БИОХИМИЯ, 2021, том 86, вып. 12, с. 1808 - 1817
УДК 57.05;57.07
ДОСТИЖЕНИЯ И ПЕРСПЕКТИВЫ ЭПИГЕНЕТИЧЕСКИХ
ИССЛЕДОВАНИЙ ДРЕВНЕЙ ДНК
Обзор
© 2021
К.В. Жур1, В.А. Трифонов2, Е.Б. Прохорчук1*
1 ФИЦ «Фундаментальные основы биотехнологии» РАН, 119071 Москва, Россия;
электронная почта: prokhortchouk@gmail.com
2 Институт истории материальной культуры РАН, 191186 Санкт*Петербург, Россия
Поступила в редакцию 29.10.2021
После доработки 17.11.2021
Принята к публикации 17.11.2021
Развитие технологий высокопроизводительного полногеномного секвенирования и совершенствование
методик пробоподготовки сделало возможным исследование ДНК из археологических образцов возрастом
более миллиона лет. В процессе изучения древней ДНК (дДНК) учёными было сделано множество крупных
открытий, касающихся миграций людей, замещений одних популяций другими, межвидовых скрещиваний
кроманьонцев с неандертальцами и денисовцами, эволюции патогенов человека и др. Не менее революци*
онным стало заявление о возможности исследовать эпигенетические модификации геномов древних лю*
дей, что позволило извлечь ранее недоступную информацию, в том числе об активности генов, позициони*
ровании нуклеосом, метилировании ДНК. Анализируя статус метилирования определенных локусов в гено*
ме, можно установить хронологический возраст человека на момент его смерти, реконструировать некото*
рые особенности фенотипа, как это было сделано для денисовца, и даже определить неблагоприятные фак*
торы внешней среды, воздействию которых подвергался древний человек. Представленный обзор посвящён
современным достижениям и перспективным направлениям изучения эпигенетических модификаций
дДНК с анализом методических подходов, применяемых в данной области научных исследований.
КЛЮЧЕВЫЕ СЛОВА: древняя ДНК, палеоэпигенетика, эпигенетические часы, редактирование эпигено*
ма, ожирение.
DOI: 10.31857/S0320972521120058
ВВЕДЕНИЕ
генетический материал не сохраняется. Пере*
смотрели теоретический предел сохранности
Стремительное развитие высокопроизводи*
ДНК после выхода статьи в журнале Nature, в
тельных методов полногеномного секвенирова*
которой были проанализированы полногеном*
ния способствовало заметному увеличению ко*
ные данные, полученные из останков мамонтов
личества и качества информации, получаемой
раннего и среднего плейстоцена [2]. В соответ*
из образцов древней ДНК (дДНК). Совершен*
ствии с данным молекулярного датирования
ствование методов пробоподготовки позволило
(байесовские молекулярные часы) [3], возраст
преодолеть множество сложностей, характер*
животного, найденного на реке Крестовка, сос*
ных для работы с археологическими образцами,
тавил 1,65 миллиона лет [2].
связанных с низким качеством и количеством
Сохранность генетического материала свы*
ДНК, а также с контаминацией генетическим
ше миллиона лет открывает огромные перспек*
материалом других организмов. Долгое время
тивы для более детального изучения эволюции
самым древним образцом, из которого были по*
человека. Анализируя последовательности ДНК,
лучены полногеномные данные, считали остан*
возможно отслеживать миграции людей, выяв*
ки лошади из среднего плейстоцена возрастом
лять замещение одних популяций другими, ус*
560-780 тысяч лет [1], в связи с чем предполага*
танавливать факт межвидового скрещивания,
ли, что в образцах старше одного миллиона лет
исследовать предрасположенность к различным
заболеваниям, предсказывать физические и не*
которые психофизиологические особенности
Принятые сокращения: дДНК - древняя ДНК;
CpG*локус - сайт, где цитозин (C) находится рядом с гуа*
древнего человека [4]. Исследуя генетический
нином (G) в последовательности ДНК.
материал из зубного налёта, можно изучать дие*
* Адресат для корреспонденции.
ту архаичных людей и их микробиом [5, 6], в
1808
ЭПИГЕНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ ДРЕВНЕЙ ДНК
1809
свою очередь, анализ ДНК древних патогенов
стрировали, что ДНК неандертальца возрастом
даёт возможность проследить происхождение и
более 38 000 лет сохраняет картину метилирова*
эволюцию возбудителей различных заболева*
ния [13]. Несколько позже, в 2014 г., была полу*
ний, например, как это было показано для бак*
чена первая карта позиционирования нуклео*
терий Yersinia pestis, возбудителя чумы [7].
сом для генома древнего человека [14]. Возмож*
За последнее десятилетие учёные значитель*
ность установить, какие участки ДНК входят в
но продвинулись в изучении дДНК, в том числе
состав нуклеосомного комплекса, а какие явля*
вымерших видов людей. Многих интересовал
ются межнуклеосомными, т.е. открытыми, поз*
вопрос: как сильно отличался архаичный чело*
воляет определить доступность генетического
век от современного, и что позволило последне*
материала для взаимодействия с регуляторными
му выжить и успешно размножиться. Первый
белками, в том числе факторами, регулирующи*
черновой вариант генома неандертальца был
ми транскрипцию, и, соответственно, предска*
опубликован в 2010 г. [8], а уже в 2012-2013 гг.
зать экспрессию тех или иных генов у древних
были прочитаны полные геномы денисовского
людей. В свою очередь, метилирование ДНК
человека [9] и неандертальца [10] с качеством,
участвует в регуляции структуры хроматина,
не уступающим образцам современной ДНК
клеточной дифференцировки и многих других
(с 30* и 52*кратным покрытием соответствен*
процессов, в том числе контролирует экспрес*
но). Сравнение последовательностей геномов
сию генов на уровне транскрипции. Несмотря
ныне живущих людей с геномами денисовского
на то что метилирование генома и составление
человека и неандертальца выявило более 30 000
карты позиционирования нуклеосом не являют*
однонуклеотидных позиций, отделяющих се*
ся единственными механизмами эпигенетичес*
годняшних людей от архаичных [10]. Тем не ме*
кой регуляции, тем не менее изучение этих про*
нее большинство из этих замен были нейтраль*
цессов даёт возможность извлечь из образцов
ными, 3117 приходились на регуляторные эле*
дДНК ранее недоступную информацию об ак*
менты, 32 предположительно влияли на сайты
тивности генов.
сплайсинга и только 96 замен приводили к ами*
Основная сложность, возникающая при ра*
нокислотным заменам. Учёные пришли к выво*
боте с дДНК - это существенная посмертная
ду, что выявленными отличиями в последова*
деградация генетического материала до фраг*
тельностях геномов современных людей и арха*
ментов длиной в
25
пар нуклеотидов под
ичных нельзя объяснить весь спектр их феноти*
действием эндогенных нуклеаз, а также в ре*
пических и адаптационных различий, а полу*
зультате случайного гидролиза и окисления [16].
ченные результаты свидетельствуют в пользу
Наиболее частым гидролитическим поврежде*
значительного влияния эпигенетических моди*
нием дДНК является дезаминирование цитози*
фикаций на эволюцию человека [11, 12].
