БИОХИМИЯ, 2019, том 84, вып. 5, с. 692 - 703
УДК 577.214.43
ГИПОКСИЯ КАК ФАКТОР РЕГУЛЯЦИИ ЭКСПРЕССИИ
ГЕНОВ apoA 1, ABCA1 И КОМПОНЕНТА КОМПЛЕМЕНТА С3
В МАКРОФАГАХ ЧЕЛОВЕКА*
© 2019
А.М. Богомолова1,2, В.С. Шавва2, А.А. Никитин1,2, Е.В. Некрасова2,
Э.Б. Диже2, Е.Е. Ларионова2, И.В. Кудрявцев2, С.В. Орлов1,2**
1 Санкт Петербургский государственный университет, 199034 Санкт Петербург,
Россия; электронная почта: s.orlov@spbu.ru, serge@iem.sp.ru
2 Институт экспериментальной медицины, 197376 Санкт Петербург, Россия
Поступила в редакцию 10.12.2018
После доработки 29.01.2019
Принята к публикации 29.01.2019
Гипоксия играет важную роль в прогрессии атеросклероза. Локальное снижение концентрации кислорода в
бляшке создает специфическое микроокружение, изменяющее траснкриптом клеток!резидентов, в част!
ности макрофагов. Одним из путей регрессии атеросклероза является обратный транспорт холестерина из
бляшки в печень. Главные участники этого процесса - АТР!связывающий кассетный транспортер А1 (АВСА1),
экспрессирующийся на поверхности всех типов клеток, и аполипопротеин А!1 (АроА!1), синтез которого
происходит главным образом в печени и тонком кишечнике. Недавно была показана экспрессия эндоген!
ного АроА!1 в макрофагах человека. В то время как функции АВСА1 и АроА!1 являются атеропротектор!
ными, роль компонента комплемента С3 спорна. Показана положительная корреляция между уровнем C3
в плазме крови и риском развития сердечно!сосудистых заболеваний у человека. С другой стороны, нокаут
по гену C3 в модели атеросклероза на мышах приводит к развитию более крупных бляшек и повышению
уровня триглицеридов в крови. В настоящей работе впервые показана активация экспрессии генов aроА 1
и С3, а также повышение содержания внутриклеточного и поверхностного белка АроА!1 в макрофагах ТНР!1
при ответе на гипоксию, вызванную СоCl2. Выявлен механизм СoCl2!зависимого повышения уровней
мРНК apoA I, АВСА 1 и С3 - в активацию генов вовлечены сигнальные каскады MEK1/2-Erk1/2,
MKK4/7-JNK1/2/3 и MKK3/6-p38, а также транскрипционный фактор NF!κB, работающий в кооперации
с HIF!1α!субъединицей регулятора адаптации к гипоксии HIF!1.
КЛЮЧЕВЫЕ СЛОВА: атеросклероз, гипоксия, макрофаги, THP!1, ген apoA 1, ген ABCA1, ген С3.
DOI: 10.1134/S0320972519050075
Атеросклероз - хроническое воспалитель!
экспрессировать на поверхностной мембране
ное заболевание, характеризующееся образова!
молекулы адгезии, интегрины, селектины и сек!
нием бляшек в интиме артериальных сосудов.
ретировать интерлейкины, интерфероны и дру!
Атеросклеротическим изменениям наиболее
гие цитокины. Параллельное увеличение про!
подвержены искривленные участки артерий,
ницаемости стенки сосуда приводит к инфильт!
испытывающие так называемый гемодинами!
рации субэндотелиального пространства липо!
ческий стресс растяжения [1]. Стресс растяже!
протеинами низкой плотности (ЛПНП). Эти час!
ния может быть вызван как ламинарным, так и
тицы задерживаются в интиме благодаря связы!
турбулентным потоком крови, именно второй
ванию с протеогликанами внеклеточного матрик!
тип приводит к травмированию сосуда с после!
са, где подвергаются различным модификациям
дующей активацией эндотелия и утолщением
(окислению, протеолитическому расщеплению
tunica media и intima. Эндотелиоциты начинают
и др.). Модифицированные ЛПНП поглощаются
Принятые сокращения: АроА!1 - аполипопротеин А!1; АВСА1 - АТР!связывающий кассетный транспортер А1;
ЛПНП и ЛПВП - липопротеины низкой и высокой плотности; миРНК - малая интерферирующая РНК; DMSO - диметил!
сульфоксид; HIF!1 - фактор 1, индуцирующийся при гипоксии; PBS - фосфатно!солевой буфер; BSA - бычий сывороточ!
ный альбумин; PMA - форбол!12!миристат!13!ацетат; MAPK - митоген!активируемые протеинкиназы; LXR - X!рецептор
печени; ОТ!ПЦР в реальном времени - полимеразная цепная реакция в реальном времени с обратной транскрипцией.
* Первоначально английский вариант рукописи опубликован на сайте «Biochemistry» (Moscow) http://protein.bio.
msu.ru/biokhimiya, в рубрике «Papers in Press», BM18!346, 01.04.2019.
** Адресат для корреспонденции.
692
ГИПОКСИЯ АКТИВИРУЕТ ГЕНЫ apoA 1, ABCA1 И C3 В МАКРОФАГАХ
693
макрофагами, привлеченными в ходе активации
макрофаги. Белок обнаруживается в везикуляр!
эндотелия, провоцируя накопление внутрикле!
ных компартментах и на поверхности клеточной
точного холестерина и, как следствие, транс!
мембраны, причем в мембране АроА!1 либо со!
формацию макрофагов в пенистые клетки [2].
локализуется с АВСА1, либо самостоятельно
Привлечение новых моноцитов и утолщение
присутствует в липидных рафтах [8]. Методом
интимы за счет пролиферации гладкомышеч!
проточной цитофлуориметрии было выявлено,
ных клеток ведет к разрастанию атеросклероти!
что количество мембранного АВСА1 выше в
ческой бляшки и увеличению расстояния между
АроА!1!богатых макрофагах, а трансфекция
капиллярами, питающими среднюю оболочку
клеток малой интерферирующей РНК (миРНК)
артерии. Повышенная потребность в кислороде
к АроА!1 приводит к снижению уровня белка
запускает васкуляризацию интимы и бляшки,
ABCA1. Таким образом, синтезируемый макро!
однако образование новых сосудов сконцентри!
фагами АроА!1 играет важную роль в стабилиза!
ровано на границах бляшки, в то время как
ции АВСА1 в плазматической мембране. Кроме
центральная часть находится в условиях посто!
того, оказалось, что АроА!1 влияет на провоспа!
янной гипоксии [3].
лительный статус макрофага: нокдаун АроА!1 с
Ключевым медиатором эффектов, опосреду!
помощью миРНК приводит к усиленной экспрес!
емых гипоксией, является транскрипционный
сии IL!1β и NOS2 в ответ на липополисахарид [8].
фактор HIF!1. Он функционирует в виде гетеро!
Если АроА!1 и АВСА1 при атеросклерозе об!
димера, состоящего из конститутивно экспрес!
ладают протективными свойствами, роль ком!
сируемой субъединицы HIF!1β и индуцибель!
понента комплемента С3 спорна. Повышенное
ной субъединицы HIF!1α [4]. В условиях нор!
содержание С3 в плазме крови является факто!
моксии белки, обладающие пролилгидрокси!
ром риска развития атеросклероза и инфаркта
лазным доменом (PHD1, PHD2, PHD3), и аспа!
миокарда, а уровень анафилатоксина С3а, про!
рагилгидрокcилаза FIH опосредуют протеасом!
дукта гидролиза С3, коррелирует с утолщением
ную деградацию HIF!1α через гидроксилирова!
интимы и средней оболочки сонной артерии и
ние. При низких концентрациях кислорода ре!
развитием острого коронарного синдрома [10, 11].
акция не катализируется, и происходит увеличе!
Компоненты системы комплемента обнаружи!
ние времени жизни белка HIF!1α [5]. HIF!1
ваются в атеросклеротической бляшке, попадая
способствует пролиферации гладкомышечных
в интиму из кровотока и синтезируясь клетка!
клеток и рекрутированию моноцитов, других
ми!резидентами. Активация каскада в бляшке
лейкоцитов и Т!лимфоцитов в бляшку [6, 7],
осуществляется по нескольким путям; к приме!
снижает миграционную активность макрофагов
ру, классический путь может быть запущен ауто!
и удерживает их в интиме [3]. Таким образом,
антителами к окисленным ЛПНП [12], альтер!
популяция макрофагов и пенистых клеток пос!
нативный - кристаллами холестерина, что, в ко!
тоянно растет.
нечном итоге, ведет к сборке NLRP3!инфлам!
В настоящее время считается, что централь!
масомы и продукции провоспалительных цито!
ным путем регрессии атеросклероза является
кинов [13]. Макрофаги бляшки экспрессируют
обратный транспорт холестерина в составе ли!
рецепторы к С3а и С5а, с которыми связываются
попротеинов высокой плотности (ЛПВП) из
анафилатоксины, что приводит к запуску окси!
макрофагов бляшки в печень. Главные участники
дативного взрыва и синтезу TNFα и IL!1. Также
этого процесса - аполипопротеин А1 (ApoA!1)
С3а и C5а служат хемоаттрактантами для эози!
и кассетный транспортер АВСА1. АроА!1 - ос!
нофилов, тучных клеток, моноцитов, Т! и В!лим!
новной белковый компонент атеропротектор!
фоцитов, увеличивают проницаемость мелких
ных ЛПВП, определяющий их концентрацию в
сосудов и вызывают сокращение гладкомышеч!
плазме крови. Основные места синтеза ApoA!1 -
ных клеток [10].
печень и тонкий кишечник. АроА!1 способен
В то же время ряд экспериментов говорит об
усиливать отток холестерина из клеток перифе!
атеропротекторной роли С3 на системном уров!
рических тканей через АВСА1. АВСА1 - повсе!
не. Атеросклеротические бляшки мышей, нока!
местно экспрессируемый кассетный транспор!
утных по LDL!R и С3, содержат больше пенис!
тер, образующий канал в мембране и обеспечи!
тых клеток [14], а у таких же мышей с дополни!
вающий «перебрасывание» липидов из клетки
тельно нокаутированным АроЕ значительно по!
энергозависимым путем. В нашей лаборатории
вышается уровень холестерина и триглицеридов
был показан синтез эндогенного АроА!1 в мак!
