БИОХИМИЯ, 2022, том 87, вып. 9, с. 1232 - 1245
УДК 57.088
ХИМИЧЕСКАЯ ПРОТЕОМИКА
НА ОСНОВЕ МАСС-СПЕКТРОМЕТРИИ
В ЗАДАЧАХ ПОИСКА ЛЕКАРСТВЕННЫХ МИШЕНЕЙ
Мини-обзор
© 2022 И.И. Федоров1,2, В.И. Линева2, И.А. Тарасова1, М.В. Горшков1*
1 ФГБУН Федеральный исследовательский центр химической физики РАН им. Н.Н. Семенова РАН,
Институт энергетических проблем химической физики им. В.Л. Тальрозе,
119334 Москва, Россия; электронная почта: mike.gorshkov@gmail.com
2 Московский физико-технический институт (национальный исследовательский университет),
141700 Долгопрудный, Московская обл., Россия
Поступила в редакцию 27.05.2022
После доработки 04.07.2022
Принята к публикации 06.07.2022
Быстроразвивающаяся в последние годы химическая протеомика представляет собой основной
метод для идентификации взаимодействий малых молекул и белков в масштабах всего протеома и
картирования сигнальных и/или метаболических путей этих взаимодействий. Метод позволяет не
только идентифицировать белки-мишени для лекарств, охарактеризовать их токсичность и выявить
возможные нецелевые белки, участвующие в побочных взаимодействиях с лекарством, но и выяснить
фундаментальные механизмы, регулирующие жизнедеятельность клеток в условиях лекарственного
воздействия или особенностей физиологического состояния самого организма. Решение этих про-
блем играет ключевую роль в биологии и клинической практике, расширяя существующие возмож-
ности для решения фундаментальных и практических проблем медицины, включая диагностику раз-
личных форм тяжелых заболеваний или предсказание эффективности терапевтического воздействия.
При этом развитие масс-спектрометрии высокого разрешения в последние годы позволило решать
задачи поиска мишеней лекарственного воздействия на уровне всех белков клеточных протеомов.
В данном обзоре рассматриваются основные задачи, стоящие перед химической протеомикой на ос-
нове масс-спектрометрии, описываются методы и подходы к их решению, а также приведены приме-
ры реализации этих методов в практике биомедицинских исследований.
КЛЮЧЕВЫЕ СЛОВА: химическая протеомика, масс-спектрометрия, лекарственные мишени.
DOI: 10.31857/S0320972522090056, EDN: BAVDEB
ВВЕДЕНИЕ
терапевтических подходов к лечению большин-
ства социально-значимых заболеваний. Персо-
Несмотря на бурное развитие знаний в об-
нализированный отклик организма на терапию,
ласти биохимии и биомедицины, поиск, раз-
а также развитие как индивидуальной, так и по-
работка и тестирование новых лекарственных
пуляционной устойчивости патологий к лекар-
препаратов до сих пор является наиболее слож-
ственному воздействию приводят к необходи-
ным этапом разработки более эффективных мости разработки новых препаратов и классов
Принятые сокращения: ВЭЖХ-МС/МС - метод анализа сложных смесей органических и биоорганических соедине-
ний, включая протеолитические пептиды, основанный на комбинации высокоэффективной жидкостной хроматографии
и тандемной масс-спектрометрии; МС - масс-спектрометрия; TMT - изобарные метки для мультиплексного количе-
ственного протеомного анализа на основе тандемной масс-спектрометрии; TPP - температурное профилирование про-
теомов; ABPP - метод профилирования белков по ферментативной активности; AS-MS - метод аффинного выделения
белков в сочетании с масс-спектрометрической идентификацией; CCMS - метод масс-спектрометрической идентифи-
кации белков с использованием фотоиндуцированной сшивки; HDX-MS - масс-спектрометрия водородно-дейтериево-
го обмена; LiP-MS - масс-спектрометрия структурно-зависимого протеолиза белков; PISA - метод интегрального изме-
рения растворимости белков; PREPL - пролилэндопептидазоподобный фермент, кодируемый у человека геном PREPL.
* Адресат для корреспонденции.
1232
ХИМИЧЕСКАЯ ПРОТЕОМИКА
1233
соединений для лечений одних и тех же забо-
определяются индивидуальными, или «персо-
леваний [1]. Тем не менее ни широкий выбор
нализированными», особенностями развития
уже имеющихся лекарств, ни разнообразие те-
патологии в конкретном организме. Однако и
рапевтических подходов на их основе зачастую
в рамках таких персонализированных подходов
не дают желаемый физиологический эффект.
проблема выработки устойчивости патологиче-
Подтверждением этому является, например,
ских клеток к воздействию остается нерешен-
непрекращающееся противостояние антибио-
ной. Кроме того, сама концепция «одно лекар-
тиков и бактерий, развивающих устойчивость к
ство-одна мишень» является в общем случае
ним [2]. Еще более характерным примером яв-
ошибочной [6]. Лекарство неизбежно начинает
ляется быстроразвивающаяся резистентность
взаимодействовать с другими молекулярными
клеток злокачественных опухолей практиче-
мишенями, причем не только в патологиче-
ски ко всему спектру зарегистрированных и
ских, но и в здоровых клетках организма, вы-
используемых в настоящее время противоопу-
зывая довольно слабопредсказуемые (и, снова,
холевых химиотерапевтических препаратов [3,
«персонализированные») последствия, вклю-
4]. Если говорить о лечении злокачественных
чая побочные эффекты. В свою очередь, по-
опухолей, то одними из наиболее перспектив-
бочные эффекты и их разнообразие могут быть
ных в последние годы рассматриваются подхо-
обусловлены в том числе взаимодействием пре-
ды на основе таргетной терапии [5]. Упрощен-
парата не только с «узаконенными» мишеня-
но, эти подходы можно охарактеризовать как
ми, но и с иными молекулярными участниками
триаду
«один организм-одна мишень-одно
процессов (рис. 1). Таким образом, создание
лекарство», в которой и мишень, и лекарство
лекарств нового поколения требует не только
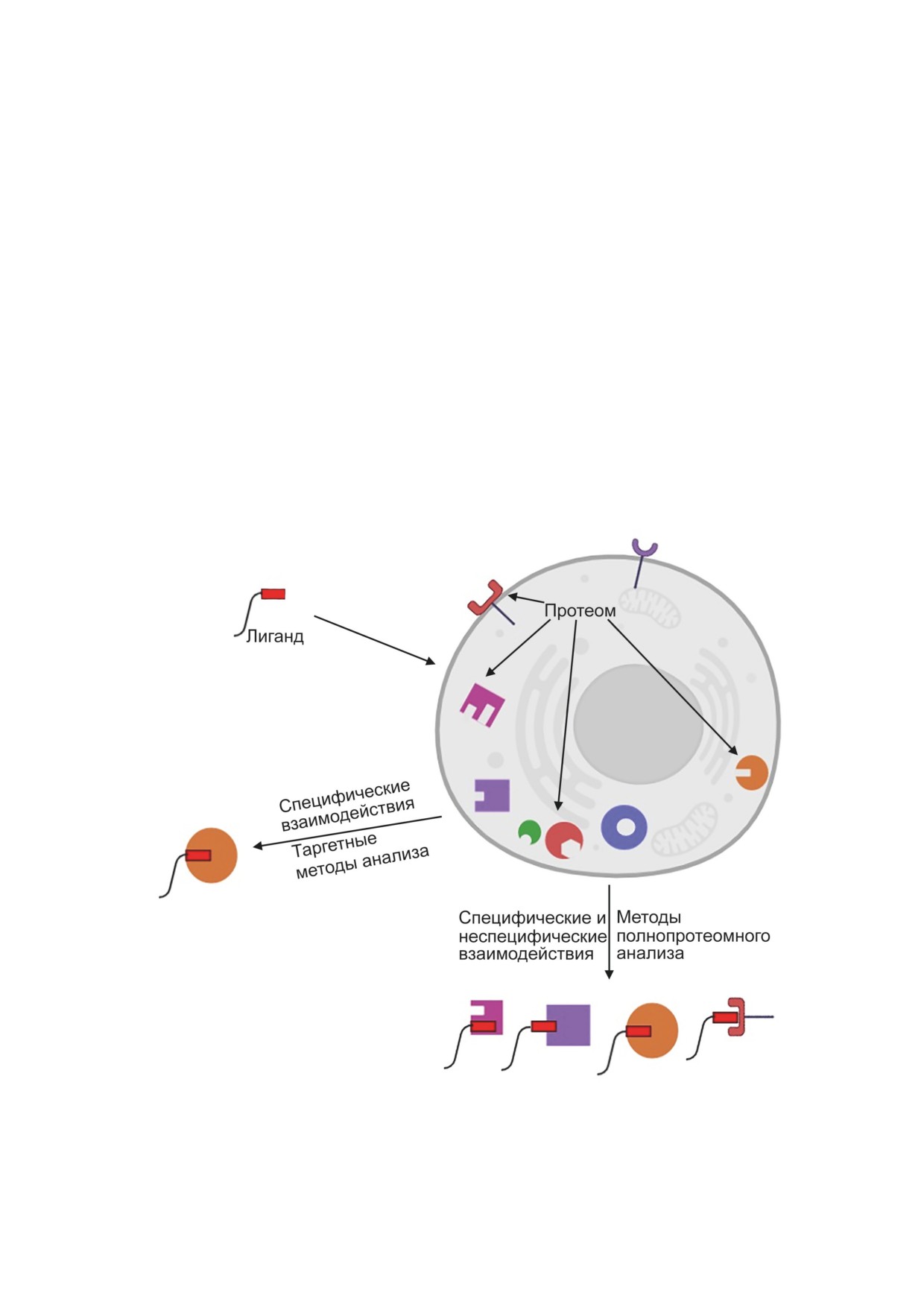
Рис. 1. Задачей химической протеомики является поиск мишеней взаимодействия, как правило, небольших химических
соединений с белками. В то время как традиционные методы химической протеомики позволяли выявлять и подтвер-
ждать специфические взаимодействия лигандов с конкретными белками, создание лекарств нового поколения требует
не только изучения фармакокинетических свойств используемых химических препаратов, но и идентификации полного
спектра их мишеней на уровне всего протеома клеток, включая те из них, которые участвуют в неспецифических взаи-
модействиях, потенциально ответственных за развитие побочных эффектов
БИОХИМИЯ том 87 вып. 9 2022
1234
ФЕДОРОВ и др.
