БИОХИМИЯ, 2020, том 85, вып. 4, с. 578 - 586
УДК 578.282;57.012.7; 577.359; 616.921.5
СТРУКТУРНАЯ ОРГАНИЗАЦИЯ И ДИНАМИЧЕСКИЕ
ХАРАКТЕРИСТИКИ УЧАСТКА СВЯЗЫВАНИЯ ИНГИБИТОРОВ
КОНФОРМАЦИОННОЙ ПЕРЕСТРОЙКИ ГЕМАГГЛЮТИНИНА
ВИРУСА ГРИППА H3N2 И H7N9*
© 2020
Д.Д. Подшивалов1**, Е.М. Кирилин1,2, С.И. Коннов1, В.К. Швядас1,2**
1 Московский государственный университет им. М.В. Ломоносова, факультет биоинженерии и биоинформатики,
119991 Москва, Россия; электронная почта: david.podshivalov@belozersky.msu.ru, vytas@belozersky.msu.ru
2 НИИ физико*химической биологии им. А.Н. Белозерского,
Московский государственный университет им. М.В. Ломоносова, 119991 Москва, Россия
Поступила в редакцию 04.02.2020
После доработки 02.03.2020
Принята к публикации 02.03.2020
Для исследования структурной организации и динамических характеристик участка связывания ингибито
ров конформационной перестройки белка разработаны компьютерные модели гемагглютинина вирусов
гриппа H3N2 и H7N9. Использование метадинамики позволило составить карту свободной энергии данных
участков и установить объемы их наиболее энергетически выгодных состояний. При молекулярном моде
лировании взаимодействия умифеновира (арбидола) с гемагглютинином показано, что для связывания ли
ганда необходимо увеличение объема и деформация энергетически наиболее выгодного состояния участка
связывания ингибиторов конформационной перестройки. Идентифицированы аминокислотные остатки,
непосредственно участвующие в связывании ингибитора и определяющие эффективность взаимодействия,
а также динамическое поведение участка связывания ингибиторов конформационной перестройки гемаг
глютинина. Выявленные свойства и особенности структурной организации участка связывания ингибито
ров конформационной перестройки гемагглютинина вируса гриппа необходимо учитывать при поиске но
вых противовирусных препаратов, способных модулировать его функциональные свойства.
КЛЮЧЕВЫЕ СЛОВА: гемагглютинин, вирус гриппа, молекулярная динамика, метадинамика.
DOI: 10.31857/S0320972520040107
Гемагглютинин (НА) - важнейший глико
женный в головной области гемагглютинина,
протеин на поверхности вируса гриппа. Он игра
ориентированной от мембраны вируса в сторону
ет ключевую роль в процессе заражения, так как
окружающей среды, что делает его доступным
распознает рецепторы на поверхности мембра
для иммунной системы. Вакцины против грип
ны клетки хозяина, при связывании претерпе
па часто направлены именно на этот участок [2].
вает существенные структурные изменения и
К сожалению, его расположение приводит к то
обеспечивает слияние вирусной и клеточной
му, что этот участок приобрел крайне высокую
мембран, а также проникновение вирусных ри
вариабельность в результате мутаций [3-6]. В
бонуклеопротеидов в здоровую клетку [1]. По
связи с этим необходим поиск противовирусных
давление процесса заражения на ранних стади
препаратов, направленных на другие, более
ях - важная задача в борьбе с вирусом гриппа,
консервативные участки связывания в структу
поэтому гемагглютинин является привлекатель
ре гемагглютинина. В последнее время особый
ной мишенью для поиска противовирусных
интерес исследователей вызывает участок, рас
препаратов. Первичной мишенью действия те
положенный в высококонсервативной стволо
рапевтических средств является участок связы
вой области [7], вблизи пептида слияния, ответ
вания олигосахаридных рецепторов, располо
ственного за структурную перестройку белка. С
помощью методов рентгеноструктурного анали
Принятые сокращения: HA — гемагглютинин; CV —
за показана способность данного участка связы
коллективная переменная.
вать такие соединения, как третичный бутил
* Первоначально английский вариант рукописи опубли
гидрохинон [7], N циклогексилтаурин [8] и
msu.ru/biokhimiya, в рубрике «Papers in Press», BM20 031,
умифеновир [9]. Подобные соединения, связы
01.04.2020.
ваясь на этом участке в структуре HA, по мне
** Адресат для корреспонденции.
нию авторов, способны индуцировать конфор
578
УЧАСТОК ИНГИБИТОРОВ ПЕРЕСТРОЙКИ ГЕМАГГЛЮТИНИНА
579
мационные изменения прилежащих остатков,
яний участков, а также определения влияния их
образуя сеть взаимодействий, стабилизирующих
динамических свойств на эффективность взаи
всю субъединицу НА и препятствующих струк
модействия с предполагаемыми ингибиторами
турным перестройкам белка. Найденный учас
на примере умифеновира. Полученные знания
ток связывания ингибиторов конформацион
могут быть полезны для понимания механизма
ной перестройки НА может быть ранее неизве
действия известных препаратов или использова
стной мишенью действия противовирусных
ны при поиске новых противовирусных препа
препаратов и альтернативой для поиска новых
ратов, способных модулировать функциональ
противогриппозных средств.
ные свойства гемагглютинина вируса гриппа.
Широко применяемым методом компьютер
ного поиска потенциальных ингибиторов бел
ков является докинг. Для эффективного исполь
МАТЕРИАЛЫ И МЕТОДЫ
зования этого метода необходимо создать биб
лиотеку подходящих низкомолекулярных пре
Молекулярная динамика. При исследовании
тендентов и определиться с областью их связы
участка связывания умифеновира использовали
вания. Однако в случаях, когда область связыва
структуры гемагглютинина H3N2 (PDB ID 5t6n)
ния не является хорошо изученным участком в
и H7N9 (PDB ID 5t6s). Для атомов белка исполь
структуре белка, целесообразно разработать
зовали силовое поле Amber ff14SB [11]. Каждый
адекватную компьютерную модель молекуляр
белок был помещен в ячейку, представленную
ной мишени, причем от качества созданной мо
прямоугольным параллелепипедом с гранями
дели в значительной степени зависит успех пос
100×100×163 Å. Минимальное расстояние между
ледующего поиска новых ингибиторов. Наряду
белком и границей ячейки составляло 12 Å, ог
с организацией статической трехмерной струк
раничение на радиус действия невалентных взаи
туры необходимо также исследовать динамичес
модействий составляло 10 Å. Расчет дальнодей
кие характеристики малоизученного участка.
