БИОХИМИЯ, 2020, том 85, вып. 1, с. 80 - 92
УДК 577.1, 57.088
МЕТОДЫ ВЫЧИСЛИТЕЛЬНОЙ ИНТЕРАКТОМИКИ
В ВОПРОСАХ ВЗАИМОДЕЙСТВИЯ ПРОТЕОФОРМ ЧЕЛОВЕКА
Обзор
© 2020
Е.В. Поверенная*, О.И. Киселева, А.С. Иванов, Е.А. Пономаренко
НИИ биомедицинской химии имени В.Н. Ореховича, 119121 Москва, Россия;
электронная почта: k.poverennaya@gmail.com
Поступила в редакцию 25.03.2019
После доработки 16.09.2019
Принята к публикации 07.10.2019
Для человека известно около 20 000 белок кодирующих генов, которые могут быть транслированы в миллио
ны уникальных видов белков (протеоформ). Протеоформы, кодируемые одним геном, зачастую отличают
ся по функции, что соответствует различиям в белковых партнерах. Взаимодействуя между собой, протео
формы образуют сеть, отражающую динамически изменяющиеся клеточные процессы в организме. Нару
шение белок белковых взаимодействий (ББВ) вызывает изменение в топологии сети, что зачастую приво
дит к возникновению патологических процессов. Изучение протеоформ - это относительно новая задача в
протеомике, а потому экспериментальных работ по взаимодействию протеоформ немного. Биоинформати
ческие инструменты позволяют решать ряд задач, комплементарно дополняя и обогащая эксперименталь
ные результаты, в частности, расширяя возможности в исследовании взаимодействия протеоформ.
КЛЮЧЕВЫЕ СЛОВА: белок белковые взаимодействия, интерактомика, биоинформатика.
DOI: 10.31857/S0320972520010066
Функционирование живой клетки осущест
дрожжевая система (yeast two hybrid, Y2H) [4] и
вляется за счет множества различных молекуляр
аффинное выделение белкового комплекса с
ных взаимодействий между белками, нуклеино
последующим масс спектрометрическим ана
выми кислотами, пептидами, метаболитами и т.д.
лизом (affinity purification-mass spectrometry,
Несмотря на то что понятие «интерактом» ком
AP MS) [5].
плексное, объединяющее все виды взаимодей
Технология Y2H основана на активации
ствий, наиболее часто его употребляют в отноше
экспрессии репортерных генов, которая возни
нии белок белковых взаимодействий (ББВ).
кает в результате взаимодействия между целе
Именно белковые взаимодействия играют клю
вым белком «наживкой» и его партнером бел
чевую роль в биологических процессах [1].
ком «добычей» и может быть детектирована.
Под взаимодействием белков подразумева
За 25 лет с момента изобретения первого ва
ется связь двух и более белковых молекул, обес
рианта метода Y2H [6] было разработано множе
печивающая выполнение биологической функ
ство альтернативных протоколов, позволяю
ции. Различают физические взаимодействия и
щих, например, использовать помимо дрожжей
взаимодействия в составе сети [2]. Физические
клетки других организмов [7], а также исследо
взаимодействия подразделяются на прямые (би
вать взаимодействия между белками и нуклеи
нарные) и комплексные. Группа белков, кото
новыми кислотами: ДНК (one hybrid, 1Н, [8]) и
рые взаимодействуют друг с другом одновре
РНК (three hybrid, 3H, [9]). Преимуществом ме
менно, называется белковым комплексом.
тода Y2H является простота и возможность ав
Существуют различные экспериментальные
томатизации анализа, что позволяет масштаби
методы выявления белок белковых взаимодей
ровать эксперименты.
ствий [3]. Основными являются двугибридная
Метод AP MS состоит в «вылавливании»
белков партнеров на иммобилизованный бе
Принятые сокращения: AP MS - аффинное выде
лок «наживку» с последующим протеолизом и
ление белкового комплекса с последующим масс спектро
масс спектрометрическим анализом пептидных
метрическим анализом, ББВ - белок белковые взаимо
действия, Y2H
- двугибридная дрожжевая система,
фрагментов образовавшихся комплексов, смы
ПТМ - посттрансляционные модификации.
ваемых с колонки [10, 11]. Масс спектрометри
* Адресат для корреспонденции.
ческая часть метода позволяет «регистрировать»
80
БИОИНФОРМАТИКА ДЛЯ ИНТЕРАКТОМИКИ
81
динамично изменяющиеся белковые комплек
белков и результатах транскриптомных и проте
сы, учитывая посттрансляционные модифика
омных экспериментов. Отдельной задачей био
ции (ПТМ), которые могут обладать биологи
информатики является верификация сведений о
ческой функцией. Следует также отметить гиб
ББВ, заключающаяся в определении взаимо
кость метода AP MS. Она выражается в возмож
действий в результатах, полученных масс спек
ности использования нативных антител к эндо
трометрически [15]. Основываясь на предполо
генным «наживкам» или в использовании эпи
жении, что физически взаимодействующие бел
топных меток, что в первом случае позволяет
ки функционально связаны между собой, вы
проводить очистку белков в естественной среде
числительные подходы используются также для
клеточного лизата и учитывать взаимодействия
предсказания функций белков [16].
сразу нескольких природных форм белка, а во
Кроме того, построение и анализ интерак
втором - позволяет проводить эксперимент да
томных сетей возможны только с помощью вы
же в отсутствие природного антитела [3]. Суще
числительных инструментов, учитывающих
ствует несколько десятков модификаций мето
комплексность данных, обработка которых тре
да, касающихся не только способа «вылавлива
бует недостижимых человеку когнитивных спо
ния» целевого белка, но и вариантов пробопод
собностей. Интеграция экспериментальных и
готовки и очистки белкового комплекса [5].
биоинформатических методов, направленная на
Оба метода не лишены недостатков, основ
детальное изучение аспектов взаимодействия
ной из которых - высокая доля ложных резуль
белков и их форм в клетке, представляет собой
татов: ложноположительных, при которых ре
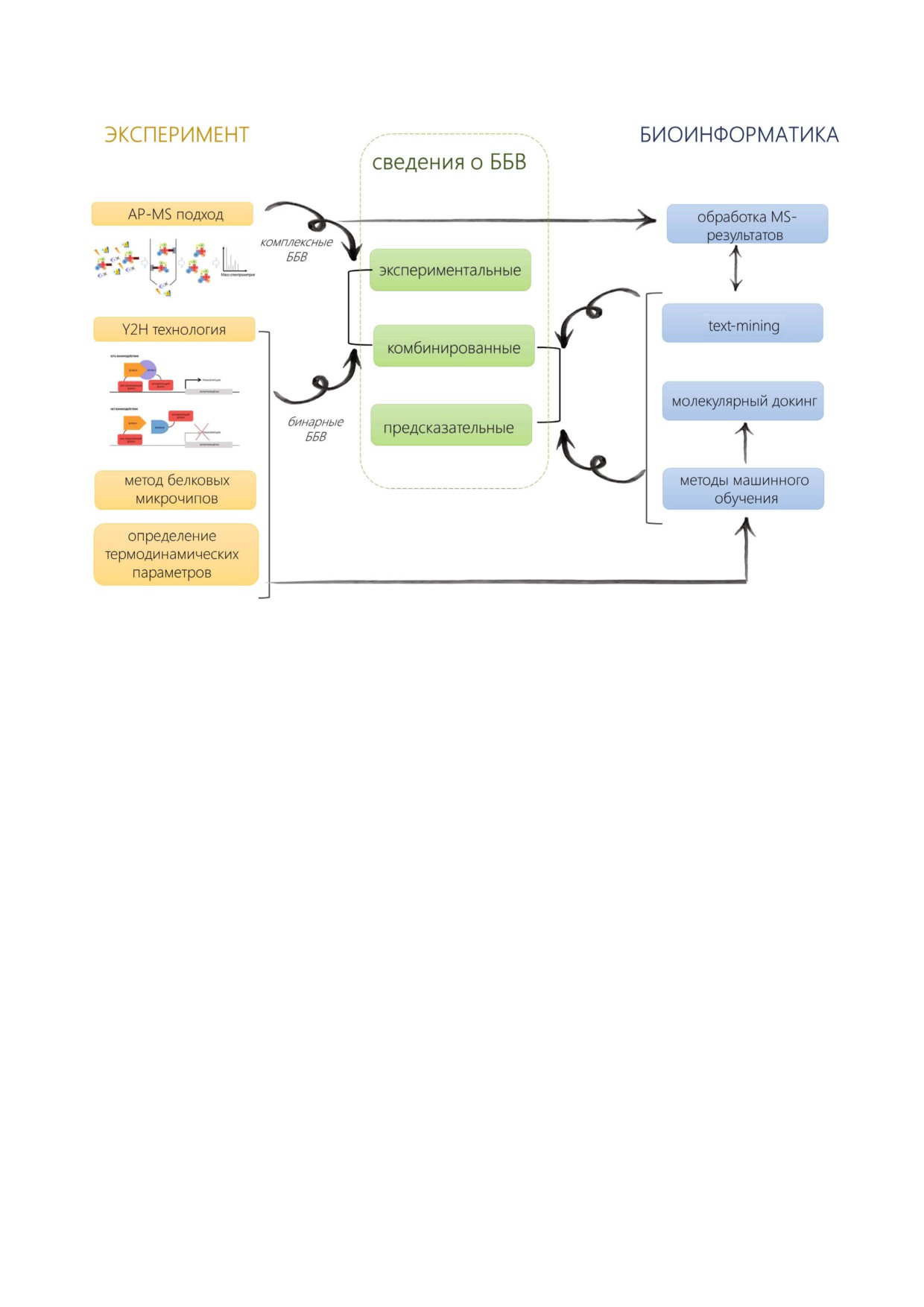
современную интерактомику (рисунок).
