БИОХИМИЯ, 2023, том 88, вып. 2, с. 171 - 198
УДК 577.21:575.853
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА
ДАННЫХ СЕКВЕНИРОВАНИЯ РНК ЕДИНИЧНЫХ КЛЕТОК
Обзор
© 2023 А.А. Хозяинова1*, А.А. Валяева2, М.С. Арбатский2,
С.В. Исаев3,4, П.С. Ямщиков1,5, Е.В. Волчков6, М.C. Сабиров7, В.Р. Зайнуллина1,
В.И. Чечехин2, Р.С. Воробьёв1, М.Е. Меняйло1, П.А. Тюрин-Кузьмин2, Е.В. Денисов1
1 Научно-исследовательский институт онкологии,
Томский национальный исследовательский медицинский центр Российской академии наук,
634050 Томск, Россия; электронная почта: khozyainova@onco.tnimc.ru
2 Московский государственный университет имени М.В. Ломоносова,
119991 Москва, Россия
3 Институт персонализированной медицины,
Национальный центр персонализированной медицины эндокринных заболеваний,
Национальный медицинский исследовательский центр эндокринологии,
117036 Москва, Россия
4 Московский физико-технический институт, физтех-школа биологической и медицинской физики,
115184 Долгопрудный, Россия
5 Национальный исследовательский Томский государственный университет,
634050 Томск, Россия
6 ФГБУ «НМИЦ ДГОИ им. Дмитрия Рогачева» Минздрава России,
117198 Москва, Россия
7 ФГБУН Институт биологии развития им. Н.К. Кольцова РАН, 119334 Москва, Россия
Поступила в редакцию 23.09.2022
После доработки 13.12.2022
Принята к публикации 13.12.2022
Секвенирование РНК единичных (отдельных/индивидуальных) клеток (single-cell RNA-sequencing,
scRNA-seq) является революционным инструментом для изучения физиологии нормальных и па-
тологически изменённых тканей. Данный подход предоставляет информацию о молекулярных осо-
бенностях (генной экспрессии, мутациях, степени открытости хроматина и др.) клеток, открывает
возможность для анализа траекторий клеточной дифференцировки/филогении и межклеточных взаи-
модействий и позволяет обнаруживать новые типы клеток и ранее неизученные процессы. В кли-
ническом аспекте scRNA-seq позволяет проводить более глубокий и детальный анализ молекулярных
механизмов развития различных заболеваний и предоставляет основу для разработки новых профи-
лактических, диагностических и терапевтических решений. В данном обзоре описываются различ-
ные подходы к анализу данных scRNA-seq, рассматриваются сильные стороны и недостатки биоин-
форматических инструментов, приводятся рекомендации и примеры их успешного использования и
предлагаются потенциальные направления в области их совершенствования. Также подчёркивается
необходимость создания новых, в том числе мультиомиксных, протоколов для подготовки библиотек
единичных клеток с целью получения более полного и системного представления о каждой клетке.
КЛЮЧЕВЫЕ СЛОВА: секвенирование РНК единичных клеток, клеточный цикл, кластеризация, диффе-
ренциальная экспрессия, клеточные типы, траектории развития, межклеточная коммуникация, генные
регуляторные сети, вариации числа копий ДНК, однонуклеотидные замены, филогенетика, эпигеномика,
пространственная транскриптомика.
DOI: 10.31857/S032097252302001X, EDN: QFSJMW
Принятые сокращения: ДЭГ - дифференциально экспрессирующиеся гены; aCGH - микроматричная
сравнительная геномная гибридизация; bulk RNA-seq - секвенирование тотальной РНК; CNV - вариа-
ции числа копий ДНК; MLPA - амплификация лигированных зондов; SNV - однонуклеотидные замены;
scRNA-seq - секвенирование РНК единичных клеток; WGCNA - анализ взвешенных сетей коэкспрессии
генов; WGS - полногеномное секвенирование.
* Адресат для корреспонденции.
171
172
ХОЗЯИНОВА и др.
ВВЕДЕНИЕ
доступны различные платформы для выпол-
нения scRNA-seq, среди которых Fluidigm C1/
Секвенирование РНК единичных клеток
Smart-seq, BD Rhapsody
(«BD Biosciences»,
(single-cell RNA-sequencing, scRNA-seq) стало
США), Chromium («10x Genomics», США) и
поистине революционным методом, позво-
другие, которые обеспечивают высокую про-
лившим в значительной степени расширить
изводительность данного типа анализа [3, 4].
понимание гетерогенности и динамики тран-
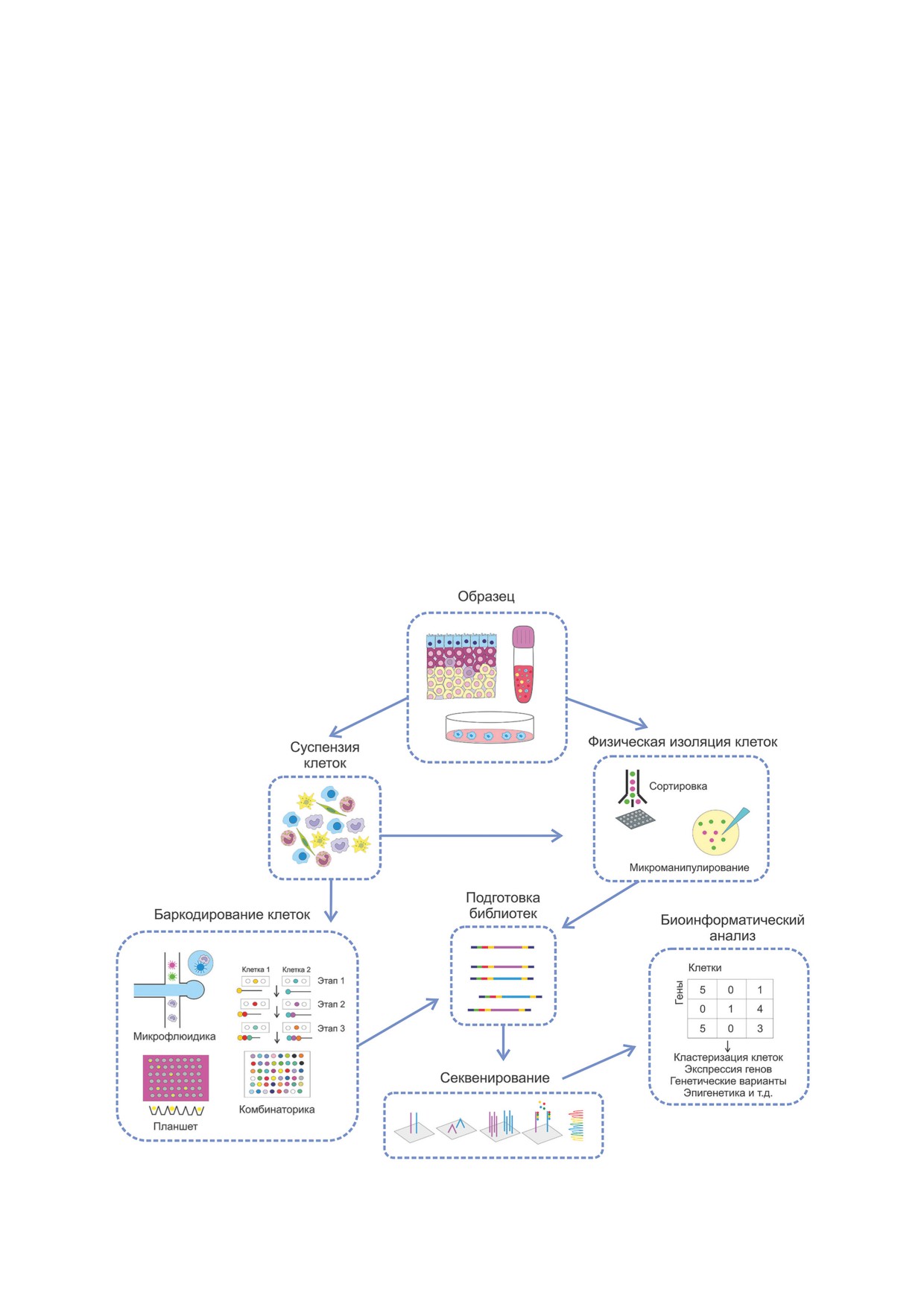
Процесс scRNA-seq схематически пред-
скриптома клеток в различных биологических
ставлен на рис. 1. Посредством гомогенизации
объектах. Впервые данный метод был приме-
из исследуемого образца получают суспензию
нён в 2009 г. для изучения бластомеров мыши
клеток, которые далее разделяются либо фи-
на стадии второго деления [1]. Именно тогда
зически, например, с помощью сортировки и
было показано, что секвенирование единич-
микроманипуляции, либо посредством барко-
ных клеток существенно превосходит техноло-
дирования с использованием олигонуклеоти-
гию микрочипов для количественного анализа
дов в составе планшетов или на основе микро-
экспрессии генов. Однако главным ограниче-
флюидики и комбинаторики [5, 6]. Образцы
нием того времени являлась невозможность
крови и клеточных культур подвергают сорти-
мультиплексирования образцов, и библиоте-
ровке и микроманипуляции без подготовки сус-
ка каждой клетки создавалась вручную в от-
пензии. Полученные клетки используют для
дельной пробирке. Однако уже в 2011 г. был
подготовки библиотек и последующего секве-
разработан первый протокол мультиплексно-
нирования, данные которого обрабатываются
го scRNA-seq [2], а в 2014 г. - первая ком-
биоинформатически.
мерчески доступная платформа автомати-
Развитие технологий в области scRNA-seq
ческой подготовки библиотек единичных
позволило охарактеризовать основные клеточ-
клеток Fluidigm C1 [2]. На настоящий момент
ные и молекулярные механизмы, вовлечённые
Рис. 1. Процесс scRNA-seq
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
173
в развитие сердечно-сосудистых [7], нейро-
цов, анализе дифференциальной экспрессии,
дегенеративных [8, 9], онкологических [10] и
оценке клеточного цикла и идентификации
других заболеваний, определить клеточную
клеточных типов [1, 16]. Хотя уже тогда было
таксономию широко используемых модельных
ясно, что возможности применения scRNA-seq
организмов, таких как резуховидка Таля [11],
намного шире, и создание биоинформати-
дрозофила обыкновенная [12] и данио-рерио
ческих методов, способных интегрировать и
[13], и расшифровать гетерогенность клеток
преобразовывать полученные нуклеотидные
иммунной системы в нормальном и патологи-
прочтения в информацию о различных состо-
ческих состояниях, включая онкологические
яниях клетки в мультимодальном измерении,
заболевания [14, 15].
было лишь вопросом времени.
В начале развития эры секвенирования
В данном обзоре мы описываем прогресс
единичных клеток исследователи в большей
в области обработки данных scRNA-seq и свя-
степени акцентировали внимание на изучении
занные с ним методы анализа для получения
популяционного состава исследуемых образ-
информации о клеточном цикле, клеточных
Рис. 2. Подходы биоинформатического анализа данных scRNA-seq
БИОХИМИЯ том 88 вып. 2 2023
174
ХОЗЯИНОВА и др.
кластерах и типах клеток, дифференциально
дифференциальной экспрессии. Однако в не-
экспрессирующихся генах (ДЭГ), траектори-
которых случаях, например, при сравнении
ях развития и скорости РНК (Cell trajectory
субпопуляций делящихся и неделящихся кле-
inference and RNA Velocity), межклеточных взаи-
ток, информация о стадии клеточного цикла
модействиях, генетических вариантах, вклю-
может оказаться важной, и данный конфаун-
чая вариации числа копий ДНК (CNV) и одно-
дер удалять не стоит.
нуклеотидные замены (SNV), филогении кле-
Популярные программные пакеты для ана-
ток, доступности хроматина, сайтах связы-
лиза данных scRNA-seq, такие как Seurat [18]
вания транскрипционных факторов и про-
и Scanpy [19], предлагают считать два параме-
странственной транскриптомике (рис. 2). Для
тра [20], основанных на усреднённой экспрес-
каждого из мультимодальных приложений
сии известных генов-маркеров стадий клеточ-
подчёркиваются сильные стороны, недостат-
ного цикла S и G2/M [21]. В зависимости от
ки, возможные пути их преодоления и биоло-
рассчитанных значений параметра клетка мо-
гические примеры использования.
жет быть проаннотирована как находящаяся
на стадии G2/M, G1 или S клеточного цикла.
Удаление эффекта, связанного с клеточным
ОЦЕНКА КЛЕТОЧНОГО ЦИКЛА
циклом, происходит с помощью линейной ре-
грессии, в которой учитываются рассчитан-
Секвенирование РНК единичных клеток,
ные параметры. Если для последующего ана-
в отличие от секвенирования тотальной РНК
лиза необходимо сохранить разделение между
(bulk RNA-seq), позволяет получить инфор-
субпопуляциями делящихся и покоящихся
мацию о транскрипционном профиле каждой
клеток и одновременно избавиться от разли-
индивидуальной клетки в исследуемом образ-
чий в стадиях клеточного цикла, то рекомен-
це. С помощью scRNA-seq возможно исследо-
дуется в линейной регрессии использовать не
вание клеточного многообразия тканей, обна-
G2/M- и S-параметры, а разницу между ними.
ружение ранее неизвестных популяций клеток
Ещё один метод Cyclone [20] также исполь-
и изучение биологических процессов на уров-
зует известные гены-маркеры стадий клеточ-
не единичных клеток. Однако увеличению
ного цикла для оценки и удаления эффектов,
разрешения метода сопутствует и повышение
связанных с клеточным делением. Однако этот
доли технического и биологического шума в
алгоритм построен на сравнении экспрессии
данных. При этом одним из главных источни-
пар маркерных генов, поскольку их отноше-
ков биологического шума в данных scRNA-seq
ние позволяет определить стадию клеточного
оказывается клеточный цикл [17]. При анали-
цикла, в которой клетка находится.
зе данных scRNA-seq стадия клеточного цик-
Другие методы позволяют проводить бо-
ла часто рассматривается как конфаундер, то
лее подробный анализ клеточного цикла на
есть та переменная, которая может исказить
основе данных scRNA-seq и реконструировать
биологический эффект, искомый исследова-
продвижение индивидуальных клеток по кле-
телем, будь то различия между клеточными
точному циклу. Некоторые из них, такие как
типами или изменения транскрипционных
peco [22] и reCAT [23], как и вышеперечислен-
программ при заболеваниях или в процессе
ные методы, используют наборы известных
лечения. Клетки исследуемого образца могут
генов, ассоциированных с клеточным циклом.