ствующих электростатических взаимодействий
Как было показано на примере нейраминида
проводили с использованием PME алгоритма
зы - другого белка вируса гриппа, участки свя
для периодических граничных условий; шаг сет
зывания лигандов могут иметь несколько воз
ки составлял 1 Å. В систему добавляли молекулы
можных конформаций, их объем может также
воды типа TIP3P и ионы Na+ Cl- в концентра
зависеть от штамма вируса [10]. Исследование
ции 0,1 М для создания ионной силы раствора и
динамического поведения участка связывания и
нейтрализации заряда. Стартовые модели опти
поиск наиболее энергетически выгодных состо
мизировали в ходе 5000 циклов минимизации
яний являются ключевыми шагами молекуляр
энергии. На подвижность атомов белков накла
ного моделирования, предшествующими ком
дывали ограничения, после чего систему посте
пьютерному поиску новых соединений. Такой
пенно нагревали от 0 до 300 К при постоянном
подход в случае участка связывания ингибито
объеме. На следующем шаге при постоянной
ров конформационной перестройки гемагглю
температуре устанавливали давление в 1 атм.
тинина является тем более актуальным, пос
Итоговый запуск молекулярной динамики НА
кольку этот участок располагается в области,
при постоянном объеме и температуре системы
наиболее подверженной структурным измене
проводили в программе pmemd.cuda програм
ниям при слиянии вируса со здоровыми клетка
много пакета Amber14 [12]. Длительность полу
ми в организме хозяина, и при поиске компле
ченных траекторий составляла 100 нс, из траек
ментарных ингибиторов необходимо учитывать
торий выделяли по 5000 кадров.
эту особенность.
Метадинамика. Для того чтобы охарактеризо
В данной работе для исследования структур
вать энергетическое состояние участков свя
ной организации и функциональных особен
зывания в гемагглютининах H3N2 и H7N9 при
ностей участка связывания ингибиторов кон
изменении их объема применяли метод метади
формационной перестройки белка были созда
намики [13], позволяющий проводить ускорен
ны компьютерные модели гемагглютинина ви
ное накопление статистики редких событий,
русов гриппа H3N2 и H7N9. Методы молекуляр
контролируя при этом различные параметры
ного моделирования были использованы для
системы с использованием т.н. коллективных
изучения динамических характеристик участков
переменных (CV). Ускоренное накопление ста
связывания ингибиторов конформационной пе
тистики (или сэмплирование) происходит за
рестройки в гемагглютининах вирусов гриппа
счет постепенного добавления к общему потен
H3N2 и H7N9, выявления аминокислотных ос
циалу системы дополнительного набора задан
татков, участвующих в этом процессе, и опреде
ных гауссовых потенциалов, зависящего от кол
ления наиболее энергетически выгодных состо
лективных переменных. При этом коллективные
БИОХИМИЯ том 85 вып. 4 2020
580
ПОДШИВАЛОВ и др.
переменные могут быть выбраны в очень широ
пендикулярный плоскости, содержащей пер
ком диапазоне от простых расстояний между
вый, второй и третий атомы, а также вектор r3,
атомами, двугранных или торсионных углов, до
ортогональный векторам r1 и r2. Первый атом и
сложных коллективных переменных, которые
проекции четвертого атома на плоскости, обра
могу являться, например, координационными
зованные парами созданных векторов, опреде
числами между группами атомов. По результа
ляют конечный прямоугольный параллелепи
там такого моделирования можно восстановить
пед, в котором рассчитывается значение какого
поверхность свободной энергии в зависимости
либо параметра, в данном случае количество
от CV, определив, таким образом, энергетичес
молекул воды. В качестве опорных атомов были
кие характеристики исследуемого процесса.
выбраны Сα атомы прилежащих к участку свя
При выборе коллективных переменных ру
зывания аминокислотных остатков Ala304 и
ководствовались тремя общепринятыми прин
Leu316 цепи A, Tpr92 цепи B и Lys310 C; объем
ципами: они должны определять начальное,
прямоугольного параллелепипеда рассчитывали
промежуточное и конечное состояния исследуе
за вычетом объема попадающего в него фраг
мого процесса, являться медленными для дан
мента белка (рис. 1, a). По созданной CV накла
ной системы, их количество должно быть мини
дывали дополнительные гауссовы потенциалы с
мально возможным, так как введение любой до
высотой 4 кДж/моль и шириной 0,1.
полнительной переменной существенно замед
В отличие от HA H3N2 в HA H7N9 различа
ляет расчеты, а также усложняет анализ резуль
ют закрытую и открытую форму исследуемого
татов. Для исследования участка в НА H3N2 в
участка, поэтому для получения более полной
качестве CV был выбран объем участка связыва
картины изменения свободной энергии при мо
ния умифеновира, заданный с помощью функ
делировании динамического поведения участка
ции CAVITY программы для моделирования ме
была введена дополнительная коллективная пе
тадинамики Plumed 2.4 [14]. Данная функция
ременная. Так как открытая и закрытая конфор
позволяет задавать объем, определяемый распо
мации определяются, главным образом, поло
ложением в пространстве четырёх атомов белка.
жением Arg54 цепи B, то в качестве CV была ис
От первого атома ко второму строится вектор r1.
