БИОХИМИЯ, 2019, том 84, вып. 2, с. 191 - 211
УДК 615.012.1
КОМПЬЮТЕРНЫЙ ДИЗАЙН
НИЗКОМОЛЕКУЛЯРНЫХ ИНГИБИТОРОВ
ФАКТОРОВ СИСТЕМЫ СВЕРТЫВАНИЯ КРОВИ
Обзор
© 2019 А.С. Кабанкин1, Е.И. Синауридзе1,2, Е.Н. Липец1,2,
Ф.И. Атауллаханов1,2,3,4*
1 Центр теоретических проблем физико химической фармакологии РАН,
119991 Москва, Россия; электронная почта: ataullakhanov.fazly@gmail.com
2 Национальный медицинский исследовательский центр детской гематологии,
онкологии и иммунологии им. Дмитрия Рогачева, Министерство
здравоохранения России, 117198 Москва, Россия
3 Московский государственный университет им. М.В. Ломоносова,
физический факультет, 119991 Москва
4 Московский физико технический институт, 141701 Долгопрудный
Московской области, Россия
Поступила в редакцию 05.09.18
После доработки 19.10.18
Принята к публикации 19.10.18
Представлен обзор основных подходов к поиску новых низкомолекулярных ингибиторов факторов сверты
вания крови IIa, Xa, IXa и XIa, а также результаты этого поиска, проведенного с 2015 по 2018 г. Для каждо
го из перечисленных факторов за последнее время найдено по несколько ингибиторов с IC50 менее 10 нМ.
Некоторые из них находятся на стадии клинических испытаний. Однако ни одно из этих соединений не
подходит на роль «идеального» антикоагулянта, поэтому дальнейший поиск таких ингибиторов является
актуальной задачей.
КЛЮЧЕВЫЕ СЛОВА: антикоагулянты, низкомолекулярные ингибиторы, факторы свертывания крови,
компьютерный дизайн ингибиторов, молекулярный докинг.
DOI: 10.1134/S0320972519020039
Система свертывания отвечает за сохране
фективных и безопасных антитромботических
ние крови в жидком текучем состоянии в физио
препаратов исключительно важна.
логических условиях, остановку кровотечений
Система свертывания крови представляет
при нарушении целостности сосудистой систе
собой сложный каскад ферментативных реак
мы и растворение тромбов, выполнивших свою
ций, который может активироваться по двум пу
функцию. Нарушения в системе гемостаза явля
тям: внутреннему и внешнему [1-3]. Белки,
ются одной из главных причин смертности и
участвующие в этих реакциях, называются фак
инвалидности в современном мире. Они могут
торами свертывания крови и нумеруются римс
вызывать как неконтролируемые кровотечения,
кими цифрами. В норме они присутствуют в
так и образование тромбов, препятствующих
плазме в неактивном виде и активируются при
кровоснабжению и вызывающих органную не
расщеплении одной или нескольких пептидных
достаточность. Тромбозы возникают при самых
связей активным фактором, стоящим выше по
различных заболеваниях (атеросклероз, ин
каскаду. Оба пути сходятся на факторе Ха и да
фаркт, инсульт, травмы, хирургические вмеша
лее идут одинаково. Остановка кровотечения
тельства и др.). Проблема становится все более
осуществляется за счет образования фибрино
актуальной в связи с ростом средней продолжи
вого сгустка при полимеризации мономеров
тельности жизни и увеличением количества по
фибрина, возникающих при расщеплении белка
жилых людей. Таким образом, разработка эф
фибриногена тромбином (фактором IIa), кото
рый образуется из протромбина под действием
* Адресат для корреспонденции.
фактора Xa. Фактор Х активируется либо факто
191
192
КАБАНКИН и др.
ром IXa, либо комплексом фактора VIIa и ткане
комолекулярные ингибиторы отдельных факто
вого фактора (VIIa-ТФ) (по внешнему пути ак
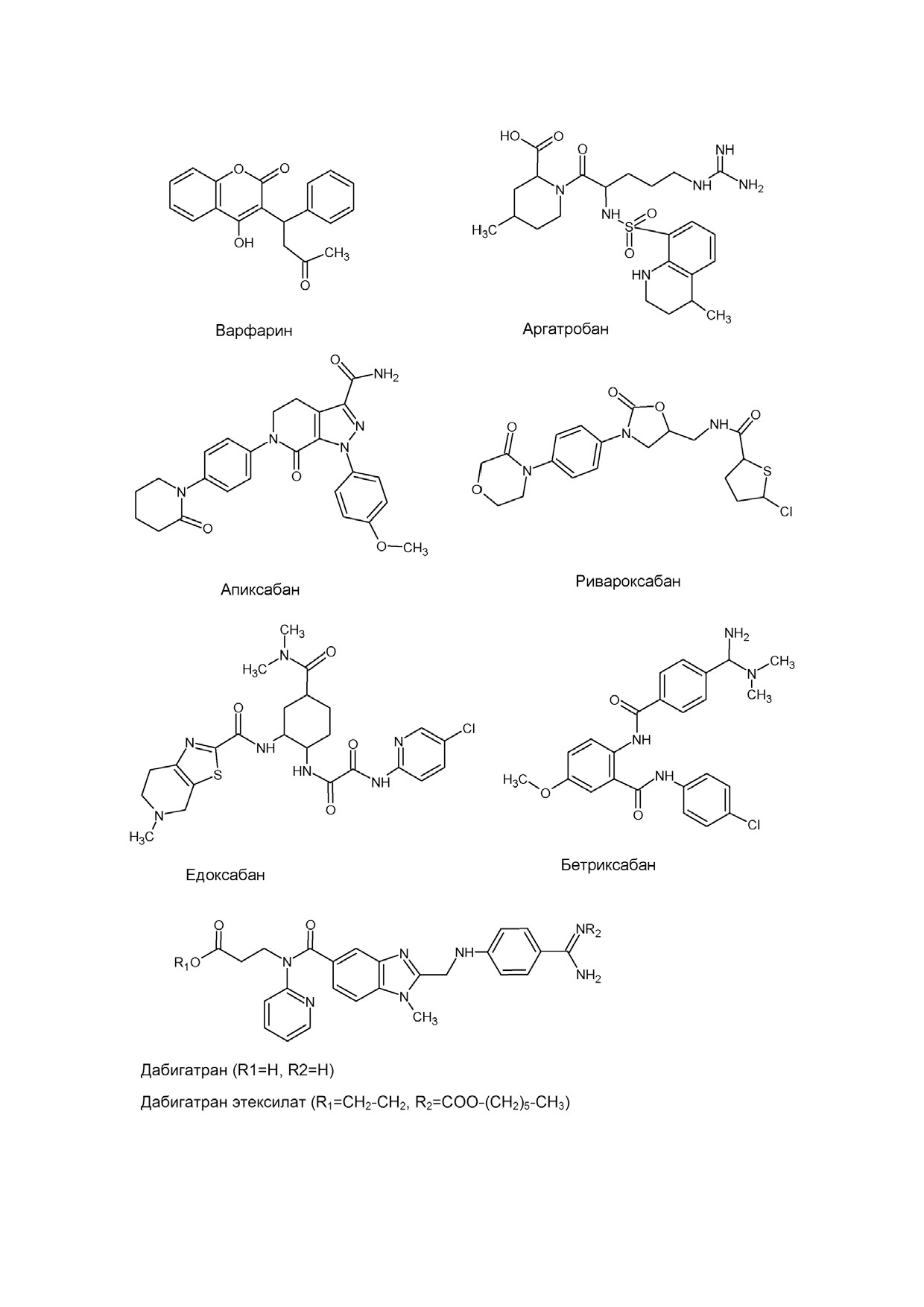
ров свертывания, такие как аргатробан, дабигат
тивации). Фактор IX также активируется комп
ран, ривороксабан и др. (рис. 1). Указанные пре
лексом VIIa-ТФ. Основной физиологический
параты достаточно эффективны, но у каждого
активатор системы свертывания - это тканевый
есть ряд серьезных ограничений, поэтому пос
фактор (ТФ), белок, содержащийся в окружаю
тоянно ведутся исследования, направленные на
щих сосуд тканях, и приходящий в контакт с
поиск и разработку новых эффективных и безо
кровью при повреждении сосуда. Этот путь ак
пасных антикоагулянтов [4, 5].
тивации называется внешним. Существует так
Гепарины и варфарин действуют на множест
же внутренний путь активации, который запус
во мишеней. Это считается одним из их недос
кается при контакте фактора XII с инородными
татков, так как не позволяет оценить и спрогно
для организма материалами. Активированный
зировать их действие на отдельные звенья коа
фактор XIIa при участии высокомолекулярного
гуляционного процесса. Поэтому сформирова
кининогена запускает дальнейший каскад реак
лась и доминирует концепция, заключающаяся
ций, в котором последовательно активируются
в том, что в первую очередь надо разрабатывать
факторы XI, IX и Х, превращаясь в XIa, IXa и Ха,
селективные ингибиторы, действующие на от
соответственно.
дельные конкретные факторы свертывания.
Тромбин является ключевым ферментом
Именно в этом ключе работает большинство ис
системы свертывания, так как он не только при
следователей.
водит к образованию фибрина, но и активирует
К настоящему времени для применения в
факторы V, VIII, XI, XIII, протеин С, и тромбо
клинической практике одобрены следующие
циты. Он участвует в основных положительных
низкомолекулярные антикоагулянты: дабигат
и отрицательных обратных связях системы.
ран этексилат (dabigatran etexilate), ривароксабан
Тромбин ускоряет свое производство на 3-5 по
(rivaroxaban), апиксабан (apixaban), эдоксабан
рядков, переводя факторы V и VIII в активные
(edoxaban) и бетриксабан (betrixaban) [6-12]
формы, которые входят в состав комплексов
(рис. 1). Последние четыре препарата в этом
протромбиназы (Ха-Va, фосфолипидная поверх
списке являются прямыми оральными ингиби
ность, Са+2) и внутренней теназы (VIIIa-IXa,
торами фактора Xa. Дабигатран этексилат явля
фосфолипидная поверхность, Са+2). В комплек
ется пролекарством, которое после перорально
се с тромбомодулином тромбин замедляет свое
го введения превращается в прямой низкомо
образование, активируя протеин С, который
лекулярный ингибитор тромбина дабигатран.
способен ингибировать Va и VIIIa.
Указанные прямые оральные антикоагулянты
Чтобы кровь в организме полностью не свер
(ПОАК) имеют некоторые преимущества перед
нулась и продолжала существовать в жидком
давно и широко используемым варфарином:
состоянии, все активные формы факторов после
удобство применения, ограниченное взаимо
окончания свертывания должны быстро и необ
действие с другими лекарствами и пищей, отсут
ратимо инактивироваться. Это осуществляется
ствие необходимости постоянного лаборатор
присутствующими в крови специфическими
ного мониторинга. Однако и эти препараты не
ингибиторами активных факторов свертывания
являются идеальными и также имеют ряд огра
(антикоагулянтами). Наиболее значимыми фи
ничений [8, 9, 12]. Отмечен повышенный, по срав
зиологическими антикоагулянтами являются
нению с варфарином, риск желудочно кишеч
антитромбин III (АТIII), гепарин, ингибитор
ных кровотечений. Не взаимодействуя с боль
пути тканевого фактора (ИПТФ), протеины С и S.
шинством лекарственных препаратов, ПОАК в
На сегодняшний день в клинике используется
то же время являются субстратами для цитохро
несколько основных антикоагулянтных препа
ма Р450 (CYP3A4) и транспортного белка Р гли
ратов с различными механизмами действия. Это
копротеина (P gp). Использование активных
полимерные гликозаминогликаны гепарины
ингибиторов этих белков повышает концентра
(нефракционированный и различные низкомо
цию ПОАК в плазме, сильно увеличивая риск
лекулярные), которые усиливают действие при
кровотечений. Рекомендуется не использовать
родного ингибитора АТIII; ингибитор агрегации
ПОАК или же применять их с большой осто
тромбоцитов аспирин; пептид гирудин, а также
рожностью для пациентов с почечной и пече
гирудиноподобный пептид бивалирудин, инги
ночной недостаточностью, с искусственным
бирующие тромбин; витамин К антагонисты
сердечным клапаном, а также для детей моложе
(ВКА), среди которых наиболее известен варфа
18 лет и беременных женщин. Для пациентов
рин, снижающие уровень витамина К, необхо
этих категорий в случае использования ПОАК
димого для синтеза полноценных факторов
необходим подбор доз и лабораторный монито
свертывания в печени; а также различные низ
ринг, с которым также есть проблемы ввиду неп
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
193
Рис. 1. Используемые и разрешенные для клинического применения низкомолекулярные антикоагулянты
4 БИОХИМИЯ том 84 вып. 2 2019
194
КАБАНКИН и др.
ригодности для ПОАК общепринятых стандарт
рамме алгоритма докинга воспроизводить ори
ных методик определения антикоагулянтной
ентацию лиганда в активном центре белка, най
активности и измерения концентрации антико
денную рентгеноструктурным анализом соотве
агулянта в плазме. Недостатком ПОАК является
тствующего комплекса белок-лиганд. Оценка
отсутствие специфических антидотов. Правда в
энергии связывания комплексов лиганд-белок
последние годы был предложен и одобрен FDA
является наиболее сложной проблемой в
(Food and Drug Administration) антидот для даби
конструировании потенциальных лигандов. Хо
гатрана - идаруцизумаб (idarucizumab) [13],
тя существуют методы расчета свободной энер
представляющий собой фрагмент моноклональ
гии с помощью молекулярной динамики [21], и
ного антитела. Недостатком ПОАК считается
в настоящее время начинается разработка прог
также высокая стоимость.
рамм для расчета свободной энергии связыва
ния лиганд рецепторных комплексов методами
статистической механики и квантовой химии
КОМПЬЮТЕРНЫЙ ДИЗАЙН И ПРОЦЕСС
[22], на практике энергия связывания чаще ап
РАЗРАБОТКИ НОВЫХ ЛЕКАРСТВЕННЫХ
проксимируется с помощью, так называемых,
ПРЕПАРАТОВ
оценочных функций (scoring functions). Исполь
зуют три вида функций: эмпирические оценоч
Начальной стадией процесса разработки лю
ные функции, оценочные функции, основан
бого лекарственного препарата, в том числе и
ные на силовых полях, и оценочные функции,
антикоагулянта, является открытие принципи
основанные на имеющихся данных о структур
ально новой базовой химической структуры
ной информации, полученной из рентгеност
(лидера), проявляющей необходимую биологи
руктурных данных для лиганд-белковых комп
ческую активность. Затем с помощью химичес
лексов. В случае эмпирических оценочных
кого синтеза различных производных структу
функций энергия связывания является суммой
ры-лидера и их модификации осуществляется
произвольного числа членов, представляющих
оптимизация новой базовой структуры, направ
вклады разных взаимодействий, важных для
ленная на усиление полезной биологической
связывания лиганда (водородные связи, ионные
активности и улучшение фармакологического
взаимодействия, липофильные и ароматические
профиля. В современных условиях для откры
взаимодействия, десольватация и др.). Весовые
тия новых базовых структур (молекулярный ди
коэффициенты для этих членов получают с по
зайн) преимущественно используют компью
мощью множественной линейной регрессии,
терный виртуальный скрининг. В зависимости
используя обучающую выборку лиганд белко
от характера имеющейся информации, возмож
вых комплексов с известными константами свя
ны различные способы реализации виртуально
зывания и экспериментально определенной
го скрининга [14-16].