тщательного изучения фармакокинетических
белковых взаимодействий и являться такими
свойств используемых химических препаратов,
маркерами [14].
но и идентификацию полного спектра их ми-
При этом количественная МС, основанная
шеней для минимизации побочных эффектов
на масс-анализаторах высокого разрешения,
и усиления эффективности и специфичности
позволяет в настоящее время выявлять такие
воздействия этих препаратов на патологиче-
изменения не только для отдельных, но и прак-
ские (и здоровые, в общем случае) клетки [7].
тически для всех белков протеомов [15].
В настоящее время сложилось не только пони-
Таким образом, методы химической про-
мание необходимости таких исследований, но
теомики являются эффективным инструмен-
и были созданы методы, позволяющие решать
том при изучении молекулярных механизмов
эти задачи как на геномном уровне, так и на
развития заболеваний, идентификации био-
уровне всего протеома.
маркеров, а также определении мишеней для
Методы химической протеомики являют-
таргетной терапии. Развитие высокопроиз-
ся эффективными инструментами для поиска
водительной количественной МС биомоле-
и идентификации путей и мишеней взаимо-
кул позволило вывести решение этих задач на
действия химических соединений с белками
уровень всех белков протеомов. В обзоре рас-
клеток [7, 8]. В то время как аффинная хрома-
сматриваются развиваемые в последние годы
тография по-прежнему является основным из
новые подходы, основанные на МС высокого
таких методов исследования биологических
разрешения, к исследованию результатов взаи-
взаимодействий и выделения белков, участвую-
модействия химических соединений, в первую
щих в специфических взаимодействиях с ли-
очередь терапевтических лекарственных пре-
гандами различной природы [9], развитие в
паратов, с белками клеточных протеомов.
последние годы протеомных технологий на ос-
нове масс-спектрометрии (МС) высокого раз-
решения позволило вывести задачу поиска ми-
МАСС-СПЕКТРОМЕТРИЧЕСКИЕ
шеней таких взаимодействий на уровень всего
МЕТОДЫ И ПОДХОДЫ
протеома клеток [10]. При этом протеомный
ХИМИЧЕСКОЙ ПРОТЕОМИКИ
эксперимент направлен либо на идентифика-
цию всех белков клетки, которые взаимодей-
Идентификация белковых мишеней био-
ствуют с исследуемым химическим соединени-
активных химических соединений уже давно
ем, либо целью анализа является определение
является одним из наиболее популярных на-
активности определенной группы белков про-
правлений исследований в области химической
теома на основе их взаимодействия с большим
биологии и поиска новых лекарств на основе
набором химических соединений, например, в
фенотипического анализа клеток. Классиче-
такой быстрорастущей теме исследований, как
ским примером такого поиска является вы-
лекарственное «репрограммирование» [11].
явление мишеней и механизмов возможного
В целом, персонализированные подходы
терапевтического действия веществ, исполь-
к терапии являются в настоящее время доми-
зуемых в традиционной медицине [16]. Если
нирующими в лечении онкологических за-
говорить о поиске белков, взаимодействую-
болеваний, когда один и тот же тип ракового
щих с тем или иным химическим соединением
заболевания может возникать у пациентов с
на уровне всего клеточного протеома, то как
различными генетическими дефектами, при
наиболее широко используемые, так и нахо-
этом проявляя различные изменения про-
дящиеся на начальной стадии своего развития
филя экспрессии белков, в том числе и в от-
современные методы химической протеомики
вет на внешнее или терапевтическое воздей-
основаны на использовании МС (рис. 2). В на-
ствие [12]. Таким образом, методы таргетной
стоящее время МС высокого разрешения явля-
медицины должны подразумевать персона-
ется основной технологией, используемой для
лизированный подход к подбору лекарствен-
структурной идентификации белков и пано-
ных препаратов и их молекулярных мишеней
рамного количественного анализа протеомов.
на клеточном уровне. Для реализации этого
В современном исполнении метод позволяет
подхода требуются молекулярные индикаторы
идентифицировать несколько тысяч белковых
или маркеры, благодаря которым можно оце-
групп в образцах протеомов клеточных линий
нить эффективность терапии
[13]. Значи-
и несколько сотен белковых групп в протео-
тельные изменения концентрации отдельных
мах, характеризующихся высоким динамиче-
белков протеома в результате воздействия хи-
ским диапазоном концентрации белков, таких
миопрепаратов могут служить источником ин-
как плазма крови. После биоинформатиче-
формации об активированных каскадах белок-
ского анализа, включающего идентификацию
БИОХИМИЯ том 87 вып. 9 2022
ХИМИЧЕСКАЯ ПРОТЕОМИКА
1235
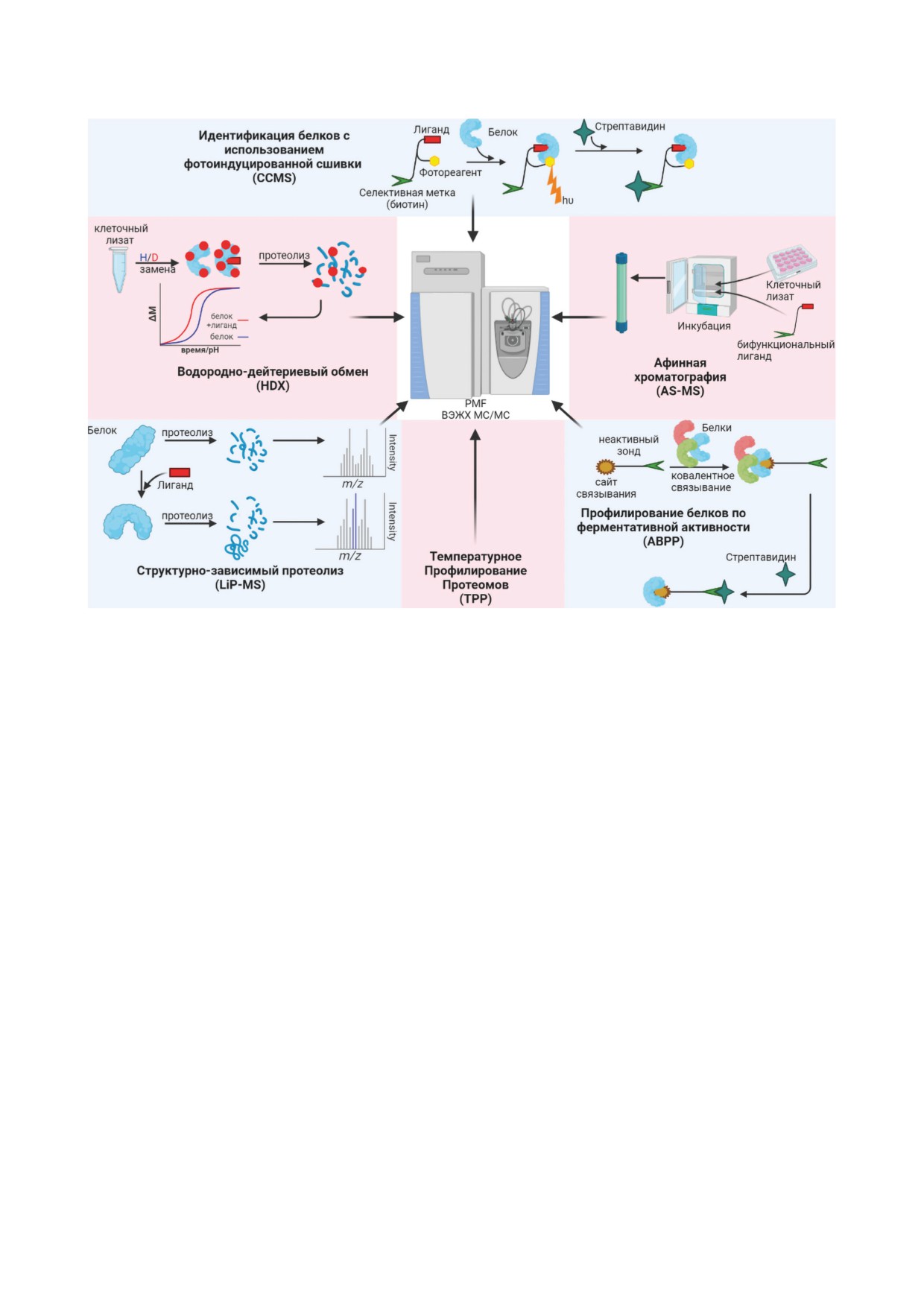
Рис. 2. Развитие технологий полнопротеомного анализа на основе МС высокого разрешения позволяет решать задачу
поиска всех мишеней лекарственного воздействия. При этом методы поиска таких белков, основанные на МС-иденти-
фикации взаимодействующих с лекарством белков, можно разделить на две группы: структурно-зависимые, в которых
МС используется для измерения структурных изменений белков, участвующих во взаимодействиях, и методы, основан-
ные на дериватизации анализируемого химического соединения с целью аффинного выделения всех провзаимодейство-
вавших белков с последующей их идентификацией. В случае одиночных мишеней для их идентификации используется
измерение точных масс их протеолитических пептидов. Для идентификации большого количества мишеней или поиска
всех возможных мишеней на уровне всего протеома клетки используется «скорострельная» протеомика на основе высо-
коэффективной жидкостной хроматографии (ВЭЖХ) в сочетании с тандемной масс-спектрометрией (МС/МС)
белков, присутствовавших в образце, из полу-
протеомных карт взаимодействия как новых,
ченных экспериментальных данных извлекают
так и уже известных онкопрепаратов с белками
количественную информацию об относитель-
протеомов клеточных линий рака различного
ном содержании белков, позволяющую опре-
типа [19, 20]. Общими проблемами таких ис-
делить биологические процессы и механизмы,
следований является не только необходимость
запускаемые в клетках в ответ на внешнее или
анализировать большое количество систем
терапевтическое воздействие [17]. Использо-
«лекарство-протеом» для получения биологи-
вание количественного панорамного полно-
чески значимой информации, но и выявлять
протеомного анализа на основе высокоэффек-
возможные мишени того или иного препара-
тивной жидкостной хроматографии (ВЭЖХ) и
та практически для каждого белка протеома,
тандемной масс-спектрометрии (МС/МС), по-
включая белки с посттрансляционными моди-
зволяющего измерять изменения концентра-
фикациями [21].