ального взаимодействия между белками нет, и
ложноотрицательных, при которых существую
щие ББВ не детектируются. Методы Y2H, сфо
ИНТЕРАКТОМ ЧЕЛОВЕКА
кусированные на выявлении бинарных белко
вых взаимодействий, характеризуются высокой
По объему накопленных сведений человек
долей ложных идентификаций в силу ряда при
является наиболее изученным организмом, сог
чин (например, неестественно высокой концен
ласно протеомному ресурсу UniProtKB (www.
трации фьюжн белков в тестовой системе и не
uniprot.org) [17]. Более 90% белок кодирующих
специфичности аффинных реагентов) [12]. Для
генов человека были подтверждены на протеом
методов, основанных на AP MS, ложноположи
ном уровне, при этом сведения о ББВ имеются
тельные результаты встречаются значительно
для 93%. Примечательно, что в ряде случаев на
реже, однако при экстракции и «отмывке» бел
личие данных о ББВ является доказательством
ковых комплексов утрачивается часть взаимо
существования белка.
действий. Суммарно доля взаимодействий, ус
Доказательство трансляции белка является
тановленных методом Y2H, достигает 20%, а ме
критичным условием для достоверности белок
тодом AP MS - 55% [11].
белковых взаимодействий. В большинстве
С физической точки зрения взаимодействие
предсказательных методов данной информаци
белков характеризуется константой диссоциа
ей пренебрегают, предполагая, что если взаи
ции. Для определения кинетики физического
модействие возможно, то оно имеет место
взаимодействия белков в большинстве случаев
быть, а текущие ограничения связаны с недос
используют оптические биосенсоры [13]. Одна
таточной чувствительностью аналитических
ко число белковых пар, для которых возможно
методов [18].
измерить константы диссоциации, весьма огра
Немаловажным аспектом является описа
ничено в силу сложности получения чистых
ние конкретного типа белковой молекулы -
белков в достаточном количестве для проведе
протеоформы, которая может быть образована
ния экспериментальных работ.
в результате альтернативного сплайсинга, неси
Накопление большого массива интерактом
нонимичных замен отдельных нуклеотидов,
ных данных, полученных опытным путем, и их
различных ПТМ и ряда других более редких со
противоречивость, а также изначальная слож
бытий [19]. Суммарное количество протеоформ
ность объектов исследования привели к актив
в организме человека окончательно не выясне
ному развитию биоинформатических методов.
но: в зависимости от используемого прогноза,
В первую очередь методы in silico используются
учитывающего возможность одновременного
для предсказания и описания новых ББВ на ос
возникновения нескольких аберраций в белко
нове ранее полученных сведений [14]. Предска
вом продукте одного гена, снизу оно ограниче
зательные алгоритмы базируются на анализе ге
но миллионами уникальных белковых молекул,
номных данных, аминокислотных последова
а сверху - может достигать астрономических
тельностей, данных о трехмерных структурах
величин [20, 21].
6 БИОХИМИЯ том 85 вып. 1 2020
82
ПОВЕРЕННАЯ и др.
Основные методы экспериментальной и вычислительной интерактомики
Важность учета индивидуальных протео
ции перехода от гено центричного к протео
форм заключается в том, что наличие модифи
форм центричному интерактому, доминирова
каций и изменений аминокислотного состава
ние сведений о ББВ для гена сохраняется.
белка зачастую критично для выполняемых им
В 2006 году было проведено исследование
функций, и профили белок белковых взаимо
интерактома человека, в котором было опреде
действий для протеоформ, кодируемых одним
лено, что среди полученного количества ББВ
геном, могут отличаться [22, 23].
(154 000-369 000) большая часть представлена
Аберрации, приводящие к возникновению
ложноположительными данными и только 10%
протеоформ и изменению их посттрансляцион
белковых взаимосвязей являются «истинными»
ного статуса, связывают с развитием не только
[32]. В 2008 году количество ББВ человека было
онкологических [24], но и других социально
оценено уже в 650 000 [33]. На текущий момент
значимых заболеваний [20]: в их числе аутоим
это самая большая оценка существующего инте
мунные [25], метаболические [26] и нейродеге
рактома, однако есть мнения, что данная вели
неративные [27] патологии.
чина выше реально существующей в гено цент
Расшифровка протеома, реализуемая в рам
ричном контексте [34, 35]. Тем не менее количе
ках международного проекта «Протеом челове
ство выявленных бинарных ББВ увеличивается
ка» [28], дала исследователям набор различных
с каждым годом [34]. В рамках 4 х крупнейших
методов по выявлению протеоформ [29]. В свя
проектов [36-38] по созданию интерактомных
зи с этим в последние годы появились работы по
карт человека было выявлено >15 000 бинарных
выявлению белковых взаимодействий для раз
взаимодействий, полученных разными экспери
личного типа протеоформ [22, 30, 31]. В боль
ментальными методами. Суммарно было выяв
шинстве случаев предсказание ББВ для протео
лено >90 000 ББВ, однако только 10% встреча
форм осуществляется на основе изменений ка
лись более чем в одном проекте [39].
нонической структуры белка либо же выявлены
Противоречивость данных может быть как
экспериментальным путем в результате AP MS
результатом технической ошибки, разной
экспериментов. Несмотря на развитие тенден
чувствительности аналитических методов, так и
БИОХИМИЯ том 85 вып. 1 2020
БИОИНФОРМАТИКА ДЛЯ ИНТЕРАКТОМИКИ
83
наблюдаемыми эффектами биологической ва
работе Scott и Barton [45], где уровень ошибки
риабельности, обусловленной анализом различ
был снижен на то время с 90 до 76%.
ных типов биоматериалов, динамичностью и ге
Деревья принятия решения используются
терогенностью протеома. Решение данной ди
как один из методов для предсказания ББВ, а
леммы возможно с помощью методов биоин
также для подтверждения взаимодействий, по
форматики, которая позволяет как предсказы
лученных экспериментальными методами.
вать ББВ для дальнейшей проверки, так и вери
Каждое дерево состоит из узлов - атрибутов, ха
фицировать получаемые экспериментальные
рактеризующих некое свойство белка, ребер -
данные для получения новых предсказаний.
«веток» дерева, на которых записаны значения
атрибутов, и «листьев», которым приписаны
значения целевой функции, т.е. взаимодейству
МЕТОДЫ МАШИННОГО
ют белки или нет [2]. Для классификации ново
ОБУЧЕНИЯ (ММО)
го случая необходимо создать такое дерево на
основе обучающей выборки, а затем «пройти от
Развитие постгеномных технологий привело
корня к листьям» для предсказания взаимодей
к накоплению данных в области протеомики,
ствия между белками.
геномики и транскриптомики. В связи с этим
В методе «случайного леса» (random forest) ис
актуальными становятся биоинформатические
пользуется множество таких «деревьев». Итого
методы анализа данных, которые позволяют по
вое решение принимается путем консенсусного
высить уровень достоверности интерактома и
голосования для совокупности всех «деревьев». В
выявить новые закономерности взаимодей
интерактомике человека метод «случайного леса»
ствий между белками [40].
получил бóльшую популярность по сравнению с
Существенную группу среди применяемых
методом дерева принятия решения [46].
алгоритмов поиска ББВ представляют методы
Методы машинного обучения широко ис
машинного обучения. Алгоритмы машинного
пользуются для разных подходов выявления
обучения, к которым относятся метод опорных
ББВ: на основе аминокислотной/нуклеотидной
векторов (support vector machine - SVM), Байе
последовательности [47], структурной инфор
совские сети (Bayesian network), деревья приня
мации [31], филогенетических данных [48], про
тия решения (decision tree), цепи Маркова
филей экспрессии [49], анализа интерактомных
(Markov chain) и др., позволяют оценить вероят
сетей и т.д. [50]. Кроме того, они получили ши
ность взаимодействия между белками по набору
рокое использование в обработке эксперимен
их признаков [41]. Обязательным условием ра
тальных данных [38] и при интеграции различ
боты таких алгоритмов является наличие обучаю
ных типов данных [51]. Модификации и вариа
щей выборки, на основе которой настраивают
ции методов различны, в некоторых случаях ис
параметры работы методов.
пользуются сразу несколько подходов [52].
