БИОХИМИЯ, 2021, том 86, вып. 5, с. 643 - 666
УДК 577.25;577.151;616.89 008.48
СИГНАЛЬНЫЕ КАСКАДЫ БЕЛКОВОГО ФОСФОРИЛИРОВАНИЯ
ПРИ АУТИЗМЕ (С АКЦЕНТОМ НА ПУТЬ mTOR)
Обзор
© 2021
И.С. Бокша1,2*, Т.А. Прохорова1, Е.Б. Терешкина1,
О.К. Савушкина1, Г.Ш. Бурбаева1
1 ФГБНУ «Научный центр психического здоровья», 115522 Москва, Россия; электронная почта: boksha_irina@mail.ru
2 Национальный исследовательский центр эпидемиологии и микробиологии имени Н. Ф. Гамалеи
Минздрава России, 123098 Москва, Россия; электронная почта: boksha_irina@gamaleya.org
Поступила в редакцию 18.12.2020
После доработки 18.03.2021
Принята к публикации 22.03.2021
Сигнальный каскад протеинкиназы mTOR является центральным регулятором клеточного метаболизма,
роста и выживания клеток в ответ на гормоны, факторы роста, питательные вещества и сигналы, вызван!
ные стрессом. Цель обзора: на основе анализа современной литературы продемонстрировать, как часто
встречаются молекулярные аномалии сигнального пути mTOR и сопряжённых с ним сигнальных путей
при расстройствах аутистического спектра (РАС), а также наметить возможные перспективы патогенети!
чески!ориентированного фармакотерапевтического воздействия, особенно при синдромных РАС. На ос!
нове анализа экспериментальных и клинических данных предполагается, что наиболее раннему выявле!
нию молекулярных аномалий в группе риска РАС могут способствовать исследования тромбоцитов пери!
ферической крови. Поиск в этой группе временнóго «окна», в котором происходят критические наруше!
ния регуляции активности описанных путей, может привести к более эффективной превентивной фарма!
котерапии аутизма.
КЛЮЧЕВЫЕ СЛОВА: mTOR, сигнальные каскады, фосфорилирование белков, расстройства аутистичес!
кого спектра.
DOI: 10.31857/S0320972521050031
ВВЕДЕНИЕ
расстройство, в общем случае характеризующе!
еся такими ключевыми симптомами, как каче!
Аутизм, или расстройство аутистического
ственное нарушение социального взаимодей!
спектра (РАС) - это нейроонтогенетическое ствия, коммуникации и ограниченное стерео!
Принятые сокращения: AMPK - АМP!активируемая протеинкиназа; APP - белок!предшественник бета!амилои!
да; КТС - комплекс туберозного склероза; MAPK - митоген!активируемая протеинкиназа (синоним ERK); MeCP2 - ме!
тил!CpG!связывающий белок 2; MEK - киназа митоген!активируемых протеинкиназ (MAPK/ERK); НФ1 - нейрофиб!
роматоз первого типа; ППСК - плюрипотентные перепрограммированные стволовые клетки; РАС - расстройство аутис!
тического спектра; И!РАС - идиопатические РАС; НС!РАС - несиндромные РАС; С!РАС - синдромные формы РАС;
СА - синдром Ангельмана; СР - синдром Ретта; ФМС - синдром Фелан-МакДермид; X!ФРА - синдром ломкой («фра!
гильной») Х!хромосомы; Akt - RAC!alpha серин!треониновая протеинкиназа; Arc - ассоциированный с цитоскелетом
белок; BDNF - нейротрофический фактор мозга; CDKL5 - киназа, подобная циклинзависимой киназе 5; eIF4 - эукарио!
тический фактор инициации трансляции 4; E/I - соотношение возбуждения/торможения; ERK - активируемая внекле!
точными сигналами киназа (синоним MAPK); 4E!BP1 - eIF4E!связывающий белок 1; FMRP - белок, ассоциируемый с
синдромом ломкой X!хромосомы; IGF!1 и IGF!2 - инсулиноподобные факторы роста; LTP - долговременная (длитель!
ная) потенциация; mGluR, mGluR1 и mGluR5 - метаботропный глутаматный рецептор и его подтипы 1 и 5; mLST8 - ас!
социированный с mTOR белковый гомолог LST8 (mammalian lethal with Sec13 protein 8); MNK - взаимодействующая с
MAPK киназа; mTOR - протеинкиназа, механистическая мишень рапамицина у млекопитающих; NF1 - белок нейро!
фибромин; NGF - фактор роста нервов; NMDA - N!метил!D!аспартат; PI3K - фосфатидилинозитол!3!киназа; PIP3 -
фосфатидилинозитол!3,4,5!трифосфат; PTEN!РАС - синдром, связанный с мутацией гомолога фосфатазы и тензина;
pAktSer473 - Akt, фосфорилированная по Ser473; p70S6K1 - киназа 1 рибосомного белка S6; Ras - семейство малых ГТФаз,
участвующих в передаче сигнала в животных клетках (от Rat sarcoma); Rheb - гомолог Ras в мозге; SGK1 - индуцируемая
сывороткой и глюкокортикоидами киназа 1; SHANK3 - содержащий SH3!домен и множественные анкириновые повто!
ры белок 3; TSC1 и TSC2 - белки комплекса туберозного склероза (гамартин и туберин); UBE3A - убиквитин!протеино!
вая лигаза E3A; Wnt - группа путей передачи сигналов, начинающихся от белков, передающих сигналы в клетку от рецеп!
торов на её поверхности.
* Адресат для корреспонденции.
643
644
БОКША и др.
типное поведение. РАС чрезвычайно гетероген!
компонентов «выше по течению», так и его ми!
ны с точки зрения их генетической основы и,
шеней («ниже по течению» - факторы белковой
соответственно, этиологии и патогенеза. Во
трансляции) [5]. В обзоре оценены результаты
всём мире диагностика и классификация РАС в
исследований сигнального каскада mTOR и
настоящее время основана на клинических наб!
сопряженных с ним биохимических путей - как
людениях (оценке тяжести симптомов психиат!
с точки зрения понимания патогенеза РАС, так
рами), в случае синдромных форм РАС выявить
и с точки зрения поиска препаратов, действую!
и идентифицировать их причину помогают ме!
щих на компоненты этого каскада и сопряжён!
дико!генетические анализы. Недавно в Рос!
ные с ним пути (его мишени), а также намечены
сийской Федерации благодаря иммунологичес!
пути исследований, которые авторы считают
ким и электрофизиологическим исследованиям
перспективными.
особенностей пациентов с разными формами
РАС появились возможности практического
применения выявленных «биомаркёров» в под!
СИГНАЛЬНЫЙ ПУТЬ mTOR -
держку клиническим диагностическим крите!
КОНВЕРГЕНЦИЯ СИНДРОМНЫХ РАС
риям РАС [1, 2]. Достижения в области генетики
И НЕ АССОЦИИРОВАННЫХ
аутизма только вступают в фазу перехода к прак!
С СИНДРОМАМИ ФОРМ РАС
тическому клиническому применению [3, 4].
За исключением синдромных форм РАС (С!
Известно, что сигнальный каскад mTOR
РАС), когда аутистическое поведение связано с
функционирует как интегратор молекулярных
определенным медицинским синдромом, обус!
систем по поддержанию взаимодействия орга!
ловленным известной генетической (хромосом!
низма и клеток с окружающей средой. Путь
ной) аномалией, в большинстве случаев генети!
mTOR регулирует гомеостаз, влияя на синтез
ческие и иные причины, обусловливающие фе!
белка, транскрипцию, метаболизм, процессы
нотипическое проявление аутистических симп!
аутофагии, биогенеза органелл, участвует в про!
томов, остаются неясными и требуют дорого!
цессах, поддерживающих иерархическую целост!
стоящего анализа генома, а для разработки «био!
ность функционирования мозга, включая диф!
маркёров» требуются дополнительные биохи!
ференцировку и пролиферацию клеток нервной
мические исследования.
системы, организацию и поддержание проводя!
Ввиду отсутствия как ясного понимания
щих путей, обеспечивающих пластичность, свя!
этиологии, патогенеза, так и классифика!
занную с обучением, и регуляцию сложного по!
ции РАС (особенно расстройств, не относящих!
ведения [6]. Поэтому дисфункцию mTOR мож!
ся к С!РАС) с биохимической точки зрения,
но рассматривать как основополагающую при!
фармакологическое воздействие сводится в
чину ряда моногенных расстройств и как важ!
большинстве непсихотических случаев к под!
ный патогенетический фактор различных нев!
держке ноотропами, а в случае психотических
рологических, нейродегенеративных и психи!
форм - к симптоматическому лечению анти!
ческих расстройств [7]. Далее будет оценено
психотиками [1, 2], хотя, безусловно, необходи!
состояние исследований mTOR при РАС.
ма разработка препаратов, воздействующих на
уже наметившиеся, открытые патогенетические
пути.
СИНДРОМНЫЕ, НЕСИНДРОМНЫЕ
Написание данного обзора вызвано необхо!
И ИДИОПАТИЧЕСКИЕ РАССТРОЙСТВА
димостью обобщения современных результатов
АУТИСТИЧЕСКОГО СПЕКТРА
исследования регуляторных сигнальных каска!
дов белкового фосфорилирования при РАС,
Фенотипические (клинические) проявления
поскольку в этом направлении достигнут опре!
патологии нервной системы, выражающиеся
деленный прогресс в изучении вклада этих пу!
как аутизм, или аутистические симптомы, силь!
тей в развитие аутистических симптомов при С!
но варьируют по выраженности и степени тя!
РАС и ведется поиск фармакологических препа!
жести. Такая вариабельность определяется раз!
ратов.
личным вкладом в развитие патологии как гене!
Обзор сфокусирован на каскаде mTOR, по!
тических нарушений, так и факторов воздей!
скольку из всех систем белкового фосфорили!
ствия внешней среды [8].
рования этот путь наиболее исследован при
В настоящее время принято разделение по
РАС, его можно считать центральным, и к нему
этиологии РАС на синдромные РАС (С!РАС),
«сходятся» данные, полученные при изучении
которые связаны с описанными клиническими
С!РАС и свидетельствующие о том, что при РАС
синдромами и обусловлены открытыми генети!
изменена как активность его регуляторов, т.е.
ческими аномалиями (так называемые генети!
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
645
ческие заболевания), несиндромные РАС (НС!
тизма фонда Simons (Simons Foundation Autism
РАС), не связанные с этими синдромами, но
обусловленные также выявленными генетичес!
щён и опубликован список генов, ассоцииро!
кими причинами, и идиопатические РАС (И!
ванных с риском РАС, ранжированный по четы!
РАС), генетические и другие причины которых
рём категориям - S (С!РАС) и 1-3: в катего!
не установлены [9]. В качестве возможных при!
рии S - гены, ассоциированные с С!РАС, в ка!
чин развития И!РАС рассматриваются прена!
тегории 1 - гены с подтверждённой ассоциаци!
тальные воздействия (например, влияние на
ей с РАС, в категории 2 - «сильные кандидаты»,
развитие плода токсинов, некоторых лекар!
в категории 3 - «предположительно гены!кан!
ственных препаратов, инфекции) и постнаталь!
дидаты». Список составлен на основе частоты
ные факторы риска нарушения развития нерв!
встречаемости сообщений об исследованиях в
ной системы (например, социальная и/или сен!
литературе и воспроизводимости/подтвержден!
сорная депривация), а также пока не выявлен!
ности данных. Но ни одна мутация в этих генах
ные генетические и/или эпигенетические фак!
не обусловливает более чем 1-2% случаев НС!
торы [8].
РАС, а все вместе формы НС!РАС, обусловлен!
С генетической и биохимической точек зре!
ные мутациями в высоко пенетрантных генах,
ния С!РАС изучены лучше, чем НС!РАС, и зна!
представляют около 10-20% всех случаев, что
чительно лучше, чем И!РАС [4].
свидетельствует о крайне высокой степени гене!
К С!РАС относят заболевания с генетически
тической гетерогенности НС!РАС.
установленными причинами (генетические бо!
Несмотря на высокую степень клинической
лезни), при которых наблюдаются симптомы
и генетической гетерогенности, описаны неко!
аутизма: редкое моногенное заболевание -
торые механизмы, общие для С!РАС и НС!РАС,
комплекс туберозного склероза (КТС) и КТС!
и наблюдается конвергенция в нескольких гене!
ассоциированная мегалэнцефалия и симптома!
тических мутациях, сходящихся к ключевым био!
тический эпилептический синдром, синдром
химическим путям. Так, по данным широко!
ломкой («фрагильной») Х!хромосомы (X!ФРА),
масштабных геномных и транскриптомных ис!
синдром Ретта (СР), синдром Ангельмана (СА),
следований, с риском РАС связаны сигнальные
синдром, связанный с аномалией гомолога фос!
каскады белкового фосфорилирования с учас!
фатазы и тензина (PTEN!РАС), нейрофиброма!
тием фосфатидилинозитол!3!киназы (PI3K),
тоз первого типа (НФ1), синдром Тимоти, Уль!
RAC!alpha серин!треониновой протеинкина!
риха-Нунан, синдром делеции 22q13.3 и ряд
зы (Akt) и протеинкиназы mTOR, т.е. пути
других относительно редких расстройств нейро!
PI3K/Akt/mTOR, а также сигнальный каскад
онтогенеза, покрываемых спектром РАС.
фактора роста нервов (NGF), нейротрофичес!
Хотя С!РАС охватывают лишь 5-10% всех
кого фактора мозга (BDNF), MAPK/ERK, сиг!
случаев РАС, их изучение внесло большой вклад
нальный путь Wnt (группа путей передачи сиг!
в понимание молекулярных основ возникнове!
налов, начинающихся от белков, передающих
ния аутистических симптомов. С другой сторо!
сигналы в клетку от рецепторов на ее поверх!
ны, в случаях РАС, не связанных с перечислен!
ности), а также Ca2+!кальмодулина [15].
ными С!РАС, разнообразные (в частности, био!
Сигнальный путь Wnt вносит вклад в клеточ!
химические) причины развития аутизма остают!
ную дифференцировку, полярность, пролифе!
ся неясными, и за прошедшие 15 лет с момента
рацию клеток, и нарушение его регуляции ведёт
публикации обзора на эту тему прогресс наме!
к развитию определенных типов РАС. Сигналь!
тился лишь в недавнее время [10, 11].
ные каскады MAPK/ERK связаны более чем с
Оценки вклада генетического компонента в
20 функциональными путями и 22 генами, ассо!
развитие НС!РАС и И!РАС варьируют от 40
циированными с РАС [16]. Активация NGF!ин!
до 90%, в зависимости от принимаемых во вни!
дуцируемого сигнального пути ведёт к сущест!
мание параметров [3, 12-14].
венному снижению концентрации белка 4E!
Геномные технологии вкупе с обследовани!
BP1 и фактора инициации трансляции eIF4E и
ем больших когорт пациентов позволили вы!
связана с развитием окислительного стресса у
явить обилие редких вариантов, имеющих отно!
пациентов с РАС. Со степенью окислительного
шение к развитию РАС, причём связанными
стресса при РАС также связана активность сиг!
с НС!РАС оказались и несколько относительно
нального каскада белкового фосфорилирования
редких мутаций de novo. На интернет!ресурсах
mTOR, которому в данном обзоре уделено осо!
рабочей группы по аутизму Психиатрического
бое внимание.
Сигнальный каскад mTOR - один из цент!
unc.edu/pgc/pgc!workgroups/autism!spectrum!
ральных биохимических путей конвергенции
disorder/) и Инициативы по исследованию ау!
при разных РАС, он служит критическим регу!
БИОХИМИЯ том 86 вып. 5 2021
646
БОКША и др.
