БИОХИМИЯ, 2022, том 87, вып. 9, с. 1203 - 1222
УДК 576.32/36
РОЛЬ СТРЕССА ЭНДОПЛАЗМАТИЧЕСКОГО РЕТИКУЛУМА
В ДИФФЕРЕНЦИРОВКЕ КЛЕТОК
МЕЗЕНХИМНОГО ПРОИСХОЖДЕНИЯ
Обзор
© 2022 Е.П. Турищева*, М.С. Вильданова, Г.Е. Онищенко, Е.А. Смирнова
Московский государственный университет имени М. В. Ломоносова, биологический факультет,
119991 Москва, Россия; электронная почта: kitten-caterina@yandex.ru
Поступила в редакцию 05.05.2022
После доработки 28.06.2022
Принята к публикации 30.06.2022
Эндоплазматический ретикулум (ЭПР) - это мультифункциональный мембранный компартмент, од-
ной из основных функций которого является котрансляционный перенос и процессинг секреторных,
лизосомных и трансмембранных белков. Неправильный процессинг белков при нарушении гомеос-
таза ЭПР приводит к состоянию, которое называется «стресс ЭПР». Для восстановления нормально-
го функционирования ЭПР активируется адаптивный механизм, который обозначают как ответ на
неправильно свёрнутые белки или UPR. Помимо контроля сворачивания белков, UPR играет ключе-
вую роль в других физиологических процессах, в частности в дифференцировке клеток соединитель-
нотканного, мышечного, эпителиального и нейрального происхождения. Однако дифференцировку
стимулирует только физиологический уровень стресса ЭПР, в то время как его повышенный уро-
вень подавляет дифференцировку и может вызвать гибель клеток. Следует отметить, что до настоя-
щего времени неизвестно, является ли активация UPR индуктором дифференцировки клеток или
же UPR запускается из-за повышенного синтеза секреторных белков в процессе дифференцировки.
Дифференцировка клеток является важным этапом в развитии многоклеточных организмов, поэтому
этот процесс строго контролируется. Подавление или, наоборот, избыточная активация дифферен-
цировки ведут к развитию патологических процессов в организме. В частности, нарушения в ходе
дифференцировки клеток соединительнотканного происхождения ведут к развитию таких заболева-
ний, как фиброз, ожирение и остеопороз. Фиброз в настоящее время вызывает особый интерес, так
как является одним из основных последствий COVID-19. В связи с этим изучение роли UPR в акти-
вации дифференцировки представляет как теоретический, так и практический интерес, так как мо-
жет привести к идентификации потенциальных молекулярных мишеней, позволяющих селективно
регулировать разные этапы дифференцировки и воздействовать на механизмы, ведущие к развитию
патологических процессов.
КЛЮЧЕВЫЕ СЛОВА: эндоплазматический ретикулум, дифференцировка, стресс эндоплазматического
ретикулума, UPR, миофибробласты, фиброз, адипогенез, миогенез, остеобластогенез, остеокластогенез.
DOI: 10.31857/S0320972522090032, EDN: AZTMDJ
Принятые сокращения: ПТГ - паратиреоидный гормон; ЭПР - эндоплазматический ретикулум; ATF4 - активирую-
щий фактор транскрипции 4 (activating transcription factor 4); ATF6 - активирующий фактор транскрипции 6 (activating
transcription factor 6); BiP - белок, связывающий иммуноглобулин (immunoglobulin binding protein); BMP2 - костный
морфогенетический белок 2 (bone morphogenetic protein 2); BSP - костный сиалопротеин (bone sialoprotein); C/EBPα -
CCAAT/энхансер-связывающий белок α (CCAAT/enhancer-binding protein α); CHOP - CCAAT/белок, гомологичный
энхансер-связывающему белку (CCAAT/enhancer-binding protein homologous protein); eIF2α - фактор инициации транс-
ляции эукариот 2α (eukaryotic translation initiation factor 2α); ERAD - ЭПР-ассоциированная деградация (ER-associated
degradation); GRP78 - белок 78 кДа, регулируемый глюкозой (glucose-regulated protein 78); IRE1 - требующий инозитол
белок 1 (inositol-requiring protein 1); MEF - эмбриональные фибробласты мыши (murine embryonic fibroblasts); MyoD -
белок детерминации миобластов 1 (myoblast determination protein 1); NFATc1 - ядерный фактор активированных Т-кле-
ток, цитоплазматический 1 (nuclear factor of activated T cells cytoplasmic 1); OCN - остеокальцин; Osx - транскрипцион-
ный фактор Osterix; 4-PBA - 4-фенилмасляная кислота (4-phenylbutyric acid); PERK - киназа ЭПР, подобная белковой
киназе RNA (protein kinase RNA-like endoplasmic reticulum kinase); PPARγ - рецептор, активируемый пролифератора-
ми пероксисом γ (peroxisome proliferator-activated receptor γ); Ppp1cc - каталитическая субъединица протеинфосфата-
зы 1 γ (protein phosphatase 1 catalytic subunit γ); RANK - рецептор-активатор ядерного фактора NF-κB (receptor activator
of nuclear factor NF-κB); RANKL - лиганд RANK; Runx2 - фактор транскрипции 2, связанный с доменом runt (runt-
related transcription factor 2); sXBP1 - сплайсированный X-box-связывающий белок 1 (spliced X-box-binding protein 1);
1203
1
204
ТУРИЩЕВА и др.
ВВЕДЕНИЕ
стресс ЭПР активируется сигнальный каскад,
известный как ответ на неправильно свёрнутые
Эндоплазматический ретикулум (ЭПР) -
белки, или UPR [2, 11]. UPR - это адаптивный
это мультифункциональный мембранный ком-
ответ, изначально нацеленный на нормализа-
партмент, одной из основных функций которого
цию гомеостаза клетки и её выживание [9, 10].
является котрансляционный перенос секретор-
Активация UPR оказывает влияние почти на
ных, лизосомных и трансмембранных белков,
каждый аспект секреторного пути в клетке,
их модификация и сворачивание [1]. В этих
модифицируя интенсивность синтеза белков
процессах участвует множество разных фермен-
и их транслокации в ЭПР, сворачивание бел-
тов и белков-шаперонов ЭПР [2]. Для нормаль-
ков, созревание и контроль качества свёрнутых
ного функционирования белков необходим их
белков, перенос белков по секреторному пути
безошибочный синтез и сворачивание, поэтому
и элиминацию неправильно свёрнутых белков
в ЭПР существует контроль качества свёрнутых
с помощью аутофагии и ERAD [11].
белков. Неправильно свёрнутые белки узна-
UPR может запускаться тремя сенсорными
ются шаперонами (например, BiP (белок, свя-
трансмембранными белками ЭПР: IRE1 (тре-
зывающий иммуноглобулин), ERdj (семейство
бующий инозитол белок 1), PERK (киназа ЭПР,
локализованных в ЭПР DnaJ-подобных бел-
подобная белковой киназе RNA) и ATF6 (акти-
ков)) и лектинами (например, OS-9 (белок, вы-
вирующий фактор транскрипции 6) [2]. Домены
сокоэкспрессирующийся в остеосаркомах-9) и
этих белков, находящиеся в просвете ЭПР (лю-
XTP3-B (XTP3-транс-активированный ген-B)),
минальные домены), в отсутствие стресса ЭПР
ретротранслоцируются из ЭПР в цитозоль, по-
связаны с шапероном ЭПР BiP или GRP78 (бе-
лиубиквитинируются и деградируют в протео-
лок 78 кДа, регулируемый глюкозой), которые
сомах [2-4]. Этот процесс называется ЭПР-ас-
за счёт этой связи не дают им активироваться.
социированной деградацией (ERAD) [2].
В условиях стресса ЭПР BiP отсоединяется от
Для нормального функционирования ЭПР
сенсоров, что приводит к их активации [9]. Та-
необходим баланс между синтезируемыми в
ким образом, IRE1, PERK и ATF6 «отслежи-
ЭПР ещё не свёрнутыми белками и активно-
вают», справляется ли свёртывающий аппа-
стью шаперонов ЭПР [1]. Различные физиоло-
рат ЭПР с имеющимся объёмом белков [2].
гические состояния и патологические факторы
Каждый сенсор стресса ЭПР (IRE1, PERK
могут нарушить этот баланс, что приводит к
и ATF6) при активации запускает свой сиг-
накоплению в ЭПР не свёрнутых и неправиль-
нальный каскад (путь). Следует отметить, что
но свёрнутых белков. Это состояние носит на-
в зависимости от индуктора стресса ЭПР и
звание «стресс ЭПР» [5]. Стресс ЭПР может
клеточного типа могут активироваться не все
индуцироваться при увеличении количества не
пути UPR, а два или один из возможных [1].
свёрнутых белков в ЭПР, например, из-за по-
Если с помощью активации UPR не удаёт-
вышения потребности в белках для секреции в
ся восстановить нормальное функционирова-
секреторных клетках, или при нарушении про-
ние ЭПР, то активируются сигнальные пути,
цесса сворачивания белков, например, из-за
запускающие гибель клетки [1, 2]. Таким обра-
мутаций или воздействия химических агентов
зом, хронический стресс ЭПР и продолжитель-
(дитиотреитола, туникамицина, тапсигаргина,
ный UPR могут приводить к нарушению функ-
брефельдина А и др.) [6-10]. Нарушение каль-
ционирования клетки и её гибели [1, 12]. Это,
циевого гомеостаза, окислительно-восстано-
в свою очередь, вовлечено в развитие хрониче-
вительного статуса ЭПР, гипоксия, глюкозное
ских заболеваний у человека, таких как нейро-
голодание, перегруженность ЭПР холестери-
дегенеративные заболевания, диабет, фиброз
ном, депривация питательных веществ и повы-
лёгких и воспалительные процессы [12].
шение температуры до 40 градусов также могут
Однако гибель или выживание клетки
приводить к нарушению сворачивания белков
не являются единственными последствиями
и стрессу ЭПР [8-10].
стресса ЭПР, так как помимо контроля сво-
Для восстановления нормального функцио-
рачивания белков UPR играет ключевую роль
нирования ЭПР и, в частности, его способно-
в различных физиологических процессах, та-
сти сворачивать белки, в клетках в ответ на
ких как врождённый иммунитет, метаболизм
Принятые сокращения (продолжение): TGF-β - трансформирующий фактор роста β (transforming growth factor-β);
TUDCA - тауро-урсодезоксихолевая кислота (tauroursodeoxycholic acid); UPR - ответ на неправильно свёрнутые белки
(unfolded protein response); XBP1 - X-box-связывающий белок 1 (X-box-binding protein 1); α-SMA - гладкомышечный
актин α (α-smooth muscle actin).
* Адресат для корреспонденции.
БИОХИМИЯ том 87 вып. 9 2022
СТРЕСС ЭПР И ДИФФЕРЕНЦИРОВКА КЛЕТОК
1205
глюкозы и липидов, а также дифференциров-
ХАРАКТЕРИСТИКА ПУТЕЙ UPR
ка клеток [11]. Физиологический стресс ЭПР
участвует в дифференцировке клеток соедини-
UPR может запускаться тремя трансмем-
тельнотканного, мышечного, эпителиального
бранными сенсорами стресса ЭПР - киназа-
и нейрального происхождения [13-16]. Так,
ми IRE1 и PERK и транскрипционным фак-
например, физиологический стресс ЭПР, вы-
тором ATF6, находящимися на мембране ЭПР.
званный растительными гормонами, гибберел-
В условиях стресса ЭПР от люминальных до-
линовой и жасмоновой кислотами, индуцирует
менов этих сенсорных белков отсоединяется
усиление дифференцировки иммортализован-
шаперон BiP, в результате чего сенсоры акти-
ных кератиноцитов линии HaCaT и появление
вируются и каждый запускает свой сигнальный
признаков дифференцировки в клетках эпи-
каскад (путь) (рис. 1).
дермоидной карциномы человека A431 [17, 18].
Путь, активируемый IRE1. Трансмембран-
Помимо этого, гиббереллиновая и абсцизовая
ная киназа IRE1 после освобождения от связи
кислоты вызывают морфологические признаки
с BiP олигомеризуется и аутофосфорилируется,
стресса ЭПР в клетках соединительнотканного
что ведёт к её активации [10]. Активированная
происхождения - дермальных фибробластах
IRE1 с помощью своего эндорибонуклеазного
человека и клетках фибросаркомы человека
домена осуществляет сплайсинг мРНК, коди-
линии HT1080 [19]. Целью данного обзора яв-
рующей транскрипционный фактор XBP1. Этот
ляется анализ данных, посвящённых участию
домен вырезает 26-нуклеотидный интрон и сме-
стресса ЭПР и UPR в дифференцировке кле-
щает рамку считывания, в результате чего синте-
ток мезенхимного происхождения, в частности
зируется активный транскрипционный фактор
фибробластов, преадипоцитов, миобластов,
sXBP1 («s» означает «сплайсированный») [11,
остеобластов и остеокластов.
