БИОХИМИЯ, 2021, том 86, вып. 3, с. 324 - 340
УДК 577.24:577.3:612.67
РОЛЬ ЕСТЕСТВЕННЫХ ПРОЦЕССОВ СТАРЕНИЯ
В ВОЗНИКНОВЕНИИ И ПАТОГЕНЕЗЕ БОЛЕЗНЕЙ, СВЯЗАННЫХ
С АНОМАЛЬНЫМ НАКОПЛЕНИЕМ БЕЛКОВЫХ АГРЕГАТОВ
Обзор
© 2021
Н.С. Ильинский1*, С.В. Нестеров1,2, Е.И. Шестопёрова1,
А.В. Фонин1,3, В.Н. Уверский1,4, В.И. Горделий1,5,6
1 Центр исследований молекулярных механизмов старения и возрастных заболеваний,
Московский физико#технический институт, 141701 Долгопрудный,
Московская обл., Россия; электронная почта: ilinsky_nick@mail.ru
2 Институт цитохимии и молекулярной фармакологии, 115404 Москва, Россия
3 Институт цитологии Российской академии наук, 194064 Санкт#Петербург, Россия
4 Отдел молекулярной медицины, Медицинский колледж им. Морсани,
Университет Южной Флориды, 33612 Тампа, США
5 Юлихский исследовательский центр, 52428 Юлих, Германия
6 Институт структурной биологии, 38000 Гренобль, Франция
Поступила в редакцию 08.08.2020
После доработки 21.08.2020
Принята к публикации 24.08.2020
Старение является системной первопричиной возрастных заболеваний, в частности протеинопатий.
Действительно, большинство болезней, связанных с неправильным сворачиванием белка, являются спора
дическими, а вероятность их возникновения растёт по мере старения организма. В данном обзоре рассмот
рен процесс образования агрегатов белков и их патогенность, устройство клеточной системы поддержания
протеостаза. Показано, как токсичность агрегатов нарушает важные клеточные процессы и приводит к про
теинопатиям. Проанализировано, как проявления старения (дисфункция митохондрий, дисбаланс сиг
нальной системы, изменения генома и эпигенома) делают возможным патогенез протеинопатий - усили
вают агрегацию напрямую и через дискоординацию стресс ответов. Проведённый анализ позволяет наме
тить перспективы поиска воздействий для лечения протеинопатий и достижения здорового долголетия.
КЛЮЧЕВЫЕ СЛОВА: протеостаз, агрегация белков, протеинопатия, старение, дисфункция митохондрий,
мутации, эпигенетические изменения.
DOI: 10.31857/S0320972521030040
ВВЕДЕНИЕ: СТАРЕНИЕ СПОСОБСТВУЕТ
ности клетки, различных систем и организма в
НАРУШЕНИЮ ПРОТЕОСТАЗА
целом. Одна из основных концепций старения
утверждает, что данный процесс является запла
Старение является процессом постепенного
нированным и, возможно, базируется на прог
нарушения гомеостаза, то есть функциональ рамме развития организма [1]. Предполагается,
Принятые сокращения: АФК - активные формы кислорода; м и тРНК - матричная и транспортная РНК; ЭПР -
эндоплазматический ретикулум; Akt - серин/треониновая протеинкиназа B; AMPK - протеинкиназа, активируемая аде
нозинмонофосфатом; ATP - аденозинтрифосфат; APOE - ген, кодирующий белок аполипопротеин E; APP - белок пред
шественник амилоида; Aβ - бета амилоид; ETC - электрон транспортная цепь; FOXO - фактор транскрипции семейства
«forkhead box» класса O; FUS - белок, ассоциированный с саркомой; Grp78 - белок, регулируемый глюкозой, 78;
hNRNPA1 - гетерогенный ядерный рибонуклеопротеин А1; HSF1 - транскрипционный фактор теплового шока; HSP -
белок теплового шока, шаперон; IR/IGFR - рецепторы инсулина и инсулиноподобных факторов роста; IIS - инсулино
вый и инсулиноподобный сигнальный каскад; mTOR - серин/треониновая протеинкиназа, мишень рапамицина млеко
питающих; mtTERT - теломераза, локализованная в митохондрии; NAD+ - никотинамидадениндинуклеотид; NADPH -
никотинамидадениндинуклеотидфосфат; NBR1 - белок, кодируемый геном, соседствующим с BRCA1; NMDA - N метил
D аспартат; NRF2 - ядерный фактор, связанный с эритроидным фактором; Pin1 - пептидил пролил цис/транс изомера
за 1; SIRT1 - сиртуин 1; SQSTM1/p62 - секвестосома 1, убиквитин связывающий белок p62; SOD1 - супероксиддисмута
за 1; TDP 43 - ДНК связывающий белок TAR; TFEB - транскрипционный фактор EB; TIA 1 - Т клеточный внутренний
антиген 1; TRiC/CCT - TCP 1 кольцевой белковый комплекс (шаперонин, содержащий TCP 1); ULK1 - серин/треони
новая протеинкиназа; UPR - стресс ответ на несвёрнутые белки; UPS - убиквитин протеасомная система.
* Адресат для корреспонденции.
324
РОЛЬ СТАРЕНИЯ В РАЗВИТИИ ПРОТЕИНОПАТИЙ
325
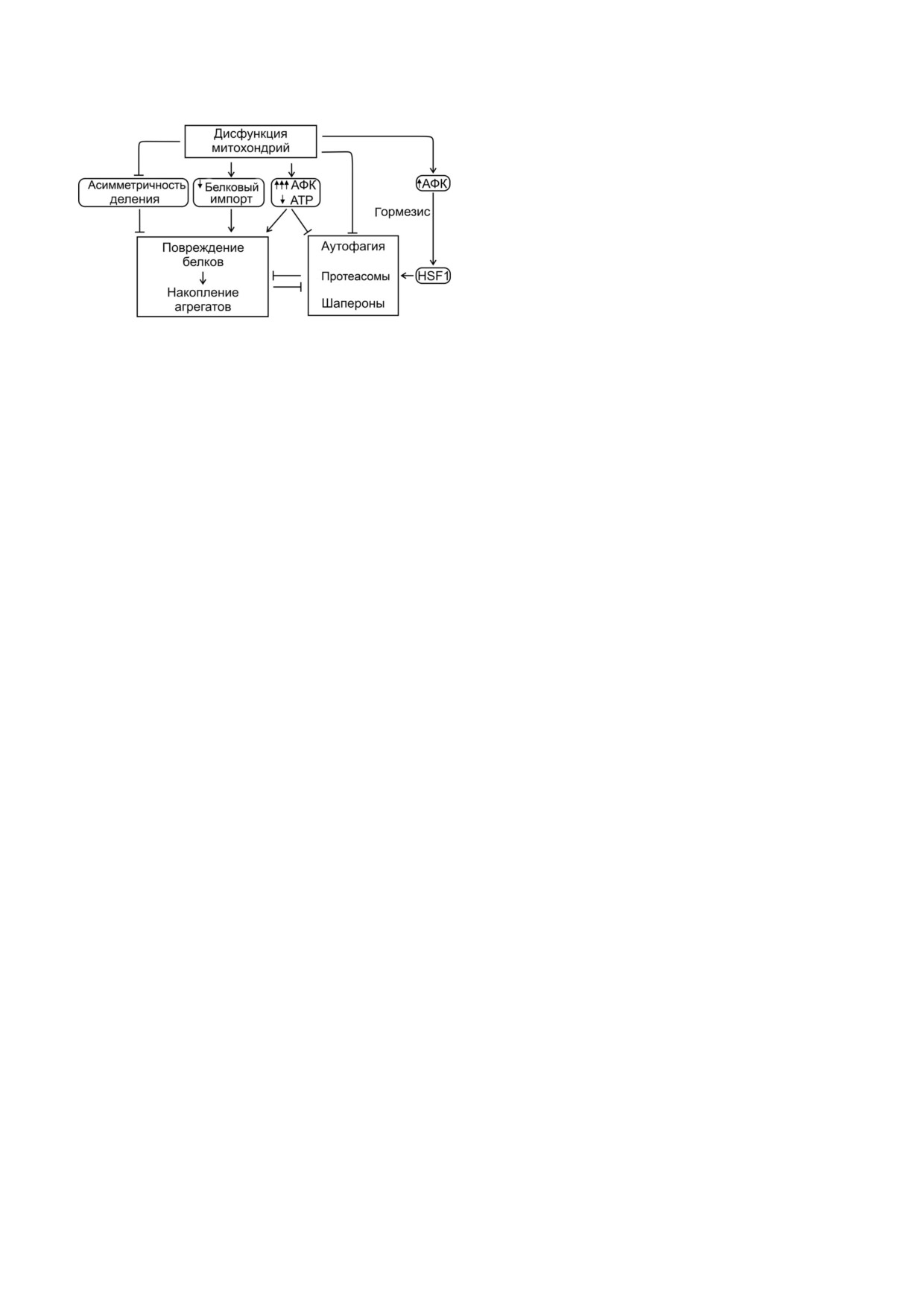
Рис. 1. Влияние дисфункции митохондрий, дисбаланса сигнальной системы, изменений генома и эпигенома на потерю
протеостаза проявляется в усугублении действия стрессов (энергетического, окислительного, подавление стресс ответов),
увеличении склонности белков к агрегации (мутантные формы и перенасыщение белком), нарушении системы поддер
жания протеостаза (например, аутофагии)
что накопление с возрастом повреждений клет
спорадический характер и увеличение частоты
ки, вызванных внешними, внутренними стрес
возникновения с возрастом [3].
сами и ослаблением защитных механизмов,
В настоящей работе представлены данные о
вместе с заложенной программой старения при
том, как поддерживается протеостаз - основы
водит к одряхлению индивида.
сворачивания и агрегации белка, защитные ме
Протеостаз - гомеостаз белков, результат
ханизмы против накопления агрегатов, возник
работы систем синтеза и контроля качества бел
новение протеинопатий как результат неэффек
ков. Протеостаз заключается в наличии функ
тивности системы поддержания протеостаза и
циональных белков в клетке и вне её в нужных
нескомпенсированной токсичности агрегатов.
концентрациях, в нужное время и на правиль
Наконец, проанализирована роль проявле
ный срок. Потеря протеостаза состоит в нару
ний старения в нарушении протеостаза и разви
шении сворачивания белка и ослабевании сис
тии протеинопатий (рис. 1). Показано, что ста
темы контроля качества белков, что ведёт к на
рение усугубляет действие стрессов и подавляет
коплению токсичных белковых агрегатов и раз
защитные механизмы, что напрямую и через ос
витию протеинопатий. Под «агрегацией» в дан
лабление системы поддержания протеостаза
ной работе подразумевается неспецифичная и
приводит к увеличению склонности белков к аг
патогенная агрегация белков, которая приводит
регации, способствует патогенезу протеинопа
к потере функциональности (в отличие от есте
тий. Изложены возможные вмешательства для
ственной олиго или полимеризации, форми
лечения заболеваний, вызванных белковой аг
рующей функциональные белковые комплек
регацией, в том числе как основа рассматрива
сы).
ется профилактика старения.
Клеточное старение характеризуется поте
рей протеостаза, дисфункцией митохондрий,
изменением генома и эпигенома, нарушением
ПРОЦЕСС АГРЕГАЦИИ БЕЛКОВ
работы сигнальной (управляющей) системы [2].
Эти повреждения усиливают друг друга, нару
В этом разделе рассмотрены основы фол
шая целостность гомеостаза клетки. Накопле
динга и агрегации белка, а также причины ток
ние большого количества повреждённых клеток
сичности агрегатов.
приводит к возрастным заболеваниям, в том
Основы фолдинга, естественная агрегация. В
числе к протеинопатиям. О вкладе старения в
геноме закодирована конечная пространствен
патогенез болезней, связанных с аномальным
ная структура белка (кроме внутренне неупоря
накоплением белковых агрегатов, свидетель
доченных белков, см. ниже) и путь его достиже
ствует их преимущественно приобретённый,
ния (фолдинг) [4]. Сворачивание многих белков
БИОХИМИЯ том 86 вып. 3 2021
326
ИЛЬИНСКИЙ и др.
может быть осуществлено только с участием ша
Внешние и внутренние стрессы, приводящие к
перонов [5]. Однако даже в физиологических ус
агрегации. Генотоксический и протеотоксичес
ловиях возможны сбои в сворачивании белков,
кий стрессы нарушают процессы транскрипции
что, в свою очередь, может привести к их агрега
и трансляции, а также сворачивания белка; они
ции [6]. Агрегация может возникнуть по ряду
могут присутствовать в острой и хронической
причин.