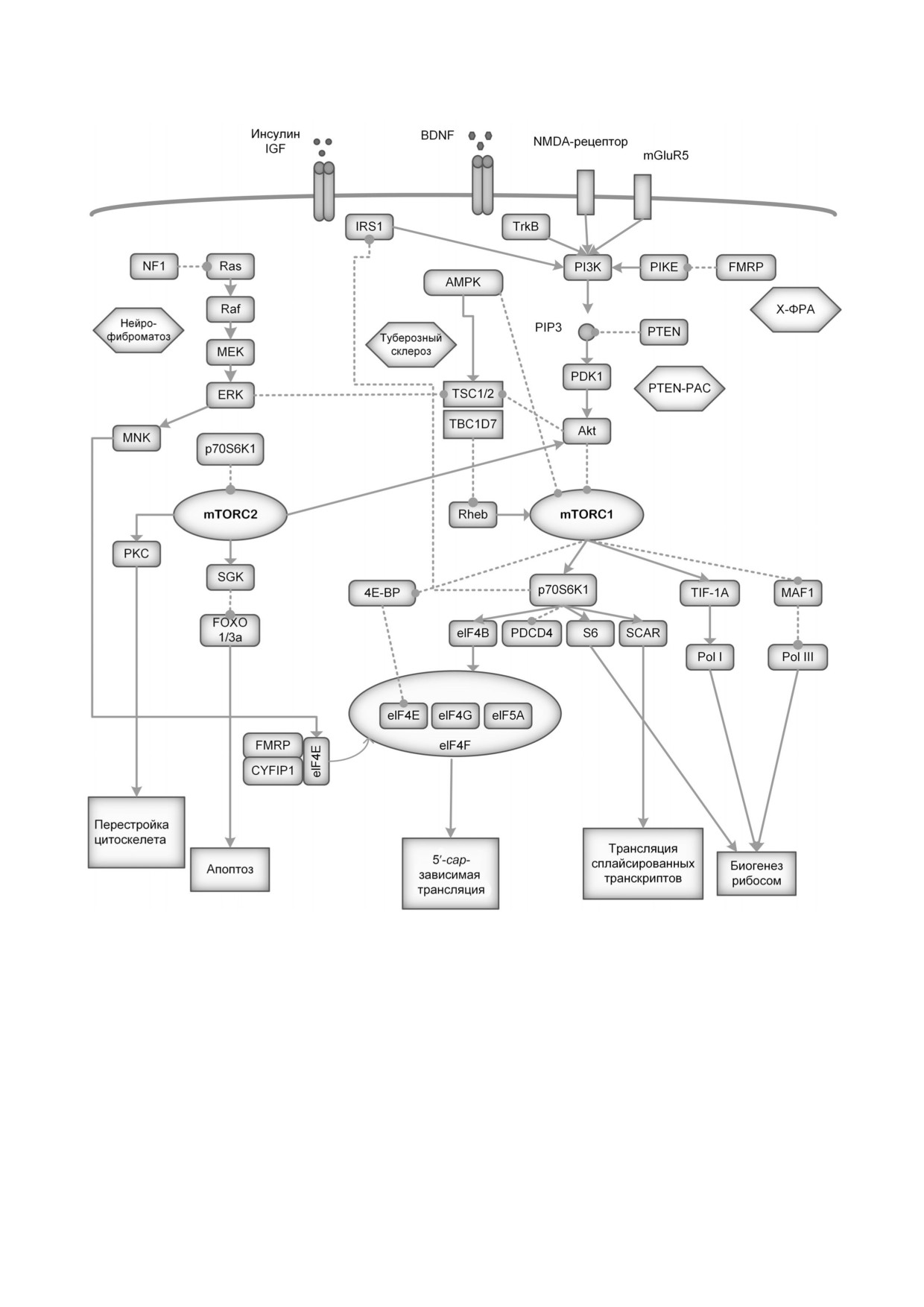
Схема сигнального каскада mTOR и сопряженных с ним путей. Схема составлена на основании вышедших недавно пуб!
ликаций [17-20]
лятором белковой трансляции и влияет на мно!
Ключевой фермент в каскаде mTOR - кина!
жество функций в развивающемся и зрелом ор!
за mTOR. Это белок, гомологичный дрожжево!
ганизме [12]. Сигнальные каскады митоген!ак!
му белку TOR, серин!треониновая (Ser/Thr)
тивируемых протеинкиназ (МАРK) и Wnt также
протеинкиназа (~ 289 кДа), член семейства
конвергируют на сигнальном каскаде mTOR
PIKK млекопитающих. mTOR входит в состав
[17] (рисунок). Приведенная на рисунке схема
двух функционально различающихся комплек!
составлена на основании вышедших недавно
сов: mTORC1 и mTORC2, подробно освещён!
публикаций [17-20].
ных в обзорах ранее [17, 19, 20].
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
647
Активность комплекса mTORC1 подавляет!
клеток реализуются посредством индуцируемой
ся рапамицином (фунгицидом микробиологи!
сывороткой и глюкокортикоидами кина!
ческого происхождения), поэтому белок mTOR
зы
1
(SGK1) (рисунок)
[23]. Кроме того,
получил название «мишень рапамицина у мле!
mTORC2 фосфорилирует Akt, активируя её и
копитающих» [17]. Механизм действия рапами!
контролируя тем самым активность mTORC1
цина подробно рассмотрен в обзоре
(рисунок).
Pupyshev et al. [21] и других цитированных пуб!
Большинство известных неврологических и
ликациях.
психических расстройств, в которых задейство!
ван mTOR, связывают с нарушением регуляции
активности mTORC1 [24].
СИГНАЛЬНЫЙ ПУТЬ mTOR
Рассмотрим ключевые моменты каскада, ле!
И СОПРЯЖЁННЫЕ С НИМ КАСКАДЫ
жащие «выше» mTORC1. Активация mTORC1
инициируется различными факторами (в т.ч.
Комплекс mTORC1 содержит, кроме фер!
NGF, PDGF, NF1 при посредстве ГТФазы Ras,
мента mTOR, регуляторные белки Raptor (свя!
BDNF и глутаматом при посредстве NMDA! и
зывающий рапамицин), mLST8 (ассоциирован!
метаботропных глутаматных mGluR5!рецепто!
ный с mTOR белковый гомолог LST8 (mam!
ров), активирующими PI3K и образование ею
malian lethal with Sec13 protein 8)), PRAS40
фосфатидилинозитол!3,4,5!трифосфата (PIP3)
(40 кДа, обогащённый пролином субстрат Akt),
(рисунок). Накопление PIP3 в цитоплазмати!
Deptor (содержащий DEP!домен белок, взаимо!
ческой мембране стимулирует рекрутинг Akt, её
действующий с mTOR) и ещё несколько бел!
фосфорилирование и активацию 3!фосфоино!
ков [19]. Raptor необходим для внутриклеточной
зитид!зависимой протеинкиназой 1 (PDK1), а
локализации mTORС1, он также рекрутирует
также фосфорилирование и активацию mTORC2.
субстраты mTORC1, связываясь с сигнальной
Будучи активированной, Akt фосфорилирует и
последовательностью mTOR, которая представ!
ингибирует TSC2, который вместе с TSC1 и
лена на канонических субстратах mTOR, таких
TBC1D7 (член 7 семейства белков, содержащих
как белки, регулирующие трансляцию (напри!
TBC!домен) образует белковый комплекс тубе!
мер, pS70S6K и 4EBP!1) и аутофагию (Ulk1). Ра!
розного склероза (TSC). Комплекс TSC функ!
памицин в присутствии FKBP12 (FK506!связы!
ционирует как ГТФаза (GAP) по отношению к
вающего белка 12) подавляет связывание Raptor
белку Rheb (гомологу Ras в мозге), стимулируя
c mTOR и подавляет катализируемое mTOR
превращение Rheb!GTP в Rheb!GDP и инакти!
фосфорилирование Raptor!зависимых субстра!
вируя этот белок. Следовательно, если ингиби!
тов, но не влияет на каталитическую актив!
руется комплекс TSC1/2 под действием Akt, то
ность mTOR. mLST8 предположительно стаби!
активируется Rheb, который далее активирует
лизирует киназный домен mTOR, а PRAS40 и
mTORC1 [25]. Активность комплекса TSC1/2
Deptor являются ингибиторами активнос!
может регулироваться несколькими сигнальны!
ти mTORC1 [19].
ми путями. Кроме пути PI3K/PDK1/Akt, TSC2
Комплекс mTORC2 содержит, кроме mTOR,
фосфорилируется и ингибируется киназа!
белки mLST8, Deptor (те же, что и mTORC1), а
ми ERK и АМP!активируемой протеинкина!
также mSIN1 (индуцируемый стрессом взаимо!
зой (AMPK) (рисунок); Wnt также подавляет
действующий с протеинкиназой белок млеко!
фосфорилирование TSC2 киназой гликоген!
питающих), Rictor (нечувствительный к рапа!
синтазы 3 альфа (GSK3α), что ведёт к стимуля!
мицину компонент) и Protor!1/2 (белок, наблю!
ции пути mTOR [26].
даемый в ассоциации с Rictor!1/2). В отличие от
Один из главных сигнальных компонентов,
mTORC1, активность mTORC2 не ингибируется
находящихся непосредственно выше mTORC1 и
рапамицином, но при хроническом воздействии
активирующих его, - это Rheb. В норме белки
рапамицина активность mTORC2 подавляется,
TSC1/2 ингибируют Rheb и его мишень -
что вызвано, как предполагают, истощением пу!
mTORC1. Когда этот процесс, происходящий
ла mTOR [22]. О комплексе mTORC2 известно
«выше по течению», подавлен (см. раздел Комп!
значительно меньше, чем о mTORC1, но
лекс туберозного склероза), происходит гипер!
он (посредством мишеней Akt, SGK1, PKC) ре!
активация mTORC1.
гулирует анаболические процессы, процессы
Ряд регуляторов действуют на активность
организации и поддержания целостности ци!
mTORC1 при посредстве сигнального пути Akt.
тоскелета, апоптоза и ионного транспорта (ри!
Так, активацию Akt, происходящую при посред!
сунок) [17, 23]. Стимуляция mTORC2 ведёт к пе!
стве PI3K, может подавлять NF1 при посред!
рестройке организации цитоскелета (при посред!
стве Ras (рисунок). Ещё один механизм актива!
стве активации PKC), а механизмы выживания
ции mTORC1 - инактивация PRAS40 либо вслед!
БИОХИМИЯ том 86 вып. 5 2021
648
БОКША и др.
ствие фосфорилирования протеинкиназой А,
дендритов) благодаря активации этого каскада
либо вследствие прямого фосфорилирования
факторами роста, включая BDNF, а также его
Akt по Thr246 и mTOR - по Ser183 [6].
стимуляции глутаматными рецепторами
Другая регуляторная молекула - PTEN -
NMDA!типа и mGluR5 [28] (рисунок). Этот кас!
участвует в управлении активностью Akt и ли!
кад и системы, с которыми он взаимодействует,
пидном сигналинге посредством регуляции
особенно пути Wnt, MAPK/ERK, играют цент!
уровня PIP3, синтезируемого PI3K. PTEN ката!
ральную роль в нейроонтогенезе и нейрональ!
лизирует реакцию дефосфорилирования PIP3
ных функциях.
до PIP2 (фосфатидилинозитол!4,5!дифосфата),
В литературе имеется много данных, указы!
отщепляя фосфатные группы от PIP3, образо!
вающих на нарушение регуляции активности
ванного PI3K. Дефосфорилирование PIP3 инги!
пути PI3K/Akt/mTOR при РАС. Наиболее убе!
бирует сигнальный путь, опосредуемый Akt, и
дительные прямые данные получены при иссле!
способствует апоптозу. Активация и фосфори!
довании моногенных С!РАС, обусловленных
лирование киназой PDK1 протеинкиназы Akt
потерей или снижением функции генов FMRP,
играет основную роль в инициации пути mTOR,
PTEN, NF1, TSC1, TSC2 и EIF4E, продукты ко!
т.к. Akt имеет мишенью белки комплекса
торых являются либо регуляторами самого кас!
TSC1/2 и тем самым регулирует активность
када mTOR, либо его мишенями, либо компо!
mTORC1 [27].
нентами путей, на которые он влияет. Hoeffer и
Кроме киназ, есть и другие модуляторы ак!
Klann [29] отмечали, что вызванные одиночны!
тивности mTORC1 и mTORC2, которые рас!
ми генами нарушения ответственны за 8-15%
смотреть в данном обзоре не представляется
всех случаев РАС, причём более половины этих
возможным. Отметим лишь, что активность
генов участвует либо в регуляции сигнального
mTOR, как и всех протеинкиназ, конечно, зави!
каскада mTOR, либо в контроле трансляции.
сит от уровня энергетического субстрата -
Изменения в активности PI3K/Akt/mTOR
АТP (точнее, комплекса АТP с ионами Mg2+
по сравнению с контролем отмечены не только
или Mn2+). То, что активность каскада mTORC1
при С!РАС, но и при некоторых НС!РАС [30].
контролируется MAPK/ERK и AMPK, еще раз
Повышение активности каскада кажется логич!
подтверждает справедливость признания mTOR
ным для объяснения таких фенотипических
в качестве «сенсора» внутренних и внешних сти!
признаков аутизма, как увеличенный размер
мулов, который поддерживает клеточный гомео!
мозга [31, 32], эпилепсия и преобладание про!
стаз, модулируя анаболические и катаболичес!
цессов возбуждения над процессами торможе!
кие процессы [19].
ния (E/I) [33], усиленный рост дендритов и их
Что касается мишеней mTORC1, то в числе
аномальное ветвление [34]. Но не всё так одно!
прочих процессов, mTORC1 стимулирует белко!
значно в вопросе активности каскада mTOR при
вую трансляцию, активируя киназу 1 рибосом!
различных формах даже С!РАС, не говоря уже о
ного белка S6 (p70S6K1) и ингибируя актив!
И!РАС: как будет отмечено далее, имеются так!
ность белков 4E!BP. В дефосфорилированном
же данные и о нарушении равновесия активнос!
состоянии 4E!BP1 подавляет трансляцию, свя!
тей комплексов mTORC1 и mTORC2, или сни!
зывая эукариотический фактор инициации
жении активности каскада mTOR, например,
трансляции 4E (eIF4E). При фосфорилирова!
при СА
[35], синдроме Фелан-МакДер!
нии киназой mTORC1 белки 4E!BP1 диссоци!
мид (ФМС) и СР. В целом отклонения (повыше!
ируют от фактора eIF4E, позволяя тому взаимо!
ние или понижение) активности сигналинга
действовать и связываться с eIF4G и eIF4A с об!
mTORC1 описаны при нескольких моногенных
разованием комплекса инициации трансляции
С!РАС: КТС, PTEN!ассоциированном РАС,
eIF4F, что является критическим этапом в cap!
НФ1, Х!ФРА, СР и СА, рассмотренных далее.
зависимой трансляции. Фосфорилирование
Кроме С!РАС, нарушение регуляции актив!
p70S6K1 стимулирует киназную активность и
ности mTORC1!зависимого сигнального каска!
усиливает фосфорилирование ею субстрата -
да (как лежащих выше путей, регулирующих
рибосомного белка S6. Предполагают, что фос!
этот каскад, так и фосфорилируемых им мише!
форилирование S6 может стимулировать тран!
ней) наблюдалось у пациентов с НС!РАС и у
скрипцию генов, вовлечённых в биогенез рибо!
животных, моделирующих НС!РАС [36-39].
сом [5].
Тем не менее до полного понимания меха!
В нейронах сигнальный каскад PI3K/Akt/
низмов, посредством которых дисбаланс пути
mTORC1 регулирует множество процессов
mTORC1 приводит к развитию аутистических
(рост, выживание клеток, синтез белка, синап!
симптомов, ещё далеко, и остается ряд нере!
тическую пластичность, зависимую от глутамат!
шенных вопросов: насколько закономерности,
ных рецепторов NMDA!типа, рост и ветвление
выясненные при изучении С!РАС, можно рас!
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
649
пространить на НС!РАС; достаточно ли свиде!
отсутствии ингибирования конститутивная ак!
тельств того, что сигнальный путь mTORC1 вно!
тивация mTORC1 представляет молекулярную
сит вклад в патогенез НС!РАС для разработки
основу КТС [63]. Однако, не слишком упрощая
фармакологических препаратов; насколько ве!
ситуацию, отметим, что TSC1 и TSC2 имеют и
лика доля случаев с измененной активностью
другие молекулярные функции: взаимодействие
сигнального пути mTORC1 среди всех НС!РАС
с актин!связывающими белками и независимая
и И!РАС; будет ли терапия, направленная на
от Rheb модуляция активности mTORC2, меха!
mTORC1 и эффективная в лечении проявлений
низм которой может включать прямое связыва!
некоторых С!РАС, также эффективна хотя бы
ние комплекса TSC1/2 с компонентами
для некоторых подгрупп пациентов с НС!РАС и
mTORC2. Активированный mTORC2 фосфори!
И!РАС; в каких случаях при РАС активность
лирует и активирует Akt (рисунок), что приво!
сигнального пути mTOR повышена, в каких -
дит к фосфорилированию ее субстратов, вклю!
понижена и каковы пределы и временные рамки
чая TSC2. Взаимодействие между mTORC1 и
этих отклонений в онтогенетическом развитии.
mTORC2 происходит ещё и при посредстве
Далее в настоящем обзоре обсуждаются ме!
p70S6K1, фосфорилирующей и ингибирующей
ханизмы и терапевтические подходы, разрабо!
Rictor. Утрата либо TSC1, либо TSC2 приводит к
танные при изучении С!РАС с доказанным
тому, что mTORC1 активируется, а mTORC2 по!
вкладом в патогенез аномалий регуляции пути
давляется. Поскольку такие наблюдения были
mTORC1, а также обсуждаются недавние наход!
сделаны не на нейрональных клетках, предстоит
ки, потенциально свидетельствующие о вкладе
важная задача: изучить, как происходит взаим!
дисфункции сигналинга mTORC1 в НС!РАС и
ная регуляция mTORC1 и mTORC2 в нервной
И!РАС.
ткани. Второе важное следствие из таких наблю!
дений - это то, что при поиске лекарственных
препаратов, воздействующих на mTOR, следует
СИГНАЛЬНЫЕ КАСКАДЫ
учитывать активность обоих комплексов -
(ВКЛЮЧАЯ mTORC1) ПРИ C6РАС
mTORC1 и mTORC2 - и их взаимодействие [26].
