
лексы II-III-IV, в то время как пары малат и
Практические аспекты. Есть несколько прак
пируват или малат и глутамат используются для
тических соображений, которые следует прини
восстановления NAD+ в матриксе, обеспечивая
мать во внимание при обсуждении данных по
перенос электронов от комплекса I далее на
измерению митохондриального дыхания в об
комплексы III-IV. Эффект ишемии/реперфу
разцах головного мозга при ишемии/реперфу
зии на митохондрии, определяемый как измене
зии. Проблема в интерпретации результатов, по
ние интенсивности дыхания во время окисле
лученных в более ранних работах, заключается в
ния митохондриями различных субстратов, не
том, что лишь немногие исследования использо
может отражать влияние только на ферменты
вали одни и те же модели ишемии in vivo, приме
дыхательной цепи (т.е. комплексы I-IV). По
няли одни и те же анестетики или даже исполь
требление кислорода митохондриями является
зовали одни и те же субрегионы мозга для отбо
функцией, определяемой несколькими пара
ра проб. Эти параметры влияют на результирую
метрами, такими как состав и целостность
щие физиологические последствия при исполь
мембраны, ее протонная проводимость, транс
зовании этих моделей, а также на биохимичес
порт субстратов, активность NAD+ зависимых
кие характеристики полученных образцов. Из
дегидрогеназ цикла трикарбоновых кислот,
мерение потребления кислорода препаратом ин
АТР синтазы и в конечном итоге комплексов
тактных митохондрий, выделенных из поражен
дыхательной цепи. Поэтому, чтобы выявить на
ной области мозга после ишемии/реперфузии,
рушения отдельных ферментов дыхательной це
действительно может служить удобным и физио
пи, оценку активности индивидуальных комп
логически значимым маркером функции мито
лексов необходимо проводить, используя пер
хондрий. Однако данные измерения дыхания
меабилизованые митохондрии или фрагменты
должны интерпретироваться с осторожностью.
внутренней мембраны. Можно предположить,
Tкань мозга состоит из различных типов клеток:
что на ранних стадиях реперфузии (до 3-4 ч) со
например, в кортексе мозга мыши имеется поч
держание митохондрий в образце существенно
ти равное количество нейронов и глиальных
не меняется [27, 74, 93]. Однако на более позд
клеток, тогда как в мозжечке соотношение
них стадиях некротическая и апоптотическая
глия/нейроны составляет 1 : 6 [90]. Поэтому пре
деградации, происходящие в тканях, могут при
параты интактных митохондрий головного моз
вести к уменьшению количества живых клеток
га, полученные стандартными методами диффе
и, следовательно, к кажущемуся падению фер
ренциального центрифугирования, скорее всего,
ментативной активности, которое происходит
представлены фракциями митохондрий из раз
из за снижения количества митохондрий. Со
личных типов клеток, и вклад каждой фракции
держание митохондрий в образце обычно оце
зависит от области мозга, взятой для выделения.
нивают по специфической активности цитрат
Поскольку абсолютное содержание митохон
синтазы или путем измерения количества ци
дрий и интенсивность окислительного фосфо
тохромов на мг общего белка. Скорее всего, наб
рилирования в нейронах и астроцитах различны
людавшееся в некоторых ранних работах вто
и эти типы клеток по разному реагируют на
ричное падение интенсивности дыхания после
ишемию/реперфузию, то данные, полученные
его восстановления во время реперфузии можно
при использовании классического препарата
отнести на счет уменьшения содержания мито
интактных митохондрий мозга, трудно отнести
хондрий вследствие лизиса ткани, иными слова
исключительно к нейрональным митохондриям.
ми, снижения количества живых клеток в участ
На практике для измерения потребления
ке, взятом для анализа. Так, снижение интен
кислорода изолированными митохондриями
сивности митохондриального дыхания через
используют несколько комбинаций субстратов
12-56 ч после возобновления кровотока в моде
для инициации дыхания различными убихи
ли полной церебральной ишемии у кошек мож
нон зависимыми дегидрогеназами дыхательной
но объяснить распадом ткани, а также относи
цепи. Например, для восстановления хинона
тельно жесткими условиями протеолиза при вы
может быть использован пальмитоилкарнитин,
делении фракции митохондрий [94].
как субстрат мембраносвязанной ЭПФ:убихи
Ишемия/реперфузия мозга и митохондриальB
нон оксидоредуктазной системы окисления
ный комплекс I. В ранних работах было показано
жирных кислот [91]. Цитоплазматический гли
прямое влияние ишемии/реперфузии на мито
церол 3 фосфат является субстратом митохон
хондриальное дыхание с использованием
БИОХИМИЯ том 84 вып. 11 2019
1750
ГАЛКИН
субстратов NAD+ зависимых дегидрогеназ (ма
работах по изучению последствий ишемии/ре
лат и пируват или малат и глутамат). Сейчас эти
перфузии мозга, то механизм инактивации фер
результаты можно интерпретировать как явные
мента до сих пор не был определен. Поэтому не
признаки повреждения комплекса I после ише
давно мы снова исследовали временную дина
мии/реперфузии. Насколько известно автору,
мику потери активности фермента во время
повреждения митохондриального комплекса I,
ишемии/реперфузии с использованием двух мо
индуцированные ишемией/реперфузией, были
делей, а именно: MCAo модели инсульта у
впервые продемонстрированы с использовани
взрослых мышей [93] и неонатальной модели
ем модели ишемии головного мозга песчанок, у
гипоксии ишемии мозга [49, 50, 99]. Нами было
которых так же, как и у человека, имеется Вил
подтверждено, что ишемия/реперфузия мозга
лизиев артериальный круг. Это наилучшая мо
вызывает ингибирование митохондриального
дель общей или локальной ишемии ткани мозга
дыхания вследствие инактивации комплекса I.
человека, по сравнению с обычно используемы
Динамика падения активности носила много
ми крысиными и мышиными моделями. Цереб
фазный характер в модели MCAo у взрослых
ральная ишемия достигалась в результате ок
мышей [93] и в то же время была двухфазной в
клюзии внутренней сонной артерии в течение
неонатальной модели гипоксии ишемии [99]. В
30 мин. Через различные промежутки времени
обеих моделях ишемия вызывала сильное инги
после реперфузии выделяли митохондрии из
бирование комплекса I. Этот результат согласу
свежей ткани и подвергали их анализу [95, 96].
ется с результатами предыдущих работ Almeida
Ишемия вызывала снижение комплекс I зави
et al. [95], Yoshimoto et al. [97] и Niatsetskaya et al.
симого дыхания (малат и пируват или малат и
[27]. После 10-15 мин реоксигенации актив
глутамат в качестве субстратов дыхания) на
ность комплекса I почти полностью восстанав
~25%, затем оно постепенно восстанавливалось
ливалась, а затем снова снижалась после 4 ч. На
в течение первых 30 мин реперфузии и через 2 ч
ми были определены по крайней мере два не
снова снижалось. Проведенный анализ отдель
описанных ранее различных механизма наруше
ных компонентов дыхательной цепи показал,
ния функционирования комплекса I, происхо
что ингибирование дыхания при ишемии было
дящих в различные моменты времени после ре
вызвано снижением NADН:убихинон редуктаз
перфузии. Один из них заключается в диссоци
ной активности комплекса I. При этом не было
ации FMN, эндогенного кофактора комплекса I
выявлено существенных изменений в скорости
[34, 93, 99], а другой механизм представляет со
окисления сукцината (комплексы II-III-IV) и
бой модификацию критически важной SH
активности цитратсинтазы. На модели кратко
групп(ы) после конформационного A/Д пере
срочной локальной ишемии у крыс также было
хода фермента [49, 50, 53, 55]. Эти механизмы
показано, что после реперфузии митохондри
более подробно обсуждаются ниже.
альное дыхание с использованием малата и глу
Высвобождение флавина. С помощью in vivo
тамата сначала восстанавливалось, а затем через
модели неонатальной гипоксии ишемии [99] и
4 ч происходило его повторное снижение [97].