находиться в различных временных точках и,
Другие алгоритмы (Cyclum [24], CYCLOPS [25]
соответственно, иметь различные профили
и CCPE [26]) основаны на обучении без учите-
экспрессии, даже если они являются клетками
ля (unsupervised learning/самообучение) и для
одного типа. Поэтому на этапе подготовки ма-
расчёта
«псевдовремени» клеточного цикла
трицы генной экспрессии для последующего
учитывают цикличность в экспрессии генов.
анализа возможно удаление из данных диспер-
При этом с помощью этих алгоритмов можно
сии, связанной с клеточным циклом. Эта про-
экстрагировать информацию о генах, наибо-
цедура представляется особо целесообразной,
лее ассоциированных с каждой из стадий кле-
когда исследователь не ожидает увидеть в сво-
точного цикла.
их данных активно пролиферирующие клет-
ки, например, при изучении апоптоза. Также
удаление генов клеточного цикла или проли-
ОПРЕДЕЛЕНИЕ КЛЕТОЧНЫХ КЛАСТЕРОВ
ферирующих клеток целесообразно в случае,
если большая часть наиболее вариабельных
Типичный анализ данных экспрессии ге-
генов представлена генами клеточного цикла,
нов в единичных клетках, полученных с по-
что отрицательно сказывается на определении
мощью scRNA-seq, начинается с определения
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
175
клеточного состава исследуемого образца.
Определение клеточных, или транскрип-
На этом этапе происходит объединение ин-
ционных кластеров в данных scRNA-seq, осу-
дивидуальных клеток в транскрипционные
ществляемое с помощью алгоритмов класте-
кластеры на основе сходства их профилей
ризации, - это одна из популярных задач
экспрессии и определение клеточного типа
обучения без учителя. Цель кластеризации за-
каждого из кластеров по уровням экспрессии
ключается в объединении похожих по своим
специфичных и дифференциально-экспресси-
транскрипционным профилям клеток в груп-
рующихся маркерных генов. Для решения этих
пы, которые затем можно охарактеризовать как
задач используются алгоритмы кластеризации
кластеры одного клеточного типа, одной стадии
данных и методы анализа дифференциальной
дифференцировки или клеточного цикла. Сто-
экспрессии генов. Однако эти алгоритмы и ме-
ит понимать, что кластеры - это математически
тоды применимы не только в контексте пер-
определяемые группы клеток, которые действи-
воначального анализа данных scRNA-seq, их
тельно могут состоять из клеток одного типа,
можно использовать и как самостоятельные
однако на практике биология зачастую имеет
инструменты для решения конкретных биоло-
малое отношение к алгоритмам кластеризации.
гических задач.
На настоящий момент существует множе-
Анализ ДЭГ применяется для исследова-
ство применимых для кластеризации scRNA-
ния влияния различных патологических или
seq методов, каждый из которых обладает
экспериментальных условий на транскрипци-
своими преимуществами и недостатками. Кон-
онные профили изучаемых популяций клеток.
сенсуса о наиболее эффективном методе до сих
С помощью такого подхода были определе-
пор нет [33]. Методы кластеризации данных
ны гены и ассоциированные с ними клеточ-
scRNA-seq основываются на широко применяе-
ные процессы, активируемые и подавляемые
мых алгоритмах, таких как иерархическая кла-
в разных типах клеток при COVID-19 [27, 28],
стеризация, метод k-средних и кластеризация
болезни Альцгеймера [29], аутизме [30] и мно-
графов. На кластеризацию клеток оказывает
гих других заболеваниях. Кроме того, диффе-
влияние не только выбор метода и его параме-
ренциальный анализ находит применение в
тров, но и увеличение количества измеряемых
идентификации генов, чья экспрессия меняет-
данных. При экспоненциальном возрастании
ся при клеточной дифференцировке или дру-
их количества происходит увеличение размер-
гом динамическом процессе, затрагивающем
ности, что сопровождается возникновением
фенотипы популяций клеток. Также данный
«проклятья размерности». Многомерность дан-
метод может применяться для отбора генов,
ных приводит к сближению индивидуальных
экспрессия которых необходима для построе-
клеток, что вызывает проблемы в определении
ния клеточных траекторий.
расстояния между клетками. Наиболее удалён-
Применение алгоритмов кластеризации
ные (различающиеся по экспрессии множества
также не ограничивается начальным этапом
генов) клетки в многомерных данных находятся
анализа данных scRNA-seq. Кластеризация
на таком же расстоянии, что и наиболее близ-
клеток как элементов набора данных может
кие (похожие по профилю РНК). Из-за этой
быть проведена несколько раз последовательно
проблемы стандартные методы кластеризации
при биоинформатическом анализе, например,
не могут разделить отличные по паттерну РНК
с целью поиска редких популяций клеток [31].
клетки. Для многомерных данных scRNA-seq
При этом повторная кластеризация клеток мо-
для снижения размерности и ускорения вы-
жет проводиться не по всему изначальному на-
числений проводят отбор значимых генов и
бору экспрессионных профилей, а, например,
применяют метод главных компонент (principal
по генной подписи - определённому набору ге-
component analysis, PCA). В качестве значимых
нов, по которым можно идентифицировать ин-
генов (признаков) могут быть выбраны высоко-
тересующий тип клеток - или по любому дру-
вариабельные гены (highly variable genes, HVG),
гому набору признаков, описывающих клетку.
чья экспрессионная изменчивость объясняет-
Аннотация клеток проводится не только для
ся преимущественно биологическими разли-
определения их типов, но и для других задач.
чиями между анализируемыми клетками, а не
Например, кластеры клеток можно аннотиро-
техническим шумом [34]. Также существуют
вать по вирусной нагрузке и их состоянию [32].
методы для моделирования технического шума
Текущий и два последующих раздела на-
и отбора значимых генов, например, на ос-
стоящего обзора рассказывают о каждой биоин-
нове построенной модели M3Drop [35]. Для
форматической задаче в отдельности: определе-
кластеризации данных затем используются
нии кластеров, поиске ДЭГ и идентификации
расстояния между клетками в пространстве
клеточных типов.
пониженной размерности.
БИОХИМИЯ том 88 вып. 2 2023
176
ХОЗЯИНОВА и др.
Алгоритм иерархической кластеризации
ток, существование которых впоследствии
подразумевает под собой объединение клеток
подтверждается дополнительными экспери-
в растущие кластеры (восходящий, или агло-
ментами, например, иммуноокрашиванием
меративный подход) или разбиение кластеров
[40, 41]. При этом для поиска редких и мало-
на подгруппы (нисходящий, или дивизионный
численных субпопуляций клеток возможно
подход) на основании матрицы расстояний
использование более сложных подходов для
между клетками. Программы, использующие
нормализации данных [41] и ре-кластериза-
этот алгоритм для идентификации клеточных
ции отобранных, одного или нескольких наи-
субпопуляций, например, pcaReduce [36] или
более интересных, кластеров [31]. Избавиться
CIDR [37], позволяют определять взаимоотно-
от биологического шума в данных scRNA-seq,
шения между кластерами с помощью дендро-
вызванного контаминацией исследуемой тка-
граммы, но по сравнению с другими методами
ни, возможно с помощью удаления кластера
кластеризации работают медленнее, что может
нецелевых клеток с последующей ре-кластери-
быть критично для больших объёмов данных
зацией данных [40].
scRNA-seq.
Метод кластеризации данных scRNA-seq
SC3 [38] использует в своей основе алгоритм
АНАЛИЗ
k-средних, идея которого заключается в ите-
ДИФФЕРЕНЦИАЛЬНОЙ ЭКСПРЕССИИ
ративном определении центров масс (центрои-
дов) заданного числа кластеров и уточнении
Анализ дифференциальной экспрессии
границ кластеров. Отличительной особенностью
генов позволяет установить различия между
методов, основанных на алгоритме k-средних,
кластерами и определить клеточные типы и
является получение кластеров примерно одного
их маркеры. Дифференциальная экспрессия
размера. В такой ситуации большие субпопу-
позволяет изучить транскрипционную дина-
ляции клеток будут разбиваться на несколько
мику в процессе дифференцировки клеток,
кластеров, а редкие клетки будут объединены
развития заболеваний или при воздействии
с другими кластерами. Кроме того, результат
каких-либо экзогенных и/или эндогенных
кластеризации с помощью k-средних во мно-
факторов.
гом зависит от случайной инициализации цен-
Несмотря на то что задачи поиска ДЭГ на
троидов и не обязательно представляет собой
основе данных bulk RNA-seq и scRNA-seq фор-
глобальный минимум.
мулируются схожим образом, методы их реше-
В случае больших данных scRNA-seq для
ния различны. Методы анализа дифференци-
более оптимального решения проблемы опреде-
альной экспрессии по данным bulk RNA-seq,
ления клеточных кластеров предлагают мето-
такие как DESeq2 [42] и edgeR [43], учитывают
ды кластеризации, основанные на поиске кла-
сложность в определении дисперсии экспрес-
стеров в графах - лувенский метод (Louvain
сии генов на основании малого количества
method) и алгоритм Лейдена (Leiden method).
повторностей - биологически различных об-
Данные методы предварительно строят граф
разцов из одной экспериментальной группы.
k ближайших соседей (kNN-граф), используя
Такой проблемы в scRNA-seq не существует,
проекцию данных в пространство пониженной
так как каждую клетку можно рассматривать
размерности, и затем определяют кластеры кле-
как повторность. Однако увеличение коли-
ток как группы наиболее плотно связанных друг
чества измерений (клеток) может привести к
с другом вершин графа. Количество определяе-
ложноположительным результатам. Например,
мых кластеров напрямую не задаётся, но на него
в одном из кластеров экспрессия гена может
влияет значение параметра разрешения, а также
быть повышена статистически значимо, но
указанное число k ближайших соседей, исполь-
всего в 1,1 раза. Если уменьшить количество
зуемое для построения графа. Графовый подход
измерений, статистическая значимость мо-
для идентификации субпопуляций реализован
жет быть полностью нивелирована. В связи с
в программе PhenoGraph [39] и программных
этим следует находить ДЭГ, повышенные до
пакетах Seurat [18] и Scanpy [19]. Более подроб-
определённого уровня, причём для каждых
но преимущества и недостатки каждой из групп
конкретных данных значение этого порога
методов описаны Kiselev et al. [33].
будет различно. Тем не менее, вышеуказан-
С помощью кластеризации данных scRNA-
ные методы оказываются применимыми и для
seq можно обнаружить уже известные типы
анализа данных scRNA-seq. Так, для анализа
или состояния клеток с характерной высокой
ДЭГ с помощью DESeq2 и edgeR из данных
экспрессией ряда генов-маркеров или опре-
scRNA-seq используют процедуру взвешива-
делить ранее не описанные популяции кле-
ния значений экспрессии для каждой клетки
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
177
и каждого гена, которая позволяет преодолеть
рого используется та же самая информация об
проблему высокой разреженности данных
экспрессии генов в индивидуальных клетках.
scRNA-seq ввиду чрезвычайно высокой доли
По этой причине анализ дифференциальной
генов с нулевой экспрессией в каждой клетке.
экспрессии, следующий за кластеризацией кле-
Ранее методы, специально разработанные для
ток, приводит к искусственно заниженным
анализа scRNA-seq, решали эту проблему за
значениям статистической значимости (p-зна-
счёт использования отрицательной биноми-
чение). Данная проблема решается с помощью
альной модели с избытком нулей (zero-inflated
теста TN (truncated normal), который учитывает
negative binomial, ZINB) [44]. Но сегодня для
уровни экспрессии генов, определяющие гра-
scRNA-seq рекомендуется использовать отри-
ницы между клеточными кластерами [49].
цательную биномиальную модель без избыт-
Идея о том, что каждая индивидуальная
ка нулей (negative binomial, NB) [45]. Кроме
клетка представляет из себя независимый об-
того, при сравнении эффективности методов
разец, в действительности является серьёзным
анализа дифференциальной экспрессии было
допущением, поскольку все клетки в образце
показано, что при простом дизайне экспери-
обычно объединены общим происхождением
мента (сравнение контроля и «эксперимента»
от одного или нескольких доноров. Поэтому
в отсутствии других переменных) лучшие ре-
при сравнении субпопуляций клеток, полу-
зультаты показывает непараметрический тест
ченных из одного организма, происходит оцен-
Манна-Уитни [46].
ка вариабельности транскрипционных профи-
Однако вышеперечисленные методы не
лей не в популяции, а только внутри одного
учитывают ряд особенностей, характерных
индивидуума. Если в наборе данных присут-
для данных scRNA-seq. Так, распределение
ствует выборка клеток, полученных от не-
экспрессии генов в единичных клетках отли-
скольких доноров, то для анализа дифферен-
чается бимодальностью. Значения экспрессии
циальной экспрессии можно использовать
либо положительны в тех клетках, где соот-
подход с подсчётом агрегированной (псевдо-
ветствующий транскрипт был детектирован,
балк, pseudobulk) экспрессии или обобщён-
либо равны нулю в клетках, где по некоторой
ную линейную смешанную модель, в кото-
биологической или технической причине экс-
рой донор указан как случайный фактор [50].
прессия не была детектирована. Отсутствие
Агрегированная экспрессия рассчитывается
экспрессии может объясняться её стохасти-
путём суммирования или усреднения экспрес-
ческой природой (биологическая причина)
сии гена среди клеток каждого из доноров, в
или потерей транскрипта на этапе подготов-
результате чего получается несколько псевдо-
ки кДНК-библиотек (техническая причина).
повторностей, соответствующих независимым
Данная особенность транскриптомики еди-
донорам. Таким образом, с помощью данного
ничных клеток принята во внимание в таких
подхода задача анализа ДЭГ на основе дан-
методах, как SCDE [47] и MAST [48]. Метод
ных scRNA-seq сводится к задаче, которая уже
SCDE использует комбинацию отрицательно-
успешно решается для bulk RNA-seq.
го биномиального распределения для положи-
Однако результаты анализа дифферен-
тельных значений экспрессии и Пуассонов-
циальной экспрессии, полученные с исполь-
ского для «нулевых» генов, в случае которых
зованием bulk RNA-seq и scRNA-seq, могут
может присутствовать также фоновый сигнал.