в плазме крови, а размер бляшки увеличивается
рофагах человека [8, 9]. Гомогенная по уровню
на 84% по сравнению с нокаутными мышами с
ApoA!1 популяция моноцитов периферической
интактным С3 [15]. Также показана важная роль
крови человека в ходе дифференцировки разде!
инактивированного С3b (iC3b) в противовоспа!
ляется на ApoA!1!бедные и ApoA!1!богатые
лительном клиренсе апоптотических клеток.
БИОХИМИЯ том 84 вып. 5 2019
694
БОГОМОЛОВА и др.
Взаимодействие макрофагов и дендритных кле!
Выделение РНК. Клетки высевали на 24!лу!
ток с iC3b подавляет NF!κB!сигналинг, приво!
ночные планшеты с плотностью 1 × 104 кл./cм2 и
дит к уменьшению секреции TGF!β и увеличе!
культивировали в атмосфере 5% СO2 при 37 °С.
нию продукции IL!10 [16].
После 16! или 24!часовой инкубации с CoCl2
Целью данной работы являлось изучение ме!
клетки лизировали коммерческим реагентом
ханизмов экспрессии важных для атерогенеза
RNA Extract («Евроген», Россия) и выделяли то!
генов apoA 1, ABCA1 и С3 в макрофагах ТНР!1
тальную РНК в соответствии с рекомендациями
человека в условиях гипоксии.
производителя. Целостность (отсутствие дегра!
дации) выделенной РНК проверяли с помощью
электрофореза в 1%!ном агарозном геле по со!
МЕТОДЫ ИССЛЕДОВАНИЯ
отношению полос, соответствующих 28S и 18S
рРНК. Концентрацию и чистоту РНК определя!
Клеточные культуры. Клеточная линия ост!
ли спектрофотометрически при помощи спект!
рой моноцитарной лейкемии человека THP1
рофотометра Avaspec!2048 («Avantes», Нидер!
получена из Коллекции клеточных культур поз!
ланды). Очищенной от примесей РНК соответ!
воночных Института цитологии РАН. Клетки
ствовали отношения оптической плотности об!
культивировали в атмосфере 5% СO2 при 37 °С в
разцов РНК при 260 и 280 нм, превышавшие
среде RPMI («Биолот», Россия) с добавлением
значение 2,0, а при 260 и 230 нм - превышавшие
10% телячьей эмбриональной сыворотки (FCS,
значение 1,7.
«HyClone», США). Дифференцировку моноци!
Обратная транскрипция и ПЦР в реальном
тов THP!1 в макрофаги индуцировали добавле!
времени. В данной работе исследовали экспрес!
нием в культуральную среду форбол!12!мирис!
сию следующих генов: референсных генов ACTB
тат!13!ацетата (PMA) до концентрации 50 нг/мл
и GAPDH, кодирующих β!актин и глицеральде!
(81 нM) на 24 ч. Затем клетки отмывали от PMA,
гидфосфатдегидрогеназу соответственно, и экс!
меняли среду на свежую RPMI с 10% FCS и про!
периментальных генов apoA 1, ABCA1, С3 и IL 8.
должали дифференцировку в течение 48 ч. Та!
Для проведения реакции обратной транскрип!
ким образом, общее время дифференцировки
ции в качестве затравки использовали обратные
макрофагов ТHР!1 составляло трое суток.
праймеры к экспериментальным генам и oligo!
Антитела. Моноклональные антитела мыши
dT18, специфические праймеры добавляли по
против β!актина человека (ab3280) были приоб!
причине малого содержания мРНК экспери!
ретены у «Abcam» (Великобритания) и исполь!
ментальных генов, а также во избежание синте!
зовались для нормализации образцов по коли!
за обратной транскриптазой протяженных 3'!не!
честву белка в экспериментах по вестерн!блот!
транслируемых участков. Реакционная смесь
тингу. Для детекции внутриклеточного белка
(25 мкл) включала в себя 2 мкг РНК, по 0,5 мкл
АроА!1 применяли поликлональные антитела козы
специфических праймеров (2 пмоль/мкл), 0,5 мкл
против АроА!1 человека, описанные ранее [17],
oligo!dT18 (50 пмоль/мкл), обратную транскрип!
и вторичные антитела кролика против IgG козы,
тазу M!MLV (50 ед), раствор нуклеотидов (по 0,5 мМ
конъюгированные с пероксидазой хрена («Sigma»,
dATP, dGTP, dCTP и dTTP), 5 мкл 5× буфера для
США; A5420). Для детекции поверхностного
обратной транскрипции (250 мM Tris!HCl, pH 8,3
белка АроА!1 использовали моноклональные
при 25 °C; 500 мM KCl; 15 мM MgCl2; 50 мM DTT).
антитела мыши против АроА!1 человека («Bio!
Реакцию обратной транскрипции проводили по
Rad», США; 0650!0050) и вторичные антитела
следующей программе: 42 °С - 1,5 ч, 70°С - 15 мин,
кролика F(ab')2 против IgG мыши, меченные
охлаждение до 4 °C. Полученную кДНК хранили
флуорофором Alexa 647 («Abcam», Великобрита!
при -20 °С и использовали для ПЦР в реальном
ния; ab169348).
времени.
Ингибиторы сигнальных путей. Ингибиторы
Для ПЦР в реальном времени использовали
MAPK и NF!κB приобретены у «Biomol» (США),
реактивы фирмы «Синтол» (Россия). Праймеры
агонист LXR - у «Sigma» (США). В среду за 1 ч
и гибридизационные пробы для генов ACTB [18],
до внесения CoCl2 добавляли одно из соедине!
GAPDH [19], apoA 1 [19], ABCA1 [20] и C3 [18] бы!
ний: ингибитор р38 SB203580 (EI!286; 12,5 мкM),
ли описаны ранее. Реакционная смесь объемом
ингибитор JNK1/2/3 SP600125 (EI!305; 10 мкM),
25 мкл включала в себя 0,5 мкл праймеров (10 мкM),
ингибитор MEK1/2 U0126 (EI!282; 10 мкM), ин!
0,2 мкл Taq!полимеразы (1,25 ед), 2,5 мкл 10×
гибитор NF!κB QNZ (EI!352; 10 нM), агонист
ПЦР!буфера (100 мM Tris!HCl, pH 8,3; 500 мM
LXR TO901317 (2,5 мкM). Поскольку все эти ве!
KCl; 15 мM MgCl2), 1 мкл кДНК (соответствует
щества были растворены в DMSO, этот агент (в со!
продуктам реакции обратной транскрипции,
ответствующих концентрациях) использовали в
полученным с 0,08 мкг тотальной РНК), деио!
качестве контроля.
низированную воду (до объема 25 мкл). В конт!
БИОХИМИЯ том 84 вып. 5 2019
ГИПОКСИЯ АКТИВИРУЕТ ГЕНЫ apoA 1, ABCA1 И C3 В МАКРОФАГАХ
695
рольные лунки добавляли смесь для ПЦР без
вторичные антитела, конъюгированные с флуо!
кДНК. Реакцию проводили в амплификаторе
рофором Alexa 647, в разведении 1 : 1000 в буфе!
CFX!96 («Bio!Rad», США). Программа для Taq!
ре (1% BSA, 0,02% Tween!20 в PBS), инкубацию
man: 95 оC - 5 мин; 50 циклов: 95 °С - 25 с,
проводили в течение 1 ч при 25 °C в защищен!
59,5 °С - 45 с. Программа для SYBR Green: 95 °C -
ном от света месте. В качестве изотипического
5 мин; 50 циклов: 95 °С - 30 с, 60 °С - 20 с, 72 °С -
контроля использовали только вторичные анти!
30 с.
тела. После отмывки в PBS клетки переносили в
Число циклов (значение Ct), необходимое
пробирки для проточного цитофлуориметра
для достижения порогового уровня флуоресцен!
Epics Altra («Beckman Coulter», США) и анали!
ции, превышающего 10 стандартных отклоне!
зировали с помощью системы Beckman Coulter
ний от уровня флуктуаций фоновой флуорес!
Navios.
ценции, определяли при помощи встроенного
Для выявления цитотоксического действия
программного обеспечения «Bio!Rad» (США).
CoCl2 THP!1 снимали с планшета раствором
Относительное количество кДНК (в процентах
Версена и без фиксации окрашивали ДНК!свя!
от контрольного образца) рассчитывали по фор!
зывающими красителями: йодидом пропидия и
муле: (2Ct контроля - Ct образца) × 100. Рассчитанные
YO!PRO!1. Йодид пропидия не проникает через
количества кДНК каждого из генов нормирова!
интактные клеточные мембраны и окрашивает
ли по геометрическому среднему уровней
мертвые клетки, флуоресцируя в красной облас!
экспрессии двух референсных генов (GAPDH,
ти спектра. YO!PRO!1 проникает через мембра!
ACTB), как описано ранее [21], и представляли
ны апоптотических, но не живых, клеток и флу!
как относительный уровень экспрессии гена,
оресцирует в зеленой области спектра. Анализ
принимая за 100% уровень экспрессии в конт!
проводили с помощью системы Beckman Coulter
рольных клетках.
Navios.
Вестерн>блоттинг. Клетки трижды отмывали
Статистическая обработка. Результаты пред!
фосфатно!солевым буфером (PBS, pH 7,6), за!
ставлены как среднее ± стандартная ошибка сред!
тем лизировали в буфере RIPA!50 (50 мМ Tris!
него. Статистический анализ различий между
HCl, 150 мМ NaCl, 1% NP!40, 1 мМ EDTA, 0,1%
сравниваемыми группами выполняли с исполь!
SDS, 0,01% NaN3, 1 мМ PMSF, pH 7,4). Концент!
зованием критерия Стьюдента (непарный t!тест).
рацию белков в пробах измеряли по методу
Множественные сравнения проводили с по!
Лоури. SDS!электрофорез белков проводили в
мощью критерия Даннета. Различия считали
12%!ном или 20%!ном ПААГ. Перенос белков
статистически значимыми при p < 0,05. Статис!
на нитроцеллюлозную мембрану осуществляли
тическую обработку данных выполняли в прог!
в течение 2 ч. В качестве блокирующего буфера
рамме Microsoft Excel.
использовали 5%!ный раствор обезжиренного
сухого молока в PBS с 0,02%!ным Tween 20. Ин!
кубацию в блокирующем буфере проводили в
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
течение 1 ч, инкубацию с первичными антите!
лами - в течение 12 ч при 4 °С, инкубацию со
CoCl2 регулирует активность генов aроА 1 и С3,
вторичными антителами, конъюгированными с
а также повышает содержание белка АроА>1 в
пероксидазой хрена, - в течение 1 ч. Визуализа!