Отметим также, что сам Rheb имеет и не зависи!
Комплекс туберозного склероза (КТС). При
мые от mTORC1 функции - регулирует сигна!
КТС наблюдается снижение интеллекта, аутизм,
линг B!Raf (серин!треониновой киназы) и
тревожные расстройства и расстройства настро!
MAPK/ERK, опосредует апоптоз и митофагию,
ения, эпилепсия, множественные опухоли и
а также выполняет другие функции [64]. Следо!
многочисленные другие расстройства. РАС при
вательно, в фенотип КТС могут вносить допол!
КТС встречаются в 20-60% случаев, причём
нительный вклад и нарушения регуляции дру!
эпилепсия, РАС и снижение интеллекта тесно
гих сигнальных путей, хотя большая часть ис!
сцеплены [20]. Структурные аномалии мозга
следований указывает на то, что в основном фе!
при КТС [40] приведены в таблице. Примеча!
нотип обусловлен гиперактивацией mTORC1.
тельно, что когнитивные нарушения и отклоне!
На животных, лабораторных моделях КТС,
ния от нормы в поведении детей с КТС и с И!
показано, что мутации с инактивацией TSC1 и
РАС очень сходны, что может свидетельствовать
TSC2 и обусловленная этим гиперактивность
о сходных молекулярных и структурных анома!
сигнального пути mTOR могут приводить к ано!
лиях при аутистических симптомах, сопровож!
малиям мозга, отмеченным у пациентов
дающих КТС, и при И!РАС. Предполагается,
с РАС при КТС.
что КТС может служить моделью РАС, и когда
Сведения о молекулярной природе нейро!
молекулярные механизмы, лежащие в его осно!
когнитивных аномалий почерпнуты также из
ве, будут полностью расшифрованы, эти наход!
исследований моделей - нокаутных мышей с
ки, вероятно, помогут также при изучении меха!
инактивированными генами TSC [65]. В экспе!
низмов, лежащих в основе И!РАС.
риментах на мышах Tsc2+/-, моделирующих по!
КТС - это классическая форма «mTOR!па!
веденческие аномалии при КТС, показано, что
тии», он вызывается мутациями генов, кодиру!
нарушения обратимы под действием рапамици!
ющих белки TSC1 или TSC2, с потерей функ!
на - ингибитора mTORC1 [41]. Производное ра!
ций. TSC1 и TSC2 вместе с TBC1D7 образуют
памицина, эверолимус (Everolimus) или афини!
комплекс, осуществляющий негативную регу!
тор, был одобрен Food and Drug Administration в
ляцию mTORC1 при посредстве Rheb (рисунок).
1999 г. для лечения субэпендимальных гиганто!
Основная функция комплекса TSC1/2 - это ин!
клеточных астроцитом, развивающихся при
теграция множественных сигналов от лежащих
КТС (при невозможности выполнения хирурги!
«выше» сигнальных путей и ингибирование ак!
ческой резекции опухоли). Рапамицин и эверо!
тивности Rheb и mTORC1, таким образом, при
лимус также проявляли противоэпилептичес!
3 БИОХИМИЯ том 86 вып. 5 2021
650
БОКША и др.
Известные генетические основы, активность mTORC1 и белков сопряженных с ним сигнальных путей, морфологические
и функциональные аномалии мозга и моделирование на животных расстройств аутистического спектра#
Генетические
Активность mTORC1 и
Морфологические и функциональные
Животные
Формы РАС
основы
белков сопряжённых с
аномалии мозга
модели
ним сигнальных путей
КТС
мутация TSC1
отсутствуют функцио!
мегалэнцефалия; нарушение синаптичес!
мыши с услов!
или TSC2
нально активные белки
кого прунинга; гипомиелинизация; сверх!
ным нокаутом
TSC1 или TSC2; консти!
возбудимость синапсов астроглиоз; дис!
гена Tsc1 (Tsc1+/-!
тутивная активация
плазии нейронов; множественные ано!
мыши); мыши с
mTORС1
мально длинные аксоны; аномальные
условным нокау!
межнейрональные связи
том гена Tsc2
(Tsc2+/-!мыши)
X!ФРА
экспансия
повышение активности
обилие незрелых форм шипиков; высокая
Fmr1 KО
повтора CGG
mTORС1 при посредстве
плотность дендритных шипиков; избыток
5′!UTR гена
PI3K; снижение концен!
синапсов; повышенная частота патологии
FMR1
трации белка FMRP
нейронов; увеличение объёма гиппокампа
PTEN
мутация с по!
конститутивно высокая
макроцефалия; мегалэнцефалия; анома!
мыши Pten KO
терей функции
активность mTORC1 при
лии миелинизации; гипертрофия тела
или нокдаун
гена протеин!
посредстве PI3K
нейронов; высокая плотность дендритных
фосфатазы
шипиков; увеличение длины дендритов;
PTEN
повышенная плотность нейронных связей
НФ1
мутации гена
повышенная активность
макроцефалия; увеличение объёмов бело!
мыши NF1 KO
NF1
RAS/MAPK (ERK); по!
го и серого вещества; нарушение целост!
вышенная активность
ности белого вещества; аномалии мигра!
PI3K; повышенная ак!
ции нейронов; астроглиоз
тивность mTORC1; ин!
гибирование TSC2;
снижение содержания
нейрофибромина
СА
делеция в
повышенная активность
микроцефалия; увеличение объёма моз!
мыши с делеци!
15q11-13 или
mTORC1; пониженная
жечка и гиппокампа; нарушение миелини!
ей UBE3A
мутация с по!
активность mTORC2;
зации; аномалии морфологии дендритных
терей функции
нарушение экспрессии
шипиков; снижение плотности шипиков;
гена UBE3A,
убиквитин!протеинлига!
снижение ветвления и длины дендритов
локализованно!
зы E3A; изменение ко!
го на 15q11-13
личества Arc
СР
мутация с по!
пониженная активность
уменьшение размеров нейронов; повы!
мыши Mecp2+/-,
терей функции
mTORC1; снижение со!
шенная плотность нейронов; астроглиоз;
Mecp2-/-
гена MECP2;
держания BDNF; сниже!
снижение ветвления дендритов; понижен!
мутация
ние содержания MECP2
ная плотность шипиков; аномалии созре!
CDKL5
вания в коре и гиппокампе; сниженное
число глутаматергических синапсов
Фелан-
микроделеция
пониженная активность
аномалии нейронов и синаптической про!
нокдаун
МакДермид
22q13, затраги!
mTORC1
водимости в кортикальных слоях и в поло!
SHANK3;
22q13
вающая локус
сатом теле; снижение плотности шипиков;
дупликация
гена SHANK3
снижение плотности нейронных связей
SHANK3
Dup15q11-13
дупликация
повышенная активность
аномалии морфологии нейронов и нейро!
дупликация
15q11-13; уве!
mTORC1
медиаторных систем
BP2!BP3
личение дозы
(patDp/+); свер!
гена CYFIP1;
хэкспрессия ге!
увеличение до!
на CYFIP1
зы гена UBE3A
eIF4E
мутации в об!
повышение активности
увеличение плотности дендритных шипи!
мыши со сверх!
НС!РАС
ласти промо!
промотора гена EIF4E
ков; нарушение синаптической пластич!
синтезом белка
тора гена
ности; снижение возбудимости; нарушение
eIF4E
EIF4E
LTP в префронтальной коре и гиппокампе
И!РАС
неизвестны
повышенная или пони!
макроцефалия; повышение плотности
мыши с индуци!
женная активность
нейронов в некоторых областях мозга;
рованными валь!
mTORС1; снижение син!
снижение численности клеток Пуркинье;
проатом анома!
теза PI3K, Akt, mTOR,
астроглиоз и активация микроглии; ано!
лиями развития;
p!mTOR, p70S6K и
малии миелинизации; изменение плот!
мыши BTBR
eIF4B
ности дендритных шипиков
T+Itpr3tf/J
# Таблица составлена на основании публикаций: [11, 37, 40-62].
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
651
кую активность у животных, лабораторных мо!
ловых клеток (ППСК), полученных от боль!
делей КТС с делецией TSC1 в нейронах или аст!
ных КТС, и клеток!онтогенетических предшест!
роцитах, и в экспериментах на таких животных,
венников нейронов, в которых изучается роль
демонстрирующих аномальное поведение и ког!
ранних событий, происходящих в сигнальных
нитивные нарушения, рапамицин уменьшал
каскадах, в онтогенезе и развитии РАС [70]. Так,
степень выраженности этих аномалий [66].
например, ППСК от пациента с КТС, несущие
В фазе III клинических испытаний эвероли!
мутацию гена TSC1, были подвергнуты диффе!
мус достоверно позволял контролировать эпи!
ренцировке в клетки!предшественники нейро!
лептические припадки у больных с КТС с реф!
нов. В гетерозиготном и нулевом вариантах кле!
рактерным эпилептическим статусом
[67].
ток наблюдались активация mTORC1 и
В настоящее время ингибиторы mTOR исполь!
MEK/ERK и усиление пролиферации и роста
зуются для лечения связанных с КТС невроло!
нейритов. Активация mTORC1 приводила к
гических проявлений у детей и взрослых, но
фосфорилированию и высвобождению ингиби!
применение рапамицина и его производных ог!
торного 4E!BP1 из комплекса с eIF4E, что поз!
раничено из!за осложнений и побочного
воляло сформироваться активному комплексу
действия. В отдельных клинических исследова!
eIF4F. Рапамицин подавлял активацию mTORC1,
ниях рапамицина и его производных сообща!
но не оказывал влияния на повышенную проли!
лось о положительной динамике аутистических
ферацию и образование отростков у клеток!
симптомов и улучшении когнитивного функци!
предшественников. Кроме того, оказалось, что
онирования при КТС, однако в других исследо!
под действием рапамицина усиливалась повы!
ваниях говорилось либо об отсутствии положи!
шенная базовая активность MEK/ ERK и акти!
тельного влияния этих препаратов на когнитив!
вировался альтернативный механизм опосредо!
ные способности детей с КТС, либо эти способ!
ванного MNK (киназой, взаимодействующей с
ности не оценивались [68]. Можно предполо!
MAPK) фосфорилирования и активации
жить, что препараты, одобренные для примене!
eIF4F (рисунок) [71]. Полностью блокировать
ния у детей с 3!летнего возраста и опробован!
эффекты, вызванные отсутствием TSC1, удава!
ные в клинике, применялись слишком поздно и
лось лишь совместной обработкой рапамици!
не были способны «обратить» патологию разви!
ном и eFT508 - ингибитором MNK.
тия ЦНС, тогда как, возможно, при более ран!
Полученные данные указывают на то, что
нем применении они могли бы её предотвра!
ранние события, вызванные отсутствием TSC1,
тить. Тем более ценными кажутся единичные
зависят не только от активации mTORC1. Регу!
обследования детей в возрасте до 2 лет [69]. Этот
ляция трансляции в нейронах, опосредованная
вопрос будет затронут далее в разделе PTEN!
MEK/ERK и MNK!eIF4E, тоже может играть
РАС (экспериментальные данные по моделиро!
роль в аспектах КТС, связанных с нарушениями
ванию патологии на мышах).
в центральной нервной системе. И, возможно,
Ограниченность применения рапамицина и
что при разработке фармакологических подхо!
его производных в терапии (в т.ч. неврологичес!
дов совместное применение ингибиторов mTOR
ких проявлений) объясняется также существо!
и MNK будет более эффективно, чем примене!
ванием обратной связи
- опосредуе!
ние одного рапамицина и его производных. Для
мой p70S6K1 петли регуляции сигналинга PI3K.
этого необходимы дальнейшие исследования
p70S6K1 - субстрат mTORC1 - фосфорилирует
протеома и транслятома (мРНК) как до, так и
субстрат рецептора инсулина (IRS!1), блокируя
после обработки ингибиторами mTOR и MNK
инсулин!опосредованную активацию сигналь!
клеток!предшественников нейронов, выделен!
ного пути PI3K/Akt (рисунок). Кроме того, ра!
ных от пациентов с КТС [70].
памицин подавляет не все функции mTORC1:
Синдром ломкой Х6хромосомы (X6ФРА, синд6
он блокирует фосфорилирование mTORC1 ки!
ром Мартина-Белл). X!ФРА - это наиболее из!
назы p70S6K1, но практически не ингибирует
вестный синдром, ассоциированный с задерж!
фосфорилирование белка 4E!BP1, что может
кой психического развития и РАС, причём диаг!
приводить к стимуляции других частей каскада
ноз РАС имеют 30-50% больных с X!ФРА [72].
mTOR (например, 4E!BP1) [21].
Чаще всего синдром обусловлен экспансией
Сигнальный путь mTOR играет критическую
повтора CGG в 5′!UTR гена FMR1 (fragile X men
роль для нормального клеточного роста и выжи!
tal retardation 1), что приводит к гиперметилиро!
вания, и все последствия, которые может ока!
ванию и снижению экспрессии гена. При пол!
зать его ингибирование и/или активация на раз!
ностью мутантном гене (>200 повторов) отсут!
вивающийся мозг, ещё до конца не изучены.
ствует продукт гена - белок, ассоциированный с
Проводятся исследования с использованием
синдромом ломкой X!хромосомы (FMRP).
плюрипотентных перепрограммированных ство!
FMRP - это РНК!связывающий белок, участву!
БИОХИМИЯ том 86 вып. 5 2021
3*
652
БОКША и др.
ющий в регуляции трансляции, зависимой от
каскада [75]. Все эти находки свидетельствуют о
BDNF и метаботропных глутаматных рецепто!
том, что помимо мРНК!связывающей актив!
ров 1 (mGluR1) - процесса, лежащего в основе
ности, FMRP также играет роль регулятора ини!
синаптической пластичности. Поэтому измене!
циации трансляции, опосредуемой PI3K/
ние интенсивности синтеза белка играет важ!
mTORC1, так как FMRP участвует в регуляции
ную роль в неврологических проявлениях X!
PI3K (рисунок) [75].
ФРА [73] (рисунок). FMRP служит репрессором
Поскольку у мышей Fmr1 KO, лабораторных
трансляции, специфически (селективно) связы!
моделей X!ФРА, при стимуляции активности
ваясь с последовательностями ряда мРНК и по!
каскада PI3K/Akt/mTOR симптомы, связанные
лирибосомами. Многие мРНК, являющиеся его
с РАС, вероятно, обусловлены усиленной транс!
мишенями, кодируют важнейшие белки, необ!
ляцией белка в синапсах и спровоцированным
ходимые для созревания, стабилизации и эли!
высоким отношением E/I в ключевых нервных
минации при перестройке синапсов, причем из!
путях, на этих мышах было опробовано фарма!
вестно, что гены, которые кодируют эти белки,
кологическое ингибирование пути PI3K [76].
ассоциированы с риском РАС: SHANK3, PTEN,
При этом наблюдалось ослабление дефицита,
TSC2, NF1, CYFIP1, NLGN3 и NRXN1 [73, 74].
связываемого с симптомами РАС. Важно, что
При X!ФРА часто развивается эпилепсия, наб!
ингибирование PI3K или mTORC1 нормализо!
людаются множественные аномалии структур
вало избыточный синтез синаптических белков
мозга (таблица), что влияет на его функции и
в нейронах мышей [76]. Кроме того, у мы!
синаптическую пластичность, а in vitro в клеточ!
шей Fmr1 KO делеция в гене, кодирующем
ных линиях нейронов, полученных из ППСК
p70S6K1, и фармакологическое или генетичес!
больных с X!ФРА, наблюдаются структурные
кое подавление фосфорилирования eIF4E (т.е.
аномалии (уменьшение размеров тела и длины
мишеней обоих путей - ERK и mTORC1) (рису!
отростков) [42].
нок) предотвращали формирование дефектов
У нокаутных мышей Fmr1 KO развиваются
дендритных шипиков, изменение синаптичес!
аномалии нервной системы, сходные с проявле!
кой пластичности, усиленный синтез белка и
ниями патологии у пациентов с X!ФРА [43].
развитие связанного с РАС поведенческого фе!
В постмортальном мозге больных с X!ФРА
нотипа [77]. Это указывает на то, что усиление
обнаружены аномалии каскадов белкового фос!
сигналинга mTORC1 и cap!зависимой трансля!
форилирования, включая сигнальный путь
ции играет роль в патофизиологии X!ФРА.
mTOR [44]. Роль каскада mTOR в X!ФРА остает!
Вместе с тем известно исследование, пока!
ся до конца не выясненной, но исследования
завшее, что гиперактивация mTOR вследствие
позволили идентифицировать механизмы, по!
утраты функции TSC2 парадоксальным образом
средством которых FMRP может регулировать
привела к снижению синтеза белка в гиппокам!
mTOR, и наоборот. Так, FMRP может подавлять
пе, и этим проявлениям (а также поведенческим
трансляцию многих мРНК, например, глута!
и электрофизиологическим аномалиям) проти!
матного рецептора mGLuR5 и белков пути
водействовали как фармакологическая актива!
PI3K/Akt/mTOR [73]. Участие этих каскадов в
ция mGluR5, так и делеция Fmr1 [78]. Следова!
