БИОХИМИЯ, 2019, том 84, вып. 5, с. 719 - 729
УДК 577.1
ХИМИЧЕСКИЕ ИНДУКТОРЫ
МЕТАБОЛИЧЕСКОГО СТРЕССА, СВЯЗАННОГО С ОЖИРЕНИЕМ,
АКТИВИРУЮТ ВОСПАЛЕНИЕ, СНИЖАЯ ЧУВСТВИТЕЛЬНОСТЬ
К ИНСУЛИНУ В АДИПОЦИТАХ 3T3 L1
© 2019 Ю.С. Стафеев1,2*, С.С. Мичурина1,3, Н.В. Подкуйченко1,3,
М.Ю. Меньшиков1, Е.В. Парфёнова1,2#, А.В. Воротников1,4#*
1 НИИ экспериментальной кардиологии, НМИЦ кардиологии, 121552 Москва,
Россия; электронная почта: yuristafeev@gmail.com, a.vorotnikov@icloud.com
2 Московский государственный университет им. М.В. Ломоносова,
факультет фундаментальной медицины, 117192 Москва, Россия
3 Московский государственный университет им. М.В. Ломоносова,
биологический факультет, 119991 Москва, Россия
4 Медицинский научно*образовательный центр, Московский государственный
университет им. М.В. Ломоносова, 119991 Москва, Россия
Поступила в редакцию 08.10.2018
После доработки 23.01.2019
Принята к публикации 23.01.2019
Ожирение сопровождается развитием дислипидемии, гипоксии, стресса эндоплазматического ретикулума
(ЭПР) и воспаления, являясь основным фактором риска развития инсулиновой резистентности и сахарно$
го диабета 2$го типа. Мы смоделировали эти состояния в культивируемых адипоцитах 3T3$L1, определив их
влияние на инсулиновую сигнализацию, транспорт глюкозы в клетки и развитие воспалительной реакции
по активации стресс$зависимых киназ JNK1/2. Во всех условиях жировой перегрузки клеток при культиви$
ровании с пальмитатом, острого воспаления при обработке клеток бактериальным липополисахаридом, ин$
дукции гипоксии солями двухвалентного кобальта или стресса ЭПР, вызванного обработкой клеток брефел$
дином А, наблюдалось снижение активации инсулином фосфорилирования компонентов инсулинового
каскада IRS, Akt и AS160. Во всех условиях, кроме острого воспаления, снижался инсулин$зависимый за$
хват глюкозы адипоцитами, а кинетика активации JNK1/2 была двухфазной, демонстрируя повышенную
активность в течение 24 ч. Напротив, в случае острого воспаления кинетика активирующего фосфорилиро$
вания JNK1/2 была транзиторной, и уровень фосфорилирования JNK возвращался к базальному уровню
через 2-3 ч стимуляции. Эти результаты указывают на ключевую роль длительного (латентного), но не
быстрого (острого) воспаления в индукции инсулиновой резистентности и в нарушении утилизации глюко$
зы жировой тканью. Таким образом, участники воспалительного сигнального каскада могут быть перспек$
тивными мишенями при разработке новых терапевтических подходов к коррекции инсулиновой резистент$
ности и сахарного диабета 2$го типа.
КЛЮЧЕВЫЕ СЛОВА: инсулиновая резистентность, сахарный диабет 2$го типа, воспаление, транспорт
глюкозы, адипоциты.
DOI: 10.1134/S0320972519050099
Ожирение является одной из ключевых
синдрома, сердечно$сосудистых осложнений и
проблем в современном обществе [1]. Ожирение
ряда онкологических заболеваний. В России
существенно повышает риск развития сахарно$
СД2Т диагностирован у 2,8% населения (~4,2 млн
го диабета 2$го типа (СД2Т), метаболического
человек). Кроме того, количество пациентов с
Принятые сокращения: БрА - брефелдин А; DМЕМ - модифицированная Дальбекко среда Игла; ИР - инсулино$
вая резистентность; TBST - Tris$HCl$буфер с добавлением 0,1% Tween 20; ЛПС - бактериальный липополисахарид; СД2Т -
сахарный диабет 2$го типа; СЖК - свободные насыщенные жирные кислоты (в данной работе - пальмитат); ЭПР - эн$
доплазматический ретикулум; Akt - протеинкиназа В; AS160 - субстрат Akt массой 160 кДа; FBS - фетальная бычья сы$
воротка; GLUT4 - глюкозный транспортер 4$го типа; HIF$1α - фактор 1α, индуцируемый гипоксией; IRS - субстрат ре$
цептора инсулина; JNK - киназа N$концевого домена белка c$Jun (с$Jun N$terminal kinase); mTORC2 - второй белковый
комплекс мишени рапамицина (mammalian target of rapamycin complex 2); NBCS - сыворотка новорожденных телят;
PI3$киназа - фосфатидилинозитол$3'$киназа.
* Адресат для корреспонденции.
# Авторы внесли равный вклад в работу.
719
720
СТАФЕЕВ и др.
избыточным весом стремительно растет как
сделаны попытки систематизировать данные о
среди взрослых, так и среди детей [2, 3]. Вместе
преимущественном фосфорилировании отдель$
с тем наблюдается существенное отставание
ных сериновых остатков IRS определенными
отечественной науки от мировой в изучении
киназами в разных условиях [13-15]. Выявлена
клеточных и молекулярных механизмов метабо$
довольно мозаичная картина, что указывает на
лических нарушений, в т.ч. связанных с СД2Т и
отсутствие строгой специфичности и вероят$
ожирением.
ность наличия более общего механизма индук$
Первичным звеном в развитии СД2Т являет$
ции ИР. Использование животных моделей с ге$
ся инсулиновая резистентность (ИР). Она выра$
нетическим нокаутом показало, что воспалитель$
жается в потере чувствительности клеток$ми$
ные киназы, такие как IKKβ/ε и JNK1/2, могут
шеней к инсулину и неспособности инсулина
играть ведущую роль в жировой ткани [16-19],
обеспечить захват глюкозы из крови [4]. В ре$
а латентное воспаление может быть тем общим
зультате развивается устойчивая гиперглике$
механизмом, который объединяет воздействие
мия, показателем которой служит рост уровня
факторов риска. Связь ожирения с воспалением
гликированного гемоглобина А1С. Несмотря на
в жировой ткани хорошо документирована [20-22],
большой прогресс, достигнутый в области меди$
хотя до сих пор нет единого мнения о том, что из
каментозной коррекции этих показателей при
них является причиной, а что - следствием [23].
лечении СД2Т [2], практически нерешенным
Однако существует несколько возможных путей
остается вопрос терапии и профилактики ИР
взаимодействия воспалительного и инсулино$
как первичного звена патогенеза СД2Т. Во мно$
вого каскадов в клетке [12], однозначности в
гом это связано с недостатком знаний о меха$
этом вопросе также нет. Тем не менее противо$
низмах развития ИР в главных клетках$мише$
воспалительная терапия восстанавливает чувстви$
нях инсулина - адипоцитах, миоцитах и гепато$
тельность к инсулину как на системном уровне
цитах.
[24, 25], так и в культуре модельных адипоцитов
Клеточные механизмы развития ИР связы$
3Т3$L1 [26].
вают с нарушением передачи сигнала инсулина
Целью данного исследования являлось мо$
внутрь клеток, что приводит к снижению выхо$
делирование патологических состояний, ассо$
да инсулин$зависимого транспортера глюкозы
циированных с ожирением, и выяснение их
GLUT4 на клеточную мембрану. Сигнальный
влияния на инсулиновую сигнализацию, захват
каскад инсулина включает активацию рецепто$
глюкозы и фосфорилирование киназы JNK в
ра инсулина, тирозиновое фосфорилирование
модели культивируемых адипоцитов 3T3$L1.
субстрата инсулинового рецептора (IRS) по ос$
татку Tyr612, активацию PI3$киназного каскада,
активацию киназы Akt путем двойного фосфо$
МЕТОДЫ ИССЛЕДОВАНИЯ
рилирования по остаткам Thr308 и Ser473 и фос$
форилирование ею белка AS160, который физи$
Культивирование и дифференцировка преади
чески регулирует перемещение GLUT4 на
поцитов 3T3 L1. Преадипоциты мыши 3T3$L1
мембрану [5-7]. Фосфорилирование Thr308 в
приобретали в АТСС (Американская коллекция
составе Akt опосредовано PI3K$зависимой фос$
стандартных клеточных культур) и культивиро$
фоинозитид$зависимой киназой PDK1, тогда
вали в среде DMEM с высоким содержанием
как за фосфорилирование Ser473 отвечает бел$
глюкозы (4,5 г/л глюкозы, 110 мг/л пирувата
ковый комплекс mTORC2, активность которо$
натрия, 2 мМ L$глутамина, 6 · 104 ед/л пеницил$
го, по крайней мере частично, также зависит от
лина, 6 · 104 ед/л стрептомицина) и добавлением
PI3$киназы [6]. Активация инсулинового каска$
10% FBS («HyClone», США или «Biological
да нарушается в результате фосфорилирования
Industries», Израиль). Адипогенную дифферен$
IRS по остаткам серина различными протеин$
цировку проводили в течение 10 дней, как опи$
киназами, что снижает сигнал$проводящую спо$
сано ранее [26, 27]. Для этого преадипоциты
собность IRS [7, 8].