структурой. В этом заключается главный недос
Если определена трехмерная структура ак
таток эмпирических оценочных функций, так
тивного центра макромолекулы мишени, как
как можно ожидать, что эти функции могут хо
правило, с помощью рентгеноструктурного ана
рошо работать лишь для белков, похожих на ис
лиза, то применяют прямые методы поиска или
пользованные в обучающей выборке. Силовые
конструирования потенциального лиганда (но
оценочные функции базируются на силовых по
вой базовой структуры), соответствующего дан
лях, применяемых в молекулярной механике.
ному активному центру как ключ замку. Этот
Вандерваальсовы взаимодействия описываются
подход, известный как конструирование ле
потенциалом Леннарда-Джонса либо другими
карств, основанное на структуре (SBDD, struc
подобными функциями, а электростатические
ture based drug design), использует достаточно
взаимодействия - законом Кулона. В последнее
сложные вычислительные процедуры молеку
время для оценки энергии связывания все боль
лярного докинга [17-20]. Цель молекулярного
шую популярность приобретают оценочные
докинга - определить наиболее предпочтитель
функции, основанные на знаниях структуры. В
ную конформацию молекулы лиганда и наибо
этом случае нет необходимости в подгонке к
лее выгодное расположение лиганда в активном
экспериментальным значениям энергии связы
центре белковой мишени, а также оценить энер
вания белок-лиганд, а используется только
гию связывания лиганда с мишенью. Современ
структурная информация, содержащаяся в уже
ные алгоритмы докинга учитывают конформа
изученных комплексах белок-лиганд. Эти дан
ционную подвижность и лиганда, и белка. Од
ные можно использовать для оценки частоты
ним из критериев выбора компьютерной прог
встречаемости атом атомных контактов опреде
раммы для проведения молекулярного докинга
ленного типа на заданных расстояниях между
является возможность используемого в прог
лигандом и белком во всех имеющихся данных
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
195
и, используя методы статистической механики,
кулярной структуры рассматриваемых соедине
определить парные потенциалы взаимодей
ний (молекулярных дескрипторов) и выбор ма
ствия атомов белка i го типа с атомами лиганда
тематического метода для построения этой за
j го типа. Тогда полная энергия взаимодействия
висимости. К настоящему времени для опреде
определяется как сумма по всем межатомным
ления количественной связи структура-актив
взаимодействиям в комплексе белок-лиганд.
ность (КССА) (QSAR, quantitative structure activi
Вклад сольватации и энтропии при этом учиты
ty relationship), предложено использовать нес
вается в неявном виде. Возможности этих оце
колько тысяч разных молекулярных дескрипто
ночных функций значительно шире по сравне
ров и множество математических методов [23,
нию с эмпирическими и силовыми оценочными
24]. Молекулярные дескрипторы - численные
функциями, и они могут быть использованы для
характеристики, отражающие структурные осо
оценки свободной энергии связывания разно
бенности соединений. Это могут быть экспери
образных комплексов белок-лиганд. Тем не ме
ментальные физико химические характеристи
нее, когда докинг применяется для скрининга
ки (например, растворимость, коэффициент
большой базы данных с большим разнообрази
распределения, температура плавления, хими
ем соединений и мишеней, которые не были ис
ческий сдвиг ЯМР и т.д.). Однако эксперимен
пользованы для калибровки оценочных функ
тальное определение физико химических свойств
ций, получаемая точность анализа весьма огра
возможно далеко не всегда. Поэтому наиболее
ничена. Дополнительные трудности при прове
широко используются теоретически рассчиты
дении молекулярного докинга связаны с реше
ваемые дескрипторы, которые могут быть вы
нием вопроса, стоит ли учитывать или игнори
числены для любых реально существующих и
ровать содержащиеся в центре связывания мо
гипотетических молекул. Теоретические деск
лекулы кристаллизационной воды, кофактор,
рипторы разделяются на несколько групп, соот
ионы металлов.
ветствующих разным уровням представления
Если данные о пространственной структуре
структуры молекул: структурные фрагменты, то
биомишени отсутствуют, но имеется достаточно
пологические дескрипторы, квантово химичес
представительная выборка лигандов, связываю
кие дескрипторы, дескрипторы пространствен
щихся с этой биомишенью, то поиск новых ли
ной структуры и дескрипторы межмолекуляр
гандов может быть выполнен непрямыми мето
ных взаимодействий.
дами. В основе этих методов лежит представле
Особо следует выделить уникальную отече
ние о существовании объективной зависимости
ственную компьютерную систему PASS (пред
между структурой химического соединения и его
сказание спектра биологической активности хи
биологической активностью. Эта зависимость
мических соединений), разработанную в Науч
может быть установлена анализом молекуляр
но исследовательском институте биомедицинс
ной структуры и биологической активности уже
кой химии им. В.Н. Ореховича РАМН и не име
изученных соединений, образующих обучающую
ющую аналогов по широте охвата разнообраз
выборку, и далее экстраполирована на новые со
ных видов биологической активности [25-27].
единения. Данный подход называется констру
На основе анализа взаимосвязей структура ак
ированием лекарств, основанным на структуре
тивность в обучающей выборке, содержащей
лиганда (LBDD, ligand based drug design). Пос
информацию о структуре и биологической ак
кольку биологическая активность соединения
тивности более 950 000 лекарственных субстан
представляет собой суммарный эффект много
ций и биологически активных соединений, сис
численных процессов, функциональная зависи
тема PASS может прогнозировать для новых не
мость биологической активности от молекуляр
тестированных органических соединений более
ной структуры сложна для описания и не может
7000 видов биологической активности со сред
быть представлена в явной форме. Для аппрок
ней точностью ~95%. При этом совершенно не
симации такой зависимости используют прос
требуется информация о пространственной
тую математическую функцию, обычно поли
конфигурации молекул рассматриваемых сое
ном, коэффициенты которого определяют с по
динений, а используется обычная структурная
мощью различных методов многомерного ста
формула для автоматического расчета ориги
тистического анализа. Процесс нахождения
нальных молекулярных дескрипторов. Важное
функции, связывающей искомую биологичес
место в компьютерном молекулярном дизайне
кую активность, выраженную в различных еди
занимают классификационные модели, исполь
ницах (например, как IC50, т.е. концентрация
зуемые при дискретном выражении биологичес
соединения, вызывающая 50% ное ингибирова
кого действия в рамках одного вида биологичес
ние активности), с молекулярной структурой,
кой активности или при дифференциации раз
включает выбор способов представления моле
личных видов биологической активности [23, 24].
БИОХИМИЯ том 84 вып. 2 2019
4*
196
КАБАНКИН и др.
В этом случае исходная обучающая выборка
ственной молекулы в организме, важно оценить
состоит из групп (классов) химических соедине
влияние самого биологического организма на
ний, взаимно исключающих друг друга. Группи
лекарственную молекулу, характеризуемое про
рование соединений осуществляется на основе
цессами абсорбции, распределения, метаболиз
качественных признаков, например, активные и
ма и экскреции. Набор параметров, описываю
неактивные соединения или высокоактивные,
щих эти процессы, в зарубежной научной лите
умеренно активные, малоактивные и неактив
ратуре обозначается как ADME свойства
ные соединения. Другие примеры - канцерогены
(ADME, absorption, distribution, metabolism, excretion).
и неканцерогены, или высокотоксичные, мало
Впоследствии сюда добавили и токсичность
токсичные и нетоксичные соединения и т.п.
(toxicity), так что соответствующая аббревиатура
Классификационные алгоритмы на основе ана
превратилась в ADMET (иначе ADME/Tox). От
лиза обучающей выборки позволяют опреде
мечалось, что отклонение не менее 50% новых
лить эффективные решающие правила для
химических структур на предклинических ис
классификации новых соединений в ту или
пытаниях, было связано с неподходящими
иную из заданных групп. Поскольку при клас
ADMET свойствами. Низкая растворимость в
сификации не нужны точные эксперименталь
воде может лимитировать степень абсорбции и
ные значения биологической активности, эти
непосредственно вызывать токсичность, если со
методы во многих случаях более удобны по
единение осаждается в почках. Лекарства долж
сравнению с регрессионной техникой. Арсенал
ны обладать достаточной липофильностью, что
используемых в настоящее время классифика
бы проникать в мембраны и далее к месту
ционных методов достаточно широк: «наив
действия. Избыточная липофильность означает
ный» Байесовский классификатор, линейный
низкую растворимость и сильное связывание с
дискриминантный анализ, классификация по
белками, накопление в тканях и клетках, увели
ближайшему соседу, искусственные нейронные
чивающее риск нежелательных токсических
сети, классификационное и регрессионное де
воздействий. Предсказание этих свойств хими
рево, метод опорных векторов (SVM, support vector
ческих соединений на как можно более ранней
machines), дерево решений (DTs, decision trees),
стадии проекта может существенно снизить фи
случайный лес (RF, random forest) и др. [23, 24].
нансовые затраты и время на разработку новых
Как следствие использования бинарной класси
лекарственных средств, поэтому уже с середины
фикации появилось эмпирическое «правило пяти»
1990 х гг. начались исследования, имеющие
Липински, позволяющее отличать лекарства от
целью построение вычислительных моделей на
«нелекарств» [28]: лекарственная молекула долж
основе физико химических и структурных ха
на удовлетворять определенным условиям: мо
рактеристик молекул для предсказания раство
лекулярный вес - не больше 500 дальтон, значе
римости в воде, липофильности, абсорбции, ме
ние логарифма коэффициента распределения
таболизма, проникновения через гемато энце
вещества в системе октан - вода (logP) - не
фалический барьер и других характеристик, оп
больше 5, число доноров водородной связи в
ределяющих транспорт лекарственных молекул
молекуле - не больше 5, число акцепторов водо
к месту действия, а также их токсичность
родной связи - не больше 10, число вращаемых
[29-31]. Итоги и перспективы этих исследова
одинарных связей, не входящих в циклы, - не
ний рассмотрены в обзорах [32, 33].
более 10, площадь полярной поверхности моле
Заканчивая этот раздел, следует подчерк
кулы - не более 140 Å2. В настоящее время это
нуть, что ни один из рассмотренных методов
(и ему подобные правила) широко используют
виртуального скрининга (SBDD и LBDD) не яв
ся в качестве фильтров для исключения ненуж
ляется исключительным и обладающим безус
ных молекул из исследуемых баз химических со
ловным преимуществом перед другим. У каждо
единений.
го из этих подходов есть свои достоинства и не
Выявление методами виртуального скри
достатки. В современных условиях наиболее ра
нинга химических соединений, обладающих
циональной представляется стратегия много
высоким сродством к некоторой мишени, т.е.
критериальной оптимизации, когда одновремен
желательной активностью и селективностью,
но с оптимизацией тем или иным способом
само по себе недостаточно для получения высо
сродства к биомишени, оптимизируются и
кокачественных кандидатов для предклиничес
ADMET свойства.
ких испытаний. Необходимо также, чтобы отоб
Методы компьютерного дизайна с успехом
ранные соединения обладали определенным
применяются и для поиска ингибиторов факто
комплексом физико химических и фармакоки
ров системы свертывания крови. Центральное
нетических свойств, обеспечивающих их био
место в каскаде коагуляционных реакций зани
доступность. Чтобы определить поведение лекар
мают фактор Xa и тромбин, поэтому поиску ин
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
197
гибиторов для этих мишеней всегда уделялось и
функциональную группу для ингибирования
продолжает уделяться большое внимание. Еще
тромбина. Встраивание в группу Р1 гетероцикл
в1990 е гг. был опубликован ряд работ, исполь
замещенного хлорфенильного фрагмента дает
зующих компьютерный дизайн для выявления
в результате активное соединение 1 (табл. 1) с
ингибиторов тромбина [34]. В настоящем обзо
IC50 = 4 нМ. Область Р2 в трипептиде важна и
ре рассмотрены работы, выполненные в послед
для ингибирования тромбина, и для проявления
ние годы, причем речь пойдет только о низко
оральной биодоступности. Внедрение 3 амино
молекулярных ингибиторах.
пиразинона в область Р2 приводит к соедине
нию 2 (табл. 1), ингибирующему тромбин с
константой ингибирования (Ki) равной 5.2 нМ
НОВЫЕ ИНГИБИТОРЫ ТРОМБИНА
путем образования водородной связи 3 амино
пиразинона с Gly216 тромбина. В обзоре [35]
Тромбин относится к семейству сериновых
(в основном ) речь идет о пептидных ингибито
протеаз. Активный центр тромбина имеет три
рах тромбина, выделенных из кровососущих на
кармана: S1, S2 и S3. На дне глубокого и узкого
секомых, клещей, пиявок, а также из яда змей,
кармана S1 расположен отрицательно заряжен
ос и секрета кожи жаб.
ный остаток аспарагиновой кислоты (Asp189).
В обзоре He et al. суммированы сведения о
Карман S2 содержит остатки пролина и глицина,
соединениях, разрабатываемых с 2010 г. в качест
а карман S3 - остатки Leu 99, Ile 174 и Trp 215.
ве орально активных ингибиторов тромбина [36].
Карман S3 - плоский и доступен растворителю.
Эти соединения получены различными синте
Молекулярные фрагменты ингибитора (называ
тическими путями, а также химической моди
емые иногда мотивами по аналогии с белками),
фикацией ингибирующих тромбин соединений,
локализующиеся при связывании в указанных
выделенных из природных флавоноидов. В табл. 1
карманах, обозначают как Р1, Р2 и Р3 соответ
приведены структурные формулы и активности
ственно. В структуре тромбина, кроме активно
наиболее активных по отношению к тромбину,
го сайта, имеются также два анион связываю
соединений (3-7), взятые из указанного обзора.