макрофагах THP>1 в зависимости от концентра>
цию пероксидазы осуществляли методом уси!
ции и времени инкубации. Для исследования ре!
ленной хемилюминесценции (ECL). Детекцию
гуляции экспрессии генов aроА 1 и С3 в макро!
люминесцентного сигнала производили с по!
фагах THP!1 использовали миметик клеточного
мощью системы мультикомплексной визуализа!
ответа на гипоксию CoCl2. CoCl2 стабилизирует
ции ChemiDoc MP System («Bio!Rad», США).
белок HIF!1α путем ингибирования PHD!со!
Проточная цитофлуориметрия. Для детекции
держащих ферментов, опосредующих протеасом!
АроА!1, связанного с поверхностной мембра!
ную деградацию HIF!1α при нормоксии [5, 22].
ной макрофагов, THP!1 фиксировали в 4%!ном
Возможные цитотоксические эффекты CoCl2
формальдегиде на PBS в течение 20 мин при 4 °С,
выявляли с помощью проточной цитометрии.
трижды отмывали PBS. Далее препараты блоки!
После инкубации с CoCl2 клетки окрашивали
ровали в течение 40 мин при 22 °С в блокирую!
ДНК!связывающими красителями йодидом
щем буфере (1% BSA, 0,02% Tween!20 в PBS).
пропидия и YO!PRO!1, после чего анализирова!
Клетки обрабатывали моноклональными анти!
ли спектры флуоресценции. CoCl2 в диапазоне
телами мыши против ApoА!1 человека в разве!
концентраций 50-300 мкМ провоцирует апоп!
дении 1 : 250 в буфере (1% BSA, 0,02% Tween!20
тоз менее чем в 10% клеток по сравнению с
в PBS) при 25 °С в течение 2 ч. После двукратной
контролем. Инкубация клеток с более высоки!
отмывки блокирующим буфером добавляли
ми концентрациями ионов кобальта приводит к
БИОХИМИЯ том 84 вып. 5 2019
696
БОГОМОЛОВА и др.
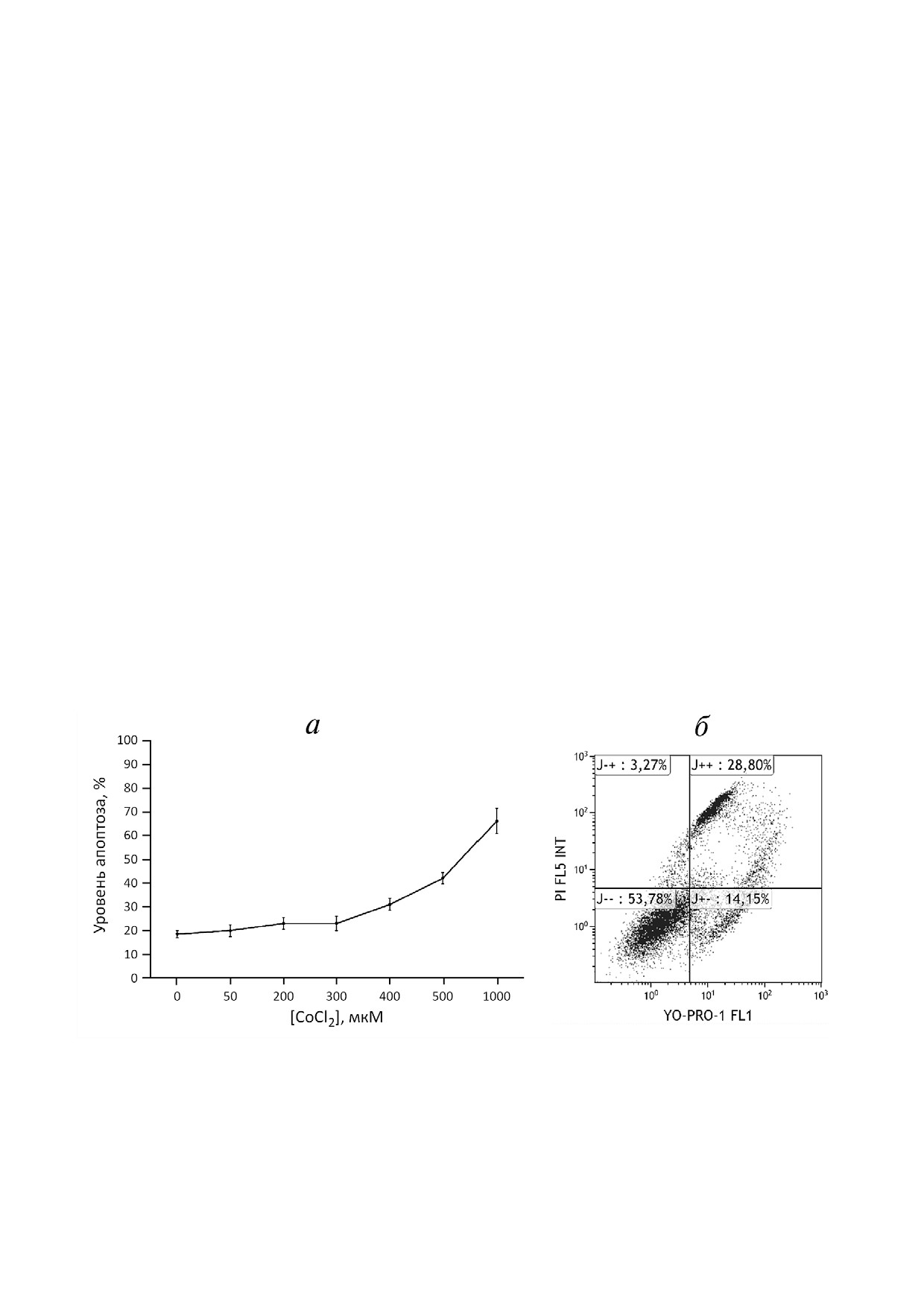
существенному возрастанию гибели клеток
АВСА1, играющий ключевую роль в оттоке хо!
(рис. 1, а). На рис. 1, б приведено распределение
лестерина из макрофагов, положительно регу!
клеток по уровням флуоресценции йодида про!
лируется HIF!1α на уровне мРНК [24], однако
пидия и YO!PRO!1 после инкубации с 500 мкМ
механизм регуляции не изучен. Одним из важ!
CoCl2. В результате только 54% клеток сохраня!
нейших участников адаптации макрофагов к ги!
ют жизнеспособность, остальные находятся на
поксии является транскрипционный фактор
тех или иных стадиях апоптоза.
NF!κВ [25]. В сигналинг, запускаемый в макро!
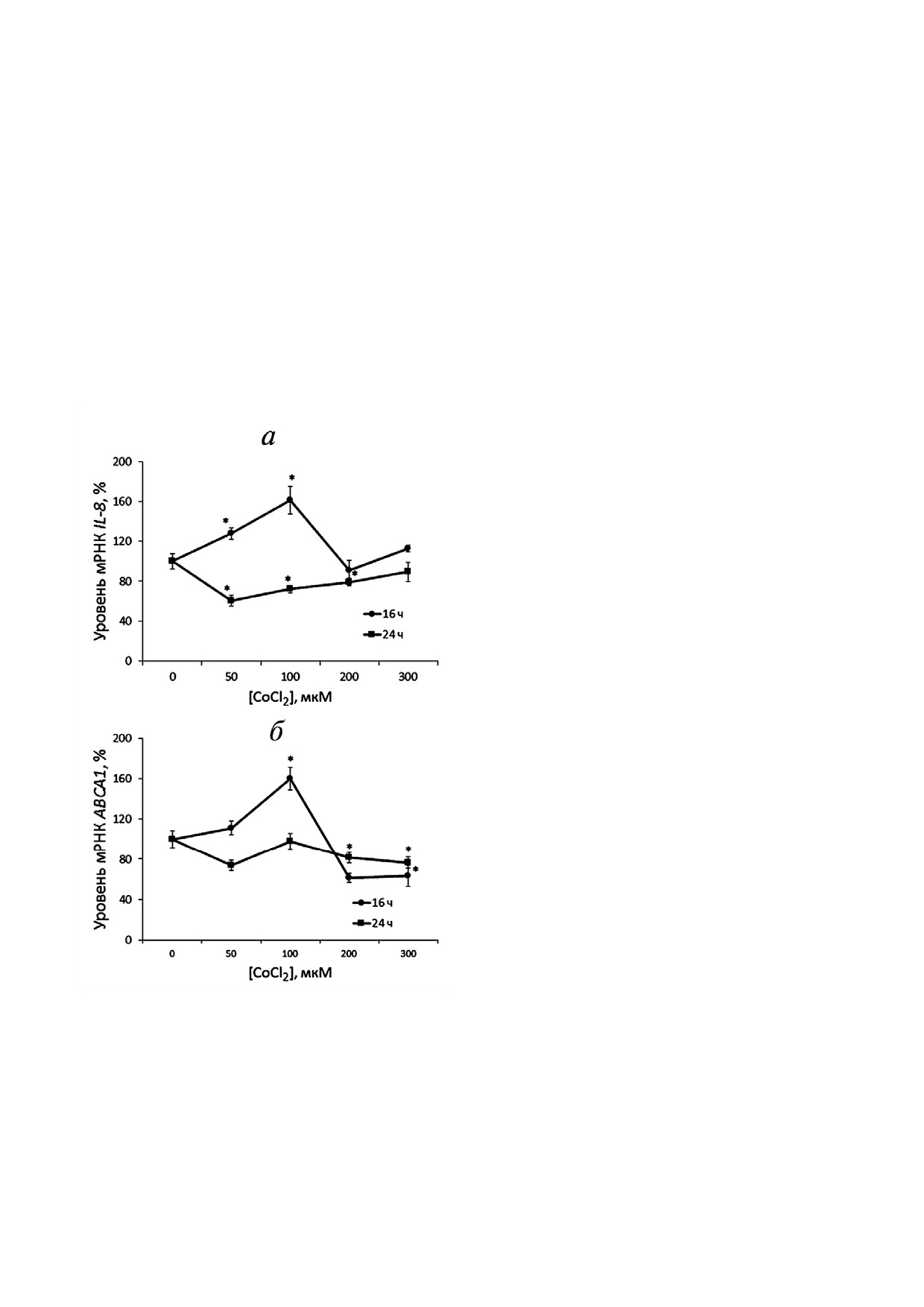
Известно, что гипоксия индуцирует экспрес!
фагах при гипоксическом окружении, вовлече!
сию генов IL 8 и АВСА1 в макрофагах [6, 23, 24].