3T3$L1 предварительно культивировали в тече$
Долгое время считалось, что участие разных
ние двух дней до конфлюэнтности ~90% и еще
сериновых киназ, подавляющих функцию IRS и
один день в среде DMEM с добавлением 10%
ведущих к развитию ИР, определяется разными
NBCS телят (newborn calf serum; «Gibco», США).
патологическими состояниями, связанными с
На 3$й день среду заменяли на DMEM с добав$
ожирением. В зависимости от типа ткани в ка$
лением 10% FBS, 0,5 мМ дексаметазона, 0,25 мкМ
честве таких состояний рассматривались дисли$
изобутилметилксантина, 2 мкМ розиглитазона
пидемия, воспаление, гипоксия, окислительный
и 1 мкг/мл инсулина (все реактивы «Sigma$
стресс, пищевая перегрузка и стресс эндоплаз$
Aldrich», США), которую меняли ежедневно в
матического ретикулума (ЭПР) [7, 9-12]. Были
течение 2-3 дней. На 5$й день среду заменяли
БИОХИМИЯ том 84 вып. 5 2019
ИНДУКТОРЫ СТРЕССА СНИЖАЮТ ЧУВСТВИТЕЛЬНОСТЬ К ИНСУЛИНУ
721
на DMEM с добавлением 10% FBS и 1 мкг/мл
содержанием глюкозы и 100 мкМ CoCl2 и анали$
инсулина. На 7$й день дифференцированные
зировали активность инсулин$зависимого сиг$
адипоциты переводили в DMEM с высоким со$
нального каскада и захвата [3H]$2$дезоксиглю$
держанием глюкозы и добавлением 10% FBS.
козы, как описано ниже.
Эксперименты проводили с 10$го по 14$й день,
Определение динамики воспалительной реак
контрольные клетки фиксировали с помощью
ции. Клетки обрабатывали СЖК, ЛПС, БрА или
4%$ного формальдегида в течение 1 ч и окраши$
CoCl2 в течение 30 мин; 1, 2, 8 или 24 ч. Концент$
вали липофильным красителем Oil Red O («Merck
рации индукторов соответствовали концентра$
Millipore», США).
циям, использованным при моделировании ИР.
Моделирование инсулиновой резистентности.
Кинетику воспалительной реакции зрелых ади$
Липид зависимая ИР. Липид$зависимую ИР ин$
поцитов 3T3$L1 в моделях ИР оценивали по
дуцировали согласно протоколу She et al. [28] с
усилению активирующего фосфорилирования
изменениями. Для зрелых адипоцитов 3T3$L1
остатков Thr183/Tyr185 в активационной петле
проводили депривацию от FBS в течение 24 ч в
стресс$зависимых киназ JNK1/2 (далее - JNK) с
DMEM с добавлением 0,5% БСА. После этого
помощью иммуноблоттинга.
на 24 ч добавляли 0,3 мМ конъюгат пальмитата с
Иммуноблоттинг. Метод иммуноблоттинга
БСА (СЖК), приготовленный по протоколу
использовали для сравнения активации фосфо$
Svedberg et al. [29]. Далее оценивали чувстви$
рилирования белков инсулинового каскада в
тельность клеток к инсулину, анализируя уро$
моделях инсулиновой резистентности (см. под$
вень активирующего фосфорилирования основ$
раздел «Моделирование инсулиновой резистент$
ных участников инсулинового каскада и влия$
ности») и для оценки кинетики фосфорилиро$
ние инсулина на GLUT4$зависимый захват
вания JNK как воспалительной реакции в дан$
клетками негидролизуемого аналога глюкозы,
ных моделях (см. подраздел «Определение дина$
[3H]$2$дезоксиглюкозы, как описано ниже.
мики воспалительной реакции»). Клетки лизи$
Воспаление. Воспаление индуцировали, обра$
ровали на льду в RIPA$буфере (RadioImmuno$
батывая зрелые адипоциты 3T3$L1 ЛПС (E. coli,
Precipitation Assay; 150 мМ NaCl, 1% Triton X$100,
серотип 0111:B4; «Sigma», США) до конечной
0,5% дезоксихолата натрия, 0,1% додецилсуль$
концентрации 50 нг/мл в течение 24 ч в среде
фата натрия, 50 мМ Tris$HCl, рН 8,0), содержав$
DMEM с добавлением 10% FBS. После этого
шем ингибиторы протеаз (cOmplete Tablets EASY$
проводили депривацию клеток от FBS в течение
pack; «Roche», Швейцария) и фосфатаз (10 мМ
4 ч в DMEM с высоким содержанием глюкозы и
глицерофосфат натрия, 20 мМ пирофосфат нат$
измеряли их чувствительность к инсулину, ана$
рия, 10 мМ фторид натрия, 1 мМ ортованадат
лизируя уровень активирующего фосфорилиро$
натрия). Клеточные лизаты разделяли с помощью
вания основных участников инсулинового кас$
SDS$электрофореза по Лэммли [32]. Электропе$
када и влияние инсулина на GLUT4$зависимый
ренос белков проводили на поливинилиденфто$
захват клетками негидролизуемого аналога глю$
ридные (PVDF) мембраны в режиме не менее
козы, [3H]$2$дезоксиглюкозы, как описано ниже.
1 А · ч. Мембраны блокировали в течение мини$
Стресс ЭПР. Стресс ЭПР вызывали согласно
мум 2 ч в 5%$ном растворе обезжиренного мо$
методике Citterio et al. [30] с изменениями. Зре$
лока («AppliChem», Германия) в TBST$буфере
лые адипоциты 3T3$L1 обрабатывали брефелди$
(50 мМ Tris$HCl, 150 мМ NaCl, 0,1% Tween 20,
ном А (БрА) в конечной концентрации 100 мкМ
pH 7,5) и последовательно обрабатывали пер$
в течение 24 ч в среде DMEM с добавлением
вичными и вторичными антителами в 1%$ном
10% FBS. После этого проводили депривацию
растворе молока в TBST$буфере в разведениях,
клеток от FBS в течение 4 ч и анализировали ак$
рекомендуемых производителем.
тивность инсулин$зависимого сигнального кас$
Активацию инсулинового каскада определя$
када и захвата [3H]$2$дезоксиглюкозы, как опи$
ли по фосфорилированию белков IRS$1, Akt и
сано ниже.
AS160. Для этого использовали антитела к фосфо$
Гипоксия. Клеточную модель ответа на ги$
Tyr612 в IRS$1 (#44816; «Thermo Fischer Scien$
поксию создавали согласно протоколу Glassford
tific», США); антитела к IRS$1 (3407; «Cell Sig$
et al. [31], стабилизируя транскрипционный
naling Technology», США), к фосфо$Thr308 в Akt
фактор HIF$1α ионами двухвалентного кобаль$
(9275; «Cell Signaling Technology», США), к фосфо$
та. Зрелые адипоциты 3T3$L1 обрабатывали
Ser473 в Akt (4060; «Cell Signaling Technology»,
CoCl2 в конечной концентрации 100 мкМ в те$
США), к Akt (ab64148; «Abcam», США); к фосфо$
чение 24 ч в среде DMEM с высоким содержа$
Ser318 в AS160 (8619; «Cell Signaling Technology»,
нием глюкозы и добавлением 10% FBS, после
США), к AS160 (2670; «Cell Signaling Technology»,
чего проводили депривацию клеток в течение
США), к винкулину (ab18058; «Abcam», США) и
4 ч в бессывороточной среде DMEM с высоким
вторичное антитело к IgG кролика, конъюгиро$
БИОХИМИЯ том 84 вып. 5 2019
722
СТАФЕЕВ и др.
ванное с пероксидазой хрена (ab6721; «Abcam»,
во белка в образце. Из полученных значений вы$
США). Активацию киназы JNK определяли с
читали значения, полученные в образцах с ци$
помощью фосфоспецифичных антител к фосфо$
тохалазином В, нивелируя таким образом вклад
Thr183/фосфо$Tyr185 в составе JNK1/2 (AF1205;
инсулин$независимого транспорта глюкозы.