20]. Этот транскрипционный фактор повышает
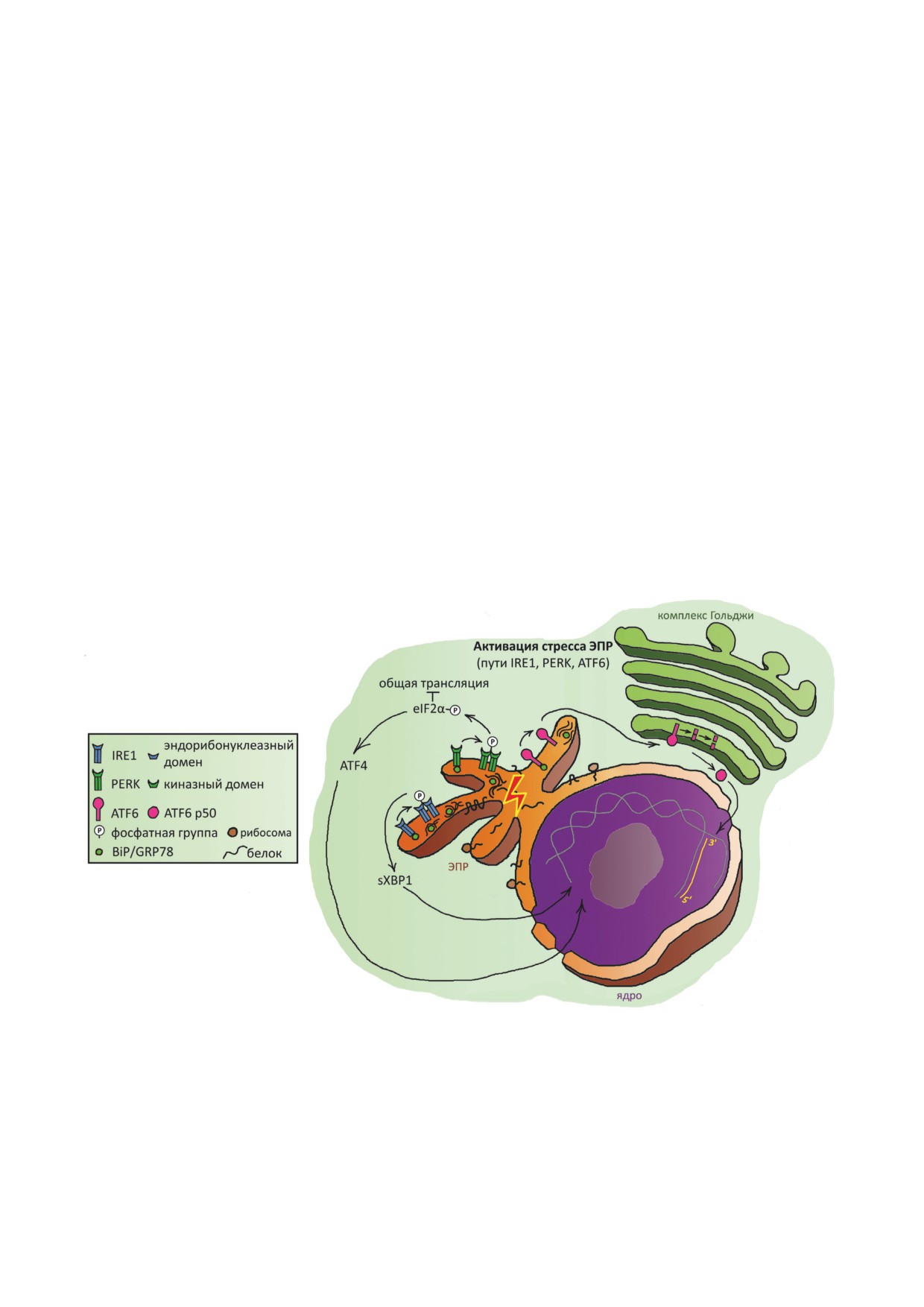
Рис. 1. Ключевые события стресса ЭПР и UPR (ответ на неправильно свёрнутые белки) после отделения BiP/GRP78
(белок, связывающий иммуноглобулин/белок 78 кДа, регулируемый глюкозой) от трёх трансмембранных сенсорных
белков ЭПР - IRE1 (требующий инозитол белок 1), PERK (киназа ЭПР, подобная белковой киназе RNA) и ATF6 (ак-
тивирующий фактор транскрипции 6). IRE1 олигомеризуется и аутофосфорилируется; его эндорибонуклеазный домен
осуществляет сплайсинг мРНК XBP1 (X-box-связывающий белок 1), в результате чего синтезируется активный тран-
скрипционный фактор sXBP1. PERK олигомеризуется, аутофосфорилируется и фосфорилирует eIF2α (фактор инициа-
ции трансляции эукариот 2α), подавляя общую трансляцию в клетке и усиливая трансляцию ATF4 (активирующий
фактор транскрипции 4) - транскрипционного фактора генов-мишеней UPR. ATF6 перемещается в комплекс/аппарат
Гольджи и разрезается протеазами. В результате высвобождается цитоплазматический фрагмент (ATF6 p50), который
перемещается в ядро, где работает как транскрипционный фактор. Расшифровка графических символов этой части
рисунка приведена в зелёном квадрате слева
БИОХИМИЯ том 87 вып. 9 2022
1
206
ТУРИЩЕВА и др.
экспрессию генов белков, вовлечённых в
рекрутируется медиатор этого сигнального
транслокацию синтезируемых белков в ЭПР,
пути - TRAF2 (ассоциированный с рецептором
их сворачивание (например, дисульфид-изоме-
TNF фактор 2) и активируется киназа ASK1 (ки-
раза PDI) и секрецию, а также деградацию не-
наза 1 сигналинга апоптоза). ASK1 активирует
правильно свёрнутых белков (например, EDEM
киназу JNK (c-Jun N-терминальная белковая
(усиливающий деградацию α-маннидозоподоб-
киназа), которая участвует в запуске апоптоза,
ный белок ЭПР), участвующий в ERAD) и про-
регулируя белки семейства BCL2 (B-клеточная
дукцию липидных компонентов ЭПР (например,
лимфома 2) [1, 10]. Кроме того, ATF4, участвую-
холин цитидилилтрансфераза) [1, 11, 21, 22].
щий в сигнальном пути PERK/eIF2α, может
Путь, активируемый PERK. Трансмембран-
активировать экспрессию проапоптотического
ная киназа PERK после освобождения от связи
фактора CHOP (CCAAT/белок, гомологичный
с BiP олигомеризуется, аутофосфорилируется
энхансер-связывающему белку). Этот фактор
и фосфорилирует Ser51 на α-субъединице ини-
регулирует белки семейства BCL-2, DR5 (ре-
циаторного фактора трансляции eIF2 [10, 23].
цептор смерти 5) и GADD34 (регуляторная
Фосфорилированный eIF2α препятствует ини-
субъединица белковой фосфатазы PP1) и сти-
циации трансляции, что ведёт к подавлению
мулирует стресс-опосредованную индукцию
трансляции большинства мРНК [23]. В резуль-
апоптоза [11].
тате в ЭПР перестают поступать синтезируе-
Таким образом, UPR - это адаптивный от-
мые белки, что снижает нагрузку на ЭПР, свя-
вет, изначально нацеленный на выживание кле-
занную с правильной укладкой белков [2]. При
ток, но запускающий их гибель в случае, если
этом мРНК транскрипционного фактора ATF4,
восстановить нормальное функционирование
наоборот, транслируются в присутствии фос-
ЭПР не удаётся [1, 2, 9, 10]. Однако, помимо
форилированного eIF2α [11, 23]. ATF4 активи-
контроля сворачивания белков, UPR играет
рует транскрипцию генов-мишеней UPR, ко-
ключевую роль в физиологических процессах,
дирующих факторы, вовлечённые в биосинтез
таких как дифференцировка клеток соедини-
аминокислот, антиоксидантный ответ, аутофа-
тельнотканного, мышечного, эпителиального
гию и апоптоз [11, 23]. Подавление трансляции
и нейрального происхождения [11, 13-16]. Дан-
с помощью eIF2α обратимо: ATF4 повышает
ный обзор посвящён роли стресса ЭПР и UPR
уровень активности регуляторной субъедини-
в дифференцировке клеток мезенхимного про-
цы белковой фосфатазы PP1 - GADD34 (бе-
исхождения, в частности фибробластов, пре-
лок, индуцируемый остановкой роста и по-
адипоцитов, миобластов, остеобластов и остео-
вреждением ДНК), таким образом участвуя в
кластов.
петле обратной связи, направленной на дефос-
форилирование eIF2α и восстановление нор-
мального уровня синтеза белка [11].
UPR И ДИФФЕРЕНЦИРОВКА
Путь, активируемый ATF6. Трансмембран-
ФИБРОБЛАСТОВ В МИОФИБРОБЛАСТЫ
ный белок ATF6 после освобождения от связи
с BiP перемещается в составе COPII-окаймлён-
Фибробласты
- клетки соединительной
ных везикул от ЭПР к аппарату Гольджи, где
ткани, синтезирующие и секретирующие ком-
он разрезается протеазами S1P и S2P [11, 24].
поненты внеклеточного матрикса, такие как
В результате высвобождается цитоплазматиче-
коллаген, фибронектин, эластин, гиалуроно-
ский фрагмент ATF6 - ATF6 p50, который пе-
вая кислота и другие гликозаминогликаны,
ремещается в ядро и работает как транскрип-
матриксные металлопротеиназы (MMPs), тка-
ционный фактор вместе с sXBP1 [11]. ATF6 p50
невые ингибиторы металлопротеиназ (TIMPs)
и sXBP1 активируют транскрипцию генов, ко-
и др. [14, 27]. Фибробласты участвуют в по-
дирующих шапероны и ферменты ЭПР, кото-
строении и ремоделировании внеклеточного
рые стимулируют транслокацию белков в ЭПР,
матрикса, заживлении ран, воспалении и ан-
их сворачивание (например, шапероны GRP78,
гиогенезе [27]. При ранении фибробласты пе-
GRP94, кальнексин), процессинг и секрецию, а
ремещаются в повреждённый участок, где
также деградацию неправильно свёрнутых бел-
пролиферируют и дифференцируются в мио-
ков [1, 11, 25, 26]. Кроме того, ATF6 p50 и sXBP1
фибробласты [14, 27]. Миофибробласты совме-
стимулируют биогенез ЭПР и аппарата Гольд-
щают в себе признаки фибробластов, напри-
жи в условиях стресса ЭПР [11].
мер, хорошо выраженный гранулярный ЭПР и
Активация гибели при хроническом стрес-
аппарат Гольджи, и признаки гладкомышечных
се ЭПР. При высоком уровне хронического
клеток - развитый сократительный аппарат,
стресса ЭПР с помощью IRE1 запускается сиг-
содержащий гладкомышечный актин (α-SMA).
нальный путь TNF (фактор некроза опухоли),
В миофибробластах, по сравнению с фибро-
БИОХИМИЯ том 87 вып. 9 2022
СТРЕСС ЭПР И ДИФФЕРЕНЦИРОВКА КЛЕТОК
1207
бластами, повышен синтез коллагена I, фиб-
Так, например, при индукции дифферен-
ронектина и трансформирующего фактора
цировки лёгочных фибробластов человека и
роста β (TGF-β), увеличено количество фокаль-
мыши в миофибробласты с помощью TGF-β
ных контактов и стресс-фибрилл, повышена
значительно повышается уровень синтеза
контрактильная способность, снижена мигра-
GRP78, sXBP1 и ATF6 [31]. При этом уровень
ционная активность [14, 28]. Миофибробласты
синтеза CHOP и фосфорилированного eIF2α
участвуют в заживлении ран, восстанавливая
не изменяется, что, возможно, связано с анти-
внеклеточный матрикс в качестве каркаса для
апоптотическим эффектом TGF-β. Напротив,
регенерации ткани, и стягивают края раны за
ингибирование стресса ЭПР с помощью хими-
счёт своей контрактильной активности [27].
ческого шаперона, 4-фенилмасляной кисло-
При нормальном заживлении ран и вос-
ты (4-PBA), подавляет не только развитие UPR,
становлении целостности тканей миофибро-
но и экспрессию генов, кодирующих α-SMA и
бласты погибают путём апоптоза [14, 28]. Если
коллаген I (основных маркёров дифференци-
процессы заживления раны нарушаются, мио-
ровки миофибробластов). Кроме того, индук-
фибробласты остаются в раневой зоне, где за-
ция синтеза α-SMA и коллагена I с помощью
тем развивается фиброз, характеризующийся
TGF-β подавляется при нокдауне GRP78, что
избыточным отложением внеклеточного мат-
свидетельствует о необходимости участия ком-
рикса и нерегулируемой контрактильной ак-
понентов UPR в дифференцировке [31]. 4-PBA
тивностью миофибробластов [28, 29]. Фиброз
также оказывает антифибротическое воздей-
также может развиваться при хроническом вос-
ствие при TGF-β-индуцированной дифферен-
палении, вызванном ядовитыми веществами,
цировке первичных синовиальных фибробла-
инфекциями и механическими повреждения-
стов крыс в миофибробласты [33]. Стресс ЭПР
ми, или как результат аутоиммунных реакций
активируется и при индукции дифференциров-
(склеродермии, язвенного колита, болезни
ки фибробластов в миофибробласты с помо-
Крона и ревматоидного артрита) [29]. Посколь-
щью эндотелина-1 и тромбина [32].
ку объём и упругость внеклеточного матрикса
Интересно, что
1%-ный экстракт сига-
играют решающую роль в структурной и функ-
ретного дыма вызывает дифференцировку
циональной целостности тканей, избыточное
лёгочных эмбриональных фибробластов че-
количество внеклеточного матрикса при фиб-
ловека (MRC-5) в миофибробласты тоже пу-
розе приводит к развитию и усугублению дис-
тём индукции стресса ЭПР [35]. При действии
функции тканей. Например, идиопатический
данного фактора в фибробластах повышает-
лёгочный фиброз характеризуется накопле-
ся синтез белков α-SMA, GRP78, IRE1, XBP1
нием миофибробластов и ремоделированием
и ATF6. Обработка 4-PBA или нокдаун GRP78
внеклеточного матрикса, что приводит к нару-
до воздействия снижает индуцированную экс-
шению строения лёгких и прогрессирующему
трактом сигаретного дыма дифференциров-
фиброзу [30]. Фиброзу подвержены почти все
ку фибробластов в миофибробласты. Однако
ткани организма, в том числе печень, почки и
остаётся неизвестным, является ли влияние
сердце [27, 29]. Присутствие миофибробластов
экстракта сигаретного дыма на дифференци-
отмечено в активно сокращающейся грануля-
ровку фибробластов результатом воздействия
ционной ткани и гипертрофических рубцах, а
одного или нескольких компонентов экстракта
также в сократительных тканях ладонной фас-
или обусловлено совместным действием всех
ции при болезни Дюпюитрена [14]. В связи с
его компонентов [35].
этим изучение механизмов дифференциров-
Ингибирование активности отдельных пу-
ки фибробластов в миофибробласты является
тей UPR показало их важность в активации
важным звеном в поиске способов предотвра-
дифференцировки фибробластов в миофибро-
щения и лечения фиброза, а также смягчения
бласты, однако пока ещё неясно, насколько рав-
его проявлений. Для моделирования фиброза
нозначно влияние каждого из них [29, 32, 36].
in vitro часто используют многофункциональ-
Роль сигнального пути IRE1. Ингибирова-
ный цитокин TGF-β, который считается основ-
ние IRE1 с помощью селективного ингибито-
ным индуктором дифференцировки фибробла-
ра 4μ8C (производное салицилового альдеги-
стов в миофибробласты [14, 28, 31].
да, подавляющее РНКазную активность IRE1)
Исследования in vitro показали, что стресс
блокирует индуцированную TGF-β диффе-
ЭПР участвует в дифференцировке фибро-
ренцировку эмбриональных лёгочных фибро-
бластов, выделенных из разных тканей, в мио-
бластов человека в миофибробласты, снижая
фибробласты [14, 29, 31, 32], а ингибирование
экспрессию и синтез α-SMA и коллагена I [29].
стресса ЭПР приводит к подавлению такой
Снижение экспрессии гена, кодирующего
дифференцировки [31, 33, 34].
α-SMA, наблюдается в эмбриональных лёгоч-
БИОХИМИЯ том 87 вып. 9 2022
1208
ТУРИЩЕВА и др.
ных фибробластах человека при подавлении
TGF-β, снижается, а при нокдауне ATF6 - по-
РНКазной активности IRE1 и в эмбриональных
вышается [36].
фибробластах мыши с нокаутом IRE1. Кроме
Влияние индукторов стресса ЭПР на диф-
того, IRE1 разрезает miRNA-150, что снима-
ференцировку фибробластов в миофибробласты.