регуляции социального поведения, обучения,
тельно, сигнальные взаимодействия FMRP и
памяти обусловлено тем, что они необходимы
mTOR сложные и разветвленные, и их наруше!
для регуляции белкового синтеза, индуцируемо!
ние потенциально может привести к РАС.
го mGluR группы 1 (Gp1), обеспечения синап!
Так, открыт ещё и такой механизм связи
тической проводимости и формирования шипи!
FMRP с mTOR, как регуляция трансляции на
ков [44, 45].
стадии инициации посредством связывания
При биохимических исследованиях нокаут!
комплекса CYFIP1/FMRP с eIF4E (рисунок).
ных мышей Fmr1 KO были получены несколько
Сигнальные каскады фосфорилирования, ини!
различающиеся результаты, что, возможно, объ!
циируемые BDNF и mGluR, активируют путь
ясняется различными экспериментальными ус!
MNK, что, в свою очередь, приводит к усиле!
ловиями. Но есть свидетельства того, что у этих
нию фосфорилирования eIF4E, ослаблению
мышей усилена активность сигнального каскада
связей в комплексе CYFIP1/FMRP/eIF4E и уси!
mTORC1 и повышено образование комплекса
лению высвобождения eIF4E из комплекса с
инициации трансляции в мозге. Это обусловле!
CYFIP1/FMRP, что позволяет взаимодейство!
но, как минимум частично, усиленной трансля!
вать eIF4E с eIF4G, и при этом осуществляется
цией мРНК, кодирующих субъединицу p110δ
трансляция связанной мРНК (рисунок) [71].
киназы PI3K и (PIKE)!S - энхансера!активато!
На сложность взаимодействий различных
ра PI3K, позитивно регулирующих активность
систем белкового фосфорилирования указывает
mTORC1 и активность всего связанного с ним
возможное вовлечение казеинкиназы 2: стало
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
653
известно, что она конститутивно фосфорилиру!
РАС!подобных поведенческих аномалий [48],
ет FMRP по остатку S499, и это стимулирует
благодаря чему, во!первых, было достигнуто по!
дальнейшее фосфорилирование FMRP по дру!
нимание связи между усиленной активностью
гим сайтам [79]. Более того, FMRP является ми!
сигнального каскада mTORC1 и неврологичес!
шенью киназы p70S6K1 - члена семейства сиг!
кими и поведенческими аномалиями мутантных
нального каскада mTOR, таким образом, транс!
мышей. Во!вторых, оказалось, что эффект от
ляция, регулируемая репрессором FMRP, зави!
применения ингибиторов наблюдается лишь
сит от активности пути mTOR (рисунок).
при наиболее раннем применении, т.е. достига!
Синдромная форма PTEN6РАС, ассоцииро6
ется при предотвращении развития аномалий
ванная с мутацией гена PTEN. PTEN!ассоцииро!
мозга, но не обращает их вспять. Однако оказа!
ванная «моногенная» форма РАС связана с гете!
лось, что рапамицин лишь частично компенси!
розиготным вариантом мутации с потерей
рует фенотип, обусловленный потерей функции
функции PTEN. Морфологические аномалии
PTEN, тогда как полный эффект достигается
головного мозга при PTEN!РАС приведены в
лишь при действии ингибитора Akt [82], следо!
таблице. Хотя умственная отсталость наблюда!
вательно, в развитие неврологических феноти!
ется примерно у 20% пациентов с мутацией
пов, связанных с утратой функции PTEN, вно!
PTEN, распространенность РАС среди этих па!
сят вклад Akt!зависимые пути.
циентов не оценивалась. Наоборот, частота
Хотя изменения роста, пролиферации, миг!
встречаемости мутации PTEN среди пациентов с
рации глиальных клеток и образования миели!
РАС с макроцефалией оценена: она варьирует в
на, наблюдаемые у животных моделей, могут
пределах 7-27% [46].
иметь отношение к патогенезу РАС, необходи!
Ген PTEN кодирует липид! и протеинфосфа!
мы дальнейшие исследования для того, чтобы
тазу PTEN (гомолог фосфатазы и тензина),
понять, в какой мере эти аномалии связаны
ключевой фермент в росте клеток, пролифера!
именно с избыточной активацией mTORC1, и
ции, выживании. PTEN противодействует
как они влияют на поведение при РАС.
функции киназы PI3K (путём дефосфорилиро!
Несмотря на то, что в мозге мышей, модели!
вания PIP3) [80] и, подобно TSC1/2, осущест!
рующих PTEN!РАС, наблюдаются невропато!
вляет негативный контроль активности каскада
логические особенности, сходные с теми, что
mTORC1 [81] (рисунок). Отсутствие контроля
описаны у пациентов с PTEN!РАС [48], и они
со стороны PTEN способствует неконтролируе!
могут быть связаны отчасти с усилением сиг!
мому образованию PIP3 под действием PI3K,
нальной активности mTORC1, окончательного
что приводит к усилению активности Akt и ги!
понимания связи не достигнуто, и вопрос требу!
перактивации mTORC1 [81]. Таким образом, де!
ет углубленного изучения.
фицит активности PTEN влечет за собой конс!
Также интересна связь пути mTOR с еще од!
титутивную активацию управляемых ею каска!
ним регуляторным фактором, влияющим на ма!
дов Akt/mTORC1.
нифестацию аутизма - белком!предшественни!
Опубликованных результатов обследований
ком бета!амилоида (APP) [83]. В синапсах при
пациентов с PTEN!РАС немного, тем не менее в
аутизме обнаружена повышенная концентра!
них отмечались патология структур мозга и на!
ция APP, что ведёт к ещё большим нарушениям
личие эпилептических приступов [47].
сигнальных путей PI3K/Akt/mTOR, которые
В более многочисленных исследованиях на
обусловлены мутацией PTEN [84].
мышиных моделях (генетический нокаут или
Нейрофиброматоз типа 1 (НФ1). НФ1 относят
нокдаун), моделирующих дефицит PTEN, были
к «RAS!патиям», т.е. генетическим заболевани!
получены ключевые данные о роли PTEN в
ям, обусловленным мутациями в генах, кодирую!
центральной нервной системе и связи между
щих белки каскадов Ras/MAPK [20, 85]. У боль!
повышенной активностью каскада mTORC1 и
шой доли пациентов с мутациями, относящими!
неврологическими и поведенческими аномали!
ся к пути Ras/MAPK, включая довольно редкие
ями мутантных мышей. Патологические изме!
синдром Ульриха-Нунан, кардио!фацио!кута!
нения в центральной нервной системе живот!
неальный синдром (КФКС) и синдром Костел!
ных включали эпилептиформные припадки,
ло, проявляются симптомы РАС. Это подтверж!
макроцефалию, гипертрофию нейронов и аст!
дает предположение о том, что в этих случаях
роцитов во всех структурах мозга и другие ано!
РАС - общий результат усиленной активности
малии [48].
Ras/MAPK [86]. Действительно, MAPK/ERK
Применение рапамицина или ингибиторов
фосфорилирует и ингибирует TSC2, что приво!
киназы p70S6K1 в раннем периоде развития мы!
дит к сверхактивации mTORC1 (рисунок).
шей, моделирующих PTEN!РАС, предотвраща!
При НФ1 приблизительно в 15-18% случаев
ло развитие нарушений нервной системы и
наблюдаются когнитивные нарушения и может
БИОХИМИЯ том 86 вып. 5 2021
654
БОКША и др.
выставляться диагноз РАС, а в целом аутисти!
Синдром Ангельмана (СА). Пациенты с СА
ческие симптомы наблюдаются значительно ча!
характеризуются существенным недоразвитием
ще. НФ1 также характеризуется предрасполо!
речи, нарушением моторных функций, и боль!
женностью к развитию опухолей (нейрофиб!
шая доля пациентов с СА соответствует диаг!
ром) [86]. НФ1 обусловлен мутациями с потерей
ностическим критериям РАС, распространён!
функции гена NF1, кодирующего нейрофибро!
ность которых при СА оценивается в
мин. Ген NF1 экспрессируется в нейронах и гли!
~34-50% [89]. Большинство случаев СА обус!
альных клетках. Нейрофибромин - негативный
ловлены мутацией аллеля на материнской хро!
регулятор Ras, активирующий Ras ГТФазу (ри!
мосоме с потерей функции гена UBE3A в нейро!
сунок), поэтому дефект NF1 запускает актива!
нах. Ген UBE3A кодирует белок убиквитин!про!
цию сигнального каскада Ras. Первые свиде!
теинлигазу E3A, которая убиквитинилирует
тельства участия mTORC1 в патологии НФ1 бы!
другие белки, помечая и определяя их в качест!
ли получены, когда было обнаружено, что Ras
ве мишеней деградации. Ген UBE3A локализован
может индуцировать активацию PI3K и после!
в кластере генов на хромосомном локу!
дующее ингибирование TSC2 киназой Akt, с
се 15q11-13 [52].
усилением активности mTORC1 в эмбриональ!
Аномалии мозга при СА приведены в табли!
ных фибробластах и астроцитах NF1!нокаутных
це [35, 53]. Нарушения развития нервной систе!
мышей и в клетках, выделенных из опухолей от
мы при СА воспроизводятся на мышиных моде!
больных с НФ1. Однако позже было показано,
лях с материнской мутацией в Ube3a [35]. В ос!
что NF1 регулирует пролиферацию глиальных
нове патологии и неврологических аспектов СА
клеток и рост глиом посредством Akt/mTORC1!
лежит дефицит развития синапсов. Более того, в
зависимого, но TSC/Rheb!независимого меха!
целом происходит более выраженное снижение
низма [87] (рисунок). У пациентов с НФ1 наб!
активности ингибиторных, чем возбуждающих
людаются общие невропатологические призна!
путей, что, возможно, объясняет подвержен!
ки [49], и у ~10% больных с НФ1 развиваются
ность эпилептическим припадкам больных СА.
эпилептиформные приступы. Эти аномалии
У мышей, моделирующих СА посредством деле!
воспроизводятся при моделировании НФ1 на
ции гена UBE3A, наблюдается гиперактивация
мышах [50].
сигнального пути mTORC1 и снижение актив!
Как в экспериментальных, так и в клиничес!
ности mTORC2 [35]. Интересно, что активация
ких работах показано, что применение ингиби!
mTORC2 восстанавливает до нормальных уров!
торов mTORC1 благоприятно сказывается, со!
ней LTP на переживающих срезах мозга, что, ве!
ответственно, как на состоянии животных, мо!
роятно, указывает на важную роль, которую иг!
делирующих НФ1, так и на состоянии больных
рает взаимное регулирование комплексов
НФ1: снижается рост нейрофибром и опухолей!
mTORC2 и mTORC1 (рисунок) [35].
глиом. Это указывает на регуляцию функций
Аномалии в центральной нервной системе
различных типов клеток нервной системы по!
при СА возникают в результате повышенного
средством пути mTOR [88]. Однако действие ин!
уровня белков!мишеней UBE3A в постсинапти!
гибиторов mTORC1 на нейрокогнитивные или
ческом пространстве, одним из которых являет!
поведенческие функции при РАС, ассоцииро!
ся ассоциированный с цитоскелетом белок Arc.
ванных с НФ1, не изучалось.
Функции белка Arc связаны с интернализацией
Заметим, что утрата функции NF1 не только
α!amino!3!hydroxy!5!methyl!4!isoxazole propionic
в астроцитах, но и в нейронах приводит к астро!
acid (AMPA), рецепторов глутамата в возбуждаю!
глиозу, что свидетельствует о включении в мозге
щих синапсах. У мышей, моделирующих СА и
автономного внеклеточного механизма [87]. Од!
мутантных по UBE3A, нарушение LTP обусловле!
нако необходимы дальнейшие исследования
но главным образом повышением уровня Arc
связи глиальных и нейрональных нарушений и
вследствие снижения его убиквитинилирования
пролиферации именно с РАС!ассоциирован!
UBE3A и, как следствие, сниженной деградаци!
ным поведением. Особенно важным было бы
ей. Поскольку трансляция Arc регулируется
проведение исследований, в которых оценива!
FMRP - белком, ответственным за формирова!
лось бы влияние ингибиторов mTORC1 на про!
ние синдрома X!ФРА, предполагалось, что гипер!
явление аутистических симптомов при НФ1.
экспрессия Arc является точкой пересечения па!
Другие невропатологические особенности
тофизиологии СА и X!ФРА, а ингибирование
НФ1 пока не связывают с нарушением актив!
mGluR5 сможет скорректировать аномалии фе!
ности mTORC1, так, нарушение миграции ней!
нотипа СА так же, как и при синдроме X!
ронов и долговременной потенциации (LTP) за!
ФРА [90]. Возможно, повышенные уровни Arc
висят от сигнального каскада регулируемой
являются результатом усиленной активности
внеклеточными сигналами киназы ERK [51].
mTORC1 и активации его мишени p70S6K1 в
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
655
мозге моделирующих СА мышей, запущенных
Кроме мутации гена MECP2 у детей с СР, вы!
усиленным ингибированием фосфорилирования
явлены мутации других генов, в частности ге!
TSC2 и снижением убиквитинилирования в от!
на CDKL5, кодирующего киназу, подобную цик!
сутствие UBE3A [35, 91], хотя данные недоста!
линзависимой киназе 5. Интересно, что актив!
точны, и точный механизм ещё неизвестен. Воз!
ность mTORC1 также снижена и у мышей, несу!
действие рапамицином или ингибитором кина!
щих мутацию CDKL5, что свидетельствует об об!
зы p70S6K1 снижало уровень Arc и нормализова!
щей причине
«типичного» и
«атипичного»
ло плотность дендритных шипиков, морфоло!
СР [93].
гию клеток Пуркинье и пирамидальных клеток, а
В отличие от большинства РАС!ассоцииро!
также LTP и, как следствие, устраняло моторную
ванных mTOR!патий, нейроны мышей!моделей
дисфункцию и дефицит обучения у мышей, мо!
Mecp2+/- и Mecp2-/- [55], а также нейроны, про!
делей СА [35, 91]. Это свидетельствует о возмож!
изводные от MeCP2!дефицитных человеческих
ности влияния на синаптическую пластичность у
ППСК [94], демонстрируют пониженные сиг!
больных с СА и общее функционирование нерв!
нальную активность mTORC1, транскрипцион!
ной системы таких пациентов посредством нор!
ную активность и скорость синтеза белка.
мализации активности mTOR. Однако, учитывая
Хотя механизм, посредством которого
факт, что связь между mTORC1, mTORC2 и де!
MeCP2 регулирует сигналинг mTORC1, еще до
фицитом UBE3A изучена недостаточно, необхо!
конца не выяснен, обнаружено, что у мышей,
димы дальнейшие исследования, направленные
моделирующих СР, снижены уровни BDNF, что,
на выяснение вопроса о том, как связаны анома!
возможно, и приводит к снижению активности
лии мозга пациентов с СА с нарушением равно!
путей PI3K/mTORC1 (рисунок) [55].
весия активностей mTORC1 и mTORC2, и можно
Воздействие на нейроны, производные из
ли возлагать надежды на mTORC1/mTORC2!
MeCP2!дефицитных ППСК, экзогенными
направленную терапию СА.
факторами роста (IGF!1 или BNDF) или гене!
Синдром Ретта (СР). Хотя при СР и ФМС
тическое «выбивание» PTEN интенсифицирует
каскад mTOR затрагивается лишь косвенно (мо!
синтез белка посредством усиления сигнальной
лекулярные «отклонения от нормы» лежат «вне»
активности PI3K/mTORC1 и нормализует раз!
каскада mTOR), эти синдромы можно считать
мер сомы и образование и ветвление отрост!
«вторичными» проявлениями нарушения ак!
ков [94]. Хотя необходимы дальнейшие иссле!
тивности каскада mTOR, которые могут вносить
дования для того, чтобы понять, как дисфунк!
вклад в развивающиеся при этих синдромах ау!
ция белка MeCP2 приводит к развитию СР, уже
тистические симптомы.
сейчас можно сказать, что все находки свиде!
СР носит характер прогрессирующего ней!
тельствуют о связи дефектов глобального конт!
роонтогенетического расстройства, наступаю!
роля транскрипции и опосредованной
щего после начального периода нормального
PI3K/mTORC1 трансляции с патогенетически!
перинатального развития. Хотя этот синдром
ми механизмами СР и, возможно, аутистичес!
исключен из списка РАС в пятой редакции Ди!
кими симптомами.
агностического и Статистического Руководства
Синдром Фелан-МакДермид (ФМС). ФМС -
по Психическим Расстройствам (DSM!5), у де!
нейроонтогенетическое расстройство, обуслов!
тей с СР часто наблюдается аутистическое пове!
ленное микроделецией 22q13, затрагивающей
дение, и встречаемость симптомов РАС при СР
локус гена SHANK3. Ген SHANK3 кодирует со!
у девочек оценивается в ~61% [89].
держащий SH3!домен и множественные анки!
СР обусловлен мутацией в гене MECP2 на Х!
риновые повторы белок 3 (SHANK3). SHANK3 -
хромосоме с утратой его функции, ген кодирует
структурный белок постсинаптической плот!
метил!CpG!связывающий белок 2, контролиру!