ции белков на уровне всего протеома клеток,
Аффинная хроматография в комбинации с
дает возможность выявлять не только извест-
масс-спектрометрией (AS-MS). Традиционным
ные и ожидаемые мишени для того или иного
методом химической протеомики является аф-
лекарства, но и дополнительные, неизвестные
финная хроматография [9], в основе которой
ранее белки, с которыми оно взаимодейству-
лежит выделение мишеней лекарственного
ет [18]. Исследования в этом направлении осо-
воздействия из клеточных экстрактов с помо-
бенно быстро развивались в последние годы в
щью специальных химических соединений -
онкопротеомике в связи с развитием МС-тех-
химических зондов, созданных на основе
нологий количественного анализа белков. Ос-
исследуемого биоактивного препарата, специ-
новной целью этих работ является составление
фически взаимодействующего с определен-
БИОХИМИЯ том 87 вып. 9 2022
1236
ФЕДОРОВ и др.
ными белками протеома. Химические зонды
привести выявление специфичности ингиби-
ковалентно иммобилизованы на стационар-
торов протеолитических ферментов методом
ной фазе, специфически связываясь с белка-
количественного профилирования белков по
ми протеома, растворенными в подвижной
ферментативной активности (Activity-Based
фазе. После промывки аффинной колонки и
Protein Profiling, ABPP [31]). Метод ABPP ос-
удаления нецелевых белков мишени, с кото-
нован на использовании специально разрабо-
рыми взаимодействует лиганд, белки-мишени
танных и синтезированных химических сое-
снимаются с фазы элюирующим буфером [22,
динений (зондов), образующих ковалентные
23]. Если целевой белок известен или суще-
связи с активными сайтами протеаз. Эти сое-
ствуют предположения о нем на основе ин-
динения, в свою очередь, представляют собой
формационных данных [24], иммуноблоттинг
аналоги лекарств, механизм действия которых
с использованием стандартных методов [25]
исследуется. При этом ковалентное связыва-
представляет собой простой, чувствительный
ние зонда с протеазой свидетельствует о свя-
и проверенный метод идентификации выде-
зывании и лекарства. Аффинные метки, такие
ленного белка. В случае, например, биотино-
как биотин, которые включают в химическую
вой метки, используется аффинное выделение
структуру зондов, позволяют выделить из про-
на стационарной фазе, иммобилизованной
теома все ковалентносвязанные протеазы с
стрептавидином за счет уникальной по специ-
последующим количественным МС-анализом
фичности и силе связывания последнего с
(рис. 2). Так, например, метод ABPP позволил
биотином способности. Однако в большин-
выявить на уровне всего протеома специфич-
стве случаев целевые белки будут неизвестны,
ность ингибиторов металлопротеаз, участвую-
по крайней мере изначально, и выделенные
щих в регулировании множества важных фи-
белки-мишени идентифицируются масс-спек-
зиологических и патологических процессов в
трометрически. Кроме того, таких белков мо-
клетках. Было показано, что металлопротеа-
жет оказаться довольно много. В этих случаях
зы могут иметь перекрывающуюся чувстви-
методы, основанные на МС, в первую очередь
тельность к ингибиторам. В связи с этим воз-
метод пептидных отпечатков масс (Peptide
никает задача оценки ингибиторов по этому
Mass Fingerprinting, PMF [26]) или «скоро-
классу ферментов в целом для последующей
стрельная» протеомика [27], используются для
разработки препаратов с требуемой избира-
идентификации всех выделенных белков-ми-
тельностью [32]. Аналогичным образом можно
шеней. На тех же принципах осуществляется
установить взаимосвязь между избытком или
и поиск мишеней химических соединений,
недостатком конкретного белка в организме
например ионов металлов, красителей, кис-
и возникновением симптомов заболевания.
лот в металл-аффинной хроматографии белков
Например, недостаток фермента PREPL, ко-
(Immobilized-Metal Affinity Chromatography,
дируемого геном PREPL, в клетках приводит к
IMAC [28]), в основе которой лежит обрати-
возникновению симптомов синдрома гипото-
мое взаимодействие аминокислотных остатков
нии-цистинурии, проявляющихся в появлении
последовательностей, выступающих в качестве
нервно-мышечных и легких когнитивных на-
доноров электронов, с ионами металлов, хела-
рушений [33]. В связи с этим возникает задача
тированными лигандами, иммобилизованны-
идентификации физиологических субстратов
ми на поверхности стационарной фазы. Метод
и путей, контролируемых PREPL, а также ин-
AS-MS играет важную роль в фосфопротеоми-
гибиторов активности этого фермента, успеш-
ке, где используется для выделения фракций
но решаемая с помощью метода ABPP. Еще
фосфорилированных пептидов в протеолити-
одним примером использования этого метода
ческих смесях для последующего МС-анали-
химической протеомики в масштабе всего кле-
за [29]. Сочетание аффинного обогащения с
точного протеома при выявлении молекуляр-
использованием меченых биоактивных малых
ных механизмов развития заболеваний может
молекул и количественной протеомики на ос-
служить исследование роли цистеиновых про-
нове МС высокого разрешения дает чувстви-
теаз в выживаемости малярийных паразитов
тельный и специфический инструмент для все-
на стадии мерозоита и выявление ингибиторов
стороннего профилирования взаимодействий
активности тех из них, которые играли ключе-
лекарств в клеточных протеомах [8, 30].
вую роль в развитии заболевания, убивающего
Масс-спектрометрическое профилирование
до 2 млн человек в год [34]. Аналогичный под-
протеомов по ферментативной активности. В ка-
ход на основе ABPP был использован в случае
честве примера использования химической
изучения вирусных заболеваний, в частности
протеомики, основанной на МС, для иссле-
вируса гепатита С (HCV), в котором основ-
дования определенного класса белков можно
ными объектами исследования были белки
БИОХИМИЯ том 87 вып. 9 2022
ХИМИЧЕСКАЯ ПРОТЕОМИКА
1237
протеома хозяина, изменение ферментатив-
под действием, например, ультрафиолетового
ной активности которых измерялось на стадии
излучения, а также метки, используемой для
патологического процесса репликации вируса.
последующего аффинного выделения таких
Белки со статистически значимыми измене-
белков, например биотина. На том же прин-
ниями активности могут отвечать за взаимо-
ципе основан поиск всех возможных мише-
действие вируса HCV c клетками хозяина и,
ней взаимодействия исследуемых химических
соответственно, диагностическими и терапев-
соединений с протеомами в методе МС-иден-
тическими мишенями [35].
тификации белков с использованием фото-
Одной из наиболее популярных областей
индуцированной сшивки (Capture Compound
исследований, в которых в последние годы ис-
Mass Spectrometry, CCMS [41]). Исследуемое
пользуются методы химической протеомики
химическое соединение модифицируется до-
в сочетании с МС, является выяснение меха-
бавлением двух функциональных групп, одна
низмов и роли фосфорилирования белков при
из которых ковалентно связывается с амино-
раковых заболеваниях. Фосфорилирование
кислотными остатками белка под действием
представляет собой модификацию белка после
УФ-излучения (фотоиндуцированная сшив-
завершения процесса трансляции путем при-
ка), а другая используется как метка, за ко-
соединения к белку фосфатной группы [36].
торую связанный с этим соединением белок
Процесс катализируется особой группой фер-
выделяется из протеомной смеси (рис. 2). Ос-
ментов, которые называются киназами. Кина-
новной целью метода CCMS является выявле-
зы фосфорилируют около 30% всех белков че-
ние не только белков-мишеней, специфически
ловека, в том числе и те, которые отвечают за
связывающихся с исследуемым лекарством,
сложные молекулярные процессы (рост, диф-
но и неспецифически связывающихся белков,
ференцировка, пролиферация и апоптоз) [37].
которые тем не менее играют роль в активиро-
Активность этих ферментов находится под
вании определенных каскадов белок-белковых
строгим контролем, однако в случае мутации
взаимодействий, ответственных за ответ кле-
киназ или других генетических изменениях
ток на лекарственное воздействие и связанных
регуляция нарушается, что приводит к возник-
с развитием, например, побочных процессов.