MCAo модели ишемического инсульта у взрос
Результаты, полученные на моделях поврежде
лых мышей [93] мы обнаружили, что ишемия/ре
ний при ишемии/реперфузии у взрослых живот
перфузия индуцировала инактивацию комплек
ных, сопоставимы с данными, полученными при
са I. Снижение активности в препарате было ас
использовании неонатальной модели гипоксии
социировано со снижением содержания FMN в
ишемии (модель Rice-Vannucci) [27]. Дефицит
ферменте. Одна молекула FMN нековалентно
кислорода в течение 15 мин снижал поддерживае
связана с NDUFV1 субъединицей (51 кДа) N мо
мое малатом и глутаматом дыхание почти в два
дуля комплекса I и непосредственно взаимодей
раза. Реоксигенация приводила к почти полному
ствует с субстратами - NADН или NAD+ [64, 65,
восстановлению дыхания после 4 ч реперфузии,
100]. Мы определили механизм высвобождения
а затем интенсивность дыхания снова начинала
FMN из фермента во время ишемии/реперфу
снижаться. Ишемия/реперфузия не оказывали
зии. В наших исследованиях [34, 93, 99] ишемия
видимого влияния на окисление сукцината [27].
индуцировала диссоциацию FMN вследствие
Недавно с помощью метода позитронно эмис
стимуляции обратного переноса электронов че
сионной томографии in situ было продемонстри
рез комплекс I. На различных моделях in vivo бы
ровано ишемическое подавление активности
ло показано, что недостаток кислорода вызывает
митохондрий вследствие ингибирования комп
значительное увеличение содержания сукцина
лекса I после 3 ч окклюзии средней церебраль
та, одного из субстратов реакции обратного пе
ной артерии мозга приматов (MCAo) [98].
реноса [18, 20-22]. В неонатальной модели ише
Если снижение активности митохондриаль
мии/реперфузии [22] было обнаружено 30× уве
ного комплекса I было показано в нескольких
личение уровня сукцината, который возвраща
БИОХИМИЯ том 84 вып. 11 2019
КОМПЛЕКС I ПРИ ИШЕМИИ/РЕПЕРФУЗИИ МОЗГА
1751
ется к исходным значения только после 30 мин
дефосфорилирована с образованием рибофлави
реперфузии. При ишемии также происходит
на или превратиться в FАD с помощью АТP за
полное ингибирование β окисления жирных
висимой FАD пирофосфатазы [106].
кислот, что приводит к быстрому накоплению в
Еще с самых первых попыток выделения
ткани различных неэтерифицированных жир
комплекса I стало ясно, что FMN легко теряет
ных кислот и ацил КоА [101, 102]. Кроме того,
ся в ходе очистки или диализа препаратов мито
во время ишемии мозга также наблюдается на
хондриальной NADH дегидрогеназы [100]. Ин
копление глицерол 3 фосфата, еще одного
дуцированная восстановлением фермента обра
субстрата, поддерживающего обратный перенос
тимая диссоциация FMN показана ранее для
[103]. Во время ишемии, когда уровень кислоро
трехсубъединичного фрагмента [107] и мембра
да в тканях очень низок (но не равен нулю), или
носвязанного препарата [108] комплекса I мле
после реперфузии эти субстраты вместе с сукци
копитающих in vitro и недавно - для бактериаль
натом могут восстанавливать убихинон и под
ного фермента [109]. Комплекс I, восстановлен
держивать условия обратного переноса, когда
ный NADH, быстро теряет FMN. Добавленный
электроны от убихинола переносятся к нуклео
экзогенно флавин предотвращает диссоциацию,
тид связывающему центру комплекса I. В ста
а также восстанавливает активность фермента.
ционарных условиях этот процесс поддерживает
Нами было показано, что в условиях in vitro до
высокий уровень восстановленности FMN в
бавление FMN во время инкубации интактных
комплексе I, что и вызывает диссоциацию ко
митохондрий клеток мозга в условиях обратного
фактора [29, 34]. Апофермент, лишенный FMN,
переноса электронов частично предотвращает
не способен катализировать физиологическое
инактивацию комплекса I [99]. Следует также
окисление NADH, снижая эффективность ды
особо отметить, что введение предшественника
хательной цепи и образования АТP.
FMN, рибофлавина (витамина B2), перед неона
Последствия диссоциации FMN не ограни
тальной ишемией/реперфузией мозга in vivo на
чиваются только нарушением функционирова
50% снижает величину церебрального инфар
ния комплекса I. Свободный восстановленный
кта, повышает активность комплекса I и умень
флавин в водной среде быстро реагирует с моле
шает неврологический дефицит по сравнению с
кулярным кислородом с образованием радикала
контрольными животными. Следует отметить,
супероксид аниона и пероксида водорода (конс
что анализ крови, проведенный у пациентов
танта скорости равна 250 M-1с-1) [104, 105]. Со
сразу после инсульта, показал дефицит рибо
держание комплекса I в препаратах митохондрий
флавина у значительной части больных, хотя
мозга мыши, выделяемых по стандартной мето
клиническая значимость этих результатов неяс
дике [29], составляет около 20-50 пкмоль на 1 мг
на [110]. Наши данные [34, 93, 99] дают меха
белка (Stepanova и Galkin, неопубликованные ре
нистическое объяснение показанного в недав
зультаты), а степень инактивации комплекса I
них клинических работах нейропротекторного
составляет около 25-35% [93, 99]. Следователь
действия препаратов рибофлавина у пациентов
но, после индуцированной ишемией диссоциа
с ишемическим инсультом [111]. Профилакти
ции FMNH2 концентрация свободного флавина
ческое введение рибофлавина беременным жен
в матриксе может достигать 15-30 мкМ (учиты
щинам может быть потенциально использовано
вая объем митохондриального матрикса, состав
в качестве дополнения к терапии против ише
ляющего не более 1 мкл на мг белка). При такой
мического повреждения некоторых патологи
высокой концентрации пара FMN/FMNH2 мо
ческих состояний плода при опасности ишемии
жет быть вовлечена в реакцию дисмутации, при
мозга.
водящую к образованию полувосстановленных
Окисление тиоловых групп. Многочисленные
радикалов флавина [105]. Кроме того, флавин
исследования показали, что окислительный
может служить в качестве редокс медиатора
стресс, вызванный реоксигенацией после репер
между донорами электронов, такими как NADH,
фузии, является одним из основных факторов
и доступными акцепторами электронов в мат
повреждения тканей при ишемии/реперфузии
риксе. Образование радикала супероксид анио
мозга [112, 113] и что эндогенный глутатион яв
на и пероксида водорода в результате нефермен
ляется основным компонентом поддержания
тативной реакции восстановленного флавина с
окислительно восстановительного баланса в ми
кислородом может способствовать кратковре
тохондриях. Вероятно, во время ишемии/репер
менному повышению уровня АФК на начальных
фузии восстановленный глутатион и другие сис
стадиях реперфузии. Дальнейшие превращения
темы тиол зависимых редуктаз предотвращают
окисленного свободного флавина предсказать
окисление или восстанавливают поврежденные
трудно. Часть FMN связывается с апоформой
в результате окислительного стресса SH группы
комплекса I, другая часть флавина может быть
белков [6, 114, 115]. На нескольких моделях in
БИОХИМИЯ том 84 вып. 11 2019
1752
ГАЛКИН
vivo была подтверждена нейропротекторная роль
Конформационный A/Д переход приводит к
введения различных антиоксидантов [116-118],
экспонированию наружу остатка Cys39 субъеди
но механизмы их действия до конца не изучены.
ницы ND3 [40, 55]. Ковалентная модификация
Используя MCAo модель инсульта [93], мы
этой ключевой SH группы блокирует процесс
обнаружили, что после восстановления актив
реактивации фермента. Окислительные агенты,
ности митохондриального комплекса I в резуль
такие как АФК и пероксинитрит, вызывают не
тате реоксигенации дополнительные 30 мин ре
обратимое ингибирование фермента, в то время
циркуляции приводили к повторному снижению
как модификация цистеина с помощью низко
активности фермента. Инкубация митохондри
молекулярных нитрозотиолов обратима [41, 47].
альных мембран, выделенных из этих образцов, с
А форма фермента не чувствительна к обработ
восстановленным глутатионом частично реакти
ке подобными SH реагентами. Следовательно,
вировала комплекс I. Поэтому нами было пред
чувствительность митохондриального комплек
положено, что вторичная инактивация фермен
са I к окислительному стрессу, вызванному ре
та, вероятно, была вызвана окислительной моди
перфузией, может быть определена степенью
фикацией критически важных тиоловых групп
экспонирования ключевого остатка цистеина,
комплекса I [93]. Нами было показано, что ло
вовлеченного в процесс A/Д перехода, индуци
кальная ишемия в течение 20 мин приводила к
рованного ишемией.