ет ингибирующий эффект этой микроРНК
Стресс ЭПР может быть индуцирован хими-
на экспрессию α-SMA, который miRNA-150
ческими агентами, такими как туникамицин и
осуществляет через подавление экспрессии
тапсигаргин. Туникамицин - это антибиотик,
транскрипционного фактора c-Myb. По это-
блокирующий N-гликозилирование белков
му механизму осуществляется снижение инду-
в ЭПР, что приводит к нарушению начальной
цированной TGF-β экспрессии α-SMA при
стадии биосинтеза гликопротеинов, накоп-
подавлении РНКазной активности IRE1. При
лению несвёрнутых гликопротеинов в ЭПР
этом sXBP1 стимулирует синтез фосфатидил-
и стрессу ЭПР [1]. Тапсигаргин - это сескви-
холина - основного фосфолипида мембраны
терпеновый лактон, который блокирует работу
ЭПР, что вносит вклад в увеличение площа-
кальциевых АТФаз (SERCA) и приводит к поте-
ди ЭПР. Это, в свою очередь, необходимо для
ре активности кальций-зависимых шаперонов,
более эффективного синтеза белков в ходе
накоплению несвёрнутых белков и стрессу ЭПР.
дифференцировки фибробластов в миофибро-
Оказалось, что туникамицин и тапсигаргин
бласты [29, 37]. Соответственно, при нокдау-
способны индуцировать дифференцировку фиб-
не XBP1 увеличение площади ЭПР и секреция
робластов в миофибробласты. Так, например,
коллагена, индуцированные с помощью TGF-β,
при действии туникамицина или тапсигаргина
подавляются [29].
на первичные лёгочные фибробласты человека
Роль сигнального пути PERK. Нокдаун PERK
наблюдается повышение уровня синтеза α-SMA
подавляет дифференцировку эмбриональных
и коллагена I, а при нокдауне GRP78 индукция
лёгочных фибробластов человека линии WI-38
синтеза α-SMA и коллагена I подавляется [31].
в миофибробласты [32]. При индукции диф-
Стресс ЭПР, индуцированный тапсигаргином,
ференцировки с помощью эндотелина-1 или
также повышает экспрессию генов, кодирую-
тромбина, но при отсутствии PERK не проис-
щих коллаген I и α-SMA, в синовиальных фиб-
ходит повышения синтеза α-SMA, коллагена I и
робластах крыс [33]. Постоянная активация
коллагена IV. При этом ингибитор киназы JNK
стресса ЭПР с помощью туникамицина в пер-
также подавляет индуцированную эндотели-
вичных фибробластах кожи мыши индуцирует
ном-1 или тромбином дифференцировку этих
дифференцировку этих клеток в миофибробла-
фибробластов в миофибробласты, в частности,
сты. В клетках повышается экспрессия α-SMA и
в клетках не повышается синтез α-SMA. Это
контрактильная активность, они приобретают
может свидетельствовать о том, что JNK акти-
более распластанную форму [14]. Туникамицин
вирует PERK, а PERK, в свою очередь, необхо-
и тапсигаргин также повышают синтез α-SMA
дим для активации синтеза α-SMA.
и коллагена IV и стимулируют дифференци-
Роль сигнального пути ATF6. Исследования
ровку эмбриональных лёгочных фибробластов
показали, что ATF6 препятствует дифферен-
человека линии WI-38 в миофибробласты [32].
цировке фибробластов желудочков сердца в
При этом нокдаун гена PERK или ингибирова-
миофибробласты. В частности, подавление ак-
ние PERK с помощью ингибитора GSK2606414
тивности ATF6 с помощью нокаута или нокда-
подавляет индуцированный тапсигаргином
уна приводит к усилению профиброзного эф-
синтез α-SMA и коллагена IV.
фекта TGF-β [36]. Напротив, дополнительная
Таким образом, сигнальные пути актива-
фармакологическая активация ATF6 с помо-
ции стресса ЭПР могут оказывать разнонаправ-
щью низкомолекулярного активатора N-(2-гид-
ленное действие на дифференцировку фибро-
рокси-5-метилфенил)-3-фенилпропанамида
бластов в миофибробласты: если необходимо
(compound 147) снижает экспрессию α-SMA
стимулировать дифференцировку, следует ак-
и подавляет профиброзное действие TGF-β.
тивировать IREI- и PERK-зависимые механиз-
Предполагается, что ATF6 подавляет диффе-
мы индукции стресса ЭПР; если же необходимо
ренцировку фибробластов в миофибробласты
подавить признаки дифференцировки, то сле-
за счёт ингибирования TGF-β-опосредован-
дует воздействовать на сигнальный путь ATF6.
ного сигналинга через посредник Smad и сни-
жения экспрессии генов, кодирующих TGF-β
и его рецепторы. В частности, при активации
UPR И АДИПОГЕНЕЗ
ATF6 с помощью compound 147 активность не-
которых профиброзных генов, например генов
Адипоциты являются основным компонен-
матриксных металлопротеаз и рецепторов к
том жировой ткани. Их функция состоит в кон-
БИОХИМИЯ том 87 вып. 9 2022
СТРЕСС ЭПР И ДИФФЕРЕНЦИРОВКА КЛЕТОК
1209
троле энергетического баланса путём хранения
при завершении дифференцировки наблюда-
триацилглицерина в периоды избытка энергии
ется при добавлении 4-PBA на более поздних
и его мобилизация во время недостатка энер-
стадиях адипогенеза. В экспериментах in vivo
гии [38]. Адипоциты являются метаболически
добавление 4-PBA в питьевую воду самкам мы-
активными клетками, способными синтезиро-
шей C57BL/6 вызывает замедление увеличения
вать и секретировать различные белки, включая
веса и снижение массы жировых прослоек, а
гормоны (например, лептин и адипонектин)
также уменьшает размер адипоцитов. Кроме
и цитокины [39]. Адипоциты дифференциру-
того, 4-PBA снижает уровень синтеза GRP78
ются из преадипоцитов [38], и ключевую роль
в жировой ткани. Такой эффект 4-PBA in vivo,
в дифференцировке адипоцитов (адипогенезе)
по-видимому, связан с тем, что 4-PBA подавля-
играют транскрипционные факторы C/EBPβ и
ет рекрутирование и дифференцировку преа-
C/EBPδ (CCAAT/энхансер-связывающие бел-
дипоцитов и препятствует гипертрофии имею-
ки β и δ), которые активируются очень рано и
щихся адипоцитов [39].
запускают экспрессию рецептора, активируе-
Ингибирование адипогенеза при подавле-
мого пролифераторами пероксисом PPARγ
нии стресса ЭПР было также обнаружено при
и CCAAT/энхансер-связывающего белка α
воздействии нетепловой атмосферной плазмы
(C/EBPα). PPARγ и C/EBPα - два ключевых
(ионизированный газ), клиническое примене-
адипогенных фактора, которые позитивно ре-
ние которой изучают в последние десятилетия.
гулируют друг друга для стимулирования и под-
Полученный авторами исследования раствор
держания дифференцировочного статуса [40].
плазмы, воздействуя в течение 4 дней, подавлял
Следует отметить, что в ходе адипогенеза пре-
адипогенную дифференцировку клеток линии
адипоциты претерпевают значительные мор-
3T3-L1, параллельно ингибируя стресс ЭПР и
фологические изменения. Так, например, они
активацию UPR в ходе адипогенеза [47].
меняют свою форму с фибробластоподобной
Исследования показали, что для успешно-
на кубическую, в них появляется большое ко-
го прохождения адипогенеза необходимы все
личество липидных капель [39-41].
три пути активации UPR [40, 43, 44, 48].
При исследовании адипогенеза in vitro ак-
Роль сигнального пути IRE1. Было по-
тивацию дифференцировки проводят с по-
казано, что ключевой ранний адипогенный
мощью специального дифференцировочного
фактор C/EBPβ напрямую связывается с
коктейля, содержащего инсулин, изобутилме-
промотором гена, кодирующего XBP1, и ин-
тилксантин, дексаметазон и троглитазон. В ка-
дуцирует его экспрессию. Затем sXBP1 свя-
честве преадипоцитов обычно используют
зывается с промотором другого критически
клетки линии 3T3-L1 или эмбриональные фиб-
важного для адипогенеза транскрипционного
робласты мыши (MEF) [39, 40, 42-47].
фактора - C/EBPα - и индуцирует его экспрес-
Как оказалось, дифференцировка адипо-
сию. Нокдаун XBP1 или IRE1 в клетках MEF
цитов in vitro и in vivo сопровождается актива-
или 3T3-L1 приводит к глубоким нарушениям
цией стресса ЭПР [39, 45-47], причём в системе
адипогенеза, в частности, в клетках почти не
in vitro уровень синтеза фосфорилированно-
формируются липидные капли. В ходе адипо-
го eIF2α и sXBP1 повышается на ранней стадии
генеза в клетках 3T3-L1 не происходит гипер-
дифференцировки и затем постепенно снижа-
фосфорилирования IRE1, хотя оно происходит
ется, а уровень синтеза CHOP, наоборот, сни-
при индукции UPR с помощью тапсигаргина.
жается на ранней стадии дифференцировки и
Интересно, что индукция ожирения у мышей
затем повышается на более поздних стадиях [39,
вызывает появление гиперфосфорилирован-
45, 46]. Ингибирование стресса ЭПР in vitro с
ного IRE1. Возможно, это связано с тем, что
помощью химических шаперонов 4-PBA или
физиологический UPR активируется в ходе
тауро-урсодезоксихолевой кислоты (TUDCA)
раннего адипогенеза и затем поддерживается
подавляет адипогенез. Это выражается в том,
на относительно низком уровне в зрелых ади-
что уменьшается количество липидных капель
поцитах, а последующее ожирение ассоцииро-
и не происходит изменение формы клеток, а
вано с повышением уровня UPR [40].
при действии 4-PBA также снижается секреция
Однако, по данным других исследователей,
адипонектина. При этом импульсное добав-
активация или делеция IRE1 не изменяла диф-
ление 4-PBA блокирует накопление липидов
ференцировку в клетках 3T3-L1 [45], что про-
на всех стадиях дифференцировки, от ранней
тиворечит результатам, описанным выше [40].
(0-2 дни) до поздней (6-10 дни), что свиде-
Роль сигнального пути PERK. Как оказа-
тельствует о важности UPR для адипогене-
лось, PERK-опосредованный механизм индук-
за на всех его стадиях. Тем не менее наиболее
ции стресса ЭПР тоже участвует в регуляции
значительное подавление накопления липидов
липогенной программы в ходе адипогенеза.
4
БИОХИМИЯ том 87 вып. 9 2022
1210
ТУРИЩЕВА и др.
Так, например, при отсутствии PERK инги-
стресса ЭПР на дифференцировку адипоци-
бируется повышение экспрессии липогенных
тов [41, 42, 45, 51]. Так, например, соедине-
ферментов SREBP-1c (белок, связывающий-
ние K-7174 (синтетический низкомолекуляр-
ся с регуляторным элементом стерола-1c),
ный ингибитор транскрипционных факторов
FAS (синтаза жирных кислот) и SCD1 (стеа-
GATA, который участвует в поддержании пре-
рил-КоА-десатураза-1), подавляется накоп-
адипоцитов в недифференцированном состоя-
ление липидных капель. При этом PERK в
нии), а также индукторы стресса ЭПР - туни-
ходе стимулирования дифференцировки ади-
камицин, A23187 и тапсигаргин, ингибировали
поцитов проявляет активность липидной ки-
адипогенез клеток линии 3T3-L1 [42, 45]. Кроме
назы по отношению к диацилглицеролу [44,
того, туникамицин и тапсигаргин подавляли
49]. Однако уровень синтеза мишени PERK -
дифференцировку бежевых адипоцитов, раз-
белка CHOP - снижается на ранней стадии
витие которых повышает термогенез путём
дифференцировки и затем повышается на
рассеивания избыточной химической энер-
более поздних стадиях. Это свидетельствует о
гии через опосредованное разобщающим
необходимости снижения активности PERK
белком 1 (UCP1) формирование тепла и по-
на ранней стадии дифференцировки адипо-
могает снизить ожирение [51]. Снижение уров-
цитов [39, 45, 46]. При этом стимуляция фос-
ня стресса ЭПР химическими шаперонами
форилирования eIF2α при отсутствии стрес-
TUDCA или 4-PBA стимулировало дифферен-
са ЭПР снижает эффективность адипогенеза в
цировку бежевых адипоцитов. У мышей с ожи-
клетках 3T3-L1, а подавление фосфорилирова-
рением, получавших TUDCA, наблюдалось
ния eIF2α, напротив, стимулирует адипогенез
побурение (browning) белого жира, возможно,
in vitro и in vivo [45]. Стимулированная продук-
вызванное транс-дифференцировкой белых
ция CHOP при отсутствии стресса ЭПР также
адипоцитов в бежевые, и значительно тормо-
ингибирует адипогенез в клетках 3T3-L1 [41,
зилось увеличение веса [51].
45]. Такое ингибирующее влияние CHOP на
По-видимому, противоречие в данных
адипогенез, возможно, связано со способ-
о роли стресса ЭПР в адипогенезе связано
ностью CHOP образовывать гетеродимер с
с тем, что разный уровень стресса ЭПР мо-
C/EBPα и β и препятствовать связыванию об-
жет по-разному влиять на адипогенез. Запуск
разовавшегося гетеродимера с классическими
стресса ЭПР выше физиологического уровня
сайтами связывания для C/EBP [41, 50]. То есть
вызывает сильный UPR, который не позво-
CHOP в этой системе функционирует как нега-
ляет клеткам уйти в дифференцировку, так
тивный регулятор адипогенеза [39, 41]. Поэто-
как ресурсы таких клеток направлены на вы-
му, возможно, снижение экспрессии CHOP на
живание. В связи с этим ряд исследователей
ранней стадии дифференцировки, показанное
предлагает выделять две формы UPR - острую
многими авторами, связано с тем, что такое
форму, также называемую «патологической»,
снижение необходимо для работы адипоген-
и хроническую форму, которую они называ-
ных генов семейства C/EBP, активности кото-
ют «физиологической» [39, 40, 45, 46]. Острая/
рых CHOP препятствует.