значительно различаться. В случае scRNA-seq
Метод SCDE возможно использовать только
дифференциальная экспрессия детектируется
для поиска ДЭГ между двумя группами кле-
преимущественно среди генов с высоким сред-
ток (контроль и «эксперимент»). Использо-
ним уровнем экспрессии, но средними значе-
вание в SCDE других переменных, таких как
ниями разницы в экспрессии между сравни-
номер группы образцов (batch-переменная)
ваемыми группами (fold change) [51]. В связи с
или временная точка, невозможно, что огра-
этим с помощью scRNA-seq-подходов оказы-
ничивает применимость данного метода толь-
вается труднее идентифицировать гены с силь-
ко для экспериментов с простым дизайном.
ным изменением уровня экспрессии в ответ на
Метод MAST учитывает более сложные дизай-
изучаемое воздействие.
ны экспериментов, например, с несколькими
Из-за особенностей протоколов подготов-
вариантами «воздействия», и использует мо-
ки scRNA-seq-библиотек, а именно исполь-
дель преодоления препятствий (hurdle model)
зования олиго(dT)-праймеров в подавляю-
для описания экспрессии генов в единичных
щем большинстве методов, в транскриптомах
клетках.
единичных клеток детектируются преимуще-
Поиск ДЭГ между субпопуляциями клеток
ственно полиаденилированные РНК - мРНК
зависит от этапа кластеризации, в ходе кото-
и некоторые длинные некодирующие РНК
БИОХИМИЯ том 88 вып. 2 2023
178
ХОЗЯИНОВА и др.
(днРНК). Для анализа экспрессии других ти-
Автоматическое типирование. Автоматиче-
пов РНК на уровне единичных клеток, на-
ское типирование осуществляется за счёт срав-
пример, микроРНК (миРНК), используются
нения клеток изучаемого образца с известными
специальные методы пробоподготовки образ-
маркерными генами, информация о которых
цов [52]. Тем не менее, судить об активности
представлена в различных базах, содержащих
микроРНК в индивидуальных клетках воз-
данные микрочипов, bulk RNA-seq или scRNA-
можно, используя в качестве такой оценки
seq для клеток определённого типа. В случае
уровень экспрессии предшественников ми-
совпадения профилей экспрессии программа
кроРНК, которые могут полиаденилироваться
автоматически определяет тип клетки. По та-
и кэпироваться, тем самым обеспечивая своё
кому принципу работает R-пакет SingleR [61],
попадание в секвенируемый транскриптом.
в состав которого входит пакет celldex, содер-
С помощью анализа дифференциальной
жащий доступ к семи клеточным базам. Для
экспрессии с использованием scRNA-seq на
типирования клеток в автоматическом режи-
клеточном уровне были определены причины
ме также возможно использовать инструмен-
нарушения регенерации эпителия альвеол лёг-
ты ScType [62], scCATCH [63], scSorter [64] и
ких при COVID-19 [27, 53] и охарактеризованы
SCINA [65]. В своей работе автоматические
особенности цитокинового шторма и иммун-
аннотаторы могут использовать ранее прове-
ного ответа на вирусную инфекцию, который
дённую кластеризацию или проводить её пере-
обеспечивается разными типами иммунных
расчёт в соответствии с клеточными типами,
клеток [54]. Выявлены маркеры, вовлечённые
которые были обнаружены в образце. Важно
в лекарственную резистентность и прогресси-
отметить, что автоматические аннотаторы спо-
рование саркомы Юинга [55]. Оценена функ-
собны распознавать ограниченное число кле-
циональная гетерогенность мультипотент-
точных типов ввиду отсутствия данных об
ных стромальных клеток человека и мыши по
экспрессионных профилях множества типов
характеру экспрессии аденилатциклаз
[56].
клеток в используемых базах.
Изменения экспрессии генов в процессе кло-
Ещё одним способом аннотировать клет-
нальной экспансии и возможного сопутствую-
ки в автоматическом режиме является ис-
щего истощения T-клеток при противоопухо-
пользование аннотированных образцов дру-
левой иммунотерапии также были изучены на
гих исследовательских групп. В биоинфор-
уровне единичных клеток с помощью методов
матике эта методика называется label trans-
дифференциальной экспрессии [57]. Эти же
ferring [66]. Суть этого метода состоит в том,
методы используются для поиска генных сиг-
что в исследуемом образце сначала находят-
натур, то есть наборов предиктивных марке-
ся клетки, совпадающие по паттерну экс-
ров, которые могли бы предсказывать ответ на
прессии с образцом-эталоном. После того
иммунотерапию с использованием ингибито-
как найдены якорные клетки (совпадающие
ров контрольных точек иммунного ответа [58].
между образцами), на определяемый обра-
зец переносится информация о типе клетки с
образца-эталона. По такому принципу работает
ИДЕНТИФИКАЦИЯ
веб-сервис Azimuth [67]. На сегодняшний день
КЛЕТОЧНЫХ ТИПОВ
в веб-сервисе доступны 11 наборов эталонных
данных.
Общепринято, что идентификация типов
Существует и промежуточный вариант ав-
клеток выполняется стандартными гистоло-
томатического типирования с созданием соб-
гическими методами, из которых основным
ственной библиотеки аннотированных образ-
является иммуноокрашивание
- связыва-
цов именно тех клеток, с которыми работает
ние антител с белковыми маркерами клеток и
исследователь. Этот подход позволяет само-
последующая визуализация. Альтернативой
стоятельно отобрать самые лучшие открытые
может быть РНК-секвенирование и последую-
данные и иметь чёткое представление о дизай-
щее биоинформатическое типирование, ког-
не эксперимента, в котором эти образцы были
да маркеры клеток детектируются на уровне
получены.
транскриптов. Однако хорошо известно, что
Ручное типирование. Необходимость ручно-
наличие мРНК в клетке не всегда коррелирует
го типирования прежде всего обусловлена на-
с продукцией соответствующего белка ввиду
хождением большей части клеток исследуемого
обилия посттранскрипционных и посттранс-
образца в промежуточных, недифференциро-
ляционных механизмов регуляции
[59,
60].
ванных формах. Такие клетки, как правило,
Типирование клеток, основанное на scRNA-
не имеют специфических маркеров, харак-
seq, может быть автоматическим или ручным.
терных для их дифференцированных форм,
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
179
и не могут быть проаннотированы системами
Типирование по переходным генам, в от-
автоматического типирования. Кроме того, в
личие от первых двух способов ручного типи-
реальной практике классических генов-мар-
рования, помимо белок-кодирующих тран-
керов может быть недостаточно для иденти-
скриптов, учитывает несплайсированные формы
фикации и дифференцированных форм. В та-
будущих мРНК. Соотношение сплайсирован-
ких случаях тип клеток может быть определён
ных и несплайсированных форм мРНК по-
вручную, на основании менее известных или
зволяет оценить, в каком состоянии находит-
заданных пользователем маркерных генов
ся экспрессия того или иного белка на момент
[68]. Также, по нашему мнению, типирова-
исследования - индуцированном или репрес-
ние клеток может быть основано на анализе
сированном, и выделить те гены, которые яв-
их вовлечённости в различные биологические
ляются ключевыми для развития клетки на
процессы, переходных генов или положения
момент анализа, например, с помощью пакета
клеток исследуемого образца относительно
scVelo [71]. Среди данных генов вручную осу-
траектории развития.
ществляется поиск ответственных за переход
Типирование по менее известным или за-
клетки в дифференцированную форму. Таким
данным пользователем специфическим мар-
образом, исследователь может предположить,
керам в большей мере подходит для опреде-
предшественником какого типа является ис-
ления типа дифференцированных клеток и
следуемая группа клеток.
осуществляется за счёт изучения списка генов
Ручное типирование клеток также мо-
каждого кластера, полученного после этапа
жет осуществляться на основании результатов
кластеризации. Исследователь визуально оце-
вывода траектории развития. В большинстве
нивает список высокопредставленных генов
случаев при выводе данной модальности кла-
на предмет наличия определённых маркеров и
стеры с отсутствием специфических маркеров
при условии их наличия аннотирует клетки к
находятся между кластерами с наличием тако-
известному типу. Ещё одной возможной реа-
вых. В таком случае можно предположить, что
лизацией данного подхода является присвое-
данный кластер является промежуточным и со-
ние кластерам клеточного типа на основании
держит клетки в переходном состоянии между
заданных пользователем маркеров в Seurat и
исходной и конечной формами.
Scanpy. Маркерные гены могут быть выбраны
пользователем на основании литературных
данных. Так, использование заранее заданной
ТРАЕКТОРИИ РАЗВИТИЯ
панели генов позволило выявить типы клеток
И СКОРОСТЬ РНК
при сравнительном анализе идентичных ре-
гионов мозга высших приматов [69]. Данный
Любой вид секвенирования является сним-
вид типирования использовался и при иден-
ком момента жизни клетки, предоставляющим
тификации субпопуляций клеток фоллику-
информацию об интересующей модальности
лярной лимфомы, которые выявляются при
на момент проведения исследования. Библио-
прогрессировании и рецидивировании забо-
тека scRNA-seq содержит информацию о тран-
левания [70].
скрипционном профиле нескольких сотен и ты-
Типирование по биологическим про-
сяч клеток, гетерогенность которых в том числе
цессам основано на выявлении групп генов,
обусловлена динамическим процессом кле-
участвующих в определённых биохимических
точного развития. Методы вывода траекторий
процессах, специфичных для некоторых кле-
развития, также называемые анализом псевдо-
ток в контексте индуцирующего воздействия.
времени, позволяют упорядочивать клетки ис-
По списку ДЭГ можно определить биологи-
следуемого образца вдоль смоделированной
ческие процессы, которые активны в дан-
временной траектории на основе сходства их
ном кластере клеток. Для этого очень удобно
паттернов экспрессии. Результатом построения
использовать веб-сервис g:Profiler, который
траектории развития в псевдовремени является
объединяет информацию о ДЭГ кластера и
графическое изображение всех клеток образ-
определяет все биологические процессы, сиг-
ца, расположенных друг за другом, начиная с
нальные пути и клеточные компоненты, за ко-
начальной/исходной клетки (root cell) и до ко-
торые ответственны белковые продукты этих
нечной или дифференцированной клетки (end
генов. С помощью данного подхода становит-
cell). С помощью вывода траекторий развития
ся возможным типирование клеток, находя-
становится возможным изучение интересую-
щихся в процессе дифференцировки на осно-
щего биологического явления, например, путей
вании детекции маркеров, ассоциированных
дифференцировки, клеточного цикла или им-
с изменением клеточного фенотипа.
мунных реакций, в динамическом контексте.
БИОХИМИЯ том 88 вып. 2 2023
180
ХОЗЯИНОВА и др.
Впервые для построения траекторий
дой клетки в пространстве со сниженной раз-
развития был предложен R-пакет Monocle.
мерностью, так формируется векторное поле,
Monocle сначала использует тест дифферен-
в котором можно видеть направление развития
циальной экспрессии для уменьшения коли-
клеток в образце. Учитывая то, что векторное
чества генов, а затем применяет анализ неза-
поле накладывается на заранее полученные
висимых компонентов для дополнительного
кластеры клеток, можно предполагать направ-
уменьшения размерности. Для построения
ление дифференцировки или восприятия клет-
траектории Monocle вычисляет минимальное
ками фактора воздействия.
остовное дерево, а затем находит самый длин-
ный соединённый путь в этом дереве. Ячейки
проецируются на ближайшую к ним точку на
МЕЖКЛЕТОЧНАЯ КОММУНИКАЦИЯ
этом пути [72]. После Monocle было предложе-
но ещё более 50 различных методов, самыми
Развитие, функционирование, регенера-
известными из которых стали TSCAN [73] и
ция и гомеостаз тканей и органов обеспечива-
Slingshot [74]. Методы отличаются друг от дру-
ются путём межклеточной коммуникации, или
га по многим параметрам: указание начальных
межклеточного сигналинга - процесса, проис-
и конечных клеток; тип визуализации графа
ходящего за счёт лиганд-рецепторного взаимо-
(прямой, линейное псевдовремя, цикличес-
действия различных клеток. В роли лигандов
кое псевдовремя, вероятность конечного со-
могут выступать цитокины, хемокины, гормо-
стояния, кластерная оценка, ортогональная
ны, факторы роста и нейромедиаторы.
проекция и клеточный граф); тип траекто-
Межклеточный сигналинг принято делить
рии (несвязный и связный граф, цикличный
на аутокринный (выделяемый клеткой лиганд
и ацикличный граф, древовидный граф) [75].
взаимодействует с рецептором той же клетки),
На сегодняшний день методов стало настоль-
паракринный (выделяемый клеткой лиганд
ко много, что появилась необходимость созда-
взаимодействует с рецепторами клеток из той
ния единой платформы, где можно анализиро-
же ткани) и эндокринный (выделяемый клет-
вать свои данные с помощью сразу нескольких
кой лиганд взаимодействует с рецепторами
методов. Одной из таких платформ является
клеток из других тканей или органов). Отдель-
dynverse, объединившая в себе 45 методов по-
но можно выделить межклеточные взаимодей-
строения траекторий развития.
ствия, то есть физический контакт двух клеток
Чтобы улучшить качество выводимых тра-
друг с другом. Межклеточные взаимодействия
екторий, в некоторых методах вместо подсчё-
могут быть как участниками межклеточной
та экспрессии генов или в дополнение к ним
коммуникации (при так называемом межкле-
используются дополнительные источники ин-
точном распознавании), так и выполнять ис-
формации, наиболее популярным из которых
ключительно структурную функцию.
на сегодняшний день является скорость РНК
Изучение межклеточной коммуникации по-
(RNA velocity) [71]. Идея RNA velocity возник-
могает понять механизмы дифференцировки
ла при изучении данных scRNA-seq, получен-
и морфогенеза клеток, этиологию заболева-
ных на различных платформах (Smart-seq2,
ний [77] и особенности формирования иммун-
STRT/C1, inDrop и 10x Genomics Chromium).
ного ответа [78]. Понимание межклеточного
Оказалось, что от 15 до 25% прочтений со-
сигналинга позволяет разрабатывать новые
держат несплайсированные интронные по-
терапевтические стратегии [79] и прогнози-
следовательности, что объясняется наличием
ровать тяжесть течения различных заболева-
поли(А)-участков не только в поли(А)-хвосте,
ний [80, 81].