ны стресс!активируемые MAP!киназы Erk1/2 [26],
В данной работе индукцию гена IL 8 в ответ на
p38 [27] и JNK [28]. Активация этих киназ в опо!
гипоксию использовали в качестве положитель!
средуемых низким содержанием кислорода про!
ного контроля. Показано, что уровни мРНК IL 8
цессах показана и на других типах клеток [29-32].
и АВСА1 максимально возрастают под действи!
Для выявления роли МАР киназ и фактора
ем CoCl2 в концентрации 100 мкМ на сроке ин!
транскрипции NF!κB в CoCl2!зависимой экспрес!
кубации 16 ч (рис. 2). Таким образом, эти усло!
сии генов АВСА1, С3 и apoA 1 в макрофагах THP!1
вия оптимальны для индукции ответа на гипо!
использовали ингибиторы киназ р38 (SB203580),
ксию в макрофагах THP!1. CoCl2 повышает уро!
JNK1/2/3 (SP600125), MEK1/2 (U0126) и факто!
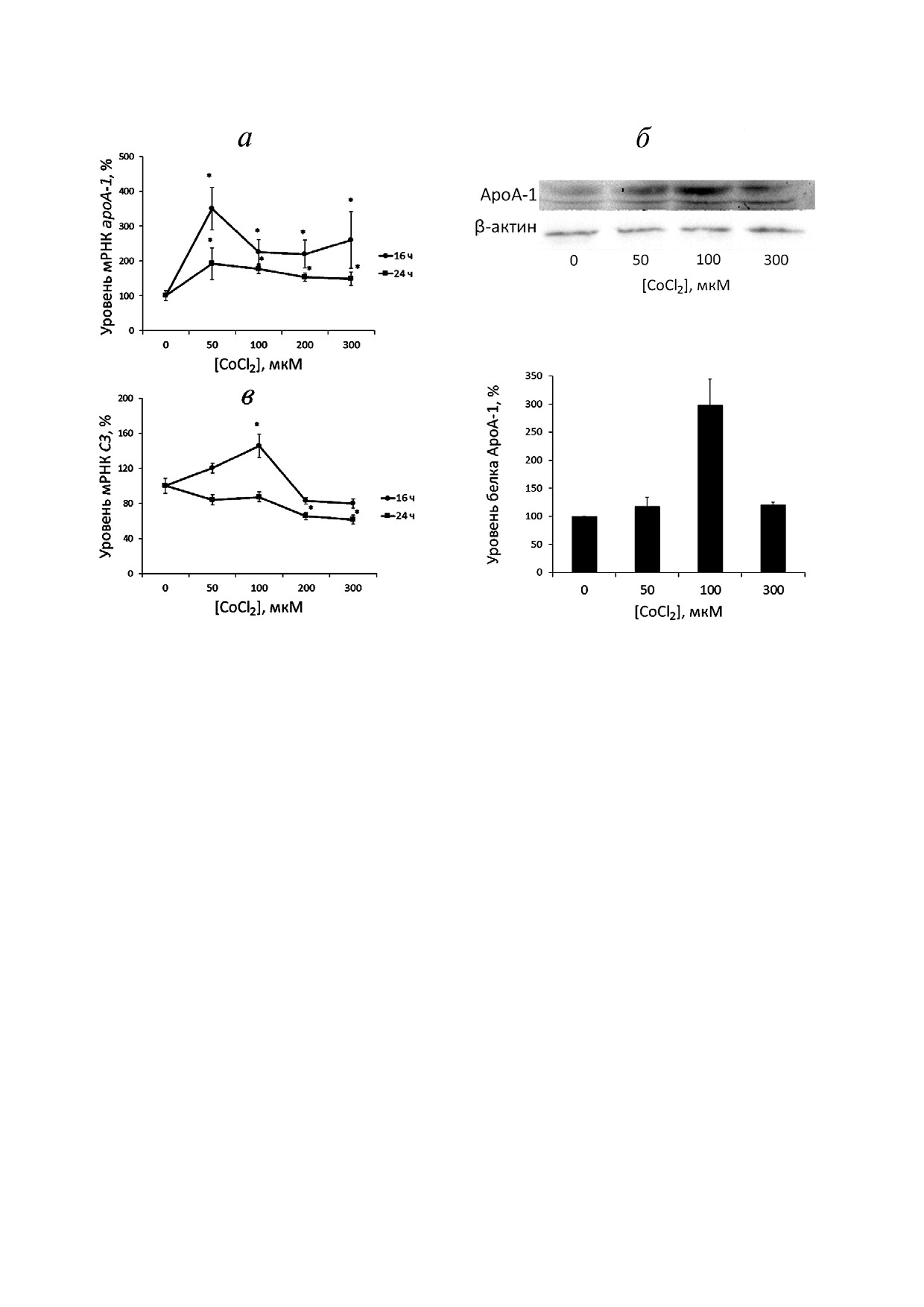
вень мРНК aроА 1 во всем диапазоне применяе!
ра транскрипции NF!κB (QNZ), которые вно!
мых концентраций на сроках инкубации 16 и 24 ч
сили в культуральную среду за 1 ч до добавления
(рис. 3, а). Увеличивается также количество
CoCl2. В качестве положительного контроля ис!
внутриклеточного (рис. 3, б) и поверхностного
пользовали агонист LXR (TO901317), приводя!
(рис. 4) белка ApoA!1. Уровень мРНК С3 возрас!
щий к значительному повышению уровня мРНК
тает при действии CoCl2 в концентрации 100 мкМ
АВСА1 [24] и аpoA 1 [9] в макрофагах.
на сроке инкубации 16 ч, но не 24 ч. При суточ!
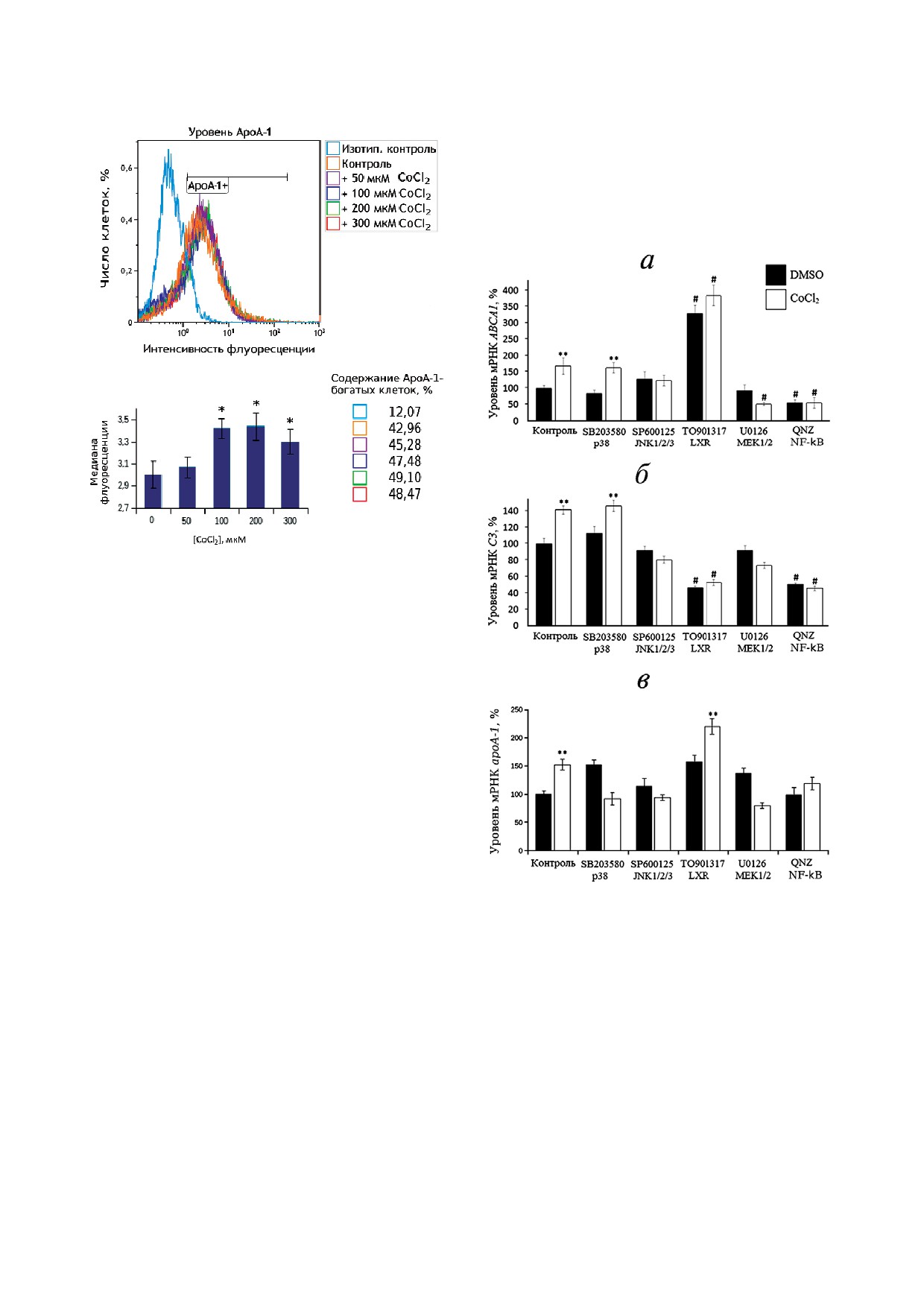
Показано, что ингибиторы МЕК1/2 и
ной инкубации активность гена снижается, на!
JNK1/2/3, но не р38, отменяют СоCl2!зависи!
чиная с концентрации 200 мкМ (рис. 3, в).
мую активацию экспрессии АВСА1 (рис. 5, а) и
Роль сигнальных каскадов MEK1/2-Erk1/2,
С3 (рис. 5, б). Ингибитор NF!κB, помимо отме!
MKK4/7-JNK1/2/3 и MKK3/6-p38, а также
ны действия CoCl2, снижает активность обоих
факторов транскрипции NF>κB и LXR в CoCl2>за>
генов в клетках, не обработанных CoCl2. Аго!
висимой активации генов АВСА1, С3 и apoA 1 в
нист LXR, как и ожидалось, сильно повышает
макрофагах THP>1. Известно, что транспортер
уровень мРНК АВСА1 самостоятельно и совмест!
Рис. 1. Выявление цитотоксических эффектов СoCl2. а - Проточная цитофлуориметрия. Каждая точка на графике пред!
ставлена как сумма квадрантов J+- и J++. График построен по данным трех независимых экспериментов. Планка погреш!