патологическая форма UPR возникает в ответ
Роль сигнального пути ATF6. Сенсор стрес-
на неблагоприятные условия, такие как гипок-
са ЭПР, ATF6, также важен для нормального
сия, гипергликемия, вирусная инфекция или
адипогенеза. Нокдаун ATF6 в мезенхимных
окислительный и механический стресс из-за
стромальных клетках линии C3H10T1/2 при-
гипертрофии адипоцитов в жировой ткани при
водит к подавлению накопления липидов,
ожирении. Эта форма стресса ЭПР может от-
значительному снижению экспрессии PPARγ,
рицательно влиять на функцию жировой ткани
липогенного транскрипционного фактора
и способствовать развитию диабета 2-го типа,
SREBP-1c, чувствительного к инсулину пере-
резистентности к инсулину и лептину и липо-
носчика глюкозы GLUT4 и связывающегося
токсичности. Напротив, хронический/физио-
с жирными кислотами белка aP2. При этом
логический тип активации UPR характерен для
разница в экспрессии PPARγ в клетках с нок-
таких клеточных процессов, как дифференци-
дауном ATF6 и контрольных клетках по мере
ровка и, по-видимому, является адаптивным
прохождения этапов адипогенеза становится
механизмом, направленным на стимуляцию
всё более значительной. Кроме того, в клетках
выживания клеток и эффективное функцио-
с нокдауном ATF6 менее выражена стимуляция
нирование ЭПР. Непосредственно в адипоци-
экспрессии раннего транскрипционного фак-
тах активация UPR, по-видимому, настраивает
тора адипогенеза C/EBPβ [48].
клетку на повышенную нагрузку в ЭПР [39].
Следует отметить, что в ряде работ при-
Подтверждение предположения о двух
сутствуют данные об ингибирующем влиянии
формах UPR было получено в работе
БИОХИМИЯ том 87 вып. 9 2022
СТРЕСС ЭПР И ДИФФЕРЕНЦИРОВКА КЛЕТОК
1211
Longo et al. [46], показавших, что стресс ЭПР
которая окружает мышечные волокна [53], и
и UPR «физиологически» активируются в ходе
плазматической мембраной мышечных воло-
адипогенеза клеток 3T3-L1, а «патологическая»
кон (сарколеммой). Они представляют собой
часть стресса ЭПР, вызванная глюкотоксиче-
популяцию сателлитных клеток (миосателли-
ским воздействием глюкозамина, ингибирует
тов) - стволовых клеток мышечной ткани [52,
их дифференцировку. Индукция дифферен-
54]. Миогенез в постнатальном периоде возоб-
цировки преадипоцитов в присутствии тапси-
новляется в ходе регенерации ткани после
гаргина вызывала значительно более высокую
повреждения мышечных волокон
[55]. При
активацию UPR, чем при физиологически
этом сателлитные клетки активируются, про-
индуцированном адипогенезе. Такая гиперак-
лиферируют и либо сливаются с мышечными
тивация UPR сопровождалась снижением экс-
волокнами, либо образуют новые мышечные
прессии генов-маркёров адипоцитов (C/EBPα,
волокна [56]. В активации дифференциров-
PPARγ2, FABP4/AP2) и ингибированием ади-
ки миобластов и поддержании их пролифе-
погенеза. Сходным эффектом обладал и другой
ративной активности участвуют белок детер-
индуктор стресса ЭПР - глюкозамин, способ-
минации миобластов 1 (MyoD) и миогенный
ный активировать гексозаминовый биосинте-
фактор 5 (Myf5) [55]. После индукции диффе-
тический путь, вовлечённый во многие отрица-
ренцировки миобласты перестают пролифери-
тельные эффекты гипергликемии. Интересно,
ровать, и в них активируется экспрессия генов,
что добавление химического шаперона 4-PBA в
кодирующих миогенин и специфичный для
концентрации 50 мкМ вместе с глюкозамином
миоцитов энхансерный фактор 2с (Mef2c), что
в дифференцировочную среду не ингибирова-
приводит к слиянию миобластов (миоцитов)
ло «физиологическое» повышение экспрессии
и их терминальной дифференцировке. Мио-
GRP78 и CHOP при дифференцировке ади-
генез сопровождается увеличением площади и
поцитов, но снимало ингибирующий эффект
объёма ЭПР, о чём свидетельствует хорошо раз-
глюкозамина на адипогенез [46]. При этом ра-
витая сеть ЭПР в миотубулах [52].
нее было показано, что 10-20 мМ 4-PBA инги-
Известно, что первичные миобласты при
бирует дифференцировку адипоцитов [39]. Как
индукции миогенеза in vitro могут дифференци-
полагают Longo et al. [46], такое различие свя-
роваться в миотубулы [54]. Оказалось, что фи-
зано с тем, что в высокой концентрации 4-PBA
зиологический стресс ЭПР стимулирует миоге-
полностью подавляет
«физиологический»
нез, причём для дифференцировки миобластов
стресс ЭПР, необходимый для нормальной
необходимы все три пути активации UPR [15,
дифференцировки адипоцитов, в частности,
52, 55, 57]. Кроме того, показано, что в ходе диф-
для адаптации ЭПР к повышенному синтезу
ференцировки миобластов транскрипционный
белка в ходе дифференцировки. В низкой кон-
фактор TEAD4 (транскрипционно усиленный
центрации этот химический шаперон ингиби-
ассоциативный домен 4), экспрессирующийся
рует только «патологический» стресс ЭПР.
в развивающейся скелетной мускулатуре эм-
Таким образом, IRE1-, PERK- и ATF6-зави-
брионов мыши и необходимый для нормальной
симые пути активации стресса ЭПР стимулируют
дифференцировки миобластов C2C12, также
адипогенез. Поэтому, например, для подавления
регулирует экспрессию генов UPR [57]. В част-
адипогенеза и смягчения развития ожирения не-
ности, при нокдауне TEAD4 снижается экспрес-
обходимо ингибировать все три пути.
сия генов шаперонов ЭПР, генов белков ERAD,
ATF6, ATF4 и CHOP, а также подавляется экс-
прессия и сплайсинг XBP1, причём экспрессия
UPR И МИОГЕНЕЗ
CHOP напрямую регулируется TEAD4.
В ходе дифференцировки миобластов про-
Миобласты представляют собой клетки-
исходит временное снижение концентрации
предшественники мышечных волокон. В ходе
кальция в ЭПР, необходимое для миогенеза [15,
ранней фазы эмбрионального развития мио-
55]. Блокирование освобождения кальция
бласты, образовавшиеся из коммитированных
из ЭПР подавляет UPR в ходе миогенеза и ин-
предшественников, сливаются и формируют
гибирует сам миогенез. Кроме того, снижение
многоядерные миотубулы, а затем миотубулы
концентрации кальция в ЭПР в дифференци-
дифференцируются в мышечные волокна [52].
рующихся миобластах in vitro и in vivo приводит
Не ушедшие в дифференцировку коммитиро-
к образованию особых структур, выявляемых с
ванные предшественники миобластов пере-
помощью ЭПР-трекера. Эти структуры имеют
ходят в состояние покоя. На поздней стадии
размер 1-4 мкм и представляют собой цистер-
эмбриогенеза и в постнатальном развитии эти
ны гранулярного ЭПР, свёрнутые в концентри-
клетки остаются между базальной мембраной,
ческие кольца. Одна структура содержит 4-10
БИОХИМИЯ том 87 вып. 9 2022
4*
1212
ТУРИЩЕВА и др.
свёрнутых в кольцо цистерн ЭПР, расположен-
отсутствие этого транскрипционного фактора
ных почти на одинаковом расстоянии друг от
компенсируется другими факторами [55].
друга. Они появляются в миобластах на третий
Роль сигнального пути PERK. PERK-опо-
день дифференцировки, а исчезают уже после
средованный механизм контролирует раннюю
слияния миобластов [15]. Сходные структуры,
дифференцировку миобластов in vitro за счёт ре-
но со свободным пространством в центре (ино-
гуляции экспрессии микроРНК, необходимых
гда заполненным органеллами), наблюдаются в
для миогенеза [52]. Нокдаун PERK в мышиных
пролиферирующих миобластах при индукции
скелетных миобластах линии C2C12 подавляет
стресса ЭПР с помощью ингибиторов кальцие-
образование миотубул, приводит к изменению
вых АТФаз - тапсигаргина и циклопиазоновой
морфологии миобластов, снижает экспрессию
кислоты. Напротив, туникамицин, не влияю-
и синтез MyoD. При этом фармакологическое
щий на концентрацию кальция в ЭПР, не вызы-
ингибирование PERK с помощью ингибитора
вает формирование таких структур [15]. Однако
фосфорилирования PERK GSK2606414 также
физиологическое значение таких конформаци-
приводит к подавлению синтеза MyoD. Более
онных изменений ЭПР остаётся неизвестным.
того, нокдаун PERK изменяет экспрессию
Роль сигнального пути IRE1. IRE1/XBP1
микроРНК, связанных с поддержанием диффе-
играет важную роль в поддержании выживае-
ренцировки и стволовых свойств клеток. При
мости клеток и экспрессии генов, связанных
нокдауне PERK экспрессия многих микроРНК,
с дифференцировкой миобластов [55]. В мио-
стимулирующих миогенез (например, miR-128),
бластах линии C2C12 нокдаун IRE1 и XBP1 по-
снижается, а экспрессия микроРНК, поддер-
давляет формирование миотубул, экспрессию
живающих стволовые свойства клеток, повы-
генов миогенеза - Mef2c, MyoD и гена, кодирую-
шается. Помимо этого, повышается активность
щего миогенин [52, 55]. Следует отметить, что
сигнальных путей, ассоциированных со ство-
ген XBP1 является прямой мишенью миогени-
ловыми свойствами клеток, таких как Nanog,
на и MyoD [58]. В популяции клеток с нокдау-
Myc и Oct4, а также повышается экспрессия
ном XBP1 повышается частота апоптозов, что
маркёра миосателлитов PAX7 [52].
подчёркивает участие пути IRE1/XBP1 в под-
Более того, сигнальный путь PERK/ATF4
держании выживания клеток путём стимуля-
активирует микроРНК, ассоциированные с мио-
ции дифференцировки [55]. Более того, sXBP1,
генезом на ранней стадии дифференцировки
связываясь с промотором гена CDK5, участвует
миобластов [52]. ATF4 связывается с промотор-
в регуляции экспрессии циклин-зависимой ки-
ными участками и стимулирует экспрессию не-
назы 5 (CDK5), которая необходима для миоге-
которых микроРНК, ассоциированных с диф-
неза. Было сделано предположение, что sXBP1
ференцировкой. При гиперэкспрессии ATF4
регулирует экспрессию генов миогенеза путём
даже без стимула к дифференцировке миобла-
индукции экспрессии CDK5 [55]. При этом ги-
сты сливаются в миотубулы, и в них повышает-
перэкспрессия sXBP1 подавляет миогенез, ак-
ся уровень синтеза MyoD.
тивируя репрессор MyoD - Mist1 [59]. В таких
Ключевой регулятор сигнального пути UPR -
миобластах подавляется экспрессия маркёров
каталитическая субъединица протеинфосфата-
миогенеза и нарушается формирование миоту-
зы 1 γ (Ppp1cc), ген которой является целевым
бул. Это может быть связано с тем, что гиперэкс-
для микроРНК miR-128, которую, в свою оче-
прессия sXBP1
«имитирует» повышенный
редь, контролирует сигнальный путь PERK [52].
уровень стресса ЭПР, в связи с чем для восста-
MiRNA-128 подавляет ингибитор миогене-
новления гомеостаза срабатывает механизм ин-
за Ppp1cc, а репрессия Ppp1cc имеет решающее
гибирования дифференцировки, то есть сраба-
значение для петли обратной связи, которая ре-
тывает «точка контроля стресса ЭПР» [55, 59].
гулирует активность сигнальных путей, связан-
Следует отметить, что нокаут XBP1 не ока-
ных с UPR. Кроме этого, подавление активности
зывает влияния на регенеративный миогенез
Ppp1cc приводит к увеличению площади ЭПР,
с участием клеток-сателлитов у взрослых мы-
миграции клеток, их слиянию и образованию
шей [56]. Нокаут XBP1 не влияет на форми-
миотубул. При этом РНК-связывающий белок
рование миотубул и мышечных волокон и на
ARPP21 (регулируемый циклическими АМФ
экспрессию маркёров миогенеза. Это может
фосфопротеин 21) противодействует активно-
свидетельствовать о том, что разные миоген-
сти miR-128, конкурируя с ней за связывание с
ные факторы вносят вклад в эмбриональный
3′-нетранслируемой областью гена, кодирующе-
миогенез и миогенез с участием клеток-са-
го Ppp1cc. В ходе позднего миогенеза ARPP21 не-
теллитов при регенерации во взрослом орга-
гативно регулирует miR-128, подавляя её блоки-
низме [55, 56]. Возможно, sXBP1 не оказыва-
рующее воздействие на Ppp1cc, и таким образом,
ет влияния на регенеративный миогенез или
препятствуя постоянной активности UPR [52].
БИОХИМИЯ том 87 вып. 9 2022
СТРЕСС ЭПР И ДИФФЕРЕНЦИРОВКА КЛЕТОК
1213
Помимо эмбрионального миогенеза, сиг-
тапсигаргином перед индукцией дифференци-
нальный путь PERK регулирует регенерацию
ровки также повышает экспрессию генов, ко-
скелетных мышц, опосредованную сателлит-
дирующих MyoD и миогенин [63]. Более того,
ными клетками [56]. При нокауте PERK in vivo
мягкий стресс ЭПР, индуцируемый тапсигарги-
нарушается регенерация мышц, что выража-
ном, устраняет подавление слияния миобластов
ется в подавлении формирования миотубул и
линии С2С12, вызываемое гиперэкспрессией
мышечных волокон, снижении экспрессии и
деубиквитинирующего фермента USP19 [64].
синтеза маркёров миогенеза MyoD и миогени-
Таким образом, все три пути активации
на. Фармакологическое ингибирование PERK
UPR необходимы для нормального прохожде-
с помощью GSK2606414 в сателлитных клетках
ния эмбрионального миогенеза, однако только
in vitro также приводит к подавлению форми-
сигнальный путь PERK стимулирует регенера-
рования миотубул [56].
тивный миогенез.
Следует отметить, что CHOP, напротив,
подавляет транскрипцию MyoD и задержива-
ет дифференцировку миобластов in vitro [60].
UPR И ОСТЕОБЛАСТОГЕНЕЗ
Экспрессия CHOP наблюдается в субпопу-
ляции миобластов, которые при инкубации
Остеобласты - это клетки, синтезирую-
с дифференцировочной средой не уходят в
щие и образующие внеклеточный матрикс, и
дифференцировку. При нокдауне CHOP на-
таким образом участвующие в формировании
блюдается более ранняя и более выраженная
костной ткани [65, 66]. Они дифференциру-
дифференцировка, в миотубулах увеличивает-
ются из преостеобластов, которые, в свою
ся количество ядер, в то время как постоянная
очередь, дифференцируются из мезенхимных
экспрессия CHOP задерживает дифференци-
стромальных (стволовых) клеток при акти-
ровку и уменьшает количество ядер в миоту-
вации мастер-регулятора остеогенеза Runx2
булах. Показано, что CHOP связывается с ре-
(фактор транскрипции 2, связанный с доме-
гуляторными участками гена MyoD и снижает
ном runt) [66]. Мишенями Runx2 являются
ацетилирование гистонов в этих участках. Та-
остеопонтин, остеокальцин (OCN) и костный
ким образом, активность CHOP, по-видимому,
сиалопротеин (BSP). Преостеобласты (незре-
препятствует преждевременной дифференци-
лые остеобласты) дифференцируются в зрелые
ровке миобластов [60].