Метод опорных векторов весьма популярен
при исследовании интерактома [2]. Суть подхо
да заключается в минимизации рисков ложных
ПРЕДСКАЗАНИЕ БЕЛКОВЫХ
идентификаций путем поиска гиперплоскости,
ВЗАИМОДЕЙСТВИЙ НА ОСНОВЕ
которая наилучшим образом разделяет два
СТРУКТУРНОЙ ИНФОРМАЦИИ
класса объектов (взаимодействующие и невзаи
модействующие белки) по набору переменных
Возрастающее внимание к изучению протео
[42].
форм способствовало открытию «второго дыха
Другой подход - использование Байесовских
ния» у методов выявления белковых взаимодей
сетей для предсказания ББВ [43]. Байесовские
ствий на основе структурной информации. Дан
сети позволяют использовать данные различно
ные методы основаны на наблюдении, что бел
го типа (к примеру, описательные и числовые ха
ки, имеющие сходную третичную структуру или
рактеристики), выражая их в виде условных ве
консервативные участки последовательности,
роятностей. При этом можно компенсировать
обладают и сходной функцией в клетке, а значит
отсутствие каких либо признаков у объектов, а
могут взаимодействовать с одинаковыми парт
также учитывать вес каждого признака в соот
нерами [53, 54].
ветствии с его надежностью [44]. Вероятность
Десятилетия применения рентгеновской
взаимодействия двух белков рассчитывается как
кристаллографии позволили получить данные о
вероятность наблюдения определенного набора
структуре белковых комплексов. Эти сведения
признаков для двух белков при условии, что они
являются богатым источником для изучения
являются взаимодействующими. Преимущества
принципов взаимодействий белков. На настоя
использования данного метода были показаны в
щий момент в PDB (Protein Data Bank) [55]
БИОХИМИЯ том 85 вып. 1 2020
6*
84
ПОВЕРЕННАЯ и др.
опубликовано >40 000 3D структур белков чело
МЕТОДЫ АВТОМАТИЧЕСКОГО
века и их комплексов, и использование принци
АНАЛИЗА ТЕКСТОВ НАУЧНЫХ
па гомологии белковых последовательностей
ПУБЛИКАЦИЙ (TEXT4MINING)
позволяет существенно расширить этот набор. В
2002 году Aloy и Russel [56] использовали дан
Данные о взаимосвязанных белках (ББВ)
ные о трехмерной структуре гомологичных бел
могут быть получены на основе автоматическо
ков, чтобы оценить вероятность взаимодей
го анализа текстов научных публикаций (text
ствия предлагаемых пар. Учитывая известные
mining) [68].
3D структуры белковых комплексов и гомоло
Методы быстрого и надежного извлечения
гичные последовательности для каждого взаи
данных из научной литературы - современная
модействующего белка, подходы, основанные
необходимость. Совершенствование экспери
на аналогах трехмерных структур, позволяют
ментальных постгеномных методов ведет к экс
оценить вероятность возможных взаимодей
поненциальному росту количества статей, пре
ствий между гомологами одного и того же вида
доставляя, с одной стороны, «большие данные»
[57] или протеоформ, к примеру, в случае одно
для анализа, а с другой - глобальный вызов раз
аминокислотных замен [58] или ПТМ [59].
работчикам биоинформатических систем для
Предсказание белок белковых взаимодей
обработки данных. По состоянию на 2019 год
ствий с помощью сопоставления информации о
наиболее широко используемая база данных био
структуре белка реализовано в программе
медицинской литературы содержит ~30 млн
PRISM [60]. При разработке этой системы ис
ссылок, при этом для 5 млн из них полнотексто
пользовали данные об известных трехмерных
вая версия статьи представлена в открытом дос
структурах, доступные в PDB (~5500 белков,
тупе в ресурсе PubMedCentral (данные на март
идентичны <50%). Межбелковые контакты вы
2019 года). Автоматический анализ накоплен
деляли как совокупность атомов, расстояние
ных в форме научных публикаций знаний в об
между ван дер ваальсовыми радиусами которых
ласти взаимодействий белков во многом являет
было в пределах 5 Å. Всего была обработана
ся основой для получения новых сведений о мо
21 000 контактных площадок и выявлено 3800
лекулярных механизмах возникновения заболе
характерных интерфейсных поверхностей,
ваний человека, а также методах диагностики,
обеспечивающих контакты между субъединица
профилактики и лечения.
ми комплекса.
Большинство исследований в области извле
Домены являются наиболее консервативны
чения информации о ББВ из текстов научных
ми участками белков и зачастую обеспечивают
публикаций основаны на анализе резюме науч
контактный интерфейс для взаимодействия
ных публикаций, поскольку это наиболее дос
между субъединицами комплекса. Метод ассо
тупная часть статьи [68]. Для автоматического
циации (association method, AS) - это один из
выявления в текстах данных о белок белковых
первых методов предсказания домен-доменных
взаимодействиях используют созданные вруч
взаимодействий, основанный на оценке коли
ную правила поиска или заранее размеченный
чества доменных пар во взаимодействующих
куратором набор текстов, на основе автомати
белках по отношению к встречаемости каждого
ческого анализа которого создаются правила
домена в паре в независимой выборке [61]. Не
для последующего поиска взаимосвязей [69].
сколько позже Deng et al. предложили алгоритм,
Наиболее простые алгоритмы, используемые
где взаимодействие доменов предсказывается
для выявления ББВ, базируются на анализе час
путем оценки максимального правдоподобия
тот совместного упоминания названий белков
данного ББВ [62]. Другими подходами к выяв
или генов в заданном фрагменте текста. Поиск
лению белковых взаимодействий на уровне до
такого рода взаимосвязей основан на гипотезе,
менов являются: метод линейного программи
что, вероятно, два белка взаимодействуют друг с
рования (linear programming, LP) и его модифи
другом, если они встречаются совместно в текс
кации для выявления бинарных и комплексных
те одной [70] или близких по смыслу публика
взаимодействий [63]; метод опорных векторов;
циях [71]. Очевидным недостатком этого подхо
метод моделирования вероятностных сетей
да является существенное количество ложнопо
(probabilistic network modeling) и др. [64].
ложительных результатов. Снижение количест
На основе домен доменных взаимодействий
ва ложноположительных результатов может дос
для сплайс форм предсказываются партнеры в
тигаться за счет использования дополнительных
виде нативных вариантов белков [31], а также в
правил, но воспроизводимость полученных ре
виде сплайс форм [65], белковые партнеры для
зультатов при этом остается невысока [72].
протеоформ с различными ПТМ [66] или одно
Следующее поколение методов анализа
аминокислотными заменами [67].
текстов для выявления ББВ основано на ма
БИОХИМИЯ том 85 вып. 1 2020
БИОИНФОРМАТИКА ДЛЯ ИНТЕРАКТОМИКИ
85
шинном обучении [69]. Показано, что исполь
ченных масс спектрометрически для комплек
зование таких методов позволяет существенно
сов после аффинного выделения. С одной сто
снизить количество ложноположительных ре
роны, особенностью таких экспериментов явля
зультатов. Методы динамического программи
ется сложность идентификации белков, заклю
рования, сходные с методами выравнивания
чающаяся в ограниченном количестве получае
последовательностей, могут быть использованы
мых уникальных пептидов, по которым возмож
для нахождения шаблонов в размеченных текс
но безошибочно определить белок. Кроме того,
тах и поиска ББВ [73].
возможна ошибочная идентификация пептида,
Перспективным считается предсказание
возникающая в результате неверной интерпре
взаимодействующих белков с использованием
тации масс спектра [15]. С другой стороны, ве
ядерных методов (kernel based approaches) -
лика вероятность получения ложноположитель
класса алгоритмов для распознавания образов
ных результатов комплексных взаимодействий,
[74]. Тем не менее недостатком использования
обусловленных неспецифическими взаимодей
таких методов является отсутствие информации
ствиями из за высокой концентрации белков в
о семантических взаимосвязях между объекта
биоматериале, или же ложноотрицательных ре
ми - названиями белков. Решением этой проб
зультатов в силу разрушения белкового комп
лемы является использование для построения
лекса при экстракции [5].
интерактомной карты триплетов (триплет сос
Для решения указанных выше проблем были
тоит из названий двух белков и связанного с ни
разработаны различные биоинформатические
ми термина, характеризующего взаимосвязь),
подходы [78] и базы данных распространенных
которые могут быть найдены с использованием
белков контаминантов (CRAPome). В первую
байесовских методов машинного обучения.
очередь используют статистические пороги
Зачастую существенного улучшения резуль
(t критерий, p value), полученные на основе
татов предсказания ББВ удается достичь за счет
масс спектров для белка «наживки» и для конт
комбинирования различных методических под
рольного образца [79].