«R&D Systems», США) и тотальных антител к
Статистический анализ. Статистическую об$
JNK1/2 (AF1387; «R&D Systems», США).
работку полученных данных проводили с по$
Белковые полосы визуализировали с ис$
мощью пакета программ Microsoft Excel 2007.
пользованием хемилюминесценции, реактивов
Результаты представлены на гистограммах как
Clarity ECL («Bio$Rad», США) и гель$докумен$
средние значения ± стандартные отклонения.
тирующей системы FusionX («Vilber Lourmat»,
Для расчета достоверности различий использо$
Франция) в накопительном режиме. Для коли$
вали двухвыборочный t$критерий с различной
чественной денситометрии использовали про$
дисперсией выборок, различия считали статис$
грамму GelAnalyzer2010a. Результаты представ$
тически значимыми при p < 0,05.
ляли в виде гистограмм с параметром «Стимуля$
ция фосфорилирования по активационному ос$
татку», который отражает возрастание уровня
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
фосфорилирования остатка исследуемого белка
в ответ на стимуляцию инсулином. Относитель$
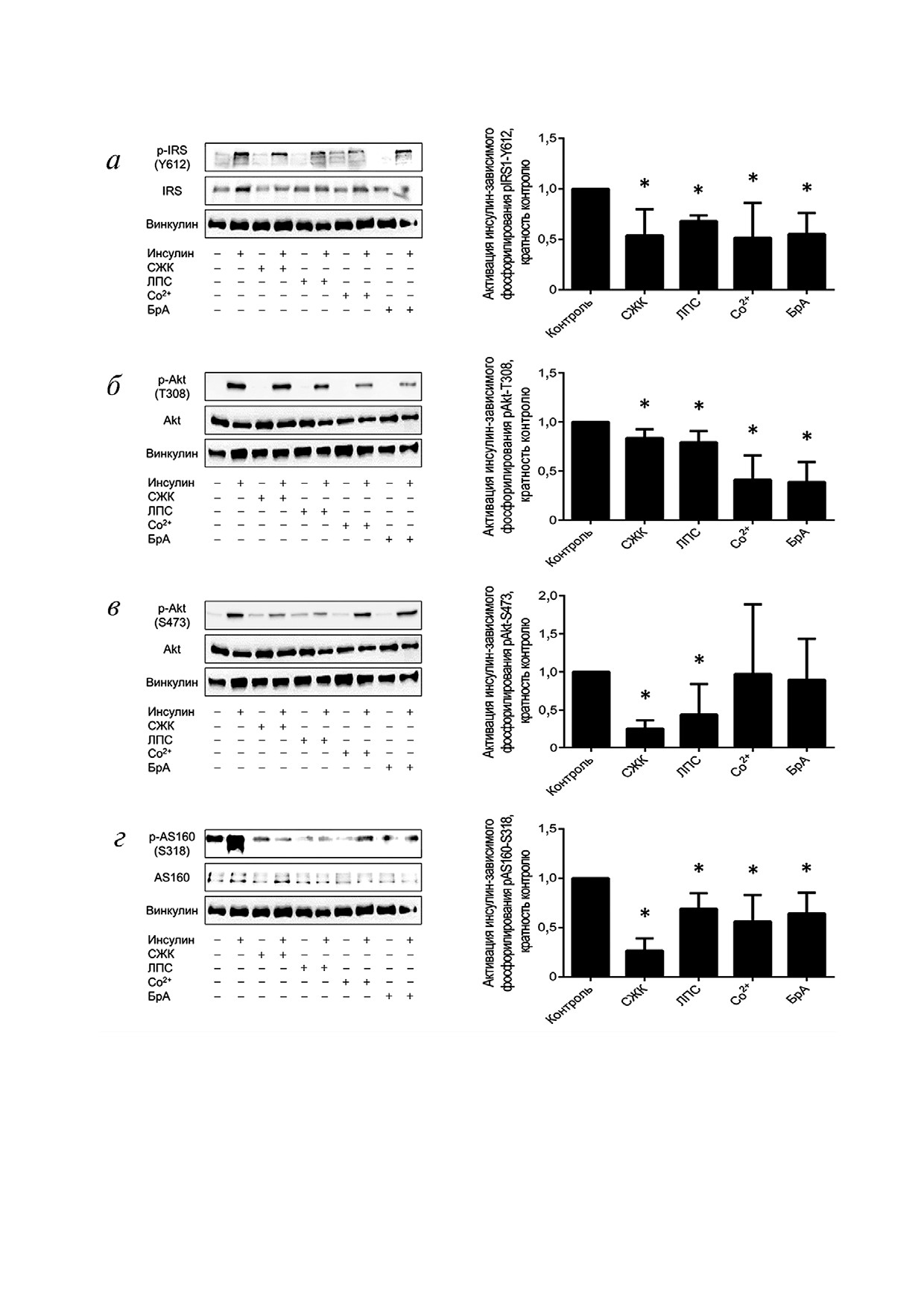
Химические индукторы ИР снижают актива
ный уровень фосфорилирования данного белка
цию инсулинового каскада в адипоцитах 3T3 L1.
вычисляли как отношение интенсивности хе$
Тирозиновое фосфорилирование IRS по остатку
милюминесценции фосфорилированной формы
Tyr612 обеспечивает начальный этап передачи
белка к интенсивности хемилюминесценции
инсулинового сигнала в направлении PI3$ки$
тотальной формы белка. После этого рассчиты$
назного каскада [5]. На рис. 1, а показано, что
вали стимуляцию фосфорилирования инсули$
стимуляция инсулином фосфорилирования
ном как отношение уровня фосфорилирования
этого остатка в IRS снижается после обработки
после стимуляции инсулином к уровню фосфо$
зрелых адипоцитов 3T3$L1 любым из химичес$
рилирования в нестимулированных клетках.
ких индукторов ИР. Эти результаты указывают
Измерение инсулин индуцируемого захвата
на инактивацию инсулинового каскада на уров$
[3H] 2 дезоксиглюкозы. После моделирования
не IRS в условиях жировой перегрузки клеток,
условий инсулиновой резистентности в течение
воспаления, гипоксии и стресса ЭПР.
24 ч в адипоцитах измеряли инсулин$зависи$
Мишенью PI3$киназного каскада является
мый вход глюкозы. Проводили депривацию
киназа Akt, активация которой требует фос$
адипоцитов от FBS в среде DMEM с высоким
форилирования по двум остаткам: Thr308 и
содержанием глюкозы и 0,1% БСА в течение 4 ч,
Ser473 [6]. На рис. 1, б, в показаны изменения
после чего промывали 3 раза DMEM без глюко$
уровня инсулин$зависимого фосфорилирова$
зы. Далее добавляли инсулин до концентрации
ния этих остатков. Все химические индукторы
100 нМ на 20 мин. Для определения захвата глю$
снижали уровень фосфорилирования Akt, но
козы через инсулин$независимые транспортеры
различным образом. В случае Thr308 (рис. 1, б)
в контрольные пробы добавляли цитохалазин В
ЛПС и СЖК незначительно снижали уровень
до 20 мкМ на 20 мин. После этого среды с инсули$
фосфорилирования Thr308. Напротив, Со2+ и
ном и цитохалазином В заменяли на среду, содер$
БрА в 2-3 раза снижали активирующее действие
жавшую 100 мкМ 2$дезоксиглюкозы + 0,5 мкКи
инсулина на фосфорилирование Thr308. В отно$
[3H]$2$дезоксиглюкозы (NET328A001MC; «Perkin$
шении Ser473 ситуация была противоположной -
Elmer», США). Через 10 мин клетки осторожно
ЛПС и СЖК почти в 3 раза снижали уровень
промывали дважды ледяным фосфатно$соле$
фосфорилирования Ser$473, тогда как Со2+ и БрА
вым буфером и замораживали при -20 °С для
были практически неэффективны (рис. 1, в).
нарушения целостности клеточной мембраны. Да$
Однако, учитывая необходимость фосфорили$
лее клетки размораживали, лизировали в 300 мкл
рования обоих остатков для активации Akt,
RIPA$буфера и отбирали 100 мкл для измерения
можно считать, что все химические индукторы
концентрации белка с использованием бицин$
нарушают активацию инсулином PI3$киназно$
хониновой киcлоты (Pierce BCA Protein Assay
го каскада и Akt как его мишени.