снижению общего содержания глутатиона в пов
Недавно нами были получены подтвержде
режденной области мозга [93]. Введение прони
ния существования конформационного A/Д пе
цаемого для клеток этилового эфира глутатиона
рехода комплекса I митохондрий мозга. Было по
на начальном этапе фазы реоксигенации восста
казано, что комбинация ишемии и гипоксии в
навливало содержание глутатиона, предотвраща
неонатальной модели ишемии/реперфузии,
ло снижение активности комплекса I, было ассо
предложенной Rice-Vannucci, приводит к повы
циировано с меньшими размерами инфаркта, а
шению содержания Д формы от базового уровня
также снижало неврологический дефицит [93].
в 10% до 35% у крыс [49] и от 5 до 40% у мышей
Похожие результаты были продемонстрированы
[50]. Была обнаружена лишь частичная деактива
ранее в крысиной модели инсульта, но механизм
ция фермента, что неудивительно из за погреш
нейропротекции не был определен [118].
ностей экстракции потенциально поврежденной
Какие остатки цистеина определяют
ткани в моделях локальной ишемии, которая вы
чувствительность фермента к окислительному
полнялась еще до развития видимого поврежде
стрессу? Учитывая результаты наших ранних ра
ния кортекса мозга. Видимо поэтому в модели
бот [41, 47] и работ группы Murphy [48, 119], бы
глобальной ишемии мозга в течение 15 мин пос
ло бы разумным ожидать, что модификация
ле остановки сердца у мышей фракция Д формы
ключевой тиоловой группы комплекса I, вовле
в мозге увеличивалась почти до 90% [120].
ченной в конформационный A/Д переход, мо
Следовательно, после ишемии значительная
жет способствовать окислительному поврежде
часть молекул фермента находится в Д форме, и
нию ткани мозга при ишемии/реперфузии. В
критически важная SH группа субъединицы
различных работах было показано, что отсут
ND3 экспонирована наружу и доступна для мо
ствие кислорода in vivo стимулирует изменение
дификации. Реперфузия способствует реактива
конформации комплекса I и способствовует его
ции Д формы и образованию A формы. Также
превращению в деактивированную Д форму
реперфузия индуцирует окислительный стресс,
[45-48, 51]. Необходимо подчеркнуть, что Д
приводящий к необратимому ингибированию
форма является как бы «спящим» состоянием
фермента. Мы обнаружили, что обратимая за
комплекса I, а не инактивированным или моди
щитная модификация критически важного ос
фицированным ферментом; Д форма может
татка цистеина в Д форме путем введения во
быть быстро «реактивирована» в присутствии
время реперфузии митохондриально направ
субстрата [41, 54]. Исторически термин «деакти
ленного нитрозилирующего агента MitoSNO
вация» был использован для того, чтобы подчерк
[48, 119] приводит к снижению уровня окисли
нуть разницу в кинетических характеристиках
тельного стресса и способствует повышению вы
двух форм in vitro. В то же время Д форма фер
живаемости нейронов при ишемии/реперфузии
мента in situ может рассматриваться как «находя
в неонатальной модели
[50]. Благотворное
щаяся в состоянии покоя», но не инактивиро
действие MitoSNO можно объяснить с помощью
ванный фермент. Если специально это не указа
двух возможных механизмов. Один заключается
но, то при проведении стандартной оценки ак
в отмеченной ранее защите тиоловой группы
тивности комплекса I фермент предварительно
критического остатка цистеина от необратимой
активируется, и обе формы вносят вклад в изме
окислительной модификации и тем самым от
ряемую скорость ферментативной реакции [54].
инактивации комплекса I. Другим механизмом
БИОХИМИЯ том 84 вып. 11 2019
КОМПЛЕКС I ПРИ ИШЕМИИ/РЕПЕРФУЗИИ МОЗГА
1753
является задержка реперфузионно индуциро
могут окисляться митохондриями в реакции об
ванного Д/A перехода, ограничивающая про
ратного переноса электронов, что резко увели
дукцию АФК А формой комплекса I [49, 50].
чивает восстановленность комплекса I. Обрат
На основании недавно полученных резуль
ный перенос, вероятно, также приводит к зна
татов нами был предложен следующий сцена
чительному повышению генерации АФК комп
рий для описания механизма повреждения
лексом I (рис. 2, б). Поддержание фермента в
комплекса I во время ишемии/реперфузии
восстановленном состоянии индуцирует диссо
(рис. 2). В условиях нормоксии фермент в ос
циацию FMNH2 (рис. 2, б). Во время реперфу
новном находится в A форме и катализирует
зии высвободившийся кофактор может подвер
физиологическое окисление NADН убихино
гаться автоокислению, приводя к нефермента
ном, способствуя образованию протондвижу
тивному образованию АФК, и после этого, воз
щей силы. При этом критически важный оста
можно, снова связываться с апоферментом
ток Cys39 субъединицы ND3 не экспонирован
(рис. 2, в). В то же самое время недостаточный
наружу и степень восстановления фермента не
поток электронов через фермент во время ише
максимальна (рис. 2, а). Недостаток кислорода
мии ткани может вызывать A/Д конформацион
вызывает накопление сукцината и глицерол 3
ный переход комплекса I. В результате измене
фосфата, которые могут диффундировать из ли
ния конформации белка происходит экспони
шенного кислорода «ядра» и становятся доступ
рование ключевого остатка Cys39 субъединицы
ными для митохондрий, находящихся в более
ND3 (рис. 2, г). Деактивация комплекса I при
оксигенированной «пенумбре». Эти субстраты
водит к блокировке обратного переноса элек
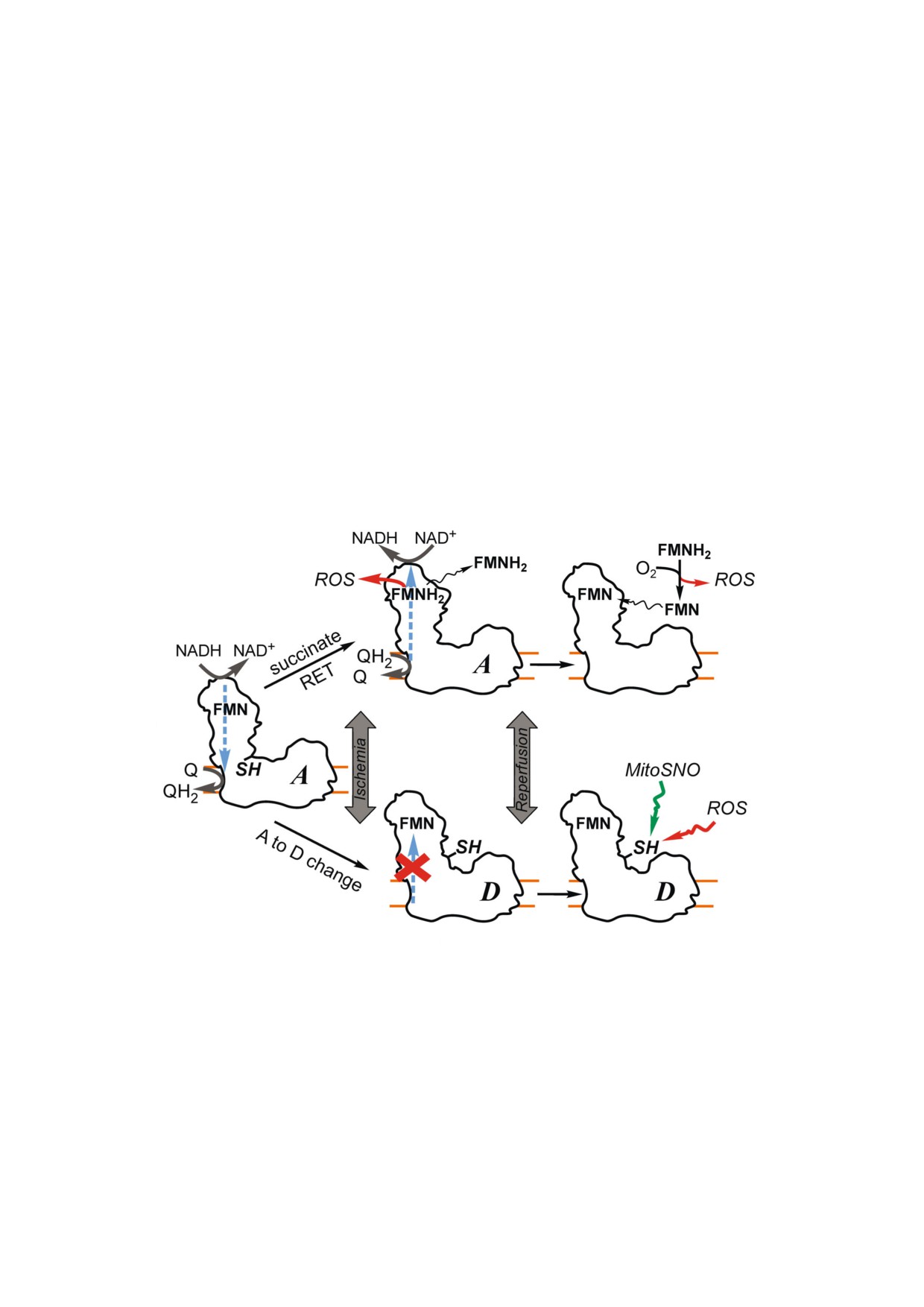
б
в
a
г
д
Рис. 2. Схематическое представление предполагаемых эффектов ишемии/реперфузии на митохондриальный комплекс I.