пользована разница в расстояниях d1 и d2, где
Затем из первого атома строится вектор r2, пер
d1 - расстояние между атомом углерода гуани
a
б
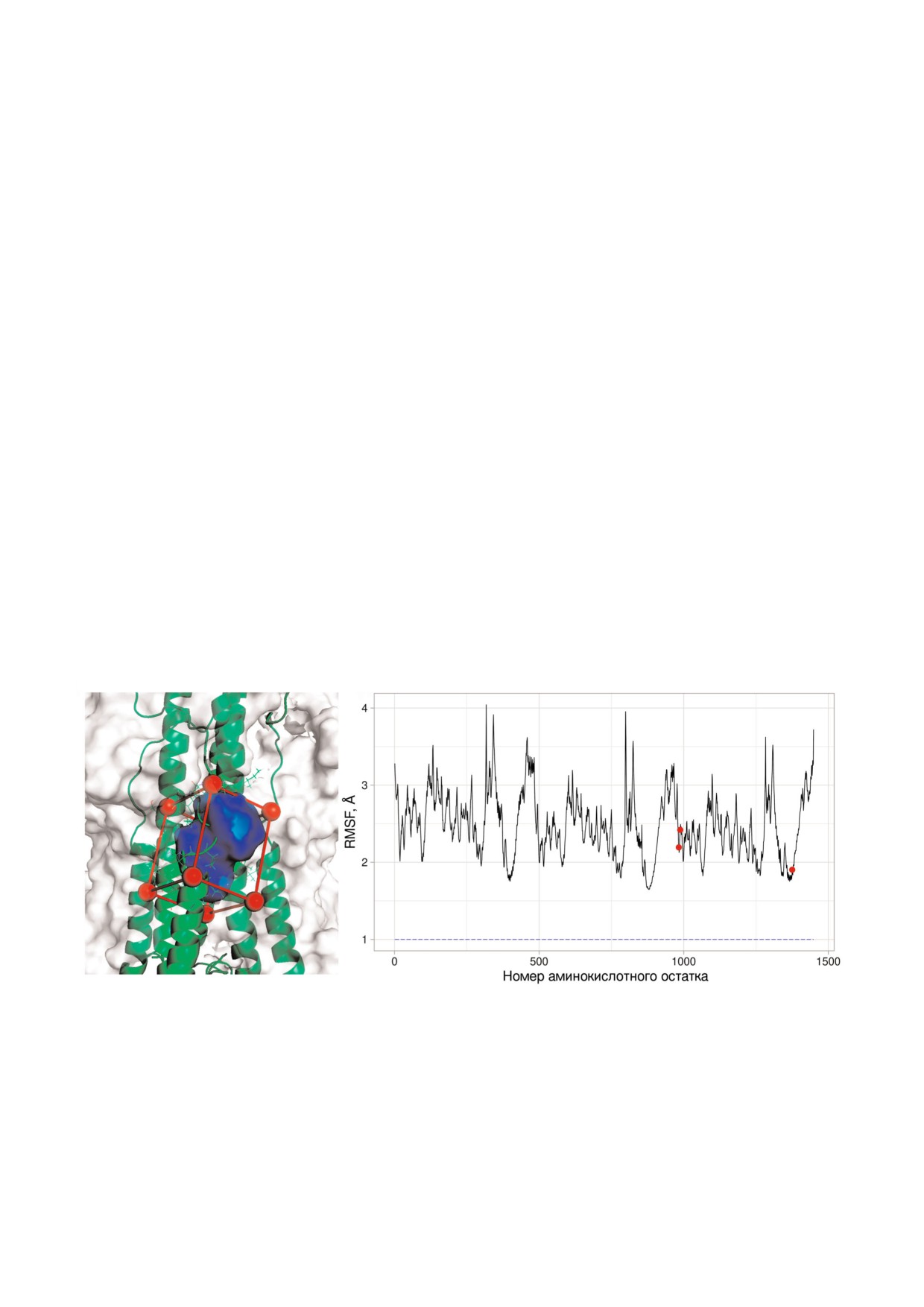
Рис. 1. a - Область связывания ингибиторов конформационной перестройки на поверхности гемагглютинина H3N2,
выбранная для исследования методом метадинамики: красным цветом показана внешняя граница области, зеленым цве
том - элементы вторичной структуры белка, а также ряд аминокислотных остатков, синим выделена область, объем ко
торой меняется и измеряется в ходе эксперимента; б - график RMSF для гемагглютинина штамма H7N9. По форме гра
фика можно различить три субъединицы белка. Красными точками отмечены аминокислотные остатки Glu97 цепи F, а
также Asn27 и Arg32 цепи E, которые были выбраны для создания дополнительной коллективной переменной. Они обла
дают низким значением RMSF и расположены вблизи исследуемой области. Синей пунктирной линией показано значе
ние RMSF (~0, 96 Å) для центра масс тяжелых атомов этих остатков.
journal/biokhsm/
БИОХИМИЯ том 85 вып. 4 2020
УЧАСТОК ИНГИБИТОРОВ ПЕРЕСТРОЙКИ ГЕМАГГЛЮТИНИНА
581
диновой группы Arg54 и центром масс трех ста
в различных программах [19]. Такой подход
бильных остатков Glu97 цепи F, а также Asn27 и
представляется перспективным при моделиро
Arg32 цепи E (рис. 1, б), а d2 - расстояние меж
вании конформационных состояний, а также
ду атомом углерода гуанидиновой группы того
энергетических характеристик участка с ис
же аргинина и атомом углерода карбоксильной
пользованием дополнительных внешних воз
группы остатка Glu103 цепи D. Такой выбор CV
действий, например, в метадинамике [13], так
обусловлен необходимостью измерять энергию
как распределение сил происходит равномерно
смещения остатка Arg54, не вводя при этом до
на все молекулы воды в исследуемом объеме, а
полнительных искусственных ограничений на
не на отдельные атомы белка, что может приво
подвижность остальных остатков. Высота доба
дить к нарушению его структуры и сэмплирова
вочных гауссовых потенциалов составляла
нию высокоэнергетических незначимых кон
4 кДж/моль, ширина равнялась 0,1 и 0,2 для
формаций. Количество вмещаемых молекул во
объема участка и расстояния соответственно.