на (C), т.е. отщепление аминогруппы от азотис*
того основания с образованием урацила (U). Де*
заминированные цитозины в основном распо*
ПОДХОДЫ К ИЗУЧЕНИЮ
лагаются на концах фрагментов дДНК, что сви*
ЭПИГЕНЕТИЧЕСКИХ МОДИФИКАЦИЙ
детельствует о наличии выступающих одноце*
ДРЕВНЕЙ ДНК
почечных концов, так как скорость дезамини*
рования цитозина в десятки раз выше в одноце*
Существуют разнообразные механизмы эпи*
почечной, чем в двухцепочечной ДНК [17-19].
генетической регуляции активности генов,
Особенность определения последовательности
включая метилирование ДНК, модификации
дДНК заключается в том, что при секвенирова*
гистонов и негистоновых белков хроматина,
нии методом синтеза на месте дезаминирован*
воздействие на экспрессию генов посредством
ных цитозинов исследователь наблюдает ти*
малых некодирующих молекул РНК и другие за*
мин (T). Это связано с тем, что в процессе при*
частую взаимосвязанные процессы. В случае с
готовления библиотек проводится обогащение с
дДНК исследователи в основном фокусируются
помощью полимеразной цепной реакции
на изучении метилирования ДНК и составле*
(ПЦР) с применением полимеразы, которая
нии карт позиционирования нуклео*
способна продолжать синтез комплементарной
сом [13-15], что в первую очередь связано с тех*
цепи в присутствие урацила. Во время синтеза
нической возможностью реконструкции данно*
комплементарной цепи урацил распознаётся
го вида эпигенетических модификаций в архео*
полимеразой как тимин, в результате чего до*
логических образцах.
страивается комплементарное тимину основа*
Первые попытки изучения метилирования
ние - аденин (A). В итоге исходная пара основа*
генома древних людей были предприняты ещё в
ний C/G (где G - гуанин) в результате дезами*
2010 г. группой С. Паабо; тогда учёные продемон*
нирования цитозина превращается в пару U/G,
БИОХИМИЯ том 86 вып. 12 2021
1810
ЖУР и др.
которая в процессе ПЦР преобразуется в T/A в
товления библиотек дДНК для секвенирования
одной из дочерних молекул. Соответственно,
обрабатывала дДНК смесью ферментов урацил*
при секвенировании библиотек фрагментов
ДНК*гликозилазы и эндонуклеазы VIII, что
дДНК исследователь наблюдает замены C → T
позволяет вырезать урацил с образованием од*
на 5′*конце молекулы ДНК или G → A - на
нонуклеотидного разрыва, оставляя неповреж*
3′*конце в зависимости от особенностей вы*
дённые части нетронутыми [13]. Такой способ
бранного протокола пробоподготовки. Важно
значительно повышает точность секвенирова*
отметить, что частота таких замен коррелирует с
ния, а также даёт возможность определить был
возрастом образца [20] и используется в качестве
ли цитозин метилирован, так как в результате
критерия, подтверждающего, что ДНК принад*
дезаминирования метилированного цитозина
лежит именно древнему организму (эндогенная
последний превращается в остаток тимина и,
ДНК), а не является результатом контаминации
соответственно, не распознаётся смесью фер*
современным генетическим материалом. В це*
ментов. Следовательно, участки генома, в кото*
лом анализ фрагментов митохондриальной
рых цитозины были прижизненно метилирова*
ДНК, выделенной из образцов возрастом от 18
ны, будут содержать больше чтений с тимином,
до 60 000 лет, продемонстрировал сильную поло*
по сравнению с неметилированными участка*
жительную корреляцию между возрастом и час*
ми, а доля замен C → T будет отражать степень
тотой замены C на T. В группе образцов возрас*
метилирования дДНК.
том 117 лет и младше частота замен C → T не
Метод подготовки библиотек для секвениро*
превышала 5%, в то время как для образцов воз*
вания с обработкой ДНК смесью ферментов ура*
растом 500 лет и старше частота замен составля*
цил*ДНК*гликозилазы и эндонуклеазы VIII
ла не менее 11%. В свою очередь, для образцов
представляет собой непрямой способ анализа ме*
ДНК, выделенных из тканей неандертальцев,
тилирования генома и предполагает применение
данный показатель обычно составляет не ме*
специальных вычислительных алгоритмов для
нее 20%, а зачастую достигает 40-50% [16, 20].
реконструкции метилирования. Существует и
Посмертная деградация дДНК, а конкретно
ещё один непрямой способ оценки степени ме*
процесс дезаминирования цитозина, усложняет
тилирования дДНК, основанный на использова*
исследование дДНК, но в то же время даёт воз*
нии двух различных полимераз для исходной
можность изучать метилирование геномов древ*
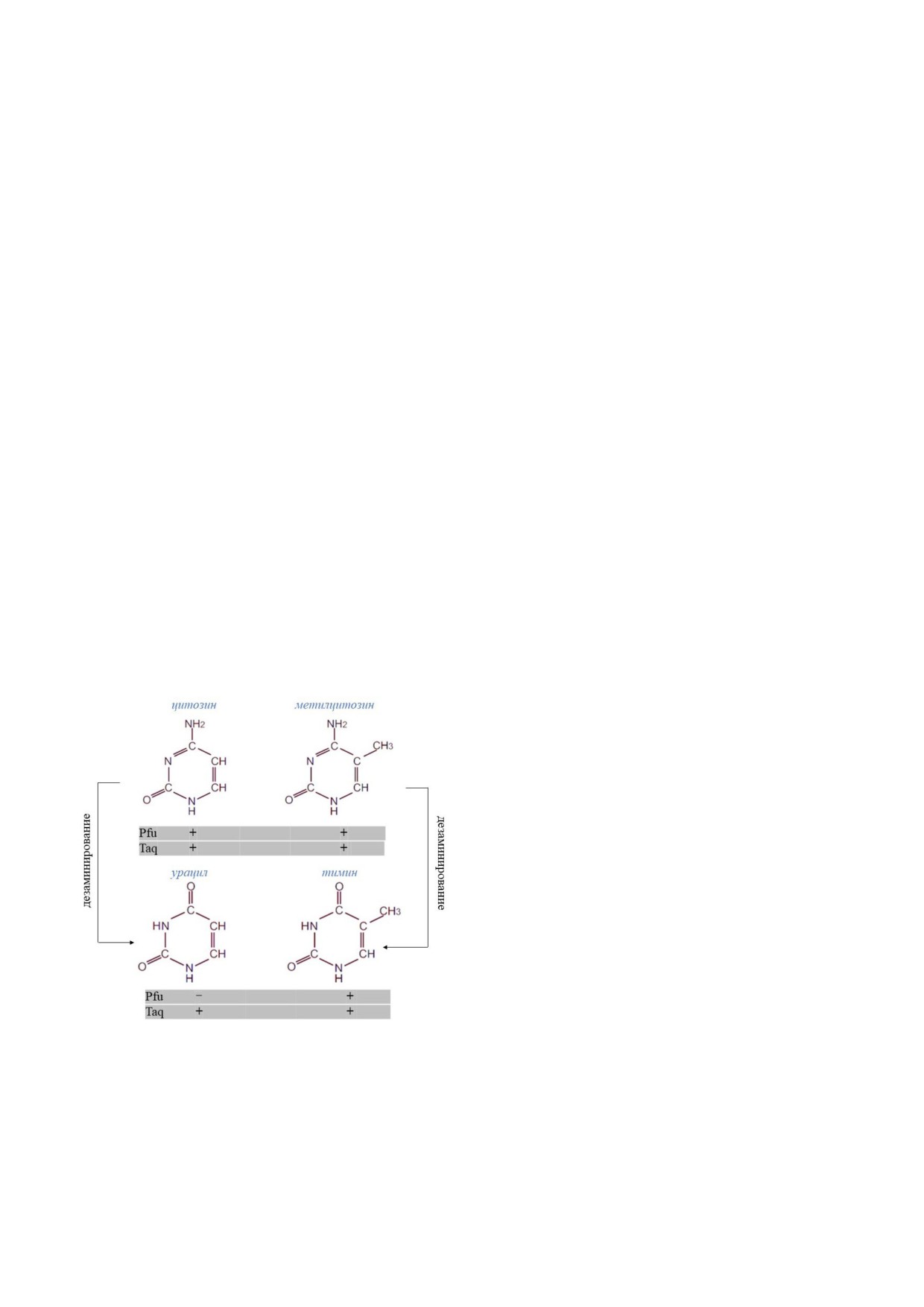
матрицы: Taq*полимеразы, не чувствительной к
них людей. Группа С. Паабо в процессе приго*
урацилу, и Pfu*полимеразы, которая в присут*
ствии урацила обрывает синтез цепи [14]. В слу*
чае дезаминирования метилированных цитози*
нов образуется тимин, следовательно, обе поли*
меразы смогут продолжить синтез цепи, и в ре*