щих центра, один из которых (экзосайт 1), рас
Селективность соединений 4, 5 к тромбину
положенный вблизи каталитического участка
была показана только относительно трипсина,
активного центра, ответственен за связывание
соединения 6 - относительно факторов сверты
фибриногена и отвечает за высокую протеоли
вания VIIa, IXa, Xa и XIIa. Соединение 3 (шифр
тическую специфичность тромбина. Со вторым
RWJ 671 818), проявившее себя как орально ак
участком (экзосайт 2), находящимся на проти
тивный и селективный относительно других
воположной стороне белковой глобулы, связы
факторов свертывания ингибитор тромбина
ваются гепарин и другие полисахариды, а также
(Ki = 1,3 нМ), передано на клинические испы
γ’ цепи фибриногена.
тания.
В обзоре Kong et al., опубликованном в 2014 г.,
В этой же работе рассмотрены аллостеричес
приведена патентная информация о прямых ин
кие ингибиторы тромбина. Известно, что при
гибиторах тромбина (ПИТ), предложенных с
соединение лигандов к экзосайтам тромбина 1
2002 по 2012 г. [35]. Отмечается, что среди низ
или 2 может индуцировать конформационные
комолекулярных ингибиторов тромбина наибо
изменения в его активном центре. Примером
лее активны соединения, содержащие аналоги
является тромбомодулин, который, взаимодей
трипептидной группировки D Phe Pro Arg (P3
ствуя с экзосайтом 1, смещает субстратную спе
P2 P1). Эти соединения могут связываться с
цифичность тромбина с фибриногена на проте
различными аминокислотами в активном цент
ин С. Поскольку существующие ингибиторы
ре тромбина. Типичные ПИТ содержат в облас
тромбина связаны с определенным риском кро
ти Р1 высокоосновные функциональные груп
вотечений, аллостерическое ингибирование
пы: гуанидин (в аргатробане), бензамид (в даби
тромбина через экзосайты 1 или 2 может быть
гатране), алкиламин или
4 аминопиридин.
альтернативой его прямому конкурентному ин
Вместе с тем эти соединения обладают низкой
гибированию, создавая возможность контроля
оральной доступностью, так как основные ами
путем неполного ингибирования экзосайта. Бы
ны сильно протонируются при рН желудочного
ли синтезированы и исследованы в качестве ал
тракта. Стратегия преодоления этого недостатка
лостерических ингибиторов тромбина, связыва
заключается в использовании пролекарств со
ющихся с экзосайтом 2, несколько десятков про
слабоосновными или неосновными группами в
изводных моносульфатированного бензофурана
области Р1, таких как дабигатран этексилат.
и его ди , три и тетрамерных гомологов. Самым
Позднее было показано, что фрагмент Р1 не
активным оказался тример с IC50 = 0,67 мкМ.
обязательно должен содержать высокоосновную
Но ни один из исследованных аллостерических
БИОХИМИЯ том 84 вып. 2 2019
198
КАБАНКИН и др.
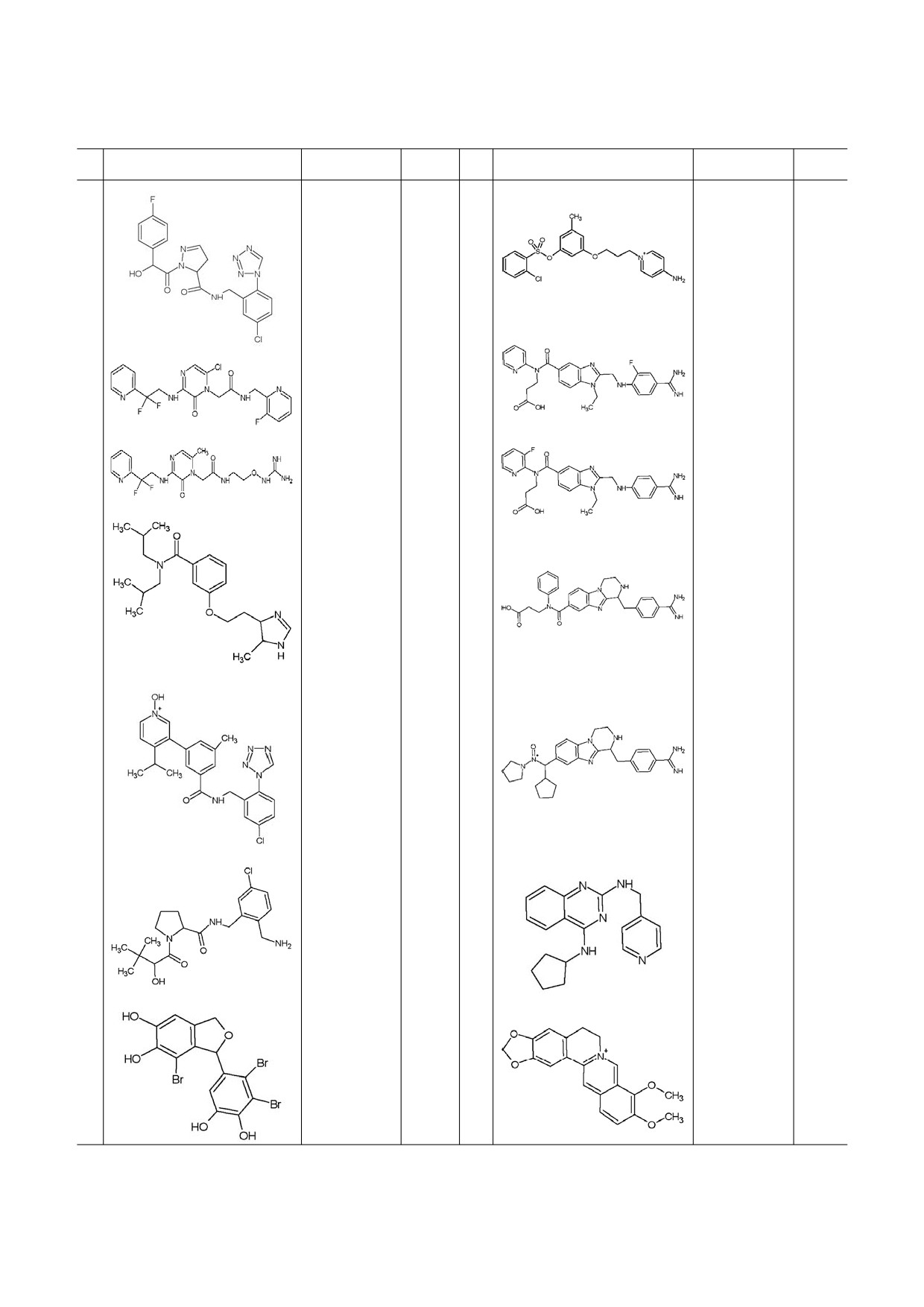
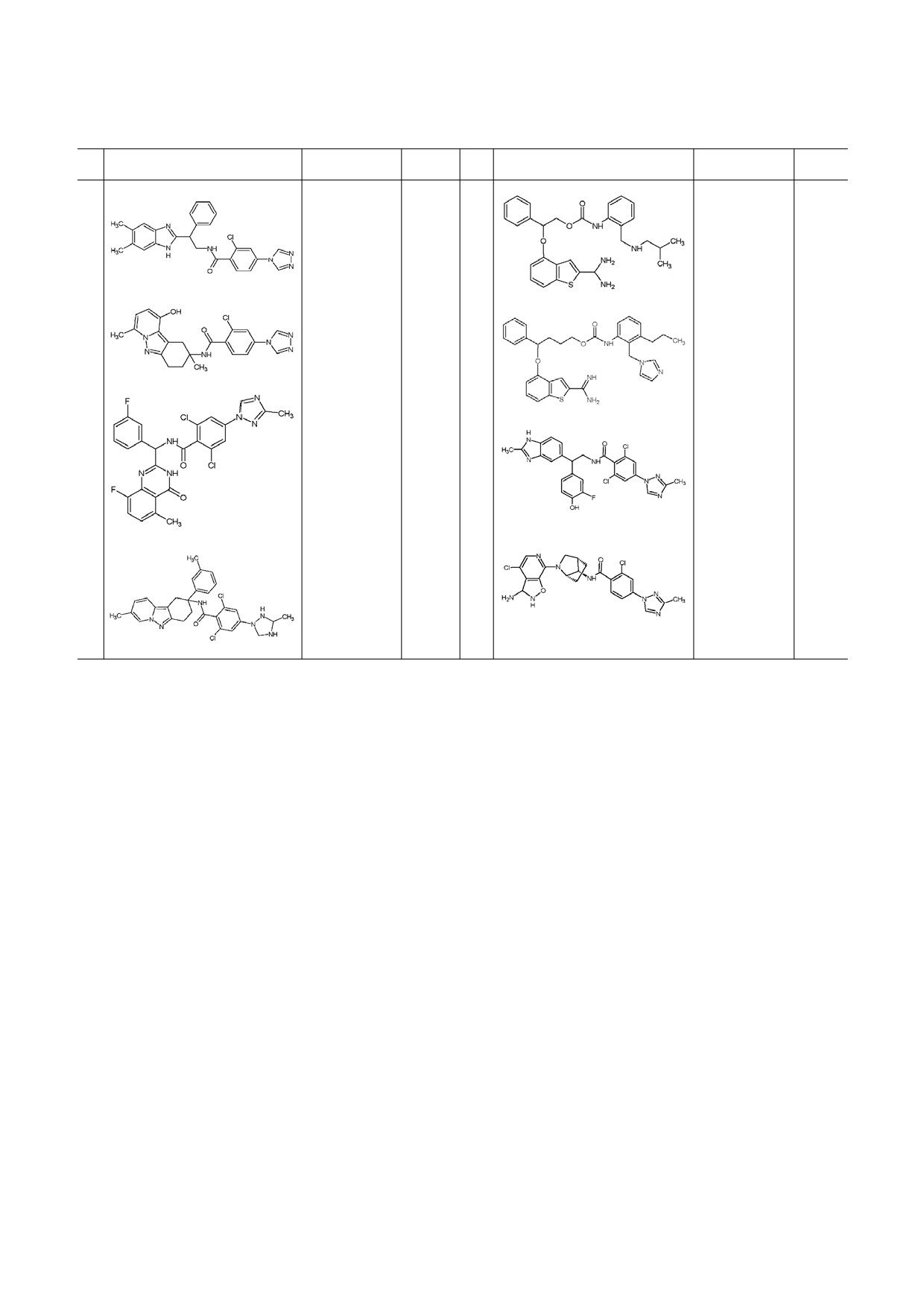
Таблица 1. Новые ингибиторы тромбина
№
Структурная формула
Активность
Ссылка
№
Структурная формула
Активость
Ссылка
1
IC50 = 4 nM
[35]
8
Ki = 0,21 nM
[39]
2
Ki = 5,2 nM
[35]
9
IC50 = 3,39 nM
[41]
3
Ki = 1,3 nM
[36]
10
IC50 = 3,52 nM
[42]
4
Ki = 3,5 nM
[36]
11
IC50 = 7,48 nM
[43]
5
Ki = 0,77 nM
[36]
12
IC50 = 82,8 nM
[44]
6
Ki = 1,5 nM
[36]
13
IC50 = 0,019 μM
[45]
7
IC50 = 1,03 nM
[36]
14
IC50 = 2,92 μM
[46]
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
199
ингибиторов не проявил оральную активность
ингибиторов в буферной системе составил от
[36].
0,21 до 5,7 нМ, а IC50 в плазме - от 0,1 до 1,24 мкМ.
К настоящему времени выявлено много хи
Наиболее эффективное соединение 8 представ
мических соединений, способных ингибировать
лено в табл. 1.
тромбин. Молекулы многих соединений имеют
В последние годы отмечается тенденция вво
сходный или достаточно близкий основной мо
да атомов фтора и фторсодержащих групп в ле
лекулярный каркас (скаффолд - scaffold) и отли
карственные молекулы [40]. Такая модифика
чаются разнообразными заместителями, причем
ция молекул изменяет их фармакокинетические
иногда замена одного или нескольких замести
свойства, приводя во многих случаях к увеличе
телей, не меняя существенно общую структуру
нию биодоступности из за увеличения прони
молекулы, приводит к резкому изменению ее
цаемости клеточных мембран для таких соеди
способности ингибировать тромбин. Таким об
нений. Фторирование может блокировать мета
разом, определение структурных молекулярных
болические пути лекарств на различных сайтах
характеристик, ответственных за проявление
связывания, изменять скорость метаболизма и
биологической активности, имеет большое зна
улучшать метаболическую стабильность.
чение для молекулярного дизайна активных со
В работе Li et al. была синтезирована серия
единений.
новых фторированных 2,5 замещенных произ
В работе Mena Ulecia et al. исследована диф
водных 1 этил 1Н бензимидазола [41]. Была
ференциальная активность 177 непептидных
экспериментально определена способность этих
ингибиторов тромбина. Молекулярные структу
соединений ингибировать тромбин in vitro.
ры, представленные восемью скаффолдами, и
Часть новых соединений оказалась более актив
активности рассмотренных соединений были
ной по сравнению с аргатробаном. Для наибо
взяты из работ, опубликованных ранее. Сначала
лее активного соединения 9 (табл. 1), молеку
была подтверждена способность используемой
лярная структура которого отличается от струк
программы молекулярного докинга Glide пра
туры дабигатрана лишь наличием атома F в бен
вильно воспроизводить кристаллические струк
зольном кольце, значение IC50 равно 3,39 нМ,
туры для пяти известных комплексов тром
что сопоставимо с активностью дабигатрана
бин-лиганд с кодами 1T4U, 1T4V, 3C27, 2R2M и
(IC50 = 2,61 нМ). В большинстве активных сое
3LDX из банка PDB. Затем с помощью програм
динений место пиридинового кольца занимает
мы Glide были определены предпочтительные
бензольное. В любом случае введение замести
конформации и энергетически наиболее выгод
телей в это кольцо сильно влияет на антикоагу
ная ориентация каждого из оставшихся 172 ин
лянтную активность. Показано, что электроно
гибиторов в активном центре тромбина. Далее,
донорные заместители снижают, а электроноак
используя оптимальные конформации лиган
цепторные немного повышают антикоагулянт
дов, определенные на стадии молекулярного до
ную активность. Положение заместителя в коль
кинга, с помощью метода CoMSIA (сравнитель
це также влияет на активность, которая умень
ный анализ индексов молекулярного сходства),
шается для разных положений заместителя в
реализованного в коммерческом пакете Sybyl 7.3,
следующем порядке: мета → орто → пара .
была построена предсказательная модель КССА.