ности, ответственный за ассоциацию и поддер!
ющий экспрессию генов и ремоделирование
жание структуры комплексов глутаматных ре!
хроматина. Структурно!функциональные ано!
цепторов в синапсах, который, как полагают,
малии мозга при СР представлены в таблице.
отвечает за развитие клинической картины за!
Аномалии структуры нейронов воспроизводят!
болевания [95]. Около 80% пациентов с ФМС
ся в моделях СР на грызунах [54] и моделях
получают диагноз РАС. У мышей, моделирую!
in vitro с использованием MECP2!дефицитных
щих генетическую аномалию
- делецию
нейронов, полученных из ППСК [92].
SHANK3 (как глобально, так и только в глутамат!
Хотя механизм, лежащий в основе наблюда!
ергических нейронах), обнаружены аномалии
емых явлений, пока не совсем ясен, в моделях с
нейронов и синаптической проводимости [56].
мутацией MECP2, вызывающей фенотипичес!
Фосфопротеомные исследования нейронов
кие проявления СР, обнаружено снижение бел!
грызунов, лабораторных моделей ФМС с нокда!
кового синтеза и активности mTORC1 [20].
уном гена SHANK3, выявили снижение фосфо!
БИОХИМИЯ том 86 вып. 5 2021
656
БОКША и др.
рилирования Akt и mTORC1, и закономерности
торого необходимо для правильного функцио!
воспроизводились на полученных из ППСК
нирования мозга, а выход за диапазон приводит
нейронах от пациентов с ФМС [57]. Это сниже!
к поведенческим аномалиям. Детально данная
ние было обусловлено накоплением CDC!по!
ситуация была смоделирована математичес!
добной киназы 2 (CLK2) - негативного регуля!
ки [100].
тора Akt, вследствие отсутствия SHANK3, а ин!
гибитор CLK2 улучшал социальные взаимодей!
ствия животных с делецией SHANK3 [57]. Важно
СИГНАЛЬНЫЕ КАСКАДЫ, ВКЛЮЧАЯ
отметить, что у мышей с дупликацией SHANK3
mTORC1, ПРИ НЕСИНДРОМНЫХ (НС6РАС)
обнаруживалось сниженное фосфорилирование
НЕ МОНОГЕННЫХ ФОРМАХ РАС
mTOR в мозге при нормальном уровне фосфо!
рилирования Akt, что является отличительной
Дупликация 15q11-13 (Dup15q). Унаследо!
чертой молекулярного механизма этой моде!
ванные по материнской линии дупликации
ли [58]. В опытах in vitro введение гена SHANK3
15q11-13 - один из наиболее часто встречаю!
дикого типа в нейроны, выращенные из инду!
щихся вариантов НС!РАС, он обнаруживается у
цированных ППСК пациентов с ФМС, восста!
~1-2% пациентов с РАС и свидетельствует о
навливало нормальный фенотип [96]. SHANK3,
том, что гены, находящиеся в этой области, бу!
вероятно, вовлечён в путь, модулирующий ак!
дучи представленными в нескольких копиях,
тивность mTOR, но подробности взаимодей!
могут приводить к развитию РАС. Делеции в
ствий предстоит изучить в будущем.
этой же области приводят к синдрому Праде!
Поскольку, в отличие от остальных С!РАС,
ра-Вилли или СА в зависимости от того, уна!
рассмотренных выше, при СР и ФМС сигналь!
следована ли делеция по отцовской или материн!
ная активность mTORC1 не повышена, а, нао!
ской линии соответственно, а, как обсуждалось
борот, снижена, непонятно, связана ли аберрант!
выше, аутистические симптомы обычны для па!
ная работа каскада mTOR с поведенческим фе!
циентов с СА. Нестабильность генома в этой об!
нотипом или же это непрямой результат нейро!
ласти опосредована наличием пяти тандемных
нальной дисфункции, обусловленной мутацией.
повторов с низким числом копий, обозначае!
Воздействие на животных, моделирующих СР,
мых BP1-BP5 (Breakpoints). Заметим, что хотя
IGF!1 приводило к увеличению продолжитель!
дупликации по отцовской линии не характери!
ности жизни, изменениям в морфологии и фи!
зуются высокой пенетрантностью, на мышиной
зиологии нейронов, восстанавливало работу
модели с унаследованной по отцовской линии
возбуждающих нейромедиаторных систем [96].
дупликацией района BP2-BP3 (мыши patDp/+)
IGF!1 в настоящее время проходит клиничес!
обнаружено расстройство поведения, напоми!
кие испытания при СР и ФМС, и известно, что
нающее симптомы, наблюдаемые при РАС, а
сигналинг IGF!1 реализуется посредством пути
также нарушения развития мозга [59].
PI3K и повышает активность mTORC1 [97], хотя
Кроме того, есть указания на то, что область
функция mTORC1 в этих исследованиях напря!
между BP1 и BP2 (15q11.2) также является «го!
мую не изучалась.
рячей точкой», критичной для проявления РАС,
Синдром Аспергера. Ещё одно расстройство,
и что гены, локализованные в этой области,
отнесённое к спектру РАС по современной меж!
важны для нормального функционирования
дународной классификации болезней МКБ!
нервной системы и поведенческих функ!
10, - это синдром Аспергера. Известны две пуб!
ций [101]. Из четырёх генов!кандидатов, лока!
ликации [98, 99] с описанием двух отдельных
лизованных между BP1 и BP2, ген CYFIP1 явля!
случаев, когда постановку такого диагноза уда!
ется наиболее важным кандидатом на роль,
лось связать с генетическими аномалиями, при!
обусловливающую РАС [37]: его продукт напря!
чём в одном случае - с мутацией гена, кодирую!
мую взаимодействует с FMRP и eIF4E и служит
щего SHANK3, а в другом - с дупликацией
посредником в репрессии трансляции посред!
22q13. В большинстве же случаев это расстрой!
ством FMRP в мозге (рисунок), а также регули!
ство, по!видимому, следует отнести к И!РАС,
рует полимеризацию актина при ремоделирова!
поскольку причины его неизвестны, и, к сожа!
нии цитоскелета посредством взаимодействия с
лению, неизвестны работы, в которых бы изуча!
ГТФазой Rac1 (из семейства малых G!белков).
лись каскады белкового фосфорилирования при
Показано, что уровни белка CYFIP1 повышены
данной патологии.
в клетках лимфобластомы и постмортальной
Все перечисленные сведения позволяют
ткани мозга пациентов с РАС, имеющих дупли!
высказать предположение о том, что может су!
кацию Dup15q [37], и сверхэкспрессия кодиру!
ществовать некий диапазон активности сиг!
ющего его гена в культивируемых нейронах че!
нального каскада mTOR, сохранение внутри ко!
ловека и мыши приводит к аномалиям структу!
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
657
ры нейронов. Сходные аномалии проявляются и
торной области гена EIF4E, которые, как пред!
у мышиных моделей с избыточным синтезом
полагается, приводят к усилению активности
белка CYFIP1 [37].
промотора [103]. У трансгенных мышей с повы!
Заметим, что сверхактивация mTORC1 свя!
шенным уровнем белка eIF4E и у мышей, у ко!
зана с увеличением размеров нейронов и уси!
торых отсутствует 4E!BP2, наблюдаются анома!
ленным ветвлением нейритов в культуре нейро!
лии мозга, подобные тем, что наблюдаются у
нальных клеток со сверхэкспрессией CYFIP1 в
других моделей РАС, включая аномально уси!
локусе 15q11.2, ассоциированном с риском
ленную cap!зависимую трансляцию в мозге.
РАС [37]. С другой стороны, в моделях СР сни!
Воздействие на мышей, у которых отсутствует
женная активность mTORC1 связана со сниже!
4E!BP (нокаут EIF4EBP2) ингибитором, подав!
нием размеров нейронов и аномалиями ветвле!
ляющим взаимодействие eIF4E и eIF4G, восста!
ния нейритов [94]. Это свидетельствует о том,
навливает синтез белка и устраняет аномалии
что для поддержания должного размера нейро!
синаптической пластичности, а также нормали!
нов и мозга и нормального паттерна ветвления
зует поведение, до воздействия напоминавшее
нейритов нужен оптимальный уровень актив!
РАС. Эти факты дают основание установить
ности сигналинга mTORC1, а вызываемые нару!
причинно!следственные связи между eIF4E!
шением регуляции активности mTORC1 анома!
НС!РАС и избыточной cap!зависимой трансля!
лии развития нервной системы могут вносить
цией, а также свидетельствуют о том, что это од!
вклад в развитие симптомов РАС.
на из мишеней, посредством которой ингибито!
Однако необходимы дальнейшие исследова!
ры mTORC1 могут восстановить синаптическую
ния, чтобы ответить на вопрос о том, проявляют
пластичность и устранить симптомы, характер!
ли трансгенные CYFIP1!мыши аутистические
ные для РАС.
черты, характерные для поведения при РАС.
Идиопатический аутизм (И6РАС). В качестве
Важно, что при изучении постмортального
возможных причин развития идиопатических
мозга носителей дупликации Dup15q больных
РАС рассматриваются воздействие факторов ок!
РАС, а также эмбрионов трансгенных мышей
ружающей среды (токсины, пестициды, инфек!
CYFIP1 и культивируемых нейронов с повышен!
ции), нарушение внутриутробного развития под
ным содержанием белка CYFIP1 были получе!
действием различных факторов, включая валь!
ны свидетельства аномального усиления сигна!
проевую кислоту (вальпроат) и случайные мута!
линга mTORC1. Воздействие рапамицином на
ции de novo.
культивируемые нейроны приводило к исчезно!
Основные используемые эксперименталь!
вению наблюдаемых аномалий в размерах кле!
ные модели исследования И!РАС - это живот!
ток, ветвлении и длине отростков [37]. Свиде!
ные (мыши и крысы), аномалии развития кото!
тельство гиперфункции сигналинга mTORC1
рых индуцированы вальпроатом [11], инбред!
также наблюдалось в культивируемых индуци!
ные мыши с фенотипическими поведенческими
рованных ППСК пациентов с НС!РАС с дупли!
проявлениями, характерными для аутизма (на!
кацией Dup15q [102]. Все вместе эти факты сви!
пример, мыши BTBR T+Itpr3tf/J [60]), лабора!
детельствуют о том, что опосредованная CYFIP1
торные модели материнской иммунной актива!
сверхактивация пути mTORC1 может вносить
ции (МИА) на животных [104] и индуцирован!
вклад в патогенез РАС при дупликации Dup15q.
ные ППСК от пациентов [10].
Учитывая данные о том, что у пациентов с
Интересно, что в индуцированной вальпроа!
Dup15q обнаружена аберрантная активность
том мышиной модели И!РАС обнаружено нару!
mTORC1 [102], представляют интерес клини!
шение регуляции 4E!BP1 [11].
ческие испытания, в которых была бы оценена
Подобно нокаутным мышиным моделям мо!
способность лекарств, действующих на каскад
ногенных С!РАС, инбредные мыши BTBR
mTORC1, облегчать поведенческие аномалии
T+Itpr3tf/J [60] с нарушениями в когнитивной
при Dup15q.
сфере и социальном поведении могут служить
НС6РАС, ассоциированные с eIF4E (eIF4E6
моделью для исследования И!РАС. При подроб!
НС6РАС). mTOR в составе комплекса mTORC1
ном исследовании модели BTBR T+Itpr3tf/J была
фосфорилирует связывающие eIF4E белки (4E!
обнаружена конвергенция многих сигнальных
BP), что приводит к высвобождению их из
путей и проявления определенных нейроанато!
комплекса с eIF4E, в результате чего eIF4E при!
мических характеристик. Также обнаружено,
обретает способность взаимодействовать с
что рапамицин ослабляет нарушения социаль!
eIF4G и eIF4A с образованием комплекса
ного поведения этих животных. Исследования
eIF4F - критический этап в cap!зависимой
мозга (гиппокампа) мышей показало значитель!
трансляции (рисунок). У некоторых пациентов с
ное повышение уровня mTOR и фосфо!mTOR,
НС!РАС обнаружены редкие мутации в промо!
р70S6K1 и фосфо!p70S6K, AMPK и фосфо!
БИОХИМИЯ том 86 вып. 5 2021
658
БОКША и др.
AMPK, Ulk1 и фосфо!Ulk1 [105]. Нарушения
сформироваться комплексу eIF4F, при этом по!
регуляции сигнального пути mTOR и социаль!
вышается общая интенсивность белкового син!
ного поведения этих мышей исчезали при сис!
теза (рисунок).
темном введении IGF!2.
Субстратами активированной киназы
Известны лишь единичные работы, посвя!
p70S6K1 служат рибосомный белок S6 на субъ!
щенные исследованиям mTOR у пациентов с И!
единице 40S, несколько факторов трансляции,
РАС. Так, компоненты сигнального пути mTOR
включая eIF4B (стимулятор геликазной актив!
и MAPK при И!РАС анализировались в клетках
ности фактора eIF4A) и eEF2K (так же, как и са!
периферической крови (лимфоцитах) детей
мого mTORC1, на рисунке не показано). Таким
3-11 лет. Уровни экспрессии mTOR, 4E!BP1,
образом, p70S6K1 фосфорилирует и инактиви!
p70S6K1, FMRP, TSC1, TSC2 и Rheb не различа!
рует фактор репрессии трансляции eEF2K, ко!
лись между И!РАС и контролем, но было повы!
торый фосфорилирует и инактивирует фактор
шено содержание рибосомной субъедини!
элонгации трансляции eEF2, стимулирующий
цы 40S (rpS6) и фосфорилированной фор!
включение аминокислот в растущую полипеп!
мы eIF4E. Интересно, что уровни экспрессии
тидную цепь. Фосфорилирование eEF2K факто!
компонентов этих сигнальных путей, а именно
ра eEF2 предотвращает его ассоциацию с рибо!
rpS6, p!eIF4E, TSC1 и p!MNK1, различались в
сомой, таким образом снижая скорость элонга!
зависимости от клинического диагноза (степени
ции, и предполагается, что данный механизм
выраженности аутистических симптомов) [106].
реализуется при И!РАС [107]. Таким образом,
Как было установлено при изучении С!РАС,
при И!РАС важна не только и не столько актив!
изменения активности mTOR влияют на множе!
ность самого mTOR, сколько сбалансирован!
ство нейроонтогенетических процессов, вклю!
ность активностей его мишеней. При этом
чая дифференцировку нервной ткани, рост ак!
предполагается, что в снижении интенсивности
сонов, миграцию клеток, формирование паттер!
синтеза белка при посредстве p70S6K1 и eIF4B
на структур мозга, а при И!РАС отмечается на!
задействован также путь NGF, который здесь
рушение всех этих процессов. В современных
играет роль дополнительного регулятора, но
работах выдвигаются гипотезы, предполагаю!
насколько весом его вклад - пока неизвестно.
щие, что нарушение регуляции именно каскада
Тем не менее предполагается, что сами белки!
mTOR - центральное событие и вероятная при!
мишени регуляции mTOR - p70S6K1, eIF4B -
чина И!РАС [107].
могут послужить новыми терапевтическими ми!
В попытках связать сведения, полученные
шенями фармакологического действия препа!
при исследовании И!РАС (а при И!РАС обнару!
ратов при РАС [107].
жен сниженный уровень синтеза таких белков,
Кроме того, по сравнению с нормальным
как различные изоформы PI3K, Akt, mTOR,
развитием, при И!РАС обнаружены различия в
p!mTOR, p70S6K1, eIF4B [38]), была выдвинута
уровнях киназы рецептора тропомиозина TrkB,
гипотеза о «разветвлении» молекулярных собы!
а TrkB регулирует путь Akt/mTOR (рисунок) [10,
тий!причин С!РАС и И!РАС на уровне факто!
11]. Заметим, что роль различных изоформ Trk
ров инициации и элонгации белковой трансля!
при РАС пока не изучена, хотя роль, которую
ции (посредством киназы p70S6K1 - мишени
TrkA играет в пути NGF, известна: TrkA является
mTOR и eIF4B - мишени p70S6K1), причём ес!
рецептором NGF, и этот путь имеет значение в
ли в большинстве случаев С!РАС синтез белка
патогенезе болезни Альцгеймера и синдро!
стимулируется, то при И!РАС - подавляет!
ма Дауна.
ся [38, 107].
В таблице суммированы наиболее часто
Поскольку предполагается, что в патогенезе
встречающиеся аномалии мозга при И!РАС.
И!РАС задействован не сам mTOR, а его мише!
Рассматривая данные относительно молеку!
ни, необходимо отдельно рассмотреть роль
лярной основы патологии дендритных шипиков
mTOR в регуляции cap!зависимой трансляции.
при РАС, можно отметить исследования
Инициация белковой трансляции регулиру!
Nicolini et al.
[38], показавшие, что путь
ется в мозге рядом белковых факторов инициа!
Akt/mTOR, регулирующий трансляцию белка в
ции трансляции (eIF3, eIF4A, eIF4B, eIF4G и
дендритных шипиках, является потенциальным
cap!связывающим eIF4E). Механизм, посред!