ды в качестве коллективной переменной было
Докинг. Кадры молекулярной динамики НА
выбрано для изучения участка связывания ин
H3N2 были выровнены на стартовую структуру,
гибиторов структурной перестройки в гемаг
а затем экспортированы в отдельные pdb фай
глютининах H3N2 и H7N9. Отличительной осо
лы. С помощью PMV [15, 16] были подготовле
бенностью участка структурной перестройки
ны pdbqt файлы НА и одного из ингибиторов
HA H7N9 по сравнению с HA H3N2 является
структурной перестройки - умифеновира. Для
показанное методами рентгеноструктурного
полученных структур был проведен докинг с по
анализа наличие открытой и закрытой форм ис
мощью SMINA, использующей для докинга
следуемого участка, определяемое конформаци
программу Vina [17]. SMINA запускали с пара
ей аминокислотного остатка Arg54 цепи B [9].
метром exhaustiveness, равным 20, а центром об
Поэтому исследование структурной организа
ласти для докинга была выбрана точка внутри
ции и динамического поведения участка в HA
исследуемого участка связывания лиганда.
H7N9 при помощи метадинамики проводили с
Дальнейший анализ проводили для 10 результа
использованием дополнительной коллективной
тов докинга, характеризуемых наименьшей ве
переменной, характеризующей положение
личиной оценочной функции. Для этих струк
Arg54 цепи B.
тур, а также для структур, полученных методом
Исследование динамических свойств участка
рентгеноструктурного анализа, была составлена
связывания ингибиторов структурной перестройки
таблица попарных RMSD. Итоговые данные
HA H3N2. Поверхность свободной энергии
были кластеризованы методом DBSCAN [18].
участка связывания ингибиторов конформаци
Значение minPts было выбрано равным 4, значе
онной перестройки НА H3N2 была определена
ние ε увеличивали до тех пор, пока кристалло
при интегрировании всех добавленных к системе
графическая конформация умифеновира не
в ходе метадинамики гауссовых потенциалов от
оказывалась в одном из формирующихся клас
носительно выбранной коллективной перемен
теров из множества выбросов. Затем проводили
ной количества молекул воды (рис. 2). Видно,
оценку значения silhouette для ε, начиная с най
что на графике присутствует один энергетичес
денного и заканчивая таким ε, при котором sil*
кий минимум участка связывания, который со
houette был максимальным. Визуализацию полу
ответствует объему, вмещающему от 23 до 34 мо
ченных кластеров методом главных компонент
лекул воды, при этом для выхода за пределы это
проводили в Rstudio с помощью функции fviz
го интервала необходимы большие (> 4 ккал/м)
cluster. Такую же процедуру применяли и для
затраты энергии. Важно понять, какие амино
траекторий метадинамики.
кислотные остатки формируют участок, опреде
ляют его динамическое поведение и специфи
ческие взаимодействия с ингибиторами.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для получения этой информации из полной
траектории динамики были выделены кадры, в
В водном растворе структура белка подвер
которых объем исследуемого участка соответ
жена естественным флуктуациям, что относится
ствовал наиболее выгодным состояниям. С по
и к объему участка ингибиторов конформаци
мощью функционала программы VMD [20] на
онной перестройки HA из за движения образую
основе выделенных кадров была построена ус
щих его остатков. Мы предлагаем определить
редненная структура гемагглютинина H3N2, со
объем участка связывания как количество вме
ответствующая наиболее энергетически выгод
щаемых молекул воды, что позволяет его оха