Chen et al. синтезировали ряд структурных
Для этого исходный набор соединений был раз
аналогов дабигатрана, в которых пиридиновое
делен случайным образом на две группы: обуча
кольцо заменено бензольным и в разные поло
ющую выборку (134 соединения) и тестовую вы
жения этого кольца введены атомы фтора. Оп
борку (34 соединения). На основе этой модели
ределена антикоагулянтная активность синте
был выполнен прогноз активности тестовых со
зированных соединений in vitro. Наиболее ак
единений, результат которого представлен в ви
тивное соединение 10 (табл. 1) с IC50 = 3,52 нМ
де графика зависимости между эксперименталь
содержит атом F в орто положении бензольно
ными и вычисленными значениями активности
го кольца. Атом F в мета положении уменьша
тестовых соединений статей [37].
ет ингибиторную активность примерно в два ра
Sinauridze et al., взяв за основу известный ра
за, а в пара положении уменьшает незначитель
нее скаффолд орцина [38], и подбирая с по
но. Введение группы CF3 в бензольное кольцо
мощью программы докинга SOL фрагмент Р1,
уменьшает ингибиторную активность: в орто
связывающийся с карманом S1, обнаружили
положении - в два раза, в мета положении - в
группу новых высокоэффективных ингибито
7 раз, в пара положении - почти в 60 раз [42].
ров тромбина. В качестве заряженного фрагмен
В работе Chen et al. проведено исследование
та Р1 молекулы этих веществ содержали изотиу
антитромбиновой активности соединений,
рониевый, 4 аминопиридиниевый или 2 ами
имитирующих дабигатран и отличающихся от
нотиазолиниевый остатки [39]. Диапазон Кi этих
него заменой бензамидиновой части молекулы
БИОХИМИЯ том 84 вып. 2 2019
200
КАБАНКИН и др.
на трициклический скаффолд. Значение IC50
цо и гидрофобную группу. В результате скри
для наиболее активного соединения 11 было
нинга с помощью этой модели фармакофора ба
равно 7,48 нМ. Затем на этой основе были син
зы данных TCMD 2009 (23 023 соединений) бы
тезированы и исследованы 22 производных, не
ли выделены 93 хита, которые были исследова
которые из которых проявили достаточно силь
ны методом молекулярного докинга (программа
ное ингибирующее действие на индуцирован
Surflex Dock) с использованием кристалличес
ную тромбином агрегацию тромбоцитов [43].
кой структуры тромбина. Для эксперименталь
Ингибиторное действие на тромбин десяти
ной оценки способности ингибировать тромбин
cинтезированных производных 4 (1,2,3,4 тетра
были отобраны и синтезированы 23 соедине
гидробензо[4,5]имидазо[1,2 а]пиразин 1 ил)
ния, наиболее удачным из которых был признан
метил)бензимидамида было оценено в работе [44].
берберин (berberine) (14) с IC50 = 2,92 мкМ [46].
Наиболее активное соединение 12 с IC50 = 82,8 нМ
Из представленных выше данных можно
было выбрано в качестве базовой структуры (ли
заключить, что, если отдельно рассматривать
дера), на основании которой был получен ряд
группы соединений из различных химических
производных, большинство из которых показа
классов, проявившие себя как ингибиторы тром
ли ингибиторный эффект в отношении индуци
бина, то внутри каждого такого класса возмож
рованной тромбином агрегации тромбоцитов.
ны определенные заключения о влиянии хими
Максимальное значение IC50 в этом случае ока
ческой природы и положения различных моле
залось равным 1,95 мкМ.
кулярных групп на проявляемую активность,
В работе Lee et al. с помощью высокопроиз
что позволяет оптимизировать поиск наиболее
водительного виртуального скрининга базы сое
активных соединений. Однако одни и те же мо
динений ZINC15 (программа докинга DOCK3.6)
лекулярные группы в различном молекулярном
с использованием структуры тромбина (PDB
окружении будут оказывать различное влияние
ID:2CF9 H), найдено новое соединение
13
на свойства и активность. Таким образом, зара
(шифр JJ1), оказавшееся активным и высокосе
нее сконструировать новую структуру активного
лективным ингибитором тромбина in vitro и in
ингибитора, основываясь на частных заключе
vivo. Соединение JJ1 представляет собой новый
ниях о влиянии различных химических групп в
скаффолд, отличающийся от ранее известных
конкретном окружении на активность, не пред
ингибиторов тромбина. Проведено сравнение
ставляется возможным. Поэтому надо либо ис
молекулярной структуры JJ1 с молекулярными
пользовать найденные ранее частные законо
структурами 2038 непептидных ингибиторов тром
мерности связи структура-активность и искать
бина, содержащихся в базе Binding DB. Средний
новые более активные соединения в этом же хи
коэффициент молекулярного сходства соедине
мическом классе, либо проводить поиск новых
ния 13 с этими ингибиторами, оцененный с по
базовых структур в имеющихся базах соедине
мощью известного коэффициента Танимото,
ний методами высокопроизводительного вирту
составил 0,12 ± 0,03, что свидетельствует о вы
ального скрининга с последующей структурной
сокой степени новизны структуры JJ1 [45].
модификацией найденных хитов для выявления
Поиск новых ингибиторов тромбина прово
и оптимизации наиболее активных соединений.
дили и среди соединений, выделенных из при
родных источников, например, из трав, исполь
зуемых в традиционной китайской медицине
НОВЫЕ ИНГИБИТОРЫ
(ТКМ). Wang et al. использовали модель фарма
ФАКТОРА Xa
кофора (совокупности стереоэлектронных ха
рактеристик, необходимых для оптимального
Фактор Ха - это белок из семейства серино
взаимодействия лиганда со специфической ми
вых протеаз, состоящий из легкой (139 а.о.) и
шенью) и молекулярный докинг для виртуаль
тяжелой (303 a.о.) цепей, связанных дисульфид
ного скрининга библиотеки, содержащей 23 023
ным мостиком. Место связывания лиганда с фер
соединений из ТКМ. С помощью модуля гене
ментом располагается в тяжелой цепи и включа
рации фармакофора на основании 6 до опреде
ет 4 кармана: S1, S2, S3 и S4, с которыми связы
ленной степени разнообразных соединений с IC50
ваются Р1, Р2, Р3 и Р4 фрагменты ингибитора
от 0,1 нМ до 73 нМ были сформированы 10 мо
соответственно. Поскольку фактор Ха предше
делей. Для их валидации использовали выборку,
ствует тромбину в коагуляционном каскаде, его
составленную из 100 известных ингибиторов
ингибиторы могут быть более эффективны в ос
тромбина и 256 неактивных соединений. Модель
лаблении коагуляции, чем ингибиторы самого
фармакофора, которая лучше всех идентифици
тромбина. В настоящее время для использова
ровала активные соединения (92%), включала
ния в клинической практике одобрены 4 инги
акцептор водородной связи, ароматическое коль
битора фактора Ха [6-10, 47]: ривароксабан
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
201
(rivaroxaban), апиксабан (apixaban), эдоксабан
вое или тиофеновое кольцо. Замена пиридоно
(edoxaban) и бетриксабан (betrixaban). Однако у
вого кольца в группе Р4 на лактамное кольцо
этих препаратов, как уже сказано во введении,
также снижала антикоагулянтную активность.
отмечены разные побочные эффекты и проти
Положение карбоксамидной группы в цент
вопоказания, что заставляет искать новые эффек
ральном кольце соединения 16 сильно влияет на
тивные и безопасные ингибиторы фактора Ха.
активность. Наиболее выгодно расположение
В объемном обзоре Patel et al. подведены
этой группы в позиции 5 (см. табл. 2), когда она
итоги выполненных в основном до 2015 г. иссле
образует водородные связи с аминокислотными
дований по разработке новых ингибиторов фак
остатками Glu146, Gly216 и Gly218. Замена кар
тора Ха. Представлены структурные формулы
боксамидной группы на сложноэфирную или
химических групп, используемых в качестве
карбоксикислотную группу уменьшает антикоа
центрального скаффолда (11 групп) и мотивов
гулянтную активность.
Р1 (25 групп) и Р4 (25 групп). Приведены также
Аналогично анализ связи структура - биоло
данные о кристаллических структурах комплек
гическая активность для синтезированных про
сов Ха с 33 различными ингибиторами, разме
изводных антраниламида привел к открытию
щенные в банке PDB с 2010 г. [48]. В настоящем
активного орального ингибитора фактора Ха (17)
обзоре рассмотрены работы по ингибиторам
с IC50 = 8,7 нМ [51]. Проанализировано влияние
фактора Ха, опубликованные, начиная с 2015 г.
химической природы и положения заместите
Виртуальный скрининг двух молекулярных
лей в группе Р1 (табл. 2) на способность ингиби
библиотек (базы данных NCI Diversity (США),
ровать фактор Ха. Показано, что 4 замещенные
содержащей 1888 соединений, и базы данных хи
производные имеют более высокую ингибирую
мического факультета Воронежского универси
щую активность по сравнению с 3 замещенны
тета, включающей 14 271 соединений) был про
ми производными. Соединения, содержащие
веден с использованием оригинальных прог
малые липофильные заместители в позиции 4
рамм молекулярного докинга SOL и постдокин
(хлор, метильная или метоксильная группы),
га DISCORE в работе [49]. Структура Ха была
были высокоактивны, тогда как ввод объемных
взята из комплекса PDB ID:3IIT. Большая часть
заместителей (этильная или изопропильная
(12 из 17) отобранных по результатам скрининга
группы) в эту позицию резко уменьшал актив
соединений показала в опытах in vitro ингибиру
ность. Замена 2 аминопиридина на другие цик
ющее действие в отношении фактора Ха с при
лические системы (3 аминопиридины, 5 член
мерными константами ингибирования на мик
ные гетероциклы, бициклические группы) при
ромолярном уровне. На основе полученных ре
водила к существенному уменьшению ингиби
зультатов было синтезировано новое соедине
торной активности.
ние 15 (табл. 2), которое при тестировании in vitro
Используя опубликованные эксперимен
ингибировало фактор Ха с IC50 равным 0,7 мкМ.
тальные данные по активности трициклических
Обсуждена физическая природа взаимодей
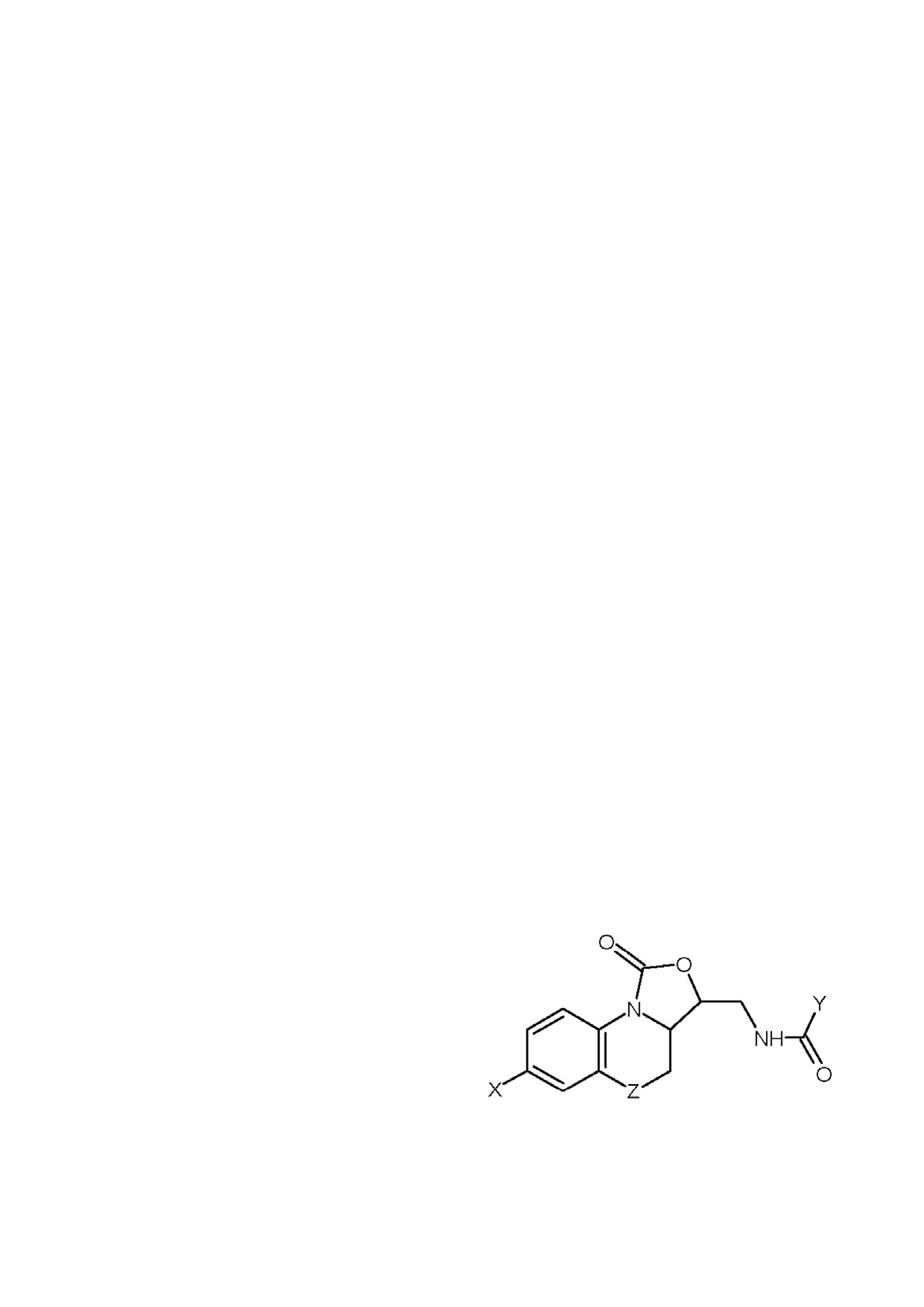
оксазолидинонов с приведенной на рис. 2 об
ствий молекул рассматриваемых соединений с
щей формулой, проявивших ингибирующее
карманами S1, S2, S3 и S4 связывающего сайта
действие в отношении фактора Ха, C. Xu и др. с
фактора Ха.