Kit, #23225; «Thermo Scientific», США). Остав$
Для того чтобы подтвердить инактивацию ин$
шиеся 200 мкл лизата растворяли в сцинтилля$
сулинового каскада далее в направлении GLUT4,
ционной жидкости Beckman Ready$Solv HP
мы проследили изменения инсулин$зависимого
(#158726; «Beckman», США), измеряли число рас$
фосфорилирования AS160, являющегося прямым
падов (c.p.m.) на счетчике RackBeta («LKB Wallac»,
регулятором GLUT4, по остатку Ser318, кото$
Швеция) и нормировали результаты на количест$
рый служит главной мишенью Akt [33, 34]. Как
БИОХИМИЯ том 84 вып. 5 2019
ИНДУКТОРЫ СТРЕССА СНИЖАЮТ ЧУВСТВИТЕЛЬНОСТЬ К ИНСУЛИНУ
723
Рис. 1. Химические индукторы ИР снижают инсулин$зависимое фосфорилирование основных участников инсулинового
сигнального каскада в адипоцитах 3T3$L1. а - Репрезентативные результаты вестерн$блоттинга и стимуляция фосфори$
лирования IRS по остатку Tyr612; б - репрезентативные результаты вестерн$блоттинга и стимуляция фосфорилирования
Akt по остатку Thr308; в - репрезентативные результаты вестерн$блоттинга и стимуляция фосфорилирования Akt по ос$
татку Ser473; г - репрезентативные результаты вестерн$блоттинга и стимуляция фосфорилирования AS160 по остатку
Ser318. Результаты статистического анализа на диаграммах представлены как среднее ± стандартное отклонение, t$крите$
рий Стьюдента; * р < 0,05; n = 3
БИОХИМИЯ том 84 вып. 5 2019
724
СТАФЕЕВ и др.
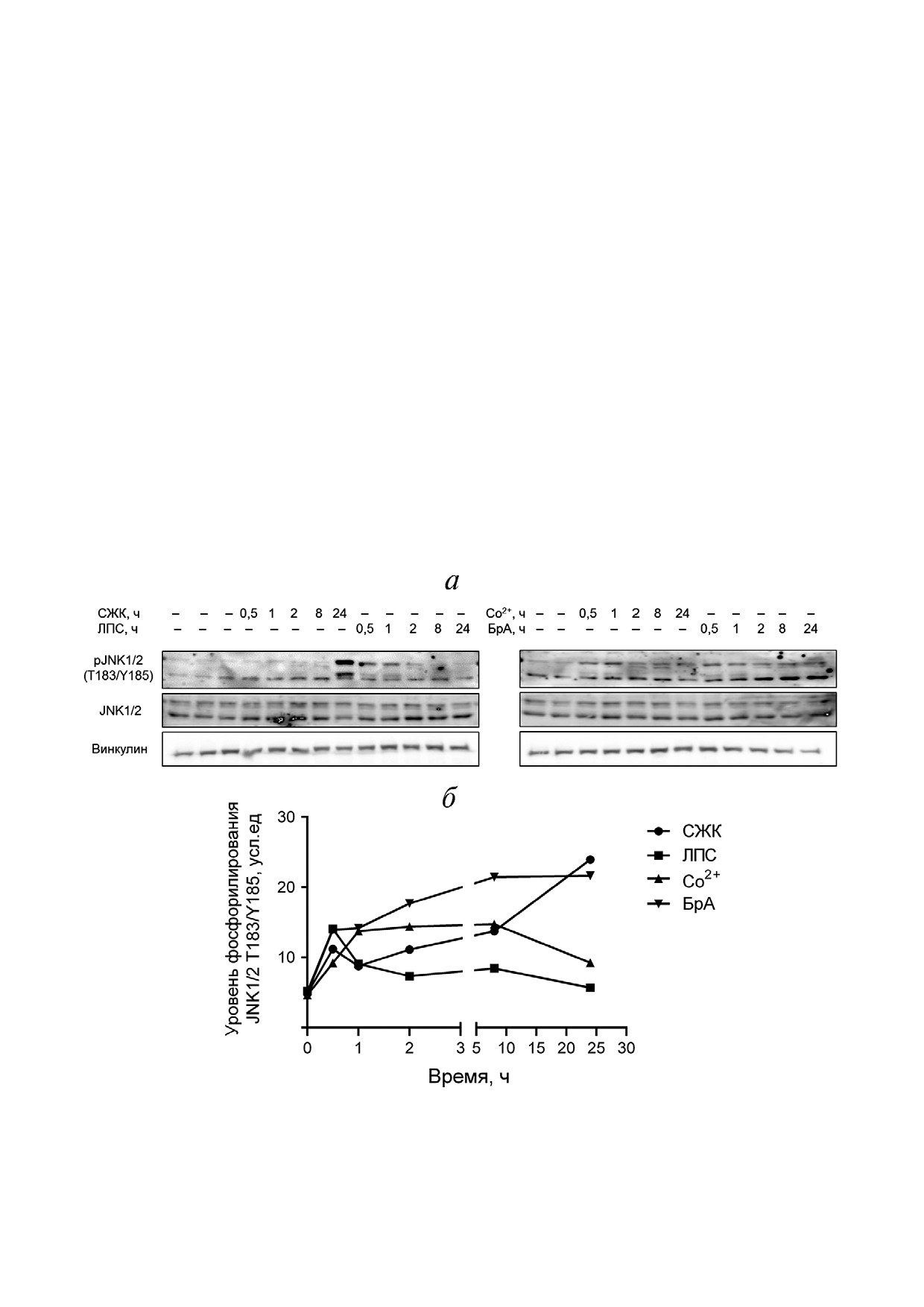
показано на рис. 1, г, все химические индукторы
фосфорилирование активирующих остатков в
ИР снижали инсулин$зависимое фосфорилиро$
JNK1/2 возрастает во всех условиях жировой пе$
вание AS160, при этом эффект СЖК был значи$
регрузки, воспаления, гипоксии и стресса ЭПР,
тельно более выраженным.
однако кинетика активации JNK1/2 существен$
Таким образом, активация Akt инсулином и
но различается (рис. 3).
дальнейшая передача сигнала к GLUT4 действи$
Уровень фосфорилирования JNK1/2 быстро
тельно были нарушены в условиях жировой пе$
возрастает после добавления к клеткам индук$
регрузки, воспаления, гипоксии и стресса ЭПР.
торов, отражая первую острую фазу воспали$
Все химические индукторы ИР, за исключением
тельного ответа (1-2 ч). Однако дальнейшая ки$
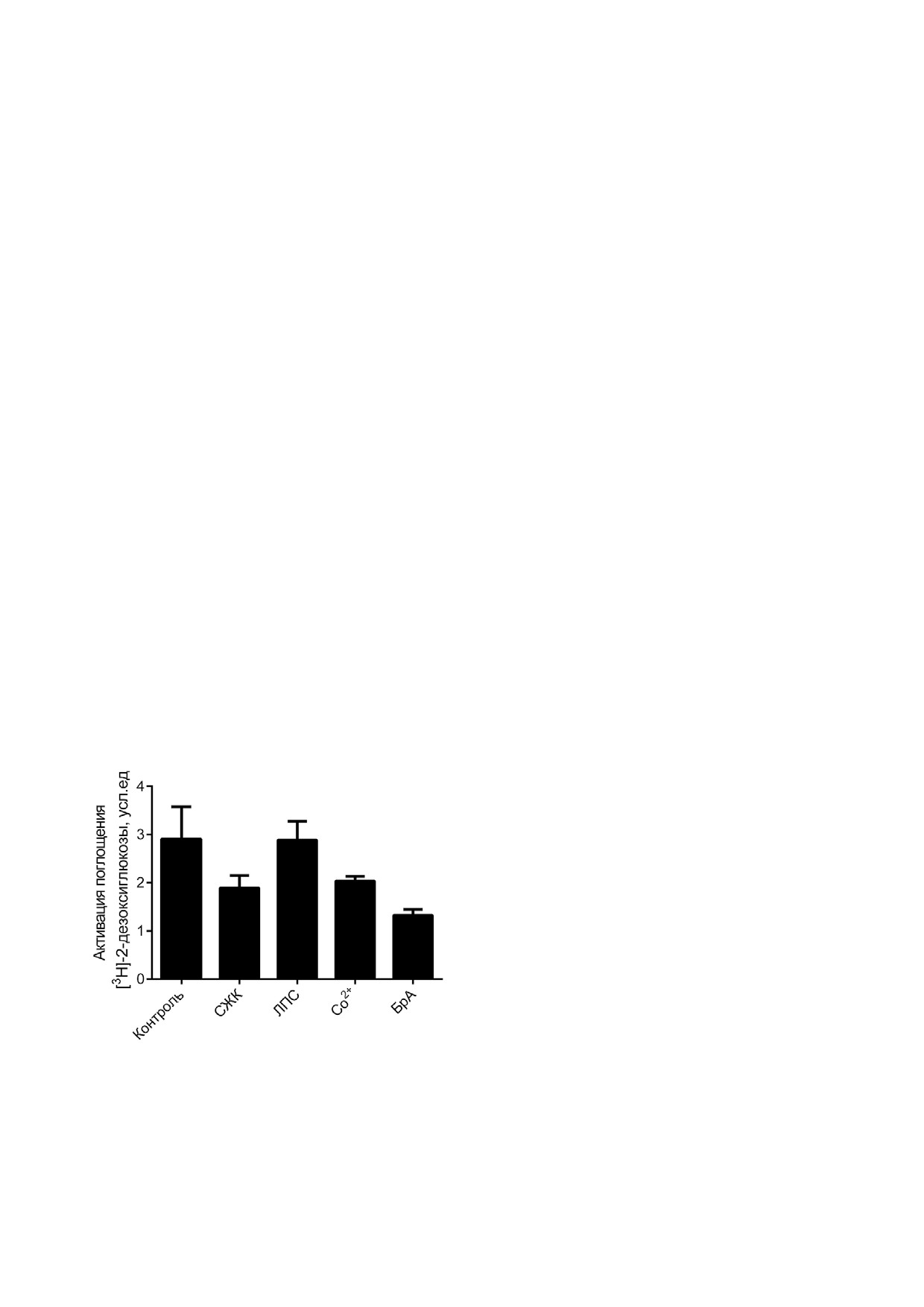
ЛПС, снижают инсулин зависимый захват [3H] 2
нетика резко отличается наличием или отсут$
дезоксиглюкозы адипоцитами 3T3 L1. Инсулин$
ствием второй продолжительной фазы (3-24 ч).