зультате секвенирования исследователь в обеих
реакциях будет наблюдать замену C → T в после*
довательности ДНК (рис. 1). В свою очередь, де*
заминирование неметилированных цитозинов
создаёт «непроходимый» для Pfu*полимеразы
урацил, и синтез цепи обрывается. Таким обра*
зом, на основании соотношения замен C → T,
выявляемых при секвенировании библиотек
фрагментов ДНК, приготовленных с помощью
Taq* и Pfu*полимераз, становится возможным
восстановление профиля метилирования дДНК.
Непрямые способы анализа метилирования
генома являются предпочтительными при рабо*
те с дДНК в отличие от прямых методов, таких
как бисульфитная конверсия [21] или обогаще*
ние молекул ДНК, содержащих метилирован*
ные CpG, с помощью белков, содержащих ме*
Рис. 1. Схематичное изображение метода оценки метили*
тил*связывающие домены (methyl*CpG*binding
рования дДНК, основанного на использовании двух раз*
domain, MBD) [22]. Например, для бисульфит*
личных полимераз для исходной матрицы. Способность
ной конверсии необходимо большое количество
Pfu*полимеразы и Taq*полимеразы продолжать синтез це*
генетического материала, что уже делает этот
пи в присутствии различных типов нуклеотидов во время
ПЦР*амплификации обозначена знаками «+» (способна)
подход не применимым для древних образцов, а
и «-» (неспособна)
обогащение с помощью белков, содержащих ме*
БИОХИМИЯ том 86 вып. 12 2021
ЭПИГЕНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ ДРЕВНЕЙ ДНК
1811
тил*связывающие домены, может вносить су*
го гена, сказываются на фенотипе человека, в
щественный сдвиг в сторону анализа более
данном случае на строении скелета. Метод был
длинных фрагментов ДНК, игнорируя корот*
предварительно проверен на видах с известной
кие, действительно эндогенные, молекулы.
морфологией, таких как неандертальцы и шим*
панзе. Точность определения расходящихся
черт составила более 85%, что позволило приме*
РЕКОНСТРУКЦИЯ ЭПИГЕНОМОВ
нить данный подход для реконструкции фено*
АРХАИЧНЫХ ЛЮДЕЙ
типа денисовца (девочки из Денисовой пеще*
ры) [25]. На текущий момент не найдено пол*
В 2014 г. научная группа Д. Гохмана (David
ных скелетов или черепов архаичных людей
Gokhman) впервые опубликовала полные рекон*
данного вида, и восстановление их внешности
струированные карты метилирования ДНК не*
на основании эпигенетических модификаций
андертальца и денисовца [23]. Исследователи
генома является единственно возможным спо*
сравнили картины метилирования ДНК, выде*
собом. Исследователям удалось предсказать
ленной из костной ткани древнего и современ*
56 отличий денисовцев от неандертальцев и/или
ного человека, и выявили порядка 2000 диффе*
современных людей. Денисовцы имели ряд об*
ренциально метилированных участков
[23].
щих черт с неандертальцами: удлинённое лицо,
На тот момент особое внимание привлекли от*
низкий лоб, выступающие челюсти, широкий
личия в метилировании кластера генов HOXD,
таз и крупную грудную клетку, также были вы*
регулирующих развитие разных частей тела у
явлены и уникальные особенности, например,
многоклеточных животных. В промоторе ге*
более длинная зубная дуга и более выраженное
на HOXD9 и в теле гена HOXD10 костной ткани
латеральное расширение черепа [25]. Важно от*
древнего человека было установлено гиперме*
метить, что полученные результаты не противо*
тилирование, в то время как у всех исследован*
речат морфологии обнаруженных на сегодняш*
ных образцов костной ткани современных лю*
ний день фрагментов скелета денисовцев.
дей для этих участков характерно гипометили*
Несмотря на впечатляющие результаты, по*
рование. Ранее, в исследовании на мышиных
лученные командой Д. Гохмана, у исследовате*
модельных системах, было показано, что изме*
лей возник ряд трудностей, связанных с отсут*
нения в экспрессии генов данного кластера
ствием полных референсных карт метилирова*
приводят к морфологическим преобразовани*
ния костной ткани современного человека, ма*
ям, напоминающим различия в строении ко*
лым количеством образцов, необходимостью
нечностей неандертальца и современного чело*
сравнивать данные метилирования, полученные
века [24]. Таким образом, эпигенетические мо*
с помощью различных технологий, а также с от*
дификации данного региона могут играть клю*
сутствием таких данных для организмов, кото*
чевую роль в эволюции конечностей современ*
рые можно было бы использовать в качестве
ного человека, что в очередной раз свидетель*
внешней группы (аутгруппы) при определении
ствует о том, что при изучении микроэволюци*
эволюционных взаимоотношений между иссле*
онных и макроэволюционных процессов необ*
дуемыми популяциями людей разных видов [25].
ходимо учитывать как генетический, так и эпи*
Для преодоления перечисленных сложнос*
генетический фактор.