новению злокачественных опухолей. Таким об-
При отсутствии ковалентной сшивки белки в
разом, киназы являются потенциальными ми-
большинстве случаев будут потеряны. Выде-
шенями для противоопухолевой терапии [38].
ленные из протеома белки-мишени идентифи-
Здесь следует отметить, что при исполь-
цируются с использованием «скорострельной»
зовании ингибиторов активности большого
протеомики на основе ВЭЖХ-МС/МС-анали-
класса белков, тех же киназ, таргетная терапия
за [27]. Метод CCMS особенно хорошо под-
сталкивается с существенной проблемой. Ин-
ходит для поиска как можно большего набора
гибиторы распознают консервативные участки,
потенциальных взаимодействий малых моле-
которые одинаковы для многих белков данно-
кул с белками. Особенностью метода является
го класса. В случае киназ в роли такого участка
возможность его реализации непосредственно
выступает так называемый ATP-связывающий
в растворе при физиологических условиях, в
«карман» [39]. Таким образом, ингибиторы,
которых белки находятся в нативной форме.
нацеленные на определенный консервативный
В отличие от аффинной хроматографии, свя-
участок, могут перекрестно взаимодействовать
занной с использованием стационарной фазы
с другими членами класса и другими белками,
с заданными свойствами, такими как размер
связанными с этим участком. Возникает задача
пор, метод не имеет каких-либо ограничений
идентификации возможных мишеней действия
на размер лигандов. Также преимуществом ме-
конкретного ингибитора среди всех белков
тода в сравнении с ABPP является то, что в слу-
клеток, которая в настоящее время может быть
чае ABPP выбор анализируемых соединений
решена методами химической протеомики,
ограничен исключительно теми, которые ко-
основанными на полнопротеомном количе-
валентно связываются с активными центрами
ственном МС-анализе [40, 18].
ферментов, как в случае, например, сериновых
Масс-спектрометрическая идентификация
протеаз. Метод CCMS может использоваться
белков с использованием фотоиндуцированной
для исследований специфичности химических
сшивки. Ингибиторы со специфической актив-
соединений, образующих слабые нековалент-
ностью по отношению к определенным белкам
ные связи с белками-мишенями. В качестве
могут быть модифицированы путем присоеди-
известного примера реализации метода CCMS
нения реакционной группы, которая может об-
в химической протеомике можно упомянуть
разовывать ковалентные связи с аминокислот-
работу Fischer et al. [42] по идентификации
ными остатками последовательностей белков
всех возможных белков, взаимодействующих
БИОХИМИЯ том 87 вып. 9 2022
1238
ФЕДОРОВ и др.
с лекарствами толкапон и энтакапон, исполь-
что потребует несколько недель затрат инстру-
зуемыми для лечения болезни Паркинсона.
ментального времени. Еще одной проблемой
Оба лекарства являются ингибиторами фер-
метода является потребность в большом коли-
мента COMP (catechol-O-methyltransferase),
честве клеток для анализа одной такой систе-
нередко проявляя при этом тяжелые гепато-
мы «лекарство-протеом» - от 105 до 106 клеток
токсические эффекты в клинической практи-
на каждую точку сканирования. Хотя процесс
ке [43-45]. В частности, в упомянутой выше
денатурации белка считается двухстадийным
работе Fischer et al. [42], методом CCMS было
переходом от свернутого нативного состоя-
идентифицировано 124 белка клеточной ли-
ния к структуре случайного клубка, часто су-
нии HepG2, которые взаимодействовали с тол-
ществуют промежуточные продукты, которые
капоном, часть из которых относилась к бел-
могут приводить к взаимодействию лекарства
кам, ответственным за возможные побочные
in vitro в процессе пробоподготовки с белками,
эффекты терапии.
которые в действительности не являются его
Температурное профилирование протеомов -
мишенями in vivo. С другой стороны, сигмои-
новый метод поиска мишеней лекарственного
дальная подгонка кривых плавления белков
воздействия на основе панорамной масс-спек-
приводит к тому, что белки, для которых такая
трометрии. Метод температурного профи-
форма не является характерной, «выпадают» из
лирования протеомов (TPP) был предложен
анализа, что увеличивает риск пропуска важ-
в 2014 г. Savitski et al. [40] и основан на опре-
ных мишеней.
делении точки денатурации белков при изме-
Структурно-зависимые методы химической
нении температуры. В результате денатурации
протеомики. Одним из следствий связывания
белки становятся нерастворимыми в соответ-
белков с лигандами является изменение их кон-
ствующем буферном растворе. Соответствен-
формационной динамики [48]. На этом свой-
но, если говорить обо всем протеоме, то проис-
стве основаны так называемые структурно-за-
ходит разделение всех белков на две фракции:
висимые методы поиска белков-мишеней, в
растворимых и нерастворимых при заданной
которых эти изменения измеряются масс-спек-
температуре. Если при этом измерять относи-
трометрически (рис. 2).
тельную концентрацию всех белков раствори-
Одним из наиболее распространенных
мой фазы методом полнопротеомного количе-
структурно-зависимых методов является МС
ственного анализа при разных температурах, то
водородно-дейтериевого обмена (Hydrogen-
для каждого белка протеома можно построить
Deuterium Exchange Mass Spectrometry, HDX-
соответствующую температурную кривую рас-
MS), который в последние годы стал рутинной
творимости, как правило, S-образной формы
технологией для исследования структуры и
(рис. 3), и определить точку денатурации. Взаи-
динамики белков в растворах. В методе изме-
модействие того или иного белка с химическим
ряется обмен дейтерия с водородом в амидной
соединением приводит к изменению точки
группе основной цепи белка [49, 50]. Водород в
температурной денатурации таких белков, что
амидной группе белковой цепи обменивается с
проявляется в смещении соответствующих
водородом в воде со скоростью, которая зави-
температурных кривых количественных изме-
сит от окружения амидной группы и связана с
нений белков в растворимой фазе протеома.
динамикой белка. При этом водород, участвую-
Таким образом, полнопротеомный анализ на
щий во взаимодействиях белок-лиганд, демон-
основе количественной МС дает возможность
стрирует более медленный обмен. Скорость ре-
отслеживания не только каждого уникального
акции обмена в основной цепи происходит в
состояния белков при взаимодействии с лекар-
экспериментально доступной временной шка-
ством или другим белком на уровне всего про-
ле, соответственно, информацию о структуре
теома, но и выявления ключевых каскадов бе-
и динамике белка можно получить, наблюдая
лок-белковых взаимодействий, активируемых
зависящее от времени включение дейтерия в
лекарственным воздействием
[46]. Следует
положение водорода в амидной группе посред-
отметить, что метод имеет ряд ограничений, в
ством инкубации в дейтерированной воде. Та-
частности, необходимость проведения большо-
ким образом, в результате связывания лиганда
го количества полнопротеомных ВЭЖХ-МС/
с белком меняется кинетика водородно-дейте-
МС-анализов систем
«лекарство-протеом»
риевого обмена, которую можно наблюдать в
при разных температурах и концентрациях.
виде сдвига кинетической кривой при измере-
Так, например, 10 точек по температурам и
нии скорости реакции обмена либо во време-
концентрациям соответствуют 100 фракциям,
ни, либо при различной кислотности раство-
для каждой из которых требуется осуществлять
ра, в котором протекает эта реакция (рис. 2).