помощью комбинации различных методов мо
Yang et al., исходя из результатов предыду
лекулярного моделирования (CoMFA, CoMSIA,
щих исследований, испытали in vitro в качестве
молекулярный докинг, молекулярная динамика)
ингибиторов фактора Ха новую серию синтези
построили и исследовали трехмерную модель
рованных ими соединений, являющихся произ
количественной связи структура - активность
водными 3,4 диаминобензоила [50]. Большин
(КССА - 3D QSAR) [52]. Исходный набор дан
ство этих соединений проявили хорошую и от
личную ингибирующую активность. Был вы
полнен качественный анализ связи структу
ра-активность, прояснивший влияние хими
ческого состава групп Р1 и Р4, связывающихся,
соответственно, с карманами S1 и S4, на актив
ность, что привело к открытию нового активного
высокоселективного прямого ингибитора Ха 16
(табл. 2) с хорошей активностью in vivo (крысы),
сравнимой с активностью ривароксабана. Ак
тивность соединений возрастала с увеличением
объема группы Р1 и уменьшалась при замене
Рис. 2. Общая формула ингибиторов фактора Xa класса
бензольного кольца в этой группе на пиридино
трициклических оксазолидинонов
БИОХИМИЯ том 84 вып. 2 2019
202
КАБАНКИН и др.
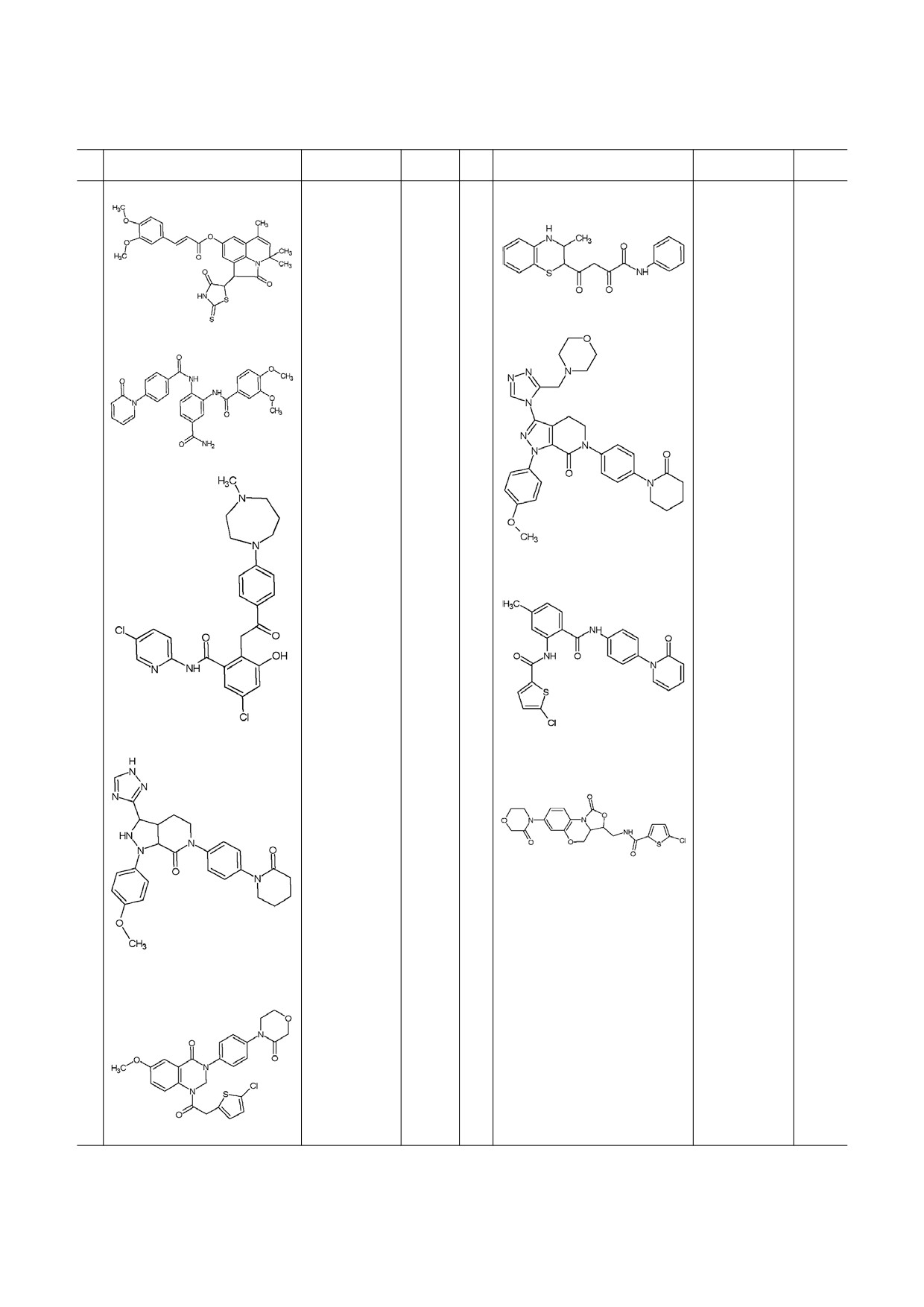
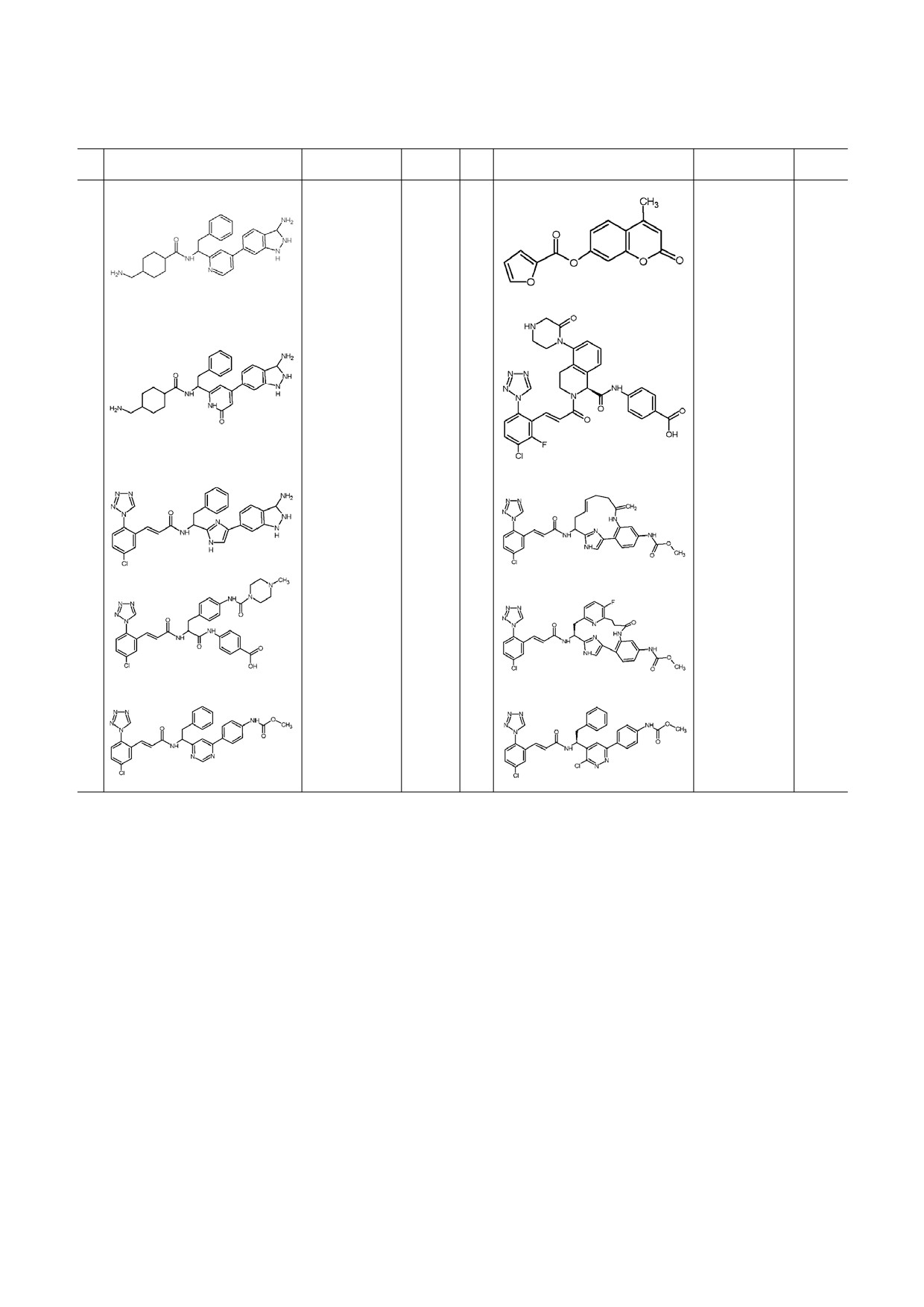
Таблица 2. Новые ингибиторы фактора Xa
№
Структурная формула
Активность
Ссылка
№
Структурная формула
Активность
Ссылка
15
IC50 = 0,7 μM
[49]
20
IC50 = 2,02 nM
[56]
16
IC50 = 17,1 nM
[50]
21
IC50 = 0,14 μM
[57]
17
IC50 = 8,7 nM
[51]
22
IC50 = 25 nM
[58]
18
IC50 = 0,15 μM
[53]
23
IC50 = 3,33 nM
[59]
19
IC50 = 21 nM
[54]
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
203
ных (38 соединений) разделили на две группы:
макофор комплементарен активному центру спе
обучающую выборку (31 соединение), на кото
цифического рецептора и представляет собой
рой строили модель, и тестовую (7 соединений).
умозрительную стереохимическую конструкцию,
Модель, построенная методом CoMFA (сравни
необходимую для оптимального взаимодей
тельный анализ молекулярных полей), имеет сле
ствия с рецептором и свойственную активным
дующие статистические характеристики: R2 = 0,984,
молекулам. Для генерации модели фармакофо
Q2 = 0,511. Модель CoMSIA (сравнительный
ра использовали фармакофорный модуль из ком
анализ индексов молекулярного сходства) дает
мерческого пакета Discovery Studio Version 4.0
R2 = 0,993, Q2 = 0,700. Здесь R2 - коэффициент
(DS 4.0). Из различных публикаций был собран
корреляции для обучающей выборки, Q2 - ко
набор из 24 ингибиторов фактора Ха. Обучаю
эффициент корреляции при скользящем конт
щую выборку составляли 10 соединений, а ос
роле с исключением по одному. Обе модели
тальные 14 рассматривались как тестовые. Было
удовлетворительно воспроизводят эксперимен
построено 10 моделей фармакофора. С по
тальные значения pIC50 ( logIC50) для соедине
мощью тестовых соединений была выбрана на
ний тестовой выборки. Методами молекулярно
илучшая модель, которая и использовалась в
го докинга и молекулярной динамики детально
виртуальном скрининге. В итоге для последую
исследованы наиболее вероятные способы
щего скрининга методом молекулярного докин
структурного связывания лигандов в активном
га (программный пакет GOLD 5.2.2) были отоб
сайте белка (структура белка взята из PDB, код
раны 10 000 соединений. После этой процедуры
2W26).
осталось 200 соединений, из которых, анализи
Замещая в областях связывания молекулы
руя способы связывания лигандов и сродство к
апиксабана с фактором Ха (Р1, Р2 и Р4) группы
активному центру фактора Xa (программы
апиксабана на различные молекулярные группы,
SYBYL, DS 4.0, Pymol), авторы, не вдаваясь в де
Wang et al. синтезировали 4 серии производных в
тали, предложили для дальнейших эксперимен
качестве новых ингибиторов фактора Ха [53].
тальных исследований 10 новых различающихся
Проведена экспериментальная оценка ингиби
базовых структур в качестве новых потенциаль
торной активности новых соединений в отно
ных ингибиторов фактора Ха. Приведены струк
шении фактора Ха in vitro в плазме человека и
турные формулы этих соединений [55].
кролика. Для наиболее активного соединения
В результате виртуального скрининга базы
18 (табл. 2) значение IC50 составляло 0,15 мкМ.
данных NCI Open (260 000 соединений) с ис
Замена метоксильной группы в области Р1 на
пользованием комбинации различных методов,
различные электроноакцепторные группы приво
идентифицированы новые базовые структуры
дила к уменьшению ингибиторной активности в
потенциальных ингибиторов фактора Ха [56].
следующем порядке: OCH3 → F → OCHF2 →
Из 30 соединений, отобранных для экспери
→ Cl → OCF3 → Br. Ввод заместителей в раз
ментального тестирования in vitro, наиболее
личные позиции триазольного кольца в области
активным оказалось соединение 20 (табл. 2) с
Р2 также, как правило, снижал активность.
IC50 = 2,02 нМ.
Синтезирован также ряд производных 2,3 ди
Для выявления новых эффективных ингиби
гидрохиназолин 4(1Н) она [54]. Большинство
торов фактора Ха на основе найденной ранее за
соединений показали в эксперименте in vitro
висимости структура - активность спроектиро
значительную ингибиторную активность в отно
вали и синтезировали серию новых производ
шении фактора Ха. Для наиболее активного со
ных тетрагидропиразолопиридона, содержащих
единения 19 (табл. 2) значение IC50 составляло
в связываемой рецептором области Р2 различ
21 нМ, что свидетельствует о его высокой селек
ные молекулярные группировки (1,3,4 триазол,
тивности, поскольку по отношению к тромбину
триазолилметил и другие) [57]. Все соединения
данный ингибитор имеет значение IC50 = 67 мкМ.
проявили ингибиторную активность в отноше
С целью выявления новых ингибиторов фак
нии фактора Ха in vitro. Для наиболее активного
тора Ха в работе Pu et al. проведен виртуальный
соединения 21 (табл. 2) значение IC50 было рав
скрининг базы данных SPECS, содержащей
но 0,14 мкМ.
~220 000 молекул [55]. Первоначальная фильт
В работе Wang et al. в качестве базовой струк
рация указанной базы была выполнена с помощью
туры был использован антраниламид [58]. Из
известного эмпирического «правила пяти» Ли
спроектированных на его основе и синтезиро
пински, отличающего лекарства от «нелекарств»
ванных производных 3 соединения показали
(см. выше). В результате число соединений,
при испытании in vitro заметное ингибирующее
отобранных для дальнейшей работы, снизилось
действие на фактор Ха и высокую селективность
до 75 671. Далее проводили скрининг этих сое
относительно тромбина. Значение IC50 для наи
динений с помощью модели фармакофора. Фар
более активного соединения 22 (табл. 2) было
БИОХИМИЯ том 84 вып. 2 2019
204
КАБАНКИН и др.
равно 25 нМ. Методом молекулярного докинга
водительного скрининга и продемонстрировав
(программа Glide) установлено, что связывание
ших микромолярную активность в отношении
этого соединения с активным центром Ха осу
фактора IХа, были синтезированы и исследова
ществляется по тому же механизму, что и связы
ны различные производные этих соединений,
вание ривароксабана. Соединение 22 может
относящиеся к замещенным бензимидазолам
быть лидером для последующей модификации
(класс А) и замещенным пиразолопиридинам
молекулы с целью усиления антикоагулянтной
(класс В) [64]. Тестирована способность этих со
активности.