формах. Стабильность (растворимость) белка
Во первых, высокая концентрация макро
зависит от кислотности, ионного баланса, окис
молекул в клетке (макромолекулярный крау
лительно восстановительного потенциала сре
динг) может привести к взаимодействию между
ды, температуры, давления, наличия сораство
сворачивающейся аминокислотной цепью и
рителей [6], присутствия химических токсинов,
другими молекулами, а также возникновению
таких как тяжёлые металлы, пестициды [17], а
ненативных контактов участков полипептидной
также токсичных метаболитов, побочных про
цепи [7].
дуктов обмена веществ [18]. Кроме того, сами
Во вторых, агрегация возникает из за боль
агрегаты стимулируют вторичную и последую
шого количества неспецифических взаимодей
щую агрегации, что обусловлено выгодностью
ствий подвижных участков полипептидной це
взаимодействия между экспонированными гид
пи, как правило, не входящих в упорядоченную
рофобными участками неправильно свёрнутых
вторичную структуру белка [8, 9].
белков и высокой конформационной стабиль
В третьих, склонность к агрегации проявля
ностью белковых агрегатов.
ют «сложные» белки: многодоменные [9], мемб
Молекулярные механизмы токсичности белH
ранные [10], нуждающиеся в посттрансляцион
ковых агрегатов. Существуют две основные ста
ных модификациях для созревания [11].
бильные формы белковых агрегатов: аморфные
В четвёртых, к агрегации могут приводить
агрегаты и амилоидные фибриллы. Несмотря на
невысокие энергетические барьеры между раз
то, что аморфные агрегаты зачастую богаты β
личными конформационными состояниями по
листами, они не имеют высокой упорядочен
липептидной цепи, присущие, в частности,
ности и реже ассоциированы с заболеваниями.
внутренне неупорядоченным белкам (англ. абб.
Примером такой болезни может являться ката
IDPs от intrinsically disordered proteins) [12].
ракта, при которой образуются аморфные агре
Внутренне неупорядоченные белки являют
гаты α кристаллина (принадлежит к семейству
ся особыми с точки зрения фолдинга. Само наз
малых шаперонов [19]). Как правило, формиро
вание подчёркивает, что неупорядоченность их
вание аморфных агрегатов является очень быст
структуры является неотъемлемым свойством
рым процессом.
этих белков. Фактически структура внутренне
Амилоидные фибриллы имеют упорядо
неупорядоченного белка находится в динами
ченную β структуру, в которой параллельные
ческом равновесии между различными конфор
или антипараллельные β нити сориентирова
мерами, разделёнными незначительными энер
ны перпендикулярно оси агрегата. Кинетика
гетическими барьерами. Вследствие этого любое
спонтанного процесса фибриллообразования,
незначительное изменение внешних условий
как правило, описывается сигмоидальной кри
может приводить к существенному изменению
вой, где начальный период соответствует мед
энергетической поверхности белка. Это обеспе
ленному процессу нуклеации, затем следует
чивает полифункциональность внутренне не
фаза экспоненциального роста фибрилл (элон
упорядоченных белков. В эукариотическом про
гации), завершающаяся фазой равновесия. Не
теоме более половины белков являются в той
которые амилоидные фибриллы обладают
или иной степени неупорядоченными [13]. Мо
функцией в клетке, например, премеланосом
заичная гетерогенная пространственно времен
ный белок PMEL17 работает как шаблон для
ная организация IDPs способствует склонности
полимеризации меланина. Однако в большин
этих белков к агрегации при изменении внеш
стве случаев амилоиды токсичны для клетки.
них условий. К внутренне неупорядоченным от
При этом совокупность данных свидетельству
носится ряд белков, агрегация которых наблю
ет о том, что растворимые протофибриллярные
дается при нейродегенеративных заболеваниях
промежуточные агрегаты существенно более
(β амилоид, тау белок, островковый амилоид
токсичны по сравнению со зрелыми фибрилла
ный полипептид, α синуклеин и др.) [14].
ми [3].
Склонность к образованию амилоидных
Можно выделить следующие основные при
фибрилл зависит от первичной структуры белка,
чины токсичности агрегатов:
однако при определённых условиях практичес
1. Ингибирование функциональности белка
ки все белки могут переходить в форму амило
при агрегации через уменьшение числа его ак
идных фибрилл [15, 16].
тивных единиц в клетке.
БИОХИМИЯ том 86 вып. 3 2021
РОЛЬ СТАРЕНИЯ В РАЗВИТИИ ПРОТЕИНОПАТИЙ
327
2. Ингибирование активности белков при их
белка и их последующего исправления (рефол
включении в состав агрегатов (факторы тран
динга), а также способствуют распознаванию
скрипции [20], шапероны [5], элементы протеа
протеолитическими системами неправильно
сом [21], факторы ядерно цитоплазматического
свернутых белков, помогают поддерживать бел
транспорта [20]). Белки, склонные к связыва
ки в пригодном к деградации состоянии [5].
нию с агрегатами, как правило, метастабильны,
Протеазы, убиквитин протеасомная систе
содержат внутренне неупорядоченные области
ма и аутофагия осуществляют регулируемую
(домены с низкой сложностью аминокислотной
деградацию неверно свёрнутых белков и агрега
последовательности) [22]. Также коагрегации
тов. Протеолитическая система также контро
способствует подобие первичных структур
лирует время функционирования белка, абсо
участвующих в ней белков. При этом опасность
лютную и относительную концентрации бел
представляют не только «аморфные» агрегиро
ков компонентов комплексов, обеспечивая осу
ванные тела, но и структурированные фибрил
ществление всех основных клеточных функ
лы [10].
ций [28].
3. Нарушение целостности мембран (поро
Только согласованное функционирование
образование растворимыми олигомерами [23],
всех систем протеостаза эффективно предотвра
деформация или пронизывание фибриллами
щает накопление неправильно свёрнутых бел
[24]), особенно критичное для митохондрий
ков и токсичных агрегатов. Например, несмотря
(приводящее к нарушению их функций при взаи
на наличие системы шаперонов, от 5 до 30%
модействии с мутантными белками гентингти
всех вновь синтезированных белков не сворачи
ном, SOD1, β амилоидом, α синуклеином, пар
ваются должным образом, имеют склонность к
кином, DJ 1, PINK1 [25]) и плазматической
агрегации и должны быть направлены на немед
мембраны нейронов.
ленную деградацию [27].
Ослабление системы поддержания протеос
Увеличение, даже незначительное, по срав
таза способствует дальнейшему усилению агре
нению с физиологической нормой агрегацион
гации (автокатализ), которое может привести к
ной способности белка и/или его внутриклеточ
одновременному появлению двух разных проте
ной концентрации ведёт к агрегации [29]. Сис
инопатий, таких как болезнь Альцгеймера и бо
тема контроля качества протеостаза, настроен
ковой амиотрофический склероз или болезнь
ная на норму для каждого белка, способна сдер
Паркинсона [26]. Наиболее выраженная роль
живать снижение растворимости лишь до опре
белковой агрегации в старении проявляется в её
деленного предела. Было обнаружено, что белки
ассоциации с клеточной дегенерацией, приво
с высоким уровнем экспрессии вносят основ
дящей к возрастным заболеваниям [3]. Ключе
ной вклад в общую агрегацию белков при дости
вые примеры таких болезней, поражающих
жении ими критической концентрации. Этот
центральную нервную систему, кратко описаны
эффект называется перенасыщением белком
в справке «Протеинопатии».
(supersaturation) [30]. Подавление производства
патологического, склонного к агрегации белка,
приводит к разрушению накопившихся агрега
СИСТЕМА ПОДДЕРЖАНИЯ ПРОТЕОСТАЗА
тов и снимает симптомы заболевания [31].
И ЕЁ ОСЛАБЛЕНИЕ ПРИ СТАРЕНИИ
Система шаперонов как основа системы подH
держания протеостаза. Протеотоксические
Стрессы и природная предрасположенность
стрессы приводят к появлению ненативных
белков к агрегации приводят к неправильному
конформаций белков. При недостаточности
фолдингу или их (частичному) разворачиванию
системы поддержания протеостаза HSF1 (тран
и образованию токсичных агрегатов. В данном
скрипционный фактор белков теплового шока)
разделе рассмотрены защитные механизмы,
освобождается от ингибирующей связи с шапе
препятствующие появлению и накоплению аг
ронами [32] и запускает синтез новых шаперо
регатов.
нов, обеспечивая стресс ответ.
Система поддержания протеостаза в клетке
Цитозольный ответ на протеотоксический
человека включает в себя около 2 000 белков
стресс выполняют ATP зависимые шапероны
[27]. Шапероны участвуют в сворачивании
HSP70 (с кошаперонами HSP40, HSP110),
вновь синтезируемых белков и в поддержании
HSP90, шаперонин TRiC/CCT (ассистирует
их нативных конформаций, в предотвращении
сворачиванию 10% протеома). Подавление агре
неправильного сворачивания (в том числе при
гации происходит через связывание шаперонов
транспортировке) и агрегации, осуществлении
со склонными к агрегации конформациями, де
дезагрегации (разборки), контролируемого раз
загрегацию, рефолдинг или деградацию. Уже
ворачивания неправильно свёрнутых молекул
сформировавшиеся агрегаты могут быть экра
БИОХИМИЯ том 86 вып. 3 2021
328
ИЛЬИНСКИЙ и др.
нированы шаперонами, что блокирует вредные
рез изомеризацию пролиновых остатков и обра
взаимодействия, позволяет удалить такие агре
зование дисульфидных связей [43]. Поскольку
гаты через разборку или аутофагию [33].
активность серин/треонин специфических про
При старении в клетке происходит измене
теинкиназ сильно зависит от изомера пролина,
ние профиля экспрессии шаперонов. Подавля
пептидил пролил цис/транс изомераза Pin1 иг
ется экспрессия ATP зависимых шаперонов,
рает важную роль в контролировании многих
повышается экспрессия ATP независимых ма
процессов, включая стресс и иммунный ответ,
лых шаперонов sHSP, что позволяет поддержи
клеточное развитие и рост, реакцию, дифферен
вать протеостаз при недостатке энергии [34]. В
цировку и выживание нейронов [44].
результате токсичные растворимые олигомеры
Система шаперонов способна ингибировать
не идут на разборку или рефолдинг, вместо это
агрегацию белков, определяющих патологию
го запускается контролируемая агрегация. Ма
нейродегенеративных заболеваний [3]. У мутант
лые шапероны объединяют агрегаты в тельца
ных мышей с пониженным уровнем белков
включения (содержат аморфные или амилоид
теплового шока наблюдается ускоренное старе
ные агрегаты), уменьшая реакционную поверх
ние, а в долгоживущих линиях наблюдается вы
ность агрегатов, сокращая таким образом требу
сокая активность шаперонов [45]. Активация
емое число связанных шаперонов [35]. Соответ
транскрипционного фактора теплового шока
ственно, гиперэкспрессия малого шаперона
HSF1, сверхэкспрессия шаперонов или приме
HSP 16 может приводить к увеличению продол
нение фармакологических шаперонов способ
жительности жизни (у нематод [36]).
ствуют увеличению продолжительности жиз
При транспортировке белка в определен
ни [46].
ный субклеточный компартмент цитозольные
Эндоплазматический стресс и ответ на неH
шапероны предотвращают преждевременное
свёрнутые белки: ингибирование трансляции. Эн
сворачивание белка. Шапероны защищают
доплазматический ретикулум обеспечивает син
трансмембранные гидрофобные области мемб
тез, сворачивание и процессирование около
ранных белков от водной среды и обеспечивают
трети от общего числа белков. В случае длитель
прохождение через мембраны органелл. Шапе
ного и неисправляемого аномального фолдинга
роны присутствуют в эндоплазматическом рети
белков возникает ЭПР стресс, на который акти
кулуме (ЭПР) (например, шаперон GRP78 [37]),
вируется ответ на несвёрнутые белки (англ. абб.