полнопротеомный количественный анализ,
Количественно эффективность обмена про-
БИОХИМИЯ том 87 вып. 9 2022
ХИМИЧЕСКАЯ ПРОТЕОМИКА
1239
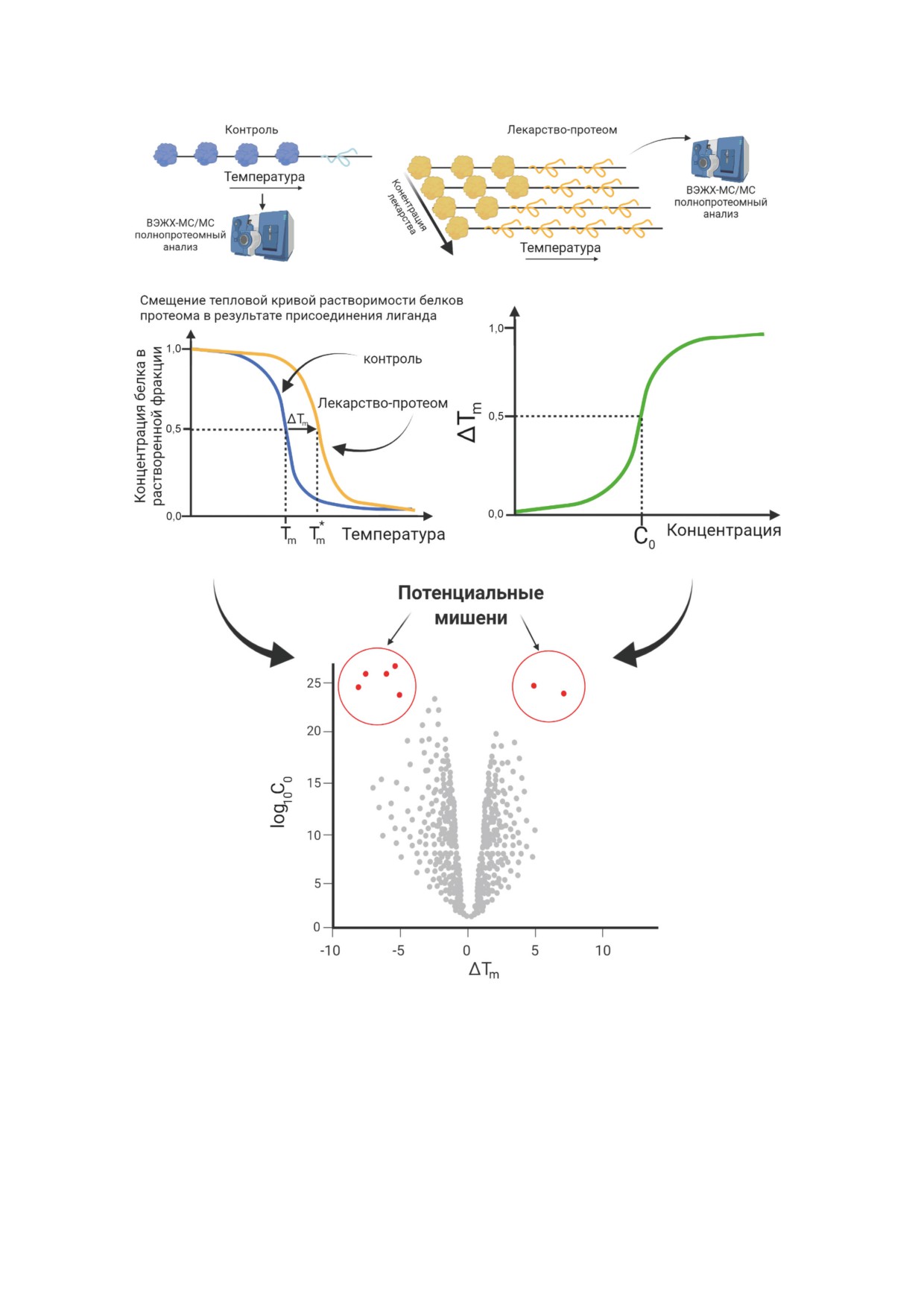
Рис. 3. Иллюстрация метода температурного профилирования протеомов. Клеточный лизат предварительно разделяют
на несколько проб, каждая из которых обрабатывается анализируемым препаратом в разных концентрациях. Клеточ-
ный лизис осуществляется методом «заморозки-разморозки» (freeze-thaw [47]) с целью сохранения нативной формы
белков. В результате обработки те из белков протеома, к которым присоединилось лекарство, изменяют свои термо-
динамические свойства. Каждую из «концентрационных» проб снова функционируют с последующей инкубацией каж-
дой из фракций при разных температурах в диапазоне 37-67 °С (как правило, до 10 температурных точек). После инку-
бации ультрацентрифугированием выделяют растворенную в буферном растворе белковую фракцию с последующим
количественным полнопротеомным анализом с использованием ВЭЖХ-МС/МС. Для каждого белка анализируемой
системы «лекарство-протеом» строится кривая количественных изменений (кривая растворимости) в координатах
(температура, концентрация), которая сравнивается с кривой растворимости этого белка в контрольной пробе, не об-
работанной лекарством. Статистический анализ на значимость сдвигов температурных кривых растворимости, пред-
ставленный в виде диаграмм рассеяния, позволяет выявить белки со статистически значимыми изменениями темпера-
турных точек денатурации, которые и являются белками-мишенями для исследуемого лекарства
БИОХИМИЯ том 87 вып. 9 2022
1240
ФЕДОРОВ и др.
является в сдвиге массы белка или суммарном
Одним из наиболее широко используемых
сдвиге массы его протеолитических пептидов
решений проблемы производительности пол-
в масс-спектрах. Таким образом, можно осу-
нопротеомного анализа является мультиплек-
ществлять измерения для всех белков протео-
синг проб на основе изобарных меток (Tandem
ма в рамках стандартного полнопротеомного
Mass Tags, TMT), позволяющий одновременно
ВЭЖХ-MC/МС-анализа и выявлять взаимо-
анализировать несколько протеомов в рамках
действия белок-лиганд [51, 52]. Одной из про-
одного ВЭЖХ-МС/МС-эксперимента
[57].
блем метода HDX-MS является его довольно
Метод заключается в присоединении к протео-
низкая чувствительность, в результате многие
литическим пептидам каждой из проб изо-
из мишеней лиганда остаются вне рамок анали-
барных меток одной массы, в состав которых
за. Для решения этой проблемы был предложен
входят так называемые «репортерные» части,
так называемый гистидин-водородно-дейтерие-
отличные по массе. Меченые таким образом
вый обмен (His-HDX). В основе этого подхода
пептиды разных проб объединяются в одну,
лежит обогащение протеолитических смесей
однако в процессе анализа при фрагментации
гистидин-содержащими пептидами с последую-
конкретного пептида происходит диссоциация
щим анализом обогащенной смеси стандарт-
не только самого пептида, но и его «репортер-
ным ВЭЖХ-МС/МС. В частности, этот подход
ной» части TMT-метки. В спектре фрагмен-
был успешно продемонстрирован для выявле-
тации пептида, на основе которого происхо-
ния взаимодействий между митоген-активируе-
дит его идентификация, присутствует также
мой протеинкиназой MARK14 и ее ингибито-
и спектр ионов-репортеров метки, относи-
ром в лизатах клеток HEK293 [53].
тельные интенсивности которых соответству-
Еще одним набирающим популярность в
ют относительным интенсивностям данного
последние годы структурно-зависимым мето-
пептида в разных пробах и, соответственно,
дом является ограниченный протеолиз в сочета-
относительным концентрациям белка, которо-
нии с МС-детектированием (Limited Proteolysis-
му данный пептид соответствует. Реализация
Coupled Mass Spectrometry, LiP-MS [54]). Метод
10-плексного протеомного анализа [58] позво-
основан на предположении о том, что в резуль-
ляет, таким образом, на порядок сократить за-
тате присоединения лиганда к белку меняется
траты инструментального времени на количе-
профиль протеолитических пептидов последне-
ственный протеомный анализ каждой пробы.
го в результате гидролиза высокоспецифичны-
С целью дальнейшего сокращения времени
мы ферментами. Важной особенностью метода
анализа в задачах химической протеомики в не-
является проведение именно ограниченного
давней работе Gaetani et al. [59] был представ-
протеолиза, не проводя гидролиз полностью,
лен новый метод, так называемого интеграль-
что привело бы к потере информации о связан-
ного изменения растворимости белков (Protein
ном с лигандом белке. После проведения огра-
Integral Solubility Alteration, PISA). В анализе
ниченного протеолиза образцы анализируют с
PISA объединяются образцы по всему темпера-
помощью МС с целью идентификации белков
турному градиенту. Содержание белка в объе-
и оценки изменений в профилях их протеоли-
диненном образце, которое представляет собой
тических пептидов [55]. Следует отметить, что
площадь под кривой плавления, используется
одним из недостатков метода LiP-MS является
для измерения влияния лиганда на термоста-
длительность пробоподготовки [54], а также не-
бильность белка. Основной метрикой в мето-
высокая специфичность [56].
де PISA является показатель разницы между
Методы и подходы ультракороткой коли-
площадями под кривыми плавления обрабо-
чественной протеомики. Как уже отмечалось
танных и контрольных образцов (ΔSm). Также
выше, решение задач химической протеомики
такой метрикой может служить кратность из-
в контексте поиска мишеней лекарственного
менений белков в объединенных обработан-
воздействия на уровне всего протеома требует
ных образцах по сравнению с интегральными
проведения большого количества полнопроте-
контрольными образцами. Как ΔSm, так и крат-
омных анализов. Так, в случае использования
ность изменения связаны со сдвигом темпера-
метода TPP речь идет о десятках и сотнях ана-
тур плавления (ΔTm) и, следовательно, с изме-
лизов только для одной системы «лекарство-
нением термостабильности белка. В анализе
протеом». Даже в случае использования альтер-
PISA экспериментальная ΔSm аппроксимиру-
нативных методов разнообразие возможных
ется путем нормализации этой разницы между
химиотерапевтических средств и подходов, а
обработанными и контрольными образцами по
также персонализированный отклик протео-
контрольным образцам, в то время как крат-
мов клеток на их использование требуют зна-
ные изменения не требуют дополнительной
чительных затрат инструментального времени.
нормализации данных. Метод PISA позволяет
БИОХИМИЯ том 87 вып. 9 2022
ХИМИЧЕСКАЯ ПРОТЕОМИКА
1241
дополнительно сократить время анализа одной
реализации быстрого количественного полно-
системы «лекарство-протеом» на порядок.
протеомного анализа за счет возможности ис-
Объединения фракций в перечисленных
пользования ультракоротких градиентов разде-
выше подходах дает двойной выигрыш: во-пер-
ления протеолитических смесей [67]. Понятие
вых, значительно увеличивается производитель-
«прямая» подразумевает исключение стадии
ность анализа одной системы «лекарство-про-
последовательного получения масс-спектров
теом» и, во-вторых, существенно сокращается
фрагментации протеолитических пептидов
расход образца. Например, использование муль-
смеси, являющейся одной из основных причин
типлексных TMT-меток в сочетании с мето-
увеличения затрат инструментального времени
дом PISA позволяет на два порядка сократить
или уменьшения глубины анализа при исполь-
затраты инструментального времени в задачах
зовании коротких градиентов разделения. При
химической протеомики на основе темпера-
этом прямая идентификация белков осущест-
турного полнопротеомного профилирования.
вляется за счет использования всей совокуп-
Недавно представленные реагенты TMTpro для
ности экспериментальных данных о пептидах,
количественного полнопротеомного анализа
комплементарных к масс-спектрометриче-
позволяют использовать 16-18 интегральных
ским, в частности, хроматографических вре-
образцов в одном эксперименте [60].
мён или ионной подвижности, а также специ-
Основной проблемой метода температур-
фических особенностей последовательностей
ного полнопротеомного профилирования и его
белков, которым эти пептиды соответствуют.