ходов [75] или интеграции многомерных данных
Вопрос о статистической значимости оцен
[76]. Для извлечения триплетов, характеризую
ки белковых взаимодействий на основе масс
щих ББВ, в 2018 году в работе, выполненной Yu
спектрометрических данных был решен в рам
et al. [69], использовали методы анализа текстов
ках программ SAINT (Significance Analysis of
на естественном языке (nature language process
INTeractome) [80] и ComPASS (Comparative
ing). Взаимосвязь может быть описана произ
Proteomics Analysis Software Suite) [81]. В качест
вольным термином или термином, присутствую
ве критерия для исключения ложноположитель
щим в заранее сформированном словаре. Полу
ных результатов в программах используется
ченные триплеты затем были разделены на «ис
распространенная в протеомике относительная
тинные» и «ложные», что позволило достичь су
количественная оценка содержания белка в
щественного преимущества в точности предло
пробе - «счетчик спектров» (spectral count). Она
женного алгоритма в сравнении с существующи
определяется путем подсчета количества масс
ми решениями. Для планирования эксперимен
спектров фрагментации, относящихся к белку
та или актуализации сведений в базах данных
пептидов. Бóльшие значения счетчика характе
точность аналитического алгоритма предсказа
ризуют более высокий уровень содержания бел
ния ББВ имеет бóльшее значение, чем высокие
ка в исследуемом образце. Близкие по идеоло
показатели воспроизводимости результата [69].
гии программы незначительно отличаются эф
В настоящее время создано внушительное
фективностью в пользу SAINT [15].
количество систем, позволяющих реконструи
С помощью SAINT можно не только подтвер
ровать интерактомную карту как в гено цент
ждать специфические бинарные взаимодей
ричном формате, так и находить взаимосвязи
ствия между белками, но и выявлять новые бел
между протеоформами. Методы анализа текстов
ковые взаимодействия. Например, при анализе
используют в основном для предсказания ББВ
результатов масс спектрометрических экспери
между фосфорилированными формами [77].
ментов для серин/треонин фосфатазы РР5 было
установлено ранее неизвестное взаимодействие
со стресс индуцированным фосфопротеином 1
ОБРАБОТКА РЕЗУЛЬТАТОВ
(STIP 1) [82].
МАСС4СПЕКТРОМЕТРИЧЕСКИХ
Другой метод анализа достоверности ББВ,
ЭКСПЕРИМЕНТОВ ПРИ AP4MS
выявленных с помощью АР МС - MiST (Mass
spectrometry interaction STatistics), основан на
Биоинформатические подходы востребова
интегрированной оценке по нескольким пара
ны и для анализа интерактомных данных, полу
метрам: содержанию белка (интенсивности пи
БИОХИМИЯ том 85 вып. 1 2020
86
ПОВЕРЕННАЯ и др.
ка), воспроизводимости (повторяемости белко
БАЗЫ ДАННЫХ
вого комплекса) и специфичности (уникальнос
ти белка «добычи» по отношению к другим бел
Многие биоинформатические алгоритмы
кам в комплексе) взаимодействия [83].
используют опубликованные сведения о ББВ
Несколько идеологически отличается алго
для настройки или определения ложных резуль
ритм SFINX (Straightforward Filtering INdeX)
татов. Полученные результаты публикуются в
[84], в рамках которого мета данные анализиру
виде баз данных. Практически каждый метод
ются автоматически - нет указания белка «на
имеет свой ресурс, однако в силу того что есть
живки» в результатах AP MS, используется не
базы данных, агрегаторы, и количество новых
счетчик спектров, а оценка интенсивности сиг
интерактомных ресурсов возрастает с каждым
налов, относящихся к пептидам. Данный метод
годом, некоторые ресурсы перестают поддержи
самостоятельно определяет пороговую величи
ваться. Данные могут обновляться постоянно
ну достоверности в каждом случае на основе би
или с некой периодичностью (версией ресурса),
номиального распределения.
а могут быть статичными.
Для оценки ложноположительных результа
Ниже приведен список актуальных и наибо
тов, возникающих в первую очередь из за конта
лее распространенных баз данных ББВ человека
минации, были разработаны методы CRAPome
(таблица). Можно выделить две группы интерак
[85] и Decontaminator [86], в основе которых ле
томных ресурсов - специализирующихся на
жит оценка частоты встречаемости в различных
конкретном организме (например, содержащих
экспериментах.
данные только для человека) и на конкретном ти
В качестве входных данных для методов вы
пе данных (предсказанные ББВ, эксперименталь
числительной интерактомики используются ре
но подтвержденные ББВ, комбинированные).
зультаты масс спектрометрических экспери
Суммарно для человека имеются данные о
ментов, выполненных после аффинного выде
нескольких миллионах белковых взаимодей
ления белкового комплекса. Большое количе
ствий. Многие ресурсы интегрируют различные
ство таких экспериментов депонируется в цент
дополнительные сведения - о клеточной лока
рализованные протеомные репозитории. Для
лизации [92], уровне экспрессии [93], свойствах
многих экспериментов при этом отсутствуют
гомологичных белков у других организмов [94] и
мета данные, например, в репозитории GPMdb
т.д. Отдельно стоит упоминать базы данных,
[87], которые бы указывали, что результаты
описывающие межорганизменные взаимодей
идентификации белков были получены в рам
ствия (в частности, человека с вирусами) [95].
ках исследования интерактома. В работе Zhang
Тем не менее данные ресурсов совпадают не бо
et al. [88] был предложен метод, позволяющий в
лее чем на 10-20% [39], в связи с чем возникает
автоматическом режиме извлечь из репозито
необходимость разработки новых алгоритмов
рия данные, которые с наибольшей вероят
предсказания и анализа ББВ.
ностью относятся к анализу белковых взаимо
действий. В основе метода лежит идея о том,
что частота совместно встречающихся белков в
ИНТЕРАКТОМНЫЕ СЕТИ
результатах AP MS будет высока для обоих бел
ковых партнеров.
Использование биоинформатических мето
Методы панорамной масс спектрометрии
дов позволяет визуализировать сети (или карты)
позволяют выявлять протеоформы без дополни
ББВ. Визуализация дает представление о роли
тельных трудозатрат - путем использования
каждого белка в биологических процессах, а
расширенных библиотек и настроек при анали
также позволяет выявлять новые белковые взаи
зе масс спектра. На сегодняшний момент экс
модействия на основе построенных карт. При
периментальная база данных ББВ IntACT [89]
мером служит работа Kovács et al. [96], где на ос
является единственным ресурсом, поддерживаю
нове структурных и эволюционных данных по
щим публикацию идентифицированных
казали, что белки, взаимодействующие через
сплайс форм и их взаимодействий. Необрабо
одного партнера, на самом деле в ряде случаев
танные масс спектры интерактомных экспери
взаимодействуют напрямую.
ментов депонируются непосредственно в проте
Перед алгоритмами визуализации интерак
омные репозитории PRIDE [90], GPMdb [87] и
томных карт стоит несколько задач: 1) совмести
PeptideAtlas [91]. В случае top down подхода в
мость с различными форматами сведений о ББВ,
масс спектрометрии был разработан биоинфор
2) воспринимаемый формат визуализации с воз
матический способ анализа белковых комплек
можностью масштабирования и 3) анализ сетей
сов, образованных в том числе различными ти
различными методами (кластеризация, анализ
пами протеоформ [30].
графа и т.д.), включая возможность подключе
БИОХИМИЯ том 85 вып. 1 2020
БИОИНФОРМАТИКА ДЛЯ ИНТЕРАКТОМИКИ
87
Интерактомные ресурсы, содержащие сведения о ББВ человека
Количество
База данных
Ссылка
По типу данных
ББВ для
Обновляемость
человека
Специализированная для человека
BioPlex
экспериментальная
56 000
периодически
обновляемая
HAPPI
комбинированная
2 922 202
статичная
HPID
комбинированная
9000
периодически
обновляемая
HPRD
комбинированная
41 327
статичная
PepCyber: P Pep
предсказательная
11 269
статичная
TissueNet
периодически
243 706
предсказательная
обновляемая
UniHI
периодически
573 995
предсказательная
обновляемая
Содержащая данные для различных организмов
APID
комбинированная
385 438
постоянно
обновляемая
BioGRID
комбинированная
496 730
постоянно
обновляемая
ComPPI
комбинированная
1 311 184
статичная
DIP
периодически
9141
комбинированная
обновляемая
IID
периодически
975 877
комбинированная
обновляемая
IntACT
экспериментальная
~455 000
постоянно
обновляемая
I2D
периодически
296 008
комбинированная
обновляемая
STRING
комбинированная
~4 000 000
постоянно
обновляемая
MINT
комбинированная
48 352
постоянно
обновляемая
3did
комбинированная
~6000
статичная
ния дополнительной информации из других ре
elegans. На основе анализа сведений о белковых
сурсов. В качестве программ построения инте
взаимодействиях модельных организмов были
рактомных карт наибольшую популярность по
выделены консервативные участки интерактома.
лучили Cytoscape, NAViGaTOR, VisAnt, ProViz и
Затем появились публикации по построению
др. [97]. Первая интерактомная карта человека
карты ББВ для человека на основе результатов,
появилась в 2004 году [98]. Она была построена
полученных высокопроизводительным методом
по гомологии с данными о белковых взаимодей
двугибридной дрожжевой системы [99]. В 2007
ствиях модельных организмов: Saccharomyces
году была сконструирована первая карта на ос
cerevisiae, Drosophila melanogaster и Caenorhabditis
нове данных метода AP MS [100]. Сеть состоит
БИОХИМИЯ том 85 вып. 1 2020
88
ПОВЕРЕННАЯ и др.