тей исследователи приняли решение дополни*
В 2019 г. команда Д. Гохмана опубликовала
тельно реконструировать профили метилирова*
работу, в которой представила новый подход в
ния ещё нескольких древних геномов, в том
изучении функциональной роли дифференци*
числе эпигеном древних людей современного
ально метилированных участков генома архаич*
анатомического облика, а также целенаправлен*
ных людей [25]. Предложенный способ позво*
но реконструировать полные карты метилиро*
лял предсказывать фенотипические особеннос*
вания в костной ткани современных людей и
ти древних людей, основываясь на допущении,
нескольких шимпанзе с применением различ*
что изменения, вызываемые сильным метили*
ных технологий. Такой подход позволил макси*
рованием промотора, похожи на изменения,
мально учесть факторы, которые могут влиять
возникающие в результате мутаций в гене, кото*
на результат поиска дифференциально метили*
рые либо существенно снижают уровень его
рованных регионов: технологию секвенирова*
экспрессии, либо полностью выключают ген.
ния, различия в покрытии генома и в степени
При разработке метода исследователи исполь*
дезаминирования, возраст и пол человека (на*
зовали базу данных «Онтология человеческого
пример, некоторые регионы генома подверже*
фенотипа» (The Human Phenotype Ontology,
ны гиперметилированию только у мужчин),
HPO) [26], где содержится информация о том,
часть скелета (бедренная кость, височная кость
как мутации, нарушающие работу того или ино*
черепа, фаланга пальца, зуб), из которой выде*
БИОХИМИЯ том 86 вып. 12 2021
1812
ЖУР и др.
лялась ДНК, а также потенциальный шум, кото*
нях мозга архаичных людей. Например, если ис*
рый возникает во время реконструкции карт ме*
следуемый локус неметилирован в костной тка*
тилирования.
ни шимпанзе и у неандертальского или денисов*
В результате командой Д. Гохмана была соз*
ского человека (сценарий 1), но метилирован в
дана уникальная платформа для изучения дина*
костной ткани современного человека и тканях
мики метилирования ДНК в процессе эволю*
мозга современных людей и шимпанзе, то дан*
ции человека, что позволило обнаружить диф*
ная эпимутация (изменением метилирования
ференциально метилированные участки в
ДНК без изменения последовательности) явля*
588 генах [25]. В том числе было установлено ги*
ется специфичной для костной ткани, которая
перметилирование в участках генов SOX9, ACAN,
возникла у современных людей, изменив состо*
COL2A1, NFIX и XYLT1, связанных с анатомией
яние локуса с неметилированного на метилиро*
лица и голосового тракта, характерное только
ванный. Следовательно, у архаичных видов лю*
для современных людей. Известно, что SOX9,
дей этот локус в тканях мозга будет скорее всего
ACAN и COL2A1 представляют собой группу ско*
метилирован. В том случае, если исследуемый
ординированно функционирующих генов, взаи*
локус неметилирован в костной ткани шимпан*
модействующих друг с другом и регулирующих
зе и у неандертальского или денисовского чело*
рост и формирование скелета, включая каркас
века (сценарий 2), но метилирован в костной
гортани и кости лица [27]. Предположительно
ткани современного человека, и при этом в тка*
гиперметилирование этих генов могло сыграть
нях мозга современных людей локус метилиро*
ключевую роль в формировании лица и голосо*
ван, а в тканях мозга шимпанзе неметилирован,
вого тракта современного человека [15].
то данная эпимутация затронула все ткани, из*
Важно отметить, что в тканях, не относя*
менив статус локуса на метилированный. Такое
щихся к скелетным (мозг, кровь и др.), профиль
изменение могло произойти, например, если
метилирования данных генов практически не
эпигенетическое состояние локуса определяется
различается у шимпанзе и современного челове*
до тканевой дифференцировки. В данном слу*
ка, что свидетельствует о том, что различия яв*
чае у архаичных видов людей этот локус в тканях
ляются специфическими именно для скелетных
мозга будет скорее всего неметилирован. Стоит
тканей. Такой вывод позволяет сделать наличие
также различать такие тканеспецифические
профиля метилирования для шимпанзе, орга*
эпимутации с эпимутациями, которые приводят
низма, выступающего в качестве внешней эво*
к противоположному состоянию метилирова*
люционной группы. Если исследователю извес*
ния гена во всех органах и тканях у людей (сов*
тен статус метилирования определенного регио*
ременных и архаичных) и шимпанзе. Последние
на у шимпанзе в костной и других тканях, то,
являются отражением генетических изменений
имея эту информацию, становится возможным
в процессе эволюции от обезьяны к человеку.
предсказать статус метилирования исследуемого
Следует также отметить, что кроме генов, ре*
локуса в тканях архаичных людей, не сохранив*
гулирующих рост и формирование скелета, диф*
шихся для исследования, в том числе в тканях
ференциально метилированные участки в гено*
мозга [28], в то время как в костной ткани уро*
мах архаичных и современных людей выявлены
вень метилирования можно определить экспе*
в генах, ассоциированных с развитием нервной,
риментально.
мышечной, сердечно*сосудистой систем и дру*
На рис. 2 представлена схема предсказания
гими важными процессами в организме, что не*
статуса метилирования геномного локуса в тка*
сомненно требует дальнейшего детального изу*
Рис. 2. Схема предсказания статуса метилирования исследуемого геномного локуса в тканях мозга архаичных людей.
«+» - Метилированное состояние геномного локуса; «-» - неметилированное состояние геномного локуса
БИОХИМИЯ том 86 вып. 12 2021
ЭПИГЕНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ ДРЕВНЕЙ ДНК
1813
чения. Таким образом, исследование эпигенети*
определения возраста палеоэскимоса исследова*
ческих модификаций древних геномов открыва*
тели использовали только два локуса (cg07533148
ет новые возможности для изучения микро* и
и cg01530101) из пяти, которые предварительно
макроэволюционных процессов живых организ*
показали наибольшую информативность для
мов, а также предоставляет уникальную возмож*
ДНК, выделенной из волос современного чело*
ность реконструировать фенотипические приз*
века, с различиями между прогнозируемым и ре*
наки архаичных людей, в том числе тех, для ко*
альным возрастом в пределах 1,7-12,4 года. Со*
торых ещё не обнаружено полных скелетов.
гласно статусу метилирования регионов вблизи
данных локусов, возраст палеоэскимоса на мо*
мент смерти составил приблизительно 40 лет.