молекулярным субстратом этой патологии, что,
ством которого mTOR регулирует трансляцию,
возможно, играет роль при И!РАС. Их исследо!
реализуется через мишени
-
4E!BP или
вания на постмортальном мозге 11 больных с И!
p70S6K1. 4E!BP связывается с eIF4E и ингиби!
РАС показали снижение активности сигналинга
рует инициацию трансляции. Когда mTORC1
mTORC1 [38]. Авторы подчеркивают, что сни!
фосфорилирует 4E!BP1, сродство 4E!BP1 к
женная активность каскада mTOR может небла!
eIF4E снижается, что позволяет эффективно
гоприятно влиять на формирование шипиков,
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
659
нарушая регуляцию кортикальных путей, задей!
также наблюдалось усиление экспрессии субъ!
ствованных в высших нервных функциях и по!
единицы p110δ киназы PI3K и синтеза белка,
ведении, что обусловливает аутистический фе!
который можно было подавить ингибитором,
нотип. Hutsler и Zhang [61] также выдвинули ги!
специфичным к p110δ [39].
потезу о том, что нарушение пути mTOR лежит в
Высокая гетерогенность И!РАС может объ!
основе аномалий формирования шипиков (их
яснять расхождения сведений относительно ак!
повышенной плотности) и вносит вклад в раз!
тивации либо снижения активности сигналинга
витие И!РАС.
mTORC1, поэтому необходимы дополнитель!
Однако разнобой в данных об активности
ные исследования для того, чтобы понять при!
mTOR и других систем фосфорилирования при
чинную связь между каскадом mTORC1 и дис!
И!РАС пока не позволяет сложить четкую кар!
функциями при И!РАС. Следовательно, хотя в
тину данных. Так, в отличие от Nicolini et al.
ряде исследований обнаружен дефект сигналин!
[38], в работе Onore et al. [30] при И!РАС обна!
га mTORC1 у субкогорт пациентов с И!РАС [38,
ружено повышение общей концентрации IRS!1
39, 102], нужны дальнейшие функциональные
и повышение уровней фосфорилирования
исследования для получения доказательств свя!
PTEN, TSC2 и mTOR у больных с аутизмом по
зи между аномалиями mTORC1 и развитием ау!
сравнению с нормой; наблюдалась повышенная
тизма в такой гетерогенной группе, как И!РАС.
активность пути Akt/mTOR, внеклеточной ре!
Исследования с выделенными от пациентов
цепторной киназы и p70S6K и сниженная ак!
не нейрональными клетками [39, 102], в том
тивность киназы гликогенсинтазы 3α (GSK3α)
числе тромбоцитами и лимфоцитами, предо!
и туберина (TSC2).
ставляют возможность широкомасштабного
Хотя описанные аномалии могут быть вы!
скрининга сигнальных дефектов mTORC1 с ис!
званы различными причинами в результате кон!
пользованием наиболее легко доступных биоло!
вергенции событий в ходе развития нервной
гических материалов от пациентов. Это помо!
системы, исследования показывают, что в об!
жет ответить на вопрос, какова доля пациентов с
ширном ряду И!РАС имеются по меньшей мере
нарушениями mTORC1 среди всех пациентов с
подгруппы с отклонением сигнальной актив!
РАС неизвестной этиологии, и будет способ!
ности mTORC1 от нормы, причем, возможно,
ствовать выявлению тех пациентов, которым,
как в сторону снижения, так и повышения.
возможно, принесут пользу лекарственные
Так, в постмортальном мозге пациентов с И!
средства с действием, направленным на каскад
РАС наблюдалась сверхактивация каскада
mTORC1 либо его мишени.
mTORC1, а также исследования показали нару!
шения аутофагии и синаптического прунинга в
детском и юношеском возрасте, приводящие к
СИГНАЛЬНЫЕ КАСКАДЫ БЕЛКОВОГО
увеличению плотности дендритных шипиков и,
ФОСФОРИЛИРОВАНИЯ В КЛЕТКАХ КРОВИ
следовательно, усилению возбуждающей актив!
ности [62]. С этим согласуется тот факт, что в
В качестве объектов исследования сигналь!
культивированных нейронах, производных от
ных каскадов белкового фосфорилирования в
индуцированных ППСК, выделенных от паци!
клетках периферической крови при РАС ис!
ентов с И!РАС, обнаружено нарушение равно!
пользовались лимфоциты и тромбоциты. В ос!
весия E/I синапсов [108].
новном клетки крови применялись при модели!
Сходные наблюдения описаны и при иссле!
ровании синдрома X!ФРА. Сравнение тромбо!
довании мышиной модели КТС (С!РАС). Это
цитов с лимфоцитами в общем говорит в пользу
подтверждает, что для постнатального прунинга
первых: их легче выделить в большем количест!
нейронов спинного мозга необходимо своевре!
ве, их протеом более стабилен при выделении и
менное снижение активности mTORC1 [62].
в течение времени в организме, они устроены
Повышенный уровень фосфорилирования
проще, и их сигнальные пути менее вариабель!
белков (уровень фосфорилированного mTOR
ны под влиянием физиологических изменений,
(p!mTORS2448) и его мишеней) и повышенная
что позволяет провести на них биохимические
активность mTORC1 наблюдались и в не нейро!
исследования, в том числе и при РАС. Период
нальных клетках, производных индуцирован!
продолжительности жизни тромбоцитов коро!
ных ППСК, выделенных от троих из 13 обследо!
че (~10 дней), чем лимфоцитов (от нескольких
ванных пациентов с И!РАС, хотя наблюдаемые
недель до лет), поэтому изменения, происходя!
изменения сильно зависели от эксперименталь!
щие в экспрессии генов в мегакариоцитах, срав!
ных условий выделения и индукции ППСК [39,
нительно быстро отражаются на протеоме тром!
102]. Интересно, что в клетках лимфобластомы
боцитов. В неактивированных тромбоцитах не
пациента с усиленной активностью mTORC1
задействованы сигнальные пути, они не реаги!
БИОХИМИЯ том 86 вып. 5 2021
660
БОКША и др.
руют на стимулы, иные, чем стимулы тромбооб!
таллопротеиназы MMP9), однако данные, полу!
разования. В отличие от тромбоцитов, лимфо!
ченные на этой модели, оказались бесполезны!
циты реагируют на значительное число внеш!
ми при клинических исследованиях в предик!
них стимулов и сравнительно более метаболи!
ции эффективности разрабатываемых лекар!
чески активны. Их центральные киназы, напри!
ственных препаратов при их апробации в конт!
мер ERK, активно участвуют во многих сигналь!
ролируемых исследованиях на пациентах. Таким
ных путях, и наблюдать не слишком выражен!
образом, потребность в модели РАС (в том чис!
ные изменения, связанные, например, с патоло!
ле, X!ФРА и И!РАС), основанной на клетках пе!
гией РАС, в них трудно. Хотя сигнальные пути в
риферических тканей человека, очевидна, и из!
тромбоцитах существенно упрощены по сравне!
вестны примеры таких разработок на лимфоци!
нию с нейронами, они происходят от различных
тах, тромбоцитах [72] и фибробластах пациен!
клеток!предшественников в эмбриональном
тов [111].
развитии, и молекулярный интерактом тромбо!
Однако, по сравнению с лимфоцитами и
цитов не такой обширный, как нейронов, тем не
фибробластами, а также по сравнению С!РАС с
менее тромбоциты признаны удачной перифе!
другими формами РАС (НС!РАС и И!РАС в це!
рической моделью исследования нейронов и ис!
лом), наиболее веские основания получены в
пользуются при изучении компонентов нейро!
пользу применения тромбоцитов как модели
медиаторных систем [109]. Интерактом тромбо!
исследования X!ФРА [72]. Основой адекватнос!
цитов содержит важнейшие сигнальные каска!
ти модели является наличие FMRP в тромбоци!
ды белкового фосфорилирования, вторичные
тах и их предшественниках - мегакариоцитах.
мессенджеры и сигнальные киназы, рассматри!
Отметим, что как в мегакариоцитах, так и в ак!
ваемые как «хабы» биохимических путей [110].
тивируемых глутаматом mGluR!содержащих
Благодаря биохимической «гомологии» тромбо!
нейронах, кроме FMRP, обнаружены и другие
цитов с нейронами тромбоциты могут служить
РНК!связывающие белки: STAU1, STAU2,
адекватной клеточной биохимической «сурро!
CASC3 [112]. Функциональная роль FMRP (ста!
гатной» моделью X!ФРА [72], их относительно
билизация и транспорт мРНК) в мегакариоци!
легко получать малоинвазивным способом за!
тах вполне отражает его роль в нейронах [112].
бора периферической крови, выделять, а в изо!
Поскольку тромбоциты происходят из мегака!
лированном виде в них сохраняется активность
риоцитов, образуясь путём выпячивания и от!
тромбоцитарных ферментов.
почковывания цитоплазмы, содержащей все
Тромбоциты при нарушениях нейроонтоге!
компоненты тромбоцитов, для доставки мРНК
неза исследованы мало, и, хотя описаны неко!
в отпочковывающиеся тромбоциты с помощью
торые общие биохимические аномалии в тром!
микротрубочек используются РНК!связываю!
боцитах при РАС, трудно понять, насколько они
щие белки, в том числе FMRP.
имеют отношение к патогенезу аутизма из!за
В настоящее время диагностика синдрома X!
слабой изученности патологии мозга при РАС,
ФРА основана на ПЦР и Саузерн!блот!анализе,
особенно И!РАС, и пока неизвестно, коррели!
но также предложены тесты, основанные на оп!
руют ли эти периферические аномалии в тром!
ределении концентрации FMRP в лимфоцитах
боцитах с процессами, происходящими в мозге.
или волосяных фолликулах иммунохимическим
Биохимические исследования тромбоцитов при
методом с антителами к FMRP. И хотя с приме!
РАС показали ряд отклонений от контроля, на!
нением этих методов была обнаружена связь
пример, в концентрации тромбоцитарного се!
концентрации FMRP с уровнем интеллекта, в
ротонина и серотонинового метаболита N!аце!
случае мозаицизма чувствительность методов
тилсеротонина, плотности серотонинового пе!
оказалась ограниченной: чувствительность и
реносчика, концентрации гамма!аминомасля!
специфичность метода, доходившие до 100%
ной кислоты (ГАМК), секреции гранул с АТP, но
у мужчин, у женщин оказались значительно ни!
ни один из перечисленных показателей не был
же и составили всего 41-43%. Результаты же из!
валидирован как биомаркёр для использования
мерения уровня тромбоцитарного FMRP в
в клинике при диагностике РАС.
контрольной группе и у больных с подтвержден!
Хотя упомянутая модель X!ФРА Fmr1 KO на
ным X!ФРА показали, что больных можно уве!
мышах позволила открыть биохимические зако!
ренно отделить от здоровых (площадь под ROC!
номерности, наблюдающиеся при утрате
кривой 0,948). Содержание FMRP в тромбоци!
FMRP (повышенная активность Gp1 mGluR!за!
тах также оказалось связанным с уровнем ин!
висимого сигналинга, сниженный уровень об!
теллекта в тесте Векслера. Что касается исследо!
разования цАМP, снижение активности ингиби!
ваний белков, компонентов каскадов белкового
торной ГАМК!ергической нейромедиаторной
фосфорилирования при X!ФРА с использова!
системы, усиление трансляции матриксной ме!
нием тромбоцитов, проведены лишь первые
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
661
эксперименты на ограниченном числе боль!
ны в мозге при X!ФРА. Некоторые лекарствен!
ных [113], при этом авторы отметили влияние
ные вещества, влияющие на метаболизм и си!
условий выделения и обработки биопроб на
наптическое высвобождение моноаминовых
оценки степени фосфорилирования белков.
нейромедиаторов в нейронах (серотонин, дофа!
Ряд ключевых молекулярных нарушений ре!
мин, норадреналин) сходным образом действу!
гуляции, обнаруженных в нейронах человека
ют и на тромбоциты. Примечательно, что инги!
при X!ФРА и у моделирующих Х!ФРА мы!
биторы ацетилхолинэстеразы, используемые
шей Fmr1 KO, воспроизводятся в тромбоцитах,
для лечения симптомов болезни Альцгеймера,
что также подтверждает возможность их ис!
нормализуют нарушенную регуляцию метабо!
пользования для моделирования X!ФРА, на!
лизма тромбоцитарного АРР параллельно со
пример гиперактивность путей MAPK/ERK и
стабилизацией снижения когнитивных функ!
PI3K/Akt/mTOR. Так, показано, что при синд!
ций: эти факты важны и для исследований в об!
роме X!ФРА в тромбоцитах усилено фосфори!
ласти аутизма, поскольку, как отмечалось выше,
лирование ERK и Akt по сравнению с нормой
APP является одним из регуляторов синапти!
примерно в 1,6 раз, и это свидетельствует о том,
ческой пластичности, связанным с сигнальным
что в норме FMRP регулирует эти сигнальные
каскадом mTOR [84].
пути [113]. Также были обнаружены отрицатель!
В будущем, для того чтобы расширить воз!
ные корреляции между уровнями фосфорили!
можности использования тромбоцитарной мо!
рованных pERK и фосфорилированной по
дели, резонно исследовать, изменен ли уровень
Ser473 Akt (pAktSer473) и когнитивными функци!
синтеза белка в тромбоцитах пациентов с X!
ями по тесту Векслера.
ФРА и других С!РАС, поскольку повышенный
Кроме того, удалось оценить уровень вариа!
уровень синтеза белка обнаружен в лимфоцитах
бельности (повышения) фосфорилирования
и фибробластах пациентов с X!ФРА.
ERK и Akt в зависимости от уровня FMRP в
Эти находки свидетельствуют о том, что изу!
тромбоцитах. Оказалось, что в подгруппе с
чение множества биохимических параметров
уровнем тромбоцитарного FMRP, не поддаю!
тромбоцитов окажется полезным в предикции
щимся обнаружению, достоверно усилено фос!
когнитивного фенотипа индивидуумов с
форилирование ERK (отношение pERK/ERK
РАС (по меньшей мере при X!ФРА), что, в свою
повышено в 2,27 раз) и Akt (pAktSer473/Akt повы!
очередь, может способствовать улучшению кли!
шено в 2 раза). А у индивидуумов!мозаиков,
нического сопровождения пациентов. А пока
сохраняющих некоторый уровень экспрессии
известны лишь отдельные попытки применения
FMRP, превышение над контролем уровней
параметров белкового фосфорилирования в ка!
фосфорилирования этих киназ было незначи!
честве биомаркёра при испытании действия
тельным и недостоверным [113]. Более того, ле!
препаратов в целях предикции терапевтическо!
чение ловастатином в течение трех месяцев кор!
го ответа на лекарства: это степень активации
ректировало гиперактивацию ERK в тромбоци!
ERK в лимфоцитах в клинических исследовани!
тах X!ФРА пациентов [72], однако ловастатин
ях лития, рилузола, и статус базового фосфори!
не снижал достоверно уровень фосфорилирова!
лирования ERK в тромбоцитах в исследовании
ния Akt (pAktSer473/Akt) по сравнению с базовым
ловастатина при X!ФРА [72].
уровнем. Есть косвенные указания на то, что на
Тромбоциты не только воспроизводят био!
активность ERK могут влиять психостимулято!
химические нарушения, обнаруженные в ней!
ры, антидепрессанты, антипсихотики и лекар!
ронах при X!ФРА, но также и «реагируют» на
ства, снижающие артериальное давление, хотя
лекарства, направленные на центральную нерв!
прямых данных пока не получено.
ную систему и на молекулярные механизмы, ле!
Для сравнения, в исследованиях белкового
жащие в основе патологии. В связи с этим такой
фосфорилирования лимфоцитов при X!ФРА
биомаркёр, как тромбоцитарный уровень фос!
обнаружено превышение над контролем фосфо!
форилирования ERK, применялся в исследова!
рилирования Akt [29, 76], но не ERK, что сильно
нии ловастатина [72]. Примечательно, что под
ставит под удар адекватность лимфоцитарной
действием лечения повышенный уровень ос!
модели как модели патологии X!ФРА. Следова!
новного фосфорилирования ERK поддался кор!
тельно, тромбоциты - более адекватная модель,
рекции у большинства обследованных [72].
которая полнее отражает события, происходя!
Кроме того, изменения фосфорилирования
щие в нейронах, по меньшей мере при X!ФРА.
ERK коррелировали с клиническим ответом,
Важно, что в тромбоцитах обнаружены ре!
судя по оценке Шкалы адаптивного поведения
цепторы нейромедиаторных систем (глутамат!
Вайнленд (VABS) [72]. Значит, параметры тром!
ной, серотониновой, дофаминовой), наруше!
боцитов могут помочь и в оценках эффектив!
ния со стороны которых были зарегистрирова!
ности лечения препаратами, нацеленными на
БИОХИМИЯ том 86 вып. 5 2021
662
БОКША и др.
путь MAPK/ERK, хотя ограничения в оценках
раннее применение разрабатываемых лекар!
улучшения состояния и исходов лечения паци!
ственных препаратов.
ентов с РАС вносят трудности в применение био!