единений ингибировать фактор IХа, а также
Исходя из структуры нескольких активных
фактор Ха для сравнения. Структуры наиболее
ингибиторов фактора Ха и структуры белка Ха,
активных веществ для каждого из этих классов
Hu et al. спроектировали и синтезировали более
соединений приведены в табл. 3 (соединения 24
100 новых соединений [59]. В результате скри
и 25). Ингибирующая активность соединений
нинга различными методами было отобрано со
24 и 25 по отношению к фактору IXa была выше,
единение 23 (табл. 2) с шифром DJT06001, анти
чем по отношению к фактору Ха в 24 и 120 раз,
коагулянтное действие которого было детально
соответственно. Проведен анализ связи струк
исследовано in vitro и in vivo. Соединение оказа
тура-активность, необходимый для дальней
лось эффективно в предотвращении образова
ших исследований при поиске ингибиторов
ния тромбов в результате прямого и специфи
фактора IХа. Удаление обеих метильных групп в
ческого ингибирования фактора Ха. Риск кро
бензимидазольном кольце соединения 24 сни
вотечений при его использовании был сходный
жало активность. Перенос триазольного кольца
или даже более низкий, чем для ривароксабана.
из пара позиции относительно амидной группы
Соединение 23 имело более высокую раствори
в орто или мета позицию в обоих соединени
мость и биодоступность чем ривароксабан. Зна
ях уменьшал активность. Исследовано влияние
чение IC50 для ингибирования свободного Ха в
длины алкильной цепочки в центральной части
плазме оказалось равным 3,33 нМ, а для инги
молекулы 24 и различных замещений в этой об
бирования Ха, связанного с протромбиназой, -
ласти на активность. Ввод заместителя в пози
2,53 нМ.
цию, смежную с бензамидом, уменьшал актив
ность, тогда как ввод заместителя в позицию,
смежную с бензимидазольным кольцом, ее уве
НОВЫЕ ИНГИБИТОРЫ
личивал. Замена фенильного кольца в этой час
ФАКТОРА IXa
ти на метильную группу снижала активность.
Урезание цепочки до одного атома С или увели
Как уже было сказано выше, при внешнем
чение ее до трех атомов С также снижало актив
повреждении сосуда образовавшийся комплекс
ность. Соединение 24 является R энантиомером,
«внешняя теназа» (TФ-VIIa) активирует факто
который в 30 раз более активен, чем S энантио
ры IХ и Х. Фактор IХа катализирует образова
мер.
ние Ха из Х, однако почти в 50 раз более эффек
Используя соединение 24 как базовую струк
тивная активация фактора Х осуществляется
туру, Zhang et al. синтезировали серию аналого
комплексом «внутренняя теназа» (IXa-VIIIa).
вых производных в качестве потенциальных
Фактор Ха нестабилен в плазме и быстро инги
кандидатов для ингибирования фактора IХа [65].
бируется антитромбином и ИПТФ. Напротив,
Более половины синтезированных соединений
фактор IХа в плазме относительно стабилен и
показали при тестировании in vitro очень высо
диффундирует от несущих тканевый фактор
кую селективность при ингибировании фактора
клеток к активированным тромбоцитам, поэто
IХа относительно ингибирования фактора Ха.
му фактор IХа рассматривается как очень перс
Наиболее активное соединение 26 (табл. 3) с
пективная мишень для разработки новых эф
Ki = 1,0 нМ было почти в 30 000 раз более селек
фективных антикоагулянтов [60].
тивно к IХа, чем к Ха.
К настоящему времени опубликовано доста
В другой работе синтезировали ~40 трицик
точно большое количество статей, в которых в
лических соединений, являющихся структурны
качестве ингибиторов фактора IХа предлагались
ми аналогами соединения 25 (табл. 3) [66]. Не
разные структуры: моноклональные антитела,
которые из них показали значительное ингиби
аптамеры ДНК, а также синтетические низко
рующее действие на фактор IХа и были при этом
молекулярные соединения (например, полиза
высоко селективными (по сравнению с действи
мещенные бензотиофены) [61-63]. Мы рас
ем на фактор Ха). Приведенная в табл. 3 струк
смотрим лишь работы, опубликованные с 2015 г.
турная формула 27 соответствует не самому ак
На основе молекулярной структуры двух хи
тивному, но одному из наиболее селективных
тов, найденных ранее с помощью высокопроиз
соединений полученного ряда (Ki = 1,8 нМ, ин
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
205
Таблица 3. Новые ингибиторы фактора IXa
№
Структурная формула
Активность
Ссылка
№
Структурная формула
Активность
Ссылка
24
Ki = 0,016 μM
[64]
28
pIC50 = 8,70
[67]
25
Ki = 0,09 μM
[64]
29
pIC50 = 10,50
(CoMFA)
[67]
10,66 CoMSIA)
26
Ki = 1,0 nM
[65]
30
IC50 = 1,86 nM
[68]
27
Ki = 1,8 nM
[66]
31
IC50 = 4,9 nM
[69]
гибирующая способность по отношению к фак
динений (тестовый набор), остальные 72 соеди
тору IXa в 300 с лишним раз больше, чем в отно
нения составили обучающую выборку. При
шении фактора Ха). В работе представлены под
построении КССС характеристикой селектив
робные таблицы, в которых показано влияние
ности (S) служил отрицательный логарифм от
на антикоагулянтную активность химической
ношения значений IC50 для ингибирования
природы заместителей, введенных в различные
факторов IХа и Ха. В этом случае исходный на
положения молекулы ингибитора. Однако сде
бор содержал 73 соединения (обучающая выбор
лать однозначные выводы сложно, так как вли
ка - 64 соединения, тестовая - 9 соединений).
яние каждого заместителя в определенном по
Наиболее активное соединение исходного набо
ложении на активность сильно зависит от хими
ра 28 (табл. 3) имело следующие характеристики:
ческой природы заместителей в других положе
pIC50 (для ингибирования фактора IXa) = 8,70,
ниях.
S = 2,56. Указанными методами для обучающих
На основании опубликованных эксперимен
выборок получены статистически значимые
тальных данных о 84 производных амидинобен
уравнения, количественно связывающие значе
зотиофена, способных ингибировать фактор IХа
ния pIC50 и S с характеристиками соединений.
[62, 63] в работе Gao et al. с помощью методов
Исходя из факта успешной проверки вычислен
CoMFA и CoMSIA были определены трехмер
ных уравнений, Gao et al. спроектировали и
ные количественные связи структура - актив
предсказали активность и селективность для 16
ность (КССА - 3D QSAR) и трехмерные коли
новых производных в рассматриваемом хими
чественные связи структура - селективность
ческом ряду. Для всех этих соединений была
(КССС - 3D QSSR) [67]. При построении
предсказана более высокая способность инги
КССА для ингибирования фактора IХа в качест
бировать фактор IXa и более высокая селектив
ве зависимой переменной использовали значе
ность, чем у наилучшего соединения из исход
ния pIC50 = lgIC50. Из исходного массива (84 со
ного набора. Для наиболее активного из новых
единения) случайным образом выбрали 12 сое
соединений 29 (табл. 3) были предсказаны сле
БИОХИМИЯ том 84 вып. 2 2019
206
КАБАНКИН и др.
дующие характеристики: pIC50 (для ингибирова
нальные антитела; 2) антисмысловые олигонук
ния фактора IXa) = 10,50, S = 4,34 (CoMFA);
леотиды (АСО) - химически модифицированные
pIC50 (для ингибирования фактора IXa) = 10,66,
одноцепочечные последовательности нуклеоти
S = 4,41 (CoMSIA). К сожалению, в работе ни
дов, которые могут комплементарно связывать
чего не сказано об экспериментальной проверке
ся со специфическими участками мРНК, влияя
этих результатов [67].
на экспрессию гена и модулируя синтез соотве
В другой работе после инкубации одного из
тствующего белка; 3) полипептиды; 4) синтети
активных ингибиторов IXa (IC50 = 9,99 нМ) с
ческие низкомолекулярные соединения (ацикли
микросомами печени (человек, крыса) с по
ческие, моноциклические, бициклические, мак
мощью специальной технологии были выделе
роциклические), связывающиеся непосредствен
ны и идентифицированы 3 метаболита, один из
но с активным центром фактора XIa; 5) синтети
которых 30 оказался более активным ингибито
ческие аллостерические ингибиторы; 6) естест
ром IXa (IC50 = 1,86 нМ), чем исходное соедине
венные соединения, выделенные из природных
ние [68].
источников (бромфенильные карбаматы) [70, 71].
Ранее [64, 66] трициклические производные
В настоящем обзоре, как и для факторов, рас
пиразинопиридина были идентифицированы
смотренных выше, речь пойдет только о синте
как ингибиторы фактора IXa. Заменив трицик
тических ингибиторах фактора XIa, предложен
лическую часть молекул на более простые хими
ных в 2015 г. Структуры наиболее эффективных
ческие группы, Sakurada et al. синтезировали се
из этих новых ингибиторов представлены в
рию производных аминобензизоксазола в каче
табл. 4.
стве потенциальных ингибиторов фактора IXa и
Как часть программы по изучению структур
испытали их в эксперименте. Наиболее актив
но разнообразных ингибиторов XIa и на основе
ное соединение 31 (табл. 3) обладало высокой
выделенных ранее соединений Corte et al. син
ингибирующей способностью относительно
тезировали и исследовали ряд новых производ
фактора IXa (IC50 = 4,9 нМ) и высокой селектив
ных, полученных введением разных заместите
ностью по сравнению с ингибированием факто
лей в имидазольное кольцо исходных соедине
ра Ха (более, чем в 6300 раз). Для этого соедине
ний [72]. В табл. 4 приведены структурные
ния характерны умеренная растворимость в во
формулы двух соединений 32 и 33, проявивших
де, хорошая мембранная проницаемость, микро
наибольшую активность при ингибировании
сомальная стабильность, разумный период по
фактора XIa. Соединение 32 является S энанти
лураспада (0,8 ч для крыс, 4,7 ч для собак). Сое
омером, который существенно более активен
динение получило шифр CFM 184 и является кан
(Ki = 8,4 нМ) по сравнению с соответствующим ра
дидатом для предклинических испытаний [69].
цематом. Для соединения 33 значение Ki = 3,7 нМ.
Для соединения 32 была исследована кристал
лическая структура комплекса S энантиомера с
НОВЫЕ ИНГИБИТОРЫ
фактором XIa. Подробно рассмотрено связыва
ФАКТОРА XIa
ние соединения с различными элементами ак
тивного центра этого фактора.
Кроме активации комплексом VIIa TF, фак
Модифицируя мотивы Р1 и Р2 в найденных
тор IX может быть активирован фактором XIa.
ранее соединениях, проявивших себя как актив
Несмотря на то, что при нормальных физиоло
ные ингибиторы фактора XIa, Pinto et al. иссле
гических условиях вклад фактора XIа в нормаль
довали серию новых производных с целью улуч
ный гемостаз минимален, активация этого фак
шения общего профиля селективности и ораль
тора играет важную роль в развитии тромботи
ной биодоступности [73]. Наиболее активным
ческих осложнений. На основании фундамен
(Ki = 6,7 нМ) и высокоселективным оказалось
тальных и эпидемиологических исследований,
соединение 34.
а также клинических наблюдений, сформирова
В работе Smith et al. было исследовано вли
лось мнение, что фактор XIa следует рассматри
яние различных замещений в мотивах Р1 и Р2
вать как главную и наиболее перспективную ми
фенилаланиндиамидов на ингибирование фак
шень для разработки антикоагулянтных ле
тора XIa, что и привело авторов к открытию со
карств со сниженным риском кровотечений [70,
единения 35 с высоким сродством к фактору XIa
71]. Начиная с 2010 г., число патентов и публи
(Ki = 2,0 нМ) [74].
каций, связанных с разработкой ингибиторов
В работе Corte et al. [75], являющейся про
фактора XIa, существенно увеличилось. В обзо
должением исследований, опубликованных в [72],
рах Bane et al. и Al Horani et al. представлены ос
был синтезирован и исследован ряд новых про
новные группы веществ, предлагавшихся в ка
изводных, способных ингибировать фактор XIa.
честве ингибиторов фактора XIa: 1) монокло
Целью исследования было улучшение оральной
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
207
Таблица 4. Новые ингибиторы фактора Xia
№
Структурная формула
Активность
Ссылка
№
Структурная формула
Активность
Ссылка
32
Ki = 8,4 nM
[72]
37
IC50 = 0,77 μM
[76]
33
Ki = 3,7 nM
[72]
38
Ki = 0,7 nM
[77]
34
Ki = 6,7 nM
[73]
39
Ki = 0,16 nM
[78]
35
Ki = 2,0 nM
[74]
40
Ki = 0,02 nM
[79]
36
Ki = 6,7 nM
[75]
41
Ki = 1,9 nM
[80]
биодоступности ингибиторов. Одним из наибо
BMS 962212. Соединение хорошо растворимо в
лее активных (Ki = 6,7 нМ) оказалось соедине
воде, имеет короткий период полураспада и по
ние 36, которое является S энантиомером с хо
своим фармакокинетическим характеристикам
рошей биодоступностью.
пригодно для парентерального введения. Пре
Показано, что производное кумарина 37 (эф
парат прошел первую фазу клинических испы
фективно (IС50 = 0,77 мкМ) ингибирует фактор
таний.
XIa и демонстрирует достаточную селективность
Исходя из ациклических производных фе
(от умеренной до хорошей) относительно панели
нилимидазола, проявивших себя как ингибито
родственных сериновых протеаз [76]. Соединение
ры фактора XIa, Corte et al. спроектировали и
37 может быть использовано как центральный
исследовали методами молекулярного модели
скаффолд для разработки новых эффективных
рования и конформационного анализа серию
ингибиторов фактора XIa с отсутствующим или
11 , 12 , 13 и 14 членных макроциклов [78]. Тео
ограниченным риском внутренних кровотечений.
ретический анализ показал, что лучше всего в
Последовательной оптимизацией серии сое
активный центр фактора XIa вписываются 12 и
динений, содержащих в качестве скаффолдов
13 членные макроциклы, которые и были син
фенилаланин и тетрагидроизохинолин [77], по
тезированы раскрытием бензильного кольца из
лучили прямой обратимый высокоселективный
области Р1 и соединением с орто позицией фе
ингибитор фактора XIa 38, обозначенный как
нила из области Р2 посредством алкильного и
БИОХИМИЯ том 84 вып. 2 2019
208
КАБАНКИН и др.