модификаций является то, что он основан на
Существенными составляющими метода яв-
сигмоидальной форме кривых плавления бел-
ляются программные средства для обработки
ков. Однако существует большая группа белков,
масс-спектров ионов пептидов и идентифика-
для которых это условие не выполняется, и кри-
ции их изотопных кластеров [68], ранжирова-
вые плавления не могут быть воспроизведены с
ние идентификаций, соотнесение их с белками
достаточно высокой точностью для измерения
в соответствующих базах данных и определе-
соответствующего сдвига в температуре дена-
ние уровня достоверности [69]. Важно, что в
турации [61], что потенциально может служить
отличие от подходов, основанных на МС/МС,
источником ложноположительных результатов
метод DirectMS1 позволяет идентифициро-
в определении мишеней лекарственного воз-
вать белки с существенно (почти на порядок)
действия. Также количество белков во фракци-
большим покрытием последовательности, что,
ях резко снижается при высоких температурах,
в свою очередь, дает более точные измерения
особенно белков с низкими температурами де-
относительного содержания белков в пробах
натурации (Tm), что потенциально затрудняет
на основе измерения интенсивностей спектров
обнаружение соответствующего сдвига
[62].
ионов, соответствующих этим белкам пептидов.
В свою очередь, использование более узкого
Как было показано в недавних работах, метод
диапазона температур для построения кривых
позволяет в настоящее время количественно
плавления также повышает риски пропуска це-
идентифицировать более 2000 белков протео-
левых белков лекарственного воздействия.
мов в рамках 5-минутного хроматомасс-спек-
Как уже отмечалось выше, при использова-
трометрического эксперимента [70].
нии мультиплексного и интегрального подходов
Еще одним методом ультракороткого пол-
затраты инструментального времени для одной
нопротеомного количественного анализа яв-
системы «лекарство-протеом» в методе TPP
ляется метод ScanningSWATH, основанный на
сокращаются на два и более порядков. Однако
одновременной фрагментации ионов пепти-
даже в этом случае разнообразие как групп ле-
дов смесей в широком окне отношений масс к
карств, так и объектов их воздействия настолько
заряду (m/z), так называемом методе слепого
велико, что проблема дальнейшего повышения
сбора данных фрагментации (Data Independent
производительности полнопротеомного анали-
Acquisition, DIA) с одновременным непре-
за по-прежнему остается критически важной в
рывным сканированием окна фрагментации.
задачах химической протеомики.
Используя непрерывное сканирование окна
Первые работы по разработке методов
фрагментации прекурсорных ионов пептидов,
ультракороткого полнопротеомного анализа
метод продемонстрировал возможности его ис-
для широкомасштабных скрининговых ис-
пользования в режиме ультракоротких, порядка
следований в области сравнительной протео-
0,5-5 мин, градиентов разделения протеолити-
мики появились буквально в последние не-
ческих смесей пептидов при высокой глубине
сколько лет [63-66]. Среди них метод прямой
анализа протеомов клеточных линий человека,
масс-спектрометрической идентификации бел-
достигающей в отдельных экспериментах не-
ков DirectMS1 является одним из подходов к
скольких тысяч белков [71, 72].
6
БИОХИМИЯ том 87 вып. 9 2022
1242
ФЕДОРОВ и др.
ЗАКЛЮЧЕНИЕ
ниже порога чувствительности измерений. Кро-
ме того, в структурно-зависимых методах ста-
Химическая протеомика на основе коли-
вится задача поиска взаимодействий лекарств с
чественного профилирования протеомов яв-
неденатурированными последовательностями
ляется в настоящее время основным методом
белков в их нативной форме, и клеточный ли-
выявления белков-мишеней лекарственного
зис осуществляется с использованием соответ-
воздействия. В то время как на ранней стадии
ствующих методик, таких как «заморозка-раз-
своего развития химическая протеомика внес-
морозка» при криогенных температурах. В этом
ла существенный вклад в понимание механиз-
случае, как правило, идет потеря мембранных
мов действия конкретных классов ферментов
белков. Решение этих проблем видится в исполь-
и выявление белков-мишеней специфического
зовании методов, основанных на фотоиндуци-
взаимодействия с различными классами хими-
рованном связывании белков, в первую очередь
ческих соединений, большая часть протеома ор-
CCMS, в котором анализируемый лиганд дери-
ганизма до сих пор оставалась за рамками этих
ватизируется добавлением фотореагирующей
работ. В то время как аффинная хроматография
группы, которая под действием УФ-излучения
продолжает широко использоваться в таких ис-
образует ковалентные связи с аминокислотны-
следованиях, доминирующими подходами в на-
ми остатками белка-мишени лиганда. В целом,
стоящее время становятся методы, основанные
высокопроизводительный полнопротеомный
на количественном полнопротеомном анализе
МС-анализ позволяет не только решать в ко-
с использованием МС высокого разрешения.
роткие сроки задачи поиска всех возможных
В первую очередь это относится к так называе-
мишеней лекарственного воздействия для бы-
мой «лекарство-центричной» области химиче-
строрастущего разнообразия разрабатываемых
ской протеомики, задачей которой является по-
химиотерапевтических препаратов, но исполь-
иск всех возможных мишеней специфического
зовать методы химической протеомики в таких
и неспецифического взаимодействия анализи-
перспективных областях исследований, как пе-
руемого соединения на уровне всего протеома
репрограммирование существующих лекарств и
клетки. Развитие новых методов ультрабыстрого
персонализированный подбор лекарств с задан-
протеомного анализа позволяет масштабиро-
ными терапевтическими свойствами.
вать решение основных задач химической про-
теомики по поиску мишеней лекарственного
Вклад авторов. И.И. Федоров, В.И. Лине-
воздействия и разработке новых химиотерапев-
ва - подбор и анализ литературы по теме обзора,
тических подходов к лечению социально-зна-
написание текста обзора; И.А. Тарасова - об-
чимых заболеваний человека. Среди методов
суждение темы обзора и описываемых методов;
химической протеомики, которые особенно
М.В. Горшков - руководство работой над обзо-
активно развивались в последние годы, следу-
ром, написание и редактирование текста.
ет выделить методы, основанные на измерении
Финансирование. Работа выполнена при
малых изменений в конформации или стабиль-
финансовой поддержке Российского научного
ности белков, с которыми связался лиганд (та-
фонда (грант № 20-14-00229).
кие как TPP, HDХ-MS и LiP-MS) и которые не
Благодарности. Авторы выражают благо-
требуют дополнительной дериватизации иссле-
дарность профессору РАН С.А. Мошковскому
дуемого химического соединения. Большинство
за плодотворное обсуждение темы обзора.
методов не решают проблему поиска мишеней
Конфликт интересов. Авторы заявляют об
лекарственного воздействия среди мембранных
отсутствии конфликта интересов.
белков, поскольку, как правило, лекарства об-
Соблюдение этических норм. Настоящая
разуют с ними слабые, нековалентные связи, и
статья не содержит описания каких-либо ис-
соответствующий эффект в изменении той же
следований с участием людей или животных в
термической стабильности белка становится
качестве объектов.
СПИСОК ЛИТЕРАТУРЫ
1. Schutte, M., Ogilvie, L. A., Rieke, D. T., Lange,
otic resistance, J. Infect. Public Health, 10, 369-378,
B. M. H., Yaspo, M. L., et al. (2017) Cancer pre-
doi: 10.1016/j.jiph.2016.08.007.
cision medicine: why more is more and DNA is
3. Vasan, N., Baselga, J., and Hyman, D. M. (2019)
not enough, Public Health Genomics,
20,
70-80,
A view on drug resistance in cancer, Nature, 575, 299-
doi:10.1159/000477157.
309, doi: 10.1038/s41586-019-1730-1.
2. Frieri, M., Kumar, K., and Boutin, A. (2017) Antibi-
4. Ramos, A., Sadeghi, S., and Tabatabaeian, H. (2021)
БИОХИМИЯ том 87 вып. 9 2022
ХИМИЧЕСКАЯ ПРОТЕОМИКА
1243
Battling chemoresistance in cancer: root causes and
as a proteome signature library of anticancer molecules
strategies to uproot them, Int. J. Mol. Sci., 22, 9451,
for functional discovery, Nat. Commun., 10, 5715,
doi: 10.3390/ijms22179451.
doi: 10.1038/s41467-019-13582-8.
5.
Ashley, E. A. (2016) Towards precision medicine. Nat.
20.
Ruprecht, B., Di Bernardo, J., Wang, Z., Mo, X.,
Rev. Genet., 17, 507-522, doi: 10.1038/nrg.2016.86.
Ursu, O., et al. (2020) A mass spectrometry-based
6.
Mroz, E. A., and Rocco, J. W. (2017) The challenges
proteome map of drug action in lung cancer cell
of tumor genetic diversity, Cancer, 123, 917-927,
lines, Nat. Chem. Biol., 16, 1111-1119, doi: 10.1038/
doi: 10.1002/cncr.30430.
s41589-020-0572-3.
7.
Rix, U., and Superti-Furga, G. (2009) Target profiling
21.
Schubert, O., Röst, H., Collins, B., Rosenberger, G.,
of small molecules by chemical proteomics, Nat.
and Aebersold, R. (2017) Quantitative proteomics:
Chem. Biol., 5, 616-624, doi: 10.1038/nchembio.216.
challenges and opportunities in basic and applied
8.
Wright, M. H., and Sieber, S. A. (2016) Chemical
research, Nat. Protoc., 12, 1289-1294, doi: 10.1038/
proteomics approaches for identifying the cellular
nprot.2017.040.
targets of natural products, Nat. Prod. Rep., 33, 681-
22.
Cuatrecasas, P. (1970) Protein purification by affinity
708, doi: 10.1039/c6np00001k.
chromatography. Derivatizations of agarose and
9.