а - В присутствии кислорода комплекс I катализирует окисление NADH убихиноном (Q) в прямой реакции, и большая
часть комплекса I находится в каталитически активной A форме, когда критический остаток Cys39 субъединицы ND3 не
доступен; б - в условиях ишемии уровень сукцината повышается, и поэтому активная фракция фермента способна ката
лизировать реакцию обратного переноса электронов (RET, reverse electron transfer) от убихинола на флавин (FMN), нахо
дящийся в нуклеотид связывающем центре комплекса I. Обратный перенос электронов поддерживает наиболее высокую
скорость образования АФК (ROS) в комплексе I. Восстановление флавина (FMNH2) в условиях обратного переноса при
водит к его диссоциации и инактивации фермента; в - свободный FMNH2 может неферментативно реагировать с моле
кулярным кислородом с образованием АФК и окисленного флавина, который может снова связаться с апо комплексом I;
г - длительная ишемия приводит к изменению конформации комплекса I, образованию Д формы и экспонированию на
ружу остатка Cys39 в ND3 субъединице; д - этот остаток цистеина является мишенью для необратимой окислительной
модификации под действием АФК или пероксинитрита во время реоксигенации, но может быть защищен путем времен
ной модификации митохондриально направленным нитирозилирующим агентом MitoSNO
БИОХИМИЯ том 84 вып. 11 2019
1754
ГАЛКИН
тронов ферментом [35], что, в свою очередь,
тельного повреждения в результате модифика
предотвращает образование АФК и диссоциа
ции остатка Cys39 АФК или пероксинитритом.
цию FMN. Степень диссоциации FMN и пре
вращения А формы фермента в Д форму зави
сят от времени и глубины ишемии («ядро» и
ПЕРСПЕКТИВЫ
«пенумбра») и метаболического статуса клетки.
Реперфузия ишемической ткани вызывает
Повреждение митохондрий во время ишемии
реактивацию комплекса I за промежуток време
мозга скорее всего происходит путем различных
ни от нескольких секунд до нескольких минут.
механизмов. Нарушение функционирования
Такая задержка активации фермента на ранних
комплекса I во время ишемии и последующей
стадиях реперфузии препятствует резкому окис
реоксигенации мозга хорошо известно, хотя ме
лению накопившихся молекул NADH в прямой
ханизм повреждения до сих пор не был установ
или сукцината в обратной реакции, что может
лен. Нами было предложены два различных ме
служить в качестве внутреннего защитного ме
ханизма инактивации фермента: диссоциация
ханизма, направленного против повышенной
флавинового кофактора, индуцированная обрат
генерации АФК (рис. 2, г). В то же время инду
ным переносом электронов при восстановлении
цированный реперфузией окислительный
комплекса I, и специфическая модификация
стресс может привести к модификации SH
критически важной SH группы одной из субъ
групп(ы) в Д форме комплекса I и потере актив
единиц фермента. Аспект нашей работы, имею
ности фермента. Если во время реперфузии
щий наибольшее значение, - это определение
присутствует MitoSNO, то тогда часть Д формы
специфичности этих механизмов для различных
фермента подвергается нитрозилированию по
типов клеток. Необходимо с помощью признан
экспонированному ключевому остатку Cys39
ных in vivo моделей выяснить степень поврежде
ND3 субъединицы (рис. 2, д). Это временно бло
ния митохондрий нейронов и астроцитов в ре
кирует комплекс I в Д форме, снижая образова
зультате ишемии/реперфузии мозга. Чтобы оп
ние АФК на ранних стадиях реоксигенации. Бо
ределить аминокислотные остатки, подвергаю
лее того, нитрозилирование защищает эту клю
щиеся окислительной модификации, приводя
чевую SH группу от необратимой модификации
щей к потере функции фермента, необходимо
во время окислительного стресса, вызванного
провести протеомные исследования посттранс
реоксигенацией. S нитрозилированный комп
ляционных модификаций аминокислотных ос
лекс I может постепенно восстанавливаться ти
татков митохондриального комплекса I после
ол редуктазами митохондриального матрикса
ишемии/реперфузии [121]. С более широкой
[48] с образованием Д формы и в конечном ито
точки зрения, для того чтобы в дальнейшем ис
ге превращаться в каталитически активную A
пользовать полученные результаты в рамках
форму фермента.
трансляционной медицины, необходимо выяс
Данные, приведенные в этом обзоре, позво
нить происходят ли эти процессы у людей во вре
ляют разработать экспериментальные подходы,
мя ишемического инсульта или неонатальной
чтобы определить, защищают ли A/Д переход и
гипоксически ишемической энцефалопатии.
потеря FMN комплексом I ткань мозга во время
ишемии/реперфузии или, наоборот, способству
ют ее повреждению. В зависимости от доступ
Финансирование. Выполнение работы в ла
ности кислорода и продолжительности ишемии
боратории автора было поддержано грантом
диссоциация флавина и A/Д переход могут быть
NS 100850 (NIH USA) и грантами G1100051 и
как вредными, так и полезными для восстанов
MR/L007339/1 (MRC UK).
ления ткани после ишемии/реперфузии. С од
Благодарности. Автор работы искренне бла
ной стороны, высвобождение FMN из комплек
годарен своим коллегам Вадиму Тену и Анне
са I и обратимый переход из активного в неак
Степановой за ценные замечания при подготов
тивное состояние приводят к снижению образо
ке данной работы. Автор выражает благодар
вания ферментом АФК на ранних стадиях реок
ность Вере Георгиевне Гривенниковой за по
сигенации. С другой стороны, восстановленный
мощь в переводе данного обзора на русский
флавин в свободном состоянии может неэнзи
язык.
матически реагировать с кислородом, приводя к
Конфликт интересов. Автор заявляет об отсут
кратковременной генерации АФК, в то время
ствии конфликта интересов.
как комплекс I не функционалeн. То же самое
Соблюдение этических норм. Настоящая
верно и для A/Д перехода: обратимая деактива
статья не содержит каких либо исследований с
ция замедляет продукцию АФК при реоксигена
участием людей или использованием животных
ции, но делает фермент уязвимым для окисли
в качестве объектов исследований.
БИОХИМИЯ том 84 вып. 11 2019
КОМПЛЕКС I ПРИ ИШЕМИИ/РЕПЕРФУЗИИ МОЗГА
1755
СПИСОК ЛИТЕРАТУРЫ
1.
Lee, A.C., Kozuki, N., Blencowe, H., Vos, T., Bahalim, A.,
Metabolomic analyses of plasma reveals new insights into
Darmstadt, G.L., Niermeyer, S., Ellis, M., Robertson,
asphyxia and resuscitation in pigs, PLoS One, 5, e9606,
N.J., Cousens, S., and Lawn, J.E. (2013) Intrapartum
doi: 10.1371/journal.pone.0009606.
related neonatal encephalopathy incidence and impair
20.
Benzi, G., Arrigoni, E., Marzatico, F., and Villa, R.F.
ment at regional and global levels for 2010 with trends from
(1979) Influence of some biological pyrimidines on the
1990, Pediatr. Res., 74, 50-72, doi: 10.1038/pr.2013.206.
succinate cycle during and after cerebral ischemia,
2.
Roger, V.L., Go, A.S., Lloyd Jones, D.M., Benjamin, E.J.,
Biochem. Pharmacol., 28, 2545-2550.
Berry, J.D., et al. (2012) Heart disease and stroke statis
21.
Chouchani, E.T., Pell, V.R., Gaude, E., Aksentijevic, D.,
tics-2012 update: a report from the American Heart
Sundier, S.Y., et al. (2014) Ischaemic accumulation of suc
Association, Circulation, 125, e2-e220, doi: 10.1161/CIR.
cinate controls reperfusion injury through mitochondrial
0b013e31823ac046.
ROS, Nature, 515, 431-435.