остеобласты в ходе трёх этапов: 1) преостео-
Роль сигнального пути ATF6. Проведённые
бласты продолжают пролиферировать и экс-
исследования показали, что нокдаун ATF6 в
прессируют маркёры коллаген I, остеопонтин
мышиных скелетных миобластах линии C2C12
и др., 2) преостеобласты выходят из клеточно-
подавляет образование миотубул, экспрессию
го цикла и начинают дифференцироваться в
и синтез MyoD [52, 61]. Однако при регене-
остеобласты, экспрессируя маркёры коллаген I
ративном миогенезе с участием сателлитных
и активную щелочную фосфатазу, 3) незрелые
клеток уровень экспрессии ATF6 не изменя-
остеобласты минерализуют матрикс и экспрес-
ется [56]. По-видимому, роли разных сигналь-
сируют маркёр OCN [66]. Таким образом, ког-
ных путей UPR в эмбриональном миогенезе и
да незрелые остеобласты дифференцируются в
постнатальном регенеративном миогенезе раз-
зрелые, они продуцируют множество белков,
личаются, однако для получения более деталь-
в первую очередь коллаген I и OCN [67-69].
ной информации необходимы дополнительные
Для дифференцировки остеобластов необ-
исследования.
ходимо множество факторов, среди которых
Влияние индукторов стресса ЭПР на мио-
ключевыми являются Osx (Osterix) и BMP2
генез. Индукторы стресса ЭПР, туникамицин
(костный морфогенетический белок 2) [66, 68].
и тапсигаргин, селективно элиминируют не
Интересно, что BMP2 стимулирует остеогенез
способные дифференцироваться миобласты
путём активации мягкого стресса ЭПР с уча-
линии С2С12, позволяя выжившим клеткам
стием IRE1, PERK и ATF6 [68, 70-72].
более эффективно дифференцироваться в мио-
Роль сигнального пути IRE1. Роль пути
тубулы [62, 63]. В частности, инкубация мио-
IRE1/XBP1 в дифференцировке остеобластов
бластов с туникамицином или тапсигаргином
в настоящее время однозначно не определе-
перед индукцией дифференцировки приводит
на. По данным одной группы исследователей,
к увеличению количества ядер в миотубулах
путь IRE1/XBP1 стимулирует дифференциров-
и увеличению размера самих миотубул [62].
ку остеобластов, причём транскрипционный
Такие миотубулы формируют саркомеры и спо-
фактор Osx, необходимый для формирования
собны сокращаться, что редко наблюдается в
костей, является мишенью sXBP1 [73]. Так,
системе in vitro [62]. Инкубация миобластов с
например, при стимулировании остеогенной
БИОХИМИЯ том 87 вып. 9 2022
1214
ТУРИЩЕВА и др.
дифференцировки с помощью BMP2 в фибро-
ных балок в ходе постнатального развития [75].
бластах мыши MEF значительно повышается
Мишенями ATF4 являются маркёры диффе-
синтез sXBP1 и экспрессия Osx, синтез коллаге-
ренцировки остеобластов OCN и BSP [68, 75].
на I и OCN, активность щелочной фосфатазы.
ATF4 активирует OCN, формируя на промо-
В то же время при нокауте IRE1 индукция диф-
торе OCN комплекс с Runx2 [68, 76]. Следу-
ференцировки с помощью BMP2 не вызывала
ет отметить, что Runx2 нужен для экспрессии
повышения активности щелочной фосфатазы,
самого ATF4, так как при отсутствии экспрес-
был снижен уровень BSP, коллагена I и OCN,
сии Runx2 экспрессия ATF4 подавлена [75].
то есть при отсутствии IRE1 дифференцировка
ATF4 является транскрипционным ак-
остеобластов ингибировалась [73].
тиватором не только для OCN, но и для гена
Противоположные результаты были полу-
транскрипционного фактора остеогенеза Osx
чены другой группой исследователей, которые
[77]. ATF4 повышает экспрессию Osx, связыва-
показали, что нокдаун IRE1 стимулирует инду-
ясь с его промотором. Нокаут ATF4 приводит к
цированную BMP2 дифференцировку мезен-
значительному снижению уровня синтеза Osx,
химных стволовых клеток линии C2C12 в остео-
хотя уровень Runx2, необходимый для экспрес-
бласты, а гиперэкспрессия IRE1 ингибирует
сии Osx, не изменяется.
дифференцировку [74]. Противоречие в полу-
Интересно, что паратиреоидный гор-
ченных данных можно объяснить тем, что в ка-
мон (ПТГ), используемый в клинике в каче-
честве модели остеогенеза были использованы
стве лекарственного препарата для лечения
разные клеточные линии, а также неполным по-
возрастного остеопороза, воздействует на сиг-
давлением экспрессии IRE1 при его нокдауне.
нальный путь PERK и стимулирует формиро-
Роль сигнального пути PERK. В отличие
вание костей [69, 77]. ПТГ активирует PERK
от сигнального пути IRE1 необходимость ак-
в преостеобластах линии MC3T3-E1 и в пер-
тивности PERK для дифференцировки остео-
вичных остеобластах голени и стимулирует
бластов не вызывает сомнений [67, 68, 75].
активацию генов-маркёров дифференцировки
Мыши с нокаутом PERK имеют тяжёлую нео-
остеобластов, таких как Runx2, OCN и гены,
натальную остеопению, которая проявляется в
кодирующие щелочную фосфатазу и коллаген
уменьшении толщины кортикальной части ко-
I [69]. Подавление активности PERK или ATF4
сти и степени минерализации, а также в сни-
с помощью ингибиторов или нокдауна подав-
жении объёма и толщины костных балок [67,
ляет экспрессию генов-маркёров дифферен-
68]. Такая остеопения вызвана недостаточным
цировки остеобластов, активность щелочной
количеством зрелых остеобластов, нарушен-
фосфатазы, минерализацию матрикса и сек-
ной дифференцировкой остеобластов и нару-
рецию OCN в этих клетках. Напротив, салу-
шением секреции проколлагена I. При нокау-
бринал, поддерживающий фосфорилирование
те PERK у мышей и в первичных остеобластах
eIF2α, усиливает дифференцировку остеобла-
мышей снижается экспрессия генов, кодирую-
стов, индуцированную с помощью ПТГ. Кроме
щих маркёры зрелых остеобластов - щелочную
того, было показано, что ПТГ усиливает взаи-
фосфатазу, коллаген I, OCN и BSP, задержива-
модействие между шапероном HSP90 (белок
ется формирование минерализованных отло-
теплового шока 90) и PERK, а ингибирование
жений, подавлена экспрессия Runx2 и Osx [67,
HSP90 снижает синтез PERK и ингибирует
68]. Напротив, при введении в первичные остео-
дифференцировку остеобластов при воздей-
бласты мышей PERK-/- вектора с ATF4 актив-
ствии ПТГ. Таким образом, HSP90 также не-
ность щелочной фосфатазы и формирование
обходим для дифференцировки остеобластов,
минерализованных отложений возвращается к
так как он усиливает стабильность PERK [69].
нормальному уровню [68].
Участие сигнального пути PERK в активации
Активность PERK необходима для акти-
остеогенеза с помощью ПТГ было подтверж-
вации критического регулятора дифференци-
дено в экспериментах in vivo. У мышей с но-
ровки остеобластов - ATF4 [68, 75], который
каутом ATF4 ПТГ не индуцировал или слабо
является транскрипционным фактором, необ-
индуцировал экспрессию маркёров дифферен-
ходимым для дифференцировки и экспрессии
цировки остеобластов [77].
специфичных генов остеобластов [75]. Кроме
Следует отметить, что путь PERK стимули-
того, ATF4 участвует в посттранскрипционной
рует остеогенную дифференцировку не только
регуляции синтеза коллагена I. У мышей с но-
остеобластов, но и других клеток мезенхимно-
каутом ATF4 развивается тяжёлая остеопения.
го происхождения. При индукции остеогенной
В частности, задерживается формирование
дифференцировки стандартной дифференци-
костей и минерализация в эмбриональный пе-
ровочной средой (питательная среда α-MEM
риод, и снижается количество и толщина кост-
с добавлением 10% FBS, 100 мкМ L-аскор-
БИОХИМИЯ том 87 вып. 9 2022
СТРЕСС ЭПР И ДИФФЕРЕНЦИРОВКА КЛЕТОК
1215
бат-2-фосфата, 10 мМ β-глицерофосфата и
в первичных остеобластах мыши и мезенхим-
10 нМ дексаметазона) стволовые клетки перио-
ных стволовых клетках костного мозга крысы
донтальной связки человека (плотная соеди-
тапсигаргин повышает уровни синтеза OCN
нительная ткань вокруг корня и альвеолярной
и BSP, а при нокауте PERK в первичных остео-
кости зуба) дифференцируются в остеобласто-
бластах и при нокдауне PERK в мезенхимных
подобные клетки, в них повышается экспрес-
стволовых клетках костного мозга крысы дан-
сия генов-маркёров остеогенной дифференци-
ный эффект не наблюдается [68, 81].
ровки [78]. Гиперэкспрессия PERK ещё больше
При этом повышенный (патологический)
повышает уровень экспрессии генов-маркёров
стресс ЭПР подавляет остеогенез и индуцирует
остеогенной дифференцировки и активность
апоптоз [80-82]. Так, тапсигаргин стимулирует
щелочной фосфатазы, а подавление PERK, на-
остеогенез в линии стромальных клеток кост-
против, ингибирует дифференцировку.
ного мозга мыши ST2 [82]. Однако следует от-
Кроме того, ATF4 играет ключевую роль
метить, что использование более высокой дозы
в индукции дифференцировки мезенхимных
тапсигаргина подавляет остеогенез [81].
стволовых клеток костного мозга в остеобла-
Таким образом, все три пути активации
сты, в том числе стимулируя синтез β-катенина,
UPR стимулируют остеогенез, что может быть
который необходим для дифференцировки в
использовано для улучшения восстановления
остеобласты [79]. Подавление экспрессии гена,
костной ткани.
кодирующего ATF4, ингибировало дифферен-
цировку в остеобласты. Нокдаун ATF4 снижал
уровень синтеза β-катенина, но не экспрессии
UPR И ОСТЕОКЛАСТОГЕНЕЗ
гена, кодирующего β-катенин, что свидетель-
ствует о преимущественно посттранскрипци-
Остеокласты - тканеспецифичные много-
онной регуляции уровня β-катенина. Помимо
ядерные макрофаги, дифференцирующиеся из
этого, ATF4 индуцирует признаки остеогенной
моноцитарных/макрофагальных предшествен-
дифференцировки в фибробластах NIH3T3,
ников на поверхности или около поверхности
миобластах C2C12 и лимфобластах S194 [65].
кости [83]. Они играют важную роль в резорб-
Роль сигнального пути ATF6. Исследования
ции и ремоделировании кости, и их активность
показали, что в остеогенезе, индуцируемом
тщательно регулируется гормонами и цитоки-
фактором дифференцировки BMP2, участвует
нами для того, чтобы обеспечить баланс меж-
и ATF6 [71]. BMP2 стимулирует экспрессию и
ду резорбцией кости и её формированием [84].
активацию ATF6 в клетках линии MC3T3-E1,
Нарушение этого баланса в сторону избыточной
усиливая связывание Runx2 с промотором
резорбции ведёт к таким патологиям, как остео-
гена ATF6. При нокауте Runx2 BMP2 не спо-
пороз, метастазирование в костях, разруше-
собен активировать ATF6. В свою очередь, ги-
ние костей при артрите и периодонтит [84, 85].
перэкспрессия ATF6 повышает активность про-
Дифференцировка остеокластов запускается
мотора гена OCN, а ингибирование активности
связыванием рецептора-активатора ядерного
ATF6 блокирует индукцию экспрессии OCN.
фактора NF-κB (RANK), расположенного на
По-видимому, BMP2 индуцирует дифферен-
поверхности предшественников остеокластов, с
цировку остеобластов через Runx2-зависимую
лигандом RANK (RANKL), расположенным на
экспрессию ATF6, который напрямую регули-
поверхности остеобластов и остеоцитов [83, 84].
рует транскрипцию OCN [71]. Интересно, что
После связывания с лигандом RANK активиру-
гомолог ATF6 - OASIS (вещество, специфи-
ет различные сигнальные пути, а также индуци-
чески индуцируемое старыми астроцитами),
рует колебания внутриклеточной концентрации
экспрессирующийся в костях, не влияет на
кальция, что ведёт к активации транскрип-
экспрессию OCN, однако является транскрип-
ционных факторов остеокластогенеза, таких
ционным активатором гена, кодирующего кол-
как протоонкоген c-Fos и ядерный фактор
лаген I, и имеет сайт связывания на его про-
активированных Т-клеток, цитоплазматиче-
моторе [70]. У мышей, нокаутных по OASIS,
ский 1 (NFATc1) [84]. Исследования последних
наблюдается сильная остеопения из-за задерж-
лет показали, что в дифференцировке остеокла-
ки дифференцировки остеобластов, характе-
стов важную роль играют сигнальные пути UPR,
ризующаяся в том числе снижением отложения
а именно: ветви IRE1/XBP1 и PERK/eIF2.
коллагена I.
Роль сигнального пути IRE1. В ходе остео-
Влияние индукторов стресса ЭПР на остео-
кластогенеза in vitro, индуцированного с помо-
генез. При запуске физиологического стрес-
щью рекомбинантного RANKL и колониести-
са ЭПР индукторы стресса ЭПР могут стиму-
мулирующего фактора 1 (CSF1) мыши, путь
лировать остеогенез [80, 81]. Так, например,
IRE1/XBP1 активируется в начале дифферен-
БИОХИМИЯ том 87 вып. 9 2022
1216
ТУРИЩЕВА и др.
цировки, достигает пика активности на вто-
стимулирующего остеокластогенез, а точнее,
рой день после индукции остеокластогенеза, а
активатором PERK, является окислительный
затем инактивируется [84]. Подавление IRE1/
стресс [85].
XBP1 значительно репрессирует остеокласто-
Влияние индукторов стресса ЭПР на остео-
генез, что ведёт к уменьшению числа остео-
кластогенез. Участие стресса ЭПР в остеокла-
кластов и увеличению костной массы in vivo.
стогенезе наводит на мысль о возможности
Это связано с тем, что sXBP1 является тран-
стимулировать дифференцировку остеокла-
скрипционным фактором мастер-регулятора
стов с помощью индукторов стресса ЭПР.
остеокластогенеза NFATc1 и стимулирует его
Действительно, показано, что индуктор стрес-
транскрипцию, непосредственно связываясь
са ЭПР, тапсигаргин, может запускать остео-
с промотором гена NFATc1. Подавление IRE1/
кластогенез, активируя экспрессию и син-
XBP1 ведёт к супрессии транскрипции NFATc1 и
тез NFATc1 и маркёров остеокластов [88, 89].