Поскольку не всегда при РАС нарушения
маркёров в целом. Тем не менее «коррекция»
сигнальных каскадов фосфорилирования затра!
уровней периферических биомаркёров свиде!
гивают именно путь mTOR, но также и располо!
тельствует о том, что наблюдаемые улучшения в
женные выше него эффекторы, и его мишени,
клинических испытаниях можно отнести к бла!
и, кроме того, сопряженные с ним пути, перс!
гоприятному действию лекарства на централь!
пективна разработка комплексных, сочетанных
ную патофизиологию X!ФРА.
препаратов, воздействующих (ингибирующих)
Гипотетически другие сигнальные пути
не только mTOR, но и эти, затронутые патоло!
тромбоцитов и белки сигнального каскада
гическими изменениями, звенья.
mTOR могут послужить биомаркёрами в клини!
Применение тромбоцитов в качестве модели
ческих испытаниях эффективности лекарств,
биохимических исследований аномалий белко!
действие которых направлено на сигнальные
вого фосфорилирования при РАС позволило бы
регуляторные пути.
проверить гипотезы и о наличии «подгрупп» па!
То, что компоненты сигнального каскада
циентов с РАС, у которых повышена либо сни!
mTOR и сопряженных с ним путей обнаружены
жена активность mTOR и сопряженных с ним
в тромбоцитах, дает возможность опробовать
сигнальных каскадов фосфорилирования, и о
эти белки в качестве претендентов на роль био!
необходимости регуляции активности сигналь!
химических биомаркёров с целью выделения из
ного каскада mTOR в определенных границах
клинических групп больных РАС тех подгрупп,
(диапазоне), и об актуальности временнóго «ок!
в которых будут обнаружены отклонения актив!
на» в регуляции активности mTOR в процессе
ности сигнального каскада mTOR от контроль!
онтогенетического развития детей как можно в
ных диапазонов, для возможных целенаправ!
более раннем возрасте, т.е. ответить на вопросы,
ленных клинических испытаний действия спе!
поднятые в начале обзора.
цифических препаратов. Учитывая современ!
ный уровень разработки проблемы, это направ!
ление исследований авторам обзора представля!
Конфликт интересов. Авторы заявляют об от!
ется наиболее перспективным и реальным по
сутствии конфликта интересов.
выполнимости в ближайшее время.
Благодарности. Авторы признательны д!ру
Так как эксперименты с моделями С!РАС
Ричарду Лозиеру за помощь в подготовке версии
показали, что ингибиторы mTOR и сопряжен!
рукописи на английском языке.
ных с ним путей способны предотвратить, но не
Соблюдение этических норм. Настоящая
обратить развитие структурных аномалий и ау!
статья не содержит описания выполненных ав!
тистического фенотипа, можно предположить,
торами исследований с участием людей или ис!
что эффективным окажется как можно более
пользованием животных в качестве объектов.
СПИСОК ЛИТЕРАТУРЫ
1.
Simashkova, N. V., Boksha, I. S., Klyushnik, T. P.,
6. Bockaert, J., and Marin, P. (2015) mTOR in brain physiol!
Iakupova, L. P., Ivanov, M. V., and Mukaetova!Ladinska,
ogy and pathologies, Physiol. Rev., 95, 1157!1187.
E. B. (2019) Diagnosis and management of autism spec!
7. Lipton, J. O., and Sahin, M. (2014) The neurology of
trum disorders in russia: clinical!biological approaches,
mTOR, Neuron, 84, 275!291.
J. Autism Dev. Disord., 49, 3906!3914.
8. Mandy, W., and Lai, M. C. (2016) Annual research review:
2.
Mukaetova!Ladinska, E. B., Simashkova, N. V.,
the role of the environment in the developmental psy!
Mukaetova, M. S., Ivanov, M. V., and Boksha, I. S. (2018)
chopathology of autism spectrum condition, J. Child
Autism spectrum disorders in children and adults: the
Psychol. Psychiatry, 57, 271!292, doi: 10.1111/jcpp.12501.
experience of reserches from different countries,
9. Miles, J. H., Takahashi, T. N., Bagby, S., Sahota, P. K.,
Zh. Nevrol. Psikhiatr. Im S. S. Korsakova, 118, 92!99.
Vaslow, D. F., et al. (2005) Essential versus complex autism:
3.
Trifonova, E. A., Khlebodarova, T. M., and Gruntenko,
definition of fundamental prognostic subtypes, Am. J. Med.
N. E. (2017) Molecular mechanisms of autism as a form of
Genet. A, 135, 171!180, doi: 10.1002/ajmg.a.30590.
synaptic dysfunction, Russ. J. Genet. Appl. Res., 7, 869!877.
10. Liu, X., Campanac, E., Cheung, H. H., Ziats, M. N.,
4.
Vorstman, J. A. S., Parr, J. R., Moreno!De!Luca, D.,
Canterel!Thouennon, L., et al. (2017) Idiopathic autism:
Anney, R. J. L., Nurnberger, J. I., Jr., and Hallmayer, J. F.
cellular and molecular phenotypes in pluripotent stem cell!
(2017) Autism genetics: opportunities and challenges for
derived neurons, Mol. Neurobiol., 54, 4507!4523.
clinical translation, Nat. Rev. Genet., 18, 362!376.
11. Nicolini, C., and Fahnestock, M. (2018) The valproic
5.
Chauvin, C., Koka, V., Nouschi, A., Mieulet, V., Hoareau!
acid!induced rodent model of autism, Exp. Neurol., 299,
Aveilla, C., et al. (2014) Ribosomal protein S6 kinase activ!
217!227.
ity controls the ribosome biogenesis transcriptional pro!
12. Trifonova, E. A., Klimenko, A. I., Mustafin, Z. S., Lashin,
gram, Oncogene, 33, 474!483.
S. A., and Kochetov, A. V. (2019) The mTOR signaling
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
663
pathway activity and vitamin d availability control the
31.
Hevner, R. F. (2015) Brain overgrowth in disorders of
expression of most autism predisposition genes, Int. J. Mol.
RTK!PI3K!AKT signaling: a mosaic of malformations,
Sci., 20, 6332.
Semin. Perinatol., 39, 36!43.
13.
Sandin, S., Lichtenstein, P., Kuja!Halkola, R., Larsson,
32.
Yeung, K. S., Tso, W. W. Y., Ip, J. J. K., Mak, C. C. Y.,
H., Hultman, C. M., and Reichenberg, A. (2014) The
Leung, G. K. C., et al. (2017) Identification of mutations
familial risk of autism, JAMA, 311, 1770!1777.
in the PI3K!AKT!mTOR signalling pathway in patients
14.
Gaugler, T., Klei, L., Sanders, S. J., Bodea, C. A.,
with macrocephaly and developmental delay and/or
Goldberg, A. P.,et al. (2014) Most genetic risk for autism
autism, Mol. Autism, 8, 66.
resides with common variation, Nat. Genet., 46, 881!885.
33.
Berdichevsky, Y., Dryer, A. M., Saponjian, Y., Mahoney,
15.
DeRosa, B. A., El Hokayem, J., Artimovich, E., Garcia!
M. M., Pimentel, C. A., et al. (2013) PI3K!Akt signaling
Serje, C., Phillips, A. W., et al. (2018) Convergent pathways
activates mTOR!mediated epileptogenesis in organotypic
in idiopathic autism revealed by time course transcriptomic
hippocampal culture model of post!traumatic epilepsy,
analysis of patient!derived neurons, Sci. Rep., 8, 8423.
J. Neurosci., 33, 9056!9067.
16.
Wen, Y., Alshikho, M. J., and Herbert, M. R. (2016)
34.
Gilbert, J., and Man, H. Y. (2017) Fundamental elements
Pathway network analyses for autism reveal multisystem
in autism: from neurogenesis and neurite growth to synap!
involvement, major overlaps with other diseases and con!
tic plasticity, Front Cell Neurosci., 11, 359.
vergence upon MAPK and calcium signaling, PLoS One,
35.
Sun, J., Liu, Y., Moreno, S., Baudry, M., and Bi, X. (2015)
11, e0153329.
Imbalanced mechanistic target of rapamycin C1 and C2
17.
Liu, G. Y., and Sabatini, D. M. (2020) mTOR at the nexus
activity in the cerebellum of Angelman syndrome mice
of nutrition, growth, ageing and disease, Nat. Rev. Mol. Cell
impairs motor function, J. Neurosci., 35, 4706!4718.
Biol., 21, 183!203.
36.
Huber, K. M., Klann, E., Costa!Mattioli, M., and Zukin,
18.
Madigan, J. P., Hou, F., Ye, L., Hu, J., Dong, A., et al.
R. S. (2015) Dysregulation of mammalian target of
(2018) The tuberous sclerosis complex subunit TBC1D7 is
rapamycin signaling in mouse models of autism,
stabilized by Akt phosphorylation!mediated 14!3!3 bind!
J. Neurosci., 35, 13836!13842.
ing, J. Biol. Chem., 293, 16142!16159.
37.
Oguro!Ando, A., Rosensweig, C., Herman, E.,
19.
Parkhitko, A. A., Favorova, O. O., Khabibullin, D. I.,
Nishimura, Y., Werling, D., et al. (2015) Increased
Anisimov, V. N., and Henske, E. P. (2014) Kinase mTOR:
CYFIP1 dosage alters cellular and dendritic morphology
regulation and role in maintenance of cellular homeostasis,
and dysregulates mTOR, Mol. Psychiatry, 20, 1069!1078.
tumor development, and aging, Biochemistry (Moscow), 79,
38.
Nicolini, C., Ahn, Y., Michalski, B., Rho, J. M., and
88!101.
Fahnestock, M. (2015) Decreased mTOR signaling path!
20.
Winden, K. D., Ebrahimi!Fakhari, D., and Sahin, M.
way in human idiopathic autism and in rats exposed to val!
(2018) Abnormal mTOR activation in autism, Annu. Rev.
proic acid, Acta Neuropathol. Commun., 3, 3.
Neurosci., 41, 1!23.
39.
Poopal, A. C., Schroeder, L. M., Horn, P. S., Bassell, G. J.,
21.
Pupyshev, A. B., Korolenko, T. A., and Tikhonova, M. A.
and Gross, C. (2016) Increased expression of the PI3K cat!
(2018) Effects and mechanisms of rapamycin action on
alytic subunit p110δ underlies elevated S6 phosphorylation
experimental neurodegeneration, Neurochem. J., 12, 347!
and protein synthesis in an individual with autism from a
358.
multiplex family, Mol. Autism, 7, 3.
22.
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H.,
40.
Ruppe, V., Dilsiz, P., Reiss, C. S., Carlson, C.,
Hsu, P. P., et al. (2006) Prolonged rapamycin treatment
Devinsky, O., Zagzag, D., et al. (2014) Developmental
inhibits mTORC2 assembly and Akt/PKB, Mol. Cell, 22,
brain abnormalities in tuberous sclerosis complex: a com!
159!168.
parative tissue analysis of cortical tubers and perituberal
23.
Gleason, C. E., Oses!Prieto, J. A., Li, K. H., Saha, B.,
cortex, Epilepsia, 55, 539!550.
Situ, G., et al. (2019) Phosphorylation at distinct subcellu!
41.
Sato, A., Kasai, S., Kobayashi, T., Takamatsu, Y.,
lar locations underlies specificity in mTORC2!mediated
Hino, O., et al. (2012) Rapamycin reverses impaired social
activation of SGK1 and Akt, J. Cell Sci., 132, jcs224931,
interaction in mouse models of tuberous sclerosis complex,
doi: 10.1242/jcs.224931.
Nat. Commun, 3, 1292.
24.
Costa!Mattioli, M., and Monteggia, L. M. (2013) mTOR
42.
Telias, M., Kuznitsov!Yanovsky, L., Segal, M., and Ben!
complexes in neurodevelopmental and neuropsychiatric
Yosef, D. (2015) Functional deficiencies in fragile X neu!
disorders, Nat. Neurosci., 16, 1537!1543.
rons derived from human embryonic stem cells,
25.
Menon, S., Dibble, C. C., Talbott, G., Hoxhaj, G.,
J. Neurosci., 35, 15295!15306.
Valvezan, A. J., et al. (2014) Spatial control of the TSC
43.
Pacey, L. K., Guan, S., Tharmalingam, S., Thomsen, C.,
complex integrates insulin and nutrient regulation of
and Hampson, D. R. (2015) Persistent astrocyte activation
mTORC1 at the lysosome, Cell, 156, 771!785.
in the fragile X mouse cerebellum, Brain Behav.,
26.
Han, J. M., and Sahin, M. (2011) TSC1/TSC2 signaling in
5, e00400.
the CNS, FEBS Lett., 585, 973!980.
44.
Wang, X., Snape, M., Klann, E., Stone, J. G., Singh, A.,
27.
Dan, H. C., Ebbs, A., Pasparakis, M., Van Dyke, T.,
et al. (2012) Activation of the extracellular signal!regulated
Basseres, D. S., and Baldwin, A. S. (2014) Akt!dependent
kinase pathway contributes to the behavioral deficit of frag!
activation of mTORC1 complex involves phosphorylation
ile x!syndrome, J. Neurochem., 121, 672!679.
of mTOR (mammalian target of rapamycin) by IκB
45.
Lugo, J. N., Smith, G. D., Arbuckle, E. P., White, J.,
kinase α (IKKα), J. Biol. Chem., 289, 25227!25240.
Holley, A. J., et al. (2014) Deletion of PTEN produces
28.
Crespi, B. J. (2019) Comparative psychopharmacology of
autism!like behavioral deficits and alterations in synaptic
autism and psychotic!affective disorders suggests new tar!
proteins, Front. Mol. Neurosci., 7, 27.
gets for treatment, Evol. Med. Public Health, 2019, 149!168.
46.
Hobert, J. A., Embacher, R., Mester, J. L., Frazier,
29.
Hoeffer, C. A., and Klann, E. (2010) mTOR signaling: at
T. W., 2nd, and Eng, C. (2014) Biochemical screening and
the crossroads of plasticity, memory and disease, Trends
PTEN mutation analysis in individuals with autism spec!
Neurosci., 33, 67!75.
trum disorders and macrocephaly, Eur. J. Hum. Genet., 22,
30.
Onore, C., Yang, H., Van de Water, J., and Ashwood, P.
273!276.
(2017) Dynamic Akt/mTOR signaling in children with
47.
Vanderver, A., Tonduti, D., Kahn, I., Schmidt, J.,
autism spectrum disorder, Front. Pediatr., 5, 43.
Medne, L., et al. (2014) Characteristic brain magnetic res!
БИОХИМИЯ том 86 вып. 5 2021
664
БОКША и др.
onance imaging pattern in patients with macrocephaly and
the PERK!eIF2α signaling cascade, Cell Rep., 10, 684!
PTEN mutations, Am. J. Med. Genet. A, 164a, 627!633.
693.
48.
Huang, W. C., Chen, Y., and Page, D. T.
(2016)
65.
Zeng, L. H., Rensing, N. R., Zhang, B., Gutmann, D. H.,
Hyperconnectivity of prefrontal cortex to amygdala projec!
Gambello, M. J., and Wong, M. (2011) Tsc2 gene inactiva!
tions in a mouse model of macrocephaly/autism syn!
tion causes a more severe epilepsy phenotype than Tsc1
drome, Nat. Commun., 7, 13421.
inactivation in a mouse model of tuberous sclerosis com!
49.
Karlsgodt, K. H., Rosser, T., Lutkenhoff, E. S., Cannon, T.
plex, Hum. Mol. Genet., 20, 445!454.
D., Silva, A., and Bearden, C. E. (2012) Alterations in
66.
Meikle, L., Pollizzi, K., Egnor, A., Kramvis, I., Lane, H.,
white matter microstructure in neurofibromatosis!1, PLoS
et al. (2008) Response of a neuronal model of tuberous scle!
One, 7, e47854.
rosis to mammalian target of rapamycin (mTOR) inhibitors:
50.
Petrella, L. I., Cai, Y., Sereno, J. V., Gonçalves, S. I., Silva,
effects on mTORC1 and Akt signaling lead to improved sur!
A. J., and Castelo!Branco, M. (2016) Brain and behaviour
vival and function, J. Neurosci., 28, 5422!5432.
phenotyping of a mouse model of neurofibromatosis type!
67.
French, J. A., Lawson, J. A., Yapici, Z., Ikeda, H., Polster, T.,
1: an MRI/DTI study on social cognition, Genes Brain
et al. (2016) Adjunctive everolimus therapy for treatment!
Behav., 15, 637!646.
resistant focal!onset seizures associated with tuberous sclero!
51.
Kim, E., Wang, Y., Kim, S. J., Bornhorst, M., Jecrois,
sis (EXIST!3): a phase 3, randomised, double!blind, place!
E. S., et al. (2014) Transient inhibition of the ERK pathway
bo!controlled study, Lancet, 388, 2153!2163.
prevents cerebellar developmental defects and improves
68.
Mizuguchi, M., Ikeda, H., Kagitani!Shimono, K.,
long!term motor functions in murine models of neurofi!