но и в поли(А)-вставке [76]. В связи с таким
Исследования межклеточного сигналинга
наблюдением было предложено при анализе
берут своё начало с определения белок-белко-
данных учитывать как сплайсированные, так и
вых взаимодействий при помощи двугибрид-
несплайсированные формы мРНК. Под скоро-
ных систем, коиммунопреципитации и иных
стью в данном случае понимается производная
методов [82]. С их помощью накоплен целый
по времени от стадии экспрессии гена. Весь
пласт экспериментально подтверждённых ли-
процессинг был поделен на три стадии: тран-
ганд-рецепторных взаимодействий, который,
скрипция, сплайсинг и деградация. Экспрессия
однако, был получен лишь для конкретных
гена констатируется в случае преобладания
типов клеток в конкретных тканях. ScRNA-
транскрипции и сплайсинга над деградацией
seq позволяет оценивать уровни экспрессии
и ингибируется, если деградация преобладает
генов лигандов и рецепторов в тысячах кле-
над транскрипцией и сплайсингом. Значение
ток за один эксперимент и не только изучать
скорости определяет направление вектора каж-
клеточный состав ткани, но и на системном
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
181
уровне оценивать возможные паракринные и
все коммуникации с силой выше некоторо-
аутокринные регуляции.
го установленного ими порогового значения.
Анализ межклеточного сигналинга по дан-
Другой алгоритм CellCall предполагает, что ком-
ным scRNA-seq ставит перед собой задачу по-
муникация значима, если ожидаемая доля лож-
нять, коммуницирует ли определённая пара
ных отклонений анализа обогащения набора ге-
типов клеток A-B по определённому каналу
нов (FDR GSEA) регулона RegB меньше 0,05.
лиганд-рецептор l-r. Простые методы, такие
Особо следует выделить алгоритм scTensor
как iTalk [83] и CellTalker, решают эту задачу
[87], в котором сначала из данных формируется
следующим образом: если ген лиганда l диф-
тензор третьего ранга размерности A × A × L,
ференциально активирован в типе клеток A,
где A - количество типов клеток, L - коли-
а ген рецептора r дифференциально активи-
чество исследуемых пар лиганд-рецептор,
рован в типе клеток B, то такие клетки счита-
а (a, b, l)-й элемент этого тензора - сила ком-
ются взаимодействующими. Эти методы ин-
муникации клеток A и B при помощи пары
туитивно понятны и легко интерпретируемы,
лиганд-рецептор l-r. Т.е. тензор состоит из
однако они нечувствительны к коммуникаци-
всех попарных сил коммуникации всеми воз-
ям, которые характерны для большого числа
можными парами лиганд-рецептор. Сила ком-
типов клеток ткани.
муникации в данном методе рассчитывается
В более сложных методах вводится поня-
как простое произведение lA и rB. Сконструи-
тие силы, или активности коммуникации S,
рованный тензор преобразуется в произведе-
которую оценивают как функцию от сред-
ние трёх матриц и нового тензора при помо-
них экспрессий l в A (lA) и r в B (rB) - от их
щи неотрицательного разложения Таккера.
суммы (метод CellPhoneDB [84]) либо от их
В результате информация о межклеточном
произведения (SingleCellSignalR [85]). Алго-
сигналинге описывается сразу для всего на-
ритм CellCall
[86] для оценки активности
бора данных, и это позволяет увидеть более
коммуникации между клетками A и B допол-
комплексные эффекты, в частности, включаю-
нительно использует информацию об экс-
щие в себя целые коммуникационные сети.
прессии регулона RegB (набор генов-мише-
Несмотря на свои достоинства, этот метод не
ней транскрипционного фактора, которые
пользуется большой популярностью в пер-
коэкспрессируются вместе с транскрипцион-
вую очередь из-за сложности интерпретации
ным фактором), находящегося под регуляци-
результатов.
ей транскрипционного фактора, который ак-
Описанные выше подходы к определе-
тивируется при воздействии на клетку через
нию межклеточного сигналинга принципи-
рецептор r. Отдельно стоит упомянуть случаи,
ально отличаются в первую очередь гипо-
когда рецептор состоит из нескольких субъ-
тезами, которые они тестируют. CellTalker,
единиц, кодируемых разными генами. В та-
iTalk и CellPhoneDB позволяют определить
ком случае за r будет взята либо минимальная
сигналинги, уникальные для некоторых ти-
экспрессия среди всех субъединиц рецептора
пов клеток в исследуемом наборе данных.
(CellPhoneDB), либо их среднее геометриче-
SingleCellSignalR, CellCall и scTensor позво-
ское (CellCall).
ляют детектировать большее число коммуни-
Однако не все клетки, коэкспрессирую-
каций, в том числе и неспецифичных, однако
щие пару лиганд-рецептор, коммуницируют
могут оказаться нечувствительными в случаях,
в реальности. Одним из способов преодоле-
когда сила коммуникации низкая [88]. Кроме
ния ложноположительных результатов яв-
того, все перечисленные выше методы го-
ляется пермутационный тест (реализован в
ворят только о возможных путях сигналин-
CellPhoneDB), в ходе которого метки клеточ-
га, которые необходимо в дальнейшем ва-
ных типов множество раз случайно переме-
лидировать экспериментально, и результат
шиваются, а сила коммуникации S считается
работы данных инструментов сильно зависит
заново, задавая нулевое распределение, по
от базы лиганд-рецепторных взаимодействий.
которому будет рассчитываться p-значение
Более значимое подтверждение коммуни-
для исходного S. Минус такого подхода схож
кации между различными клетками можно
с минусами подходов, основанных на диф-
получить при помощи бурно развивающих-
ференциальной экспрессии: широко распро-
ся методов пространственной транскрипто-
странённые в исследуемом наборе данных
мики
[89], которые, по всей видимости,
коммуникации могут оказаться статистически
позволят точно ответить на множество во-
незначимыми. Иное решение этой проблемы
просов о том, каким образом формирует-
реализовано в SingleCellSignalR: авторы это-
ся и поддерживается архитектура различных
го алгоритма предлагают считать значимыми
тканей.
БИОХИМИЯ том 88 вып. 2 2023
182
ХОЗЯИНОВА и др.
ГЕННЫЕ РЕГУЛЯТОРНЫЕ СЕТИ
связей между регуляторами и генами-мише-
нями, в которых гены-мишени имеют предпо-
Регуляция экспрессии генов внутри клет-
лагаемый сайт связывания с соответствующи-
ки осуществляется за счёт сложного сочетания
ми транскрипционным факторами. При этом
процессов синтеза и сплайсинга РНК, а так-
транскрипционный фактор вместе с активи-
же деградации уже зрелой мРНК. В основном
руемыми генами-мишенями называют регуло-
уровень экспрессии генов тесно связан с ак-
ном (regulon).
тивностью транскрипции мРНК. Транскрип-
Построение генных сетей, основанных
ция, в свою очередь, регулируется за счёт воз-
на корегуляторных взаимодействиях, под-
действия на клетки различного рода сигналов.
разумевает подсчёт корреляции экспрессии
Например, гормоны, воздействуя на специ-
генов в единичных клетках с помощью коэф-
фические рецепторы, запускают сигнальные
фициента Пирсона и рангового коэффици-
каскады, локализующиеся в основном в ци-
ента Спирмена и реализуется посредством
топлазме клетки. Сигнальные каскады запу-
анализа взвешенных сетей коэкспрессии ге-
скают транскрипционные факторы, которые
нов (WGCNA) [95]. Коэкспрессионные мо-
взаимодействуют с сайтами связывания на
дули соотносятся с функциями генов с помо-
генах-мишенях. Эти взаимодействия осуще-
щью метода GSEA [96] и баз данных, таких
ствляются в ядре клетки и называются ген-
как STRING [97] и HumanNet [98]. Примене-
ными регуляторными сетями (gene regulatory
ние WGCNA на данных scRNA-seq позволяет
networks). Именно генные регуляторные сети
идентифицировать функциональные модули и
осуществляют поддержание клеточного го-
составляющие их ключевые гены для каждо-
меостаза, формирование клеточной гетеро-
го типа клеток, которые могут быть связаны
генности, а их нарушение может приводить к
с конкретным физиологическим или патофи-
развитию различных патологических состоя-
зиологическим состоянием
[99]. Ключевые
ний и утяжелять течение заболеваний [90, 91].
гены имеют наибольшее количество корре-
Изучение генных регуляторных сетей улучша-
ляционных связей в плане коэкспрессии и в
ет понимание механизмов различных биоло-
большей степени определяют функциональную
гических процессов в живых организмах и по-
принадлежность модулей. Выявление таких ге-
зволяет разрабатывать новые терапевтические
нов позволяет обнаруживать, например, факто-
стратегии для борьбы с заболеваниями.
ры, связанные с устойчивостью к химиотера-
Построение генных регуляторных сетей из
пии [100], или прогностические маркеры [101].
данных scRNA-seq может осуществляться на
Анализ траекторий развития позволяет
основании регрессионных моделей, корегуля-
рассмотреть данные секвенирования единич-
торных взаимодействий и вывода траектории
ных клеток как динамическую систему, что
развития.
даёт возможность выйти за пределы статиче-
Подходы на основе регрессии работают
ской природы транскриптома и получить псев-
с конкретными списками генов и позволяют
довремя для последующего построения генных
оценивать связь между регуляторами и гена-
сетей с помощью метода обычных дифферен-
ми-мишенями, а также делают вывод об ин-
циальных уравнений. Такие сети отражают
тенсивности этого взаимодействия. Первона-
генные взаимодействия в динамике, т.е. изме-
чально для построения генных регуляторных
нение экспрессии генов в течение непрерыв-
сетей на основе регрессии был разработан ме-
ного псевдовремени характеризуется функцией,
тод GENIE3 [92]. Этот метод широко исполь-
которая включает активирующее или подавляю-
зуется для построения генных сетей из данных
щее влияние других генов в качестве перемен-
bulk RNA-seq и scRNA-seq. Однако примене-
ных [102]. Данный подход наиболее точно опи-
ние GENIE3 невозможно в случаях scRNA-
сывает генные взаимодействия в непрерывных
seq, когда количество исследуемых клеток ис-
процессах, таких как дифференцировка, и реа-
числяется тысячами. Данная проблема была
лизован в инструменте SCODE [103].
успешно решена с помощью градиентного бу-
стинга в методе GRNBoost2 [93]. Тем не менее
значительным недостатком анализа регуля-
АНАЛИЗ CNV
торных сетей, выведенных из регрессионного
анализа отдельных клеток, является большее
CNV вносят важный вклад в генетическую
количество ложноположительных связей по
изменчивость живых организмов и определя-
сравнению с анализом bulk RNA-seq. Исполь-
ют предрасположенность к различным забо-
зование инструмента SCENIC [94] позволяет
леваниям. К основным критериям определе-
преодолеть данный недостаток за счёт отбора
ния структурного варианта как CNV относят
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
183
повторяемость, числовую изменчивость и «зна-
об относительной нормализованной мере со-
чительную» длину. Несмотря на заданные кри-
отношения измеренных интенсивностей двух
терии, границы между типами структурных ва-
аллелей (BAF), что приводит к повышенному
риантов формируются по-разному в различных
количеству ложноположительных результатов.
работах, поэтому некоторые CNV соответству-
В других инструментах для анализа CNV
ют одновременно нескольким категориям [104].
реализован подход объединения генетической
В настоящее время многие исследователи опре-
и транскрипционной информации. Напри-
деляют CNV как несбалансированные хро-
мер, метод HoneyBADGER [109], использую-
мосомные перестройки - делеции и вставки
щий байесовский подход с интегрированной
участков ДНК, размеры которых варьируют от
скрытой марковской моделью, рассчитывает
нескольких килобаз до целых хромосом и могут
отклонение доли аллелей гетерозиготных ва-
включать мобильные элементы и некодирую-
риантов от ожидаемой и определяет регионы
щие последовательности [105]. Соответствен-
CNV. Для защиты от ложноположительных
но, в зависимости от размера CNV могут быть
результатов для предсказанных регионов оце-
фокальными и полнохромосомными. Послед-
нивается апостериорная вероятность принад-
ние генерируются анеуплоидными клетками с
лежности области CNV заданному состоянию.
аномальным числом хромосом и ведут к изме-
При использовании HoneyBADGER необ-
нению уровня транскрипции большого количе-
ходимо предварительно определять SNV, так
ства генов. CNV могут быть представлены как
как инструмент подтверждает наличие CNV в
нейтральными, так и патогенными формами.
регионах-кандидатах на основании моноал-
Патогенность определяется прямым влиянием
лельного характера экспрессии SNV в данных
CNV на экспрессию генов и/или образованием
регионах. Другой метод, CaSpER [110], исполь-
новых белковых продуктов [106].
зует многомасштабную декомпозицию для сгла-
Классическими методами для идентифи-
живания сигналов экспрессии и аллельного
кации CNV являются микроматричная срав-
сдвига (allelic shift), благодаря чему большая
нительная геномная гибридизация (aCGH),
часть шума удаляется. Ввиду того, что данный
мультиплексная амплификация лигированных
инструмент генерирует профиль сигнала ал-
зондов (MLPA) и секвенирование следующе-
лельного сдвига из выровненных прочтений,
го поколения (NGS), главным образом пол-
определение SNV не требуется. Однако по-
ногеномное секвенирование (WGS). Однако
скольку сигнал сдвига частоты альтернатив-
aCGH и MLPA ограничены разрешением чипа
ного аллеля вычисляется путём объединения
(связанным с охватом и плотностью флуорес-
всех прочтений, клетки, имеющие большое
центных зондов) и неспособны детектировать
количество прочтений, могут доминировать
копий-нейтральные потери гетерозиготности.
в сигнале сдвига и быть основным фактором
Стоимость, продолжительность обработки
искажения результата. Перечисленные методы
данных и высокие вычислительные требова-
были разработаны для анализа полноразмер-
ния усложняют проведение анализа CNV с по-
ных транскриптов, однако были валидирова-
мощью WGS [107].
ны для данных секвенирования одноконцевых
Существует лишь небольшое количество
транскриптов [109, 110]. Для последнего был
методов, созданных для идентификации CNV
разработан инструмент CopyKAT [111] c инте-
по данным scRNA-seq. Все эти методы основа-
гративным байесовским подходом и иерархи-
ны на предположении, что дифференциальная
ческой кластеризацией. Данный метод в боль-
экспрессия генов коррелирует с CNV [108].
шей степени подходит для анализа опухолевых
Метод inferCNV основан на усреднении уров-
клеток, которые часто являются анеуплоид-
ня экспрессии генов и сравнении профи-
ными. Так, данный метод показал свою эф-
ля CNV изучаемого образца с эталонным.