ному состоянию участка связывания, и по всей
рактеризовать, не прибегая при этом к сложным
траектории динамики для аминокислотных ос
алгоритмическим процедурам, как это делается
татков в радиусе 8 Å от центра участка были рас
БИОХИМИЯ том 85 вып. 4 2020
582
ПОДШИВАЛОВ и др.
считаны отклонения от усредненной структуры.
Карта зависимости значений RMSD выбранных
остатков от объема участка представлена на
рис. 3.
Значение RMSD остатков цепи A Phe294,
цепи B Lys51, Glu57, Asn95, Ala96, Leu99,
Glu103, цепи C Lys27, Thr28, Ile29, Asp32, Val309,
Lys310, Gln311, Leu314 и цепи D Ile89, Asp90,
Leu91, Trp92, Ser93, Tyr94, Asn95, Ala96, Glu97,
Leu98 оставалось малым при оптимальном объ
еме участка связывания (т.е. в наиболее энерге
тически выгодном состоянии участка положе
ние таких остатков стабильно) и сильно возрас
тало за его пределами. Это говорит о том, что
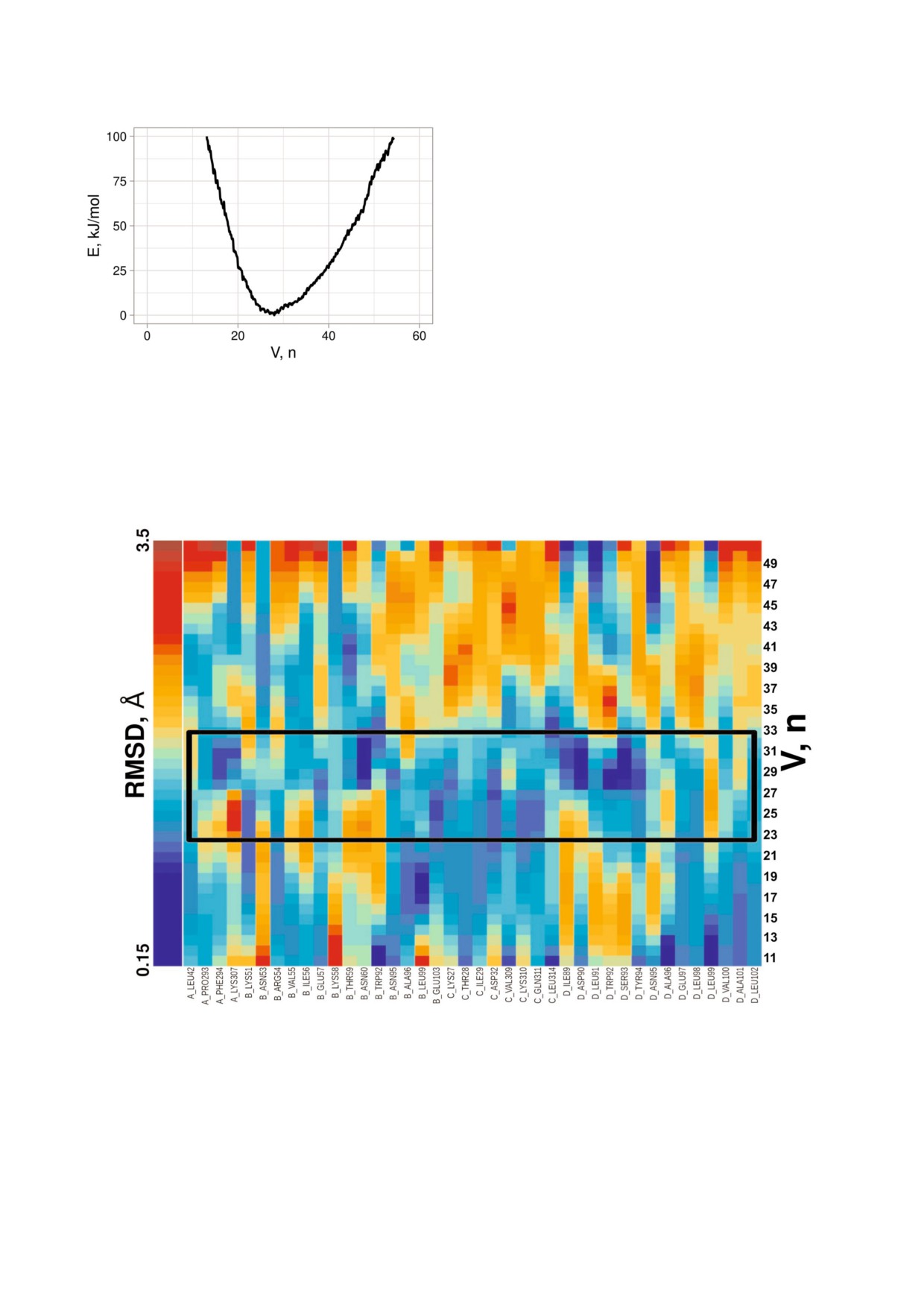
Рис. 2. Поверхность свободной энергии участка связыва
количество молекул воды в участке связывания
ния ингибиторов конформационной перестройки гемаг
глютинина H3N2 от объема (объем участка связывания
зависит именно от этих остатков. Для остальных
указан в количестве вмещаемых молекул воды), опреде
остатков этот параметр не имеет ярко выражен
ленная при моделировании с использованием метадина
ных минимумов в зависимости от объема, сле
мики.
довательно, они не влияют на количество моле
С цветным вариантом рисунка можно ознакомиться в
кул воды в участке. RMSD остатков цепи B
journal/biokhsm/
Arg54, Asn60, Glu103 и остатка цепи C Lys310 ос
Рис. 3. Карта зависимости RMSD аминокислотных остатков, формирующих участок связывания в гемагглютинине
H3N2, от его объема. Черной рамкой выделена область наиболее энергетически выгодных состояний участка. Синий цвет
ячейки соответствует самому низкому значению, красный - самому высокому значению RMSD аминокислотного остат
ка при данном состоянии участка. График позволяет определить остатки, оказывающие наиболее сильное влияние на
объем участка связывания - их RMSD остается низким для наиболее энергетически выгодных состояний участка (внут
ри черной рамки) и растет при дестабилизации участка (увеличении или уменьшении объема за пределы оптимального),
тогда как RMSD остальных аминокислотных остатков изменяется вне зависимости от объема участка.
journal/biokhsm/
БИОХИМИЯ том 85 вып. 4 2020
УЧАСТОК ИНГИБИТОРОВ ПЕРЕСТРОЙКИ ГЕМАГГЛЮТИНИНА
583
a
б
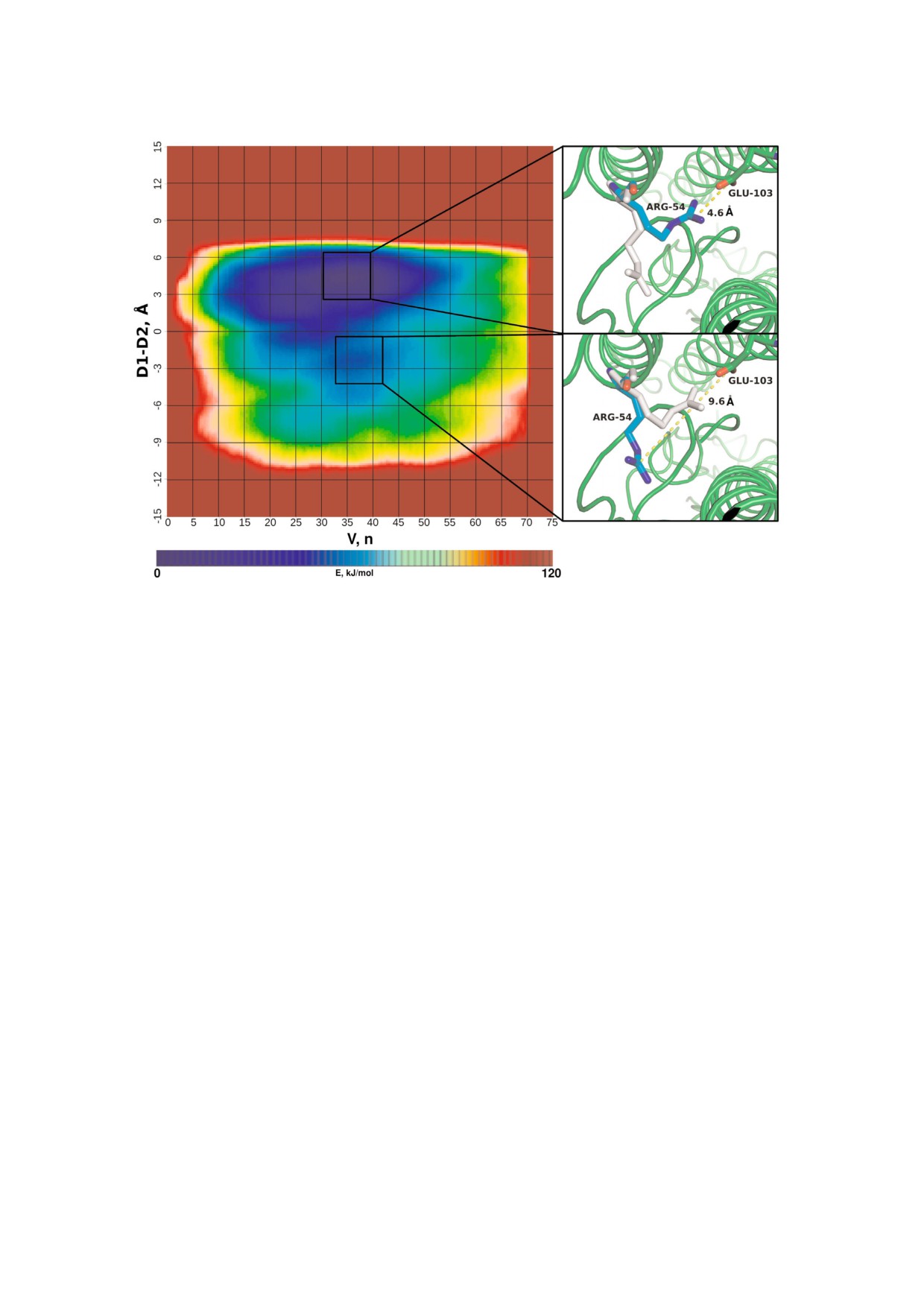
Рис. 4. Динамические характеристики участка связывания ингибиторов конформационной перестройки НА H7N9 при
различных энергетических состояниях. а - Поверхность свободной энергии (значения представлены цветом по указан
ной шкале) при изменении объема участка, представленного как количество вмещаемых молекул воды. По вертикальной
оси отложена разница в расстояниях d1 и d2, где d1 — расстояние между атомом углерода гуанидиновой группы Arg54 и
центром масс трех стабильных остатков Glu97 цепи F и Asn27, Arg32 цепи E (не представлены на рисунке), а d2 — рассто
яние между атомом углерода гуанидиновой группы того же аргинина и атомом углерода карбоксильной группы остатка
Glu103 цепи D (см. «Материалы и методы»); б - положение ключевого аминокислотного остатка Arg54 при закрытом кон
формационном состоянии участка (верхняя картинка) и изменение положения при переходе из закрытого состояния в
открытое (нижняя картинка), зеленым цветом показаны элементы вторичной структуры HA, желтым штрихом - рассто