Yoshinaga, H., Suzuki, Y., et al. (2019) Everolimus for
bromatosis type
1, Elife,
3, e05151, doi:
10.7554/
epilepsy and autism spectrum disorder in tuberous sclerosis
eLife.05151.
complex: EXIST!3 substudy in Japan, Brain Dev., 41, 1!10.
52.
Buiting, K., Williams, C., and Horsthemke, B. (2016)
69.
Saffari, A., Brösse, I., Wiemer!Kruel, A., Wilken, B.,
Angelman syndrome ! insights into a rare neurogenetic dis!
Kreuzaler, P., et al. (2019) Safety and efficacy of mTOR
order, Nat. Rev. Neurol., 12, 584!593.
inhibitor treatment in patients with tuberous sclerosis com!
53.
Maranga, C., Fernandes, T. G., Bekman, E., and da
plex under 2 years of age - a multicenter retrospective
Rocha, S. T. (2020) Angelman syndrome: a journey
study, Orphanet J. Rare Dis., 14, 96.
through the brain, FEBS J, 287, 2154!2175.
70.
Martin, P., Wagh, V., Reis, S. A., Erdin, S., Beauchamp,
54.
Castro, J., Garcia, R. I., Kwok, S., Banerjee, A.,
R. L., et al. (2020) TSC patient!derived isogenic neural
Petravicz, J., et al. (2014) Functional recovery with recom!
progenitor cells reveal altered early neurodevelopmental
binant human IGF1 treatment in a mouse model of Rett
phenotypes and rapamycin!induced MNK!eIF4E signal!
Syndrome, Proc. Natl. Acad. Sci. USA, 111, 9941!9946.
ing, Mol. Autism, 11, 2.
55.
Ricciardi, S., Boggio, E. M., Grosso, S., Lonetti, G.,
71.
Bramham, C. R., Jensen, K. B., and Proud, C. G. (2016)
Forlani, G., et al. (2011) Reduced AKT/mTOR signaling
Tuning specific translation in cancer metastasis and synap!
and protein synthesis dysregulation in a Rett syndrome ani!
tic memory: control at the MNK!eIF4E axis, Trends
mal model, Hum. Mol. Genet., 20, 1182!1196.
Biochem. Sci., 41, 847!858.
56.
Yoo, T., Cho, H., Park, H., Lee, J., and Kim, E. (2019)
72.
Pellerin, D., Çaku, A., Fradet, M., Bouvier, P., Dubé, J.,
Shank3 Exons 14!16 Deletion in glutamatergic neurons
and Corbin, F. (2016) Lovastatin corrects ERK pathway
leads to social and repetitive behavioral deficits associated
hyperactivation in fragile X syndrome: potential of
with increased cortical layer 2/3 neuronal excitability,
platelet’s signaling cascades as new outcome measures in
Front. Cell Neurosci., 13, 458.
clinical trials, Biomarkers, 21, 497!508.
57.
Bidinosti, M., Botta, P., Krüttner, S., Proenca, C. C.,
73.
Darnell, J. C., and Klann, E. (2013) The translation of
Stoehr, N., et al. (2016) CLK2 inhibition ameliorates
translational control by FMRP: therapeutic targets for
autistic features associated with SHANK3 deficiency,
FXS, Nat. Neurosci., 16, 1530!1536.
Science, 351, 1199!1203.
74.
Sledziowska, M., Galloway, J., and Baudouin, S. J. (2020)
58.
Lee, Y., Kim, S. G., Lee, B., Zhang, Y., Kim, Y., et al.
Evidence for a contribution of the Nlgn3/Cyfip1/Fmr1
(2017) Striatal Transcriptome and interactome analysis of
pathway in the pathophysiology of autism spectrum disor!
Shank3!overexpressing mice reveals the connectivity
ders, Neuroscience, 445, 31!41.
between Shank3 and mTORC1 signaling, Front. Mol.
75.
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T.,
Neurosci., 10, 201.
McBride, S. M., et al. (2010) Dysregulation of mTOR sig!
59.
Piochon, C., Kloth, A. D., Grasselli, G., Titley, H. K.,
naling in fragile X syndrome, J. Neurosci., 30, 694!702.
Nakayama, H., et al. (2014) Cerebellar plasticity and
76.
Gross, C., Banerjee, A., Tiwari, D., Longo, F., White,
motor learning deficits in a copy!number variation mouse
A. R., et al. (2019) Isoform!selective phosphoinositide 3!
model of autism, Nat. Commun., 5, 5586.
kinase inhibition ameliorates a broad range of fragile X syn!
60.
Meyza, K. Z., and Blanchard, D. C. (2017) The BTBR
drome!associated deficits in a mouse model,
mouse model of idiopathic autism - current view on
Neuropsychopharmacology, 44, 324!333.
mechanisms, Neurosci. Biobehav. Rev., 76, 99!110.
77.
Bhattacharya, A., Mamcarz, M., Mullins, C., Choudhury, A.,
61.
Hutsler, J. J., and Zhang, H. (2010) Increased dendritic
Boyle, R. G., et al. (2016) Targeting translation control with
spine densities on cortical projection neurons in autism
p70 S6 kinase 1 inhibitors to reverse phenotypes in fragile X
spectrum disorders, Brain Res., 1309, 83!94.
syndrome mice, Neuropsychopharmacology, 41, 1991!2000.
62.
Tang, G., Gudsnuk, K., Kuo, S. H., Cotrina, M. L.,
78.
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011)
Rosoklija, G., et al. (2014) Loss of mTOR!dependent
Mutations causing syndromic autism define an axis of
macroautophagy causes autistic!like synaptic pruning
synaptic pathophysiology, Nature, 480, 63!68.
deficits, Neuron, 83, 1131!1143.
79.
Bartley, C. M., O’Keefe, R. A., Blice!Baum, A.,
63.
Overwater, I. E., Rietman, A. B., Mous, S. E., Bindels!de
Mihailescu, M. R., Gong, X., et al. (2016) Mammalian
Heus, K., Rizopoulos, D., et al. (2019) A randomized con!
FMRP S499 is phosphorylated by CK2 and promotes sec!
trolled trial with everolimus for IQ and autism in tuberous
ondary phosphorylation of FMRP, eNeuro, 3, ENEU!
sclerosis complex, Neurology, 93, e200!e209.
RO0092!16.2016, doi: 10.1523/ENEURO.0092!16.2016.
64.
Tyagi, R., Shahani, N., Gorgen, L., Ferretti, M., Pryor, W.,
80.
Papa, A., and Pandolfi, P. P. (2019) The PTEN!PI3K axis
et al. (2015) Rheb Inhibits protein synthesis by activating
in cancer, Biomolecules, 9, 153.
БИОХИМИЯ том 86 вып. 5 2021
СИГНАЛЬНЫЕ КАСКАДЫ ПРИ АУТИЗМЕ
665
81.
Song, M. S., Salmena, L., and Pandolfi, P. P. (2012) The
treatment of childhood!onset neurodevelopmental disor!
functions and regulation of the PTEN tumour suppressor,
ders, Front. Neurosci., 10, 450.
Nat. Rev. Mol. Cell Biol., 13, 283!296.
98. Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann,
82.
Nikolaeva, I., Kazdoba, T. M., Crowell, B., and
J., Chaste, P., et al. (2007) Mutations in the gene encoding
D’Arcangelo, G. (2017) Differential roles for Akt and
the synaptic scaffolding protein SHANK3 are associated
mTORC1 in the hypertrophy of Pten mutant neurons, a
with autism spectrum disorders, Nat. Genet., 39, 25!27.
cellular model of brain overgrowth disorders, Neuroscience,
99. Okamoto, N., Kubota, T., Nakamura, Y., Murakami, R.,
354, 196!207.
Nishikubo, T., et al. (2007) 22q13 Microduplication in two
83.
Sokol, D. K., Maloney, B., Westmark, C. J., and Lahiri,
patients with common clinical manifestations: a recogniz!
D. K. (2019) Novel contribution of secreted amyloid!β
able syndrome? Am. J. Med. Genet. A, 143a, 2804!2809.
precursor protein to white matter brain enlargement in
100. Khlebodarova, T. M., Kogai, V. V., Trifonova, E. A., and
autism spectrum disorder, Front. Psychiatry, 10, 165.
Likhoshvai, V. A. (2018) Dynamic landscape of the local
84.
Lahiri, D. K., Sokol, D. K., Erickson, C., Ray, B., Ho,
translation at activated synapses, Mol Psychiatry, 23, 107!
C. Y., and Maloney, B. (2013) Autism as early neurodevel!
114. doi: 10.1038/mp.2017.245.
opmental disorder: evidence for an sAPPα!mediated ana!
101. Burnside, R. D., Pasion, R., Mikhail, F. M., Carroll, A. J.,
bolic pathway, Front. Cell Neurosci., 7, 94.
Robin, N. H., et al. (2011) Microdeletion/microduplica!
85.
Kaczorowski, J. A., Smith, T. F., Shrewsbury, A. M.,
tion of proximal 15q11.2 between BP1 and BP2: a suscep!
Thomas, L. R., Knopik, V. S., and Acosta, M. T. (2020)
tibility region for neurological dysfunction including devel!
Neurofibromatosis type 1 implicates Ras pathways in the
opmental and language delay, Hum. Genet., 130, 517!528.
genetic architecture of neurodevelopmental disorders,
102. Suzuki, A. M., Griesi!Oliveira, K., de Oliveira Freitas
Behav. Genet., 50, 191!202.
Machado, C., Vadasz, E., Zachi, E. C., et al. (2015)
86.
Garg, S., Brooks, A., Burns, A., Burkitt!Wright, E.,
Altered mTORC1 signaling in multipotent stem cells from
Kerr, B., et al. (2017) Autism spectrum disorder and other
nearly 25% of patients with nonsyndromic autism spec!
neurobehavioural comorbidities in rare disorders of the
trum disorders, Mol. Psychiatry, 20, 551!552.
Ras/MAPK pathway, Dev. Med. Child Neurol., 59, 544!549.
103. Neves!Pereira, M., Müller, B., Massie, D., Williams, J. H.,
87.
Banerjee, S., Crouse, N. R., Emnett, R. J., Gianino, S.
O’Brien, P. C., et al. (2009) Deregulation of EIF4E: a novel
M., and Gutmann, D. H. (2011) Neurofibromatosis!1 reg!
mechanism for autism, J. Med. Genet., 46, 759!765.
ulates mTOR!mediated astrocyte growth and glioma for!
104. Meyer, U. (2013) Prenatal poly(i:C) exposure and other
mation in a TSC/Rheb!independent manner, Proc. Natl.
developmental immune activation models in rodent sys!
Acad. Sci. USA, 108, 15996!16001.
tems, Biol. Psychiatry,
75,
307!315, doi:
10.1016/
88.
Weiss, B., Widemann, B. C., Wolters, P., Dombi, E.,
j.biopsych.2013.07.011.
Vinks, A., et al. (2015) Sirolimus for progressive neurofi!
105. Steinmetz, A. B., Stern, S. A., Kohtz, A. S., Descalzi, G.,
bromatosis type 1!associated plexiform neurofibromas: a
and Alberini, C. M. (2018) Insulin!like growth factor II
neurofibromatosis Clinical Trials Consortium phase II
targets the mtor pathway to reverse autism!like phenotypes
study, Neuro Oncol., 17, 596!603.
in mice, J. Neurosci., 38, 1015!1029.
89.
Richards, C., Jones, C., Groves, L., Moss, J., and
106. Rosina, E., Battan, B., Siracusano, M., Di Criscio, L.,
Oliver, C. (2015) Prevalence of autism spectrum disorder
Hollis, F., et al. (2019) Disruption of mTOR and MAPK
phenomenology in genetic disorders: a systematic review
pathways correlates with severity in idiopathic autism,
and meta!analysis, Lancet Psychiatry, 2, 909!916.
Transl. Psychiatry, 9, 50.
90.
Zoghbi, H. Y., and Bear, M. F. (2012) Synaptic dysfunction
107. Ganesan, H., Balasubramanian, V., Iyer, M.,
in neurodevelopmental disorders associated with autism
Venugopal, A., Subramaniam, M. D., et al. (2019) mTOR
and intellectual disabilities, Cold Spring Harb Perspect Biol,
signalling pathway ! A root cause for idiopathic autism?
4, a009886.
BMB Rep., 52, 424!433.
91.
Sun, J., Liu, Y., Tran, J., O’Neal, P., Baudry, M., and Bi, X.
108. Mariani, J., Coppola, G., Zhang, P., Abyzov, A.,
(2016) mTORC1!S6K1 inhibition or mTORC2 activation
Provini, L., et al. (2015) FOXG1!dependent dysregulation
improves hippocampal synaptic plasticity and learning in
of GABA/glutamate neuron differentiation in autism spec!
Angelman syndrome mice, Cell. Mol Life Sci., 73, 4303!4314.
trum disorders, Cell, 162, 375!390.
92.
Zhang, Z. N., Freitas, B. C., Qian, H., Lux, J., Acab, A.,
109. Baier, P. C., Koch, J. M., Seeck!Hirschner, M., Ohlmeyer,
et al. (2016) Layered hydrogels accelerate iPSC!derived
K., Wilms, S., et al. (2009) A flow!cytometric method to
neuronal maturation and reveal migration defects caused
investigate glutamate!receptor!sensitivity in whole blood
by MeCP2 dysfunction, Proc. Natl. Acad. Sci. USA, 113,
platelets - results from healthy controls and patients with
3185!3190.
schizophrenia, J. Psychiatr. Res., 43, 585!591.
93.
Wang, H., Xu, H., Wang, X., Zhou, A., Wu, M., et al.
110. Boyanova, D., Nilla, S., Birschmann, I., Dandekar, T., and
(2016) Amyloid precursor protein associates with autism
Dittrich, M. (2012) PlateletWeb: a systems biologic analy!
spectrum disorder: a potential candidate biomarker for
sis of signaling networks in human platelets, Blood, 119,
early screening, Int. J. Clin. Exp. Med., 9, 22259!22266.
e22!34.
94.
Li, Y., Wang, H., Muffat, J., Cheng, A. W., Orlando, D. A.,
111.
Kumari, D., Bhattacharya, A., Nadel, J., Moulton, K.,
et al. (2013) Global transcriptional and translational
Zeak, N. M., et al. (2014) Identification of fragile X syn!
repression in human!embryonic!stem!cell!derived Rett
drome specific molecular markers in human fibroblasts: a
syndrome neurons, Cell Stem Cell, 13, 446!458.
useful model to test the efficacy of therapeutic drugs, Hum.
95.
Monteiro, P., and Feng, G. (2017) SHANK proteins: roles
Mutat., 35, 1485!1494.
at the synapse and in autism spectrum disorder, Nat. Rev.
112. McCoy, M., Poliquin!Duchesneau, D., and Corbin, F.
Neurosci., 18, 147!157.
(2016) Molecular dynamics of FMRP and other RNA!
96.
Shcheglovitov, A., Shcheglovitova, O., Yazawa, M.,
binding proteins in MEG!01 differentiation: the role of
Portmann, T., Shu, R., et al. (2013) SHANK3 and IGF1
mRNP complexes in non!neuronal development,
restore synaptic deficits in neurons from 22q13 deletion
Biochem. Cell Biol., 94, 597!608.
syndrome patients, Nature, 503, 267!271.
113. Pellerin, D., Lortie, A., and Corbin, F. (2018) Platelets as a
97.
Vahdatpour, C., Dyer, A. H., and Tropea, D. (2016)
surrogate disease model of neurodevelopmental disorders:
Insulin!like growth factor 1 and related compounds in the
insights from fragile X syndrome, Platelets, 29, 113!124.
4 БИОХИМИЯ том 86 вып. 5 2021
666
БОКША и др.
PROTEIN PHOSPHORYLATION SIGNALING CASCADES
IN AUTISM WITH EMPHASIS ON THE mTOR PATHWAY
Review
I. S. Boksha1,2*, T. A. Prokhorova1, E. B. Tereshkina1,
O. K. Savushkina1, and G. Sh. Burbaeva1
1 Mental Health Research Center, 115522 Moscow, Russia; E mail: boksha_irina@mail.ru
2 Gamaleya Research Center of Epidemiology and Microbiology, Ministry of Health of the Russian Federation,
123098 Moscow, Russia; E mail: boksha_irina@gamaleya.org
The mammalian target of rapamycin (mTOR) signaling pathway is a central regulator of cell metabolism, growth, and
survival in response to hormones, growth factors, nutrients, and stress!induced signals. For this review, we analyzed
the available literature that describes how molecular abnormalities of signaling cascades associated with mTOR are
encountered in autism spectrum disorders (ASDs), and we outline prospects for pathogenetically!targeted pharma!
cotherapeutic approaches to ASDs, especially in syndromic ASDs. Considering the available experimental and clini!
cal data, we suggest that very early detection of molecular abnormalities in ASD risk groups can be facilitated from
studies of peripheral blood platelets. Also, the determination of a time window in which critical dysregulations in the
activity of the described pathways in risk groups might suggest promising directions for further studies that could lead
to more effective pharmacotherapeutic interventions in ASDs.
Keywords: mTOR, signaling cascades, protein phosphorylation, autism spectrum disorders
БИОХИМИЯ том 86 вып. 5 2021