3.
Siesjo, B.K., Elmer, E., Janelidze, S., Keep, M., Kristian, T.,
22.
Sahni, P.V., Zhang, J., Sosunov, S., Galkin, A.,
Ouyang, Y.B., and Uchino, H. (1999) Role and mecha
Niatsetskaya, Z., Starkov, A., Brookes, P. S., and Ten, V.S.
nisms of secondary mitochondrial failure, Acta Neurochir.
(2017) Krebs cycle metabolites and preferential succinate oxi
Suppl., 73, 7-13.
dation following neonatal hypoxic ischemic brain injury in
4.
Vannucci, R.C., Towfighi, J., and Vannucci, S.J. (2004) Sec
mice, Pediatr. Res., 83, 491-497, doi: 10.1038/pr.2017.277.
ondary energy failure after cerebral hypoxia ischemia in the
23.
Hinkle, P.C., Butow, R.A., Racker, E., and Chance, B.
immature rat, J. Cereb. Blood Flow Metab., 24, 1090-1097.
(1967) Partial resolution of the enzymes catalyzing oxidative
5.
Hertz, L. (2008) Bioenergetics of cerebral ischemia: a cel
phosphorylation. XV. Reverse electron transfer in the flavin
lular perspective, Neuropharmacology,
55,
289-309,
cytochrome beta region of the respiratory chain of beef heart
doi: 10.1016/j.neuropharm.2008.05.023.
submitochondrial particles, J. Biol. Chem., 242, 5169-5173.
6.
Sims, N.R., and Muyderman, H. (2010) Mitochondria, oxida
24.
Turrens, J.F., and Boveris, A. (1980) Generation of super
tive metabolism and cell death in stroke, Biochim. Biophys. Acta,
oxide anion by the NADH dehydrogenase of bovine heart
1802, 80-91, doi: 10.1016/j.bbadis.2009.09.003.
mitochondria, Biochem. J., 191, 421-427.
7.
Mracek, T., Drahota, Z., and Houstek, J. (2013) The func
25.
Grivennikova, V.G., and Vinogradov, A.D. (2006) Genera
tion and the role of the mitochondrial glycerol 3 phosphate
tion of superoxide by the mitochondrial complex I,
dehydrogenase in mammalian tissues, Biochim. Biophys.
Biochim. Biophys. Acta, 1757, 553-561.
Acta, 1827, 401-410, doi: 10.1016/j.bbabio.2012.11.014.
26.
Pryde, K.R., and Hirst, J. (2011) Superoxide is produced
8.
Watmough, N.J., and Frerman, F.E. (2010) The electron trans
by the reduced flavin in mitochondrial complex I: a single,
fer flavoprotein: ubiquinone oxidoreductases, Biochim. Biophys.
unified mechanism that applies during both forward and
Acta, 1797, 1910-1916, doi: 10.1016/j.bbabio.2010.10.007.
reverse electron transfer, J. Biol. Chem., 286, 18056 18065.
9.
Galkin, A.S., Grivennikova, V.G., and Vinogradov, A.D.
27.
Niatsetskaya, Z.V., Sosunov, S.A., Matsiukevich, D.,
(1999) H+/2e- stoichiometry in NADH quinone reductase
Utkina Sosunova, I.V., Ratner, V.I., Starkov, A.A., and
reactions catalyzed by bovine heart submitochondrial par
Ten, V.S. (2012) The oxygen free radicals originating from
ticles, FEBS Lett., 451, 157-161.
mitochondrial complex I contribute to oxidative brain
10.
Hirst, J. (2013) Mitochondrial complex I, Annu. Rev.
injury following hypoxia ischemia in neonatal mice,
Biochem., 82, 551-575.
J. Neusorci., 32, 3235-3244.
11.
Brandt, U. (2006) Energy converting NADH:quinone oxi
29.
Quinlan, C.L., Perevoshchikova, I.V., Hey Mogensen, M.,
doreductases, Annu. Rev. Biochem., 75, 69-92.
Orr, A.L., and Brand, M.D. (2013) Sites of reactive oxygen
12.
Burbaev, D.S., Moroz, I.A., Kotlyar, A.B., Sled, V.D., and
species generation by mitochondria oxidizing different
Vinogradov, A.D. (1989) Ubisemiquinone in the NADH
substrates, Redox. Biol., 1, 304-312.
ubiquinone reductase region of the mitochondrial respira
29.
Stepanova, A., Konrad, C., Manfredi, G., Springett, R.,
tory chain, FEBS Lett., 254, 47-51.
Ten, V., and Galkin, A. (2018) The dependence of brain
13.
De Jong, A.M., and Albracht, S.P. (1994) Ubisemiquinones
mitochondria reactive oxygen species production on oxy
as obligatory intermediates in the electron transfer from
gen level is linear, except when inhibited by antimycin A,
NADH to ubiquinone, Eur. J. Biochem., 222, 975-982.
J. Neurochem., doi: 10.1111/jnc.14654.
14.
Cabrera Orefice, A., Yoga, E.G., Wirth, C., Siegmund, K.,
30.
Kudin, A.P., Bimpong Buta, N.Y., Vielhaber, S., Elger, C.E.,
Zwicker, K., Guerrero Castillo, S., Zickermann, V., Hunte, C.,
and Kunz, W.S. (2004) Characterization of superoxide
and Brandt, U. (2018) Locking loop movement in the
producing sites in isolated brain mitochondria, J. Biol.
ubiquinone pocket of complex I disengages the proton pumps,
Chem., 279, 4127-4135.
Nat. Commun., 9, 4500, doi: 10.1038/s41467 018 06955 y.
31.
Vinogradov, A.D., and Grivennikova, V.G. (2005) Generation
15.
Vinogradov, A.D., Gavrikova, E.V., Grivennikova, V.G.,
of superoxide radical by the NADH:ubiquinone oxidoreduc
Zharova, T.V., and Zakharova, N.V. (1999) Catalytic prop
tase of heart mitochondria, Biochemistry (Moscow), 70, 120-127.
erties of mitochondrial NADH ubiquinone reductase
32.
Galkin, A., and Brandt, U. (2005) Superoxide radical forma
(Complex I), Biochemistry (Moscow), 64, 136-152.
tion by pure complex I (NADH:ubiquinone oxidoreductase)
16.
Chance, B., and Hollunger, G. (1960) Energy linked
from Yarrowia lipolytica, J. Biol. Chem., 280, 30129-30135.
reduction of mitochondrial pyridine nucleotide, Nature,
33.
Kussmaul, L., and Hirst, J. (2006) The mechanism of
185, 666-672.
superoxide production by NADH:ubiquinone oxidoreduc
17.
Klingenberg, M., and Slenczka, W. (1959) Pyridine
tase (complex I) from bovine heart mitochondria, Proc.
nucleotide in liver mitochondria. An analysis of their redox
Natl. Acad. Sci. USA, 103, 7607-7612.
relationships, Biochemische Zeitschrift, 331, 486-517.
34.
Stepanova, A., Kahl, A., Konrad, C., Ten, V., Starkov, A.S.,
18.
Folbergrova, J., Ljunggren, B., Norberg, K., and Siesjo, B.K.
and Galkin, A. (2017) Reverse electron transfer results in a loss
(1974) Influence of complete ischemia on glycolytic
of flavin from mitochondrial complex I: potential mechanism
metabolites, citric acid cycle intermediates, and associated
for brain ischemia reperfusion injury, J. Cereb. Blood Flow
amino acids in the rat cerebral cortex, Brain Res., 80,
Metab., 37, 3649-3658, doi: 10.1177/0271678X17730242.
265-279, doi: 10.1016/0006 8993(74)90690 8.
35.
Kotlyar, A.B., and Vinogradov, A.D. (1990) Slow active/
19.
Solberg, R., Enot, D., Deigner, H.P., Koal, T., Scholl
inactive transition of the mitochondrial NADH ubiquinone
Burgi, S., Saugstad, O.D., and Keller, M.
(2010)
reductase, Biochim. Biophys. Acta, 1019, 151-158.
БИОХИМИЯ том 84 вып. 11 2019
1756
ГАЛКИН
36.
Maklashina, E.O., Sled, V.D., and Vinogradov, A.D.
Tello, D., Buendia, I., Marina, A., Egea, J., Lopez, M.G.,
(1994) Hysteresis behavior of complex I from bovine heart
Bogdanova, A., and Martinez Ruiz, A.
(2017)
mitochondria: kinetic and thermodynamic parameters of
Mitochondrial complex I deactivation is related to super
retarded reverse transition from the inactive to active state,
oxide production in acute hypoxia, Redox Biol., 12,
Biochemistry (Moscow), 59, 946-957.