в митохондриях (например, HSP60 [38]), где
UPR от unfolded protein response). UPR реализу
способствуют функциональному рефолдингу в
ется в трех направлениях. Во первых, временно
месте назначения. Органельные шапероны так
останавливается трансляция белков, характер
же участвуют в нейтрализации цитозольных аг
ных для бесстрессового режима, что уменьшает
регатов. При тепловом стрессе определённые
производство дефектных белков, снижает на
неправильно свёрнутые белки, не имеющие ми
грузку на шапероны и системы деградации.
тохондриальной сигнальной последовательнос
Во вторых, увеличивается производство шапе
ти, с помощью HSP104 направляются в митохон
ронов, запускается протеасомная деградация.
дрии [39]. Более того, шапероны органелл игра
В третьих, при отсутствии снижения количест
ют критическую роль в обеспечении асиммет
ва агрегатов в течение определённого времени
ричности деления в дрожжах [40] и стволовых
UPR может вызвать апоптоз, потерю дифферен
клетках [41], способствующей уменьшению ко
цировки (дедифференцировку; показано для
личества агрегатов в дочерней или стволовой
секретирующих клеток), пироптоз, некроп
клетке соответственно.
тоз [37].
Отдельный набор шаперонов действует для
Нужно отметить, что временное снижение
поддержания функциональных конформаций
уровня трансляции является ключевым спосо
секретируемого протеома во внеклеточном
бом поддержания протеостаза и противодей
пространстве (внеклеточная система контроля
ствия старению в целом [2]. Например, для
качества). Наличие такого стресс ответа осо
уменьшения количества агрегатов с возрастом
бенно важно при прионном течении болезни
происходит адаптационное ингибирование
(см. справку «Протеинопатии»), поскольку он
трансляции (у нематод [47]). Изменение окис
способствует поглощению клеткой неверно
лительно восстановительного потенциала, дис
свёрнутых или агрегированных белков (в том
функция шаперонов изомераз в ЭПР нарушают
числе прионных) для дальнейшей деграда
образование дисульфидных связей, подавляют
ции [42].
UPR, приводят к накоплению неправильно
Необходимо также упомянуть о существова
свёрнутых белков и агрегации [37]. Устойчивая
нии шаперонов изомераз, обеспечивающих пра
сверхактивация UPR (наблюдается при старе
вильное сворачивание и стабильность белков че
нии, избыточности питания и малой активности
БИОХИМИЯ том 86 вып. 3 2021
РОЛЬ СТАРЕНИЯ В РАЗВИТИИ ПРОТЕИНОПАТИЙ
329
организма) играет роль в развитии заболеваний.
токсических стрессов появляются неверно свер
При хроническом подавлении трансляции про
нутые белки и агрегаты, которые должны быть
исходит гибель нейронов из за потери крити
подвержены избирательной деградации. Рас
ческих белков и активации апоптоза. Ингиби
смотрим роль убиквитин протеасомной и ауто
рование UPR может стать средством лечения
фаго лизосомальной систем в патогенезе проте
болезней Крейтцфельдта-Якоба, Альцгеймера,
инопатий.
Паркинсона и Гентингтона [48].
Убиквитин протеасомная система (англ.
СтрессHгранулы. Защита от протеотоксич
абб. UPS от ubiquitin proteasome system) состоит
ности через подавление трансляции может осу
из системы маркировки подлежащих деграда
ществляться независимо от шаперонов при об
ции белков убиквитином и протеасом, больших
разовании стресс гранул. Стресс гранулы
белковых комплексов, содержащих разрезаю
представляют собой цитоплазматические не
щие субстрат протеазы [28]. Синхронизирован
мембранные компартменты, появляющиеся в
ная сборка протеасом является шаперон зави
клетке в ответ на стресс и распадающиеся после
симой. Протеасомы деградируют белки, экспор
перехода клетки в нормальное состояние. Ос
тируемые из ЭПР, и цитозольные белки [37].
новная функция этих RNP (RNA/protein)
При этом деградация возможна только для раст
структур состоит в регулировании стресс ответа
воримой формы белков, для чего требуются ша
клетки и предотвращении повреждения генети
пероны. С другой стороны, компоненты убик
ческого материала в неблагоприятных услови
витиновой системы имеют шаперонные функ
ях. Регуляция состава и свойств стресс гранул
ции [50]. Таким образом, шаперонная система и
обеспечивается посттрансляционными моди
UPS взаимозависимы.
фикациями каркасных белков этих образова
Количество протеасомных субъединиц уве
ний, изменением pH, концентрации солей,
личивается с возрастом, что, по видимому, от
температуры окружающей среды и другими
ражает попытку организма удалить аберрантные
факторами. Стрессовое воздействие на клетку
белки и компенсировать сниженную при старе
вызывает арест трансляции и диссоциацию по
нии активность протеасом [51]. Решающая роль
лирибосомального комплекса. Согласно совре
UPS в протеинопатиях подтверждается повы
менным представлениям, диссоциация поли
шением токсичности агрегатов белков при ин
рибосомы и не успевшей подвергнуться транс
гибировании протеасом [52]. Усиление экспрес
ляции мРНК параллельно сопровождается пе
сии компонентов убиквитин протеасомной
реходом каркасных внутренне неупорядочен
системы и ингибирование деубиквитилаз повы
ных белков стресс гранул в жидкокапельную
шает устойчивость к протеотоксическому стрес
фазу с последующим привлечением свободной
су, продлевает срок жизни дрожжей, нематод и
мРНК и других компонентов этих немембран
клеток человека [51].
ных органелл.
Нерастворимые агрегаты могут быть под
Ряд нейродегенеративных заболеваний, в
вергнуты деградации с помощью аутофаго ли
том числе боковой амиотрофический склероз,
зосомальной системы. Аутофагия заключается в
сопровождается деградацией стресс гранул
деградации субстрата, попадающего в лизосому
вследствие включения в их состав мутантных
напрямую (микроаутофагия), с помощью шапе
форм белков FUS, TDP 43, hNRNPA1 с после
ронов (шаперон опосредованная и шаперон
дующим образованием этими белками амилои
управляемая аутофагия) или при слиянии ауто
доподобных фибрилл. Образуемые мутантными
фагосом и лизосом (макроаутофагия). Также су
белками стресс гранул амилоидоподобные фиб
ществует эндосомная микроаутофагия, которая
риллы токсичны для клеток, однако конкрет
осуществляется в поздних эндосомах [3].
ный механизм их действия неизвестен. Транс
Процесс макроаутофагии, позволяющий
формация стресс гранул в амилоидные фибрил
деградировать большие агрегаты и целые орга
лы при нейродегенеративных заболеваниях мо
неллы (митохондрии, пероксисомы), происхо
жет быть обусловлена включением в состав
дит в несколько этапов. Белки, кодируемые ге
стресс гранул мутантных белков FUS, TDP 43,
нами, связанными с аутофагией (англ. абб. ATG
hNRNPA1 и TIA 1. Нарушение деградации
от autophagy related genes), в частности бек
стресс гранул также может привести к форми
лин 1, координируют образование и функцио
рованию в клетке упорядоченных амилоидных
нирование фагофора - места сбора субстратов
фибрилл [49].
для деградации. Белки SQSTM1/p62, NBR1 за
Протеолитические системы как последняя
хватывают и заносят в фагофор убиквитиниро
стадия UPR. Несмотря на согласованную работу
ванный субстрат, фагофор закрывается, образуя
шаперонов и других защитных механизмов,
аутофагосому. Далее она сливается с плазмати
из за естественных причин и наличия протео
ческой мембраной (для выброса содержимого во
БИОХИМИЯ том 86 вып. 3 2021
330
ИЛЬИНСКИЙ и др.
внеклеточную среду, нетрадиционный путь сек
между стволовой и дочерней клеткой, предназ
реции белков) или с лизосомой (для деградации
наченной для дифференцировки [41].
субстратов). Эффективность расщепления зави
Асимметричность деления достигается при
сит от количества и функциональности лизо
согласованном действии цитоскелета (центро
сом. Синтез лизирующих ферментов и мемб
сомы, актиновых, промежуточных, септиновых
ранных белков лизосомы контролируется тран
филаментов), мембраны клетки и органелл (яд
скрипционным фактором TFEB, для эффектив
ра, лизосомы, ЭПР, митохондрий), создающих
ной работы ферментов внутри лизосомы под
диффузионные барьеры, локальные каркасы
держивается pH 4,5-5,0 (ATP зависимый меха
для заякоривания шаперонами агрегатов и те
низм) [53]. Кроме описанной селективной мак
лец включения, их направленного транспорта в
роаутофагии существует неизбирательный зах
одну из клеток [40].
ват части цитоплазмы с последующим расщеп
Деление клеток и асимметричность деления
лением [54], как происходит, например, при
стволовых клеток, прогениторных клеток при
клеточной смерти через аутофагию.
образовании долгоживущих, постмитотических
В целом, лизосомы поддерживают правиль
клеток являются важными механизмами защи
ное функционирование многих клеточных про
ты от накопления агрегатов белков и поддержа
цессов через перераспределение внутренних ре
ния гомеостаза в целом.
зервов аминокислот, ионов, вносят критический
вклад в старение [54]. Возрастное снижение эф
фективности аутофагии (подавление экспрессии
СПРАВКА. ПРОТЕИНОПАТИИ:
генов, связанных с аутофагией, неэффективная
ТОКСИЧНОСТЬ АГРЕГАТОВ
митофагия при мутантном белке паркине, заще
И НЕДОСТАТОЧНОСТЬ СИСТЕМЫ
лачивание лизосом и их загрузка липофусци
ПОДДЕРЖАНИЯ ПРОТЕОСТАЗА
ном) определяет развитие протеинопатий [51].
Активация аутофагии (гиперэкспрессия тран
В данном разделе рассмотрены распростра
скрипционного фактора TFEB [55], усиление
нённые нейродегенеративные заболевания, воз
шаперон опосредованной аутофагии [56], при
никающие из за токсичности агрегатов опреде
менение рапамицина, ограничение калорий
лённых белков (см. подробный обзор [60]).
[57]) является одним из способов борьбы с про
Болезнь Альцгеймера является наиболее
теинопатиями.
распространённым хроническим нейродегене
Таким образом, шаперонная, убиквитин
ративным заболеванием, симптомы которого
протеасомная и аутофаго лизосомальная систе
включают потерю памяти и деменцию. Насле
мы взаимосвязаны и образуют целостную систе
дуемые формы заболевания (которые составля
му поддержания протеостаза.
ют ~5% случаев) вызываются мутациями в бел
Деление клеток как механизм уменьшения
ке предшественнике амилоида (APP), пресени
концентрации агрегатов. Долгоживущие клетки,
линах 1 и 2. Полиморфизм генов, таких как ва
такие как нейроны, наиболее уязвимы к протеи
рианты ε4 и ε2 гена APOE, могут влиять на пред
нопатиям [3]. Нарушение описанных выше ме
расположенность к спорадической болезни
ханизмов защиты протеостаза в этих клетках не
Альцгеймера (обнаружены в ~50% случаев). Эти
компенсируется уменьшением концентрации
четыре гена ответственны за 30-50% наслед
агрегатов (или склонных к агрегации белков)
ственных случаев болезни. Аутосомно доминант
при делении, как это происходит в митотичес
ная форма болезни Альцгеймера очень редка
ких клетках. Восстанавливать функции клеток
(<1%). Физиологически заболевание характе
головного мозга помогают стволовые клетки,
ризуется амилоидными отложениями в виде се
известные особой устойчивостью протеостаза.
нильных бляшек бета амилоида (пептидов
Например, в стволовых клетках пациентов с
Aβ40 и Aβ42, дефектных продуктов разрезания
атаксией третьего типа не обнаружены полиглу
APP) в нейритах и сосудах головного мозга.
таминовые (polyQ) агрегаты, в отличие от других
Другим маркёром болезни Альцгеймера являет
клеток [16]. Это происходит благодаря высокой
ся наличие нейрофибриллярных клубков из
эффективности системы поддержания протеос
гиперфосфорилированных тау белков в ней
таза (например, протеасомная активность [58],
ронных и глиальных клетках. Бета амилоид
эффективная сборка комплекса шаперонина
подавляет мембранные функции (активность
TRiC/CCT [59]). Уникальными защитными ме
глутаматного NMDA рецептора, функциони
ханизмами стволовых клеток являются возмож
рование митохондрий, гомеостаз кальция), ак
ность существования в неактивном (quiescent)
тивирует NADPH оксидазу. Ингибирование
состоянии с низким уровнем метаболизма,
NADPH оксидазы защищает астроциты и ней
асимметричное разделение агрегатов белков
роны [61].