зависимый транспорт глюкозы в клетки измеря$
В то время как СЖК и БрА устойчиво повыша$
ли с использованием радиоактивно$меченого
ли уровень фосфорилирования JNK1/2, в случае
аналога глюкозы. Вклад инсулин$независимых
ЛПС вторая фаза фактически отсутствовала, а в
транспортеров глюкозы определяли в присут$
случае Со2+ вторую фазу наблюдали, но уровень
ствии цитохалазина В, который блокирует экс$
фосфорилирования JNK1/2 снижался к 24 ч об$
понирование GLUT4 на плазматическую мемб$
работки.
рану. Как показано на рис. 2, все химические
индукторы, за исключением ЛПС, снижали ин$
сулин$индуцируемый захват [3H]$2$дезоксиглю$
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
козы. Эти результаты лишь частично согласуют$
ся с изменениями активности инсулинового
В данной работе мы проверили гипотезу о
каскада, представленными выше. Несмотря на
том, что моделирование in vitro различных пато$
подавление активации инсулинового каскада,
логических ситуаций, характерных для ожире$
ЛПС фактически не влиял на инсулин$зависи$
ния, вызывает ИР и нарушает транспорт глюко$
мый захват клетками [3H]$2$дезоксиглюкозы.
зы в адипоциты. Во всех случаях (жировая пе$
Все химические индукторы ИР, за исключением
регрузка, острое воспаление, гипоксия и стресс
ЛПС, вызывают длительную активацию стресс
ЭПР) снижение активирующего действия инсу$
зависимых киназ JNK1/2 в адипоцитах 3T3 L1.
лина происходило на: 1) начальном уровне IRS;
Стресс$зависимые киназы JNK1/2 активирова$
2) промежуточном уровне мишени PI3$киназ$
ны при ожирении и играют важную роль в раз$
ного каскада, киназы Akt; 3) конечном этапе
витии ИР [12]. Мы проследили кинетику акти$
мишени инсулинового каскада, AS160, регули$
вации JNK1/2 в адипоцитах 3T3$L1 в процессе
рующего активность инсулин$зависимого транс$
химической индукции ИР и обнаружили, что
портера глюкозы GLUT4. При этом наблюда$
лось повышенное фосфорилирование стресс$
зависимых киназ JNK1/2, которые активируют$
ся при воспалении, однако кинетика активации
JNK1/2 была различной. Поскольку JNK1/2 из$
вестны как основные сериновые киназы IRS,
наши данные указывают на единый воспали$
тельный механизм, обеспечивающий развитие
ИР различной этиологии.
Механизмы снижения активности инсулино
вого каскада под действием СЖК. Хорошо из$
вестно, что насыщенные СЖК подавляют захват
глюкозы из крови инсулин$зависимыми тканя$
ми [35, 36]. Этот физиологический феномен, ле$
жащий в основе глюкозо$жирнокислотного
цикла Рэндла [35], был подробно описан для
мышечных клеток как основных потребителей
циркулирующей глюкозы в организме. В мень$
шей степени он был изучен для адипоцитов, где
ключевым событием считалось подавление ли$
Рис. 2. Влияние химических индукторов ИР на инсулин$
зависимый захват [3H]$2$дезоксиглюкозы адипоцитами
полиза под действием избыточно поступающей
3T3$L1. Приведенные значения показывают, во сколько
в клетки глюкозы. Лишь позже было показано,
раз инсулин усиливает транспорт глюкозы в клетки. Дан$
ные представлены как среднее ± стандартное отклонение,
что избыток СЖК (алиментарного или липолити$
t$критерий Стьюдента; * р < 0,05; n = 6
ческого происхождения) также снижает транс$
БИОХИМИЯ том 84 вып. 5 2019
ИНДУКТОРЫ СТРЕССА СНИЖАЮТ ЧУВСТВИТЕЛЬНОСТЬ К ИНСУЛИНУ
725
порт глюкозы в адипоциты [37, 38]. Существует
на животных и клеточных моделях стресса ЭПР
огромное число работ, подтверждающих этот
и воспаления [16, 18].
феномен [36], но его клеточно$биохимический
Полученные нами результаты согласуются с
механизм долгое время был неизвестен.
этим механизмом, т.к. в модели жировой пере$
Исследования 1990$х и 2000$х гг. показали,
грузки мы наблюдали снижение инсулин$зави$
что в мышечных клетках избыток липидов вы$
симого фосфорилирования IRS по Tyr612, необ$
зывает активацию атипичных форм протеинки$
ходимого для дальнейшей передачи сигнала к
назы С, которые прерывают передачу сигнала от
PI3$киназному каскаду [5], а также фосфорили$
рецептора инсулина к транспортеру глюкозы
рования его мишеней Akt и AS160 (рис. 1) и
GLUT4 [39, 40]. Однако высокий липидный фон
транспорта глюкозы в адипоциты (рис. 2). При
в адипоцитах предполагает наличие иного меха$
обработке клеток СЖК или ЛПС базальный
низма нарушения инсулинового каскада. Пони$
уровень фосфорилирования Ser473 в Akt был
мание данного механизма пришло после иден$
повышен (рис. 1, в). Его могут обеспечивать ки$
тификации СЖК как активаторов Toll$подоб$
назы JNK1/2, которые либо напрямую фосфори$
ных рецепторов 4$го типа (TLR4) [41, 42]. Эти
лируют Ser473 [18], либо активируют mTORC2
рецепторы сопряжены с активацией воспалитель$
[15, 16]. Действительно, мы наблюдали продол$
ных каскадов клетки и «воспалительных» киназ
жительную активацию JNK1/2 под действием
IKK и JNK1/2. Участие этих киназ в прерывании
СЖК (рис. 3). Мы также не исключаем других ме$
инсулинового сигнала на уровне IRS к настоя$
ханизмов действия насыщенных СЖК, т.к. ме$
щему времени хорошо документировано [15].
ханизмы действия СЖК на адипоциты разно$
Наконец, ключевая роль JNK в сериновом фос$
образны [41, 42].
форилировании IRS и развитии ИР в жировой
Механизмы снижения активности инсулино
ткани была убедительно продемонстрирована вого каскада при гипоксии. Известно, что разви$
Рис. 3. Кинетика фосфорилирования киназы JNK1/2 при обработке адипоцитов 3T3$L1 химическими индукторами ИР.
а - Репрезентативные иммуноблоты; б - график зависимости уровня фосфорилирования JNK1/2 по Thr183 и Tyr185 от
времени
БИОХИМИЯ том 84 вып. 5 2019
726
СТАФЕЕВ и др.
тие ожирения и ИР связано с гипертрофией жи$
вой особенностью этого механизма развития ИР
ровой ткани, а развитие гипоксии может быть
является длительный (латентный) характер вос$
причиной ИР в адипоцитах [10]. Транскрипци$
паления [9, 12].