эфирного линкеров разной длины. Один из
Таким образом, несмотря на определенные
13 членных макроциклов (соединение 39), явля
достижения в разработке методов антикоагулянт
ющийся Е изомером, показал хорошую активность
ной терапии, поиск новых антикоагулянтных
при ингибировании фактора XIa (Ki = 0,16 нМ)
препаратов продолжается, так как в настоящее
и высокую селективность. Вместе с тем этот
время нет идеального антикоагулянта, доступ
макроцикл обладал очень низкой оральной био
ного в клинике для предотвращения и терапии
доступностью, что обусловлено, по мнению ав
тромбоэмболических заболеваний.
торов, высокой полярностью молекулы. В 2017 г.
Поскольку тромбоз представляет собой
тот же коллектив авторов, используя другую схе
сложный процесс, реализующийся по множест
му макроциклизации, получил еще более актив
ву механизмов, нельзя не упомянуть об исследо
ное (Ki = 0,02 нМ), высокоселективное вещест
ваниях, в которых для терапии тромбозов ис
во 40, но его оральная биодоступность также
пользовали комбинацию двух или более антит
оказалась низкой [79].
ромботических лекарств. В обзоре Neves et al.
В поиске активных и орально биодоступных
[81] обсуждены связи структура-активность и
ингибиторов фактора XIa Hu et al. синтезирова
приведены структурные формулы для 163 хими
ли и исследовали производные пиридазина [80].
ческих соединений, являющихся дуальными
В целом, полученные соединения были иденти
ингибиторами различных факторов свертыва
фицированы как активные и селективные инги
ния крови: тромбина и Xa, тромбина и VIIa, Xa
биторы фактора XIa. Фармакокинетические ха
или IXa, Xa и VIIa, XIa или калликреина плазмы
рактеристики данных соединений были оцене
и др. Например, соединение с шифром ЕР217609
ны на собаках. Наиболее активное из получен
и самой громоздкой структурной формулой сре
ных соединений 41 (Ki = 1,9 нМ) обладало уме
ди представленных структур является прямым ин
ренной оральной биодоступностью.
гибитором тромбина (IC50 = 11,5 нМ) и непря
мым ингибитором фактора Xa (IC50 = 11,5 нМ).
После рассмотрения всех представленных в
Это соединение передано на клинические ис
обзоре структур можно сделать вывод, что ни
пытания. Ключевая проблема при поиске таких
одно из этих соединений не подходит на роль
ингибиторов заключается в достижении комп
«идеального» антикоагулянта. В работе R.A. Al
ромисса между активностями соединения,
Horani и др. [71] сформулированы основные ха
действующего одновременно на несколько ми
рактеристики, которыми должен обладать такой
шеней, представляющих интерес, и его прием
идеальный антикоагулянт: 1) предсказуемая
лемым фармакокинетическим профилем.
фармакокинетика; 2) отсутствие токсичности
при действии на печень (гепатотоксичность),
Финансирование
кости (остеопороз), тромбоциты (тромбоцитопе
ния); 3) быстрая обратимость при использова
Работа поддержана грантом по программе
нии эффективных и недорогих антидотов; 4) от
фундаментальных исследований Президиума
сутствие необходимости непрерывного монито
РАН «Фундаментальные основы технологии фи
ринга, подбора и коррекции дозы; 5) безопас
зиологических адаптаций», раздел «Новые инги
ность для особых популяций (беременные жен
биторы тромбина» - грантом РНФ 16 14 00224.
щины, раковые пациенты, дети, пожилые люди
и т.п.); 6) доступность в сравнительно недорогой
Конфликт интересов
оральной и/или парентеральной форме; 7) эф
фективность и безопасность действия при любом
Авторы работы декларируют отсутствие
повышении риска внутреннего кровотечения.
конфликта интересов.
СПИСОК ЛИТЕРАТУРЫ
1.
Пантелеев М.А., Атауллаханов Ф.И. (2008) Свертыва
approaches and recent developments, Blood Coagul.
ние крови: биохимические основы, Клиническая онко
Fibrinolysis, 23, 482-493.
гематология, 1, 50-62.
5.
Broussalis, E., Anna, W., Trinka, E., Mutzenbach, S., and
2.
Пантелеев М.А., Васильев С.А., Синауридзе Е.И., Во
Killer, M. (2014) Latest developments in anticoagulant
робьев А.И., Атауллаханов Ф.И. (2012) Практическая
drug discovery, Drug Discov. Today, 19, 921-935.
коагуология, Практическая медицина, Москва.
6.
Ahrens, I., Peter, K., Lip, G.Y.H., and Bode, C. (2012) Deve
3.
Пантелеев М., Котова Я., Токарев А. (2008) Механиз
lopment and clinical applications of novel oral anticoagulants.
мы регуляции свертывания крови, Терапевтический
Part I. Clinically approved drugs, Discov. Med., 13, 433-443.
архив, 7, 88-91.
7.
Roca, B., and Roca, M. (2015) The new oral anticoagu
4.
Sinauridze, E.I., Panteleev, M.A., and Ataullakhanov, F.I.
lants: reasonable alternatives to warfarin, Cleve Clin. J. Med.,
(2012) Anticoagulant therapy: basic principles, classic
82, 847-854.
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
209
8.
Adcock, D.M., and Gosselin, R. (2015) Direct oral antico
ков В.В. (2014) Предсказание спектров биологической
agulants (DOACs) in the laboratory: 2015 review, Thromb.
активности органических соединений с помощью
Res., 136, 7-12.
веб ресурса PASS Online, Химия гетероциклических со
9.
Mekaj, Y.H., Mekaj, A.Y., Duci, S.B., and Miftari, E.I.
единений, 3, 483-499.
(2015) New oral anticoagulants: their advantages and dis
28.
Lipinski, C.A., Lombardo, F., Dominy, B.W., and Feeney, P.J.
advantages compared with vitamin K antagonists in the
(2001) Experimental and computational approaches to
prevention and treatment of patients with thromboembolic
estimate solubility and permeability in drug discovery and
events, Ther. Clin. Risk. Manag., 11, 967-977.
development settings, Adv. Drug Deliv. Rev., 46, 3-26.
10.
Gomez Outes, A., Suarez Gea, M.L., Lecumberri, R.,
29.
Shen, J., Cheng, F., Xu, Y., Li, W., and Tang, Y. (2010)
Terleira Fernandez, A.I., and Vargas Castrillon, E. (2015)
Estimation of ADME properties with substructure pattern
Direct acting oral anticoagulants: pharmacology, indica
recognition, J. Chem. Inf. Model., 50, 1034-1041.
tions, management, and future perspectives, Eur. J. Haematol.,
30.
Khakar, P.S. (2010) Two dimensional (2D) in silico models
95, 389-404.
for absorption, distribution, metabolism, excretion and
11.
Синауридзе Е., Вуймо Т., Атауллаханов Ф. (2017) Да
toxicity (ADME/T) in drug discovery, Curr. Top. Med.
бигатран этексилат: новый антикоагулянт для перо
Chem., 10, 116-126.
рального введения, Вопросы гематологии/онкологии и
31.
Kujawski, J., Bernard, M.K., Janusz, A., and Kuzma, W.
иммунологии в педиатрии, 16, 1-15.
(2012) Prediction of log P?: ALOGPS application in medi
12.
Joppa, S.A., Salciccioli, J., Adamski, J., Patel, S.,
cinal chemistry education, J. Chem. Educ., 89, 64-67.
Wysokinski, W., McBane, R., Al Saffar, F., Esser, H., and
32.
Matter, H., and Schmider, W. (2006) In silico ADME
Shamoun, F. (2018) A practical review of the emerging
modelling. in Drug discovery and evaluation, safety and
direct anticoagulants, laboratory monitoring, and reversal
pharmacokinetic assays (Vogel, H.G. ed.), Springer,
agents, J. Clin. Med., 7, 29.
Heidelberg, pp. 409-436.
13.
Pollack, C.V., Reilly, P.A., van Ryn, J., Eikelboom, J.W.,
33.
Wang, Y., Xing, J., Xu, Y., Zhou, N., Peng, J., Xiong, Z.,
Glund, S., Bernstein, R.A., Dubiel, R., Huisman, M.V.,
Liu, X., Luo, X., Luo, C., Chen, K., Zheng, M., and Jiang, H.
Hylek, E.M., Kam, C.W., Kamphuisen, P.W., Kreuzer, J.,
(2015) In silico ADME/T modelling for rational drug
Levy, J.H., Royle, G., Sellke, F.W., Stangier, J., Steiner, T.,
design, Q Rev. Biophys., 48, 488-515.
Verhamme, P., Wang, B., Young, L., and Weitz, J.I. (2017)
34.
Obst, U., Banner, D.W., Weber, L., and Diederich, F.
Idarucizumab for dabigatran reversal — full cohort analysis,
(1997) Molecular recognition at the thrombin active site:
N. Engl. J. Med., 377, 431-441.
structure based design and synthesis of potent and selective
14.
Hung, C. L., and Chen, C. C. (2014) Computational
thrombin inhibitors and the X ray crystal structures of two
approaches for drug discovery, Drug Dev. Res., 75,
thrombin inhibitor complexes., Chem. Biol., 4, 287-295.
412-418.
35.
Kong, Y., Chen, H., Wang, Y. Q., Meng, L., and Wei, J. F.
15.
Lill, M. (2013) Virtual screening in drug design, Methods
(2014) Direct thrombin inhibitors: patents 2002 2012
Mol. Biol., 993, 1-12.
(Review), Mol. Med. Rep., 9, 1506-1514.
16.
Baron, R. (Ed.) (2012) Computational drug discovery and
36.
He, L. W., Dai, W. C., and Li, N. G. (2015) Development
design, Springer New York, NY.
of orally active thrombin inhibitors for the treatment of
17.
Хельтье Х. Д., Зиппль В., Роньян Д., Фолькерс Г.
thrombotic disorder diseases, Molecules, 20, 11046-11062.
(2015) Молекулярное моделирование. Теория и практика,
37.
Mena Ulecia, K., Tiznado, W., and Caballero, J. (2015)
БИНОМ. Лаборатория знаний, Москва.
Study of the differential activity of thrombin inhibitors
18.
De Ruyck, J., Brysbaert, G., Blossey, R., and Lensink, M.F.
using docking, QSAR, molecular dynamics, and MM
(2016) Molecular docking as a popular tool in drug design,
GBSA, PLoS One, 10, e0142774. doi: 10.1371/journal.
an in silico travel, Adv. Appl. Bioinform. Chem., 9, 1-11.
pone.0142774.
19.
Chen, Y. C. (2015) Beware of docking! Trends Pharmacol.
38.
Lu, T., Tomczuk, B., Illig, C.R., Bone, R., Murphy, L.,
Sci., 36, 78-95.
Spurlino, J., Salemme, F.R., and Soll, R.M. (1998) In vitro
20.
Сулимов В.Б., Сулимов А.В. (2017) Докинг: молекуляр
evaluation and crystallographic analysis of a new class of
ное моделирование для разработки лекарств, ИИнтелл,
selective, non amide based thrombin inhibitors, Bioorg.
Москва, 348 с.
Med. Chem. Lett., 8, 1595-1600.
21.
Klimovich, P.V., Shirts, M.R., and Mobley, D.L. (2015)
39.
Sinauridze, E.I., Romanov, A.N., Gribkova, I.V, Konda
Guidelines for the analysis of free energy calculations,
kova, O.A., Surov, S.S., Gorbatenko, A.S., Butylin, A.A.,
J. Comput. Aided. Mol. Des., 29, 397-411.
Monakov, M.Y., Bogolyubov, A.A., Kuznetsov, Y.V,
22.
Sulimov, A.V, Kutov, D.C., Katkova, E.V, Ilin, I.S., and
Sulimov, V.B., and Ataullakhanov, F.I. (2011) New synthe
Sulimov, V.B. (2017) New generation of docking programs:
tic thrombin inhibitors: molecular design and experimental
Supercomputer validation of force fields and quantum che
verification, PLoS One, 6, e19969, doi: 10.1371/journal.
mical methods for docking, J. Mol. Graph. Model., 78, 139-147.
pone.0019969.
23.
Раевский О. (2013) Свойства химических соединений и
40.
Hagmann, W. K. (2008) The many roles for fluorine in
лекарств как функции их структуры, Добрсовет, КДУ,
medicinal chemistry, J. Med. Chem., 51, 4359-4369.
Москва.
41.
Li, M., and Ren, Y. (2015) Synthesis and biological evalu
24.
Раевский О. (2015) Моделирование соотношений «струк
ation of some new 2,5 substituted 1 ethyl 1H benzoimi
тура-свойство», Добрсовет, КДУ, Москва.
dazole fluorinated derivatives as direct thrombin inhibitors,
25.
Филимонов Д., Поройков В. (2006) Прогноз спектра
Arch. Pharm. (Weinheim), 348, 353-365.
биологической активности органических соедине
42.
Chen, H., and Ren, Y. (2015) Design, synthesis, and anti
ний, Российский химический журнал, 50, 66-75.
thrombotic evaluation of some novel fluorinated thrombin
26.
Поройков В., Филимонов Д., Глориозова Т., Лагунин А.,
inhibitor derivatives, Arch. Pharm. (Weinheim), 348,
Дружиловский Д., Степанчикова А. (2009) Компью
408-420.
терное предсказание биологической активности хи
43.
Chen, D., Wang, S., Diao, X., Zhu, Q., Shen, H., Han, X.,
мических веществ: виртуальная хемогеномика, Ин
Wang, Y., Gong, G., and Xu, Y. (2015) Design, synthesis
формационный вестник ВОГиС, 13, 137-142.
and antithrombotic evaluation of novel dabigatran etexilate
27.
Филимонов Д.А., Лагунин А.А., Глориозова Т.А., Ру
analogs, a new series of non peptides thrombin inhibitors,
дик А.В., Дружиловский Д.С., Погодин П.В., Порой
Bioorg. Med. Chem., 23, 7405-7416.
5 БИОХИМИЯ том 84 вып. 2 2019
210
КАБАНКИН и др.
44.
Chen, D., Shi, J., Liu, J., Zhang, X., Deng, X., Yang, Y.,
59.
Hu, X., Xiao, Y., Yu, C., Zuo, Y., Yang, W., Wang, X., Gu, B.,
Cui, S., Zhu, Q., Gong, G., and Xu, Y. (2017) Design, syn
and Li, J. (2018) Characterization of a novel selective fac
thesis and antithrombotic evaluation of novel non peptide
tor Xa inhibitor, DJT06001, which reduces thrombus for
thrombin inhibitors, Bioorg. Med. Chem., 25, 458-470.
mation with low risk of bleeding, Eur. J. Pharmacol., 825,
45.