БИОХИМИЯ том 86 вып. 3 2021
РОЛЬ СТАРЕНИЯ В РАЗВИТИИ ПРОТЕИНОПАТИЙ
331
Болезнь Паркинсона является вторым наи
ков и нарушении системы поддержания протео
более распространённым нейродегенеративным
стаза.
заболеванием, характеризуется двигательной
Нефункциональные митохондрии: гормезис,
дисфункцией и, во многих случаях, деменцией и
окислительный и энергетический стрессы, митоH
депрессией. Основным белком, формирующим
хондриальноHлизосомальная ось. Митохондрии
характерные для этой болезни аномальные ами
являются ключевыми регуляторами выживания,
лоидные фибриллы (тельца Леви), является
старения и смерти клеток [25]. При старении у
α синуклеин. Для наследственных форм этой
пациентов с болезнями Альцгеймера и Паркин
болезни характерны мутации белков паркин,
сона наблюдается накопление делеций и точеч
LRRK2, PINK1, DJ 1 и ATP13A2. Олигомерный
ных мутаций в митохондриальной ДНК (из за
α синуклеин действует через повреждение
ошибок репликации и окислительного повреж
мембранных функций (стабильное увеличение
дения), изменение морфологии митохондрий,
проницаемости, приводящее к выбросу ионов
дисбаланс в работе электрон транспортной це
кальция) [62], изоляцию агрегатами важных бел
пи (англ. абб. ETC от electron transport chain),
ков (например, компонентов протеасомы [63]).
ATP синтазы, разобщающих и антиоксидант
Болезнь Гентингтона - это наследственное
ных белков. В результате снижается митохонд
нейродегенеративное заболевание, характеризу
риальный потенциал, производство ATP, гене
ющееся деменцией, двигательными и психичес
рируются активные формы кислорода (АФК), в
кими расстройствами. Вызывается увеличен
том числе и в ответ на окислительный стресс [2].
ным числом повторов CAG в ДНК, приводящих
Болезни Альцгеймера, Паркинсона, Гентингто
к образованию удлинённого полиглутаминового
на, боковой амиотрофический склероз характе
домена на N конце белка гентингтина, что оп
ризуются снижением активности комплексов
ределяет образование стабильных амилоидных
ETC. Дисфункция митохондрий усиливается
агрегатов. Фрагменты гентингтина нарушают
из за возрастного нарушения циклов слияния и
митохондриальный транспорт, усиливают окис
деления митохондрий [66], что приводит к уве
лительный и энергетический стрессы [64], нару
личению их массы и размера [67], блокирует от
шают функции ключевых элементов внутрикле
щепление повреждённых частей митохондрий,
точной сигнализации и системы поддержания
нарушая процесс митофагии [68]. Механизмы,
протеостаза [20].
по которым дисфункция митохондрий приво
Боковой амиотрофический склероз харак
дит к нарушению протеостаза, показаны на
теризуется двигательными нарушениями. Воз
рис. 2.
никает при мутациях в SOD1, гене каркасных
Мягкий окислительный стресс: гормезис, ак
белков стресс гранул TDP 43 (TARDBP), FUS,
тивация HSF1. Митохондрии являются одним
амплификации гексануклеотидного повтора в
из основных источников активных форм кисло
последовательности гена C9orf72 [60]. В резуль
рода, которые в низких концентрациях выпол
тате накапливаются агрегаты из SOD1, затвер
няют сигнальную функцию, направленную в
девших стресс гранул, нарушающих функцио
первую очередь на восстановление энергетики
нальность ядерной, митохондриальной мембран
клетки и активацию стрессовых защитных отве
[25], подавляющих протеасомную актив
тов (эффект гормезиса). Такой мягкий окисли
ность [21].
тельный стресс в том числе вызывает ремодели
Прионные болезни вызываются накоплени
рование хроматина, активацию HSF1 и митохон
ем и амилоидной агрегацией аномальных изо
дриального ответа на неправильно свёрнутые
форм прионного белка (PrP), возникающих
белки, восстанавливает протеостаз и повышает
внутри клетки либо попадающих извне (2% ин
продолжительность жизни (показано на нема
фекционных случаев для трансмиссивных губ
тодах [69]).
чатых энцефалопатий) [60]. Прионоподобное
Нефункциональные митохондрии подавляют
распространение агрегатов в мозге вносит вклад
аутофагию, подчёркивая тем самым наличие ми
в патогенез болезней Альцгеймера, Паркинсо
тохондриально лизосомальной оси. Нарушение
на, Гентингтона [65].
работы комплекса I ETC ослабляет эффектив
ность макроаутофагии из за энергетического
стресса [70]. Нефункциональные митохондрии
ВОЗРАСТНЫЕ КЛЕТОЧНЫЕ ИЗМЕНЕНИЯ
могут блокировать работу лизосом, заполняя их
КАК ПРИЧИНА УСИЛЕНИЯ
липофусцином, который образуется в результате
НАРУШЕНИЙ ПРОТЕОСТАЗА
реакции железа Fe(II) и перекиси водорода.
Возникающие при этом АФК способствуют по
В данном разделе рассмотрена роль процес
явлению сшивок в белках (особенно гликопро
сов естественного старения в повреждении бел
теинах, конечных продуктах гликирования),
БИОХИМИЯ том 86 вып. 3 2021
332
ИЛЬИНСКИЙ и др.
сия генов стресс ответа, антиоксидантной за
щиты и репарации ДНК [25]. Повреждения
ДНК и митохондрий усугубляются при недоста
точности компенсаторных механизмов, что
приводит к формированию положительной об
ратной связи, гибели клеток и воспалительным
процессам [73].
Ковалентные модификации аминокислот в
белках меняют поверхностный заряд и тополо
гию, что может приводить к неверному сворачи
ванию полипептидных цепей или (частичному)
разворачиванию уже свёрнутых белков. Окис
Рис. 2. Дисфункция митохондрий нарушает протеостаз.
Умеренный синтез АФК митохондриями приводит к адап
лительный стресс усиливает фосфорилирование
тивному стресс ответу (эффект гормезиса). Избыточный
тау белка, сшивку гликопротеинов. Наиболь
синтез АФК (окислительный стресс) способствует повреж
ший урон протеостазу наносят модификации
дению белков, в том числе защитной системы. Снижение
шаперонов, которые особенно чувствительны к
синтеза ATP (энергетический стресс) нарушает работу
АФК. Усиление работы антиоксидантных фер
энергозависимой системы поддержания протеостаза. На
рушения импорта приводят к агрегации митохондриаль
ментов снижает окислительный стресс и агрега
ных белков в цитозоле, недоступности митохондрии как
цию белков [74].
вспомогательного места деградации цитозольных агрега
Нарушение импорта белков. В митохондриях
тов. Снижение токсичности агрегатов путём разбавления и
с пониженным потенциалом (один из призна
асимметричного распределения при делении клетки также
ков старения) нарушается система митохондри
ослабляется дисфункцией митохондрий
ального импорта. В результате в цитозоле про
исходит накопление и агрегация белков, пред
жирных кислотах мембран митохондрии и лизо
назначавшихся для митохондрии [75]. При этом
сомы [71]. Нарушение лизосомальной актив
также не может осуществляться защитная функ
ности препятствует дальнейшей митофагии и
ция митохондрий по деградации неправильно
обновлению митохондрий. Восстановление ра
свёрнутых цитозольных белков [39]. Некоторые
боты лизосом может способствовать нормализа
агрегирующие белки, например, мутантный
ции функционирования митохондрий (через
гентингтин, могут встраиваться во внешнюю
эффективную митофагию), восстановлению
мембрану митохондрии и мешать импорту белка
протеостаза и обращению процесса старения
[64]. Это создаёт положительную обратную
отдельной клетки. Так проявляется необходи
связь, усугубляющую нарушения протеостаза.
мость согласованной работы митохондрий и ли
Нефункциональные митохондрии препят
зосом [72].
ствуют асимметричному разделению агрегатов.
Энергетический и острый окислительный
Митохондрии заякоривают на себе агрегаты (с
стрессы: повреждение ДНК и вредные посттранс
помощью HSP104), обеспечивая их несиммет
ляционные модификации белков. Митохондриаль
ричное распределение между материнской и до
ная дисфункция усугубляет окислительный
черней клетками (показано на дрожжах [40]),
стресс, вызывающий повреждения ДНК и бел
что нарушается при старении из за изменения
ков. Одновременный дефицит ATP приводит к
морфологии митохондрий. На культивируемых
недостаточности репарации ДНК и работы
клетках млекопитающих было показано, что
ATP зависимых шаперонов.
митохондрии связаны с агресомами (специфи
Окислительное повреждение ДНК затраги
ческий вид контролируемой агрегации неверно
вает в основном промоторы генов и теломеры
свёрнутых белков для аутофагосомной деграда
(см. раздел «Теломеры и теломераза»), которые
ции, возникающий при загруженности убикви
по своей нуклеотидной последовательности бо
тин протеасомной системы), которые неравно
лее чувствительны к окислительному стрессу
мерно распределяются во время митоза. Белки,
(богаты гуанином) и мало способны к восста
которые образуют агрегаты при некоторых деге
новлению (не подвергаются транскрипции,
неративных заболеваниях, также связаны с
сопровождающейся репарацией, или закрыты
мембраной митохондрий. Хотя эта связь потен
от репарации в шелтерине соответственно). За
циально может помочь ограничить распростра
счёт такой запрограммированной уязвимости к
нение болезнетворных белков, она способствует
окислению снижается экспрессия генов, под
дисфункции митохондрий [41].
держивающих синаптическую пластичность, ве
Дисбаланс сигнальной системы. При сбалан
зикулярный транспорт и митохондриальную
сированном метаболизме происходит периоди
функцию, одновременно повышается экспрес
ческое чередование режимов анаболизма и ката
БИОХИМИЯ том 86 вып. 3 2021
РОЛЬ СТАРЕНИЯ В РАЗВИТИИ ПРОТЕИНОПАТИЙ
333
болизма, контролируемое переключением меж
ULK1) и SIRT1 (активируя AMPK, через взаи
ду mTORC1 (у млекопитающих, далее для крат
модействие c PGC 1α/TFEB) активируют ауто
кости mTOR) и AMPK (рис. 3). Периодическая
фагию. Возрастное снижение чувствительности
активация катаболических процессов критичес
AMPK и SIRT1 способствует дисрегуляции ауто
ки важна для всех неспособных к неограничен
фагии [2].
ному росту многоклеточных организмов, не
Подавление анаболических процессов (через
имеющих возможности для всех клеток снижать
ингибирование mTOR) даёт возможность проте
количество агрегатов и других нарушений путём
канию катаболических процессов (протеасомной
деления [76]. При старении из за хронического
и лизосомной деградации). Сенсор достаточности
избытка питания и низкого использования
уровня аминокислот mTOR запускает трансля
энергии человеком нарушается периодичность
цию, на транскрипционном уровне усиливает
переключения катаболизм-анаболизм, пони
биосинтез нуклеиновых кислот, липидов, окис
жается чувствительность сенсоров энергии
лительное фосфорилирование и гликолиз. С
(AMPK, SIRT1) и питательных веществ (mTOR,
другой стороны, mTOR подавляет деградацию
IR/IGFR (рецепторов инсулина и инсулинопо
белков, органелл и других компонентов клетки
добных факторов роста)). Синтез сложных со
(ингибируя TFEB, ULK1). Активность mTOR
единений и рост клетки происходят при подав
увеличивается в процессе старения, в том числе
лении стресс ответов, что приводит ко множе
из за хронической избыточности питания при
ству клеточных дефектов. Рассмотрим, как пов
малой активности организма. Длительная акти
реждения метаболизма нарушают протеостаз
вация анаболических систем может приводить к
(рис. 3).
хроническим эндоплазматическому и окисли
Сигнальный путь инсулина и инсулиноподобно
тельному стрессам, развитию нейродегенерации
го фактора роста 1 (анаболизм) нарушает про
и возрастного ожирения. Клетка может проти
теостаз. Каскад инсулина и инсулиноподобных
водействовать стрессам за счёт включения отве
лигандов (англ. абб. IIS от insulin/insulin like sig
та на несвёрнутые белки. Блокирование избы
naling) состоит из рецептора IGF 1 (DAF 2 у не
матод), PI3K (AGE 1) и Akt белков, консерва
тивен от червей к человеку, ускоряет старение,
снижает устойчивость к стрессу и способность
поддержания протеостаза (рис. 3). Эффект сиг
нального каскада определяется инактивацией
факторов транскрипции FOXO (DAF 16, конт
ролирующего синтез белков, управляющего
продолжительностью жизни клеток, в том числе
стресс ответом), HSF1 (синтез шаперонов),
NRF2 (SKN 1, синтез антиоксидантных бел
ков), митохондриальной функции теломеразы
TERT (см. раздел «Теломеры и теломераза»).