ЭПИГЕНЕТИЧЕСКИЕ «ЧАСЫ»
Основная цель разработчиков моделей для
ДЛЯ ОПРЕДЕЛЕНИЯ ВОЗРАСТА
прогнозирования хронологического возраста
ДРЕВНЕГО ЧЕЛОВЕКА
состоит в том, чтобы научиться определять био*
логический возраст человека, который не всегда
Ещё одним интересным направлением ис*
совпадает с хронологическим, что может отра*
следований является возможность определения
жать интенсивность процессов старения для
хронологического возраста древнего человека
каждого организма индивидуально. В свою оче*
на основе оценки статуса метилирования опре*
редь, более детальное изучение эпигенетичес*
деленных CpG*локусов (сайтов, где цитозин (C)
ких механизмов старения поможет разрабаты*
находится рядом с гуанином (G) в последова*
вать стратегии по замедлению этого процесса и
тельности ДНК), степень метилирования кото*
увеличению продолжительности и качества
рых изменяется по мере взросления и старения
жизни. На сегодняшний момент разработано не
организма. Повышенный интерес к такому под*
менее 15 эпигенетических «часов» старения [31],
ходу связан с тем, что установление возраста
в одних моделях для оценки возраста достаточно
древнего человека на момент его смерти как
3 CpG*локуса [32], в других - необходимо про*
правило вызывает трудности, так как останки
анализировать несколько сотен [33, 34]. В свою
древних людей зачастую представлены в виде
очередь, каждая модель характеризуется опреде*
отдельных фрагментов скелета. Например, в
ленной погрешностью и типом ткани, для кото*
случае денисовского человека были найдены
рой прогнозирование работает наилучшим об*
всего несколько мелких костей и зубов [9, 29],
разом. Например, модель Horvath использует
что не позволяет достоверно определить возраст
353 CpG*локуса, работает с большим количест*
классическими подходами, изучая морфологию
вом тканей человека и имеет среднюю ошибку
костей черепа или других частей скелета. Кроме
предсказания 3-5 лет [33], что является весьма
того, традиционные методы основаны на визу*
неплохим показателем. Тем не менее подавляю*
альной, зачастую довольно субъективной, оцен*
щее большинство имеющихся сегодня эпигене*
ке морфологических особенностей скелета, что
тических калькуляторов прогнозируют возраст
может приводить к ошибкам.
по профилю метилирования ДНК, выделенной
Практическая возможность определения
из цельной крови (наиболее доступного материа*
возраста древнего человека на момент его смер*
ла), что абсолютно неприменимо для образцов
ти на основании данных метилирования ДНК
дДНК. Следовательно, разработка новых моде*
была впервые показана на примере 4000*летне*
лей или изучение возможности применения су*
го палеоэскимоса, принадлежащего к культуре
ществующих моделей для прогнозирования хро*
Saqqaq [14]. ДНК палеоэскимоса была выделена
нологического возраста древнего человека по
из стержня волос, хорошо сохранившихся в
данным метилирования ДНК, выделенной из
вечной мерзлоте, что позволило произвести
костной ткани, костей черепа и зубов, является
широкомасштабное секвенирование образца с
актуальной задачей и требует дополнительных
20*кратным покрытием. Для определения воз*
исследований в этом направлении.
раста исследователи применили линейную мо*
дель Koch и Wagner 2011 года [30], описываю*
щую связь возраста и статуса метилирования пя*
ИССЛЕДОВАНИЕ НЕДОСТУПНЫХ РАНЕЕ
ти CpG*локусов, из которых четыре, ассоцииро*
АСПЕКТОВ ЖИЗНИ ДРЕВНИХ ЛЮДЕЙ
ванные с генами NPTX2 (cg12799895),
TRIM58 (cg07533148), GRIA2 (cg25148589) и
Отдельным перспективным направлением в
KCNQ1DN (cg01530101), подвергаются гиперме*
палеоэпигенетике является изучение локусов
тилированию с возрастом и один CpG*локус в ге*
генома, характеризующихся чувствительностью
не BIRC4BP (cg23571857) по мере взросления ор*
к условиям окружающей среды [35]. Такие локу*
ганизма претерпевает гипометилирование. Для
сы, выступая в качестве «посредников» между
БИОХИМИЯ том 86 вып. 12 2021
1814
ЖУР и др.
внешней средой и ДНК, модифицируют свой
ный эмбриональный период развития. В связи с
паттерн метилирования в ответ на определен*
тем, что изменение в метилировании, возник*
ные воздействия, такие как изменения в пита*
шее на ранней стадии развития, ещё до диффе*
нии, смена климатических условий, взаимодей*
ренцировки, будет перенесено на все дочерние
ствие с химическими веществами и др. На се*
клетки организма, это позволяет изучать эпиге*
годняшний день опубликовано большое коли*
нетические локусы, чувствительные к воздей*
чество работ, подтверждающих влияние различ*
ствиям внешней среды, в тканях, доступных для
ных внешних факторов на эпигенетические мо*
исследования в палеоэпигенетике (зубы, волосы
дификации генома, в том числе на эпигеном че*
и кости скелета) [35].
ловека. Например, в исследовании под руковод*
Особый интерес для учёных представляет
ством Т. Пауса (T. Paus) было показано, что воз*
изучение эпигенетических модификаций гено*
действие сигаретного дыма на плод во время
мов древних людей для исследования механиз*
внутриутробного развития изменяет статус ме*
мов таких заболеваний, как ожирение и ассоции*
тилирования целого ряда генов [36]. В работе
рованные с ним патологии. Различают моноген*
М. Росарио (М. Rosario) с соавторами установ*
ную тяжёлую форму ожирения с ранней мани*
лено, что у детей, рождённых от матерей с диа*
фестацией, вызванную мутацией в одном гене, и
бетом второго типа, выявляется более 4000 диф*
полигенную форму. При моногенной форме
ференциально метилированных регионов в ге*
внешние факторы не оказывают существенного
номе при сравнении с детьми, рождёнными от
влияния на развитие заболевания. В то время как
женщин без данного заболевания [37]. Интерес*
полигенная форма относится к мультифактор*
ные данные были получены Б. Хейманс
ным гетерогенным заболеваниям и развивается в
(B. Heijmans) в ходе исследования детей, выно*
результате дисбаланса между потребляемой и
шенных в период массового голода, поразивше*
расходуемой энергией, в основе которого лежат
го население Нидерландов под конец Второй
нарушения метаболизма, генетическая предрас*
мировой войны, известного как «Голодная зима
положенность, нарушение поведенческих реак*
1944 года». Исследователями было показано,
ций и влияние внешних факторов. Полигенная
что в группе людей, подвергшихся воздействию
форма ожирения является наиболее распростра*
голода на самых ранних этапах своего развития,
нённой в современном обществе [42].
наблюдается снижение уровня метилирования
C развитием технологий секвенирования в
гена IGF2 [38]. Данный ген представляет особый
ходе исследований методом полногеномного
интерес, поскольку кодирует инсулиноподоб*
поиска ассоциаций (genome*wide association
ный фактор роста 2 и играет важную роль в регу*
studies, GWAS) выявлено более 1000 локусов, ас*
ляции процессов роста и развития организма.
социированных с признаками ожирения, тем не
Хорошо известно, что активность данного гена
менее совместно они могут объяснить только
регулируется по механизму геномного имприн*
8,4% вариабельности индекса массы тела чело*
тинга, в том числе посредством метилирования
века [43]. Большинство обнаруженных вариан*
ДНК [39], соответственно, снижение уровня ме*
тов, выявленных в результате полногеномного
тилирования гена IGF2 может приводить к изме*
анализа ассоциаций, по отдельности имеют сла*
нению активности инсулиноподобного фактора
бую связь с ожирением и связанными с ним па*
роста 2, оказывая существенное влияние на про*
тологиями и зачастую расположены в некодиру*
цессы роста и развития организма, а также на
ющей части генов или в межгенном простран*
развитие патологических состояний во взрос*
стве [44-46].