1040-1051, doi: 10.1016/j.redox.2017.04.025.
37.
Gavrikova, E.V., and Vinogradov, A.D. (1999) Active/de
52.
Lopez Fabuel, I., Le Douce, J., Logan, A., James, A.M.,
active state transition of the mitochondrial complex I as
Bonvento, G., Murphy, M.P., Almeida, A., and Bolanos, J.P.
revealed by specific sulfhydryl group labeling, FEBS Lett.,
(2016) Complex I assembly into supercomplexes deter
455, 36-40.
mines differential mitochondrial ROS production in neu
38.
Grivennikova, V.G., Kapustin, A.N., and Vinogradov, A.D.
rons and astrocytes, Proc. Natl. Acad. Sci. USA, 113,
(2001) Catalytic activity of NADH ubiquinone oxidore
13063-13068, doi: 10.1073/pnas.1613701113.
ductase (complex I) in intact mitochondria. Evidence for
53.
Vinogradov, A.D., and Grivennikova, V.G. (2001) The
the slow active/inactive transition, J. Biol. Chem., 276,
mitochondrial complex I: progress in understanding of cat
9038-9044.
alytic properties, IUBMB Life, 52, 129-134.
39.
Roberts, P.G., and Hirst, J. (2012) The deactive form of
54.
Babot, M., Birch, A., Labarbuta, P., and Galkin, A. (2014)
respiratory complex I from mammalian mitochondria is a
Characterisation of the active/de active transition of mito
Na+/H+ antiporter, J. Biol. Chem., 287, 34743-34751.
chondrial complex I, Biochim. Biophys. Acta, 1837,
40.
Galkin, A., Meyer, B., Wittig, I., Karas, M., Schagger, H.,
1083-1092, doi: 10.1016/j.bbabio.2014.02.018.
Vinogradov, A., and Brandt, U. (2008) Identification of the
55.
Dröse, S., Stepanova, A., and Galkin, A. (2016) Ischemic
mitochondrial ND3 subunit as a structural component
A/D transition of mitochondrial complex I and its role in
involved in the active/deactive enzyme transition of respi
ROS generation, Biochim. Biophys. Acta, 1857, 946-957,
ratory complex I, J. Biol. Chem., 283, 20907-20913.
doi: 10.1016/j.bbabio.2015.12.013.
41.
Galkin, A., and Moncada, S. (2007) S nitrosation of mito
56.
Kotlyar, A.B., Sled, V.D., and Vinogradov, A.D. (1992)
chondrial complex I depends on its structural conforma
Effect of Ca2+ ions on the slow active/inactive transition of
tion, J. Biol. Chem., 282, 37448-37453.
the mitochondrial NADH ubiquinone reductase, Biochim.
42.
Maklashina, E., Kotlyar, A.B., and Cecchini, G. (2003)
Biophys. Acta, 1098, 144-150.
Active/de active transition of respiratory complex I in bac
57.
Babot, M., and Galkin, A. (2013) Molecular mechanism
teria, fungi, and animals, Biochim. Biophys. Acta, 1606,
and physiological role of active deactive transition of mito
95-103.
chondrial complex I, Biochem. Soc. Trans., 41, 1325-1330.
43.
Matsuzaki, S., and Humphries, K.M. (2015) Selective
58.
Loskovich, M.V., Grivennikova, V.G., Cecchini, G., and
inhibition of deactivated mitochondrial complex I by
Vinogradov, A.D. (2005) Inhibitory effect of palmitate on
biguanides, Biochemistry, 54, 2011-2021.
the mitochondrial NADH:ubiquinone oxidoreductase
44.
Babot, M., Labarbuta, P., Birch, A., Kee, S., Fuszard, M.,
(complex I) as related to the active de active enzyme tran
Botting, C.H., Wittig, I., Heide, H., and Galkin, A. (2014)
sition, Biochem. J., 387, 677-683.
ND3, ND1 and 39kDa subunits are more exposed in the
59.
Stepanova, A., Valls, A., and Galkin, A. (2015) Effect of
de active form of bovine mitochondrial complex I,
monovalent cations on the kinetics of hypoxic conforma
Biochim. Biophys. Acta, 1837, 929-939.
tional change of mitochondrial complex I, Biochim. Biophys.
45.
Galkin, A., Abramov, A.Y., Frakich, N., Duchen, M.R.,
Acta, 1847, 1085-1092, doi: 10.1016/j.bbabio.2015.05.012.
and Moncada, S. (2009) Lack of oxygen deactivates mito
60.
Ciano, M., Fuszard, M., Heide, H., Botting, C.H., and
chondrial complex I: implications for ischemic injury? J.
Galkin, A. (2013) Conformation specific crosslinking of
Biol. Chem., 284, 36055-36061.
mitochondrial complex I, FEBS Lett., 587, 867-872.
46.
Maklashina, E., Sher, Y., Zhou, H.Z., Gray, M.O.,
61.
Blaza, J.N., Vinothkumar, K.R., and Hirst, J. (2018) Struc
Karliner, J.S., and Cecchini, G.
(2002) Effect of
ture of the deactive state of mammalian respiratory complex I,
anoxia/reperfusion on the reversible active/de active tran
Structure, 26, 312-319, e3, doi: 10.1016/j.str.2017.12.014.
sition of NADH ubiquinone oxidoreductase (complex I)
62.
Parey, K., Brandt, U., Xie, H., Mills, D.J., Siegmund, K.,
in rat heart, Biochim. Biophys. Acta, 1556, 6-12.
Vonck, J., Kuhlbrandt, W., and Zickermann, V. (2018)
47.
Gorenkova, N., Robinson, E., Grieve, D., and Galkin, A.
Cryo EM structure of respiratory complex I at work, Elife,
(2013) Conformational change of mitochondrial complex I
7, doi: 10.7554/eLife.39213.
increases ROS sensitivity during ischaemia, Antioxid.
63.
Agip, A.A., Blaza, J.N., Bridges, H.R., Viscomi, C.,
Redox Signal., 19, 1459-1468.
Rawson, S., Muench, S.P., and Hirst, J. (2018) Cryo EM
48.
Chouchani, E.T., Methner, C., Nadtochiy, S.M., Logan, A.,
structures of complex I from mouse heart mitochondria in
Pell, V.R., Ding, S., James, A.M., Cocheme, H.M.,
two biochemically defined states, Nat. Struct. Mol. Biol.,
Reinhold, J., Lilley, K.S., Partridge, L., Fearnley, I.M.,
25, 548-556, doi: 10.1038/s41594 018 0073 1.
Robinson, A.J., Hartley, R.C., Smith, R.A., Krieg, T.,
64.
Fiedorczuk, K., Letts, J.A., Degliesposti, G., Kaszuba, K.,
Brookes, P.S., and Murphy, M.P. (2013) Cardioprotection
Skehel, M., and Sazanov, L.A. (2016) Atomic structure of
by S nitrosation of a cysteine switch on mitochondrial
the entire mammalian mitochondrial complex I, Nature,
complex I, Nat. Med., 19, 753-759.
537, 644-648, doi: 10.1038/nature19794.
49.
Stepanova, A., Konrad, C., Guerrero Castillo, S.,
65.
Zhu, J., Vinothkumar, K.R., and Hirst, J. (2016) Structure
Manfredi, G., Vannucci, S., Arnold, S., and Galkin, A. (2018)
of mammalian respiratory complex I, Nature, 536,
Deactivation of mitochondrial complex I after hypoxia
354-358, doi: 10.1038/nature19095.
ischemia in the immature brain, J. Cereb. Blood Flow Metab.,
66.
Zickermann, V., Wirth, C., Nasiri, H., Siegmund, K.,
39, 1790-1802, doi: 10.1177/0271678X18770331.
Schwalbe, H., Hunte, C., and Brandt, U.
(2015)
50.
Kim, M., Stepanova, A., Niatsetskaya, Z., Sosunov, S.,
Mechanistic insight from the crystal structure of mito
Arndt, S., Murphy, M.P., Galkin, A., and Ten, V.S. (2018)
chondrial complex I, Science, 347, 44-49.
Attenuation of oxidative damage by targeting mitochon
67.
Koopman, W.J., Willems, P.H., and Smeitink, J.A. (2012)
drial complex I in neonatal hypoxic ischemic brain injury,
Monogenic mitochondrial disorders, N. Engl. J. Med., 366,
Free Radic. Biol. Med., 124, 517-524, doi: 10.1016/
1132-1141, doi: 10.1056/NEJMra1012478.
j.freeradbiomed.2018.06.040.