Нарушение протеостаза определяется также
тем, что IIS активирует mTOR (см. ниже) [2].
Ограничение калорий, активация AMPK и
SIRT1 (переход на катаболизм) способствуют
поддержанию протеостаза. Ограничение рацио
на питания (сокращение потребления пищи без
недоедания) является наиболее действенным
вмешательством для увеличения продолжитель
ности жизни и задержки возрастной дисфунк
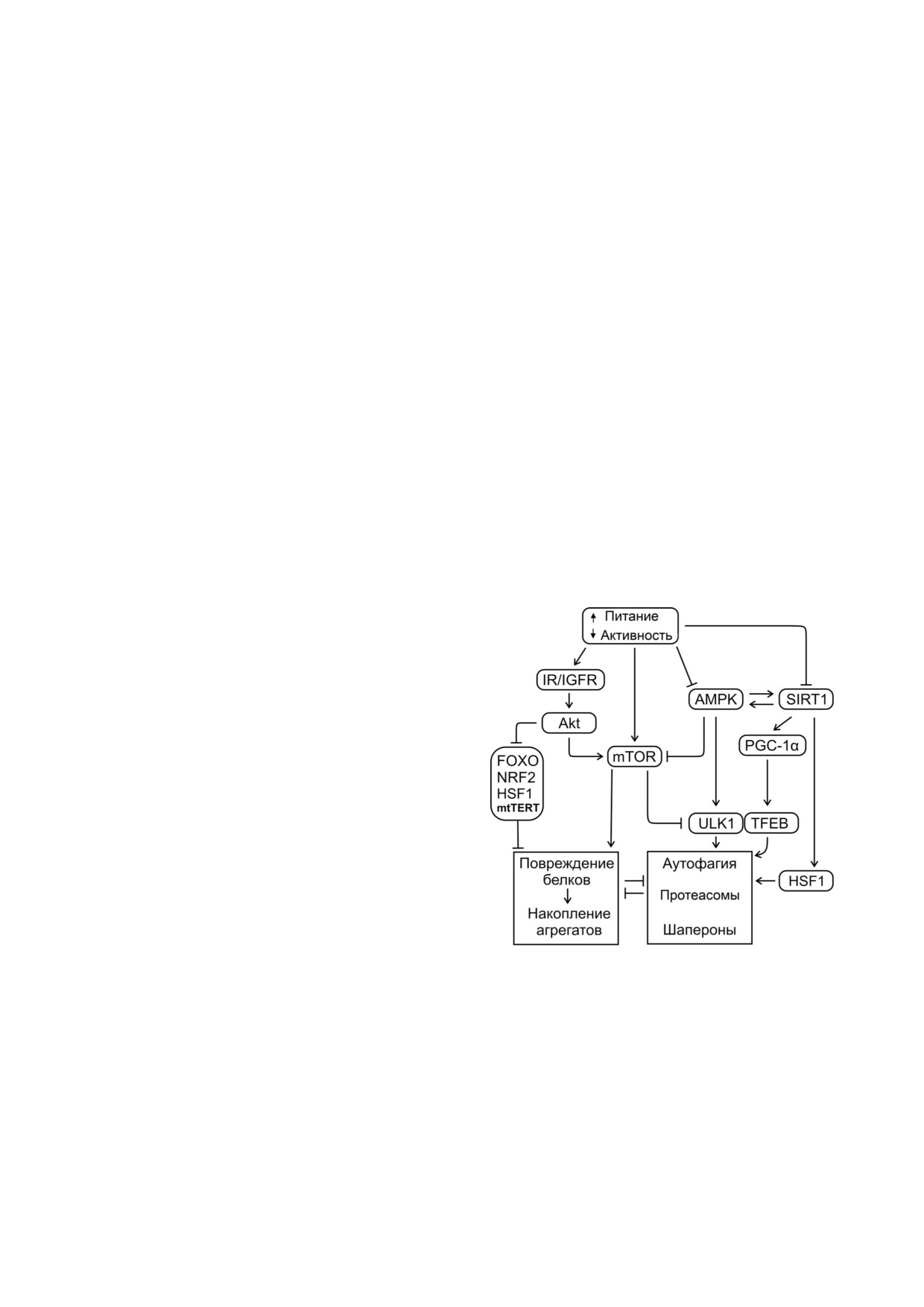
Рис. 3. Дисбаланс сигнальной системы приводит к нару
ции в организмах от дрожжей до млекопитаю
шению протеостаза. Сигнальный каскад инсулина и инсу
щих. Ограничение калорий напрямую и через
линоподобных факторов, действуя через ряд звеньев, в том
числе IR/IGFR (рецепторы инсулина и инсулиноподоб
AMPK и SIRT1 ингибирует mTOR. AMPK (че
ных факторов роста), киназу Akt, активирует mTOR (пере
рез отношение AMP/ATP) и SIRT1 (через отно
водит клетку в режим анаболизма). Киназа Akt подавляет
шение NAD+/NADH) детектируют низкоэнер
факторы стресс ответа (FOXO, NRF2, HSF1, митохондри
гетическое состояние клетки, являются взаимо
альную функцию теломеразы (mtTERT)). SIRT1, AMPK,
детектируя недостаток энергии, определяют переключение
активируемыми. SIRT1 активирует факторы
метаболизма на катаболизм (ингибируют mTOR), в том
транскрипции стресс ответа FOXO (DAF 16 у
числе активируя аутофагию (ULK1, TFEB). Деацетилаза
нематод), HSF1 и NRF2 (через PGC 1α). AMPK
SIRT1 активирует фактор биогенеза митохондрии
(через ингибирование mTOR и активацию
(PGC 1α) и факторы стресс ответа (FOXO, HSF1)
БИОХИМИЯ том 86 вып. 3 2021
334
ИЛЬИНСКИЙ и др.
точных анаболических сигналов может быть ре
позволяет смягчить симптоматику нейродегене
ализовано в подавлении IIS (отрицательная об
ративных заболеваний [80].
ратная связь), что может приводить к инсулино
Ингибирование mTOR через IIS. При отсут
резистентности и развитию диабета 2 типа [77].
ствии сигналов инсулина/IGF1 происходит час
В результате инсулинорезистентности может
тичное ингибирование mTOR, подавляется
возникнуть гиперинсулинемия, усиливающая
трансляция, усиливается аутофагия и уменьша
окислительный стресс и накопление окислен
ется количество агрегатов белков. В результате
ных белков [78].
возрастные изменения протеома существенно
Таким образом, подавление mTOR является
менее выражены у долгоживущих DAF 2 мутан
важным терапевтическим вмешательством для
тов C. elegans, дефектных по IIS [2]. Однако не
поддержания протеостаза и продления жизни.
достаток ATP может привести к неэффектив
Рассмотрим возможности сохранения протео
ности аутофагии, поэтому одновременно усили
стаза через воздействия на метаболическую сиг
вается контролируемое малыми шаперонами
нальную систему клетки.
(ATP независимыми) образование телец вклю
Активация стресс ответа независимо от
чения из префибриллярных олигомеров. Так,
mTOR. Активация транскрипционных факторов
под контролем инсулинового каскада реализу
FOXO через ингибирование IIS (Akt не фосфо
ется отказоустойчивый защитный механизм
рилирует FOXO) и усиление работы SIRT1 (де
поддержания протеостаза [35].
ацетилирует FOXO) индуцирует стресс ответ, в
Ингибирование mTOR через SIRT1 и AMPK.
том числе активирует протеасомную деграда
При ингибировании хронического анаболизма
цию. Активация сиртуинов может быть достиг
и активации катаболизма происходит восста
нута добавлением NAD+ (или усилением его
новление активности инсулинового каскада,
синтеза через добавление никотинамидмоно
ликвидация гиперинсулинемии. Снижение
нуклеотида NMN и активацию никотинамид
окислительного стресса и активация аутофагии
фосфорибозилтрансферазы NAMPT), что спо
играют важную роль в восстановлении протеос
собствует сохранению протеостаза (деацетили
таза. Это обстоятельство позволяет рассматри
рование HSF1 и снижение уровня карбонилиро
вать применение сенсибилизаторов инсулина в
вания белков), усилению аутофагии и антиокси
качестве перспективных стратегий для восста
дантной защиты [79]. Другим независимым от
новления протеостаза. Было показано, что рес
mTOR способом усиления аутофагии является
вератрол (активатор SIRT1) [81] и метформин
влияние на сборку фагофора, например, через
(активатор AMPK, антидиабетическое средство)
подавление сигнального пути инозитола (ис
обладают нейропротекторными свойства
пользуя препараты лития и карбамазепин), что
ми [82].
Прямое ингибирование mTOR. Ингибирова
ние mTOR может производиться ограничением
калорий, а также фармакологически, при помо
щи рапамицина и ряда других веществ. Рапами
цин показал способность увеличивать продол
жительность жизни мышей, был применён для
усиления аутофагии в in vivo моделях болезней
Альцгеймера, Паркинсона, Гентингтона и дру
гих заболеваний. Предпринимаются меры по
снижению побочного действия рапамицина для
возможности его широкого применения [83].
Изменения генома и эпигенома как причина
нарушения протеостаза. ДНК претерпевает изме
нения с увеличением возраста организма. При
этом происходит нарушение стабильности гено
ма, изменение эпигенетического профиля ДНК
Рис. 4. Изменения генома и эпигенома как причина нару
и укорочение теломер (рис. 4).
шения протеостаза. Геномная нестабильность увеличивает
Рассмотрим, как изменения генома и эпиге
склонность белка к агрегации и его общую концентрацию.
Эпигенетические изменения усиливают окислительный
нома приводят к нарушению протеостаза.
стресс через подавление экспрессии генов ключевых ком
Генетическая нестабильность: мутации,
понентов митохондрий и системы поддержания протео
приводящие к нарушению протеостаза. Неста
стаза, способствуют повышению экспрессии белков,
бильность генома напрямую связана с возраст
склонных к агрегации. Укорочение теломер подавляет
фактор биогенеза митохондрий PGC 1α, способствуя
ными протеинопатиями, вызывая наследствен
окислительному стрессу
ные и приобретённые (в большинстве случаев)
БИОХИМИЯ том 86 вып. 3 2021
РОЛЬ СТАРЕНИЯ В РАЗВИТИИ ПРОТЕИНОПАТИЙ
335
формы болезней [3]. Система поддержания про
бильные белки отвлекают на себя компоненты
теостаза способствует исправлению или сглажи
системы поддержания протеостаза. В результате
ванию результатов ошибок даже при наличии
появляются белки (мутантные или неправильно
соответствующих мутаций в геноме. Нарушение
процессированные), приводящие к развитию
защитных механизмов с возрастом объясняет
нейродегенеративных заболеваний [25].
позднее проявление семейных форм протеино
Зачастую увеличение уровня экспрессии
патий [1]. Повреждение генома приводит к ус
выше критической концентрации (мутация в
коренному старению и ряду возрастных заболе
промоторе, амплификация гена), а не наличие
ваний [2], хотя не все мутации ДНК определяют
патогенных аминокислотных мотивов, служит
патологическое изменение белка [84].
причиной амилоидных заболеваний. Так, трип
Данные крупномасштабного секвенирова
ликация гена α синуклеина приводит к наслед
ния указывают на присутствие в человеческой
ственной форме болезни Паркинсона [90].
популяции большого количества однонуклео
Перспективным способом лечения нейро
тидных полиморфизмов (точечных замен) (англ.
дегенеративных заболеваний, вызванных мута
абб. SNP от single nucleotide polymorphism) в об
циями в отдельных генах, является антисмысло
ластях генома, кодирующих белок, которые
вая РНК интерференция (подавление трансля
должны влиять на его стабильность и сворачива
ции мРНК с мутацией) [3]. Ещё одним перспек
ние [85]. Мутации возникают из за особенностей
тивным подходом является увеличение внут
химической структуры ДНК, неидеальности и
риклеточной концентрации никотинамидаде
ослабления процессов репликации, рекомбина
ниндинуклеотида (NAD+), который необходим
ции, репарации при старении, длительной экс
для защиты от окислительного стресса и обеспе
позиции организма генотоксичным стрессовым
чения репарации ДНК [79].