фективность в идентификации опухолевых и
Несмотря на то что такой метод с высокой
гибридных клеток среди циркулирующих эпи-
точностью выявляет клональные изменения
телиальных клеток у больных раком молочной
на уровне плеч хромосом, inferCNV с трудом
железы [112].
удается идентифицировать субклональные из-
В то время как инструменты по поиску
менения. Результаты, получаемые с помощью
CNV, используемые при WGS, основаны на
inferCNV, также высокочувствительны к вы-
равномерном покрытии генома прочтения-
бору эталонных клеток. Ввиду этого необхо-
ми, при scRNA-seq сигнал концентрируется
дима независимая нормализация различных
только на экзонных участках. В этом плане ре-
клеточных типов с помощью соответствующих
комендуется проведение анализа аллельного
эталонных клеток [109]. На конечные резуль-
дисбаланса для понимания корреляции между
таты оказывает влияние и отсутствие данных
геномом и транскриптомом. Однако отличие
БИОХИМИЯ том 88 вып. 2 2023
184
ХОЗЯИНОВА и др.
настоящих генетических вариантов от тех-
поли(А)-хвост мРНК и конверсия образовав-
нических артефактов достаточно осложнено
шихся фрагментов в цепь кДНК, на основе ко-
из-за выпадения аллелей, неоднородности
торых и будет происходить подготовка библио-
и низких показателей глубины секвениро-
теки. Другим ограничением при SNV-анализе
вания [113]. Таким образом, на данный мо-
данных scRNA-seq может быть выпадение од-
мент анализ плоидности ДНК и идентифи-
ного исследуемого аллеля (allelic dropout), в
кация анеуплоидии в scRNA-seq даёт более
частности, при использовании технологии мас-
корректные результаты, чем нахождение фо-
ляных капель для изоляции отдельных клеток
кальных CNV.
перед баркодированием и амплификацией
целевых молекул, что затрудняет идентифи-
кацию гетерозиготных субпопуляций клеток.
ИДЕНТИФИКАЦИЯ
Таким образом, при планировании исследо-
ОДНОНУКЛЕОТИДНЫХ ЗАМЕН
вания SNV на основе данных scRNA-seq не-
обходимо учитывать указанные ограничения
Как и CNV, однонуклеотидные варианты
и по возможности использовать двуконцевое
составляют генетическую изменчивость жи-
прочтение при секвенировании с последую-
вых организмов, влияют на протекание биоло-
щей верификацией находок классическими
гических процессов и могут выступать в роли
молекулярно-генетическими методами.
генетических факторов предрасположенности
Идентификация SNV на основе scRNA-
к заболеваниям. Идентификация SNV возмож-
seq в большинстве своём осуществляется ме-
на посредством использования различных мо-
тодами, разработанными для анализа дан-
лекулярно-генетических методов, основными
ных секвенирования ДНК: SAMtools, GATK,
из которых являются полимеразная цепная ре-
CTAT, FreeBayes, MuTect2, Strelka2, VarScan2
акция, микроматричный анализ, секвенирова-
и др. Общая схема работы данных алгоритмов
ние по Сэнгеру и NGS. Для обнаружения SNV
заключается в четырёх последующих опера-
на уровне отдельных клеток классическим ме-
циях: картирование на референсный геном,
тодом является секвенирование ДНК. Наибо-
предобработка, идентификация вариантов и
лее информативным и концептуально верным
фильтр ложноположительных вариантов. Для
для этой цели является scDNA-seq, реали-
картирования чаще всего используют алго-
зованный в платформе Tapestri (Mission Bio).
ритм STAR, рекомендованный GATK Best
Однако анализ SNV также возможно прово-
Practices [114]. Для анализа данных scRNA-seq
дить на основе данных scRNA-seq, тем самым
может дополнительно использоваться инстру-
получая одновременно информацию и об экс-
мент GSNAP, позволяющий работать с корот-
прессии генов. Основным ограничением явля-
кими и сложнокартируемыми последователь-
ется анализ SNV только в белок-кодирующих
ностями [115]. Предобработка предназначена
участках (экзонах), поскольку в качестве ис-
для удаления дубликатов, повторного вырав-
ходного материала для scRNA-seq чаще всего
нивания и базовой оценки качества прочте-
выступает матричная РНК. Более того, различ-
ний. Выявление генетических вариантов про-
ные паттерны экспрессии генов и альтерна-
водится на основе расхождения нуклеотидных
тивный сплайсинг существенно ограничивают
последовательностей с референсом и удаления
доступную для анализа белок-кодирующую
вариантов с низким качеством или недоста-
область генома. Другим важным моментом яв-
точным покрытием. Хотя MuTect2, Strelka2 и
ляется то, что при анализе экспрессии генов
VarScan2 применяются в основном для сек-
чаще всего применяется короткое однокон-
венирования ДНК, а также и bulk RNA-seq,
цевое секвенирование с 5′- или 3′-конца, что
выявленные с помощью данных алгоритмов
опять же исключает из анализа значительную
варианты могут быть соотнесены с кластера-
часть финальной библиотеки. Эта проблема
ми единичных клеток на основе других ин-
наиболее остро возникает в случае с 3′-секве-
струментов, например, VarTrix, с целью выво-
нированием, где прочтение затрагивает только
да связи генотип-фенотип. Стоит отметить,
небольшой участок с поли(А)-хвоста мРНК,
что большинство описанных алгоритмов, за
и большая часть экзонных последователь-
исключением SAMtools [116], в той или иной
ностей, наиболее интересных для SNV-ана-
степени работают на основе GATK. Более де-
лиза, теряется. В случае 5′-секвенирования
тальное сравнение описанных пайплайнов
при достаточной экспресcии интересующего
представлено в обзоре Liu et al. [117]. Что ка-
участка экзома проблема частично решается,
сается SAMtools, то в литературе имеется со-
если в процессе пробоподготовки происхо-
общение о применения инструмента Pysam,
дит случайная фрагментация захваченной за
функционирующего на основе SAMtools, для
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
185
детекции вариантов в митохондриальной ДНК
бильность [120]. Генетические нарушения, в
методом scRNA-seq [118].
частности однонуклеотидные замены и абер-
При использовании разных платформ для
рации числа копий ДНК, являются драйвера-
подготовки библиотек при scRNA-seq необхо-
ми клональной эволюции опухолевых клеток,
димо учитывать их разную «пропускную спо-
приводя к формированию клонов и субкло-
собность», то есть количество клеток, которое
нов, устойчивых к противоопухолевому лече-
можно проанализировать за один запуск и,
нию и обладающих высоким потенциалом к
как следствие, число прочтений на одну клет-
метастазированию и рецидивированию. Иссле-
ку. Так, например, для Fluidigm C1 (1000 кле-
дование клонального состава опухолей, особен-
ток за запуск) значение глубины секвениро-
но в динамике терапии, позволяет не только
вания может достигать
1 миллиона ридов
понять механизмы появления и прогрессиро-
на клетку, а для 10х Genomics Chromium (до
вания злокачественных новообразований, но
10 000 клеток за запуск) глубина секвениро-
и разработать эффективные методы лечения,
вания в реальной практике редко превышает
в том числе адаптированные под конкретного
10-20 тысяч. Это приводит к тому, что суще-
пациента.
ствует вероятность не обнаружить варианты
Как правило, для изучения генетической
со слабой экспрессией и субклональные SNV.
гетерогенности и клональной эволюции опу-
Дальнейшее увеличение количества прочтений
холи используют bulk DNA-seq. Однако при
может быть малоэффективным в случае малой
смешивании ДНК нескольких тысяч или мил-
«сложности» библиотеки и большой скоро-
лионов клеток информация о редких событиях
сти «насыщения» секвенирования (sequencing
зачастую теряется. Использование scDNA-seq
saturation rate). В частности, это связано с тем,
в полной степени способно решить проблему
что подавляющее число ридов будет картиро-
поиска редких вариантов и анализа клональ-
ваться на ограниченную группу сильно пред-
ной структуры опухолей из-за баркодирования
ставленных транскриптов, а детекция слабо
каждой клетки. Однако на сегодняшний день
экспрессирующихся генов и вариантов в них
применение данного метода в значительной
потребует сильного увеличения глубины про-
степени ограничено. Единственная коммерче-
чтения. Такая ситуация, например, описана
ски доступная технология scDNA-seq Tapestri
для 10х Genomics scRNA-seq мононуклеаров
позволяет судить лишь о структуре заранее вы-
периферической крови, где показатель «насы-
бранной пользователем или предоставленной
щения» секвенирования составлял более 90%.
производителем панели генов. Подходы, ос-
Повысить вероятность детекции SNV в та-
нованные на полногеномной амплификации,
ких случаях можно путём анализа дубликатов
страдают от ряда проблем, в числе которых не-
ПЦР, образующихся в результате многократ-
достаточный процент охвата генома либо си-
ной амплификации малого числа исходных
стематическая ошибка амплификации, кото-
молекул. В стандартных биоинформатических
рая может привести к высокой зашумлённости
алгоритмах такие дубликаты удаляются из по-
данных [121]. В связи с этим особо привлека-
следующего анализа как источник ложнополо-
тельной задачей становится анализ клональ-
жительных вариантов. Однако в статье Wilson
ной эволюции на основе данных scRNA-seq
et al. описан пайплайн scSNV, позволяющий
ввиду возможности совместной оценки ге-
анализировать подобные дубликаты с низким
нетической и транскрипционной гетероген-
процентом ложноположительных SNV [119].
ности. Однако такой подход для построения
Суть метода заключается в «слиянии» дубли-
филогении опухолей осложнён рядом ограни-
рующих прочтений в длинные молекулы после
чений, в частности, невозможностью поиска
выравнивания на референс и последующий
генетических вариантов в нетранскрибируе-
анализ. При этом риды с низкой «сложно-
мых регионах, наличием аллель-специфичной
стью» и артефакты из неправильно картиро-
экспрессии, низкими показателями глубины
ванных прочтений, являющихся основным
scRNA-seq и высоким уровнем шума в полу-
источником ложноположительных вариантов,
ченных данных [117, 122, 123]. В связи с этим
удаляются.
биоинформатический анализ данных scRNA-
seq для понимания клональной архитектуры
опухолей является вызовом и пока реализован
ФИЛОГЕНЕТИКА ЗЛОКАЧЕСТВЕННЫХ
только в некоторых инструментах: DENDRO,
НОВООБРАЗОВАНИЙ
Cardelino, Trisicell и SASC.
DENDRO позволяет учитывать транскрип-
Одним из основных признаков онкологи-
ционные всплески (transcriptional bursting), вы-
ческих заболеваний является геномная неста-
падение SNV и ошибки секвенирования [124].
2
БИОХИМИЯ том 88 вып. 2 2023
186
ХОЗЯИНОВА и др.
С использованием DENDRO была оценена мута-
Trisicell-PartF вычисляет вероятность содержа-
ционная нагрузка, определены неоантигены для
ния каждого узла консенсусного древа в иссле-
каждого опухолевого субклона и выявлена связь
дуемых клетках. Trisicell-Cons, в свою очередь,
между транскриптомными изменениями и гене-
предназначен для вывода консенсусных фи-
тической дивергенцией опухолевых клеток [124].
логенетических древ, полученных с помощью
Байесовский метод Cardelino позволя-
различных инструментов и/или из данных
ет интегрировать информацию о филогении,
scDNA и scRNA-seq. Trisicell-Cons миними-
построенной на основе bulk или scDNA-seq с
зирует количество ветвей двух или более древ,
данными об аллельных вариантах, полученных
выводя более достоверную историю прогрес-
с помощью scRNA-seq [125]. Данный подход
сирования опухоли.
учитывает стохастические выпадения SNV в
Стоит отметить, что перечисленные выше
транскриптомных данных и систематический
инструменты в большей степени применимы
аллельный дисбаланс ввиду моноаллельного
для обработки данных секвенирования пол-
характера экспрессии или влияния регулятор-
норазмерных транскриптов (например, Smart-
ных факторов. Кроме того, Cardelino может
seq, NuGen Solo и др.), обеспечивающего наи-
работать только на данных scRNA-seq, предо-
более равномерное покрытие и относительно
ставляя информацию о субклональной иерар-
низкий уровень шума [128]. Анализ данных
хии опухолевых клеток.
секвенирования одноконцевых транскриптов
Большинство инструментов для вывода
(10x Genomics Chromium, BD Rhapsody и др.)
филогении основаны на предположении о бес-
ввиду низкой глубины секвенирования мо-
конечных участках (infinite sites assumption),
жет приводить к ошибкам в идентификации
согласно которому каждая мутация возникает
генетических вариантов и, как следствие, по-
не более одного раза и не элиминируется в про-
строению некорректных филогенетических
цессе филогенеза. Использование такой теории
деревьев. В этом плане секвенирование одно-
значительно упрощает вычислительный про-
концевых транскриптов целесообразно ком-
цесс и приемлемо для построения филогении
бинировать с bulk DNA-seq или scDNA-seq и
нормальных клеток, но не злокачественных,
проводить совместный биоинформатический
ввиду высокой скорости накопления мутаций,
анализ, например, с помощью Cardelino или
а также их элиминации за счёт возникновения
Trisicell.
CNV. Авторы инструмента для анализа внут-
риопухолевой прогрессии SASC отходят от
модели совершенной филогении и использу-
ЭПИГЕНОМИКА:
ют филогенетическую Dollo-k, допускающую
ДОСТУПНОСТЬ ХРОМАТИНА,
элиминацию мутаций на протяжении филоге-
ИДЕНТИФИКАЦИЯ
неза [126]. Использование данной модели при-
САЙТОВ СВЯЗЫВАНИЯ
ближает выводимое in silico филогенетическое
ТРАНСКРИПЦИОННЫХ ФАКТОРОВ
древо к реальному. Кроме того, инструмент
учитывает различия в частоте ложноотрица-
Пространственно-временная динамика экс-
тельных результатов для каждой мутации ввиду
прессии генов обусловлена работой различных
разницы в уровне экспрессии генов.