из вершин (белков), связанных ребрами (белок
в выявлении новых механизмов развития забо
белковые взаимодействия). По мере накопления
леваний ключевым фактором становится изме
сведений об интерактоме возник интерес к ис
нение взаимодействий аберрантных протеоформ
следованию структур сетей.
[109]. Именно анализ сетей ББВ способствовал
В зависимости от расстояния между любыми
развитию изучения протеоформ, которые уже
двумя узлами в сети выделяют три структуры
рассматриваются и в клиническом аспекте [110].
интерактомных сетей: «безразмерные» (scale
free network), «маленького мира» (small word
Несмотря на активно развивающиеся подхо
network) и «рандомные» (random network). В се
ды геномного и постгеномного анализа совре
тях «маленького мира» расстояния между узла
менная интерактомика невозможна без биоин
ми небольшие, в отличие от «безразмерных».
форматических методов. Высокопроизводи
«Рандомная» структура интерактомных сетей
тельные технологии генерируют огромные мас
была предложена первой, но оказалась не самой
сивы молекулярных данных, но расшифровка
удачной моделью, поскольку не включала в себя
этих данных подразумевает анализ их взаимо
узловые белки, или белки «центры» («hub»)
действий между собой. Построение интерак
[101]. Особенностью узловых белков является
томных карт, равно как и их трактовка, возмож
повышенное количество взаимодействий по от
ны только с помощью методов биоинформати
ношению к другим белкам, участвующим в сети.
ки. Переход к прецизионной медицине требует
Ключевая роль узловых белков проявляется
выявлять особенности «нормы» для каждого ин
в изменении биологического состояния [102].
дивидуума. При сравнительно небольшом коли
Взаимодействия одного или нескольких белков
честве белок кодирующих генов гетерогенность
«центров» с компонентами белковой сети быва
на уровне протеома у человека значительна.
ет достаточно для переключения клеточного
Для человека, согласно данным UniProt [17],
процесса. Основная часть белков, участвующих
вариатив протеоформ, кодируемых с одного ге
в динамических процессах, экспрессируется не
на, наибольший среди всех организмов, однако
зависимо от узловых белков, образование кото
количество «нейтральных» и «аномальных» бел
рых происходит в течение всего процесса рабо
ковых молекул только предстоит узнать. На фо
ты комплекса (статистические узловые белки)
не перехода к прецизионной медицине выявле
или обусловлено определенными условиями
ние особенности «нормы» для каждого индиви
(динамические узловые белки) [103]. Динамич
дуума - это вызов как для протеомики (детек
ная модульная структура сети присуща различ
ция всех транслируемых протеоформ, в том чис
ным видам биологических процессов, включая
ле органоспецифичных), так и для интеракто
экспрессию генов, посттрансляционную моди
мики (выявление критических протеоформ, ха
фикацию и субклеточную организацию.
рактерных для развития патологических состоя
Анализ сетей ББВ вычислительными и ста
ний). В данных вопросах биоинформатика явля
тистическими методами позволяет получать ин
ется универсальным инструментом анализа на
формацию об организации биологических сис
всех уровнях получения и интерпретации ре
тем [104]. Все больше внимания уделяется ис
зультатов, позволяя, в том числе, компенсиро
следованию интерактомных карт в медицин
вать ограничения экспериментальных методов.
ском аспекте. Центральная гипотеза, лежащая в
основе связи карт ББВ и фенотипа заболеваний,
заключается в нарушении локальной или гло
Финансирование. Работа выполнена при под
бальной структуры сетей, приводящей к патоло
держке Российского фонда фундаментальных
гическим изменениям [102].
исследований по теме «Построение интерак
С помощью исследований интерактомных
томной карты человека» (грант № 18 34 00879).
сетей возможно выявление новых аспектов кле
Конфликт интересов. Авторы заявляют об от
точных процессов: не только при заболеваниях,
сутствии конфликта интересов.
включающих в том числе наркологические зави
Соблюдение этических норм. Настоящая
симости [105] и взаимодействия с вирусами
статья не содержит описания выполненных ав
[106], но и при поиске новых лекарств [107], в
торами исследований с участием людей или ис
том числе антибиотиков [108]. Непосредственно
пользованием животных в качестве объектов.
СПИСОК ЛИТЕРАТУРЫ
1.
Braun, P., and Gingras, A. C. (2012) History of pro
2.
Qi, Y., Bar Joseph, Z., and Klein Seetharaman, J. (2006)
tein-protein interactions: from egg white to complex net
Evaluation of different biological data and computational
works, Proteomics, 12, 1478-1498, doi: 10.1002/pmic.
classification methods for use in protein interaction predic
201100563.
tion, Proteins, 63, 490-500, doi: 10.1002/prot.20865.
БИОХИМИЯ том 85 вып. 1 2020
БИОИНФОРМАТИКА ДЛЯ ИНТЕРАКТОМИКИ
89
3.
Snider, J., Kotlyar, M., Saraon, P., Yao, Z., Jurisica, I., and
forms are there? Nat. Chem. Biol.,
14,
206-214,
Stagljar, I. (2015) Fundamentals of protein interaction net
doi: 10.1038/nchembio.2576.
work mapping, Mol. Syst. Biol., 11, 848, doi: 10.15252/
21.
Ponomarenko, E.A., Poverennaya, E.V., Ilgisonis, E.V.,
msb.20156351.
Pyatnitskiy, M.A., Kopylov, A.T., Zgoda, V.G., and
4.
Brückner, A., Polge, C., Lentze, N., Auerbach, D., and
Archakov, A.I. (2016) The size of the human proteome: the
Schlattner, U. (2009) Yeast two hybrid, a powerful tool for
width and depth, Int. J. Anal. Chem., doi: 10.1155/
systems biology, Int. J. Mol. Sci.,
10,
2763-2788,
2016/7436849.
doi: 10.3390/ijms10062763.
22.
Uversky, V.N. (2016) p53 proteoforms and intrinsic disor
5.
Hakhverdyan, Z., Domanski, M., Hough, L.E., Oroskar, A.A.,
der: an illustration of the protein structure function con
Oroskar, A.R., Keegan, S., and LaCava, J. (2015) Rapid,
tinuum concept, Int. J. Mol. Sci., 17, doi: 10.3390/
optimized interactomic screening, Nat. Methods, 12,
ijms17111874.
553-560, doi: 10.1038/nmeth.3395.
23.
Kelemen, O., Convertini, P., Zhang, Z., Wen, Y., Shen, M.,
6.
Fields, S., and Song, O. (1989) A novel genetic system to
Falaleeva, M., and Stamm, S. (2013) Function of alterna
detect protein-protein interactions, Nature,
340,
tive splicing, Gene, 514, 1-30, doi: 10.1016/j.gene.
245-246, doi: 10.1038/340245a0.
2012.07.083.
7.
Riegel, E., Heimbucher, T., Höfer, T., and Czerny, T. (2017)
24.
Pyatnitskiy, M., Karpov, D., Poverennaya, E., Lisitsa, A.,
A sensitive, semi quantitative mammalian two hybrid assay,
and Moshkovskii, S. (2015) Bringing down cancer aircraft:
BioTechniques, 62, 206-214, doi: 10.2144/000114544.
searching for essential hypomutated proteins in skin
8.
Gaudinier, A., Tang, M., Bågman, A. M., and Brady, S.M.
melanoma, PLoS One, 10, e0142819, doi: 10.1371/journal.
(2017) Identification of protein-DNA interactions using
pone.0142819.
enhanced yeast one hybrid assays and a semiautomated
25.
Plymire, D.A., Wing, C.E., Robinson, D.E., and Patrie, S.M.
approach, Methods Mol. Biol.,
1610,
187-215,
(2017) Continuous elution proteoform identification of
doi: 10.1007/978 1 4939 7003 2_13.
myelin basic protein by superficially porous reversed phase
9.
Glass, F., and Takenaka, M. (2018) The yeast three hybrid
liquid chromatography and fourier transform mass spec
system for protein interactions, Methods Mol. Biol., 1794,
trometry, Anal. Chem., 89, 12030-12038, doi: 10.1021/
195-205, doi: 10.1007/978 1 4939 7871 7_12.
acs.analchem.7b02426.
10.
Dunham, W.H., Mullin, M., and Gingras, A. C. (2012)
26.
Nedelkov, D. (2017) Mass spectrometric studies of
Affinity purification coupled to mass spectrometry: basic
apolipoprotein proteoforms and their role in lipid metabo
principles and strategies, Proteomics, 12, 1576-1590,
lism and type 2 diabetes, Proteomes, 5, 27, doi: 10.3390/
doi: 10.1002/pmic.201100523.
proteomes5040027.
11.
Morris, J.H., Knudsen, G.M., Verschueren, E., Johnson, J.R.,
27.
Lacovich, V., Espindola, S.L., Alloatti, M., Pozo Devoto, V.,
Cimermancic, P., Greninger, A.L., and Pico, A.R. (2014)
Cromberg, L.E., Čarná, M.E., Giancarlo, F., Gallo, J.M.,
Affinity purification-mass spectrometry and network
Bruno, L., Stokin, J.B., Avale, M.E., and Falzone, T.L.
analysis to understand protein-protein interactions, Nat.