факторам [2]. Особенно подвержен мутациям
Эпигенетические изменения как причина на
митохондриальный геном (см. раздел «Нефунк
рушения протеостаза. Существуют свидетель
циональные митохондрии»). Возрастное увели
ства об эпигенетическом подавлении с возрас
чение числа подвижных элементов генома (рет
том уровня экспрессии фактора транскрипции
робиом, таких как ретротранспозон LINE 1),
NRF2 (антиоксидантная защита) [91], шаперо
возникающее из за иммуносенесцентности [2],
нов (HSP70, что способствует развитию болезни
также может нарушить структуру генов.
Паркинсона [92]), белков аутофагии [93], проте
Нестабильность генома приводит к появле
аз, способных к деградации амилоидов [94],
нию белков, ассоциированных с нейродегенера
ключевых компонентов митохондрий, лизосом
тивными заболеваниями. Мутации, меняющие
и протеасом, что в совокупности способствует
рамку считывания гена (например, PTV - англ.
нарушению протеостаза [2]. Также возрастные
абб. от protein truncating variant), приводят к по
эпигенетические изменения приводят к повы
явлению дефектных безостановочных, укоро
шению экспрессии белков, склонных к агрега
ченных мРНК. Их трансляция приводит к бло
ции [95].
кировке рибосом полипептидными цепями,
Эпигенетическое репрограммирование спо
неспособными к сворачиванию, либо к получе
собствует восстановлению профиля экспрессии
нию усечённой версии белка с неправильным
генов, характерного для молодого организма.
сворачиванием [84]. Для противодействия рибо
Контролируемая экспрессия факторов Яманаки
сомальному стрессу существует механизм конт
достаточна для замедления старения модельных
роля качества рибосом (англ. абб. RQC от ribo
животных и клеточных культур, в том числе для
some quality control), нарушения которого связа
нормализации протеостаза путём активации
ны с возрастной нейродегенерацией [86].
протеасом и уменьшения окислительного стрес
Мутации, дестабилизирующие свёрнутое
са [96].
состояние белка или влияющие на кинетику его
Было показано, что обработка спермидином
сворачивания, приводят к повышению склон
вызывает ингибирование гистонацетилтрансфе
ности белков к агрегации [87]. Сайленс мутации
раз и деацетилирование гистона H3 (по лизинам
(при которых замены аминокислоты в синтези
K9, K14, K18), гиперацетилирование которого
руемом белке не происходит, меняется лишь нук
происходит с возрастом вследствие истощения
леотидная последовательность) могут также вли
полиаминов. Изменённый статус ацетилирова
ять на процесс трансляции и скорость сворачи
ния хроматина приводит у дрожжей, мух, червей
вания [88]. Мутации антикодона тРНК вызыва
и клеток человека к подавлению окислительно
ют замедление трансляции [89]. В результате за
го стресса, некроза, значительной активации
рождающаяся полипептидная цепь может занять
аутофагии [97].
неверно свёрнутую промежуточную конформа
Гиперэкспрессия SIRT6 через деацетилиро
цию, ведущую к агрегации белка. Такие метаста
вание лизина гистона H3K9 подавляет инсули
БИОХИМИЯ том 86 вып. 3 2021
336
ИЛЬИНСКИЙ и др.
новый каскад, способствует переходу на катабо
протеостазе из за усиления трансляции, подав
лизм и нормализации протеостаза, в том числе в
ления деградации белков и ослабления стресс
случае ожирения, вызванного диетой [2].
ответов. Возрастные изменения генома и эпиге
Теломеры и теломераза, стволовые клетки,
нома увеличивают склонность белков к агрега
сенесцентность. Истощение запасов нейрональ
ции (мутантные формы и перенасыщение бел
ных стволовых клеток, накопление сенесцент
ком), подавляют систему поддержания протеос
ных глиальных клеток, усугубляемое иммуносе
таза, усиливают окислительный стресс. Наруше
несцентностью, вносят важный вклад в патоге
ния протеостаза в совокупности с дисфункцией
нез болезней Альцгеймера и Паркинсона [98].
митохондрий, дисбалансом сигнальной систе
Дисфункция стволовых клеток во многом про
мы, изменениями генома и эпигенома вызывают
исходит из за нарушения активности теломера
истощение запасов стволовых клеток, что на ор
зы, сенесцентности клеток ниши, то есть из за
ганизменном уровне приводит к ослаблению ре
укорачивания теломер.
генерации нейронов и других клеток мозга.
Активация теломеразы улучшает общее со
В результате накопления нарушений и, воз
стояние организма и увеличивает продолжи
можно, согласно заложенной программе, насту
тельность жизни [2]. Обеспечение протеостаза с
пает дисбаланс появления и удаления агрегатов.
помощью теломеразы может проходить по двум
Их токсическое взаимодействие с мембранами и
механизмам. Во первых, при поддержании дли
включение важных белков в состав агрегатов уча
ны теломер не уменьшается экспрессия
щаются и приводят к развитию протеинопатий.
PGC 1α/β, что способствует рециркуляции ми
Таким образом, нарушение протеостаза и
тохондрий и снижению окислительного стресса
старение являются взаимосвязанными и взаи
[2]. Во вторых, неканоническое митохондри
моусиливающимися процессами, синергия ко
альное функционирование теломеразы снижает
торых приводит к катастрофическому, нелиней
окислительный стресс. Стоит отметить, что ог
ному во времени нарастанию дисфункций орга
раничение калорий снижает побочный эффект
низма. Для профилактики протеинопатий важ
активации теломеразы (риск развития рака) и,
ны воздействия, замедляющие старение - хоро
за счёт подавления Akt киназы, должно усили
шая экология, поддержание здорового образа
вать теломеразный антиоксидантный стресс от
жизни, иммунитета, развитие устойчивости к
вет (см. рис. 3) [99]. Баланс протеостаза способ
эмоциональным стрессам, лечение хронических
ствует активности теломеразы за счёт
заболеваний (таких как диабет). В целом, пока
TRiC/CCT опосредованной сборки функцио
занное воздействие старения на протеостаз
нальной теломеразы [100].
подтверждает необходимость поддержания го
меостаза организма для противодействия возра
стным заболеваниям и достижения здорового
ВЫВОДЫ
долголетия.
Белковая агрегация является результатом
Финансирование. Работа выполнена при фи
естественной нестабильности, действия протео
нансовой поддержке Российского фонда фунда
токсических стрессов на белки, особенно на те,
ментальных исследований в рамках научного
которые входят в систему поддержания протео
проекта № 19 14 50506.
стаза. Дисбаланс клеточных процессов и исто
Благодарности. Ильинский Н. С. выражает
щение ресурсов для восстановления, происхо
благодарность Министерству науки и высшего
дящие при старении, усиливают нарушения
образования Российской Федерации за под
протеостаза.
держку (соглашение № 075 00337 20 03, проект
Увеличивающаяся со старением дисфунк
FSMG 2020 0003).
ция митохондрий вызывает усиление действия
Конфликт интересов. Авторы заявляют об
внешнего окислительного стресса. При этом ра
отсутствии конфликта интересов.
бота систем репарации ДНК и защиты белков
Соблюдение этических норм. Настоящая
подавлена недостатком производства ATP. Пре
статья не содержит описания каких либо иссле
обладание анаболизма над катаболизмом, харак
дований с участием людей или животных в каче
терное для старения, негативно сказывается на
стве объектов.
СПИСОК ЛИТЕРАТУРЫ
1.
Taylor, R. C., and Dillin, A. (2011) Aging as an event of
2.
López Ot n, C., Blasco, M. A., Partridge, L., Serrano, M.,
proteostasis collapse, Cold Spring Harb. Perspect. Biol., 3,
and Kroemer, G. (2013) The hallmarks of aging, Cell, 153,
a004440, doi: 10.1101/cshperspect.a004440.
1194 1217, doi: 10.1016/j.cell.2013.05.039.
БИОХИМИЯ том 86 вып. 3 2021
РОЛЬ СТАРЕНИЯ В РАЗВИТИИ ПРОТЕИНОПАТИЙ
337
3.
Hipp, M. S., Kasturi, P., and Hartl, F. U. (2019) The pro
22.
Olzscha, H., Schermann, S. M., Woerner, A. C.,
teostasis network and its decline in ageing, Nat. Rev. Mol.
Pinkert, S., Hecht, M. H., et al. (2011) Amyloid like
Cell Biol., 20, 421 435, doi: 10.1038/s41580 019 0101 y.
aggregates sequester numerous metastable proteins with
4.
Onuchic, J. N., Luthey Schulten, Z., and Wolynes, P G.
essential cellular functions, Cell,
144,
6778,
(1997) Theory of protein folding: the energy landscape per
doi: 10.1016/j.cell.2010.11.050.
spective, Annu. Rev. Phys. Chem.,
48,
545600,
23.
Anguiano, M., Nowak, R. J., and Lansbury, P. T., Jr. (2002)
doi: 10.1146/annurev.physchem.48.1.545.
Protofibrillar islet amyloid polypeptide permeabilizes syn
5.
Hartl, F. U., Bracher, A., and Hayer Hartl, M. (2011)
thetic vesicles by a pore like mechanism that may be rele
Molecular chaperones in protein folding and proteostasis,
vant to type II diabetes, Biochemistry, 41, 11338 11343,
Nature, 475, 324 332, doi: 10.1038/nature10317.
doi: 10.1021/bi020314u.
6.
Chiti, F., and Dobson, C. M. (2009) Amyloid formation by
24.
Milanesi, L., Sheynis, T., Xue, W. F., Orlova, E. V.,
globular proteins under native conditions, Nat. Chem. Biol.,
Hellewell, A. L., et al. (2012) Direct three dimensional
5, 15 22, doi: 10.1038/nchembio.131.
visualization of membrane disruption by amyloid fibrils,
7.
Ellis, R. J., and Minton, A. P. (2006) Protein aggregation in
Proc. Natl. Acad. Sci. USA,
109,
2045520460,
crowded environments, Biol. Chem.,
387,
485497,
doi: 10.1073/pnas.1206325109.
doi: 10.1515/BC.2006.064.
25.
Lin, M. T., and Beal, M. F. (2006) Mitochondrial dysfunc
8.
Vendruscolo, M., Paci, E., Karplus, M., and Dobson,
tion and oxidative stress in neurodegenerative diseases,
C. M. (2003) Structures and relative free energies of par
Nature, 443, 787 795, doi: 10.1038/nature05292.
tially folded states of proteins, Proc. Natl. Acad. Sci. USA,
26.
Tsigelny, I. F., Crews, L., Desplats, P., Shaked, G. M.,
100, 14817 14821, doi: 10.1073/pnas.2036516100.
Sharikov, Y., et al. (2008) Mechanisms of hybrid oligomer
9.
Wright, C. F., Teichmann, S. A., Clarke, J., and Dobson,
formation in the pathogenesis of combined Alzheimer’s
C. M. (2005) The importance of sequence diversity in the
and Parkinson’s diseases, PLoS One,
3, e3135,
aggregation and evolution of proteins, Nature, 438, 878
doi: 10.1371/journal.pone.0003135.
881, doi: 10.1038/nature04195.
27.
Klaips, C. L., Jayaraj, G. G., and Hartl, F. U. (2018)
10.
Gusach, A., Luginina, A., Marin, E., Brouillette, R. L.,
Pathways of cellular proteostasis in aging and disease,
Besserer Offroy, É., et al. (2019) Structural basis of ligand
J. Cell Biol., 217, 51 63, doi: 10.1083/jcb.201709072.
selectivity and disease mutations in cysteinyl leukotriene
28.
Dikic, I. (2017) Proteasomal and autophagic degradation
receptors, Nat. Commun., 10, 5573, doi: 10.1038/s41467
systems, Annu. Rev. Biochem.,
86,
193224,
019 13348 2.
doi: 10.1146/annurev biochem 061516 044908.
11.
Walsh, G., and Jefferis, R. (2006) Post translational modi
29.
Tartaglia, G. G., Pechmann, S., Dobson, C. M., and
fications in the context of therapeutic proteins, Nat.
Vendruscolo, M. (2007) Life on the edge: a link between
Biotechnol., 24, 1241 1252, doi: 10.1038/nbt1252.
gene expression levels and aggregation rates of human pro
12.