яние между атомами углерода гуанидиновой и карбоксильной групп Arg54 и Glu103.
journal/biokhsm/
таются наиболее стабильными в своих положе
коллективных переменных: 1) количества моле
ниях при флуктуациях объема участка и пред
кул воды в участке структурной перестройки и
ставляют собой особый интерес в качестве точек
2) положения остатка Arg54. Анализ поверхнос
взаимодействия с потенциальными ингибито
ти позволяет определить различие между откры
рами. Отдельного внимания заслуживают три
той и закрытой формами структуры участка свя
остатка цепи D Ile89, Leu91 и Asn95: значение их
зывания ингибиторов конформационной пере
RMSD быстро растет с увеличением объема, а
стройки НА H7N9 по энергии и значению объе
затем быстро падает. Это свидетельствует о
ма: оптимальному состоянию участка соответ
сложной зависимости объема участка связыва
ствует его закрытая конформация, которую
ния от конформационной подвижности и ре
можно характеризовать объемом 30-39 вмещае
лаксационных перестроек этих остатков.
мых молекул воды, тогда как энергетически ме
Исследование динамических свойств участка
нее выгодная открытая конформация выше по
связывания ингибиторов структурной перестройки
энергии на 8 ккал/м и характеризуется увеличе
HA H7N9. Поверхность свободной энергии
нием объема участка связывания до 43 молекул
участка связывания ингибиторов конформаци
воды, а также изменением положения остатка
онной перестройки НА H7N9 (рис. 4, а) была
цепи B Arg54 (рис. 4, б).
определена путем интегрирования всех добав
Особенности связывания HA H3N2 и H7N9 с
ленных к системе в ходе метадинамики гауссо
умифеновиром. Широкий набор конформаций
вых потенциалов относительно двух выбранных
белков, получаемый при использовании мета
БИОХИМИЯ том 85 вып. 4 2020
584
ПОДШИВАЛОВ и др.
a
б
Рис. 5. Изучение расположения молекулы умифеновира на участке связывания ингибиторов конформационной пере
стройки гемагглютинина H3N2. а - Кластеризация результатов докинга в структуры HA траектории классической моле
кулярной динамики: обособленнного кластера, соответствующего кристаллографической структуре не образуется; б -
кластеризация результатов докинга в структуры HA с расширенным состоянием участка связывания из метадинамики:
красной границей показан кластер, соответствующий положению умифеновира в кристаллографической структуре. Цвет
соответствует различным кластерам, Dim1 и Dim2 формируют пространство двух первых главных компонент, вычислен
ных для пространства конформаций и расположений умифеновира в результатах докинга.
journal/biokhsm/
Рис. 6. Положение молекулы умифеновира в участке связывания ингибиторов структурной перестройки в HA H3N2.
Оранжевыми толстыми линиями показана молекула умифеновира. Толстыми линиями показаны остатки, значения
RMSD которых существенно увеличиваются с увеличением объема. Зеленым представлены остатки, отвечающие за не
посредственное связывание умифеновира [9].
journal/biokhsm/
БИОХИМИЯ том 85 вып. 4 2020
УЧАСТОК ИНГИБИТОРОВ ПЕРЕСТРОЙКИ ГЕМАГГЛЮТИНИНА
585
динамики, создает основу для изучения связы
случае для связывания умифеновира необходим
вания ингибиторов. При оценке взаимодей
переход в менее энергетически выгодное состо
ствий в рентгенографических структурах бе
яние, связанный с перемещением остатка Arg54
лок-ингибиторных комплексов, как правило,
и сопутствующим небольшим изменением объ
не учитывается энергетическое состояние само
ема участка.