Lee, W., Lee, S., Choi, J., Park, J. H., Kim, K. M., Jee, J. G.,
85-91.
and Bae, J. S. (2017) Antithrombotic properties of JJ1, a
60.
Smiley, D.A., and Becker, R.C. (2014) Factor IXa as a tar
potent and novel thrombin inhibitor, Sci. Rep., 7, 14862,
get for anticoagulation in thrombotic disorders and condi
doi: 10.1038/s41598 017 13868 1.
tions, Drug Discov. Today, 19, 1445-1453.
46.
Wang, X., Zhang, Y., Yang, Y., Wu, X., Fan, H., and Qiao, Y.
61.
Choudhari, P., and Bhatia, M. (2012) 3D QSAR, pharma
(2017) Identification of berberine as a direct thrombin
cophore identification studies on series of 4 substituted
inhibitor from traditional Chinese medicine through struc
benzothiophene analogs as factor IXa inhibitors,
tural, functional and binding studies, Sci. Rep., 7, 44040,
Pharmacophore, 3, 189-198.
doi: 10.1038/srep44040.
62.
Wang, S., Beck, R., Blench, T., Burd, A., Buxton, S.,
47.
Levy, J.H., Spyropoulos, A.C., Samama, C.M., and
Malic, M., Ayele, T., Shaikh, S., Chahwala, S., Chander, C.,
Douketis, J. (2014) Direct oral anticoagulants: new drugs
Holland, R., Merette, S., Zhao, L., Blackney, M., and
and new concepts, JACC Cardiovasc. Interv., 7, 1333-1351.
Watts, A. (2010) Studies of benzothiophene template as
48.
Patel, N.R., Patel, D.V, Murumkar, P.R., and Yadav, M.R.
potent factor IXa (FIXa) inhibitors in thrombosis, J. Med.
(2016) Contemporary developments in the discovery of
Chem., 53, 1465-1472.
selective factor Xa inhibitors: a review, Eur. J. Med. Chem.,
63.
Wang, S., Beck, R., Burd, A., Blench, T., Marlin, F.,
121, 671-698.
Ayele, T., Buxton, S., Dagostin, C., Malic, M., Joshi, R.,
49.
Sulimov, V.B., Gribkova, I.V, Kochugaeva, M.P., Katkova, E.V,
Barry, J., Sajad, M., Cheung, C., Shaikh, S., Chahwala, S.,
Sulimov, A.V, Kutov, D.C., Shikhaliev, K.S., Medvedeva, S.M.,
Chander, C., Baumgartner, C., Holthoff, H.P., Murray, E.,
Krysin, M.Y., Sinauridze, E.I., and Ataullakhanov, F.I.
Blackney, M., and Giddings, A. (2010) Structure based
(2015) Application of molecular modeling to development
drug design: development of potent and selective factor IXa
of new factor Xa inhibitors, Biomed Res. Int., 120802,
(FIXa) inhibitors, J. Med. Chem., 53, 1473-1482.
doi: 10.1155/2015/120802.
64.
Parker, D.L. Jr., Walsh, S., Li, B., Kim, E., and Sharipour, A.,
50.
Yang, J., Su, G., Ren, Y., and Chen, Y. (2015) Synthesis of
Smith, C., Chen, Y.H., Berger, R., Harper, B., Zhang, T.,
3,4 diaminobenzoyl derivatives as factor Xa inhibitors,
Park, M., Shu, M., Wu, J., Xu, J., Dewnani, S., Sherer, E.C.,
Eur. J. Med. Chem., 101, 41-51.
Hruza, A., Reichert, P., Geissler, W., Sonatore, L.,
51.
Ishihara, T., Koga, Y., Iwatsuki, Y., and Hirayama, F.
Ellsworth, K., Balkovec, J., Greenlee, W., and Wood, H.B.
(2015) Identification of potent orally active factor Xa
(2015) Rapid development of two factor IXa inhibitors
inhibitors based on conjugation strategy and application of
from hit to lead, Bioorg. Med. Chem. Lett., 25, 2321-2325.
predictable fragment recommender system, Bioorg. Med.
65.
Zhang, T., Andre, P., Bateman, T. J., Chen, Y. H., and
Chem., 23, 277-289.
Desai, K., Ellsworth, K., Geissler, W.M., Guo, L., Hruza, A.,
52.
Xu, C., and Ren, Y. (2015) Molecular modeling studies of
Jian, T., Meng, D., Parker, D.L., Jr., Qian, X., et al. (2015)
[6,6,5] tricyclic fused oxazolidinones as fXa inhibitors
Development of a novel class of potent and selective FIXa
using 3D QSAR, topomer CoMFA, molecular docking
inhibitors, Bioorg. Med. Chem. Lett., 25, 4945-4949.
and molecular dynamics simulations, Bioorg. Med. Chem.
66.
Meng, D., Andre, P., Bateman, T.J., Berger, R., and
Lett., 25, 4522-4528.
Chen, Y.H., Desai K2, Dewnani, S., Ellsworth, K., Feng, D.,
53.
Wang, Y., Sun, X., Yang, D., Guo, Z., Fan, X., Nie, M.,
Geissler, W.M., Guo, L., Hruza, A., Jian, T., Li, H., et al.
Zhang, F., Liu, Y., Li, Y., Wang, Y., Gong, P., and Liu, Y.
(2015) Development of a novel tricyclic class of potent and
(2016) Design, synthesis, and structure activity relation
selective FIXa inhibitors, Bioorg. Med. Chem. Lett., 25,
ship of novel and effective apixaban derivatives as FXa
5437-5443.
inhibitors containing 1,2,4 triazole/pyrrole derivatives as
67.
Gao, J. S., Tong, X. P., Chang, Y. Q., He, Y. X., Mei, Y. D.,
P2 binding element, Bioorg. Med. Chem., 24, 5646-5661.
Tan, P. H., Guo, J. L., Liao, G. C., Xiao, G. K., Chen, W. M.,
54.
Xing, J., Yang, L., Yang, Y., Zhao, L., Wei, Q., Zhang, J.,
Zhou, S. F., and Sun, P. H. (2015) Design and prediction
Zhou, J., and Zhang, H. (2017) Design, synthesis and bio
of new anticoagulants as a selective Factor IXa inhibitor via
logical evaluation of novel 2,3 dihydroquinazolin
4(1H)
three dimensional quantitative structure property rela
one derivatives as potential fXa inhibitors, Eur. J. Med.
tionships of amidinobenzothiophene derivatives, Drug Des.
Chem., 125, 411-422.
Devel. Ther., 9, 1743-1759.
55.
Pu, Y., Liu, H., Zhou, Y., Peng, J., Li, Y., Li, P., Li, Y., Liu, X.,
68.
Zhang, T., Liu, Y., Yang, X., Martin, G.E., Yao, H., Shang, J.,
and Zhang, L. (2017) In silico discovery of novel FXa
Bugianesi, R.M., Ellsworth, K.P., Sonatore, L.M., Nizner, P.,
inhibitors by pharmacophore modeling and molecular
Sherer, E.C., Hill, S.E., Knemeyer, I.W., Geissler, W.M.,
docking, Nat. Products Bioprospect., 7, 249-256.
Dandliker, P.J., Helmy, R., and Wood, H.B. (2016) Defini
56.
Lagos, C.F., Segovia, G.F., Nunez Navarro, N., Faun
tive metabolite identification coupled with automated ligand
dez, M.A., and Zacconi, F.C. (2017) Novel FXa inhibitor
identification system (ALIS) technology: a novel approach
identification through integration of ligand and structure
to uncover structure-activity relationships and guide drug
based approaches, Molecules, 22, pii: E1588, doi: 10.3390/
design in a factor IXa inhibitor program, J. Med. Chem.,
molecules22101588.
59, 1818-1829.
57.
Sun, X., Hong, Z., Liu, M., Guo, S., Yang, D., Wang, Y.,
69.
Sakurada, I., Endo, T., Hikita, K., Hirabayashi, T., Hosa
Lan, T., Gao, L., Qi, H., Gong, P., and Liu, Y. (2017)
ka, Y., Kato, Y., Maeda, Y., Matsumoto, S., Mizuno, T., Naga
Design, synthesis, and biological activity of novel tetrahy
sue, H., Nishimura, T., Shimada, S., et al. (2017) Discovery
dropyrazolopyridone derivatives as FXa inhibitors with
of novel aminobenzisoxazole derivatives as orally available fac
potent anticoagulant activity, Bioorg. Med. Chem., 25,
tor IXa inhibitors, Bioorg. Med. Chem. Lett., 27, 2622-2628.
2800-2810.
70.
Bane, C.E., and Gailani, D. (2014) Factor XI as a target for
58.
Wang, W., Yuan, J., Fu, X., Meng, F., Zhang, S., Xu, W.,
antithrombotic therapy, Drug Discov. Today, 19, 1454-1458.
Xu, Y., and Huang, C. (2016) Novel anthranilamide based
71.
Al Horani, R.A., and Desai, U.R. (2016) Factor XIa
FXa inhibitors: drug design, synthesis and biological evalua
inhibitors: a review of the patent literature, Expert. Opin.
tion, Molecules, 21, doi: 10.3390/molecules21040491.
Ther. Pat., 26, 323-345.
БИОХИМИЯ том 84 вып. 2 2019
НОВЫЕ ИНГИБИТОРЫ ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
211
72. Corte, J.R., Fang, T., Hangeland, J.J., Friends, T.J.,
77. Pinto, D.J.P., Orwat, M.J., Smith, L.M., 2nd, Quan, M.L.,
Rendina, A.R., Luettgen, J.M., Bozarth, J.M., Barbera, F.A.,
Lam. P.Y.S., et al. (2017) Discovery of a parenteral small
Rossi, K.A., Wei, A., Ramamurthy, V., Morin, P.E.,
molecule coagulation factor XIa inhibitor clinical candi
Seiffert, D.A., Wexler, R.R., and Quan, M.L.
(2015)
date (BMS 962212), J. Med. Chem., 60, 9703-9723.
Pyridine and pyridinone based factor XIa inhibitors,
78. Corte, J.R., Fang, T., Osuna, H., Pinto, D.J.P., Rossi, K.A.,
Bioorg. Med. Chem. Lett., 25, 925-930.
Myers, J.E., Sheriff, S., Lou, Z., Zheng, J.J., Harper, T.W.,
73. Pinto, D.J.P., Smallheer, J.M., Corte, J.R., Austin, E.J.D.,
Bozarth, J.M., Wu, Y., Luettgen, J.M., Seiffert, D.A.,
and Wang, C., et al. (2015) Structure based design of
Decicco, C.P., Wexler, R.R., and Quan, M.L. (2017)
inhibitors of coagulation factor XIa with novel P1 moieties,
Structure based design of macrocyclic factor XIa inhi
Bioorg. Med. Chem. Lett., 25, 1635-1642.
bitors: discovery of the macrocyclic amide linker, J. Med.
74. Smith, L.M., Orwat, M.J., Hu, Z., Han, W., and Wang, C.,
Chem., 60, 1060-1075.
et al. (2016) Novel phenylalanine derived diamides as fac
79. Wang, C., Corte, J.R., Rossi, K.A., Bozarth, J.M., Wu, Y.,
tor XIa inhibitors, Bioorg. Med. Chem. Lett., 26, 472-478.
Sheriff, S., Myers, J.E., Luettgen, J.M., Seiffert, D.A.,
75. Corte, J.R., Fang, T., Pinto, D.J.P.P., Orwat, M.J.,
Wexler, R.R., and Quan, M.L. (2017) Macrocyclic factor
Rendina, A.R., Luettgen, J.M., Rossi, K.A., Wei, A.,
XIa inhibitors, Bioorg. Med. Chem. Lett., 27, 4056-4060.
Ramamurthy, V., Myers, J.E., Sheriff, S., Narayanan, R.,
80. Hu, Z., Wang, C., Han, W., Rossi, K.A., Bozarth, J.M.,
Harper, T.W., Zheng, J.J., Li, Y. X.X., Seiffert, D.A.,
Wu, Y., Sheriff, S., Myers, J.E., Luettgen, J.M., Seiffert, D.A.,
Wexler, R.R., and Quan, M.L. (2016) Orally bioavailable
Wexler, R.R., and Quan, M.L. (2018) Pyridazine and pyri
pyridine and pyrimidine based factor XIa inhibitors: dis
dazinone derivatives as potent and selective factor XIa
covery of the methyl N phenyl carbamate P2 prime group,
inhibitors, Bioorg. Med. Chem. Lett., 28, 987-992.
Bioorg. Med. Chem., 24, 2257-2272.
81. Neves, A.R., Correia da Silva, M., Sousa, E., and
76. Obaidullah, A.J., and Al Horani, R.A. (2017) Discovery of
Pinto, M. (2016) Structure activity relationship studies for
chromen 7 yl furan 2 carboxylate as a potent and selective
multitarget antithrombotic drugs, Future Med. Chem., 8,
factor XIa inhibitor, Cardiovasc. Hematol. Agents Med.
2305-2355.
Chem., 15, 40-48.
COMPUTER DESIGN OF LOW MOLECULAR
WEIGHT INHIBITORS OF COAGULATION
FACTORS
A. S. Kabankin1*, E. I. Sinauridze1,2, E. N. Lipets1,2,
and F. I. Ataullakhanov1,2,3,4
1 Center for Theoretical Problems of Physicochemical Pharmacology,
Russian Academy of Sciences, 119991 Moscow, Russia;
E mail: rla2001@mail.ru
2 Dmitry Rogachev National Medical Research Center of Pediatric
Hematology, Oncology and Immunology, 1179971 Moscow, Russia
3 Lomonosov Moscow State University, Faculty of Physics,
119991 Moscow, Russia
4 Moscow Institute of Physics and Technology, 141701 Dolgoprudny,
Moscow Region, Russia
Received September 5, 2018
Revision received October 19, 2018
Accepted October 19, 2018
The work presents an overview of the main approaches to the search for new low molecular weight inhibitors of blood
coagulation factors IIa, Xa, IXa, and XIa and the results of this search conducted from 2015 to 2018. For each of these
factors, several inhibitors with an IC50 below 10 nM have been found in recent years. Some of them are on the stage
of clinical trial. Nevertheless, none of these substances meets the requirements of «ideal» anticoagulant, and the fur
ther studies are relevant.
Keywords: anticoagulants, low molecular weight inhibitors, coagulation factors, computer molecular design, molecu
lar docking
БИОХИМИЯ том 84 вып. 2 2019
5*