(2017) Tau isoforms imbalance impairs the axonal trans
Prot., 9, 2539-2554, doi: 10.1038/nprot.2014.164.
port of the amyloid precursor protein in human neurons,
12.
Serebriiskii, I.G., and Golemis, E.A. (2001) Two hybrid
J. Neurosci., 37, 58-69, doi: 10.1523/JNEUROSCI.
system and false positives. Approaches to detection and
2305 16.2017.
elimination, Methods Mol. Biol.,
177,
123-134,
28.
Пономаренко Е.А., Поверенная Е.В., Ильгисонис Е.В.,
doi: 10.1385/1 59259 210 4:123.
Копылов А.Т., Згода В.Г., Лисица А.В., Арчаков А.И.
13.
Florinskaya, A., Ershov, P., Mezentsev, Y., Kaluzhskiy, L.,
(2017) Перспективы исследования протеома человека,
Yablokov, E., Medvedev, A., and Ivanov, A. (2018) SPR
Вестник Российской академии наук, 87, 599-604,
biosensors in direct molecular fishing: implications for pro
doi: 10.1134/S1019331617040049.
tein interactomics, Sensors, 18, 1616, doi: 10.3390/
29.
Киселева О.И., Лисица А.В., Поверенная Е.В. (2018)
s18051616.
Протеоформы: методы исследования и клинические
14.
Иванов А.С., Згода В.Г., Арчаков А.И. (2011) Техноло
перспективы, Молекулярная биология, 52, 394-410,
гии белковой интерактомики, Биоорганическая химия,
doi: 10.7868/S0026898418030047.
37, 8-21, doi: 10.1134/s1068162011010092.
30.
Skinner, O.S., Havugimana, P.C., Haverland, N.A.,
15.
Nesvizhskii, A.I. (2012) Computational and informatics
Fornelli, L., Early, B.P., Greer, J.B., and Kelleher, N.L.
strategies for identification of specific protein interaction
(2016) An informatic framework for decoding protein
partners in affinity purification mass spectrometry experi
complexes by top down mass spectrometry, Nat. Methods,
ments, Proteomics,
12,
1639-1655, doi:
10.1002/
13, 237-240, doi: 10.1038/nmeth.3731.
pmic.201100537.
31.
Ghadie, M.A., Lambourne, L., Vidal, M., and Xia, Y.
16.
Teng, Z., Guo, M., Liu, X., Tian, Z., and Che, K. (2017)
(2017) Domain based prediction of the human isoform
Revealing protein functions based on relationships of inter
interactome provides insights into the functional impact of
acting proteins and GO terms, J. Biomed. Semantics, 8, 27,
alternative splicing, PLoS Comput. Biol., 13, e1005717,
doi: 10.1186/s13326 017 0139 8.
doi: 10.1371/journal.pcbi.1005717.
17.
The UniProt Consortium (2008) The Universal Protein
32.
Hart, G.T., Ramani, A.K., and Marcotte, E.M. (2006)
Resource (UniProt), Nucleic Acids Res., 36, Suppl. 1,
How complete are current yeast and human protein inter
D190-D195, doi: 10.1093/nar/gkm895.
action networks? Genome Biol., 7, 120, doi: 10.1186/gb
18.
Medvedev, A., Kopylov, A., Buneeva, O., Zgoda, V., and
2006 7 11 120.
Archakov, A. (2012) Affinity based proteomic profiling:
33.
Stumpf, M.P.H., Thorne, T., de Silva, E., Stewart, R., An, H.J.,
problems and achievements, Proteomics, 12, 621-637,
Lappe, M., and Wiuf, C. (2008) Estimating the size of the
doi: 10.1002/pmic.201100373.
human interactome, Proc. Natl. Acad. Sci. USA, 105,
19.
Kiseleva, O., Poverennaya, E., Shargunov, A., and Lisitsa, A.
6959-6964, doi: 10.1073/pnas.0708078105.
(2017) Proteomic cinderella: customized analysis of bulky
34.
Kotlyar, M., Pastrello, C., Malik, Z., and Jurisica, I. (2019)
MS/MS data in one night, J. Bioinform. Comput. Biol., 16,
IID 2018 update: context specific physical protein-pro
doi: 10.1142/S021972001740011X.
tein interactions in human, model organisms and domesti
20.
Aebersold, R., Agar, J.N., Amster, I.J., Baker, M.S.,
cated species, Nucleic Acids Res., 47, D581-D589,
Bertozzi, C.R., et al. (2018) How many human proteo
doi: 10.1093/nar/gky1037.
БИОХИМИЯ том 85 вып. 1 2020
90
ПОВЕРЕННАЯ и др.
35.
Vidal, M. (2016) How much of the human protein interac
functional annotation for the human proteome, eLife, 5,
tome remains to be mapped? Sci. Signal., 9, eg7,
doi: 10.7554/eLife.18715.
doi: 10.1126/scisignal.aaf6030.
52.
Dick, K., and Green, J.R. (2018) Reciprocal perspective
36.
Wan, C., Borgeson, B., Phanse, S., Tu, F., Drew, K., Clark, G.,
for Improved protein-protein interaction prediction, Sci.
and Emili, A. (2015) Panorama of ancient metazoan
Rep., 8, 11694, doi: 10.1038/s41598 018 30044 1.
macromolecular complexes, Nature,
525,
339-344,
53.
Gromiha, M.M., Yugandhar, K., and Jemimah, S. (2017)
doi: 10.1038/nature14877.
Protein-protein interactions: scoring schemes and binding
37.
Hein, M.Y., Hubner, N.C., Poser, I., Cox, J., Nagaraj, N.,
affinity, Curr. Opin. Struct. Biol., 44, 31-38, doi: 10.1016/
Toyoda, Y., and Mann, M. (2015) A human interactome in
j.sbi.2016.10.016.
three quantitative dimensions organized by stoichiometries
54.
Gemovic, B., Sumonja, N., Davidovic, R., Perovic, V., and
and abundances, Cell, 163, 712-723, doi: 10.1016/j.cell.
Veljkovic, N. (2018) Mapping of protein-protein interac
2015.09.053.
tions: web based resources for revealing interactomes,
38.
Huttlin, E.L., Bruckner, R.J., Paulo, J.A., Cannon, J.R.,
Curr. Med. Chem.,
26,
3890-3910, doi:
10.2174/
Ting, L., Baltier, K., and Harper, J.W. (2017) Architecture
0929867325666180214113704.
of the human interactome defines protein communities
55.
Velankar, S., and Kleywegt, G.J. (2011) The Protein Data
and disease networks, Nature,
545,
505-509,
Bank in Europe (PDBe): bringing structure to biology,
doi: 10.1038/nature22366.
Acta Crystallogr. D,
67,
324-330, doi:
10.1107/
39.
Luck, K., Sheynkman, G.M., Zhang, I., and Vidal, M.
S090744491004117X.
(2017) Proteome scale human interactomics, Trends
56.
Aloy, P., and Russell, R.B. (2002) The third dimension for
Biochem. Sci., 42, 342-354, doi: 10.1016/j.tibs.2017.
protein interactions and complexes, Trends Biochem. Sci.,
02.006.
27, 633-638.
40.
Kotlyar, M., Rossos, A.E.M., and Jurisica, I.
(2017)
57.
Fang, Y., Sun, M., Dai, G., and Ramain, K. (2016) The
Prediction of protein-protein interactions, Curr. Protoc.
intrinsic geometric structure of protein-protein interac
Bioinformatics, 60, 8.2.1-8.2.14, doi: 10.1002/cpbi.38.
tion networks for protein interaction prediction,
41.
Zhang, M., Su, Q., Lu, Y., Zhao, M., and Niu, B. (2017)
IEEE/ACM Trans. Comput. Biol. Bioinform., 13, 76-85,
Application of machine learning approaches for pro
doi: 10.1109/TCBB.2015.2456876.
tein-protein interactions prediction, Med. Chem., 13,
58.
Tuncbag, N., Keskin, O., Nussinov, R., and Gursoy, A.
506-514, doi: 10.2174/1573406413666170522150940.
(2017) Prediction of protein interactions by structural
42.
Horvatovich, P., Lundberg, E.K., Chen, Y. J., Sung, T. Y.,
matching: prediction of PPI networks and the effects of
He, F., et al. (2015) Quest for missing proteins: update
mutations on PPIs that combines sequence and structural
2015 on chromosome centric human proteome project, J.
information, Methods Mol. Biol.,
1558,
255-270,
Proteome Res., 14, 3415-3431, doi: 10.1021/pr5013009.
doi: 10.1007/978 1 4939 6783 4_12.
43.
Bradford, J.R., Needham, C.J., Bulpitt, A.J., and
59.
Su, M. G., Weng, J.T. Y., Hsu, J.B. K., Huang, K. Y.,
Westhead, D.R. (2006) Insights into protein-protein inter
Chi, Y. H., and Lee, T. Y. (2017) Investigation and identi
faces using a Bayesian network prediction method, J. Mol.
fication of functional post translational modification sites
Biol., 362, 365-386, doi: 10.1016/j.jmb.2006.07.028.
associated with drug binding and protein-protein interac
44.