остеокластогенеза. При этом подавление IRE1
При этом химический шаперон и ингибитор
не влияет на экспрессию других транскрипци-
стресса ЭПР, 4-PBA, подавляет остеокласто-
онных факторов дифференцировки остеокла-
генез и активацию NFATc1 как в присутствии
стов [84]. Предполагается, что путь IRE1/XBP1
тапсигаргина, так и без него [89]. Кроме того,
при остеокластогенезе активируется из-за фи-
4-PBA подавляет остеокластогенез, индуци-
зиологического стресса ЭПР, косвенно вызы-
рованный частицами полиэтилена (средний
ваемого колебаниями концентрации кальция,
диаметр частиц 65 нм), которые также активи-
как следствие сигналинга RANKL-RANK. Ак-
руют стресс ЭПР [90]. Такролимус (иммуносу-
тивация IRE1/XBP1, в свою очередь, стимули-
прессирующее соединение, блокирующее ак-
рует транскрипцию NFATc1 [84].
тивацию T-клеток при ревматоидном артрите)
Следует отметить, что внеклеточные ве-
ингибирует стимулированный тапсигаргином
зикулы из клеток множественной миеломы
стресс ЭПР и остеокластогенез, подавляя экс-
индуцируют остеокластогенез в кости путём
прессию NFATc1 [91]. Другой индуктор стрес-
активации механизма IRE1/XBP1, что ведёт
са ЭПР, туникамицин, также стимулирует
к заболеваниям костей, которыми страдают
остеокластогенез, индуцируя экспрессию мар-
более 70% пациентов с множественной мие-
кёров остеокластов [88].
ломой [86]. Сигнальные молекулы UPR (на-
Примечательно то, что 0,05 и 0,1 нМ тапси-
пример, GRP94 и GRP78) являются карго для
гаргин стимулируют остеокластогенез, в то
внеклеточных везикул клеток множественной
время как тапсигаргин в концентрации 0,2 нМ
миеломы, и поглощение таких везикул мак-
не оказывает влияния на остеокластогенез [85].
рофагами мыши Raw264.7 быстро запускает
Это свидетельствует о том, что остеокласто-
фосфорилирование IRE1, сплайсинг XBP1 и
генез стимулируется только определёнными
активацию транскрипции NFATc1. Соответ-
уровнями стресса ЭПР. Это предположение
ственно, подавление в макрофагах мыши пути
подтверждают данные о влиянии салубрина-
IRE1/XBP1 с помощью селективного ингиби-
ла на дифференцировку остеокластов
[92].
тора IRE1 GSK2850163 при одновременном
Салубринал повышает фосфорилирование и,
введении в культуральную среду внеклеточных
как следствие, инактивацию eIF2α, что сни-
везикул из клеток множественной миеломы
жает общий синтез белков, включая NFATc1,
ингибирует экспрессию NFATc1 и препятствует
и подавляет дифференцировку остеокластов
дифференцировке макрофагов в остеокласты.
из клеток костного мозга мыши [92, 93]. Кро-
При этом подавляется и резорбционная актив-
ме того, салубринал смягчает признаки остео-
ность таких макрофагов [86].
пороза в мышиной модели остеопороза [93].
Роль сигнального пути PERK. RANKL-ин-
Возможно, салубринал индуцирует слишком
дуцированный остеокластогенез сопровожда-
сильный стресс ЭПР, что приводит не к стиму-
ется активацией пути PERK. Ингибирова-
ляции дифференцировки, а к её подавлению.
ние PERK с помощью GSK2606414 подавляет
Таким образом, сигнальные пути, активируе-
формирование остеокластов и их резорбци-
мые IRE1 и PERK, стимулируют остеокластоге-
онную активность, в том числе при модели-
нез. В связи с этим для подавления остеокласто-
ровании остеопороза у мышей [85]. Кроме
генеза необходимо ингибировать оба этих пути.
того, ATF4 является транскрипционным ак-
тиватором для NFATc1. Остеокластогенез по-
давляется при нокауте ATF4 in vitro и in vivo и
ЗАКЛЮЧЕНИЕ
усиливается при искусственном повышении
экспрессии ATF4 [87]. Предположительно, ак-
Активация стресса ЭПР и UPR являются не-
тиватором физиологического стресса ЭПР,
обходимыми условиями для дифференцировки
БИОХИМИЯ том 87 вып. 9 2022
СТРЕСС ЭПР И ДИФФЕРЕНЦИРОВКА КЛЕТОК
1217
фибробластов в миофибробласты, а также для
детельствует о различии в регуляции миогенеза
дифференцировки клеток в ходе адипогенеза,
в эмбриональном и постнатальном периоде и
миогенеза, остеобластогенеза и остеокласто-
потенциальной возможности контролировать
генеза (рис. 2). По-видимому, именно хро-
регенерацию мышечной ткани, регулируя ак-
нический/физиологический стресс ЭПР сти-
тивность этого сенсора стресса ЭПР. Интерес-
мулирует дифференцировку этих клеток, в то
но, что некоторые компоненты UPR являются
время как острый/патологический стресс ЭПР
транскрипционными факторами, активиру-
ингибирует дифференцировку и может запу-
ющими экспрессию генов-маркёров диффе-
скать гибель клеток. Как видно из рис. 2, все
ренцировки. Например, sXBP1 связывается с
три сигнальных пути UPR стимулируют ади-
промотором транскрипционного фактора ади-
погенез, эмбриональный миогенез и остео-
погенеза C/EBPα и фактора остеобластогене-
бластогенез. Дифференцировка фибробластов
за Osx, а ATF4 является транскрипционным
в миофибробласты стимулируется сигнальны-
фактором для Osx и маркёра остеобластогенеза
ми путями IRE1 и PERK, а сигнальный путь с
OCN. Пока неясно, является ли UPR индукто-
участием ATF6 подавляет дифференцировку.
ром дифференцировки клеток мезенхимного
В регенеративном миогенезе обнаружено уча-
происхождения или же UPR запускается в про-
стие только сигнального пути PERK, что сви-
цессе дифференцировки, например, из-за по-
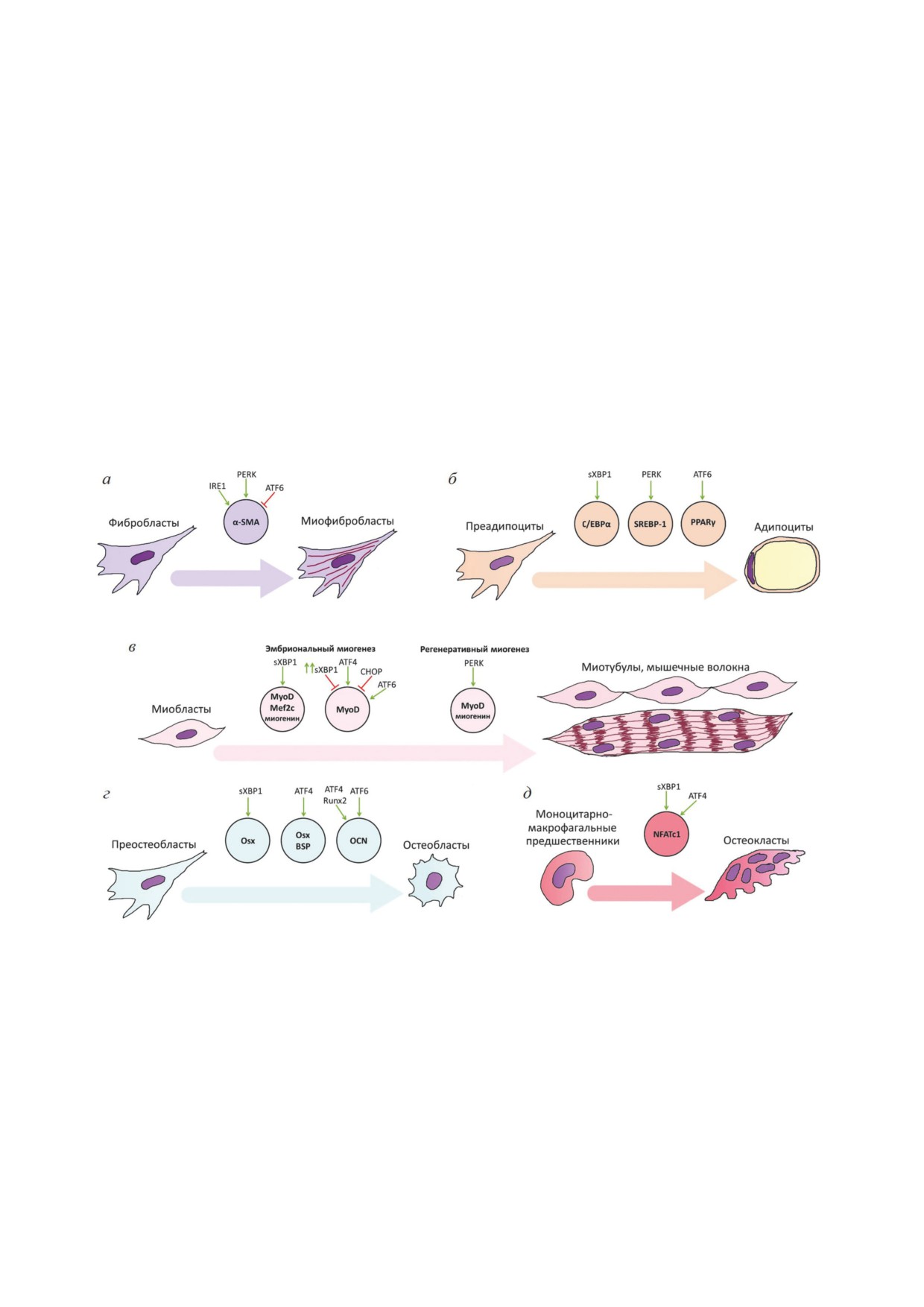
Рис. 2. Роль различных путей стресса ЭПР (IRE1, PERK, ATF6) в дифференцировке клеток мезенхимного происхож-
дения. Дифференцировка фибробластов, преадипоцитов, миобластов, преостеобластов и моноцитарно-макрофагаль-
ных предшественников в более зрелые формы сопровождается активацией стресса ЭПР и обозначена стрелками соот-
ветствующих цветов. В кружках таких же цветов приведены названия маркёров дифференцировки для каждого типа
клеток. Зелёными стрелками обозначена стимуляция синтеза маркёров дифференцировки сенсорами или участниками
сигнальных каскадов UPR, красными линиями - подавление синтеза. а - Дифференцировка фибробластов в мио-
фибробласты; б - дифференцировка преадипоцитов в адипоциты; в - дифференцировка миобластов в миотубулы и
мышечные волокна (различия в эмбриональном и регенеративном миогенезах); г - дифференцировка преостеобластов
в остеобласты; д - дифференцировка моноцитарно-макрофагальных предшественников в остеокласты. Сокращения:
ATF4 - активирующий фактор транскрипции 4; ATF6 - активирующий фактор транскрипции 6; BSP - костный сиа-
лопротеин; C/EBPα - CCAAT/энхансер-связывающий белок α; CHOP - CCAAT/белок, гомологичный энхансер-свя-
зывающему белку; IRE1 - требующий инозитол белок 1; Mef2c - специфичный для миоцитов энхансерный фактор 2с;
Myf5 - миогенный фактор 5; MyoD - белок детерминации миобластов 1; NFATc1 - ядерный фактор активированных
Т-клеток, цитоплазматический 1; OCN - остеокальцин; Osx - транскрипционный фактор Osterix; PERK - киназа ЭПР,
подобная белковой киназе RNA; PPARγ - рецепторы, активируемые пролифераторами пероксисом γ; Runx2 - фактор
транскрипции 2, связанный с доменом runt; α-SMA - гладкомышечный актин α; SREBP-1 - белок, связывающийся с
регуляторным элементом стерола-1; sXBP1 - сплайсированный X-box-связывающий белок 1
БИОХИМИЯ том 87 вып. 9 2022
1218
ТУРИЩЕВА и др.
вышенного синтеза секреторных белков. Сле-
Вклад авторов. Е.П. Турищева - рабо-
дует отметить, что усиление или подавление
та с литературой и написание текста ста-
дифференцировки может играть критическую
тьи; М.С. Вильданова - авторство рисунков;
роль, внося вклад в развитие многих заболева-
Е.А. Смирнова, Г.Е. Онищенко - редактиро-
ний. Так, например, избыточная дифференци-
вание текста статьи.
ровка фибробластов, адипоцитов и остеокла-
Финансирование. Работа выполнена при
стов вызывает фиброз, ожирение и остеопороз.
финансовой поддержке Российского фон-
Фиброз в настоящее время вызывает особый
да фундаментальных исследований (гранты
интерес, так как является одним из основных
№№ 19-015-00233 и 20-315-90118) в рамках на-
последствий COVID-19. Изучение механиз-
учного проекта государственного задания МГУ
мов участия UPR в дифференцировке клеток
№ 121032300098-5.
и способов влияния на сигнальные пути UPR
Конфликт интересов. Авторы заявляют об
открывает перспективы для поиска препара-
отсутствии конфликта интересов.
тов, позволяющих тонко регулировать и даже
Соблюдение этических норм. Настоящая
перенастраивать процессы, разбалансирован-
статья не содержит описания выполненных ав-
ные при нарушениях развития организма или
торами исследований с участием людей или ис-
возникновении патологических состояний.
пользованием животных в качестве объектов.
СПИСОК ЛИТЕРАТУРЫ
1.
Oslowski, C. M., and Urano, F. (2011) Measuring ER
signalling kinetics determine cell survival outcome
stress and the unfolded protein response using mam-
through activation of MKP-1, Cell. Signal., 23, 35-45,
malian tissue culture system, Method. Enzymol., 490,
doi: 10.1016/j.cellsig.2010.07.019.
71-92, doi: 10.1016/B978-0-12-385114-7.00004-0.
9.
Corazzari, M., Gagliardi, M., Fimia, G. M., and
2.
Sicari, D., Delaunay-Moisan, A., Combettes,
Piacentini, M. (2017) Endoplasmic reticulum stress,
L., Chevet, E., and Igbaria, A. (2020) A guide to
unfolded protein response, and cancer cell fate, Front.
assessing endoplasmic reticulum homeostasis and
Oncol., 7, 78, doi: 10.3389/fonc.2017.00078.
stress in mammalian systems, FEBS J., 287, 27-42,
10.
Almanza, A., Carlesso, A., Chintha, C.,
doi: 10.1111/febs.15107.
Creedican, S., Doultsinos, D., et al.
(2019)
3.
Hwang, J., and Qi, L. (2018) Quality control in the
Endoplasmic reticulum stress signalling - from basic
endoplasmic reticulum: crosstalk between ERAD
mechanisms to clinical applications, FEBS J., 286,
and UPR pathways, TiBS, 43, 593-605, doi: 10.1016/
241-278, doi: 10.1111/febs.14608.
j.tibs.2018.06.005.