го участка. Характерным примером могут быть
Использование методов молекулярного мо
кристаллографические структуры комплексов
делирования позволило охарактеризовать наи
гемагглютининов с умифеновиром. Докинг
более энергетически выгодные состояния участ
умифеновира был проведен с использованием
ков связывания ингибиторов конформацион
наиболее энергетически выгодных состояний
ной перестройки гемагглютининов двух штам
участка связывания отобранных из траекторий
мов вируса гриппа H3N2 и H7N9 и установить,
классической молекулярной динамики и мета
что в обоих случаях для взаимодействия с уми
динамики HA H3N2. При кластеризации ре
феновиром необходима существенная перест
зультатов докинга в выбранный ансамбль кон
ройка участка связывания по сравнению со
формационных состояний HA (рис. 5, a) не уда
структурой свободного белка. Выявлены амино
лось обнаружить обособленного кластера со
кислотные остатки, формирующие участок свя
связанным ингибитором, положение которого
зывания, а также непосредственно участвующие
соответствует кристаллографической структуре
в связывании лиганда и определяющие эффек
комплекса [9]. Образование комплекса было об
тивность этих взаимодействий. В обоих случаях
наружено только при кластеризации результа
перестройка участка связывания белка требует
тов докинга в структуры HA с расширенным
затрат энергии, но протекает по разным меха
состоянием участка связывания: обособленный
низмам: для HA H3N2 необходимо увеличение
кластер (значение ε = 2,6), соответствующий ре
объема участка связывания за счет смещения це
зультатам рентгеноструктурного анализа, пред
лого ряда аминокислотных остатков, тогда как
ставлен на рис. 5, б. Это также подтверждается
для HA H7N9 достаточен переход в энергетичес
тем фактом, что все аминокислотные остатки, с
ки менее выгодную открытую форму за счет пе
которыми умифеновир взаимодействует напря
ремещения остатка Arg54 цепи B. Полученные
мую в структуре комплекса - Phe294, Lys307 це
знания могут быть полезны как для понимания
пи A и Leu98 цепи D - главным образом вовле
механизма действия известных препаратов, так
чены в дестабилизацию наиболее выгодного
и при поиске новых лидерных соединений, нап
энергетического состояния участка и увеличе
равленных на модуляцию функциональных
ния его объема (см. рис. 3).
свойств гемагглютинина вирусов гриппа.
В HA H7N9 функционирование участка свя
зывания ингибиторов структурной перестройки
отличается от HA H3N2. В этом случае участок
Финансирование. Исследование выполнено
существует в двух конформационных состояни
при финансовой поддержке РФФИ (проект 18
ях - открытом и закрытом. Докинг показал, что
315 00390).
связывание умифеновира имеет место лишь с
Благодарности. Исследование выполнено с
открытым состоянием, при этом, в отличие от
использованием оборудования Центра коллек
HA H3N2, нет необходимости в значительном
тивного пользования сверхвысокопроизводи
увеличении объема участка. Положение инги
тельными вычислительными ресурсами МГУ
битора соответствует положению умифеновира
им. М.В. Ломоносова [21].
в кристаллографической структуре. Особен
Конфликт интересов. Авторы заявляют об от
ность взаимодействия в случае HA H7N9 заклю
сутствии конфликта интересов.
чается в том, что открытое состояние участка не
Соблюдение этических норм. Настоящая
является наиболее энергетически выгодным и
статья не содержит исследований с участием
на 8 ккал/моль выше по сравнению с закрытым
людей или использованием животных в качест
состоянием (рис. 6). Таким образом, и в этом
ве объектов.
СПИСОК ЛИТЕРАТУРЫ
1.
Skehel, J. J., and Wiley, D. C. (2000) Receptor binding and
3.
Hensley, S. E., Das, S. R., Bailey, A. L., Schmidt, L. M.,
membrane fusion in virus entry: the influenza hemagglu
Hickman, H. D., Jayaraman, A., Viswanathan, K.,
tinin, Ann. Rev. Biochem., 69, 531 569, doi: 10.1146/
Raman, R., Sasisekharan, R., Bennink, J. R., and
annurev.biochem.69.1.531.
Yewdell, J. W. (2009) Hemagglutinin receptor binding avid
2.
Hensley, S. E. (2014) Challenges of selecting seasonal
ity drives influenza A virus antigenic drift, Science, 326,
influenza vaccine strains for humans with diverse pre
734 736, doi: 10.1126/science.1178258.
exposure histories, Curr. Opin. Virology,
8,
8589,
4.
Heider, A., Mochalova, L., Harder, T., Tuzikov, A., Bovin, N.,
doi: 10.1016/j.coviro.2014.07.007.
Wolff, T., Matrosovich, M., and Schweiger, B. (2015)
9 БИОХИМИЯ том 85 вып. 4 2020
586
ПОДШИВАЛОВ и др.
Alterations in hemagglutinin receptor binding specificity
ing the accuracy of protein side chain and backbone para
Accompany the emergence of highly pathogenic avian
meters from ff99SB, J. Chem. Theory Comput., 11, 3696
influenza viruses, J. Virology, 89, 5395 5405, doi: 10.1128/
3713, doi: 10.1021/acs.jctc.5b00255.
JVI.03304 14.
12. Case, D. A., Babin, V., Berryman, J., Betz, R. M., Cai, Q.,
5.
Tharakaraman, K., Raman, R., Viswanathan, K.,
Cerutti, D. S., Cheatham III, T. E., Darden, T. A., et al.
Stebbins, N. W., Jayaraman, A., Krishnan, A.,
(2014) Amber 14, University of California, San Francisco.