ности соответствует стандартной ошибке среднего; б - точечный график распределения макрофагов THP!1 по уровням
флуоресценции YO!PRO!1 и йодида пропидия при обработке 500 мкМ CoCl2. Ось YO!PRO!1 FL1 соответствует уровню
флуоресценции YO!PRO!1, ось PI FL5 INT - уровню флуоресценции йодида пропидия. Квадранты соответствуют процент!
ному содержанию клеток в разных состояниях: живых клеток (J--), клеток в состоянии раннего (J+-) и позднего (J++)
апоптоза, мертвых клеток (обломки клеток; J-+)
БИОХИМИЯ том 84 вып. 5 2019
ГИПОКСИЯ АКТИВИРУЕТ ГЕНЫ apoA 1, ABCA1 И C3 В МАКРОФАГАХ
697
но с CoCl2. Противоположный эффект агониста
тов связывания для фактора NF!κB, NF!κB!за!
наблюдается в отношении гена С3, уровень
висимая регуляция гена apoA 1 в макрофагах
мРНК падает относительно контроля в присут!
была описана ранее [9]. Вероятно, NF!κB регу!
ствии и в отсутствии CoCl2. Опосредованная ио!
лирует активность гена apoA 1, взаимодействуя
нами Co2+ индукция экспрессии гена apoA 1 за!
с лиганд!активированным ядерным рецептором
висит от всех трех основных MAP!киназных
PPARα (негативный регулятор apoA 1 в макро!
каскадов (рис. 5, в). При этом блокирование ки!
фагах), что приводит к инактивации обоих фак!
наз JNK приводит к отмене воздействия гипоксии
торов транскрипции.
на уровень активности гена apoA 1, тогда как
Таким образом, индукция клеточного ответа
ингибирование киназ p38 и MEK1/2 сопровож!
на гипоксию повышает уровень экспрессии ге!
дается инверсией эффекта ионов Co2+ (рис. 5, в).
нов aроА 1, АВСА1 и С3, а также уровень внут!
Стимулирующее действие ионов Co2+ отменя!
риклеточного и поверхностного белка АроА!1 в
лось также ингибитором NF!κB, но не агонис!
макрофагах ТНР!1. В регуляцию CoCl2!зависи!
том факторов транскрипции LXR. Хотя 5'!регу!
мой экспрессии генов АВСА1 и С3 вовлечены
ляторная область гена apoA 1 не содержит сай!
сигнальные каскады NF!кB, MKK4/7-JNK1/2/3
и MEK1/2-Erk1/2, но не MKK3/6-р38. CoCl2!за!
висимая индукция экспрессии гена apoA 1 зави!
сит от всех трех протестированных MAP!киназ!
ных каскадов и фактора транскрипции NF!κB.
Активация ядерного рецептора LXRα приводит
к возрастанию уровня мРНК АВСА1 и apoA 1 в
присутствии и в отсутствии CoCl2, но оказывает
противоположное влияние на экспрессию С3.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Присутствие локальной гипоксии в атероскле!
ротической бляшке описано в литературе [33].
Гипоксическое микроокружение является од!
ним из стимулов дифференцировки моноцитов
в макрофаги, в частности через активацию про!
воспалительных генов [34]. Сигналинг, запуска!
емый в условиях гипоксии, во многом пересека!
ется с воспалительным. Особенно четко это про!
слеживается на многоуровневом взаимодействии
факторов транскрипции NF!κB и HIF!1α, основ!
ного модулятора приспособления клетки к ги!
поксии. NF!κB не только опосредует цитокин!
зависимое увеличение уровня транскрипции ге!
на HIF1А при гипоксии, но и принимает участие
в конститутивной экспрессии фактора [35]. В свою
очередь, HIF!1α повышает уровень мРНК p65!
субъединицы NF!κB и IKKα [36]. Интересно,
что активация NF!κB в условиях гипоксии мо!
жет частично опосредоваться тем же механиз!
мом, что и стабилизация HIF!1α. Киназа IKKβ,
приводящая к диссоциации комплекса NF!κB/
IκBα, содержит консервативный мотив LxxLAP
регулирует экспрессию генов IL 8 (а) и ABCA1 (б)
Рис. 2. CoCl2
для гидроксилирования пролина. Такой же мо!
в макрофагах THP!1 в зависимости от концентрации и вре!
тив в HIF!1α при нормоксии гидроксилируется
мени инкубации; приведены результаты ОТ!ПЦР в реаль!
ном времени. Клетки инкубировали с CoCl2 в указанных
PHD!cодержащими ферментами. Ингибирование
концентрациях в течение 16 и 24 ч. Планки погрешности
PHD1 и PHD2 приводит к увеличению пула IKKβ
соответствуют стандартной ошибке среднего. Приведены
в клетке и последующей активации NF!κB [37].
данные по трем независимым экспериментам; * p < 0,05
В данной работе мы показали, что ингибиро!
(статистически значимые различия между уровнями мРНК
в контрольных и обработанных CoCl2 клетках, критерий
вание NF!κB не только инвертирует CoCl2!за!
Стьюдента)
висимое повышение экспрессии АВСА1 и С3, но
7 БИОХИМИЯ том 84 вып. 5 2019
698
БОГОМОЛОВА и др.
Рис. 3. CoCl2 регулирует экспрессию генов aроА 1 (а) и С3 (в) и увеличивает уровень внутриклеточного белка АроА!1 в
макрофагах THP!1 в зависимости от концентрации и времени инкубации (б); а, в - результаты ОТ!ПЦР в реальном вре!
мени. Клетки инкубировали с CoCl2 в указанных концентрациях в течение 16 и 24 ч. Планки погрешности соответствуют
стандартной ошибке среднего. Приведены данные по трем независимым экспериментам. Значения по оси ординат соот!
ветствуют уровню мРНК (%), за 100% принят уровень мРНК в необработанных клетках. * p < 0,05 (статистически значи!
мые различия между уровнями мРНК в сравниваемых группах, критерий Стьюдента); б - вестерн!блоттинг. Клетки ин!
кубировали с CoCl2 в указанных концентрациях в течение 16 ч. На диаграмме представлены данные, полученные в ходе
оцифровки трех иммуноблотов с помощью денситометрии. Значения по оси Y соответствуют относительному уровню
белка ApoA!1 (%), за 100% принят уровень белка ApoA!1 в необработанных клетках. Проведено выравнивание уровня
ApoA!1 по уровню β!актина
и приводит к снижению уровня мРНК этих ге!
HIF!1 и NF!κB. Известно, что активация HIF!1
нов без индукции ответа на гипоксию. На мак!
требует рекрутирования комплекса p300/CBP.
рофагах мыши выявлено, что уровень мРНК
Было показано, что ингибирование MAPK на!
АВСА1 повышается при обработке клеток TNFα
рушает взаимодействие HIF!1α/p300. Cверх!
в NF!κB!зависимой манере [38]. Также показано,
экспрессия MEK1 приводит к трансактивации
что нокдаун NF!κB в клетках эпителия тонкого
обоих белков [40]. Помимо взаимодействия с
кишечника предотвращает повышение уровня
коактиваторами, транскрипционная активность
транскрипции гена С3 в ответ на IL!1β [39]. Оче!
HIF!1α зависит от ряда посттрансляционных
видно, что TNFα! и IL!1β!сигналинг можно со!
модификаций. Фосфорилирование HIF!1α по
отнести с гипоксическим, т.к. в обоих случаях
остаткам Ser!641/643 киназой Erk1/2 предотвра!
основным модулятором служит NF!κB.
щает его выход из ядра с участием экспортина
Еще одним важным звеном сигнальных кас!
СRM1, таким образом достигается накопление
кадов, активируемых при гипоксии, являются
активного HIF!1α в ядрах клеток [41]. Кроме то!
киназы семейства МАР. Мы выявили, что кина!
го, Erk1/2, но не р38, участвует в трансактива!
зы MEK1/2 и JNK1/2/3, но не р38, вовлечены в
ции p65!субъединицы NF!κB. Это взаимодей!
регуляцию экспрессии генов АВСА1 и С3 в мак!
ствие вовлечено в положительную регуляцию
рофагах ТНР!1 при индукции ответа на гипо!
экспрессии гена MIP 2 в макрофагах мыши при
ксию. Существует множество данных, указыва!
гипоксии [42]. Активация JNK1/2/3 в условиях
ющих на кооперацию путей MEK1/2-Erk1/2,
низкого содержания кислорода показана как в
БИОХИМИЯ том 84 вып. 5 2019
ГИПОКСИЯ АКТИВИРУЕТ ГЕНЫ apoA 1, ABCA1 И C3 В МАКРОФАГАХ
699
взаимодействующих с сайтами B и C гепатоци!
тарного энхансера гена apoA 1 [45]. Более того,
разрушение сайтов B или C в гепатоцитарном
энхансере гена apoA 1 приводило к инверсии
воздействия перекиси водорода (оксидативный
стресс) на активность 5'!регуляторной области
Рис. 4. CoCl2 увеличивает уровень поверхностного белка
АроА!1 в макрофагах THP!1: проточная цитофлуоримет!
рия. Графики отражают изменение уровня мембранного
АроА!1 при обработке клеток CoCl2 в указанных концент!
рациях. ApoA!1+ - ApoA!1!богатые макрофаги. На диаг!
рамме представлены медианы распределений клеток по
уровню поверхностного ApoA!1 при различных концент!
рациях CoCl2. * p < 0,01 (статистически значимые отличия
от контроля (нулевая концентрация CoCl2), критерий
Стьюдента). Процент клеток соответствует проценту
ApoA!1!богатых макрофагов при различных концентра!
циях CoCl2 (отмечены соответствующими цветами).
С цветным вариантом рис. 4 можно ознакомиться в элект!
макрофагах [28], так и в других типах клеток
[30]. Индукция экспрессии гена apoA 1 в макро!
фагах в условиях гипоксии зависит также и от
активации провоспалительного MAP!киназно!
го каскада p38 помимо каскадов Erk1/2 и
JNK1/2/3. Известно, что гипоксия повышает
уровень фосфорилированных p38 (активная фор!
Рис. 5. Роль MAP!киназных каскадов MEK1/2-Erk1/2,
MKK4/7-JNK1/2/3 и MKK3/6-p38, а также факторов
ма) в нейронах коры гиппокампа [43]. В макро!
транскрипции NF!κB и LXR в CoCl2!зависимой активации
фагах гипоксия усиливает активацию p38 в ответ
генов АВСА1 (а), С3 (б) и apoA 1 (в) в макрофагах THP!1;
на пальмитиновую кислоту (вызывает стресс эн!