Jansen, R., Yu, H., Greenbaum, D., Kluger, Y., Krogan, N.J.,
tions, BMC Systems Biology, 11, 132, doi: 10.1186/s12918
Chung, S., and Gerstein, M. (2003) A Bayesian networks
017 0506 1.
approach for predicting protein-protein interactions from
60.
Keskin, O., Nussinov, R., and Gursoy, A. (2008) PRISM:
genomic data, Science, 302, 449-53, doi: 10.1126/
protein-protein interaction prediction by structural
science.1087361.
matching, Methods Mol. Biol., 484, 505-521, doi: 10.1007/
45.
Scott, M.S., and Barton, G.J. (2007) Probabilistic predic
978 1 59745 398 1_30.
tion and ranking of human protein-protein interactions.,
61.
Sprinzak, E., and Margalit, H.
(2001) Correlated
BMC bioinformatics, 8, 239, doi: 10.1186/1471 2105 8
sequence signatures as markers of protein-protein inter
239.
action, J. Mol. Biol., 311, 681-692, doi: 10.1006/
46.
Chatterjee, P., Basu, S., Kundu, M., Nasipuri, M., and
jmbi.2001.4920.
Plewczynski, D. (2011) PPI_SVM: prediction of pro
62.
Deng, M., Mehta, S., Sun, F., and Chen, T. (2002)
tein-protein interactions using machine learning,
Inferring domain-domain interactions from protein-pro
domain-domain affinities and frequency tables, Cell. Mol.
tein interactions, Genome Res.,
12,
1540-1548,
Biol. Lett., 16, 264-278, doi: 10.2478/s11658 011 0008 x.
doi: 10.1101/gr.153002.
47.
Guo, Y., Sheng, Q., Li, J., Ye, F., Samuels, D.C., and Shyr, Y.
63.
Hayashida, M., Ueda, N., and Akutsu, T. (2004) A simple
(2013) Large scale comparison of gene expression levels by
method for inferring strengths of protein-protein interac
microarrays and RNAseq using TCGA data, PLoS One, 8,
tions, Genome Inform., 15, 56-68.
e71462, doi: 10.1371/journal.pone.0071462.
64.
Raghavachari, B., Tasneem, A., Przytycka, T.M., and
48.
Zahiri, J., Bozorgmehr, J., and Masoudi Nejad, A. (2013)
Jothi, R. (2008) DOMINE: a database of protein domain
Computational prediction of protein-protein interaction
interactions, Nucleic Acids Res.,
36, D656-D661,
networks: algorithms and resources, Curr. Genomics, 14,
doi: 10.1093/nar/gkm761.
397-414, doi: 10.2174/1389202911314060004.
65.
Tseng, Y. T., Li, W., Chen, C. H., Zhang, S., Chen, J.J.,
49.
Bartoli, L., Martelli, P.L., Rossi, I., Fariselli, P., and
Zhou, X., and Liu, C. C. (2015) IIIDB: a database for iso
Casadio, R. (2010) The prediction of protein-protein
form isoform interactions and isoform network modules,
interacting sites in genome wide protein interaction net
BMC Genomics, 16, S10, doi: 10.1186/1471 2164 16 S2
works: the test case of the human cell cycle, Curr. Prot. Pept.
S10.
Sci., 11, 601-608, doi: 10.2174/138920310794109157.
66.
Tay, A.P., Pang, C.N.I., Winter, D.L., and Wilkins, M.R.
50.
McDowall, M.D., Scott, M.S., and Barton, G.J. (2009)
(2017) PTMOracle: a cytoscape app for covisualizing and
PIPs: human protein-protein interaction prediction data
coanalyzing post translational modifications in protein
base, Nucleic Acids Res., 37, D651-D656, doi: 10.1093/
interaction networks, J. Proteome Res., 16, 1988-2003,
nar/gkn870.
doi: 10.1021/acs.jproteome.6b01052.
51.
Garzón, J.I., Deng, L., Murray, D., Shapira, S., Petrey, D.,
67.
Ivanov, A.A., Revennaugh, B., Rusnak, L., Gonzalez
and Honig, B. (2016) A computational interactome and
Pecchi, V., Mo, X., Johns, M.A., and Fu, H. (2018) The
БИОХИМИЯ том 85 вып. 1 2020
БИОИНФОРМАТИКА ДЛЯ ИНТЕРАКТОМИКИ
91
OncoPPi Portal: an integrative resource to explore and pri
(2011) Label free quantitative proteomics and SAINT
oritize protein-protein interactions for cancer target dis
analysis enable interactome mapping for the human
covery, Bioinformatics, 34, 1183-1191, doi: 10.1093/
Ser/Thr protein phosphatase
5, Proteomics,
11,
bioinformatics/btx743.
1508-1516, doi: 10.1002/pmic.201000770.
68.
Skusa, A., Rüegg, A., and Köhler, J. (2005) Extraction of
83.
Verschueren, E., Von Dollen, J., Cimermancic, P.,
biological interaction networks from scientific literature,
Gulbahce, N., Sali, A., and Krogan, N.J. (2015) Scoring
Briefings Bioinform., 6, 263-276.
large scale affinity purification mass spectrometry datasets
69.
Yu, K., Lung, P. Y., Zhao, T., Zhao, P., Tseng, Y. Y., and
with MiST, Curr. Protoc. Bioinformatics,
49,
Zhang, J. (2018) Automatic extraction of protein-protein
8.19.1-8.19.16, doi: 10.1002/0471250953.bi0819s49.
interactions using grammatical relationship graph, BMC
84.
Titeca, K., Meysman, P., Gevaert, K., Tavernier, J.,
Med. Inform. Decis. Mak., 18, 42, doi: 10.1186/s12911
Laukens, K., Martens, L., and Eyckerman, S. (2016)
018 0628 4.
SFINX: straightforward filtering index for affinity purifica
70.
Stapley, B.J., and Benoit, G. (2000) Biobibliometrics:
tion-mass spectrometry data analysis, J. Proteome Res.,
information retrieval and visualization from co occur
15, 332-338, doi: 10.1021/acs.jproteome.5b00666.
rences of gene names in Medline abstracts, Pac. Symp.
85.
Mellacheruvu, D., Wright, Z., Couzens, A.L., Lambert, J. P.,
Biocomput., 2000, 529-540.
St Denis, N.A., Li, T., and Nesvizhskii, A.I. (2013) The
71.
Пономаренко Е.А., Лисица А.В., Ильгисонис Е.В.,
CRAPome: a contaminant repository for affinity purifica
Арчаков А.И. (2010) Создание семантических сетей
tion mass spectrometry data, Nat. Methods, 10, 730-736,
белков с использованием Pubmed/Medline, Молеку'
doi: 10.1038/nmeth.2557.
лярная биология,
44,
152-161, doi:
10.1134/
86.
Lavallée Adam, M., Cloutier, P., Coulombe, B., and
S0026893310010176.
Blanchette, M. (2011) Modeling contaminants in AP
72.
Lee, J., Kim, S., Lee, S., Lee, K., and Kang, J. (2013) On
MS/MS experiments, J. Proteome Res., 10, 886-895,
the efficacy of per relation basis performance evaluation
doi: 10.1021/pr100795z.
for PPI extraction and a high precision rule based
87.
Craig, R., Cortens, J.P., and Beavis, R.C. (2004) Open
approach, BMC Med. Inform.Decis. Mak., 13 Suppl 1, S7,
source system for analyzing, validating, and storing protein
doi: 10.1186/1472 6947 13 S1 S7.
identification data, J. Proteome Res., 3, 1234-1242,
73.
Huang, M., Zhu, X., Hao, Y., Payan, D.G., Qu, K., and
doi: 10.1021/pr049882h.
Li, M. (2004) Discovering patterns to extract protein-pro
88.
Zhang, C., Rogalski, J.C., Evans, D.M., Klockenbusch, C.,
tein interactions from full texts, Bioinformatics, 20,
Beavis, R.C., and Kast, J. (2011) In silico protein interac
3604-3612, doi: 10.1093/bioinformatics/bth451.
tion analysis using the global Proteome Machine Database
74.
Murugesan, G., Abdulkadhar, S., and Natarajan, J. (2017)
Research articles, J. Proteome Res., 10, 656-668.
Distributed smoothed tree kernel for protein-protein
89.
Kerrien, S., Aranda, B., Breuza, L., Bridge, A., Broackes
interaction extraction from the biomedical literature, PLoS
Carter, F., Chen, C., and Hermjakob, H. (2012) The
One, 12, e0187379, doi: 10.1371/journal.pone.0187379.
IntAct molecular interaction database in 2012, Nucleic
75.
Niu, Y., and Wang, Y. (2015) Protein-protein interaction
Acids Res., 40, D841-D846, doi: 10.1093/nar/gkr1088.
identification using a hybrid model, Artif. Intell. Med., 64,
90.
Jones, P., Côté, R.G., Cho, S.Y., Klie, S., Martens, L.,
185-193, doi: 10.1016/j.artmed.2015.05.003.
Quinn, A.F., and Hermjakob, H. (2008) PRIDE: new
76.