Rodriguez, E. L., Poddar, S., Iftekhar, S.,
polyacrylamide beads, J. Biol. Chem., 245, 3059-3065,
Suh, K., Woolfork, A. G., et al. (2020) Affinity
doi: 10.1016/S0021-9258(18)63022-4.
chromatography: a review of trends and developments
23.
Lolli, G., Thaler, F., Valsasina, B., Roletto, F.,
over the past 50 years, J. Chromatogr. B. Analyt.
Knapp, S., et al.
(2003) Inhibitor affinity
Technol. Biomed. Life Sci., 1157, 122332,doi: 10.1016/
chromatography: profiling the specific reactivity of the
j.jchromb.2020.122332.
proteome with immobilized molecules, Proteomics, 3,
10.
Huang, F., Zhang, B., Zhou, S., Zhao, X., Bian, C.,
1287-1298, doi: 10.1002/pmic.200300431.
et al. (2012), Chemical proteomics: terra incognita for
24.
McMasters, D. R.
(2018)
Knowledge-based
novel drug target profiling, Chinese J. Cancer, 31, 507-
approaches to off-target screening, Methods Enzymol.,
518, doi: 10.5732/cjc.011.10377.
610, 311-323, doi: 10.1016/bs.mie.2018.09.023.
11.
Pfab, C., Schnobrich, L., Eldnasoury, S., Gessner, A.,
25.
Kurien, B. T., and Scofield, R. H. (2015) Western
and El-Najjar, N. (2021) Repurposing of antimicrobial
blotting: an introduction, Methods Mol. Biol., 1312,
agents for cancer therapy: what do we know? Cancers
17-30, doi: 10.1007/978-1-4939-2694-7_5.
(Basel), 13, 3193, doi: 10.3390/cancers13133193.
26.
Dudley, E. (2019) MALDI profiling and applications
12.
Kamel, H. F. M., and Al-Amodi, H. S. A. B.
in medicine, Adv. Exp. Med. Biol., 1140, 27-43,
(2017) Exploitation of gene expression and cancer
doi: 10.1007/978-3-030-15950-4_2.
biomarkers in paving the path to era of personalized
27.
Zhang, Y., Fonslow, B. R., Shan, B., Baek, M. C., and
medicine, Genom.Proteom. Bioinform., 15, 220-235,
Yates, J. R. 3rd (2013) Protein analysis by shotgun/
doi: 10.1016/j.gpb.2016.11.005.
bottom-up proteomics, Chem. Rev.,113, 2343-2394,
13.
Sneha, P., and Doss, C. G.
(2016) Molecular
doi: 10.1021/cr3003533.
dynamics: new frontier in personalized medicine, Adv.
28.
Porath, J., Carlsson, J., Olsson, I., and Belfrage, G.
Protein Chem. Struct. Biol., 102, 181-224, doi: 10.1016/
(1975) Metal chelate affinity chromatography, a new
bs.apcsb.2015.09.004.
approach to protein fractionation, Nature, 258, 598-
14.
Siwy, J., Mischak, H., and Zürbig, P.
(2019)
599,doi: 10.1038/258598a0.
Proteomics and personalized medicine: a focus on
29.
Sun, X., Chiu, J.-F., and He, Q.-Y. (2005) Application
kidney disease, Exp. Rev. Proteomics, 16, 773-782,
of immobilized metal affinity chromatography in
doi: 10.1080/14789450.2019.1659138.
proteomics, Expert Rev. Proteomics,
2,
649-657,
15.
Meissner, F., Geddes-McAlister, J., Mann, M., and
doi: 10.1586/14789450.2.5.649.
Bantscheff, M. (2022) The emerging role of mass
30.
Ong, S. E., Schenone, M., Margolin, A. A., Li, X.,
spectrometry-based proteomics in drug discovery, Nat.
Do, K., et al. (2009) Identifying the proteins to which
Rev. Drug Discov., doi: 10.1038/s41573-022-00409-3,
small-molecule probes and drugs bind in cells, Proc.
in press.
Natl. Acad. Sci. USA, 106, 4617-4622, doi: 10.1073/
16.
Corson, T. W., and Crews, C. M. (2007) Molecular
pnas.0900191106.
understanding and modern application of traditional
31.
Sanman, L. E., and Bogyo, M. (2014) Activity-based
medicines: triumphs and trials, Cell, 130, 769-774,
profiling of proteases, Annu. Rev.Biochem., 83, 249-
doi: 10.1016/j.cell.2007.08.021.
273, doi: 10.1146/annurev-biochem-060713-035352.
17.
Aebersold, R., and Mann, M. (2016) Mass-spectro-
32.
Saghatelian, A., Jessani, N., Joseph, A., Humphrey,
metric exploration of proteome structure and func-
M., and Cravatt, B. F. (2004) Activity-based probes
tion, Nature, 537, 347-355, doi: 10.1038/nature19949.
for the proteomic profiling of metalloproteases, Proc.
18.
Chernobrovkin, A., Marin-Vicente, C., Visa, N., and
Natl. Acad. Sci. USA, 101, 10000-10005, doi: 10.1073/
Zubarev, R. A. (2015) Functional identification of tar-
pnas.0402784101.
get by expression proteomics (FITExP) reveals pro-
33.
Lone, A. M., Bachovchin, D. A., Westwood, D.,
tein targets and highlights mechanisms of action of
Speers, A. E., Timothy, P., et al. (2012) A sub-
small molecule drugs, Sci. Rep., 5, 11176, doi: 10.1038/
strate-free activity-based protein profiling screen
srep11176.
for the discovery of selective PREPL inhibitors,
19.
Saei, A. A., Beusch, C. M., Chernobrovkin, A.,
J. Am. Chem. Soc., 133, 11665-11674, doi: 10.1021/
Sabatier, P., Zhang, B., et al. (2019) ProTargetMiner
ja2036095.
БИОХИМИЯ том 87 вып. 9 2022
6*
1
244
ФЕДОРОВ и др.
34.
Greenbaum, D. C., Baruch, A., Grainger, M.,
48.
De Souza, N., and Picotti, P. (2020) Mass spectrom-
Bozdech, Z., Medzihradszky, K. F., et al.
(2002)
etry analysis of the structural proteome, Curr. Opin.
A role for the protease falcipain 1 in host cell invasion
Struct. Biol., 60, 57-65, doi: 10.1016/j.sbi.2019.10.006.
by the human malaria parasite, Science, 298, 2002-
49.
James, E. I., Murphree, T. A., Vorauer, C., Engen, R.,
2006, doi: 10.1126/science.1077426.
and Guttman, M. (2022) Advances in hydrogen/
35.
Singaravelu, R., Blais, D. R., McKay, C. S.,
deuterium exchange mass spectrometry and the
and Pezacki, J. P.
(2010) Activity-based pro-
pursuit of challenging biological systems, Chem. Rev.,
tein profiling of the hepatitis C virus replication
122, 7562-7623, doi: 10.1021/acs.chemrev.1c00279.
in Huh-7 hepatoma cells using a non-directed ac-
50.
Kaltashov, I. A., Bobst, C. E., and Abzalimov, R. R.
tive site probe, Proteome Sci., 8, 5, doi: 10.1186/
(2013) Mass spectrometry-based methods to study
1477-5956-8-5.
protein architecture and dynamics, Protein Sci., 22,
36.
Torkamani, A., and Schork, N. J. (2007) Distribution
530-544, doi: 10.1002/pro.2238.
analysis of nonsynonymous polymorphisms within
51.
Kaltashov, I. A., Bobst, C. E., and Abzalimov, R. R.
the human kinase gene family, Genomics, 90, 49-58,
(2009) H/D exchange and mass spectrometry in the
doi: 10.1016/j.ygeno.2007.03.006.
studies of protein conformation and dynamics: is there
37.
Hubbard, M. J., and Cohen, P. (1993) On target
a need for a top-down approach? Anal. Chem., 81,
with a new mechanism for the regulation of protein
7892-7899, doi: 10.1021/ac901366n.
phosphorylation, Trends Biochem. Sci., 18, 172-177,
52.
Campobasso, N., and Huddler, D. (2015) Hydrogen
doi: 10.1016/0968-0004(93)90109-z.
deuterium mass spectrometry in drug discovery,
38.
Blume-Jensen, P., and Hunter, T.
(2001)
Bioorg. Med. Chem. Lett., 25, 3771-3776, doi: 10.1016/
Oncogenic kinase signalling, Nature, 411, 355-365,
j.bmcl.2015.07.007.
doi: 10.1038/35077225.
53.
Miyagi, M., Tanaka, K., Watanabe, S., Kondo, J.,
39.
Valsasina, B., Kalisz, H. M., and Isacchi, A. (2004)
and Kishimoto, T. (2021) Identifying protein-drug
Kinase selectivity profiling by inhibitor affinity
interactions in cell lysates using histidine hydrogen
chromatography, Expert Rev. Proteomics, 1, 303-315,
deuterium exchange, Anal. Chem., 93, 14985-14995,
doi: 10.1586/14789450.1.3.303.
doi: 10.1021/acs.analchem.1c02283.
40.
Savitski, M. M., Reinhard, F. B. M., Franken, H.,
54.
Schopper, S., Kahraman, A., Leuenberger, P.,
Werner, T., Savitski, M. F., et al. (2014) Tracking
Feng, Y., Piazza, I., et al. (2017) Measuring protein
cancer drugs in living cells by thermal profiling of
structural changes on a proteome-wide scale using
the proteome, Science, 346, 1255784, doi: 10.1126/
limited proteolysis-coupled mass spectrometry, Nat.
science.1255784.