Tokuriki, N., and Tawfik, D. S. (2009) Protein dynamism
teins,
Trends Biochem. Sci.,
32,
204206,
and evolvability, Science, 324, 203 207, doi: 10.1126/sci
doi: 10.1016/j.tibs.2007.03.005.
ence.1169375.
30.
Ciryam, P., Tartaglia, G. G., Morimoto, R. I., Dobson,
13.
Uversky, V. N., and Dunker, A. K. (2010) Understanding
C. M., and Vendruscolo, M. (2013) Widespread aggrega
protein non folding, Biochim. Biophys. Acta, 1804, 1231
tion and neurodegenerative diseases are associated with
1264, doi: 10.1016/j.bbapap.2010.01.017.
supersaturated proteins, Cell Rep.,
5,
781790,
14.
Uversky, V. N., and Fink, A. L. (2004) Conformational
doi: 10.1016/j.celrep.2013.09.043.
constraints for amyloid fibrillation: the importance of
31.
Yamamoto, A., Lucas, J. J., and Hen, R. (2000) Reversal of
being unfolded, Biochim. Biophys. Acta, 1698, 131 153,
neuropathology and motor dysfunction in a conditional
doi: 10.1016/j.bbapap.2003.12.008.
model of Huntington’s disease, Cell,
101,
5766,
15.
Lang, L., Kurnik, M., Danielsson, J., and Oliveberg, M.
doi: 10.1016/S0092 8674(00)80623 6.
(2012) Fibrillation precursor of superoxide dismutase 1
32.
Gomez Pastor, R., Burchfiel, E. T., and Thiele, D. J.
revealed by gradual tuning of the protein folding equilibri
(2018) Regulation of heat shock transcription factors and
um, Proc. Natl. Acad. Sci. USA, 109, 1786817873,
their roles in physiology and disease, Nat. Rev. Mol. Cell
doi: 10.1073/pnas.1201795109.
Biol., 19, 4 19, doi: 10.1038/nrm.2017.73.
16.
Lindner, A. B., and Demarez, A. (2009) Protein aggrega
33.
Behrends, C., Langer, C. A., Boteva, R., Böttcher, U. M.,
tion as a paradigm of aging, Biochim. Biophys. Acta, 1790,
Stemp, M. J., et al. (2006) Chaperonin TRiC promotes the
980 996, doi: 10.1016/j.bbagen.2009.06.005.
assembly of polyQ expansion proteins into nontoxic
17.
Coppedè, F., Mancuso, M., Siciliano, G., Migliore, L.,
oligomers, Mol. Cell, 23, 887 897, doi: 10.1016/j.mol
and Murri, L. (2006) Genes and the environment in neu
cel.2006.08.017.
rodegeneration,
Biosci.
Rep.,
26,
341367,
34.
Brehme, M., Voisine, C., Rolland, T., Wachi, S., Soper,
doi: 10.1007/s10540 006 9028 6.
J. H., et al. (2014) A chaperome subnetwork safeguards
18.
Golubev, A., Hanson, A. D., and Gladyshev, V. N. (2017)
proteostasis in aging and neurodegenerative disease, Cell
Non enzymatic molecular damage as a prototypic driver of
Rep., 9, 1135 1150, doi: 10.1016/j.celrep.2014.09.042.
aging, J. Biol. Chem., 292, 6029 6038, doi: 10.1074/
35.
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W.,
jbc.R116.751164.
and Dillin, A. (2006) Opposing activities protect against
19.
Horwitz, J. (1992) Alpha crystallin can function as a mol
age onset proteotoxicity, Science,
313,
16041610,
ecular chaperone, Proc. Natl. Acad. Sci. USA, 89, 10449
doi: 10.1126/science.1124646.
10453, doi: 10.1073/pnas.89.21.10449.
36.
Walker, G. A., and Lithgow, G. J. (2003) Lifespan exten
20.
Kim, Y. E., Hosp, F., Frottin, F., Ge, H., Mann, M., et al.
sion in C. elegans by a molecular chaperone dependent
(2016) Soluble oligomers of PolyQ expanded huntingtin
upon insulin like signals, Aging Cell,
2,
131139,
target a multiplicity of key cellular actors, Mol. Cell, 63,
doi: 10.1046/j.1474 9728.2003.00045.x.
951 964, doi: 10.1016/j.molcel.2016.07.022.
37.
Hetz, C., and Papa, F. P. (2018) The unfolded protein
21.
Guo, Q., Lehmer, C., Mart nez Sánchez, A., Rudack, T.,
response and cell fate control, Mol. Cell, 69, 169 181,
Beck, F., et al. (2018) In situ structure of neuronal C9orf72
doi: 10.1016/j.molcel.2017.06.017.
Poly GA aggregates reveals proteasome recruitment, Cell,
38.
Shpilka, T., and Haynes, C. M. (2018) The mitochondrial
172, 696 705.e12, doi: 10.1016/j.cell.2017.12.030.
UPR: mechanisms, physiological functions and implica
4 БИОХИМИЯ том 86 вып. 3 2021
338
ИЛЬИНСКИЙ и др.
tions in ageing, Nat. Rev. Mol. Cell Biol., 19, 109 120,
synuclein toxicity, Proc. Natl. Acad. Sci. USA, 110, E1817
doi: 10.1038/nrm.2017.110.
1826, doi: 10.1073/pnas.1305623110.
39.
Ruan, L., Zhou, C., Jin, E., Kucharavy, A., Zhang, Y.,
56.
Xilouri, M., Brekk, O. R., Landeck, N., Pitychoutis, P. M.,
et al. (2017) Cytosolic proteostasis through importing of
Papasilekas, T., et al. (2013) Boosting chaperone mediated
misfolded proteins into mitochondria, Nature, 543, 443
autophagy in vivo mitigates α synuclein induced neurode
446, doi: 10.1038/nature21695.
generation, Brain, 136, 2130 2146, doi: 10.1093/brain/
40.
Miller, S. B. M., Mogk, A., and Bukau, B. (2015) Spatially
awt131.
organized aggregation of misfolded proteins as cellular
57.
Leeman, D. S., Hebestreit, K., Ruetz, T., Webb, A. E.,
stress defense strategy, J. Mol. Biol., 427, 1564 1574,
McKay, A., et al. (2018) Lysosome activation clears aggre
doi: 10.1016/j.jmb.2015.02.006.
gates and enhances quiescent neural stem cell activation
41.
Moore, D. L., Pilz, G. A., Araúzo Bravo, M. J., Barral, Y.,
during aging, Science, 359, 1277 1283, doi: 10.1126/sci
and Jessberger, S. (2015) A mechanism for the segregation
ence.aag3048.
of age in mammalian neural stem cells, Science, 349, 1334
58.
Vilchez, D., Boyer, L., Morantte, I., Lutz, M.,
1338, doi: 10.1126/science.aac9868.
Merkwirth, C., et al. (2012) Increased proteasome activity
42.
Wyatt, A. R., Yerbury, J. J., Ecroyd, H., and Wilson, M. R.
in human embryonic stem cells is regulated by PSMD11,
(2013) Extracellular chaperones and proteostasis, Annu.
Nature, 489, 304 308, doi: 10.1038/nature11468.
Rev. Biochem.,
82,
295322, doi:
10.1146/annurev
59.
Noormohammadi, A., Khodakarami, A., Gutierrez
biochem 072711 163904.
Garcia, R., Lee, H. J., Koyuncu, S., et al. (2016) Somatic
43.
Wilkinson, B., and Gilbert, H. F. (2004) Protein disulfide
increase of CCT8 mimics proteostasis of human pluripo
isomerase, Biochim. Biophys. Acta,
1699,
3544,
tent stem cells and extends C. elegans lifespan, Nat.
doi: 10.1016/j.bbapap.2004.02.017.
Commun., 7, 13649, doi: 10.1038/ncomms13649.
44.
Lu, K. P., Finn, G., Lee, T. H., and Nicholson, L. K.
60.
Shelkovnikova, T. A., Kulikova, A. A., Tsvetkov, F. O.,
(2007) Prolyl cis trans isomerization as a molecular timer,
Peters, O., Bachurin, S. O., et al. (2012) Proteinopathies,
Nat. Chem. Biol., 3, 619629, doi: 10.1038/nchem
neurodegenerative disorders with protein aggregation
bio.2007.35.
based pathology, Mol. Biol., 46, 362 374.
45.
Min, J. N., Whaley, R. A., Sharpless, N. E., Lockyer, P.,
61.
Angelova, P. R., and Abramov, A. Y. (2017) Alpha synucle
Portbury, A. L., and Patterson, C. (2008) CHIP deficiency
in and beta amyloid - different targets, same players: cal
decreases longevity, with accelerated aging phenotypes
cium, free radicals and mitochondria in the mechanism of
accompanied by altered protein quality control, Mol. Cell.
neurodegeneration, Biochem. Biophys. Res. Commun., 483,
Biol., 28, 4018 4025, doi: 10.1128/mcb.00296 08.
1110 1115, doi: 10.1016/j.bbrc.2016.07.103.
46.
Calamini, B., Silva, M. C., Madoux, F., Hutt, D. M.,
62.
Ludtmann, M. H. R., Angelova, P. R., Horrocks, M. H.,
Khanna, S., et al. (2012) Small molecule proteostasis reg
Choi, M. L., Rodrigues, M., et al. (2018) α synuclein
ulators for protein conformational diseases, Nat. Chem.
oligomers interact with ATP synthase and open the perme
Biol., 8, 185 196, doi: 10.1038/nchembio.763.
ability transition pore in Parkinson’s disease, Nat.
47.
Walther, D. M., Kasturi, P., Zheng, M., Pinkert, S.,
Commun., 9, 2293, doi: 10.1038/s41467 018 04422 2.
Vecchi, G., et al. (2015) Widespread proteome remodeling
63.
Snyder, H., Mensah, K., Theisler, C., Lee, J.,
and aggregation in aging C. elegans, Cell, 161, 919 932,
Matouschek, A., and Wolozin, B. (2003) Aggregated and
doi: 10.1016/j.cell.2015.03.032.
monomeric alpha synuclein bind to the S6′ proteasomal
48.
Moreno, J. A., Halliday, M., Molloy, C., Radford, H.,
protein and inhibit proteasomal function, J. Biol. Chem.,
Verity, N., et al. (2013) Oral treatment targeting the unfold
278, 11753 11759, doi: 10.1074/jbc.M208641200.
ed protein response prevents neurodegeneration and clini
64.
Orr, A. L., Li, S., Wang, C. E., Li, H., Wang, J., et al.
cal disease in prion infected mice, Sci. Transl. Med., 5,
(2008) N terminal mutant huntingtin associates with mito
206ra138, doi: 10.1126/scitranslmed.3006767.
chondria and impairs mitochondrial trafficking,
49.
Alberti, S., and Hyman, A. A. (2016) Are aberrant phase
J. Neurosci.,
28,
27832792, doi:
10.1523/JNEU
transitions a driver of cellular aging? Bioessays, 38, 959
ROSCI.0106 08.2008.
968, doi: 10.1002/bies.201600042.
65.
Brundin, P., Melki, R., and Kopito, R. (2010) Prion like
50.
Esser, C., Alberti, S., and Höhfeld, J. (2004) Cooperation
transmission of protein aggregates in neurodegenerative
of molecular chaperones with the ubiquitin/proteasome
diseases, Nat. Rev. Mol. Cell Biol.,
11,
301307,
system, Biochim. Biophys. Acta,
1695,
171188,
doi: 10.1038/nrm2873.
doi: 10.1016/j.bbamcr.2004.09.020.
66.
Ozawa, T. (1997) Genetic and functional changes in mito
51.
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F.,
chondria associated with aging, Physiol. Rev., 77, 425 464,
Andrews, S. P., et al. (2017) Autophagy and neurodegener
doi: 10.1152/physrev.1997.77.2.425.
ation: pathogenic mechanisms and therapeutic opportuni
67.
Lee, H. C., Yin, P. H., Chi, C. W., and Wei, Y. H. (2002)
ties, Neuron,
93,
10151034, doi:
10.1016/j.neu
Increase in mitochondrial mass in human fibroblasts under
ron.2017.01.022.
oxidative stress and during replicative cell senescence,
52.
Kitamura, A., Inada, N., Kubota, H., Matsumoto, G.,
J. Biomed. Sci., 9, 517 526, doi: 10.1007/BF02254978.
Kinjo, M., et al. (2014) Dysregulation of the proteasome
68.