Sasisekharan, V., and Sasisekharan, R. (2013) Structural
13. Barducci, A., Bonomi, M., and Parrinello, M. (2011)
determinants for naturally evolving H5N1 hemagglutinin
Metadynamics, WIREs Comput. Mol. Sci., 1, 826 843,
to switch its receptor specificity, Cell, 153, 1475 1485,
doi: 10.1002/wcms.31.
doi: 10.1016/j.cell.2013.05.035.
14. Tribello, G. A., Bonomi, M., Branduardi, D., Camilloni,
6.
Львов Д. К., Богданова В. С., Кириллов И. М.,
C., and Bussi, G. (2014) PLUMED 2: new feathers for an
Щелканов М. Ю., Бурцева Е. И., Бовин Н. В., Федя
old bird, Com. Phys. Commun., 185, 604 613, doi: 10.1016/
кина И. Т., Прилипов А.Г., Альховский С. В., Самох
j.cpc.2013.09.018.
валов Е. И., Прошина Е. С., Кириллова Е. С., Сыро
15. Sanner, M. (1999) Python: a programming language for
ешкин А. В. (2019) Эволюция пандемического вируса
software integration and development, J. Mol. Graph.
гриппа A(H1N1)pdm09 в 2009 2016 гг.: динамика ре
Model, 17, 57 61.
цепторной специфичности первой субъединицы ге
16. Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F.,
магглютинина (НA1), Вопросы вирусологии, 64, 63 72,
Belew, R. K., Goodsell, D. S. and Olson, A. J. (2009)
doi: 10.18821/0507 4088 2019 64 2 63 72.
AutoDock4 and autoDocktools4: automated docking with
7.
Russell, R. J., Kerry, P. S., Stevens, D. J., Steinhauer, D. A.,
selective receptor flexiblity. J. Comput. Chem., 16, 2785
Martin, S. R., Gamblin, S. J., and Skehel, J. J. (2008)
2791, doi: 10.1002/jcc.21256.
Structure of influenza hemagglutinin in complex with an
17. Trott, O., and Olson, A. J. (2010) AutoDock Vina: improv
inhibitor of membrane fusion, Proc. Natl. Acad. Sci. USA,
ing the speed and accuracy of docking with a new scoring
105, 17736 17741, doi: 10.1073/pnas.0807142105.
function, efficient optimization, and multithreading, J.
8.
Kadam, R. U., and Wilson, I. A. (2018) A small molecule frag
Comput. Chem., 31, 455 461, doi: 10.1002/jcc.21334.
ment that emulates binding of receptor and broadly neutraliz
18. Ester, M., Kriegel, H. P., Sander, J., and Xu, X. (1996) in
ing antibodies to influenza A hemagglutinin, Proc. Natl. Acad.
Proc. Sec. Intern. Conf. Knowl. Discov. Data Min. AAAI
Sci. USA, 115, 4240 4245, doi: 10.1073/pnas.1801999115.
Press, Portland, Oregon, pp. 226 231.
9.
Kadam, R. U., and Wilson, I. A. (2017) Structural basis of
19. Durrant, J. D., Votapka, L., Sørensen, J., and Amaro, R. E.
influenza virus fusion inhibition by the antiviral drug
(2014) POVME 2.0: an enhanced tool for determining
Arbidol, Proc. Natl. Acad. Sci. USA, 114, 206214,
pocket shape and volume characteristics, J. Chem. Theory
doi: 10.1073/pnas.1617020114.
Comput., 10, 5047 5056, doi: 10.1021/ct500381c.
10.
Han, N., Mu, Y., Miao, H., Yang, Y., Wu, Q., Li, J., Ding, J.,
20. Humphrey, W., Dalke, A., and Schulten, K. (1996) VMD:
Xu, B., and Huang, Z. (2016) The 340 cavity in neu
visual molecular dynamics, J. Mol. Graph. Model., 14, 33
raminidase provides new opportunities for influenza drug
38, doi: 10.1016/0263 7855(96)00018 5.
development: A molecular dynamics simulation study,
21. Sadovnichy, V., Tikhonravov, A., Voevodin, V., and
Biochem. Biophys. Res. Commun.,
470,
130136,
Opanasenko, V. (2013) “Lomonosov”: supercomputing at
doi: 10.1016/j.bbrc.2016.01.007.
Moscow State University, in Contem. High Perform. Comp.,
11.
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L.,
Boca Raton, USA, pp.
283307, doi:
10.1201/
Hauser, K. E., and Simmerling, C. (2015) ff14SB: improv
9781351104005 11.
STRUCTURAL ORGANIZATION AND DYNAMIC CHARACTERISTICS
OF THE BINDING SITE OF CONFORMATIONAL REARRANGEMENT
INHIBITORS OF HEMAGGLUTININ FROM H3N2
AND H7N9 INFLUENZA VIRUS*
D. D. Podshivalov1**, E. M. Kirilin1,2, S. I. Konnov1, and V. K. Švedas1,2**
1 Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics,
119991 Moscow, Russia; E*mail: david.podshivalov@belozersky.msu.ru, vytas@belozersky.msu.ru
2 Belozersky Institute of Physicochemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
Received February 4, 2020
Revised March 2, 2020
Accepted March 2, 2020
Computer models of the H3N2 and H7N9 influenza virus hemagglutinin have been developed to study structural
organization and dynamic characteristics of the binding site of conformational rearrangement inhibitors. The use of
metadynamics made it possible to map the free energy of these binding sites and define the volume of their most ener
getically favorable states. At simulation of the interaction of umifenovir with hemagglutinin it was shown that ligand
binding requires an increase of the volume and deformation of the energetically most favorable state of the confor
mational rearrangement inhibitors binding site. Amino acid residues that are directly involved in ligand binding and
determine the effectiveness of the interaction, as well as the dynamic behavior of the binding site of hemagglutinin
inhibitors were identified. The revealed properties and peculiarities of the structural organization of the binding site
of conformational rearrangement inhibitors of influenza virus hemagglutinin should be taken into account when
searching for new antiviral drugs capable to modulate its functional properties.
Keywords: hemagglutinin, influenza virus, molecular dynamics, metadynamics
БИОХИМИЯ том 85 вып. 4 2020