лом возрасте [40, 41]. Таким образом, существу*
В последние годы появляется всё больше ра*
ет множество доказательств того, что воздей*
бот, свидетельствующих о том, что ключевая
ствие неблагоприятных факторов на ранних ста*
роль в патогенезе ожирения может принадле*
диях внутриутробного развития может изменять
жать эпигенетическим механизмам [47-50].
эпигенетический статус определённых локусов
Широкое распространение ожирения за послед*
генома и сохраняться на протяжении жизни че*
ние десятилетия в странах с низким и средним
ловека, оказывая влияние на его здоровье.
уровнем доходов, в первую очередь в районах с
Исследование локусов в геноме древнего че*
большим количеством ресторанов быстрого пи*
ловека, профиль метилирования которых ассо*
тания, может быть следствием изменений в про*
циирован с тем или иным воздействием внеш*
филе метилирования ДНК в результате мало*
ней среды, может служить новым подходом в
подвижного современного образа жизни и нес*
изучении условий проживания архаичных лю*
балансированного питания. Тем не менее моле*
дей, их диеты, заболеваний, а также токсичес*
кулярный механизм патогенеза ожирения до
ких веществ, действию которых они подверга*
конца неясен, и непонятно: являются ли выяв*
лись в первую очередь в наиболее чувствитель*
ленные эпигенетические изменения причиной
БИОХИМИЯ том 86 вып. 12 2021
ЭПИГЕНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ ДРЕВНЕЙ ДНК
1815
развития заболевания или его следствием. За*
него и выявить, какие гены изменяют свою ак*
частую данные об уровне и спектре дифферен*
тивность, в том числе с учётом их функциональ*
циально метилированных генов противоречи*
ных связей между собой.
вы, а число таких работ ограниченно.
Работы такого рода довольно трудоёмкие, но
Исследование дДНК может улучшить наше
в то же время могут способствовать получению
понимание эпигенетической составляющей мо*
новых сведений об эпигенетической составляю*
лекулярной природы ожирения и ассоцииро*
щей молекулярной природы ожирения и свя*
ванных с ним патологий. Отличный от совре*
занных с ним болезней. В свою очередь, выяв*
менного образ жизни древних людей позволяет
ленные с использованием такого оригинального
использовать их профиль метилирования гено*
подхода эпигенетические локусы, ассоцииро*
ма в качестве эпигенетической «нормы». Пред*
ванные с исследуемым заболеванием, могут
полагая, что древние люди современного типа в
быть использованы в качестве перспективных
меньшей степени были подвержены таким не*
мишеней для поиска новых лекарственных пре*
благоприятным факторам, как малоподвижный
паратов.
образ жизни и высококалорийное питание с вы*
сокой долей жиров и простых углеводов, следу*
ет ожидать выявления различий в профиле ме*
ЗАКЛЮЧЕНИЕ
тилирования геномов древних людей и ныне
живущих. Логично предположить, что обнару*
Исследование последовательностей и эпиге*
женная эпигенетическая вариабельность опре*
нетических модификаций геномов древних лю*
делённых участков генома может играть важную
дей является довольно дорогим и трудозатрат*
роль в развитии ожирения и ассоциированных с
ным направлением, требующим специальных
ним заболеваний. С этой точки зрения интерес*
условий для работы, большого объёма секвени*
но сравнить профили метилирования современ*
рования, вычислительных мощностей и владе*
ных людей и, например, людей бронзового века,
ния специфическими инструментами анализа
живших порядка 5000 лет назад. Учитывая, что
данных. Тем не менее поддержание и развитие
такой промежуток времени довольно короткий
таких исследований является задачей государ*
с точки зрения эволюции (приблизительно 200
ственной важности не только с точки зрения био*
поколений), можно предположить, что измене*
логических наук, например, для изучения эво*
ния в метилировании вследствие изменения са*
люции человека и его заболеваний, но и для ис*
мой нуклеотидной последовательности ДНК
тории и политологии. Зачастую это единствен*
маловероятны. Следовательно, адаптация на
ный метод, способный объективно и наглядно
малых временных масштабах возможна преиму*
продемонстрировать и обосновать бездоказа*
щественно за счёт эпигенетических изменений.
тельность экстремистских теорий о превосход*
На сегодняшний момент исследователям
стве одних народов над другими. Ярким истори*
доступны способы проверки функциональной
ческим примером служат идеи немецкого архео*
значимости выявленных различий в профиле
лога Kossinna [53], которые позже были подхва*
метилирования современных и древних людей:
чены нацистами, что привело к известным со*
например, на клеточных линиях путём модели*
бытиям и огромному числу жертв.
рования статуса метилирования исследуемого
На сегодняшний день исследования дДНК
локуса с помощью лентивирусных конструкций,
перестали быть исключительно фундаменталь*
экспрессирующих каталитически неактивный
ными и приобрели очевидный прикладной ас*
белок dCas9 слитый с каталитическим доме*
пект. Например, изучение ДНК древних людей
ном TET1CD (вызывает таргетное деметилиро*
способно улучшить наше понимание генетичес*
вание) или Dnmt3a (вызывает таргетное метили*
кой и эпигенетической составляющей молеку*
рование) [51, 52]. Такие конструкции позволяют
лярной природы различных заболеваний, в том
изменять метилирование регионов генома, за*
числе таких как ожирение и ассоциированные с
ранее заданных исследователем с помощью
ним патологии. Исследование эволюции мозга
РНК*гидов, каждый из которых направит dCas9
архаичного и современного человека, а конк*
к собственной мишени. В случае с ожирением
ретно генетических и эпигенетических механиз*
моделирование статуса метилирования изучае*
мов регуляции нервной системы, может пролить
мых локусов в индуцированных плюрипотент*
свет на механизмы патогенеза психических и
ных стволовых клетках с последующей направ*
нейродегенеративных заболеваний. Анализ
ленной их дифференцировкой в адипогенном
ДНК патогенов древних людей даёт возмож*
направлении позволит сравнить транскрипци*
ность изучать происхождение и эволюцию воз*
онные профили адипоцитов с модифицирован*
будителей заболеваний современных людей, что
ным метилированием заданных локусов и без
позволит оценить шансы появления ещё более
БИОХИМИЯ том 86 вып. 12 2021
1816
ЖУР и др.
опасных патогенов и поможет избежать новых
исследования ценных археологических образ*
вспышек эпидемий в будущем.
цов, обнаруженных на территории страны, по*
Таким образом, палеогенетика и недавно вы*
высит конкурентоспособность российской нау*
делившееся её направление, палеоэпигенетика,
ки на мировом уровне, а, следовательно, позво*
являются важными приоритетными направле*
лит на равных участвовать в интерпретации ис*
ниями для страны, требующими поддержки со
торических событий прошлого.
стороны государства. До недавнего времени
полногеномное секвенирование уникальных
образцов из местных археологических памятни*
Финансирование. Работа выполнена при фи*
ков мирового значения в большинстве случаев
нансовой поддержке Минобрнауки России, грант
проводилось в зарубежных лабораториях, что не
№ 13.1902.21.0023 (согл. № 075*10*2020*116).
способствовало развитию исследований дДНК в
Конфликт интересов. Авторы заявляют об от*
России и не позволяло местным археологам на
сутствии конфликта интересов.