Bernhardt, D., Müller, M., Reichert, A. S., and Osiewacz,
increases the toxicity of ALS linked mutant SOD1, Genes
H. D. (2015) Simultaneous impairment of mitochondrial
Cells, 19, 209 224, doi: 10.1111/gtc.12125.
fission and fusion reduces mitophagy and shortens replica
53.
Parkitko, A. A., Favorova, O. O., and Henske, E. P. (2013)
tive lifespan, Sci. Rep., 5, 7885, doi: 10.1038/srep07885.
Autophagy: mechansms, regulation, and its role in tumori
69.
Labbadia, J., Brielmann, R. M., Neto, M. F., Lin, Y. F.,
genesis, Biochemistry (Moscow), 78, 355 367.
Haynes, C. M., and Morimoto, R. I. (2017) Mitochondrial
54.
Carmona Gutierrez, D., Hughes, A. L., Madeo, F., and
stress restores the heat shock response and prevents pro
Ruckenstuhl, C. (2016) The crucial impact of lysosomes in
teostasis collapse during aging, Cell Rep., 21, 1481 1494,
aging and longevity, Ageing Res. Rev.,
32,
2 12,
doi: 10.1016/j.celrep.2017.10.038.
doi: 10.1016/j.arr.2016.04.009.
70.
Thomas, H. E., Zhang, Y., Stefely, J. A., Veiga, S. R.,
55.
Decressac, M., Mattsson, B., Weikop, P., Lundblad, M.,
Thomas, G., et al. (2018) Mitochondrial complex I activi
Jakobsson, J., and Björklund, A. (2013) TFEB mediated
ty is required for maximal autophagy, Cell Rep., 24, 2404
autophagy rescues midbrain dopamine neurons from α
2417.e8, doi: 10.1016/j.celrep.2018.07.101.
БИОХИМИЯ том 86 вып. 3 2021
РОЛЬ СТАРЕНИЯ В РАЗВИТИИ ПРОТЕИНОПАТИЙ
339
71.
Brunk, U. T., and Terman, A. (2002) The mitochondrial
86. Choe, Y. J., Park, S. H., Hassemer, T., Körner, R.,
lysosomal axis theory of aging: accumulation of damaged
Vincenz Donnelly, L., et al. (2016) Failure of RQC
mitochondria as a result of imperfect autophagocytosis,
machinery causes protein aggregation and proteotoxic
Eur. J. Biochem., 269, 1996 2002, doi: 10.1046/j.1432
stress, Nature, 531, 191 195, doi: 10.1038/nature16973.
1033.2002.02869.x.
87. Boucher, J. I., Bolon, D. N. A., and Tawfik, D. S. (2016)
72.
Park, J. T., Lee, Y. S., Cho, K. A., and Park, S. C. (2018)
Quantifying and understanding the fitness effects of protein
Adjustment of the lysosomal mitochondrial axis for con
mutations: laboratory versus nature, Protein Sci., 25, 1219
trol of cellular senescence, Ageing Res. Rev., 47, 176 182,
1226, doi: 10.1002/pro.2928.
doi: 10.1016/j.arr.2018.08.003.
88. Kimchi Sarfaty, C., Oh, J. M., Kim, I. W., Sauna, Z. E.,
73.
Nesterov, S. V., Yaguzhinsky, L. S., Podoprigora, G. I., and
Calcagno, A. M., et al. (2007) A “silent” polymorphism in
Nartsissov, Ya. R. (2018) Autocatalytic cycle in the patho
the MDR1 gene changes substrate specificity, Science, 315,
genesis of diabetes mellitus: biochemical and pathophysio
525 528, doi: 10.1126/science.1135308.
logical aspects of metabolic therapy with natural amino
89. Nedialkova, D. D., and Leidel, S. A. (2015) Optimization
acids in the example of glycine, Diabetes Mellitus, 21, 283
of codon translation rates via tRNA modifications main
292, doi: 10.14341/DM9529.
tains proteome integrity, Cell,
161,
16061618,
74.
Korovila, I., Hugo, M., Castro, J. P., Weber, D., Höhn, A.,
doi: 10.1016/j.cell.2015.05.022.
et al. (2017) Proteostasis, oxidative stress and aging, Redox
90. Singleton, A. B., Farrer, M., Johnson, J., Singleton, A.,
Biol., 13, 550 567, doi: 10.1016/j.redox.2017.07.008.
Hague, S., et al. (2003) alpha synuclein locus triplication
75.
Wrobel, L., Topf, U., Bragoszewski, P., Wiese, S.,
causes Parkinson’s disease, Science,
302,
841,
Sztolsztener, M. E., et al. (2015) Mistargeted mitochondr
doi: 10.1126/science.1090278.
ial proteins activate a proteostatic response in the cytosol,
91. Silva Palacios, A., Ostolga Chavarr a, M., Zazueta, C.,
Nature, 524, 485 488, doi: 10.1038/nature14951.
and Königsberg, M. (2018) Nrf2: Molecular and epigenet
76.
Nesterov, S. V., Yaguzhinsky, L. S., Podoprigora, G. I., and
ic regulation during aging, Ageing Res. Rev., 47, 31 40,
Nartsissov, Ya. R. (2020) Amino acids as regulators of cell
doi: 10.1016/j.arr.2018.06.003.
metabolism, Biochemistry (Moscow),
85,
393408,
92. Harries, L. W. (2014) MicroRNAs as mediators of the age
doi: 10.1134/S000629792004001X.
ing process, Genes (Basel), 5, 656 670, doi: 10.3390/
77.
Özcan, U., Cao, Q., Yilmaz, E., Lee, A. H., Iwakoshi,
genes5030656.
N. N., et al. (2004) Endoplasmic reticulum stress links
93. Lapierre, L. R., Kumsta, C., Sandri, M., Ballabio, A., and
obesity, insulin action, and type 2 diabetes, Science, 306,
Hansen, M. (2015) Transcriptional and epigenetic regula
457 461, doi: 10.1126/science.1103160.
tion of autophagy in aging, Autophagy, 11, 867880,
78.
Anisimov, V. N. (2003) Insulin/IGF 1 signaling pathway
doi: 10.1080/15548627.2015.1034410.
driving aging and cancer as a target for pharmacological
94. Nalivaeva, N. N., Belyaev, N. D., Kerridge, C., and
intervention, Exp. Gerontol., 38, 1041 1049, doi: 10.1016/
Turner, A. J. (2014) Amyloid clearing proteins and their
s0531 5565(03)00169 4.
epigenetic regulation as a therapeutic target in Alzheimer’s
79.
Oka, S. I., Hsu, C. P., and Sadoshima, J.
(2012)
disease, Front. Aging Neurosci.,
6,
235, doi:
Regulation of cell survival and death by pyridine
10.3389/fnagi.2014.00235.
nucleotides, Circ. Res., 111, 611 627, doi: 10.1161/CIR
95. Konsoula, Z., and Barile, F. A. (2012) Epigenetic histone
CRESAHA.111.247932.
acetylation and deacetylation mechanisms in experimental
80.
Sarkar, S. (2013) Regulation of autophagy by mTOR
models of neurodegenerative disorders, J. Pharmacol.
dependent and mTOR independent pathways: autophagy
Toxicol. Methods, 66, 215220, doi: 10.1016/j.vascn.
dysfunction in neurodegenerative diseases and therapeutic
2012.08.001.
application of autophagy enhancers, Biochem. Soc. Trans.,
96. Sarkar, T. J., Quarta, M., Mukherjee, S., Colville, A.,
41, 1103 1130, doi: 10.1042/BST20130134.
Paine, P., et al. (2020) Transient non integrative expression
81.
Wu, Y., Li, X., Zhu, J. X., Xie, W., Le, W., et al. (2011)
of nuclear reprogramming factors promotes multifaceted
Resveratrol activated AMPK/SIRT1/autophagy in cellu
amelioration of aging in human cells, Nat. Commun., 11,
lar models of Parkinson’s disease, Neurosignals, 19, 163
1545, doi: 10.1038/s41467 020 15174 3.
174, doi: 10.1159/000328516.
97. Eisenberg, T., Knauer, H., Schauer, A., Büttner, S.,
82.
Anisimov, V. N., Semenchenko, A. V., and Yashin, A. I.
Ruckenstuhl, C., et al. (2009) Induction of autophagy by
(2003) Insulin and longevity: antidiabetic biguanides as
spermidine promotes longevity, Nat. Cell Biol., 11, 1305
geroprotectors, Biogerontology, 4, 297 307, doi: 10.1023/
1314, doi: 10.1038/ncb1975.
a:1026299318315.
98. Bussian, T. J., Aziz, A., Meyer, C. F., Swenson, B. L., van
83.
Martinez Vicente, M. (2015) Autophagy in neurodegener
Deursen, J. M., and Baker, D. J. (2018) Clearance of
ative diseases: from pathogenic dysfunction to therapeutic
senescent glial cells prevents tau dependent pathology and
modulation, Semin. Cell Dev. Biol.,
40,
115126,
cognitive
decline,
Nature,
562,
578582,
doi: 10.1016/j.semcdb.2015.03.005.
doi: 10.1038/s41586 018 0543 y.
84.
Kosmicki, J. A., Samocha, K. E., Howrigan, D. P.,
99. Rosen, J., Jakobs, P., Ale Agha, N., Altschmied, J., and
Sanders, S. J., Slowikowski, K., et al. (2017) Refining the
Haendeler, J. (2020) Non canonical functions of telom
role of de novo protein truncating variants in neurodevel
erase reverse transcriptase - impact on redox homeostasis,
opmental disorders by using population reference samples,
Redox Biol., 34, 101543, doi: 10.1016/j.redox.2020.101543.
Nat. Genet., 49, 504 510, doi: 10.1038/ng.3789.
100. Freund, A., Zhong, F. L., Venteicher, A. S., Meng, Z.,
85.
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K.
Veenstra, T. D., et al. (2014) Proteostatic control of
E., Banks, E., et al. (2016) Analysis of protein coding
telomerase function through TRiC mediated folding of
genetic variation in 60,706 humans, Nature, 536, 285 291,
TCAB1, Cell, 159, 1389 1403, doi: 10.1016/j.cell.2014.
doi: 10.1038/nature19057.
10.059.
БИОХИМИЯ том 86 вып. 3 2021
4*
340
ИЛЬИНСКИЙ и др.
ON THE ROLE OF NORMAL AGING PROCESSES IN THE EMERGENCE
AND PATHOGENESIS OF THE DISEASES ASSOCIATED
WITH THE ABNORMAL ACCUMULATION OF PROTEIN AGGREGATES
Review
N. S. Ilyinsky1*, S. V. Nesterov1,2, E. I. Shestoperova1,
A. V. Fonin1,3, V. N. Uversky1,4, and V. I. Gordeliy1,5,6
1 Research Center for Molecular Mechanisms of Aging and Age#related Diseases,
Moscow Institute of Physics and Technology, 141701 Dolgoprudny,
Moscow Region, Russia; E#mail: ilinsky_nick@mail.ru
2 Institute of Cytochemistry and Molecular Pharmacology, 115404 Moscow, Russia
3 Institute of Cytology, Russian Academy of Sciences, 194064 Saint#Petersburg, Russia
4 Morsani College of Medicine, University of South Florida, 33612 Tampa, USA
5 Forschungszentrum Juelich, 52428 Juelich, Germany
6 Institut de biologie structurale, 38000 Grenoble, France
Aging is a prime systemic cause of various age related diseases, in particular, proteinopathies. In fact, most diseases
associated with protein misfolding are sporadic, and their incidence increases with aging. This review examines the
process of protein aggregate formation, the toxicity of such aggregates, the organization of cellular systems involved
in proteostasis, and the impact of protein aggregates on important cellular processes leading to proteinopathies. We
also analyze how manifestations of aging (mitochondrial dysfunction, dysfunction of signaling systems, changes in the
genome and epigenome) facilitate pathogenesis of various proteinopathies either directly, by increasing the propensi
ty of key proteins for aggregation, or indirectly, through dysregulation of stress responses. Such analysis might help in
outlining approaches for treating proteinopathies and extending healthy longevity.
Keywords: proteostasis, protein aggregation, proteinopathy, aging, mitochondrial dysfunction, mutations, epigenetic
changes
БИОХИМИЯ том 86 вып. 3 2021