транскрипционных факторов и регулируется
Для уточнения результатов, повышения
со стороны разного рода эпигенетических ме-
производительной мощности существующих
ханизмов. Доступность и транскрипционная
инструментов вывода филогении, а также срав-
активность хроматина на регуляторных эле-
нения древ, полученных с помощью различ-
ментах генома является одним из ключевых
ных инструментов и/или из различных на-
факторов регуляции экспрессии генов. Регу-
боров данных, был разработан инструмент
ляторные элементы, в частности промоторы
Trisicell [127]. Trisicell состоит из трёх вычисли-
и энхансеры, представляют собой последова-
тельных методов: Trisicell-Boost, Trisicell-PartF
тельности ДНК (от нескольких сотен до тысяч
и Trisicell-Cons. Trisicell-Boost увеличивает про-
пар оснований), которые состоят из уникаль-
изводительность и точность других инструмен-
ных сайтов связывания для транскрипцион-
тов за счёт многократного отбора случайных
ных факторов [129]. Сравнительный анализ
подмножеств мутаций, для каждого из которых
мотивов связывания на регуляторных элемен-
строится филогенетическое мутационное дре-
тах в сочетании с информацией об экспрессии
во. После этого Trisicell-Boost проводит срав-
транскрипционных факторов даёт возмож-
нение различных деревьев для одного образца
ность пролить свет на механизмы клеточных
и осуществляет построение результирующего
процессов и причины возникновения различ-
дерева на основе механизма консенсуса. Затем
ных заболеваний.
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
187
Клеточно-специфичная активность про-
Ещё одним свойством функционально
моторов обычно определяется энхансерами. В
активных промоторов и энхансеров является
многоклеточном организме энхансеры в пер-
транскрипция. С промоторов транскрибирует-
вую очередь ответственны за точный контроль
ся РНК соответствующего им гена, а с энхан-
экспрессии генов [130]. Таким образом, изме-
серов - энхансерная РНК (эРНК). Изучение
нение экспрессии одного и того же гена в раз-
активности регуляторных элементов показало,
ных типах клеток или в одном и том же типе
что эРНК транскрибируется в ходе контакта
клеток в разных состояниях может быть объ-
энхансера с промотором-мишенью, коррели-
яснено изменениями в этих цис-регуляторных
рует с гистоновыми модификациями актив-
элементах [131].
ных энхансеров и ассоциирована с активацией
В последнее время общепринятым под-
транскрипции [129].
ходом для определения открытых участков
Большое количество накопленных к настоя-
ДНК стал анализ доступности хроматина для
щему времени данных RNA-seq и ATAC-seq
транспозазы с последующим секвенировани-
позволило выявить корреляцию между про-
ем (ATAC-seq). Данный подход получил осо-
филем доступности хроматина и транскрип-
бую популярность из-за относительно простой
цией. Современные методы анализа данных
экспериментальной процедуры, а также не-
позволяют использовать модели машинного
большого количества исходного материала - от
обучения с целью предсказания ландшафта
единичных клеток до нескольких сотен [132].
хроматина на основе данных RNA-seq. Осно-
Однако без информации о транскрипционных
ванный на регрессионных моделях инструмент
факторах, которые связывают тот или иной
BIRD (Big data Regression for predicting DNase I
открытый участок хроматина, а также харак-
hypersensitivity) использует данные RNA-seq
терного профиля пост-трансляционных моди-
для предсказания открытых участков хрома-
фикаций гистонов, фланкирующих доступную
тина. Изначально разработанный для метода
ДНК нуклеосом, анализ только доступности
DNase-seq в качестве тренировочного набо-
хроматина не является достаточным для опре-
ра, он был успешно применён и с использо-
деления типа регуляторного элемента. Метод
ванием данных ATAC-seq. BIRD применим
иммунопреципитации хроматина (ChIP-seq) с
для предсказания открытого хроматина как в
использованием антител к транскрипционным
данных bulk RNA-seq, так и в scRNA-seq [134].
факторам или специфичным пост-трансляци-
Полученные результаты можно использовать
онным модификациям гистонов позволил кар-
для анализа обогащения мотивов связывания
тировать расположение различных регулятор-
специфичных транскрипционных факторов на
ных элементов в активном и репрессированном
промоторах ДЭГ с помощью широко приме-
состояниях [131]. Не так давно был разработан
няемого набора инструментов MEME, осно-
метод single-cell CUT&TAG для изучения пол-
ванных на скрытых марковских моделях [135]
ногеномного распределения различных гисто-
и разработанных для пакета ChromVAR в
новых модификаций на уровне единичных
среде R [136]. Однако данный подход неприме-
клеток [133]. Эффективное картирование ре-
ним для аналогичного анализа на энхансерах,
гуляторных элементов генома достигается за
которыми являются далеко не все дистальные
счёт совместного применения вышеуказанных
от гена открытые участки хроматина. Один
методик. Однако это накладывает серьёзные
из последних инструментов, разработанных
ограничения, связанные со стоимостью, а так-
для анализа регуляторных элементов на ос-
же с потребностью в большом количестве био-
нове данных scRNA-seq, SCAFE (Single Cell
логического материала, так как данные методы
Analysis of Five-prime Ends), позволяет решить
нельзя выполнять в одних и тех же клетках од-
эту проблему. В своей основе он использу-
новременно. Помимо этого, данные scATAC-
ет факт транскрипции с активных регулятор-
seq и single-cell CUT&TAG по своей природе
ных элементов. Использование 5′-концевого
дискретны, поскольку каждый геномный ло-
scRNA-seq позволяет идентифицировать сай-
кус имеет только две копии хроматина, кото-
ты старта транскрипции (TSS) как с промото-
рые можно проанализировать внутри клетки.
ров (для оценки транскрипции гена), так и с
Данные, полученные с помощью этих методов,
энхансеров (для оценки транскрипции эРНК).
немногочисленны (~104 прочтений на клетку)
Для поиска транскрибирующихся цис-регуля-
и, таким образом, имеют узкий динамический
торных элементов SCAFE, как и ранее описан-
диапазон. Это отличает их от данных scRNA-
ный метод BIRD, использует пакет референс-
seq, которые являются более непрерывными,
ного генома, который содержит информацию
поскольку ген может иметь несколько поддаю-
о существующих открытых участках хромати-
щихся анализу транскриптов в клетке.
на для соответствующего вида, полученных
БИОХИМИЯ том 88 вып. 2 2023
2*
188
ХОЗЯИНОВА и др.
с помощью ATAC-seq. Идентифицировав TSS,
при различных злокачественных новообразо-
данный инструмент позволяет получить на-
ваниях, где клетки опухолевого микроокруже-
бор активных регуляторных элементов. Помимо
ния вносят вклад в клиническое течение и от-
анализа мотивов связывания транскрипцион-
вет на противоопухолевую терапию [139-141].
ных факторов, SCAFE позволяет оценивать
ScRNA-seq позволяет определять клеточ-
изменения динамики транскрипции активных
ный состав исследуемых образцов, транскрип-
регуляторных элементов в разных состояниях
ционные особенности клеток, траектории их
и выявлять котранскрибирующиеся энхансеры
дифференцировки и другие показатели, рас-
и промоторы для предсказания их физическо-
смотренные выше. Однако пространствен-
го взаимодействия [137]. Такой анализ можно
ное расположение клеток в структуре тканей
проводить, используя R-пакет Cicero, разра-
оказывается утерянным ввиду диссоциации
ботанный для предсказания взаимодействий
образцов во время подготовки кДНК-библио-
между цис-регуляторной ДНК на основе дан-
тек и может быть предсказано лишь прибли-
ных доступности хроматина [138].
зительно. Алгоритм реконструкции простран-
Используя один тип экспериментальных
ственной организации novoSpaRc основан
данных, стало возможным получать инфор-
на теории схожести транскрипционного про-
мацию о природе ДЭГ, оценивая активность
филя клеток, расположенных в физической
регуляторных элементов всех интересующих
близости друг от друга [142], т.е. соседние
генов. Серьёзным ограничением является не-
клетки демонстрируют большее сходство в
обходимость наличия данных ATAC-seq как
транскрипционном профиле, чем клетки, на-
части тренировочного набора при обучении
ходящиеся далеко друг от друга. Однако при
модели, что сильно затрудняет использова-
реконструкции пространственной организа-
ние BIRD и SCAFE при работе с немодельны-
ции novoSpaRc использует предопределён-
ми организмами. В отличие от классических
ную геометрическую форму в качестве эта-
экспериментальных подходов, информация
лона, и, таким образом, все расчёты строятся
о доступных участках хроматина на основе
на геометрических особенностях выбранно-
scRNA-seq формируется только за счёт генов,
го пространства. Кроме того, экспрессион-
чьи транскрипты удалось проанализировать.
ная схожесть клеток действительно может
Поэтому невозможно составить полноценной
быть следствием их близости друг к другу, но
эпигеномной картины для каждой клетки. Од-
никак не предопределять её. Другой инстру-
нако полученных данных достаточно, чтобы
мент, CSOmap, предсказывает координаты
оценить различия между разными клетками в
каждой клетки в трёхмерном псевдо-про-
исследуемом образце. Дальнейшее развитие
странстве, не ограниченном заданной фор-
экспериментальных методик для увеличения
мой [143]. CSOmap построен на предполо-
глубины секвенирования каждой отдельной
жении, что пространственное расположение
клетки, а также усовершенствование вычис-
клеток связано с их взаимодействиями по типу
лительных подходов повысит эффективность
лиганд-рецептор. В частности, данный ин-
предсказания динамики доступности хрома-
струмент комбинирует профили экспрессии
тина и активности регуляторных элементов на
генов единичных клеток и общедоступную
основе данных scRNA-seq.
информацию о лиганд-рецепторных взаимо-
действиях [144, 145] для создания матрицы
аффинности, которая переводится в трёхмер-
РЕКОНСТРУКЦИЯ
ное пространство. Такой подход позволяет не
ПРОСТРАНСТВЕННОЙ
только реконструировать пространственную
ТРАНСКРИПТОМИКИ
организацию de novo, но и оценивать статисти-
ческую значимость межклеточных взаимодей-
Пространственное расположение клеток в
ствий и вклад отдельных пар лиганд-рецептор
пределах тканей и органов тесно взаимосвяза-
в формирование таких коммуникаций. К не-
но с их биологическими функциями. Хотя все
достаткам данного инструмента можно отнести
клетки имеют один и тот же геном, их мор-
вариабельность конечных результатов: обще-
фология и паттерны экспрессии генов силь-
доступная информация о лиганд-рецепторных
но различаются в зависимости от принадлеж-
взаимодействиях может различаться среди до-
ности к типу тканей и месторасположению.
ступных источников, тем самым влияя на вы-
Такая клеточная гетерогенность связана как с
водимые данные.
регуляторными механизмами внутри клеток,
Иммуногистохимическое окрашивание, раз-
так и с влиянием внеклеточного микроокру-
личные варианты гибридизации in situ и экс-
жения. Последнее наиболее ярко выражено
прессионное профилирование, совмещённое с
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
189
лазерной микродиссекцией, не являются иде-
Тем не менее получение информации о
альными методами для изучения простран-
CNV/SNV, филогении и доступности хрома-
ственной транскриптомики. Для первых ха-
тина в значительной степени зависит от ка-
рактерна большая площадь захвата, но малый
чества и глубины секвенирования. Наиболее
охват транскриптов. Для третьего, наоборот,
корректным в этом плане решением может
свойствен большой спектр анализируемых ге-
быть коммерчески доступный, но трудоёмкий
нов, но низкая исследуемая область. В послед-
Smart-seq, основанный на захвате полнораз-
ние годы разработаны различные экспери-
мерных транскриптов, или интегративный
ментальные методы, позволяющие проводить
анализ данных scRNA-seq, bulk DNA-seq и
анализ большого количества транскриптов на
scDNA-seq. Другим потенциальным решением
больших участках ткани [146]. Коммерчески
может стать усовершенствование протоколов
доступными являются Visium («10x Genomics»),
подготовки библиотек полноразмерных тран-
GeoMx
«NanoString Technologies», Molecular
скриптов. Так, в 2022 г. был представлен про-
Cartography от «Resolve Biosciences», Stereo-seq
токол FLASH-seq, превосходящий по скорости
от «BGI STOmics» и другие методы простран-
и чувствительности любой из существующих
ственной транскриптомики. Однако в данный
протоколов scRNA-seq [147]. В основе разра-
момент Visium и GeoMx не способны предоста-
ботки лежит протокол Smart-seq 2, однако для
вить разрешение на уровне единичной клетки.
уменьшения временных затрат и повышения
Размер ячейки с пространственным штрих-ко-
разрешающей способности авторы внесли в
дом на слайде Visium составляет 55 мкм, что
него несколько ключевых модификаций: объ-
создаёт вероятность попадания в одну ячейку
единили обратную транскрипцию и предва-
нескольких клеток. Технически GeoMx спо-
рительную амплификацию кДНК; заменили
собен проводить захват на уровне единичной
обратную транскриптазу Superscript II на бо-
клетки, однако высокое отношение шум/сигнал
лее эффективную Superscript IV; увеличили
ограничивает эту возможность. Кроме того,
количество дезоксицитидинтрифосфата для
применение данных методов ограничивается
индукции С-хвостовой активности SSRTIV и
их высокой стоимостью и, как следствие, отно-
усиления реакции переключения матрицы и
сительной недоступностью. В 2021 г. компания
разместили рибогуанозин в позиции 3′ олиго-
«10x Genomics» анонсировала Visium HD - тех-
нуклеотида, необходимого для инвазии цепи
нологию пространственной транскриптомики c
ДНК и смены матрицы. Кроме того, много-
разрешением, в 400 раз превосходящим таковое
обещающей перспективой могут быть платфор-
у классического Visium и, соответственно, воз-
мы для мультиомиксного анализа единичных
можностью анализа на уровне единичной клетки.
клеток. Подобные протоколы уже разработа-
ны и предоставляют возможность комбиниро-
вания оценки доступности хроматина и тран-
ВЫВОДЫ
скриптома единичных клеток (sci-CAR [148] и
И ДАЛЬНЕЙШИЕ ПЕРСПЕКТИВЫ
SNARE-seq [149]) и совместного проведения
полногеномного секвенирования и профили-
Прогресс в области мультиплексирования
рования экспрессии генов (DNTR-seq) [150].
кДНК-библиотек единичных клеток и в раз-
Однако данные методы чрезвычайно трудо-
работке вычислительных методов биоинфор-
ёмки, дороги в исполнении и характеризуют-
матического анализа позволил в значительной
ся высоким процентом ложноположительных
степени расширить спектр информации, ко-
результатов.
торую возможно извлечь, используя данные
Стоит также уделить внимание нюансам в
scRNA-seq. Помимо классических для scRNA-
определении межклеточных взаимодействий
seq приложений, таких как определение кле-
на основе анализа пар лиганд-рецептор. Ин-
точного цикла, идентификация клеточных
струменты, позволяющие выводить данную
кластеров, анализ дифференциальной экс-
модальность из данных scRNA-seq, опира-
прессии и сигнальных путей и типирование
ются на информацию о лиганд-рецепторных
клеток, стало возможным исследовать предоп-
взаимодействиях из различных источников.