Protocols, 12, 2391-2410, doi: 10.1038/nprot.2017.100.
41.
Köster, H., Little, D. P., Luan, P., Muller, R.,
55.
Cheng, K. W., Wong, C. C., Wang, M., He, Q. Y., and
Siddiqi, S. M., et al. (2007) Capture compound mass
Chen, F. (2010) Identification and characterization
spectrometry: a technology for the investigation of
of molecular targets of natural products by mass
small molecule protein interactions, Assay Drug Dev.
spectrometry, Mass Spectrom. Rev.,
29,
126-155,
Technol., 5, 381-390, doi: 10.1089/adt.2006.039.
doi: 10.1002/mas.20235.
42.
Fischer, J. J., Michaelis, S., Schrey, A. K., Graebner,
56.
Pepelnjak, M., de Souza, N., and Picotti, P. (2020)
O. G., Glinski, M.,et al. (2010) Capture compound
Detecting protein-small molecule interactions Using
mass spectrometry sheds light on the molecular mech-
limited proteolysis-mass spectrometry (LiP-MS),
anisms of liver toxicity of two Parkinson drugs, Toxicol.
Trends Biochem. Sci.,
45,
919-920, doi:
10.1016/
Sci., 113, 243-253, doi: 10.1093/toxsci/kfp236.
j.tibs.2020.05.006.
43.
Assal, F., Spahr, L., Hadengue, A., Rubbia-
57.
Thompson, A., Schafer, J., Kuhn, K., Kienle, S.,
Brandt, L., and Burkhard, P. R. (1998) Tolcapone and
Schwarz, J., et al. (2003) Tandem mass tags: a novel
fulminant hepatitis, Lancet, 352, 958, doi: 10.1016/
quantification strategy for comparative analysis of
s0140-6736(05)61511-5.
complex protein mixtures by MS/MS, Anal. Chem.,
44.
Silva, T. B., Borges, F., Serrão, M. P., and Soares-
75, 1895-1904, doi: 10.1021/ac0262560.
da-Silva, P. (2020) Liver says no: the ongoing search
58.
Werner, T., Sweetman, G., Savitski, M. F.,
for safe catechol O-methyltransferase inhibitors to
Mathieson, T., Bantscheff, M., et al. (2014) Ion
replace tolcapone, Drug Discov. Today, 25, 1846-1854,
coalescence of neutron encoded TMT 10-plex reporter
doi: 10.1016/j.drudis.2020.07.015.
ions, Anal. Chem.,
86,
3594-3601, doi:
10.1021/
45.
Artusi, C. A., Sarro, L., Imbalzano, G., Fabbri,
ac500140S.
M., and Lopiano, L. (2021) Safety and efficacy of
59.
Gaetani, M., Sabatier, P., Saei, A. A., Beusch, C. M.,
tolcapone in Parkinson’s disease: systematic review,
Yang, Z., et al. (2019) Proteome integral solubility
Eur. J. Clin.Pharmacol., 77, 817-829, doi: 10.1007/
alteration: a high-throughput proteomics assay for
s00228-020-03081-x.
target deconvolution, J. Proteome Res., 18, 4027-4037,
46.
Mateus, A., Kurzawa, N., Becher, I., Sridharan, S.,
doi: 10.1021/acs.jproteome.9b00500.
Helm, D., et al. (2020) Thermal proteome profiling for
60.
Li, J., Van Vranken, J. G., Pontano Vaites, L.,
interrogating protein interactions, Mol. Syst. Biol., 16,
Schweppe, D. K., Huttlin, E. L., et al.
(2020)
e9232, doi: 10.15252/msb.20199232.
TMTproreagents: a set of isobaric labeling mass tags
47.
Tansey, W.P. (2006) Freeze-thaw lysis for extraction
enables simultaneous proteome-wide measurements
of proteins from mammalian cells, CSH Protoc., 2006,
across
16 samples, Nat. Methods,
17,
399-404,
pdb.prot4614, doi: 10.1101/pdb.prot4614.
doi: 10.1038/s41592-020-0781-4.
БИОХИМИЯ том 87 вып. 9 2022
ХИМИЧЕСКАЯ ПРОТЕОМИКА
1245
61. Dai, L., Prabhu, N., Yu, L. Y., Bacanu, S., Ramos,
67. Ivanov, M. V., Tarasova, I. A., Levitsky, L. I.,
A. D.,et al. (2019) Horizontal cell biology: monitoring
Solovyeva, E. M., Pridatchenko, M. L., et al. (2017)
global changes of protein interaction states with
MS/MS-free protein identification in complex
the proteome-wide Cellular Thermal Shift Assay
mixtures using multiple enzymes with complementary
(CETSA), Annu. Rev. Biochem.,
88,
383-408,
specificity, J. Proteome Res.,
16,
3989-3999,
doi: 10.1146/annurev-biochem-062917-012837.
doi: 10.1021/acs.jproteome.7b00365.
62. Li, J., Van Vranken, J. G., Paulo, J. A., Huttlin,
68. Teleman, J., Chawade, A., Sandin, M., Levander, F.,
E. L., and Gygi, S. P. (2020) Selection of heating
and Malmstrom, J. (2016) Dinosaur: a refined open-
temperatures improves the sensitivity of the proteome
source peptide MS feature detector, J. Proteome Res.,
integral solubility alteration assay, J. Proteome Res., 19,
15, 2143-2151, doi: 10.1021/acs.jproteome.6b00016.
2159-2166, doi: 10.1021/acs.jproteome.0c00063.
69. Zhang, B., Pirmoradian, M., Zubarev, R., and
63. Bekker-Jensen, D. B., Kelstrup, C. D., Batth, T. S.,
Käll, L. (2017) Covariation of peptide abundances
Larsen, S. C., Haldrup, C., et al. (2017) An optimized
accurately reflects protein concentration differences,
shotgun strategy for the rapid generation of
Mol. Cell Proteomics, 16, 936-948, doi: 10.1074/mcp.
comprehensive human proteomes, Cell Syst., 4, 587-
O117.067728.
599.e4, doi: 10.1016/j.cels.2017.05.009.
70. Ivanov, M. V., Bubis, J. A., Gorshkov, V., Abdrakhimov,
64. Meier, F., Geyer, P. E., Virreira Winter, S., Cox, J.,
D. A., Kjeldsen, F., et al. (2021) Boosting MS1-only
and Mann, M. (2018) BoxCar acquisition method
proteomics with machine learning allows 2000 protein
enables single-shot proteomics at a depth of 10,000
identifications in single-shot human proteome analysis
proteins in 100 minutes, Nat. Methods, 15, 440-448,
using 5 min HPLC gradient, J. Prot. Res., 20, 1864-
doi: 10.1038/s41592-018-0003-5.
1873, doi: 10.1021/acs.jproteome.0c00863.
65. Bache, N., Geyer, P. E., Bekker-Jensen, D. B.,
71. Messner, C. B., Demichev, V., Bloomfield, N., Yu,
Hoerning, O., Falkenby, L., et al. (2018) A novel
J. S. L., White, M., et al. (2021) Ultra-fast proteomics
LC system embeds analytes in pre-formed gradients
with scanning SWATH, Nat. Biotechnol., 39, 846-854,
forrapid, ultra-robust proteomics, Mol. Cell Proteomics,
doi: 10.1038/s41587-021-00860-4.
17, 2284-2296, doi: 10.1074/mcp.TIR118.000853.
72. Demichev, V., Messner, C. B., Vernardis, S. I.,
66. Meier, F., Brunner, A. D., Koch, S., Koch, H.,
Lilley, K. S., and Ralser, M.
(2020) DIA-NN:
Lubeck, M., et al. (2018) Online parallel accumulation-
neural networks and interference correction en-
serial fragmentation (PASEF) with a novel trapped ion
able deep proteome coverage in high through-
mobility mass spectrometer, Mol. Cell Proteomics, 17,
put, Nat. Methods,
17,
41-44, doi:
10.1038/
2534-2545, doi: 10.1074/mcp.TIR118.000900.
s41592-019-0638-x.
MASS SPECTROMETRY-BASED CHEMICAL PROTEOMICS
FOR DRUG TARGET DISCOVERIES
Mini-Review
I. I. Fedorov1,2, V. I. Lineva2, I. A. Tarasova1, and M. V. Gorshkov1*
1 Talrose Institute for Energy Problems of Chemical Physics,
Semenov Federal Research Center for Chemical Physics, Russian Academy of Sciences,
119334 Moscow, Russia; E-mail: mike.gorshkov@gmail.com
2 Moscow Institute of Physics and Technology (National University), 141700 Dolgoprudny, Moscow Region, Russia
Chemical proteomics, emerging rapidly in recent years, has become a main approach to identifying interactions
between the small molecules and proteins in the cells on a proteome scale and mapping the signaling and/or
metabolic pathways activated and regulated by these interactions. The methods of chemical proteomics allow
not only identifying proteins targeted by drugs, characterizing their toxicity and discovering possible off-target
proteins, but also elucidation of the fundamental mechanisms of cell functioning under conditions of drug
exposure or due to the changes in physiological state of the organism itself. Solving these problems is essential
for both basic research in biology and clinical practice, including approaches to early diagnosis of various
forms of serious diseases or prediction of the effectiveness of therapeutic treatment. At the same time, recent
developments in high-resolution mass spectrometry have provided the technology for searching the drug targets
across the whole cell proteomes. This review provides a concise description of the main objectives and problems
of mass spectrometry-based chemical proteomics, the methods and approaches to their solution, and examples
of implementation of these methods in biomedical research.
Keywords: chemical proteomics, mass-spectrometry, drug targets
БИОХИМИЯ том 87 вып. 9 2022