онный фактор HIF$1α обеспечивает реакцию
Классическая индукция воспаления с по$
клеток на гипоксию. Ингибирование белков
мощью ЛПС приводила к подавлению инсулин$
фон Хиппель-Линдау ионами Со2+ предотвра$
зависимой сигнализации в адипоцитах (рис. 1),
щает протеасомную деградацию и стабилизиру$
но не вызывала статистически значимого сни$
ет HIF$1α [43, 44]. Одним из целевых генов
жения инсулин$индуцируемого захвата глюко$
HIF$комплекса является фактор транскрипции
зы (рис. 2). ЛПС быстро активировал JNK1/2,
NF$κB. Стабилизация HIF$1α способствует
но эта активация уменьшалась уже через 4 ч, и
усилению воспалительных механизмов, зависи$
вторая фаза продолжительной активации JNK1/2
мых от NF$κB [45, 46]. Кроме того, Со2+ может
фактически отсутствовала (рис. 3). Такая карти$
стимулировать продукцию в клетках активных
на соответствует острой воспалительной реак$
форм кислорода и развитие окислительного
ции при активации сигнального каскада IKK/
стресса [47]. Таким образом, гипоксия стимули$
NF$κB через рецептор TLR4 на поверхности
рует активность воспалительных и стресс$зави$
адипоцитов. Возможно, отсутствие эффекта
симых киназ, включая IKK и JNK1/2.
ЛПС на инсулин$зависимый захват глюкозы
Мы показали, что Со2+ снижает инсулин$
(рис. 2) связано именно с отсутствием фазы
стимулируемое фосфорилирование IRS, Akt (по
продолжительной активации JNK1/2, а также с
Thr308) и AS160 (рис. 1). Как отмечено выше, за
различной реактивностью изоформ JNK1/2. В то
усиление фосфорилирования Akt по Ser473
время как СЖК, Со2+ и БрА влияли преимущест$
(рис. 1, в) могут отвечать киназы JNK1/2 [15, 16,
венно на фосфорилирование JNK2, ЛПС изме$
18]. Действительно, Со2+ вызывал продолжи$
нял фосфорилирование JNK1 (рис. 3). Интерес$
тельную активацию JNK1/2 (рис. 3), указывая
но, что изоформы JNK1/2 могут быть по$разно$
на запуск реакции воспаления. Несмотря на то,
му задействованы в регуляции метаболизма.
что активация JNK1/2 падала к концу экспери$
Так, реакция инсулинового каскада на воспале$
мента, инсулин$индуцируемый захват глюкозы
ние полностью отсутствовала у мышей, нокаут$
достоверно снижался (рис. 2). Таким образом, в
ных по JNK1, но не JNK2 [16]. Однако этот эф$
условиях экспериментальной гипоксии в адипо$
фект требовал наличия JNK2 и нарушался в слу$
цитах развивается ИР и нарушается захват глю$
чае нокаута JNK2 [18]. Таким образом, обе изо$
козы, по$видимому, вследствие активации вос$
формы киназы JNK играют важные, но разные
палительного ответа с участием JNK1/2.
роли в развитии ИР воспалительного генеза [50].
Механизмы снижения активности инсулино
Являясь фактором острого воспаления, ЛПС
вого каскада при стрессе ЭПР. БрА нарушает
лишь кратковременно активирует JNK1/2, и
доставку СОРI$окаймленных везикул из ЭПР в
этого, видимо, недостаточно для критических
аппарат Гольджи, вызывая белковую перегрузку
нарушений функций AS160 и GLUT4. Тот факт,
и стресс ЭПР [48]. Механизм действия БрА свя$
что ЛПС снижает уровень фосфорилирования
зан с участием воспалительного транскрипци$
AS160 по Ser318 (рис. 1, г), однозначно не ука$
онного фактора NF$κB [49].
зывает на изменение активности AS160. Хотя
БрА снижал инсулин$зависимое фосфори$
этот остаток является основной мишенью Akt,
лирование IRS, Akt (по Thr308) и AS160 (рис. 1),
AS160 несет несколько других остатков, узнава$
показывая тенденцию к росту фосфорилирова$
емых Akt [33, 34]. Кроме того, активность AS160
ния Akt по Ser473 относительно других индукто$
регулируется другими киназами. В частности, в
ров ИР (рис. 1, в). Стимуляция инсулином захвата
миоцитах АМР$зависимая киназа (АМРК) регу$
глюкозы также достоверно снижалась (рис. 2).
лирует транспорт глюкозы, фосфорилируя дру$
Кинетика активации JNK1/2 показывала нали$
гой набор остатков в AS160 [51-53].
чие продолжительной фазы, что согласуется с
В данной работе мы показали, что при моде$
ролью JNK1/2 в сериновом фосфорилировании
лировании клеточных патологий, характерных
IRS и индукции ИР в адипоцитах. Таким обра$
для ожирения, а именно дислипидемии, гипо$
зом, моделирование стресса ЭПР также приво$
ксии и стресса ЭПР, в адипоцитах 3T3$L1 разви$
дит к развитию воспалительной реакции и нару$
вается длительная воспалительная реакция, вы$
шению транспорта глюкозы в адипоциты.
ражающаяся в активации стресс$зависимых ки$
Механизмы снижения активности инсулино
наз JNK1/2, снижении активации инсулинового
вого каскада при воспалении. К настоящему вре$
каскада и транспорта глюкозы в клетки. Напро$
мени сформировалась довольно четкая картина,
тив, моделирование острого воспаления с по$
показывающая связь ожирения, воспаления и
мощью ЛПС приводило к кратковременной ак$
развития ИР в жировой ткани [19, 50]. Ключе$
тивации JNK1/2, снижению активации инсули$
БИОХИМИЯ том 84 вып. 5 2019
ИНДУКТОРЫ СТРЕССА СНИЖАЮТ ЧУВСТВИТЕЛЬНОСТЬ К ИНСУЛИНУ
727
нового каскада, но не влияло на транспорт глю$
№ 14$35$00026) и Фонда поддержки научно$про$
козы в адипоциты. Полученные результаты ука$
ектной деятельности студентов, аспирантов и мо$
зывают на связь инсулиновой резистентности с
лодых ученых «Национальное интеллектуальное
хроническим (латентным) воспалением, что де$
развитие» (грант № 17$34$80026 «мол_эв_а»).
лает воспаление перспективной мишенью для
Конфликт интересов. Авторы заявляют об от$
поиска новых терапевтических подходов к кор$
сутствии явных и потенциальных конфликтов
рекции инсулиновой резистентности и сахарно$
интересов, связанных с публикацией настоящей
го диабета 2$го типа.
статьи.
Соблюдение этических норм. Настоящая
статья не содержит описания каких$либо иссле$
Финансирование. Работа выполнена при под$
дований с участием людей и использованием
держке Российского научного фонда (грант животных в качестве объектов исследований.
СПИСОК ЛИТЕРАТУРЫ
1.
and Hotamisligil, G.S. (2004) Endoplasmic reticulum
las.org/resources/2017$atlas.html.
stress links obesity, insulin action, and type 2 diabetes,
2.
Дедов И.И., Шестакова М.В., Викулова О.К., Желез$
Science, 306, 457-461, doi: 10.1126/science.1103160.
някова А.В., Исаков М.А. (2018) Сахарный диабет в
12.
Stafeev, I.S., Vorotnikov, A.V., Ratner, E.I., Menshikov, M.Y.,
Российской Федерации: распространенность, заболе$
and Parfyonova, Ye.V. (2017) Latent inflammation and
ваемость, смертность, параметры углеводного обмена
insulin resistance in adipose tissue, Int. J. Endocrinol.,
и структура сахароснижающей терапии по данным
2017, 1-12, doi: 10.1155/2017/5076732.
федерального регистра сахарного диабета, статус 2017 г.,
13.
Zick, Y.
(2004) Uncoupling insulin signalling by
Сахарный диабет, 21, 144-159, doi: 10.14341/DM9686.
serine/threonine phosphorylation: a molecular basis for
3.
Дедов И.И., Шестакова М.В., Петеркова В.А., Вику$
insulin resistance, Biochem. Soc. Trans., 32, 812-816,
лова О.К., Железнякова А.В., Исаков М.А., Лаптев Д.Н.,
doi: 10.1042/BST0320812.
Андрианова Е.А., Ширяева Т.Ю. (2017) Сахарный ди$
14.
Morino, K., Petersen, K.F., and Shulman, G.I. (2006)
абет у детей и подростков по данным Федерального
Molecular mechanisms of insulin resistance in humans and
регистра Российской Федерации: динамика основных
their potential links with mitochondrial dysfunction,
эпидемиологических характеристик за 2013-2016 гг.,
Diabetes, 55, S9-S15, doi: 10.2337/db06$S002.