ределяющие фенотип генетические и эпигене-
Соответственно, при использовании различ-
тические характеристики клеток (CNV/SNV и
ных эталонных данных конечный результат бу-
состояние хроматина), предсказывать направ-
дет отличаться. Информация о лиганд-рецеп-
ление их дифференцировки, получать инфор-
торных взаимодействиях также используется
мацию о межклеточных взаимодействиях и
и при de novo реконструкции пространствен-
филогении, в том числе в контексте простран-
ной организации в инструменте CSOmap. Из-
ственной организации тканей и органов.
за потенциальной вариабельности конечных
БИОХИМИЯ том 88 вып. 2 2023
190
ХОЗЯИНОВА и др.
результатов данные методы могут дать лишь ряд
Вклад авторов. A.A. Хозяинова, Е.В. Де-
гипотез, которые необходимо валидировать в
нисов - концепция обзора; A.A. Хозяинова,
других экспериментах, например, с помощью
А.А. Валяева, М.С. Арбатский, С.В. Исаев,
методов пространственной транскриптомики.
П.С. Ямщиков, Е.В. Волчков, М.C. Сабиров,
В целом, мы ожидаем, что бурное развитие
В.Р. Зайнуллина, В.И. Чечехин, Р.С. Воробьёв,
инструментов биоинформатического анализа
М.Е. Меняйло, П.А. Тюрин-Кузьмин, Е.В. Де-
совместно с усовершенствованием протоколов
нисов - сбор информации, анализ публика-
по подготовке библиотек РНК/ДНК единич-
ций, написание и редактирование разделов
ных клеток и разработкой платформ для муль-
обзора.
тиомиксного анализа в значительной степени
Финансирование. Работа выполнена при
увеличит качество биомедицинских исследо-
финансовой поддержке Российского научного
ваний. Технический прогресс в области тех-
фонда (грант № 19-75-30016).
нологий на уровне единичных клеток поможет
Благодарности. Мы благодарим А.А. Щего-
расшифровать клеточную гетерогенность, об-
леву за графическое сопровождение статьи.
условленную совокупностью конститутивных
Конфликт интересов. Авторы заявляют об
и функциональных особенностей, что, в свою
отсутствии конфликта интересов.
очередь, позволит расширить понимание био-
Соблюдение этических норм. Настоящая
логических процессов в норме и патологии и
статья не содержит экспериментов и каких-ли-
сформировать принципиально новые подходы
бо исследований с участием людей или живот-
к персонализированной терапии заболеваний.
ных в качестве объектов.
СПИСОК ЛИТЕРАТУРЫ
1. Tang, F., Barbacioru, C., Wang, Y., Nordman, E.,
van der Kraak, P. H., Kuster, D. W., van der
Lee, C., Xu, N., Wang, X., Bodeau, J., Tuch, B. B.,
Velden, J., and Michels, M.
(2022) Single-cell
Siddiqui, A., Lao, K., and Surani, M. A.
(2009)
transcriptomics provides insights into hypertrophic
mRNA-Seq whole-transcriptome analysis of a
cardiomyopathy, Cell Rep., 39, 110809, doi: 10.1016/
single cell, Nat. Methods, 6, 377-382, doi: 10.1038/
j.celrep.2022.110809.
nmeth.1315.
8. Olah, M., Menon, V., Habib, N., Taga, M. F., Ma, Y.,
2. Islam, S., Kjällquist, U., Moliner, A., Zajac, P.,
Yung, C. J., Cimpean, M., Khairallah, A., Coronas-
Fan, J. B., Lönnerberg, P., and Linnarsson, S. (2011)
Samano, G., and Sankowski, R. (2020) Single cell
Characterization of the single-cell transcriptional
RNA sequencing of human microglia uncovers a
landscape by highly multiplex RNA-seq, Genome Res.,
subset associated with Alzheimer’s disease, Nat.
21, 1160-1167, doi: 10.1101/gr.110882.110.
Commun., 11, 1-18, doi: 10.1038/s41467-020-19737-2.
3. Ke, M., Elshenawy, B., Sheldon, H., Arora, A., and
9. Kamath, T., Abdulraouf, A., Burris, S., Langlieb, J.,
Buffa, F. M. (2022) Single cell RNA-sequencing:
Gazestani, V., Nadaf, N. M., Balderrama, K.,
A powerful yet still challenging technology to study
Vanderburg, C., and Macosko, E. Z. (2022) Single-
cellular heterogeneity, BioEssays, 44, 2200084, doi:
cell genomic profiling of human dopamine neurons
10.1002/bies.202200084.
identifies a population that selectively degenerates
4. Luo, G., Gao, Q., Zhang, S., and Yan, B. (2020)
in Parkinson’s disease, Nat. Neurosci., 25, 588-595,
Probing infectious disease by single-cell RNA
doi: 10.1038/s41593-022-01061-1.
sequencing: progresses and perspectives, Comput.
10. Zhou, S., Huang, Y.-E., Liu, H., Zhou, X., Yuan, M.,
Struct. Biotechnol. J., 18, 2962-2971, doi: 10.1016/
Hou, F., Wang, L., and Jiang, W. (2021) Single-cell
j.csbj.2020.10.016.
RNA-seq dissects the intratumoral heterogeneity of
5. Yifan, C., Fan, Y., and Jun, P. (2020) Visualization of
triple-negative breast cancer based on gene regulatory
cardiovascular development, physiology and disease
networks, Mol. Ther. Nucleic Acids, 23, 682-690,
at the single-cell level: opportunities and future
doi: 10.1016/j.omtn.2020.12.018.
challenges, J. Mol. Cell. Cardiol., 142, 80-92, doi:
11. Zhang, T.-Q., Chen, Y., and Wang, J.-W. (2021) A
10.1016/j.yjmcc.2020.03.005.
single-cell analysis of the Arabidopsis vegetative shoot
6. Pan, Y., Cao, W., Mu, Y., and Zhu, Q.
(2022)
apex, Dev. Cell, 56, 1056-1074.e1058, doi: 10.1016/
Microfluidics facilitates the development of single-cell
j.devcel.2021.02.021.
RNA sequencing, Biosensors, 12, 450, doi: 10.3390/
12. Fu, Y., Huang, X., Zhang, P., van de Leemput, J., and
bios12070450.
Han, Z. (2020) Single-cell RNA sequencing identifies
7. Wehrens, M., de Leeuw, A. E., Wright-Clark, M.,
novel cell types in Drosophila blood, J. Genet.
Eding, J. E., Boogerd, C. J., Molenaar, B.,
Genomics, 47, 175-186, doi: 10.1016/j.jgg.2020.02.004.
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
191
13. Jiang, M., Xiao, Y., Weigao, E., Ma, L., Wang, J.,
Chen, T. (2017) Reconstructing cell cycle pseudo
Chen, H., Gao, C., Liao, Y., Guo, Q., and Peng, J.
time-series via single-cell transcriptome data, Nat.
(2021) Characterization of the zebrafish cell landscape
Commun., 8, 1-9, doi: 10.1038/s41467-017-00039-z.
at single-cell resolution, Front. Cell Dev. Biol., 9,
24. Liang, S., Wang, F., Han, J., and Chen, K. (2020)
743421, doi: 10.3389/fcell.2021.743421.
Latent periodic process inference from single-cell
14. Ho, D. W.-H., Tsui, Y.-M., Chan, L.-K., Sze, K. M.-F.,
RNA-seq data, Nat. Commun., 11, 1-8, doi: 10.1038/
Zhang, X., Cheu, J. W.-S., Chiu, Y.-T., Lee, J. M.-F.,
s41467-020-15295-9.
Chan, A. C.-Y., and Cheung, E. T.-Y. (2021) Single-
25. Anafi, R. C., Francey, L. J., Hogenesch, J. B.,
cell RNA sequencing shows the immunosuppressive
and Kim, J.
(2017) CYCLOPS reveals human
landscape and tumor heterogeneity of HBV-associated
transcriptional rhythms in health and disease, Proc.
hepatocellular carcinoma, Nat. Commun., 12, 1-14,
Natl. Acad. Sci. USA, 114, 5312-5317, doi: 10.1073/
doi: 10.1038/s41467-021-24010-1.
pnas.1619320114.
15. Zhao, J., Zhang, S., Liu, Y., He, X., Qu, M., Xu, G.,
26. Liu, J., Yang, M., Zhao, W., and Zhou, X. (2022)
Wang, H., Huang, M., Pan, J., and Liu, Z. (2020)
CCPE: cell cycle pseudotime estimation for single
Single-cell RNA sequencing reveals the heterogeneity
cell RNA-seq data, Nucleic Acids Res., 50, 704-716,
of liver-resident immune cells in human, Cell Discov.,
doi: 10.1093/nar/gkab1236.
6, 1-19, doi: 10.1038/s41421-020-0157-z.
27. Melms, J. C., Biermann, J., Huang, H., Wang, Y.,
16. Macosko, E. Z., Basu, A., Satija, R., Nemesh, J.,
Nair, A., Tagore, S., Katsyv, I., Rendeiro, A. F.,
Shekhar, K., Goldman, M., Tirosh, I., Bialas, A. R.,
Amin, A. D., Schapiro, D., et al. (2021) A molecular
Kamitaki, N., and Martersteck, E. M. (2015) High-
single-cell lung atlas of lethal COVID-19, Nature, 595,
ly parallel genome-wide expression profiling of
114-119, doi: 10.1038/s41586-021-03569-1.
individual cells using nanoliter droplets, Cell, 161,
28. Delorey, T. M., Ziegler, C. G., Heimberg, G.,
1202-1214, doi: 10.1016/j.cell.2015.05.002.
Normand, R., Yang, Y., Segerstolpe, Å., Abbondan-
17. Kowalczyk, M. S., Tirosh, I., Heckl, D., Rao, T. N.,
za, D., Fleming, S. J., Subramanian, A., Monto-
Dixit, A., Haas, B. J., Schneider, R. K., Wagers, A. J.,
ro, D. T., et al. (2021) COVID-19 tissue atlases reveal
Ebert, B. L., and Regev, A. (2015) Single-cell RNA-
SARS-CoV-2 pathology and cellular targets, Nature,
seq reveals changes in cell cycle and differentiation
595, 107-113, doi: 10.1038/s41586-021-03570-8.
programs upon aging of hematopoietic stem
29. Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F.,
cells, Genome Res.,
25,
1860-1872, doi:
10.1101/
Mohammadi, S., Young, J. Z., Menon, M., He, L.,
gr.192237.115.
Abdurrob, F., Jiang, X., et al. (2019) Single-cell
18. Butler, A., Hoffman, P., Smibert, P., Papalexi, E., and
transcriptomic analysis of Alzheimer’s disease, Nature,
Satija, R. (2018) Integrating single-cell transcriptomic
570, 332-337, doi: 10.1038/s41586-019-1195-2.
data across different conditions, technologies, and
30. Velmeshev, D., Schirmer, L., Jung, D., Haeussler, M.,
species, Nat. Biotechnol., 36, 411-420, doi: 10.1038/
Perez, Y., Mayer, S., Bhaduri, A., Goyal, N.,
nbt.4096.
Rowitch, D. H., and Kriegstein, A. R. (2019) Single-
19. Wolf, F. A., Angerer, P., and Theis, F. J.
(2018)
cell genomics identifies cell type-specific molecular
SCANPY: large-scale single-cell gene expression data
changes in autism, Science, 364, 685-689, doi: 10.1126/
analysis, Genome Biol., 19, 1-5, doi: 10.1186/s13059-
science.aav8130.
017-1382-0.
31. Kathiriya, J. J., Brumwell, A. N., Jackson, J. R.,
20. Scialdone, A., Tanaka, Y., Jawaid, W., Moignard, V.,
Tang, X., and Chapman, H. A. (2020) Distinct airway
Wilson, N. K., Macaulay, I. C., Marioni, J. C.,
epithelial stem cells hide among club cells but mobilize
and Göttgens, B. (2016) Resolving early mesoderm
to promote alveolar regeneration, Cell Stem Cell, 26,
diversification through single-cell expression profiling,
346-358.e344, doi: 10.1016/j.stem.2019.12.014.
Nature, 535, 289-293, doi: 10.1038/nature18633.
32. Steuerman, Y., Cohen, M., Peshes-Yaloz, N.,
21. Tirosh, I.,
Izar,
B.,
Prakadan, S. M.,
Valadarsky, L., Cohn, O., David, E., Frishberg, A.,
Wadsworth, M. H., Treacy, D., Trombetta, J. J.,
Mayo, L., Bacharach, E., Amit, I., and Gat-Viks, I.
Rotem, A., Rodman, C., Lian, C., and Murphy, G.
(2018) Dissection of influenza infection in vivo by
(2016) Dissecting the multicellular ecosystem of
single-cell RNA sequencing, Cell Systems, 6, 679-691.
metastatic melanoma by single-cell RNA-seq, Science,
e674, doi: 10.1016/j.cels.2018.05.008.
352, 189-196, doi: 10.1126/science.aad0501.
33. Kiselev, V. Y., Andrews, T. S., and Hemberg, M.
22. Hsiao, C. J., Tung, P., Blischak, J. D., Burnett, J. E.,
(2019) Challenges in unsupervised clustering of single-
Barr, K. A., Dey, K. K., Stephens, M., and Gilad, Y.
cell RNA-seq data, Nat. Rev. Genet., 20, 273-282, doi:
(2020) Characterizing and inferring quantitative cell
10.1038/s41576-018-0088-9.
cycle phase in single-cell RNA-seq data analysis,
34. Brennecke, P., Anders, S., Kim, J. K., Koło-
Genome Res., 30, 611-621, doi: 10.1101/gr.247759.118.
dziejczyk, A. A., Zhang, X., Proserpio, V., Baying, B.,
23. Liu, Z., Lou, H., Xie, K., Wang, H., Chen, N.,
Benes, V., Teichmann, S. A., Marioni, J. C., and
Aparicio, O. M., Zhang, M. Q., Jiang, R., and
Heisler, M. G. (2013) Accounting for technical noise
БИОХИМИЯ том 88 вып. 2 2023
192
ХОЗЯИНОВА и др.
in single-cell RNA-seq experiments, Nat. Methods, 10,
46. Soneson, C., and Robinson, M. D. (2018) Bias,
1093-1095, doi: 10.1038/nmeth.2645.
robustness and scalability in single-cell differential
35. Andrews, T. S., and Hemberg, M. (2019) M3Drop:
expression analysis, Nat. Methods,
15,
255-261,
dropout-based feature selection for scRNASeq,
doi: 10.1038/nmeth.4612.