11.
Hetz, C.
(2012) The unfolded protein response:
4.
Fregno, I., and Molinari, M. (2019) Proteasomal
controlling cell fate decisions under ER stress
and lysosomal clearance of faulty secretory proteins:
and beyond, Nat. Rev. Mol. Cell Biol., 13, 89-102,
ER-associated degradation (ERAD) and ER-
doi: 10.1038/nrm3270.
to-lysosome-associated
degradation
(ERLAD)
12.
Oakes, S. A., and Papa, F. R. (2015) The role of
pathways, Crit. Rev. Biochem. Mol., 54, 153-163,
endoplasmic reticulum stress in human patholo-
doi: 10.1080/10409238.2019.1610351.
gy, Annu. Rev. Pathol., 10, 173-194, doi: 10.1146/
5.
Chadwick, S. R., and Lajoie, P. (2019) Endoplasmic
annurev-pathol-012513-104649.
reticulum stress coping mechanisms and lifespan
13.
Celli, A., Mackenzie, D. S., Crumrine, D. S., Tu,
regulation in health and diseases, Front. Cell Dev.
C. L., Hupe, M., et al. (2011) Endoplasmic retic-
Biol., 7, 84, doi: 10.3389/fcell.2019.00084.
ulum Ca2+ depletion activates XBP1 and controls
6.
Liu, E. S., Ou, J. H., and Lee, A. S.
(1992)
terminal differentiation in keratinocytes and epi-
Brefeldin A as a regulator of grp78 gene expression
dermis, Brit. J. Dermatol., 164, 16-25, doi: 10.1111/
in mammalian cells, J. Biol. Chem., 267, 7128-7133,
j.1365-2133.2010.10046.x.
doi: 10.1016/S0021-9258(19)50547-6.
14.
Matsuzaki, S., Hiratsuka, T., Taniguchi, M.,
7.
Yoshida, I., Monji, A., Tashiro, K. I., Nakamura,
Shingaki, K., Kubo, T., et al. (2015) Physiological ER
K. I., Inoue, R., et al.
(2006) Depletion of
stress mediates the differentiation of fibroblasts, PLoS
intracellular Ca2+ store itself may be a major factor in
One, 10, e0123578, doi: 10.1371/journal.pone.0123578.
thapsigargin-induced ER stress and apoptosis in PC12
15.
Nakanishi, K., Kakiguchi, K., Yonemura, S.,
cells, Neurochem. Int., 48, 696-702, doi: 10.1016/
Nakano, A., and Morishima, N. (2015) Transient Ca2+
j.neuint.2005.12.012.
depletion from the endoplasmic reticulum is critical
8.
Li, B., Yi, P., Zhang, B., Xu, C., Liu, Q., et al.
for skeletal myoblast differentiation, FASEB J., 29,
(2011) Differences in endoplasmic reticulum stress
2137-2149, doi: 10.1096/fj.14-261529.
БИОХИМИЯ том 87 вып. 9 2022
СТРЕСС ЭПР И ДИФФЕРЕНЦИРОВКА КЛЕТОК
1219
16.
Murao, N., and Nishitoh, H. (2017) Role of the
membrane-bound ATF6 by the same proteases
unfolded protein response in the development of
that process SREBPs, Mol. Cell,
6,
1355-1364,
central nervous system, J. Biochem., 162, 155-162,
doi: 10.1016/S1097-2765(00)00133-7.
doi: 10.1093/jb/mvx047.
27.
Kendall, R. T., and Feghali-Bostwick, C. A. (2014)
17.
Vildanova, M., Saidova, A., Fokin, A.,
Fibroblasts in fibrosis: novel roles and mediators, Front.
Potashnikova, D., Onishchenko, G., et al.
(2019)
Pharmacol., 5, 123, doi: 10.3389/fphar.2014.00123.
Jasmonic acid induces endoplasmic reticulum stress
28.
Desai, V. D., Hsia, H. C., and Schwarzbauer, J. E.
with different outcome in cultured normal and tumor
(2014) Reversible modulation of myofibroblast differen-
epidermal cells, Biochemistry (Moscow), 84, 1289-
tiation in adipose-derived mesenchymal stem cells, PLoS
1300, doi: 10.1134/S0320972519090070.
One, 9, e86865, doi: 10.1371/journal.pone.0086865.
18.
Vildanova, M., Vishnyakova, P., Saidova, A.,
29.
Heindryckx, F., Binet, F., Ponticos, M., Rombouts, K.,
Konduktorova, V., Onishchenko, G., et al.
(2021)
Lau, J., et al. (2016) Endoplasmic reticulum stress en-
Gibberellic acid initiates ER stress and activation
hances fibrosis through IRE 1α-mediated degradation
of differentiation in cultured human immortalized
of miR-150 and XBP-1 splicing, EMBO Mol. Med., 8,
keratinocytes HaCaT and epidermoid carcinoma
729-744, doi: 10.15252/emmm.201505925.
cells A431, Pharmaceutics, 13, 1813, doi: 10.3390/
30.
Zhong, Q., Zhou, B., Ann, D. K., Minoo, P., Liu, Y.,
pharmaceutics13111813.
et al. (2011) Role of endoplasmic reticulum stress in
19.
Турищева Е. П., Вильданова М. С., Поташникова
epithelial-mesenchymal transition of alveolar epi-
Д. М., Смирнова Е. А. (2020) Различная реакция
thelial cells: effects of misfolded surfactant protein,
биосинтетической системы дермальных фибро-
Am. J. Resp. Cell Mol., 45, 498-509, doi: 10.1165/
бластов и клеток фибросаркомы человека на
rcmb.2010-0347OC.
действие растительных гормонов, Цитология, 62,
31.
Baek, H. A., Kim, D. S., Park, H. S., Jang, K. Y.,
566-580, doi: 10.31857/S0041377120080088.
Kang, M. J., et al. (2012) Involvement of endoplasmic
20.
Calfon, M., Zeng, H., Urano, F., Till, J. H.,
reticulum stress in myofibroblastic differentiation of
Hubbard, S. R., et al. (2002) IRE1 couples endo-
lung fibroblasts, Am. J. Resp. Cell Mol., 46, 731-739,
plasmic reticulum load to secretory capacity by pro-
doi: 10.1165/rcmb.2011-0121OC.
cessing the XBP-1 mRNA, Nature,
415,
92-96,
32.
Chen, Y. C., Chen, B. C., Huang, H. M., Lin,
doi: 10.1038/415092a.
S. H., and Lin, C. H. (2019) Activation of PERK in
21.
Lee, A. H., Iwakoshi, N. N., and Glimcher, L. H.
ET-1-and thrombin-induced pulmonary fibroblast
(2003) XBP-1 regulates a subset of endoplasmic
differentiation: Inhibitory effects of curcumin, J. Cell.
reticulum resident chaperone genes in the unfolded
Physiol., 234, 15977-15988, doi: 10.1002/jcp.28256.
protein response, Mol. Cell. Biol., 23, 7448-7459,
33.
Jiang, S., He, R., Zhu, L., Liang, T., Wang, Z., et al.
doi: 10.1128/MCB.23.21.7448-7459.2003.
(2018) Endoplasmic reticulum stress-dependent
22.
Lam, W. Y., and Bhattacharya, D. (2018) Metabol-
ROS production mediates synovial myofibroblastic
ic links between plasma cell survival, secretion, and
differentiation in the immobilization-induced rat knee
stress, Trends Immunol., 39, 19-27, doi: 10.1016/
joint contracture model, Exp. Cell Res., 369, 325-334,
j.it.2017.08.007.
doi: 10.1016/j.yexcr.2018.05.036.
23.
Teske, B. F., Wek, S. A., Bunpo, P., Cundiff, J. K.,
34.
Qin, X., Lin, X., Liu, L., Li, Y., Li, X., et al. (2021)
McClintick, J. N., et al. (2011) The eIF2 kinase
Macrophage-derived exosomes mediate silica-induced
PERK and the integrated stress response facilitate
pulmonary fibrosis by activating fibroblast in an
activation of ATF6 during endoplasmic reticulum
endoplasmic reticulum stress-dependent manner,
stress, Mol. Biol. Cell, 22, 4390-4405, doi: 10.1091/
J. Cell. Mol. Med., 25, 4466-4477, doi: 10.1111/
mbc.e11-06-0510.
jcmm.16524.
24.
Gardner, B. M., Pincus, D., Gotthardt, K., Gal-
35.
Song, M., Peng, H., Guo, W., Luo, M., Duan, W.,
lagher, C. M., and Walter, P. (2013) Endoplasmic
et al. (2019) Cigarette smoke extract promotes human
reticulum stress sensing in the unfolded protein re-
lung myofibroblast differentiation by the induction of
sponse, Cold Spring Harb. Perspect. Biol., 5, a013169,
endoplasmic reticulum stress, Respiration, 98, 347-
doi: 10.1101/cshperspect.a013169.
356, doi: 10.1159/000502099.
25.
Yoshida, H., Haze, K., Yanagi, H., Yura, T., and
36.
Stauffer, W. T., Blackwood, E. A., Azizi, K.,
Mori, K. (1998) Identification of the cis-acting en-
Kaufman, R. J., and Glembotski, C. C.
(2020)
doplasmic reticulum stress response element respon-
The ER unfolded protein response effector, ATF6,
sible for transcriptional induction of mammalian
reduces cardiac fibrosis and decreases activation
glucose-regulated proteins: involvement of basic leu-
of cardiac fibroblasts, Int. J. Mol. Sci., 21, 1373,
cine zipper transcription factors, J. Biol. Chem., 273,
doi: 10.3390/ijms21041373.
33741-33749, doi: 10.1074/jbc.273.50.33741.
37.
Sriburi, R., Jackowski, S., Mori, K., and Brewer, J. W.
26.
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Davé,
(2004) XBP1: a link between the unfolded protein
U. P., et al. (2000) ER stress induces cleavage of
response, lipid biosynthesis, and biogenesis of the
БИОХИМИЯ том 87 вып. 9 2022
1220
ТУРИЩЕВА и др.
endoplasmic reticulum, J. Cell Biol., 167, 35-41,
response in adipogenesis, Int. J. Obesity, 36, 1248-
doi: 10.1083/jcb.200406136.
1251, doi: 10.1038/ijo.2011.233.
38.
Ali, A. T., Hochfeld, W. E., Myburgh, R., and Pepper,
49.
Mohan, S., Brown, L., and Ayyappan, P. (2019) En-
M. S. (2013) Adipocyte and adipogenesis, Eur. J. Cell
doplasmic reticulum stress: a master regulator of met-
Biol., 92, 229-236, doi: 10.1016/j.ejcb.2013.06.001.
abolic syndrome, Eur. J. Pharmacol., 860, 172553, doi:
39.
Basseri, S., Lhoták, Š., Sharma, A. M., and Austin,
10.1016/j.ejphar.2019.172553.
R. C. (2009) The chemical chaperone 4-phenylbu-
50.
Carlson, S. G., Fawcett, T. W., Bartlett, J. D.,
tyrate inhibits adipogenesis by modulating the un-
Bernier, M., and Holbrook, N. J. (1993) Regulation
folded protein response, J. Lipid Res., 50, 2486-2501,
of the C/EBP-related gene gadd153 by glucose
doi: 10.1194/jlr.M900216-JLR200.
deprivation, Mol. Cell. Biol.,
13,
4736-4744,
40.
Sha, H., He, Y., Chen, H., Wang, C., Zenno, A.,
doi: 10.1128/mcb.13.8.4736-4744.1993.
et al. (2009) The IRE1α-XBP1 pathway of the
51.
Lee, J. M., Park, S., Lee, D., Ginting, R. P., Lee,
unfolded protein response is required for adipo-
M. R., et al. (2021) Reduction in endoplasmic re-
genesis, Cell Metab.,
9,
556-564, doi:
10.1016/
ticulum stress activates beige adipocytes differenti-
j.cmet.2009.04.009.
ation and alleviates high fat diet-induced metabol-
41.
Batchvarova, N., Wang, X. Z., and Ron, D. (1995)
ic phenotypes, BBA-Mol. Basis Dis., 1867, 166099,
Inhibition of adipogenesis by the stress-induced
doi: 10.1016/j.bbadis.2021.166099.
protein CHOP (Gadd153), EMBO J., 14, 4654-4661,
52.
Tan, Y. Y., Zhang, Y., Li, B., Ou, Y. W., Xie, S. J., et al.
doi: 10.1002/j.1460-2075.1995.tb00147.x.
(2021) PERK signaling controls myoblast differentia-
42.
Shimada, T., Hiramatsu, N., Okamura, M.,
tion by regulating microRNA networks, Front. Cell Dev.
Hayakawa, K., Kasai, A., et al. (2007) Unexpected
Biol., 9, 670435, doi: 10.3389/fcell.2021.670435.
blockade of adipocyte differentiation by K-7174:
53.
Chal, J., and Pourquié, O. (2017) Making muscle:
implication for endoplasmic reticulum stress,
skeletal myogenesis in vivo and in vitro, Development,
Biochem. Biophys. Res. Commun.,
363,
355-360,
144, 2104-2122, doi: 10.1242/dev.151035.
doi: 10.1016/j.bbrc.2007.08.167.
54.
Sincennes, M. C., Brun, C. E., and Rudnicki, M. A.
43.
Bobrovnikova-Marjon, E., Hatzivassiliou, G.,
(2016) Concise review: epigenetic regulation of
Grigoriadou, C., Romero, M., Cavener, D. R., et al.
myogenesis in health and disease, Stem Cells Transl.
(2008) PERK-dependent regulation of lipogenesis
Med., 5, 282-290, doi: 10.5966/sctm.2015-0266.
during mouse mammary gland development and
55.
Tokutake, Y., Yamada, K., Hayashi, S., Arai, W.,
adipocyte differentiation, Proc. Natl. Acad. Sci. USA,
Watanabe, T., et al. (2020) IRE1-XBP1 pathway of
105, 16314-16319, doi: 10.1073/pnas.0808517105.
the unfolded protein response is required during early
44.
Bobrovnikova-Marjon, E., Pytel, D., Riese, M. J.,
differentiation of C2C12 myoblasts, Int. J. Mol. Sci.,
Vaites, L. P., Singh, N., et al. (2012) PERK utilizes
21, 182, doi: 10.3390/ijms21010182.
intrinsic lipid kinase activity to generate phosphatidic
56.
Xiong, G., Hindi, S. M., Mann, A. K., Gallot,
acid, mediate Akt activation, and promote adipocyte
Y. S., Bohnert, K. R., et al. (2017) The PERK arm
differentiation, Mol. Cell. Biol.,
32,
2268-2278,
of the unfolded protein response regulates satellite
doi: 10.1128/MCB.00063-12.
cell-mediated skeletal muscle regeneration, Elife, 6,
45.