Сахарный диабет, 20, 392-402, doi: 10.14341/DM9460.
15.
Taniguchi, C.M., Emanuelli, B., and Kahn, C.R. (2006)
4.
Wilcox, G. (2005) Insulin and insulin resistance, Clin.
Critical nodes in signalling pathways: insights into insulin
Biochem. Rev., 26, 19-39.
action, Nat. Rev. Mol. Cell. Biol., 7, 85-96, doi: 10.1038/
5.
Esposito, D.L., Li, Y., Cama, A., and Quon, M.J. (2001)
nrm1837.
Tyr612 and Tyr632 in human insulin receptor substrate$1
16.
Hirosumi, J., Tuncman, G., Chang, L., Gorgun, C.Z.,
are important for full activation of insulin$stimulated phos$
Uysal, K.T., Maeda, K., Karin, M., and Hotamisligil, G.S.
phatidylinositol 3$kinase activity and translocation of
(2002) A central role for JNK in obesity and insulin resis$
GLUT4 in adipose cells, Endocrinology, 142, 2833-2840,
tance, Nature, 420, 333-336, doi: 10.1038/nature01137.
doi: 10.1210/endo.142.7.8283.
17.
Arkan, M.C., Hevener, A.L., Greten, F.R., Maeda, S.,
6.
Guertin, D.A., Stevens, D.M., Thoreen, C.C., Burds, A.A.,
Li, Z.W., Long, J.M., Wynshaw$Boris, A., Poli, G.,
Kalaany, N.Y., Moffat, J., Brown, M., Fitzgerald, K.J.,
Olefsky, J., and Karin, M. (2005) IKK$β links inflamma$
and Sabatini, D.M. (2006) Ablation in mice of the mTORC
tion to obesity$induced insulin resistance, Nat. Med., 11,
components raptor, rictor, or mLST8 reveals that mTORC2
191-198, doi: 10.1038/nm1185.
is required for signaling to Akt$FOXO and PKCα, but not
18.
Tuncman, G., Hirosumi, J., Solinas, G., Chang, L., Karin, M.,
S6K1, Dev. Cell, 11, 859-871, doi: 10.1016/j.devcel.
and Hotamisligil, G.S. (2006) Functional in vivo interac$
2006.10.007.
tions between JNK1 and JNK2 isoforms in obesity and
7.
Ткачук В.А., Воротников А.В. (2014) Молекулярные
insulin resistance, Proc. Natl. Acad. Sci. USA, 103,
механизмы развития резистентности к инсулину, Са*
10741-10746, doi: 10.1073/pnas.0603509103.
харный диабет, 17, 29-41, doi: 10.14341/DM2014229$40.
19.
Saltiel, A.R., and Olefsky, J.M. (2017) Inflammatory
8.
Boura$Halfon, S., and Zick, Y. (2009) Phosphorylation of
mechanisms linking obesity and metabolic disease, J. Clin.
IRS proteins, insulin action, and insulin resistance, Am. J.
Invest., 127, 1-4, doi: 10.1172/JCI92035.
Physiol. Endocrinol. Metab.,
296, E581-E591, doi:
20.
Donath, M.Y., and Shoelson, S.E. (2011) Type 2 diabetes
10.1152/ajpendo.90437.2008.
as an inflammatory disease, Nat. Rev. Immunol., 11, 98-107,
9.
Stafeev, I.S., Menshikov, M.Y., Tsokolaeva, Z.I.,
doi: 10.1038/nri2925.
Shestakova, M.V., and Parfyonova, Ye.V. (2015) Molecular
21.
Saltiel, A.R. (2016) Insulin signaling in the control of glu$
mechanisms of latent inflammation in metabolic syn$
cose and lipid homeostasis, Handb. Exp. Pharmacol., 233,
drome. Possible role of sirtuins and peroxisome proliferator
51-71, doi: 10.1007/164_2015_14.
activated receptor type gamma, Biochemistry (Moscow), 80,
22.
Lackey, D.E., and Olefsky, J.M. (2016) Regulation of
1217-1226, doi: 10.1134/S0006297915100028.
metabolism by the innate immune system, Nat. Rev.
10.
Trayhurn, P. (2013) Hypoxia and adipose tissue function
Endocrinol., 12, 15-28, doi: 10.1038/nrendo.2015.189.
and dysfunction in obesity, Physiol. Rev., 93, 1-21, doi: 10.
23.
Shimobayashi, M., Albert, V., Woelnerhanssen, B., Frei, I.C.,
1152/physrev.00017.2012.
Weissenberger, D., Meyer$Gerspach, A.C., Clement, N.,
11.
Ozcan, U., Cao, Q., Yilmaz, E., Lee, A.H., Iwakoshi, N.N.,
Moes, S., Colombi, M., Meier, J.A., Swierczynska, M.M.,
Ozdelen, E., Tunchman, G., Gorgun, C., Glimcher, L.H.,
Jeno, P., Beglinger, C., Peterli, R., and Hall, M.N. (2018)
БИОХИМИЯ том 84 вып. 5 2019
728
СТАФЕЕВ и др.
Insulin resistance causes inflammation in adipose tissue,
39.
Samuel, V.T., Petersen, K.F., and Shulman, G.I. (2010)
J. Clin. Invest., 128, 1538-1550, doi: 10.1172/JCI96139.
Lipid$induced insulin resistance: unravelling the mecha$
24.
Yuan, M., Konstantopoulos, N., Lee, J., Hansen, L., Li, Z.W.,
nism, Lancet, 375, 2267-2277, doi: 10.1016/S0140$6736
Karin, M., and Shoelson, S.E. (2001) Reversal of obesity$
(10)60408$4.
and diet$induced insulin resistance with salicylates or tar$
40.
Shulman, G.I. (2014) Ectopic fat in insulin resistance, dys$
geted disruption of Ikkв, Science, 293, 1673-1677, doi: 10.
lipidemia, and cardiometabolic disease, N. Engl. J. Med.,
1126/science.1061620.
371, 1131-1141, doi: 10.1056/NEJMra1011035.
25.
Donath, M.Y. (2014) Targeting inflammation in the treat$
41.
Oeckinghaus, A., Hayden, M.S., and Ghosh, S. (2011)
ment of type 2 diabetes: time to start, Nat. Rev. Drug. Disc.,
Crosstalk in NF$κB signaling pathways, Nat. Immunol., 12,
13, 465-476, doi: 10.1038/nrd4275.
695-708, doi: 10.1038/ni.2065.
26.
Stafeev, I.S., Michurina, S.S., Podkuychenko, N.V.,
42.
Lancaster, G.I., Langley, K.G. Berglund, N.A., Kam$
Vorotnikov, A.V., Menshikov, M.Y., and Parfyonova, Ye.V.
moun, H.L. Reibe, S., Estevez, E., Weir, J., Mellett, N.A.,
(2018) Interleukin$4 restores insulin sensitivity in lipid$
Pernes, G., Conway, J.R.W., Lee, M.K.S., Timpson, P.,
induced insulin resistant adipocytes, Biochemistry (Moscow),
Murphy, A.J., Masters, S.L., Gerondakis, S., Bartonicek, N.,
83, 498-506, doi: 10.1134/S0006297918050036.
Kaczorowski, D.C., Dinger, M.E., Meikle, P.J., Bond, P.J.,
27.
Zebisch, K., Voight, V., Wabitsch, M., and Brandsch, M.
and Febbraio, M.A. (2018) Evidence that TLR4 is not a
(2012) Protocol for effective differentiation of 3T3L1 cells
receptor for saturated fatty acids but mediates lipid$
to adipocytes, Anal. Biochem., 425, 88-90, doi: 10.1016/
induced inflammation by reprogramming macrophage
j.ab.2012.03.005.
metabolism, Cell. Metab., 27, 1096-1110, doi: 10.1016/j.
28.
She, M., Hou, H., Wang, Z., Zhang, C., Laudon, M., and
cmet.2018.03.014.
Yin, W. (2014) Melatonin rescues 3T3$L1 adipocytes from
43.
Weidemann, A., and Johnson, R.S. (2008) Biology of
FFA$induced insulin resistance by inhibiting phosphoryla$
HIF$1α, Cell. Death. Differ., 15, 621-627, doi: 10.1038/
tion of IRS$1 on Ser307, Biochimie, 103, 126-130, doi: 10.
cdd.2008.12.
1016/j.biochi.2014.05.001.
44.
Greer, S.N., Metcalf, J.L., Wang, Y., and Ohh, M. (2012)
29.
Svedberg, J., Bjorntorp, P., Smith, U., and Lonnroth, P.