68.
Stefanatos, R., and Sanz, A. (2011) Mitochondrial com
51.
Hernansanz Agustin, P., Ramos, E., Navarro, E., Parada, E.,
plex I: a central regulator of the aging process, Cell Cycle,
Sanchez Lopez, N., Pelaez Aguado, L., Cabrera Garcia, J.D.,
10, 1528-1532.
БИОХИМИЯ том 84 вып. 11 2019
КОМПЛЕКС I ПРИ ИШЕМИИ/РЕПЕРФУЗИИ МОЗГА
1757
69.
Breuer, M.E., Koopman, W.J., Koene, S., Nooteboom, M.,
87. Nordstrom, C.H., Rehncrona, S., and Siesjo, B.K. (1978)
Rodenburg, R.J., Willems, P. H., and Smeitink, J.A. (2013)
Effects of phenobarbital in cerebral ischemia. Part II: resti
The role of mitochondrial OXPHOS dysfunction in the
tution of cerebral energy state, as well as of glycolytic
development of neurologic diseases, Neurobiol. Dis., 51,
metabolites, citric acid cycle intermediates and associated
27-34, doi: 10.1016/j.nbd.2012.03.007.
amino acids after pronounced incomplete ischemia,
70.
Ndubuizu, O., and LaManna, J.C. (2007) Brain tissue oxy
Stroke, 9, 335-343.
gen concentration measurements, Antioxid. Redox Signal.,
88. Sims, N.R., and Pulsinelli, W.A. (1987) Altered mitochon
9, 1207-1219.
drial respiration in selectively vulnerable brain subregions
71.
Madsen, P.L., Holm, S., Herning, M., and Lassen, N.A.
following transient forebrain ischemia in the rat,
(1993) Average blood flow and oxygen uptake in the human
J. Neurochem., 49, 1367-1374.
brain during resting wakefulness: a critical appraisal of the
89. Sims, N.R. (1991) Selective impairment of respiration in
Kety Schmidt technique, J. Cereb. Blood Flow Metab., 13,
mitochondria isolated from brain subregions following
646-655, doi: 10.1038/jcbfm.1993.83.
transient forebrain ischemia in the rat, J. Neurochem., 56,
72.
Reneau, D.D., Guilbeau, E.J., and Null, R.E. (1977)
1836-1844.
Oxygen dynamics in brain, Microvasc. Res., 13, 337-344.
90. Herculano Houzel, S., Ribeiro, P., Campos, L., Valotta da
73.
Lowry, O.H., Passonneau, J.V., Hasselberger, F.X., and
Silva, A., Torres, L.B., Catania, K.C., and Kaas, J.H.
Schulz, D.W. (1964) Effect of ischemia on known sub
(2011) Updated neuronal scaling rules for the brains of
strates and cofactors of the glycolytic pathway in brain, J.
Glires (rodents/lagomorphs), Brain Behav. Evol., 78,
Biol. Chem., 239, 18-30.
302-314, doi: 10.1159/000330825.
74.
Hillered, L., Siesjo, B.K., and Arfors, K.E.
(1984)
91. Panov, A., Orynbayeva, Z., Vavilin, V., and Lyakhovich, V.
Mitochondrial response to transient forebrain ischemia
(2014) Fatty acids in energy metabolism of the central ner
and recirculation in the rat, J. Cereb. Blood Flow Metab., 4,
vous system, Biomed. Res. Int.,
2014,
472459,
438-446, doi: 10.1038/jcbfm.1984.63.
doi: 10.1155/2014/472459.
75.
Kristian, T. (2004) Metabolic stages, mitochondria and
92. Tretter, L., Takacs, K., Hegedus, V., and Adam Vizi, V.
calcium in hypoxic/ischemic brain damage, Cell Calcium,
(2007) Characteristics of alpha glycerophosphate evoked
36, 221-233, doi: 10.1016/j.ceca.2004.02.016.
H2O2 generation in brain mitochondria, J. Neurochem.,
76.
Sugawara, T., Lewen, A., Noshita, N., Gasche, Y., and
100, 650-663.
Chan, P.H. (2002) Effects of global ischemia duration on
93. Kahl, A., Stepanova, A., Konrad, C., Anderson, C.,
neuronal, astroglial, oligodendroglial, and microglial reac
Manfredi, G., Zhou, P., Iadecola, C., and Galkin, A.
tions in the vulnerable hippocampal CA1 subregion in rats, J.
(2018) Critical role of flavin and glutathione in complex I
Neurotrauma, 19, 85-98, doi: 10.1089/089771502753460268.
mediated bioenergetic failure in brain ischemia/reperfu
77.
Astrup, J., Siesjo, B.K., and Symon, L. (1981) Thresholds
sion injury, Stroke,
49,
1223-1231, doi:
10.1161/
in cerebral ischemia - the ischemic penumbra, Stroke, 12,
STROKEAHA.117.019687.
723-725.
94. Linn, F., Paschen, W., Ophoff, B.G., and Hossmann, K.A.
78.
Becker, N.H. (1961) The cytochemistry of anoxic and
(1987) Mitochondrial respiration during recirculation after
anoxioischemic encephalopathy in rats. II. Alterations in
prolonged ischemia in cat brain, Exp. Neurol., 96,
neuronal mitochondria indentified by diphosphopyridine
321-333.
and triphosphopyridine nucleotide diaphorases, Am. J.
95. Almeida, A., Allen, K.L., Bates, T.E., and Clark, J.B.
Pathol., 38, 587-597.
(1995) Effect of reperfusion following cerebral ischaemia
79.
Def Webster, H., and Ames, A. (1965) Reversible and irre
on the activity of the mitochondrial respiratory chain in the
versible changes in the fine structure of nervous tissue during
gerbil brain, J. Neurochem., 65, 1698-1703.
oxygen and glucose deprivation, J. Cell Biol., 26, 885-909.
96. Allen, K.L., Almeida, A., Bates, T.E., and Clark, J.B.
80.
Zeman, W. (1963) Histochemical and metabolic changes in
(1995) Changes of respiratory chain activity in mitochondr
brain tissue after hypoxaemia. in Selective Vulnerability of the
ial and synaptosomal fractions isolated from the gerbil brain
Brain in Hypoxemia (Schade, J. P., and McMenemey, W. H.
after graded ischaemia, J. Neurochem., 64, 2222-2229.
eds) Blackwell Publishing Co, London, pp. 327-348.
97. Yoshimoto, T., Kristian, T., Hu, B., Ouyang, Y.B., and
81.
Ozawa, K., Seta, K., Araki, H., and Handa, H. (1967) The
Siesjo, B.K. (2002) Effect of NXY 059 on secondary mito
effect of ischemia on mitochondrial metabolism, J.
chondrial dysfunction after transient focal ischemia; com
Biochem., 61, 512-514.
parison with cyclosporin A, Brain Res., 932, 99-109.
82.
Ozawa, K., Itada, N., Kuno, S., Seta, K., and Handa, H.
98. Tsukada, H., Ohba, H., Nishiyama, S., Kanazawa, M.,
(1966) Biochemical studies on brain swelling. II. Influence
Kakiuchi, T., and Harada, N. (2014) PET imaging of
of brain swelling and ischemia on the formation of an
ischemia induced impairment of mitochondrial complex I
endogenous inhibitor in mitochondria, Folia Psychiatr.
function in monkey brain, J. Cereb. Blood Flow Metab., 34,
Neurol. Jpn., 20, 73-84.
708-714, doi: 10.1038/jcbfm.2014.5.
83.
Schutz, H., Silverstein, P.R., Vapalahti, M., Bruce, D.A.,
99. Stepanova, A., Sosunov, S., Niatsetskaya, Z., Konrad, C.,
Mela, L., and Langfitt, T.W. (1973) Brain mitochondrial
Starkov, A.A., Manfredi, G., Wittig, I., Ten, V., and
function after ischemia and hypoxia. I. Ischemia induced
Galkin, A. (2019) Redox dependent loss of flavin by mito
by increased intracranial pressure, Arch. Neurol., 29,
chondrial complex I in brain ischemia/reperfusion injury,
408-416.
Antioxid. Redox Signal., doi: 10.1089/ars.2018.7693.
84.
Ljunggren, B., Schutz, H., and Siesjo, B.K.
(1974)
100. Rao, N.A., Felton, S.P., Huennekens, F.M., and Mackler, B.
Changes in energy state and acid base parameters of the rat
(1963) Flavin mononucleotide: the coenzyme of reduced
brain during complete compression ischemia, Brain Res.,
diphosphopyridine nucleotide dehydrogenase, J. Biol.