приведены результаты ОТ!ПЦР в реальном времени.
доплазматического ретикулума) [44]. Блокиро!
Клетки до внесения в среду 100 мкМ CoCl2 инкубировали в
вание каскадов p38 и Erk1/2 приводило к усиле!
течение 1 ч с одним из соединений: 12,5 мкM SB203580,
10 мкM SP600125, 10 мкM U0126, 10 нM QNZ, 2,5 мкM
нию базовой экспрессии apoA 1 и к инверсии
TO901317. Приведены данные по трем независимым экс!
стимулирующего воздействия ионов кобальта.
периментам. Значения по оси ординат соответствуют
В предыдущих работах [45, 46] мы наблюдали
уровню мРНК (%), за 100% принят уровень мРНК в клет!
усиление базовой экспрессии apoA 1 при блоки!
ках, обработанных DMSO. Планка погрешности соответ!
ствует стандартной ошибке среднего. ** p < 0,05 (статисти!
ровании сигнального каскада Erk1/2 в клетках
чески значимые различия между уровнями мРНК в срав!
гепатомы человека HepG2. Установлено, что этот
ниваемых группах, критерий Даннета); # p < 0,05 (статис!
эффект обусловлен фосфорилированием комп!
тически значимые отличия от уровня мРНК в клетках, об!
лекса факторов транскрипции FOXO1/LXRβ, работанных DMSO; критерий Стьюдента)
БИОХИМИЯ том 84 вып. 5 2019
7*
700
БОГОМОЛОВА и др.
следованиях показано ТО901317!зависимое по!
вышение уровня мРНК С3 в макрофагах ТНР!1
5!дневной дифференцировки [18]. В настоящей
работе срок дифференцировки макрофагов
ТНР!1 составил 3 дня. Такой эффект можно со!
поставить c механизмом регуляции экспрессии
aроА 1 под действием TNFα, выявленным в на!
шей лаборатории ранее. В моноцитах ТНР!1
TNFα приводит более чем к пятикратному по!
вышению уровня мРНК aроА 1, в то время как в
макрофагах THP!1 наблюдается повышение
уровня мРНК aроА 1 лишь в 1,5 раза. Показана
также и зависимая от времени экспрессия aроА 1
в моноцитах ТНР!1. Пятикратная индукция
экспрессии наблюдается при 24!часовой инку!
бации с TNFα, 48!часовая инкубация приводит
к падению уровня мРНК aроА 1 до контрольно!
го значения [9].
Обобщая приведенные выше литературные
и полученные нами данные, можно рассмотреть
механизм положительной регуляции экспрес!
сии генов aроА 1, АВСА1 и С3 в макрофагах при
индукции ответа на гипоксию (рис. 6). Повы!
шенный уровень мРНК и, по!видимому, белка
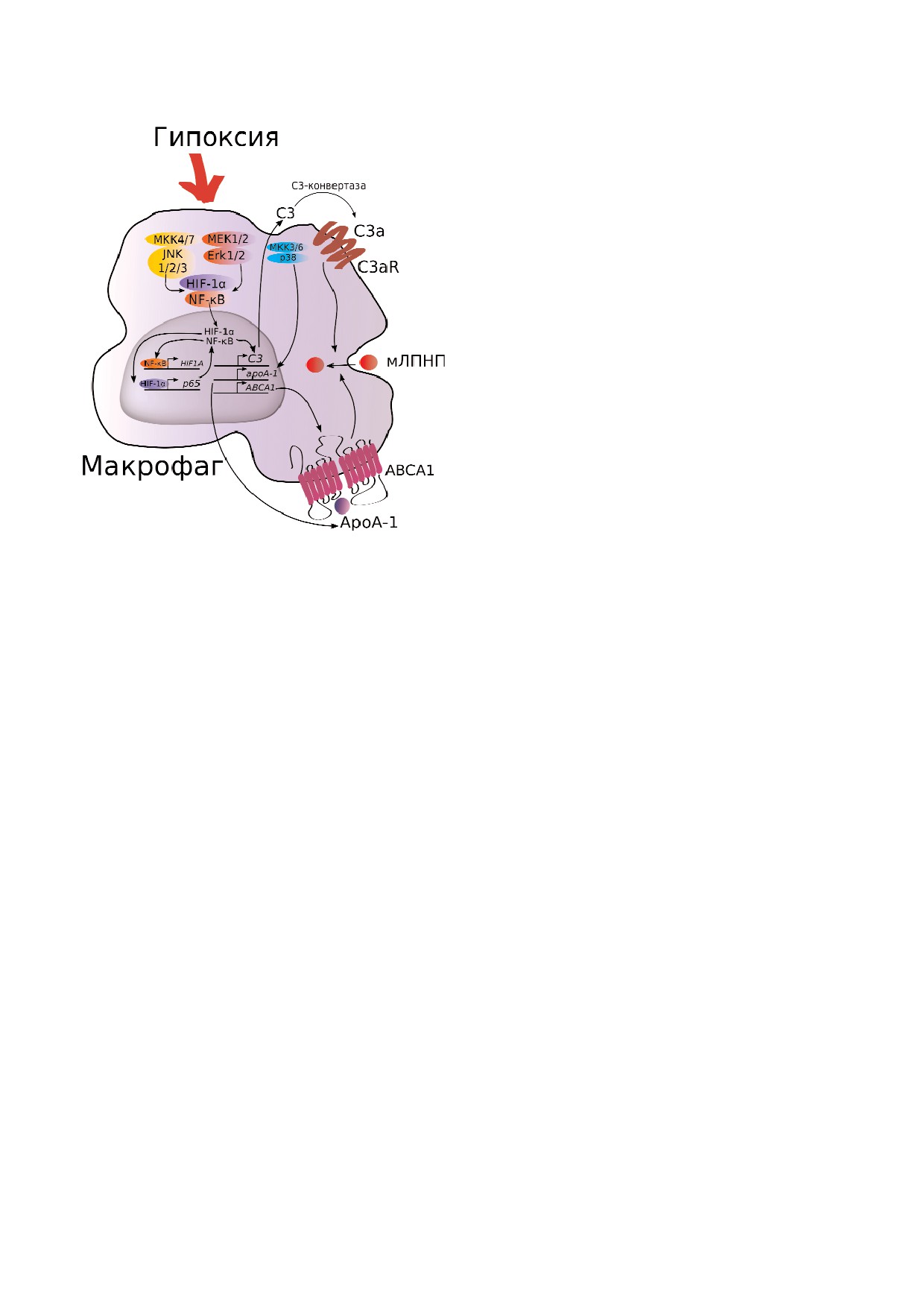
Рис. 6. Гипотетическая схема регуляции генов ABCA1, C3 и
C3 приводит к увеличению продукции анафила!
apoA 1 в макрофагах человека в условиях гипоксии (пояс!
токсина C3a. Связываясь со своим рецептором
нения см. в разделе «Обсуждение результатов»).
С цветным вариантом рис. 6 можно ознакомиться в элект!
на поверхности макрофагов, С3а индуцирует
повышение уровня экспрессии С3 и усиленный
захват модифицированных ЛПНП (мЛПНП) [18].
Хотя эндогенный АроА!1 стабилизирует АВСА1
гена apoA 1 в клетках HepG2 [46]. Учитывая вы!
и проявляет противовоспалительные свойства,
сокое сходство сигнальных путей, активируе!
его содержание не коррелирует с уровнем обрат!
мых провоспалительными цитокинами, при ги!
ного транспорта холестерина [8]. С другой сто!
поксии и в условиях оксидативного стресса [25,
роны, известно, что перенос гена aроА 1 челове!
31-37, 44], а также тот факт, что факторы транс!
ка в макрофаги мышей, нокаутных по apoA 1, с по!
крипции FOXO1 и LXRβ взаимодействуют с
мощью ретровирусных векторов приводит к зна!
сайтами B и С гепатоцитарного энхансера гена
чительному уменьшению размера бляшек [47].
apoA 1 не только в клетках HepG2, но и в макро!
Кроме того, показано, что нокаут по транспор!
фагах человека [9], можно предположить, что
терам АВСА1 и АВСG1 ведет к ухудшению хе!
обнаруженная в данной работе инверсия воз!
мотаксиса макрофагов [48]. Принимая во вни!
действия ионов кобальта на активность гена
мание нарушение миграционной активности
apoA 1 при блокировании MAP!киназных кас!
макрофагов при гипоксии, можно предполо!
кадов p38 и Erk1/2 обусловлена изменением
жить нивелирование этого эффекта эндогенным
уровня фосфорилирования факторов транскрип!
АроА!1 через стабилизацию АВСА1. Таким об!
ции LXRβ и FOXO1. Необходимы дальнейшие
разом, если индукцию экспрессии С3 при ги!
исследования для прояснения данного вопроса.
поксии можно рассматривать как атерогенный
В качестве положительного контроля в на!
фактор, роль повышения уровней мРНК aроА 1
шей системе использовался синтетический аго!
и АВСА1 остается спорной и требует дальнейше!
нист LXR TO901317, индуцирующий значитель!
го изучения.
ное увеличение уровня экспрессии гена АВСА1.
Как и ожидалось, агонист повысил уровень
мРНК АВСА1 в макрофагах THP!1 как самосто!
Финансирование. Работа выполнена при фи!
ятельно, так и совместно с CoCl2, что предпола!
нансовой поддержке Российского научного
гает независимое действие LXR и NF!κB/HIF!1,
фонда (грант № 17!15!01326; исследования ре!
описанное в данных Gerbod!Giannone et al. [38].
гуляции экспрессии генов ABCA1 и С3 в услови!
Интересно обратное действие ТО901317 на
ях гипоксии; рис. 1 и 2; рис. 3, б; рис. 5, а, б) и
экспрессию гена С3. В наших предыдущих ис!
Российского фонда фундаментальных исследо!
БИОХИМИЯ том 84 вып. 5 2019
ГИПОКСИЯ АКТИВИРУЕТ ГЕНЫ apoA 1, ABCA1 И C3 В МАКРОФАГАХ
701
ваний (грант № 17!04!01947; исследование экс!
Соблюдение этических норм. Настоящая статья
прессии гена apoA 1; рис. 3, а, в; рис. 4; рис. 5, в).
не содержит описания каких!либо исследова!
Конфликт интересов. Авторы заявляют об от!
ний с участием людей и использованием живот!
сутствии конфликта интересов.
ных в качестве объектов.