равных участвовать в интерпретации получен*
Соблюдение этических норм. Настоящая
ных результатов. Полноценное становление и
статья не содержит описания каких*либо иссле*
развитие данного направления предоставит воз*
дований с участием людей или животных в каче*
можность учёным самостоятельно проводить стве объектов.
СПИСОК ЛИТЕРАТУРЫ
1.
Orlando, L., Ginolhac, A., Zhang, G., Froese, D.,
some map and cytosine methylation levels of an ancient
Albrechtsen, A., et al. (2013) Recalibrating Equus evolu*
human genome, Genome Res., 24, 454*466.
tion using the genome sequence of an early Middle
15.
Gokhman, D., Nissim*Rafinia, M., Agranat*Tamir, L.,
Pleistocene horse, Nature, 499, 74*78.
Housman, G., Garc a Pérez, R., et al. (2020) Differential
2.
Van der Valk, T., Pečnerová, P., D ez Del Molino, D.,
DNA methylation of vocal and facial anatomy genes in
Bergström, A., Oppenheimer, J., et al. (2021) Million*
modern humans, Nat. Commun., 11, 1189.
year*old DNA sheds light on the genomic history of mam*
16.
Rohland, N., Glocke, I., Aximu*Petri, A., and Meyer, M.
moths, Nature, 591, 265*269.
(2018) Extraction of highly degraded DNA from ancient
3.
Reis, M., Donoghue, P., and Yang, Z. (2016) Bayesian
bones, teeth and sediments for high*throughput sequenc*
molecular clock dating of species divergences in the
ing, Nat. Protoc., 13, 2447*2461.
genomics era, Nat. Rev. Genet., 17, 71*80.
17.
Lindahl, T. (1993) Instability and decay of the primary
4.
Krause, J., and Pääbo, S. (2016) Genetic time travel,
structure of DNA, Nature, 362, 709*715.
Genetics, 203, 9*12.
18.
Kavli, B., Otterlei, M., Slupphaug, G., and Krokan, H.
5.
Weyrich, L. S., Duchene, S., Soubrier, J., Arriola, L.,
(2007) Uracil in DNA - general mutagen, but normal inter*
Llamas, B., et al. (2017) Neanderthal behaviour, diet, and
mediate in acquired immunity, DNA Repair, 6, 505*516.
disease inferred from ancient DNA in dental calculus,
19.
Srivastava, S., and Singh, N. (2011) The probability analy*
Nature, 544, 357*361.
sis of opening of DNA, J. Chem. Phys., 134, 115102.
6.
Jensen, T. Z. T., Niemann, J., Iversen, K. H., Fotakis,
20.
Sawyer, S., Krause, J., Guschanski, K., Savolainen, V., and
A. K., Gopalakrishnan, S., et al. (2019) A 5700 year*old
Pääbo, S. (2012) Temporal patterns of nucleotide misin*
human genome and oral microbiome from chewed birch
corporations and DNA fragmentation in ancient DNA,
pitch, Nat. Commun., 10, 5520.
PLoS One, 7, e34131.
7.
Rasmussen, S., Allentoft, M. E., Nielsen, K., Orlando, L.,
21.
Llamas, B., Holland, M. L., Chen, K., Cropley, J. E.,
Sikora, M., et al. (2015) Early divergent strains of Yersinia
Cooper, A., et al. (2012) High*resolution analysis of cyto*
pestis in Eurasia 5,000 years ago, Cell, 163, 571*582.
sine methylation in ancient DNA, PLoS One, 7, e30226.
8.
Green, R. E., Krause, J., Briggs, A. W., Maricic, T.,
22.
Seguin*Orlando, A., Gamba, C., Sarkissian, C. D.,
Stenzel, U., et al. (2010) A draft sequence of the
Ermini, L., Louvel, G., et al. (2015) Pros and cons of
Neandertal genome, Science, 328, 710*722.
methylation*based enrichment methods for ancient DNA,
9.
Meyer, M., Kircher, M., Gansauge, M.*T., Li, H.,
Sci. Rep., 5, 11826.
Racimo, F., et al. (2012) A high*coverage genome
23.
Gokhman, D., Lavi, E., Prüfer, K., Fraga, M. F., Riancho,
sequence from an archaic Denisovan individual, Science,
J. A., et al. (2014) Reconstructing the DNA methylation
338, 222*226.
maps of the Neandertal and the Denisovan, Science, 344,
10.
Prüfer, K., Racimo, F., Patterson, N., Jay, F.,
523*527.
Sankararaman, S., et al. (2014) The complete genome
24.
Zakany, J., and Duboule, D. (2007) The role of Hox genes
sequence of a Neanderthal from the Altai Mountains,
during vertebrate limb development, Curr. Opin. Gen. Dev.,
Nature, 505, 43*49.
17, 359*366.
11.
Jablonka, E. (2013) Epigenetic inheritance and plasticity: the
25.
Gokhman, D., Mishol, N., de Manuel, M., de Juan, D.,
responsive germline, Progr. Biophys. Mol. Biol., 111, 99*107.
Shuqrun, J., et al. (2019) Reconstructing Denisovan anato*
12.
Luo, X., Song, R., Moreno, D. F., Ryu, H.*Y.,
my using DNA methylation maps, Cell, 179, 180*192.e10.
Hochstrasser, M., et al. (2020) Epigenetic mechanisms
26.
Köhler, S., Gargano, M., Matentzoglu, N., Carmody, L.
contribute to evolutionary adaptation of gene network
C., Lewis*Smith, D., et al. (2021) The human phenotype
activity under environmental selection, Cell Rep., 33,
ontology in 2021, Nucleic Acids Res., 49, D1207*D1217.
108306.
27.
Lee, Y.*H., and Saint*Jeannet, J.*P. (2011) Sox9 function in
13.
Briggs, A. W., Stenzel, U., Meyer, M., Krause, J.,
craniofacial development and disease, Genesis, 49, 200*208.
Kircher, M., et al. (2010) Removal of deaminated cytosines
28.
Gokhman, D., Meshorer, E., and Carmel, L. (2016)
and detection of in vivo methylation in ancient DNA,
Epigenetics: it’s getting old. Past meets future in paleoepi*
Nucleic Acids Res., 38, e87.
genetics, Trends Ecol. Evol., 31, 290*300.
14.
Pedersen, J. S., Valen, E., Velazquez, A. M. V., Parker,
29.
Reich, D., Green, R. E., Kircher, M., Krause, J.,
B. J., Rasmussen, M., et al. (2014) Genome*wide nucleo*
Patterson, N., et al. (2010) Genetic history of an archaic
БИОХИМИЯ том 86 вып. 12 2021
ЭПИГЕНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ ДРЕВНЕЙ ДНК
1817
hominin group from Denisova Cave in Siberia, Nature,
42.
Loos, R. J. F., and Yeo, G. S. H. (2021) The genetics of
468, 1053*1060.
obesity: from discovery to biology, Nat. Rev. Genet., 23,
30.
Koch, C. M., and Wagner, W. (2011) Epigenetic*aging*sig*
1*14.
nature to determine age in different tissues, Aging, 3, 1018*
43.