73, 277-289, doi: 10.1016/0006 8993(74)91049 X.
Chem., 238, 449-455.
85.
Ginsberg, M.D., Mela, L., Wrobel Kuhl, K., and Reivich, M.
101. Yoshida, S., Abe, K., Busto, R., Watson, B.D., Kogure, K.,
(1977) Mitochondrial metabolism following bilateral cere
and Ginsberg, M.D. (1982) Influence of transient ischemia
bral ischemia in the gerbil, Ann. Neurol., 1, 519-527,
on lipid soluble antioxidants, free fatty acids and energy
doi: 10.1002/ana.410010603.
metabolites in rat brain, Brain Res., 245, 307-316.
86.
Rehncrona, S., Mela, L., and Siesjo, B.K. (1979) Recovery
102. Deutsch, J., Kalderon, B., Purdon, A.D., and Rapoport, S.I.
of brain mitochondrial function in the rat after complete
(2000) Evaluation of brain long chain acylcarnitines dur
and incomplete cerebral ischemia, Stroke, 10, 437-446.
ing cerebral ischemia, Lipids, 35, 693-696.
БИОХИМИЯ том 84 вып. 11 2019
1758
ГАЛКИН
103. Nguyen, N.H., Gonzalez, S.V., and Hassel, B. (2007)
ischemia and reperfusion injury to brain, Neurosci. Lett.,
Formation of glycerol from glucose in rat brain and cul
88, 233-238.
tured brain cells. Augmentation with kainate or ischemia,
114. Anderson, M.F., and Sims, N.R. (2002) The effects of focal
J. Neurochem., 101, 1694-1700, doi: 10.1111/j.1471
ischemia and reperfusion on the glutathione content of
4159.2006.04433.x.
mitochondria from rat brain subregions, J. Neurochem., 81,
104. Massey, V. (1994) Activation of molecular oxygen by flavins
541-549.
and flavoproteins, J. Biol. Chem., 269, 22459-22462.
115.
Mizui, T., Kinouchi, H., and Chan, P.H. (1992) Depletion of
105. Gibson, Q.H., Massey, V., and Atherton, N.M. (1962) The
brain glutathione by buthionine sulfoximine enhances cere
nature of compounds present in mixtures of oxidized and
bral ischemic injury in rats, Am. J. Physiol., 262, H313-H317.
reduced flavin mononucleotides, Biochem. J.,
85,
116. Folbergrova, J., Zhao, Q., Katsura, K., and Siesjo, B.K.
369-383.
(1995) N tert butyl alpha phenylnitrone improves recov
106. Barile, M., Brizio, C., Valenti, D., De Virgilio, C., and
ery of brain energy state in rats following transient focal
Passarella, S. (2000) The riboflavin/FAD cycle in rat liver
ischemia, Proc. Natl. Acad. Sci. USA, 92, 5057-5061.
mitochondria, Eur. J. Biochem., 267, 4888-4900.
117. Khan, M., Sekhon, B., Jatana, M., Giri, S., Gilg, A.G.,
107. Sled, V.D., and Vinogradov, A.D. (1993) Reductive inacti
Sekhon, C., Singh, I., and Singh, A.K.
(2004)
vation of the mitochondrial three subunit NADH dehydro
Administration of N acetylcysteine after focal cerebral
genase, Biochim. Biophys. Acta, 1143, 199-203.
ischemia protects brain and reduces inflammation in a rat
108. Gostimskaya, I.S., Grivennikova, V.G., Cecchini, G., and
model of experimental stroke, J. Neurosci. Res., 76,
Vinogradov, A.D. (2007) Reversible dissociation of flavin
519-527, doi: 10.1002/jnr.20087.
mononucleotide from the mammalian membrane bound
118. Anderson, M.F., Nilsson, M., Eriksson, P.S., and Sims,
NADH:ubiquinone oxidoreductase (complex I), FEBS
N.R. (2004) Glutathione monoethyl ester provides neuro
Lett., 581, 5803-5806.
protection in a rat model of stroke, Neurosci. Lett., 354,
109. Holt, P.J., Efremov, R.G., Nakamaru Ogiso, E., and
163-165.
Sazanov, L.A. (2016) Reversible FMN dissociation from
119. Prime, T.A., Blaikie, F.H., Evans, C., Nadtochiy, S.M.,
Escherichia coli respiratory complex I, Biochim. Biophys.
James, A.M., Dahm, C.C., Vitturi, D.A., Patel, R.P.,
Acta, 1857, 1777-1785, doi: 10.1016/j.bbabio.2016.08.008.
Hiley, C.R., Abakumova, I., Requejo, R., Chouchani, E.T.,
110. Gariballa, S., and Ullegaddi, R. (2007) Riboflavin status in
Hurd, T.R., Garvey, J.F., Taylor, C.T., Brookes, P.S.,
acute ischaemic stroke, Eur. J. Clin. Nutr., 61, 1237-1240,
Smith, R.A., and Murphy, M.P. (2009) A mitochondria
doi: 10.1038/sj.ejcn.1602666.
targeted S nitrosothiol modulates respiration, nitrosates
111.
da Silva Candal, A., Perez Diaz, A., Santamaria, M.,
thiols, and protects against ischemia reperfusion injury,
Correa Paz, C., Rodriguez Yanez, M., Arda, A., Perez
Proc. Natl. Acad. Sci. USA, 106, 10764-10769.
Mato, M., Iglesias Rey, R., Brea, J., Azuaje, J., Sotelo, E.,
120. Stepanova, A., Shurubor, Y., Valsecchi, F., Manfredi, G.,
Sobrino, T., Loza, M. I., Castillo, J., and Campos, F. (2018)
and Galkin, A. (2016) Differential susceptibility of mito
Clinical validation of blood/brain glutamate grabbing in
chondrial complex II to inhibition by oxaloacetate in brain
acute ischemic stroke, Ann. Neurol., doi: 10.1002/ana.25286.
and heart, Biochim. Biophys. Acta, 1857, 1561-1568,
112. Kalogeris, T., Bao, Y., and Korthuis, R.J.
(2014)
doi: 10.1016/j.bbabio.2016.06.002.
Mitochondrial reactive oxygen species: a double edged
121. Kang, P.T., Chen, C.L., Lin, P., Zhang, L., Zweier, J.L.,
sword in ischemia/reperfusion vs preconditioning, Redox
and Chen, Y.R. (2018) Mitochondrial complex I in the
Biol., 2, 702-714, doi: 10.1016/j.redox.2014.05.006.
post ischemic heart: reperfusion mediated oxidative injury
113. Cao, W., Carney, J.M., Duchon, A., Floyd, R.A., and
and protein cysteine sulfonation, J. Mol. Cell. Cardiol.,
Chevion, M. (1988) Oxygen free radical involvement in
121, 190-204, doi: 10.1016/j.yjmcc.2018.07.244.
BRAIN ISCHEMIA/REPERFUSION INJURY
AND MITOCHONDRIAL COMPLEX I DAMAGE
Review
A. Galkin*
Division of Neonatology, Department of Pediatrics, Columbia University William Black Building,
10032 New York, NY, USA; E mail: ag4003@cumc.columbia.edu
Received June 7, 2019
Revised July 8, 2019
Accepted July 9, 2019
Ischemic stroke and neonatal hypoxic ischemic encephalopathy are two of the leading causes of disability in adults
and infants. The energy demands of the brain are provided by mitochondrial oxidative phosphorylation.
Ischemia/reperfusion (I/R) affects production of ATP in brain mitochondria leading to energy failure and death of
the affected tissue. Among the enzymes of the mitochondrial respiratory chain mitochondrial complex I is the most
sensitive to I/R, however the mechanisms of inhibition are poorly understood. This article reviews some of the exist
ing data on mitochondria impairment during I/R and also proposes two distinct mechanisms of complex I damage
emerging from recent studies. One mechanism is a reversible dissociation of natural flavin mononucleotide (FMN)
cofactor from the enzyme I after ischemia. Another mechanism is a modification of critical cysteine residue of com
plex I involved into the active/deactive (A/D) conformational transition of the enzyme. Here I describe the potential
effect of these two processes in the development of mitochondrial I/R injury and briefly discuss possible neuropro
tective strategies to ameliorate I/R brain injury.
Keywords: stroke, ischemia reperfusion injury, mitochondria, complex I, flavin, thiols, nitrosation
БИОХИМИЯ том 84 вып. 11 2019