The updated biology of hypoxia inducible factor, EMBO J.,
(1990) Free$fatty acid inhibition of insulin binding, degra$
31, 2448-2460, doi: 10.1038/emboj.2012.125.
dation, and action in isolated rat hepatocytes, Diabetes, 39,
45.
Melillo, G. (2011) Hypoxia: jump$starting inflammation,
570-574, doi: 10.2337/diab.39.5.570.
Blood, 117, 2561-2562, doi: 10.1182/blood$2010$12$
30.
Citterio, C., Vichi, A., Pacheco$Rodriguez, G., Aponte, A.M.,
324913.
Moss, J., and Vaughan, M. (2008) Unfolded protein response
46.
Eltzschig, H.K., and Carmeliet, P. (2011) Hypoxia and
and cell death after depletion of brefeldin A$inhibited guanine
inflammation, N. Engl. J. Med., 364, 656-665, doi: 10.
nucleotide$exchange protein GBF1, Proc. Natl. Acad. Sci.
1056/NEJMra0910283.
USA, 105, 2877-2882, doi: 10.1073/pnas.0712224105.
47.
Kamiya, T., Hara, H., Inagaki, N., and Adachi, T. (2010)
31.
Glassford, A.J., Yue, P., Sheikh, A.Y., Chun, H.J.,
The effect of hypoxia mimetic cobalt chloride on the
Zarafshar, S., Chan, D.A., Reaven, G.M., Quertermous, T.,
expression of EC$SOD in 3T3$L1 adipocytes, Redox Rep.,
and Tsao, P.S. (2007) HIF$1 regulates hypoxia$ and
15, 131-137, doi: 10.1179/174329210X12650506623483.
insulin$induced expression of apelin in adipocytes, Am. J.
48.
Anadu, N.O., Davisson, V.J., and Cushman, M. (2006)
Physiol. Endocrinol. Metab., 293, E1590-E1596, doi: 10.
Synthesis and anticancer activity of brefeldin A ester deriv$
1152/ajpendo.00490.2007.
atives, J. Med. Chem., 49, 3897-3905, doi: 10.1021/
32.
Laemmli, U.K. (1970) Cleavage of structural proteins during
jm0602817.
the assembly of the head of bacteriophage T4, Nature, 227,
49.
Pahl, H.L., and Baeuerle, P.A. (1995) A novel signal trans$
680-685, doi: 10.1038/227680a0.
duction pathway from the endoplasmic reticulum to the
33.
Sano, H., Kane, S., Sano, E., Miinea, C.P., Asara, J.M.,
nucleus is mediated by transcription factor NF$κB, EMBO J.,
Lane, W.S., Garner, C.C., and Lienhard, G.E. (2003)
14, 2580-2588.
Insulin$stimulated phosphorylation of Rab GTPase$acti$
50.
Hotamisligil, G.S. (2006) Inflammation and metabolic
vating protein regulates GLUT4 translocation, J. Biol.
disorders, Nature, 444, 860-867, doi: 10.1038/nature
Chem., 278, 14599-14602, doi: 10.1074/jbc.C300063200.
05485.
34.
Kane, S., Sano, H., Liu, S.C.H., Asara, J.M., Lane, W.S.,
51.
Treebak, J.T., Glund, S., Deshmukh, A., Klein, D.K.,
Garner, C.C., and Lienhard, G.E. (2002) A method to
Long, Y.C., Jensen, T.E., Jorgensen, S.B., Viollet, B., An$
identify serine kinases substrates. Akt phosphorylates a
dersson, L., Neumann, D., Wallimann, T., Richter, E.A.,
novel adipocyte protein with a Rab GTPase$activating pro$
Chibalin, A.V., Zierath, J.R., and Wojtaszewski, J.F. (2006)
tein (GAP) domain, J. Biol. Chem., 277, 22115-22118,
AMPK$mediated AS160 phosphorylation in skeletal muscle
doi: 10.1074/jbc.C200198200.
is dependent on AMPK catalytic and regulatory subunits,
35.
Randle, P.J., Garland, P.B., Hales, C.N., and Newsholme, E.A.
Diabetes, 55, 2051-2058, doi: 10.2337/db06$0175.
(1963) The glucose fatty$acid cycle. Its role in insulin sen$
52.
Kramer, H.F., Witczak, C.A., Fujii, N., Jessen, N., Taylor, E.B.,
sitivity and the metabolic disturbances of diabetes mellitus,
Arnolds, D.E., Sakamoto, K., Hirshman, M.F., and
Lancet, 1, 785-789, doi: 10.1016/S0140$6736(63)91500$9.
Goodyear, L.J. (2006) Distinct signals regulate AS160
36.
Randle, P.J. (1998) Regulatory interactions between lipids
phosphorylation in response to insulin, AICAR, and con$
and carbohydrates: the glucose fatty acid cycle after 35
traction in mouse skeletal muscle, Diabetes,
55,
years, Diabetes. Metab. Rev., 14, 263-283.
2067-2076, doi: 10.2337/db06$0150.
37.
Cole, T.G., Patsch, W., Kuisk, I., Gonen, B., and
53.
Treebak, J.T., Birk, J.B., Rose, A.J., Kiens, B., Richter, E.A.,
Schonfeld, G. (1983) Increases in dietary cholesterol and
and Wojtaszewski, J.F. (2007) AS160 phosphorylation is
fat raise levels of apoprotein E$containing lipoproteins in
associated with activation of α2β2γ1$ but not α2β2γ3$AMPK
the plasma of man, J. Clin. Endocrinol. Metab., 56,
trimeric complex in skeletal muscle during exercise in
1108-1115, doi: 10.1210/jcem$56$6$1108.
humans, Am. J. Physiol. Endocrinol. Metab.,
292,
38.
Golay, A., and Bobbioni, E. (1997) The role of dietary fat
E715-E722, doi: 10.1152/ajpendo.00380.2006.
in obesity, Int. J. Obes. Relat. Metab. Disord., 21, S2-S11.
БИОХИМИЯ том 84 вып. 5 2019
ИНДУКТОРЫ СТРЕССА СНИЖАЮТ ЧУВСТВИТЕЛЬНОСТЬ К ИНСУЛИНУ
729
CHEMICAL INDUCERS OF OBESITY ASSOCIATED
METABOLIC STRESS ACTIVATE INFLAMMATORY
PATHWAYS REDUCING INSULIN SENSITIVITY
IN 3T3 L1 ADIPOCYTES
I. S. Stafeev1,2*, S. S. Michurina1,3, N. V. Podkuychenko1,3,
M. Y. Menshikov1, Ye. V. Parfyonova1,2, and A. V. Vorotnikov1,4*
1 Institute of Experimental Cardiology, National Medical Research Centre
for Cardiology, 121552 Moscow, Russia; E*mail: yuristafeev@gmail.com,
a.vorotnikov@icloud.com
2 Lomonosov Moscow State University, Faculty of Fundamental Medicine,
117192 Moscow, Russia
3 Lomonosov Moscow State University, Faculty of Biology,
119991 Moscow, Russia
4 Lomonosov Moscow State University, Medical Center,
119991 Moscow, Russia
Received October 8, 2018
Revised January 23, 2019
Accepted January 23, 2019
Obesity associated with dyslipidemia, inflammation, hypoxia, and endoplasmic reticulum (ER) stress is the major risk
for insulin resistance and type 2 diabetes development. We modelled these conditions in cultured 3T3$L1 adipocytes
to determine their effects on insulin signaling, glucose uptake, and inflammatory response using the value of activa$
tion of stress$dependent JNK1/2 kinases for quantitative assessment of effects. Under conditions of lipid (palmitate)
overload of cells, acute lipopolysaccharide (LPS)$induced inflammation, Co2+$induced hypoxia, and brefeldin
A$caused ER stress, we detected a decrease in insulin stimulation of IRS, Akt, and AS160 phosphorylation. In all
these conditions except for acute inflammation, the insulin$dependent glucose uptake by adipocytes was reduced, and
the activation kinetics of JNK1/2 was bi$phasic demonstrating an increased activity over 24 h. In contrast, in case of
acute inflammation, the kinetics of JNK1/2 activation by LPS was transient, and the level of JNK phosphorylation
returned to the basal level at 2-3 h of stimulation. These results suggest a critical role for sustained (latent) vs.
transient (acute) inflammation in induction of adipose insulin resistance and glucose uptake impairment. Thus,
inflammatory signaling cascade is a potential target for new therapeutic approaches to prevent insulin resistance and
type 2 diabetes development.
Keywords: insulin resistance, type 2 diabetes, inflammation, glucose uptake, adipocytes
9 БИОХИМИЯ том 84 вып. 5 2019