Khera, A. V., Chaffin, M., Wade, K. H., Zahid, S.,
1027.
Brancale, J., et al. (2019) Polygenic prediction of weight
31.
Bergsma, T., and Rogaeva, E. (2020) DNA methylation
and obesity trajectories from birth to adulthood, Cell, 177,
clocks and their predictive capacity for aging phenotypes
587*596.e9. doi: 10.1016/j.cell.2019.03.028.
and healthspan, Neurosci. Insights, 15, 2633105520942221.
44.
Goodarzi, M. O. (2018) Genetics of obesity: what genetic
32.
Bocklandt, S., Lin, W., Sehl, M. E., Sánchez, F. J.,
association studies have taught us about the biology of obe*
Sinsheimer, J. S., et al. (2011) Epigenetic predictor of age,
sity and its complications, Lancet Diab. Endocrinol., 6,
PLoS One, 6, e14821.
223*236.
33.
Horvath, S. (2013) DNA methylation age of human tissues
45.
Müller, M. J., Geisler, C., Blundell, J., Dulloo, A.,
and cell types, Genome Biol., 14, R115.
Schutz, Y., et al. (2018) The case of GWAS of obesity: does
34.
Zhang, Q., Vallerga, C. L., Walker, R. M., Lin, T.,
body weight control play by the rules? Int. J. Obesity, 42,
Henders, A. K., et al. (2019) Improved precision of epige*
1395*1405.
netic clock estimates across tissues and its implication for
46.
Locke, A. E., Kahali, B., Berndt, S. I., Justice, A. E.,
biological ageing, Genome Med., 11, 54.
Pers, T. H., et al. (2015) Genetic studies of body mass index
35.
Gokhman, D., Malul, A., and Carmel, L. (2017) Inferring
yield new insights for obesity biology, Nature, 518, 197*
past environments from ancient epigenomes, Mol. Biol.
206.
Evol., 34, 2429*2438.
47.
Cheng, Z., Zheng, L., and Almeida, F. A.
(2018)
36.
Toledo*Rodriguez, M., Lotfipour, S., Leonard, G.,
Epigenetic reprogramming in metabolic disorders: nutri*
Perron, M., Richer, L., et al. (2010) Maternal smoking
tional factors and beyond, J. Nutr. Biochem., 54, 1*10.
during pregnancy is associated with epigenetic modifica*
48.
Pietiläinen, K. H., Ismail, K., Järvinen, E., Heinonen, S.,
tions of the brain*derived neurotrophic factor*6 exon in
Tummers, M., et al. (2016) DNA methylation and gene
adolescent offspring, Am. J. Med. Genet. B Neuropsychiatr.
expression patterns in adipose tissue differ significantly
Genet., 7, 1350*1354.
within young adult monozygotic BMI*discordant twin
37.
Rosario, M. C., Ossowski, V., Knowler, W. C.,
pairs, Int. J. Obesity, 40, 654*661.
Bogardus, C., Baier, L. J., et al. (2014) Potential epigenet*
49.
Muniandy, M., Heinonen, S., Yki*Järvinen, H.,
ic dysregulation of genes associated with MODY and type
Hakkarainen, A., Lundbom, J., et al. (2017) Gene expres*
2 diabetes in humans exposed to a diabetic intrauterine
sion profile of subcutaneous adipose tissue in BMI*discor*
environment: an analysis of genome*wide DNA methyla*
dant monozygotic twin pairs unravels molecular and clini*
tion, Metabolism, 63, 654*660.
cal changes associated with sub*types of obesity, Int. J.
38.
Heijmans, B. T., Tobi, E. W., Stein, A. D., Putter, H.,
Obesity, 41, 1176*1184.
Blauw, G. J., et al. (2008) Persistent epigenetic differences
50.
Perfilyev, A., Dahlman, I., Gillberg, L., Rosqvist, F.,
associated with prenatal exposure to famine in humans,
Iggman, D., et al. (2017) Impact of polyunsaturated and
Proc. Natl. Acad. Sci. USA, 105, 17046*17049.
saturated fat overfeeding on the DNA*methylation pattern
39.
Smith, F. M., Garfield, A. S., and Ward, A.
(2006)
in human adipose tissue: a randomized controlled trial,
Regulation of growth and metabolism by imprinted genes,
Am. J. Clin. Nutr., 105, 991*1000.
Cytogenet. Genome Res., 113, 279*291.
51.
Morita, S., Noguchi, H., Horii, T., Nakabayashi, K.,
40.
Cui, H., Cruz*Correa, M., Giardiello, F. M., Hutcheon,
Kimura, M., et al. (2016) Targeted DNA demethylation
D. F., Kafonek, D. R., et al. (2003) Loss of IGF2 imprint*
in vivo using dCas9*peptide repeat and scFv*TET1 catalyt*
ing: a potential marker of colorectal cancer risk, Science,
ic domain fusions, Nat. Biotechnol., 34, 1060*1065.
299, 1753*1755.
52.
Nuñez, J. K., Chen, J., Pommie, G. C., Cogan, J. Z.,
41.
Ravenel, J. D., Broman, K. W., Perlman, E. J., Niemitz,
Replogle, J. M., et al. (2021) Genome*wide programmable
E. L., Jayawardena, T. M., et al. (2001) Loss of imprinting
transcriptional memory by CRISPR*based epigenome
of insulin*like growth factor*II (IGF2) gene in distinguish*
editing, Cell, 184, 2503*2519.e17.
ing specific biologic subtypes of Wilms tumor, J. Nat.
53.
Kossinna, G.
(1919) Das Weichselland, ein Uralter
Cancer Inst., 93, 1698*1703.
Heimatboden der Germanen, Kafermann, Leipzig.
ACHIEVEMENTS AND PROSPECTS FOR EPIGENETIC STUDIES
OF ANCIENT DNA
Review
K. V. Zhur1, V. A. Trifonov2, and E. B. Prokhortchouk1*
1 Federal Research Centre “Fundamentals of Biotechnology”, Russian Academy of Sciences,
119071 Moscow, Russia; E*mail: prokhortchouk@gmail.com
2 Institute for the History of Material Culture Russian Academy of Sciences, 191186 St.*Petersburg, Russia
The development of high*throughput whole*genome sequencing technology and improvement of sample preparation
techniques make it possible to study ancient DNA (aDNA) from archaeological samples that are over a million years
old. Study of aDNA sequences shed the light on a history of human migrations, clarified a chain of populations
changes, interbreeding of Cro*Magnons with Neanderthals and Denisovans, evolution of human pathogens, etc.
Equally important was the announcement of the possibility to study epigenetic modifications of ancient genomes,
which allows receiving previously inaccessible information, including gene expression data, nucleosome positioning
and DNA methylation. Cytosine methylation maps of Denisovan genome opens a road for chronological age predic*
tion, phenotype reconstruction and even elucidating a list of unfavorable environmental factors that have influenced
the ancient person. In this review we discuss actual progress in epigenetics of ancient DNA including methodological
approaches and promising directions of the studies.
Keywords: ancient DNA, paleoepigenetics, epigenetic clock, epigenome editing, obesity
6 БИОХИМИЯ том 86 вып. 12 2021