СПИСОК ЛИТЕРАТУРЫ
1.
Heo, K.!S., Fujiwara, K., and Abe, J. (2014) Shear stress
12.
Binder, C.J., Chang, M.!K., Shaw, P.X., Miller, Y.I.,
and atherosclerosis, Mol. Cells, 37, 435-440, doi: 10.14348/
Hartvigsen, K., Dewan, A., and Witztum, J.L. (2002)
molcells.2014.0078.
Innate and acquired immunity in atherogenesis, Nat. Med.,
2.
Bonomini, F., Tengattini, S., Fabiano, A., Bianchi, R., and
8, 1218-1226, doi: 10.1038/nm1102!1218.
Rezzani, R. (2008) Atherosclerosis and oxidative stress,
13.
Samstad, E.O., Niyonzima, N., Nymo, S., Aune, M.H.,
Histol. Histopathol., 23, 381-390, doi: 10.14670/HH!23.381.
Ryan, L., Bakke, S.S., Lappegard, K.T., Brekke, O.!L.,
3.
Fong, G.!H. (2015) Potential contributions of intimal and
Lambris, J.D., Damas, J.K., Latz, E., Mollnes, T.E., and
plaque hypoxia to atherosclerosis, Curr. Atheroscler. Rep.,
Espevik, T. (2014) Cholesterol crystals induce comple!
17, 32-41, doi: 10.1007/s11883!015!0510!0.
ment!dependent inflammasome activation and cytokine
4.
Parathath, S., Mick, S.L., Feig, J.E., Joaquin, V., Grauer, L.,
release, J. Immunol., 192, 2837-2845, doi: 10.4049/jim!
Habiel, D.M., Gassmann, M., Gardner, L.B., and Fisher, E.A.
munol.1302484.
(2011) Hypoxia is present in murine atherosclerotic
14.
Buono, C., Come, C.E., Witztum, J.L., Maguire, G.F.,
plaques and has multiple adverse effects on macrophage
Connelly, P.W., Carroll, M., and Lichtman, A.H. (2002)
lipid metabolism, Circ. Res., 109, 1141-1152, doi: 10.1161/
Influence of C3 deficiency on atherosclerosis, Circulation,
CIRCRESAHA.111.246363.
105, 3025-3031.
5.
Epstein, A.C., Gleadle, J.M., McNeill, L.A. Hewitson, K.S.,
15.
Persson, L., Boren, J., Robertson, A.!K., Wallenius, V.,
O’Rourke, J., Mole, D.R., Mukherji, M., Metzen, E.,
Hansson, G.K., and Pekna, M. (2004) Lack of comple!
Wilson, M.I., Dhanda, A., Tian, Y.M., Masson, N.,
ment factor C3, but not factor B, increases hyperlipidemia
Hamilton, D.L., Jaakkola, P., Barstead, R., Hodgkin, J.,
and atherosclerosis in apolipoprotein E-/- low!density
Maxwell, P.H., Pugh, C.W., Schofield, C.J., and Ratcliffe, P.J.
lipoprotein receptor-/- mice, Arterioscler. Thromb. Vasc. Biol.,
(2001) C. elegans EGL!9 and mammalian homologs define
24, 1062-1067, doi: 10.1161/01.ATV.0000127302.24266.40.
a family of dioxygenases that regulate HIF by prolyl
16.
Amarilyo, G., Verbovetski, I., Atallah, M., Grau, A., Wiser, G.,
hydroxylation, Cell, 107, 43-54, doi: 10.1016/S0092!
Gil, O., Ben!Neriah, Y., and Mevorach, D. (2010) iC3b!
8674(01)00507!4.
opsonized apoptotic cells mediate a distinct anti!inflam!
6.
Hirani, N., Antonicelli, F., Strieter, R.M., Wiesener, M.S.,
matory response and transcriptional NF!κB!dependent
Ratcliffe, P.J., Haslett, C., and Donnelly, S.C. (2001) The
blockade, Eur. J. Immunol., 40, 699-709, doi: 10.1002/
regulation of interleukin!8 by hypoxia in human
eji.200838951.
macrophages - a potential role in the pathogenesis of the
17.
McVicar, J.P., Kunitake, S.T., Hamilton, R.L., and Kane, J.P.
acute respiratory distress syndrome (ARDS), Mol. Med.
(1984) Characteristics of human lipoproteins isolated by
Camb. Mass, 7, 685-697.
selected!affinity immunosorption of apolipoprotein A!I,
7.
Murdoch, C., Muthana, M., and Lewis, C.E. (2005) Hypoxia
Proc. Natl. Acad. Sci. USA, 81, 1356-1360.
regulates macrophage functions in inflammation, J. Im
18.
Mogilenko, D.A., Kudriavtsev, I.V., Trulioff, A.S., Shavva, V.S.,
munol., 175, 6257-6263, doi: 10.4049/jimmunol.175.
Dizhe, E.B., Missyul, B.V., Zhakhov, A.V., Ischenko, A.M.,
10.6257.
Perevozchikov, A.P., and Orlov, S.V. (2012) Modified low
8.
Mogilenko, D.A., Orlov, S.V., Trulioff, A.S., Ivanov, A.V.,
density lipoprotein stimulates complement C3 expression
Nagumanov, V.K., Kudriavtsev, I.V., Shavva, V.S.,
and secretion via liver X receptor and toll!like receptor 4
Tanyanskiy, D.A., and Perevozchikov, A.P.
(2012)
activation in human macrophages, J. Biol. Chem., 287,
Endogenous apolipoprotein A!I stabilizes ATP!binding
5954-5968, doi: 10.1074/jbc.M111.289322.
cassette transporter A1 and modulates Toll!like receptor 4
19.
Orlov, S.V., Mogilenko, D.A., Shavva, V.S., Dizhe, E.B.,
signaling in human macrophages, FASEB J.,
26,
Ignatovich, I.A., and Perevozchikov, A.P. (2010) Effect of
2019-2030, doi: 10.1096/fj.11!193946.
TNFα on activities of different promoters of human
9.
Shavva, V.S., Mogilenko, D.A., Nekrasova, E.V., Trulioff, A.S.,
apolipoprotein A!I gene, Biochem. Biophys. Res. Commun.,
Kudriavtsev, I.V., Larionova, E.E., Babina, A.V., Dizhe, E.B.,
398, 224-230, doi: 10.1016/j.bbrc.2010.06.064.
Missyul, B.V., and Orlov, S.V. (2018) Tumor necrosis factor
20.
Mogilenko, D.A., Shavva, V.S., Dizhe, E.B., Orlov, S.V.,
alpha stimulates endogenous apolipoprotein A!I expres!
and Perevozchikov, A.P. (2010) PPARγ activates ABCA1
sion and secretion by human monocytes and macrophages:
gene transcription but reduces the level of ABCA1 protein
role of MAP!kinases, NF!κB, and nuclear receptors
in HepG2 cells, Biochem. Biophys. Res. Commun., 402,
PPARα and LXRs, Mol. Cell. Biochem., 448, 211-223,
477-482, doi: 10.1016/j.bbrc.2010.10.053.
doi: 10.1007/s11010!018!3327!7.
21.
Mogilenko, D.A., Kudriavtsev, I.V., Shavva, V.S., Dizhe, E.B.,
10.
Speidl, W.S., Kastl, S.P., Huber, K., and Wojta, J. (2011)
Vilenskaya, E.G., Efremov, A.M., Perevozchikov, A.P., and
Complement in atherosclerosis: friend or foe? J. Thromb.
Orlov, S.V. (2013) Peroxisome proliferator!activated recep!
Haemost., 9, 428-440, doi: 10.1111/j.1538!7836.2010.04172.x.
tor alpha positively regulates complement C3 expression
11.
Hertle, E., van Greevenbroek, M.M., Arts, I.C., van der
but inhibits TNFα!mediated activation of C3 gene in
Kallen, C.J., Geijselaers, S.L., Feskens, E.J., Jansen, E.H.,
mammalian hepatic derived cells, J. Biol. Chem., 288,
Schalkwijk, C.G., and Stehouwer, C.D. (2014) Distinct
1726-1738, doi: 10.1074/jbc.M112.437525.
associations of complement C3a and its precursor C3 with
22.
Vengellur, A., and LaPres, J.J. (2004) The role of hypoxia
atherosclerosis and cardiovascular disease. The CODAM
inducible factor 1α in cobalt chloride induced cell death in
study, Thromb. Haemost., 111, 1102-1111, doi: 10.1160/
mouse embryonic fibroblasts, Toxicol. Sci., 82, 638-646,
TH13!10!0831.
doi: 10.1093/toxsci/kfh278.
БИОХИМИЯ том 84 вып. 5 2019
702
БОГОМОЛОВА и др.
23.
Rydberg, E.K., Salomonsson, L., Hulten, L. M., Noren, K.,
hydroxylase!1 negatively regulates IкB kinase!β, giving
Bondjers, G., Wiklund, O., Bjornheden, T., and Ohlsson, B.G.
insight into hypoxia!induced NF!кB activity, Proc. Natl.
(2003) Hypoxia increases 25!hydroxycholesterol!induced
Acad. Sci. USA, 103, 18154-18159, doi: 10.1073/pnas.
interleukin!8 protein secretion in human macrophages,
0602235103.
Atherosclerosis,
170,
245-252, doi:
10.1016/S0021!
38.
Gerbod!Giannone, M.!C., Li, Y., Holleboom, A., Han, S.,
9150(03)00302!2.
Hsu, L.!C., Tabas, I., and Tall, A.R. (2006) TNFα induces
24.
Schmitz, G., and Langmann, T. (2005) Transcriptional
ABCA1 through NF!κB in macrophages and in phagocytes
regulatory networks in lipid metabolism control ABCA1
ingesting apoptotic cells, Proc. Natl. Acad. Sci. USA, 103,
expression, Biochim. Biophys. Acta, 1735, 1-19, doi: 10.1016/
3112-3117, doi: 10.1073/pnas.0510345103.
j.bbalip.2005.04.004.
39.
Moon, M.R., Parikh, A.A., Pritts, T.A., Fischer, J.E.,
25.
Marsch, E., Sluimer, J.C., and Daemen, M.J. (2013)
Cottongim, S., Szabo, C., Salzman, A.L., and Hasselgren, P.O.
Hypoxia in atherosclerosis and inflammation, Curr.
(1999) Complement component C3 production in IL!1β!
Opin. Lipidol., 24, 393-400, doi: 10.1097/MOL.0b013e!
stimulated human intestinal epithelial cells is blocked by
32836484a4.