Chang, J. W., Zhou, Y. Q., Ul Qamar, M.T., Chen, L. L.,
developments and new datasets, Nucleic Acids Res., 36,
and Ding, Y. D. (2016) Prediction of protein-protein
D878-D883, doi: 10.1093/nar/gkm1021.
interactions by evidence combining methods, Int. J. Mol.
91.
Deutsch, E.W. (2010) The PeptideAtlas Project, Methods
Sci., 17, E1946, doi: 10.3390/ijms17111946.
Mol. Biol., 604, 285-296, doi: 10.1007/978 1 60761 444
77.
Wang, Q., Ross, K.E., Huang, H., Ren, J., Li, G., Vijay
9_19.
Shanker, K., and Arighi, C.N. (2017) Analysis of protein
92.
Veres, D.V., Gyurkó, D.M., Thaler, B., Szalay, K.Z.,
phosphorylation and its functional impact on protein-pro
Fazekas, D., Korcsmáros, T., and Csermely, P. (2015)
tein interactions via text mining of the scientific literature,
ComPPI: a cellular compartment specific database
Methods Mol. Biol., 1558, 213-232, doi: 10.1007/978 1
for protein-protein interaction network analysis,
4939 6783 4_10.
Nucleic Acids Res., 43, D485-D493, doi: 10.1093/nar/
78.
Armean, I.M., Lilley, K.S., and Trotter, M.W.B. (2013)
gku1007.
Popular computational methods to assess multiprotein
93.
Basha, O., Barshir, R., Sharon, M., Lerman, E., Kirson, B.F.,
complexes derived from label free affinity purification and
Hekselman, I., and Yeger Lotem, E. (2017) The TissueNet
mass spectrometry (AP MS) experiments, Mol. Cell.
v.2 database: a quantitative view of protein-protein inter
Proteomics, 12, 1-13, doi: 10.1074/mcp.R112.019554.
actions across human tissues, Nucleic Acids Res., 45,
79.
Fernández, E., Collins, M.O., Uren, R.T., Kopanitsa, M.V.,
D427-D431, doi: 10.1093/nar/gkw1088.
Komiyama, N.H., Croning, M. D.R., and Grant, S.G.N.
94.
Brown, K.R., and Jurisica, I. (2005) Online Predicted
(2009) Targeted tandem affinity purification of PSD 95
Human Interaction Database, Bioinformatics,
21,
recovers core postsynaptic complexes and schizophrenia
2076-2082, doi: 10.1093/bioinformatics/bti273.
susceptibility proteins, Mol. Syst. Biol.,
5,
269,
95.
Rozenblatt Rosen, O., Deo, R.C., Padi, M., Adelmant, G.,
doi: 10.1038/msb.2009.27.
Calderwood, M.A., Rolland, T., and Vidal, M. (2012)
80.
Choi, H., Larsen, B., Lin, Z. Y., Breitkreutz, A.,
Interpreting cancer genomes using systematic host network
Mellacheruvu, D., Fermin, D., and Nesvizhskii, A.I.
perturbations by tumour virus proteins, Nature, 487,
(2011) SAINT: probabilistic scoring of affinity purifica
491-495, doi: 10.1038/nature11288.
tion mass spectrometry data, Nat. Methods, 8, 70-73,
96.
Kovács, I.A., Luck, K., Spirohn, K., Wang, Y., Pollis, C.,
doi: 10.1038/nmeth.1541.
Schlabach, S., and Barabási, A. L. (2019) Network based
81.
Sowa, M.E., Bennett, E.J., Gygi, S.P., and Harper, J.W.
prediction of protein interactions, Nat. Commun., 10,
(2009) Defining the human deubiquitinating enzyme inter
1240, doi: 10.1038/s41467 019 09177 y.
action
landscape,
Cell,
138,
389-403,
97.
Agapito, G., Guzzi, P.H., and Cannataro, M. (2013)
doi: 10.1016/j.cell.2009.04.042.
Visualization of protein interaction networks: problems
82.
Skarra, D.V., Goudreault, M., Choi, H., Mullin, M.,
and solutions, BMC Bioinformatics, 14, Suppl. 1, S1,
Nesvizhskii, A.I., Gingras, A. C., and Honkanen, R.E.
doi: 10.1186/1471 2105 14 S1 S1.
БИОХИМИЯ том 85 вып. 1 2020
92
ПОВЕРЕННАЯ и др.
98. Lehner, B., and Fraser, A.G. (2004) A first draft human
ing of protein interaction networks, Nat. Commun., 10,
protein interaction map, Genome Biol.,
5, R63,
1118, doi: 10.1038/s41467 019 09123 y.
doi: 10.1186/gb 2004 5 9 r63.
105. Chen, S. J., Liao, D. L., Chen, C. H., Wang, T. Y., and
99. Stelzl, U., Worm, U., Lalowski, M., Haenig, C.,
Chen, K. C. (2019) Construction and analysis of pro
Brembeck, F.H., Goehler, H., and Wanker, E.E. (2005) A
tein-protein interaction network of heroin use disorder,
human protein-protein interaction network: a resource for
Sci. Rep., 9, 4980, doi: 10.1038/s41598 019 41552 z.
annotating the proteome, Cell,
122,
957-968,
106. Ackerman, E.E., Kawakami, E., Katoh, M., Watanabe, T.,
doi: 10.1016/j.cell.2005.08.029.
Watanabe, S., Tomita, Y., and Kawaoka, Y.
(2018)
100. Ewing, R.M., Chu, P., Elisma, F., Li, H., Taylor, P.,
Network guided discovery of influenza virus replication
Climie, S., and Figeys, D. (2007) Large scale mapping of
host factors, mBio, 9, doi: 10.1128/mBio.02002 18.
human protein-protein interactions by mass spectrometry,
107. Macalino, S.J.Y., Basith, S., Clavio, N.A.B., Chang, H.,
Mol. Syst. Biol., 3, 89, doi: 10.1038/msb4100134.
Kang, S., and Choi, S. (2018) Evolution of in silico strate
101. Taylor, I.W., and Wrana, J.L. (2012) Protein interaction
gies for protein-protein interaction drug discovery,
networks in medicine and disease, Proteomics,
12,
Molecules, 23, 1963, doi: 10.3390/molecules23081963.
1706-1716, doi: 10.1002/pmic.201100594.
108. Miho, E., Roškar, R., Greiff, V., and Reddy, S.T. (2019)
102. Li, Q., Chen, W., Song, M., Chen, W., Yang, Z., and Yang, A.
Large scale network analysis reveals the sequence space
(2019) Weighted gene co expression network analysis and
architecture of antibody repertoires, Nat. Commun., 10,
prognostic analysis identifies hub genes and the molecular
1321, doi: 10.1038/s41467 019 09278 8.
mechanism related to head and neck squamous cell carci
109. Soetkamp, D., Raedschelders, K., Mastali, M., Sobhani, K.,
noma, Cancer Biol. Ther., 20, 750-759, doi: 10.1080/
Bairey Merz, C.N., and Van Eyk, J. (2017) The continuing
15384047.2018.1564560.
evolution of cardiac troponin I biomarker analysis: from
103. Taylor, I.W., Linding, R., Warde Farley, D., Liu, Y.,
protein to proteoform, Expert Rev. Proteomics,
14,
Pesquita, C., Faria, D., and Wrana, J.L. (2009) Dynamic
973-986, doi: 10.1080/14789450.2017.1387054.
modularity in protein interaction networks predicts breast
110. Van der Burgt, Y.E.M., and Cobbaert, C.M. (2018)
cancer outcome, Nat. Biotechnol.,
27,
199-204,
Proteoform analysis to fulfill unmet clinical needs and
doi: 10.1038/nbt.1522.
reach global standardization of protein measurands in clin
104. Sardiu, M.E., Gilmore, J.M., Groppe, B.D., Dutta, A.,
ical chemistry croteomics, Clin. Lab. Med., 38, 487-497,
Florens, L., and Washburn, M.P. (2019) Topological scor
doi: 10.1016/j.cll.2018.05.001.
METHODS OF COMPUTATIONAL INTERACTOMICS
FOR INVESTIGATION OF INTERACTIONS OF HUMAN PROTEOFORMS
Review
E. V. Poverennaya*, O. I. Kiseleva, A. S. Ivanov, and E. A. Ponomarenko
Institute of Biomedical Chemistry, 119121 Moscow, Russia; E'mail: k.poverennaya@gmail.com
Received March 25, 2019
Revised September 16, 2019
Accepted October 7, 2019
The human genome contains ca. 20,000 protein coding genes, which could be translated into millions of unique pro
tein species (proteoforms). Proteoforms coded by a single gene often differ in function, which implies differences in
their protein partners. By interacting with each other, proteoforms create a network reflecting dynamics of cellular
processes in an organism. Perturbations of protein-protein interactions cause changes in the network topology, which
often trigger pathological processes. The study of proteoforms is a relatively new research area in proteomics, that is
why there are relatively few experimental studies on interactions of proteoforms. Bioinformatics tools allow facilitat
ing various tasks providing valuable complementary information to experimental data such as, for example, expand
ing the possibilities in investigation of interactions of proteoforms.
Keywords: protein-protein interactions, interactomics, bioinformatics
БИОХИМИЯ том 85 вып. 1 2020