Han, J., Murthy, R., Wood, B., Song, B., Wang, S.,
e22871, doi: 10.7554/eLife.22871.
et al. (2013) ER stress signalling through eIF2α and
57.
Benhaddou, A., Keime, C., Ye, T., Morlon,
CHOP, but not IRE1α, attenuates adipogenesis
A., Michel, I., et al. (2012) Transcription factor
in mice, Diabetologia, 56, 911-924, doi: 10.1007/
TEAD4 regulates expression of myogenin and the
s00125-012-2809-5.
unfolded protein response genes during C2C12
46.
Longo, M., Spinelli, R., D’Esposito, V., Zatterale,
cell differentiation, Cell Death Differ., 19, 220-231,
F., Fiory, F., et al. (2016) Pathologic endoplasmic
doi: 10.1038/cdd.2011.87.
reticulum stress induced by glucotoxic insults
58.
Blais, A., Tsikitis, M., Acosta-Alvear, D., Sharan, R.,
inhibits adipocyte differentiation and induces an
Kluger, Y., et al. (2005) An initial blueprint for
inflammatory phenotype, BBA-Mol. Cell Res., 1863,
myogenic differentiation, Gene Dev., 19, 553-569,
1146-1156, doi: 10.1016/j.bbamcr.2016.02.019.
doi: 10.1101/gad.1281105.
47.
Kang, S. U., Kim, H. J., Kim, D. H., Han, C. H.,
59.
Acosta-Alvear, D., Zhou, Y., Blais, A., Tsikitis, M.,
Lee, Y. S., et al. (2018) Nonthermal plasma treated
Lents, N. H., et al. (2007) XBP1 controls diverse cell
solution inhibits adipocyte differentiation and lipo-
type-and condition-specific transcriptional regulatory
genesis in 3T3-L1 preadipocytes via ER stress sig-
networks, Mol. Cell,
27,
53-66, doi:
10.1016/
nal suppression, Sci. Rep., 8, 1-12, doi: 10.1038/
j.molcel.2007.06.011.
s41598-018-20768-5.
60.
Alter, J., and Bengal, E. (2011) Stress-induced C/
48.
Lowe, C. E., Dennis, R. J., Obi, U., O’Rahilly, S.,
EBP homology protein (CHOP) represses MyoD
and Rochford, J. J. (2012) Investigating the involve-
transcription to delay myoblast differentiation, PLoS
ment of the ATF6α pathway of the unfolded protein
One, 6, e29498, doi: 10.1371/journal.pone.0029498.
БИОХИМИЯ том 87 вып. 9 2022
СТРЕСС ЭПР И ДИФФЕРЕНЦИРОВКА КЛЕТОК
1221
61.
Nakanishi, K., Sudo, T., and Morishima, N. (2005)
BMP2 on osteoblast cells, Life Sci., 193, 34-39, doi:
Endoplasmic reticulum stress signaling transmitted
10.1016/j.lfs.2017.12.008.
by ATF6 mediates apoptosis during muscle devel-
73.
Tohmonda, T., Miyauchi, Y., Ghosh, R., Yoda,
opment, J. Cell Biol., 169, 555-560, doi: 10.1083/
M., Uchikawa, S., et al. (2011) The IRE1α-XBP1
jcb.200412024.
pathway is essential for osteoblast differentiation
62.
Nakanishi, K., Dohmae, N., and Morishima, N.
through promoting transcription of Osterix, EMBO
(2007) Endoplasmic reticulum stress increases
Rep., 12, 451-457, doi: 10.1038/embor.2011.34.
myofiber formation in vitro, FASEB J., 21, 2994-
74.
Guo, F. J., Jiang, R., Xiong, Z., Xia, F., Li, M., et al.
3003, doi: 10.1096/fj.06-6408com.
(2014) IRE1a constitutes a negative feedback loop with
63.
Wei, Y., Tao, X., Xu, H., Chen, Y., Zhu, L.,
BMP2 and acts as a novel mediator in modulating os-
et al. (2016) Role of miR-181a-5p and endoplas-
teogenic differentiation, Cell Death Dis., 5, e1239,
mic reticulum stress in the regulation of myogen-
doi: 10.1038/cddis.2014.194.
ic differentiation, Gene, 592, 60-70, doi: 10.1016/
75.
Yang, X., Matsuda, K., Bialek, P., Jacquot, S.,
j.gene.2016.07.056.
Masuoka, H. C., et al. (2004) ATF4 is a substrate of
64.
Wiles, B., Miao, M., Coyne, E., Larose, L., Cybulsky,
RSK2 and an essential regulator of osteoblast biology:
A. V., et al. (2015) USP19 deubiquitinating enzyme
implication for Coffin-Lowry syndrome, Cell, 117,
inhibits muscle cell differentiation by suppressing
387-398, doi: 10.1016/S0092-8674(04)00344-7.
unfolded-protein response signaling, Mol. Biol. Cell,
76.
Xiao, G., Jiang, D., Ge, C., Zhao, Z., Lai, Y., et al.
26, 913-923, doi: 10.1091/mbc.E14-06-1129.
(2005) Cooperative interactions between activating tran-
65.
Yang, X., and Karsenty, G.
(2004) ATF4, the
scription factor 4 and Runx2/Cbfa1 stimulate osteo-
osteoblast accumulation of which is determined post-
blast-specific osteocalcin gene expression, J. Biol. Chem.,
translationally, can induce osteoblast-specific gene
280, 30689-30696, doi: 10.1074/jbc.M500750200.
expression in non-osteoblastic cells, J. Biol. Chem.,
77.
Yu, S., Franceschi, R. T., Luo, M., Fan, J., Jiang, D.,
279, 47109-47114, doi: 10.1074/jbc.M410010200.
et al. (2009) Critical role of activating transcription
66.
Rutkovskiy, A., Stensløkken, K. O., and Vaage, I. J.
factor
4 in the anabolic actions of parathyroid
(2016) Osteoblast differentiation at a glance, Med.
hormone in bone, PLoS One, 4, e7583, doi: 10.1371/
Sci. Monit. Basic Res., 22, 95-106, doi: 10.12659/
journal.pone.0007583.
MSMBR.901142.
78.
Yang, S., Hu, L., Wang, C., and Wei, F.
(2020)
67.
Wei, J., Sheng, X., Feng, D., McGrath, B., and
PERK-eIF2α-ATF4 signaling contributes to
Cavener, D. R.
(2008) PERK is essential for
osteogenic differentiation of periodontal ligament
neonatal skeletal development to regulate osteoblast
stem cells, J. Mol. Histol., 51, 125-135, doi: 10.1007/
proliferation and differentiation, J. Cell. Physiol., 217,
s10735-020-09863-y.
693-707, doi: 10.1002/jcp.21543.
79.
Yu, S., Zhu, K., Lai, Y., Zhao, Z., Fan, J., et al. (2013)
68.
Saito, A., Ochiai, K., Kondo, S., Tsumagari, K.,
ATF4 promotes β-catenin expression and osteoblastic
Murakami, T., et al. (2011) Endoplasmic reticulum
differentiation of bone marrow mesenchymal stem cells,
stress response mediated by the PERK-eIF2α-ATF4
Int. J. Biol. Sci., 9, 256-266, doi: 10.7150/ijbs.5898.
pathway is involved in osteoblast differentiation
80.
Jang, W. G., Kim, E. J., and Koh, J. T.
(2011)
induced by BMP2, J. Biol. Chem., 286, 4809-4818,
Tunicamycin negatively regulates BMP2-induced
doi: 10.1074/jbc.M110.152900.
osteoblast differentiation through CREBH expression
69.
Zhang, K., Wang, M., Li, Y., Li, C., Tang, S., et al.
in MC3T3E1 cells, BMB Rep., 44, 735-740, doi:
(2019) The PERK-EIF2α-ATF4 signaling branch
10.5483/BMBRep.2011.44.11.735.
regulates osteoblast differentiation and proliferation
81.
Shi, M., Song, W., Han, T., Chang, B., Li, G., et al.
by PTH, Am. J. Physiol. Endocrinol. Metab., 316,
(2017) Role of the unfolded protein response in
E590-E604, doi: 10.1152/ajpendo.00371.2018.
topography-induced osteogenic differentiation in rat
70.
Murakami, T., Saito, A., Hino, S. I., Kondo, S.,
bone marrow mesenchymal stem cells, Acta Biomater.,
Kanemoto, S., et al. (2009) Signalling mediated by
54, 175-185, doi: 10.1016/j.actbio.2017.03.018.
the endoplasmic reticulum stress transducer OASIS is
82.
Tanaka, K. I., Yamaguchi, T., Kaji, H., Kanazawa, I.,
involved in bone formation, Nat. Cell Biol., 11, 1205-
and Sugimoto, T. (2013) Advanced glycation end
1211, doi: 10.1038/ncb1963.
products suppress osteoblastic differentiation of
71.
Jang, W. G., Kim, E. J., Kim, D. K., Ryoo, H. M.,
stromal cells by activating endoplasmic reticulum
Lee, K. B., et al. (2012) BMP2 protein regulates
stress, Biochem. Bioph. Res. Commun., 438, 463-467,
osteocalcin expression via Runx2-mediated Atf6
doi: 10.1016/j.bbrc.2013.07.126.
gene transcription, J. Biol. Chem., 287, 905-915,
83.
Boyle, W. J., Simonet, W. S., and Lacey, D. L. (2003)
doi: 10.1074/jbc.M111.253187.
Osteoclast differentiation and activation, Nature, 423,
72.
Son, H. E., Kim, E. J., and Jang, W. G. (2018)
337-342, doi: 10.1038/nature01658.
Curcumin induces osteoblast differentiation through
84.
Tohmonda, T., Yoda, M., Iwawaki, T., Matsumoto,
mild-endoplasmic reticulum stress-mediated such as
M., Nakamura, M., et al. (2015) IRE1α/XBP1-medi-
БИОХИМИЯ том 87 вып. 9 2022
1222
ТУРИЩЕВА и др.
ated branch of the unfolded protein response regulates
osteoclastogenesis by interleukin-1β and endoplasmic
osteoclastogenesis, J. Clin. Invest., 125, 3269-3279,
reticulum stress, Joint Bone Spine,
81,
520-526,
doi: 10.1172/JCI76765.
doi: 10.1016/j.jbspin.2014.04.012.
85. Guo, J., Ren, R., Sun, K., Yao, X., Lin, J., et al.
90. Zhang, L., Bao, D., Li, P., Lu, Z., Pang, L., et al.
(2020) PERK controls bone homeostasis through
(2018) Particle-induced SIRT1 downregulation pro-
the regulation of osteoclast differentiation and
motes osteoclastogenesis and osteolysis through ER
function, Cell Death Dis., 11, 1-16, doi: 10.1038/
stress regulation, Biomed. Pharmacother, 104, 300-
s41419-020-03046-z.
306, doi: 10.1016/j.biopha.2018.05.030.
86. Raimondi, L., De Luca, A., Fontana, S., Amodio,
91. Lee, W. S., Jeong, J. H., Lee, E. G., Choi, Y., Kim,
N., Costa, V., et al. (2020) Multiple myeloma-de-
J. H., et al. (2017) Tacrolimus regulates endoplas-
rived extracellular vesicles induce osteoclastogenesis
mic reticulum stress-mediated osteoclastogenesis
through the activation of the XBP1/IRE1α axis, Can-
and inflammation: in vitro and collagen-induced ar-
cers, 12, 2167, doi: 10.3390/cancers12082167.
thritis mouse model, Cell Biol. Int., 42, 393-402,
87. Cao, H., Yu, S., Yao, Z., Galson, D. L., Jiang, Y.,
doi: 10.1002/cbin.10861.
et al. (2010) Activating transcription factor 4 regulates
92. He, L., Lee, J., Jang, J. H., Sakchaisri, K., Hwang, J.,
osteoclast differentiation in mice, J. Clin. Invest., 120,
et al.
(2013) Osteoporosis regulation by salubri-
2755-2766, doi: 10.1172/JCI42106.
nal through eIF2α mediated differentiation of os-
88. Wang, K., Niu, J., Kim, H., and Kolattukudy, P. E.
teoclast and osteoblast, Cell. Signal., 25, 552-560,
(2011) Osteoclast precursor differentiation by MCPIP
doi: 10.1016/j.cellsig.2012.11.015.
via oxidative stress, endoplasmic reticulum stress, and
93. Li, J., Li, X., Liu, D., Hamamura, K., Wan, Q.,
autophagy, J. Mol. Cell Biol., 3, 360-368, doi: 10.1093/
et al. (2019) eIF2α signaling regulates autophagy of
jmcb/mjr021.
osteoblasts and the development of osteoclasts in
89. Lee, E. G., Sung, M. S., Yoo, H. G., Chae, H. J.,
OVX mice, Cell Death Dis., 10, 1-15, doi: 10.1038/
Kim, H. R., et al. (2014) Increased RANKL-mediated
s41419-019-2159-z.
ROLE OF ENDOPLASMIC RETICULUM STRESS
IN DIFFERENTIATION OF MESENCHYMAL CELLS
Review
E. P. Turishcheva*, M. S. Vildanova, G. E. Onishchenko, and E. A. Smirnova
Faculty of Biology, Lomonosov Moscow State University,
119991 Moscow, Russia; e-mail: kitten-caterina@yandex.ru
The endoplasmic reticulum (ER) is a multifunctional membrane compartment, which is responsible for
the synthesis and processing of secretory, lysosomal and transmembrane proteins. Incorrect processing of
proteins during disturbed ER homeostasis leads to the development of the ER stress. To restore the normal
ER function, cells utilize the specific adaptive mechanism — unfolded protein response (UPR). In addition
to controlling of protein folding, UPR plays a key role in other physiological processes, such as differentiation
of connective tissue, muscle, epithelial and neural cells. Moreover, only the physiological level of ER stress
stimulates differentiation, while the increased level of ER stress suppresses differentiation and may cause the
cell death. It is currently unknown whether UPR activation acts as an inducer of cell differentiation or UPR is
triggered by increased synthesis of secretory proteins during differentiation. Cell differentiation is an important
step in the development of multicellular organisms, so this process must be effectively controlled. Suppression
or, conversely, excessive activation of differentiation leads to the development of pathological processes in
the organism. For instance, disbalance in the level of differentiation of the connective tissue may lead to the
development of obesity, osteoporosis, and fibrosis. The latter is currently a subject of thorough investigation,
because appeared to be one of the consequences of COVID-19 infection. Therefore, further investigations of
the crosstalk between UPR and cellular differentiation may lead to the identification of potential molecular
targets that allow selective regulation of different stages of differentiation and influence the mechanisms leading
to the development of pathological processes.
Keywords: endoplasmic reticulum, differentiation, endoplasmic reticulum stress, UPR, myofibroblasts, fibrosis,
adipogenesis, myogenesis, osteoblastogenesis, osteoclastogenesis
БИОХИМИЯ том 87 вып. 9 2022