NF!κB inhibitors and by transfection with ser 32/36
26.
Li, R.C., Haribabu, B., Mathis, S.P., Kim, J., and Gozal, D.
mutant IκBα, J. Surg. Res., 82, 48-55, doi: 10.1006/jsre.
(2011) Leukotriene B4 receptor!1 mediates intermittent
1998.5503.
hypoxia!induced atherogenesis, Am. J. Respir. Crit. Care
40.
Sang, N., Stiehl, D. P., Bohensky, J., Leshchinsky, I.,
Med., 184, 124-131, doi: 10.1164/rccm.201012!2039OC.
Srinivas, V., and Caro, J. (2003) MAPK signaling up!regu!
27.
Ortiz!Masia, D., Diez, I., Calatayud, S., Hernandez, C.,
lates the activity of hypoxia!inducible factors by its effects
Cosin!Roger, J., Hinojosa, J., Esplugues, J.V., and
on p300, J. Biol. Chem., 278, 14013-14019, doi: 10.1074/
Barrachina, M.D. (2012) Induction of CD36 and throm!
jbc.M209702200.
bospondin!1 in macrophages by hypoxia!inducible factor 1
41.
Mylonis, I., Chachami, G., Samiotaki, M., Panayotou, G.,
and its relevance in the inflammatory process, PLoS One,
Paraskeva, E., Kalousi, A., Georgatsou, E., Bonanou, S.,
7, e48535, doi: 10.1371/journal.pone.0048535.
and Simos, G. (2006) Identification of MAPK phosphory!
28.
Fuhrmann, D.C., Tausendschon, M., Wittig, I., Steger, M.,
lation sites and their role in the localization and activity of
Ding, M.G., Schmid, T., Dehne, N., and Brune, B. (2015)
hypoxia!inducible factor!1α, J. Biol. Chem.,
281,
Inactivation of tristetraprolin in chronic hypoxia provokes
33095-33106, doi: 10.1074/jbc.M209702200.
the expression of cathepsin B, Mol. Cell. Biol., 35,
42.
Zampetaki, A., Mitsialis, S. A., Pfeilschifter, J., and
619-630, doi: 10.1128/MCB.01034!14.
Kourembanas, S. (2004) Hypoxia induces macrophage
29.
Li, Q., Yu, B., and Yang, P. (2015) Hypoxia!induced
inflammatory protein!2 (MIP!2) gene expression in
HMGB1 in would tissues promotes the osteoblast cell pro!
murine macrophages via NF!κB: the prominent role of
liferation via activating ERK/JNK signaling, Int. J. Clin.
p42/p44 and PI3 kinase pathways, FASEB J.,
18,
Exp. Med., 8, 15087-15097.
1090-1092, doi: 10.1096/fj.03!0991fje.
30.
Chiu, C.!Z., Wang, B.!W., and Shyu, K.!G. (2014)
43.
Haddad, J.J., and Hanbali, L.B. (2014) Hypoxia upregu!
Angiotensin II and the JNK pathway mediate urotensin II
lates MAPK(p38)/MAPK(ERK) phosphorylation in vitro:
expression in response to hypoxia in rat cardiomyocytes,
neuroimmunological differential time!dependent expres!
J. Endocrinol., 220, 233-246, doi: 10.1530/JOE!13!0261.
sion of MAPKs, Protein Pept. Lett., 21, 444-451, doi: 10.
31.
Zhang, J., Liu, Q., Fang, Z., Hu, X., Huang, F., Tang, L.,
2174/092986652105140218112521.
and Zhou, S. (2016) Hypoxia induces the proliferation of
44.
Snodgrass, R.G., Boss, M., Zezina, E., Weigert, A.,
endothelial progenitor cells via upregulation of Apelin/
Dehne, N., Fleming, I., Brune, B., and Namgaladze, D.
APLNR/MAPK signaling, Mol. Med. Rep., 13, 1801-1806,
(2016) Hypoxia potentiates palmitate!induced pro!inflam!
doi: 10.3892/mmr.2015.4691.
matory activation of primary human macrophages, J. Biol.
32.
Wu, Y., Yang, Y., Yang, P., Gu, Y., Zhao, Z., Tan, L., Zhao, L.,
Chem., 291, 413-424, doi: 10.1074/jbc.M115.686709.
Tang, T., and Li, Y. (2013) The osteogenic differentiation
45.
Shavva, V.S., Bogomolova, A.M., Nikitin, A.A., Dizhe, E.B.,
of PDLSCs is mediated through MEK/ERK and p38
Tanyanskiy, D.A., Efremov, A.M., Oleinikova, G.N.,
MAPK signalling under hypoxia, Arch. Oral Biol., 58,
Perevozchikov, A.P., and Orlov, S.V. (2017) Insulin!medi!
1357-1368, doi: 10.1016/j.archoralbio.2013.03.011.
ated downregulation of apolipoprotein A!I gene in human
33.
Mayr, M., Sidibe, A., and Zampetaki, A. (2008) The para!
hepatoma cell line HepG2: the role of interaction between
dox of hypoxic and oxidative stress in atherosclerosis,
FOXO1 and LXRβ transcription factors, J. Cell. Biochem.,
J. Am. Coll. Cardiol., 51, 1266-1267, doi: 10.1016/j.jacc.
118, 382-396, doi: 10.1002/jcb.25651.
2008.01.005.
46.
Shavva, V.S., Bogomolova, A.M., Nikitin, A.A., Dizhe, E.B.,
34.
Strehl, C., Fangradt, M., Fearon, U., Gaber, T., Buttgereit, F.,
Oleinikova, G.N., Lapikov, I.A., Tanyanskiy, D.A.,
and Veale, D.J. (2014) Hypoxia: how does the monocyte!
Perevozchikov, A.P., and Orlov, S.V. (2017) FOXO1 and
macrophage system respond to changes in oxygen avail!
LXRα downregulate the apolipoprotein A!I gene expres!
ability? J. Leukoc. Biol., 95, 233-241, doi: 10.1189/jlb.
sion during hydrogen peroxide!induced oxidative stress in
1212627.
HepG2 cells, Cell Stress Chaperones, 22, 123-134, doi: 10.
35.
Taylor, C.T., and Cummins, E.P. (2009) The role of NF!κB
1007/s12192!016!0749!6.
in hypoxia!induced gene expression, Ann. N. Y. Acad. Sci.,
47.
Ishiguro, H., Yoshida, H., Major, A.S., Zhu, T., Babaev, V.R.,
1177, 178-184, doi: 10.1111/j.1749!6632.2009.05024.x.
Linton, M.F., and Fazio, S. (2001) Retrovirus!mediated
36.
Walmsley, S.R., Print, C., Farahi, N., Peyssonnaux, C.,
expression of apolipoprotein A!I in the macrophage pro!
Johnson, R.S., Cramer, T., Sobolewski, A., Condliffe, A.M.,
tects against atherosclerosis in vivo, J. Biol. Chem., 276,
Cowburn, A.S., Johnson, N., and Chilvers, E.R. (2005)
36742-36748, doi: 10.1074/jbc.M106027200.
Hypoxia!induced neutrophil survival is mediated by HIF!
48.
Pagler, T.A., Wang, M., Mondal, M., Murphy, A.J.,
1α!dependent NF!κB activity, J. Exp. Med.,
201,
Westerterp, M., Moore, K.J., Maxfield, F.R., and Tall, A.R.
105-115, doi: 10.1084/jem.20040624.
(2011) Deletion of ABCA1 and ABCG1 impairs macro!
37.
Cummins, E.P., Berra, E., Comerford, K.M., Ginouves, A.,
phage migration because of increased Rac1 signaling, Circ.
Fitzgerald, K.T., Seeballuck, F., Godson, C., Nielsen, J.E.,
Res., 108, 194-200, doi: 10.1161/CIRCRESAHA.110.
Moynagh, P., Pouyssegur, J., and Taylor, C.T. (2006) Prolyl
228619.
БИОХИМИЯ том 84 вып. 5 2019
ГИПОКСИЯ АКТИВИРУЕТ ГЕНЫ apoA 1, ABCA1 И C3 В МАКРОФАГАХ
703
HYPOXIA AS A FACTOR INVOLVED IN REGULATION
OF EXPRESSION apoA 1, ABCA1 AND COMPLEMENT
COMPONENT C3 GENES IN HUMAN MACROPHAGES
A. M. Bogomolova1,2, V. S. Shavva2, A. A. Nikitin1,2, E. V. Nekrasova2,
E. B. Dizhe2, E. E. Larionova2, I. V. Kudriavtsev2, and S. V. Orlov1,2*
1 St. Petersburg State University, 199034 St. Petersburg, Russia;
E mail: s.orlov@spbu.ru, serge@iem.sp.ru
2 Institute of Experimental Medicine, 197376 St. Petersburg, Russia
Received December 10, 2018
Revised January 29, 2019
Accepted January 29, 2019
Hypoxia plays a crucial role in atherosclerosis progression. Local decrease of oxygen availability in a plaque creates
specific microenvironment that alters transcriptome in resident cells, in particular, macrophages. Reverse cholesterol
transport from plaque to liver is considered to be one of the ways of atherosclerosis regression. Ubiquitously expressed
ATP!binding cassette transporter A1 (ABCA1) and liver!derived apolipoprotein A!1 (ApoA!1) are two main partici!
pants of this process. Recently we have shown endogenous apoA 1 gene expression in human macrophages. While
ABCA1 and ApoA!1 possess antiatherogenic properties, the role of a complement factor C3 is controversial. Plasma
C3 level positively correlates with the risk of cardiovascular diseases in humans. On the other side, C3 gene knockout
in murine atherosclerosis model results in increase both in plaque size and triglycerides content in blood. In the pre!
sent work we have shown for the first time that hypoxia!mimicking agent, CoCl2, caused the upregulation of apoA 1
and C3 genes along with the increase in ApoA!1 intracellular and membrane content in THP!1 macrophages.
MEK1/2-Erk1/2 and MKK4/7-JNK1/2/3 cascades are involved in ABCA1 and C3 gene upregulation via activation
of NF!κB transcription factor, which synergies with HIF!1α subunit of HIF!1 (hypoxia!inducible factor 1). All three
main MAP!kinase cascades (Erk1/2, JNK1/2/3, and p38) as well as NF!κB transcription factor are involved in the
hypoxia!induced expression of apoA 1 gene in THP!1 macrophages.
Keywords: atherosclerosis, hypoxia, macrophages, THP!1, apoA 1 gene, ABCA1 gene, C3 gene
БИОХИМИЯ том 84 вып. 5 2019