Bioinformatics,
35,
2865-2867, doi:
10.1093/
47. Kharchenko, P. V., Silberstein, L., and Scadden, D. T.
bioinformatics/bty1044.
(2014) Bayesian approach to single-cell differential
36. Yau, C. (2016) pcaReduce: hierarchical clustering of
expression analysis, Nat. Methods,
11,
740-742,
single cell transcriptional profiles, BMC Bioinformatics,
doi: 10.1038/nmeth.2967.
17, 1-11, doi: 10.1186/s12859-016-0984-y.
48. Finak, G., McDavid, A., Yajima, M., Deng, J., Ger-
37. Lin, P., Troup, M., and Ho, J. W. (2017) CIDR:
suk, V., Shalek, A. K., Slichter, C. K., Miller, H. W.,
Ultrafast and accurate clustering through imputation
McElrath, M. J., Prlic, M., et al. (2015) MAST: a
for single-cell RNA-seq data, Genome Biol., 18, 1-11,
f lexible statistical framework for assessing transcrip-
doi: 10.1186/s13059-017-1188-0.
tional changes and characterizing heterogeneity in sin-
38. Kiselev, V. Y., Kirschner, K., Schaub, M. T., Andr-
gle-cell RNA sequencing data, Genome Biol., 16, 1-13,
ews, T., Yiu, A., Chandra, T., Natarajan, K. N.,
doi: 10.1186/s13059-015-0844-5.
Reik, W., Barahona, M., Green, A. R., and Hem-
49. Zhang, J. M., Kamath, G. M., and David, N. T.
berg, M. (2017) SC3: consensus clustering of single-
(2019) Valid post-clustering differential analysis for
cell RNA-seq data, Nat. Methods,
14,
483-486,
single-cell RNA-Seq, Cell Systems, 9, 383-392.e386,
doi: 10.1038/nmeth.4236.
doi: 10.1016/j.cels.2019.07.012.
39. Levine, J. H., Simonds, E. F., Bendall, S. C.,
50. Zimmerman, K. D., Espeland, M. A., and Lange-
Davis, K. L., El-ad, D. A., Tadmor, M. D., Litvin, O.,
feld, C. D. (2021) A practical solution to pseudo-
Fienberg, H. G., Jager, A., Zunder, E. R., et al. (2015)
replication bias in single-cell studies, Nat. Commun.,
Data-driven phenotypic dissection of AML reveals
12, 1-9, doi: 10.1038/s41467-021-21038-1.
progenitor-like cells that correlate with prognosis,
51. Denninger, J. K., Walker, L. A., Chen, X., Turkoglu, A.,
Cell, 162, 184-197, doi: 10.1016/j.cell.2015.05.047.
Pan, A., Tapp, Z., Senthilvelan, S., Rindani, R.,
40. Montoro, D. T., Haber, A. L., Biton, M., Vinarsky, V.,
Kokiko-Cochran, O. N., and Bundschuh, R. (2022)
Lin, B., Birket, S. E., Yuan, F., Chen, S., Leung, H. M.,
Robust transcriptional profiling and identification
and Villoria, J. (2018) A revised airway epithelial
of differentially expressed genes with low input RNA
hierarchy includes CFTR-expressing ionocytes,
sequencing of adult hippocampal neural stem and
Nature, 560, 319-324, doi: 10.1038/s41586-018-0393-7.
progenitor populations, Front. Mol. Neurosci., 15,
41. Plasschaert, L. W., Žilionis, R., Choo-Wing, R.,
810722, doi: 10.3389/fnmol.2022.810722.
Savova, V., Knehr, J., Roma, G., Klein, A. M., and
52. Hücker, S. M., Fehlmann, T., Werno, C., Weidele, K.,
Jaffe, A. B. (2018) A single-cell atlas of the airway
Lüke, F., Schlenska-Lange, A., Klein, C. A.,
epithelium reveals the CFTR-rich pulmonary
Keller, A., and Kirsch, S. (2021) Single-cell microRNA
ionocyte, Nature, 560, 377-381, doi: 10.1038/s41586-
sequencing method comparison and application to cell
018-0394-6.
lines and circulating lung tumor cells, Nat. Commun.,
42. Love, M. I., Huber, W., and Anders, S.
(2014)
12, 1-13, doi: 10.1038/s41467-021-24611-w.
Moderated estimation of fold change and dispersion
53. Valyaeva, A. A., Zharikova, A. A., Kasianov, A. S.,
for RNA-seq data with DESeq2, Genome Biol., 15,
Vassetzky, Y. S., and Sheval, E. V. (2020) Expression
1-21, doi: 10.1186/s13059-014-0550-8.
of SARS-CoV-2 entry factors in lung epithelial stem
43. Robinson, M. D., McCarthy, D. J., and Smyth, G. K.
cells and its potential implications for COVID-19, Sci.
(2010) edgeR: a Bioconductor package for differential
Rep., 10, 1-8, doi: 10.1038/s41598-020-74598-5.
expression analysis of digital gene expression
54. Liao, M., Liu, Y., Yuan, J., Wen, Y., Xu, G., Zhao, J.,
data, Bioinformatics,
26,
139-140, doi:
10.1093/
Cheng, L., Li, J., Wang, X., Wang, F., et al. (2020)
bioinformatics/btp616.
Single-cell landscape of bronchoalveolar immune cells
44. Van den Berge, K., Perraudeau, F., Soneson, C.,
in patients with COVID-19, Nat. Med., 26, 842-844,
Love, M. I., Risso, D., Vert, J.-P., Robinson, M. D.,
doi: 10.1038/s41591-020-0901-9.
Dudoit, S., and Clement, L. (2018) Observation
55. Yakushov, S., Menyailo, M., Denisov, E., Karlina, I.,
weights unlock bulk RNA-seq tools for zero inflation
Zainullina, V., Kirgizov, K., Romantsova, O.,
and single-cell applications, Genome Biol., 19, 1-17,
Timashev, P., and Ulasov, I. (2022) Identification of
doi: 10.1186/s13059-018-1406-4.
factors driving doxorubicin-resistant ewing tumor
45. Tang, W., Bertaux, F., Thomas, P., Stefanelli, C.,
cells to survival, Cancers, 14, 5498, doi: 10.3390/
Saint, M., Marguerat, S., and Shahrezaei, V. (2020)
cancers14225498.
bayNorm: Bayesian gene expression recovery,
56. Tyurin-Kuzmin, P. A., Karagyaur, M. N., Kulebya-
imputation and normalization for single-cell RNA-
kin, K. Y., Dyikanov, D. T., Chechekhin, V. I., Iva-
sequencing data, Bioinformatics,
36,
1174-1181,
nova, A. M., Skryabina, M. N., Arbatskiy, M. S.,
doi: 10.1093/bioinformatics/btz726.
Sysoeva, V. Y., Kalinina, N. I., and Tkachuk, V. A.
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
193
(2020) Functional heterogeneity of protein kinase a
68. Pasquini, G., Arias, J. E. R., Schäfer, P., and
activation in multipotent stromal cells, Int. J. Mol. Sci.,
Busskamp, V. (2021) Automated methods for cell
21, 4442, doi: 10.3390/ijms21124442.
type annotation on scRNA-seq data, Computat.
57. Bassez, A., Vos, H., Van Dyck, L., Floris, G., Arijs, I.,
Struct. Biotechnol. J.,
19,
961-969, doi:
10.1016/
Desmedt, C., Boeckx, B., Vanden Bempt, M.,
j.csbj.2021.01.015.
Nevelsteen, I., Lambein, K., et al. (2021) A single-
69. Khrameeva, E., Kurochkin, I., Han, D., Guijarro, P.,
cell map of intratumoral changes during anti-PD1
Kanton, S., Santel, M., Qian, Z., Rong, S., Mazin, P.,
treatment of patients with breast cancer, Nat. Med., 27,
Sabirov, M., et al. (2020) Single-cell-resolution tran-
820-832, doi: 10.1038/s41591-021-01323-8.
scriptome map of human, chimpanzee, bonobo,
58. Bi, K., He, M. X., Bakouny, Z., Kanodia, A.,
and macaque brains, Genome Res.,
30,
776-789,
Napolitano, S., Wu, J., Grimaldi, G., Braun, D. A.,
doi: 10.1101/gr.256958.119.
Cuoco, M. S., Mayorga, A., et al. (2021) Tumor and
70. Han, G., Deng, Q., Marques-Piubelli, M. L., Dai, E.,
immune reprogramming during immunotherapy in
Dang, M., Ma, M. C. J., Li, X., Yang, H., Hender-
advanced renal cell carcinoma, Cancer Cell, 39, 649-
son, J., Kudryashova, O., et al. (2022) Follicular lym-
661.e645, doi: 10.1016/j.ccell.2021.02.015.
phoma microenvironment characteristics associat-
59. Hoernes, T. P., Hüttenhofer, A., and Erlacher, M. D.
ed with tumor cell mutations and MHC class II
(2016) mRNA modifications: Dynamic regulators
expression, Blood Cancer Discov.,
3,
428-443,
of gene expression? RNA Biol., 13, 760-765, doi:
doi: 10.1158/2643-3230.BCD-21-0075.
10.1080/15476286.2016.1203504.
71. La Manno, G., Soldatov, R., Zeisel, A., Braun, E.,
60. Maier, T., Güell, M., and Serrano, L. (2009) Correlation
Hochgerner, H., Petukhov, V., Lidschreiber, K.,
of mRNA and protein in complex biological samples,
Kastriti, M. E., Lönnerberg, P., Furlan, A., et al.
FEBS Lett.,
583,
3966-3973, doi:
10.1016/j.
(2018) RNA velocity of single cells, Nature, 560,
febslet.2009.10.036.
494-498, doi: 10.1038/s41586-018-0414-6.
61. Aran, D., Looney, A. P., Liu, L., Wu, E., Fong, V.,
72. Trapnell, C., Cacchiarelli, D., Grimsby, J., Pokhar-
Hsu, A., Chak, S., Naikawadi, R. P., Wolters, P. J.,
el, P., Li, S., Morse, M., Lennon, N. J., Livak, K. J.,
Abate, A. R., et al. (2019) Reference-based analysis
Mikkelsen, T. S., and Rinn, J. L. (2014) The dynamics
of lung single-cell sequencing reveals a transitional
and regulators of cell fate decisions are revealed
profibrotic macrophage, Nat. Immunol., 20, 163-172,
by pseudotemporal ordering of single cells, Nat.
doi: 10.1038/s41590-018-0276-y.
Biotechnol., 32, 381-386, doi: 10.1038/nbt.2859.
62. Ianevski, A., Giri, A. K., and Aittokallio, T. (2022)
73. Ji, Z., and Ji, H.
(2016) TSCAN: Pseudo-time
Fully-automated and ultra-fast cell-type identification
reconstruction and evaluation in single-cell RNA-
using specific marker combinations from single-cell
seq analysis, Nucleic Acids Res.,
44, e117-e117,
transcriptomic data, Nat. Commun., 13, 1-10, doi:
doi: 10.1093/nar/gkw430.
10.1038/s41467-022-28803-w.
74. Street, K., Risso, D., Fletcher, R. B., Das, D.,
63. Shao, X., Liao, J., Lu, X., Xue, R., Ai, N., and Fan, X.
Ngai, J., Yosef, N., Purdom, E., and Dudoit, S. (2018)
(2020) scCATCH: automatic annotation on cell types
Slingshot: cell lineage and pseudotime inference for
of clusters from single-cell RNA sequencing data,
single-cell transcriptomics, BMC Genomics, 19, 1-16,
iScience, 23, 100882, doi: 10.1016/j.isci.2020.100882.
doi: 10.1186/s12864-018-4772-0.
64. Guo, H., and Li, J. (2021) scSorter: assigning cells to
75. Saelens, W., Cannoodt, R., Todorov, H., and Saeys, Y.
known cell types according to marker genes, Genome
(2019) A comparison of single-cell trajectory inference
Biol., 22, 1-18, doi: 10.1186/s13059-021-02281-7.
methods, Nat. Biotechnol., 37, 547-554, doi: 10.1038/
65. Zhang, Z., Luo, D., Zhong, X., Choi, J. H., Ma, Y.,
s41587-019-0071-9.
Wang, S., Mahrt, E., Guo, W., Stawiski, E. W.,
76. Nam, D. K., Lee, S., Zhou, G., Cao, X., Wang, C.,
Modrusan, Z., Seshagiri, S., Kapur, P., Hon, G. C.,
Clark, T., Chen, J., Rowley, J. D., and Wang, S. M.
Brugarolas, J., and Wang, T. (2019) SCINA: a semi-
(2002) Oligo (dT) primer generates a high frequen-
supervised subtyping algorithm of single cells and bulk
cy of truncated cDNAs through internal poly (A)
samples, Genes, 10, 531, doi: 10.3390/genes10070531.
priming during reverse transcription, Proc. Natl.
66. Xu, C., Lopez, R., Mehlman, E., Regier, J., Jordan, M. I.,
Acad. Sci. USA, 99, 6152-6156, doi: 10.1073/pnas.
and Yosef, N. (2021) Probabilistic harmonization and
092140899.
annotation of single-cell transcriptomics data with
77. Perrimon, N., Pitsouli, C., and Shilo, B. Z. (2012)
deep generative models, Mol. Syst. Biol., 17, e9620,
Signaling mechanisms controlling cell fate and
doi: 10.15252/msb.20209620.
embryonic patterning, Cold Spring Harb. Perspect.
67. Hao, Y., Hao, S., Andersen-Nissen, E., Mauck III,
Biol., 4, a005975, doi: 10.1101/cshperspect.a005975.
W. M., Zheng, S., Butler, A., Lee, M. J., Wilk, A. J.,
78. Gaud, G., Lesourne, R., and Love, P. E. (2018)
Darby, C., Zager, M., et al. (2021) Integrated analy-
Regulatory mechanisms in T cell receptor signalling,
sis of multimodal single-cell data, Cell, 184, 3573-
Nat. Rev. Immunol.,
18,
485-497, doi:
10.1038/
3587.e3529, doi: 10.1016/j.cell.2021.04.048.
s41577-018-0020-8.
БИОХИМИЯ том 88 вып. 2 2023
194
ХОЗЯИНОВА и др.
79. Yeung, T. L., Sheng, J., Leung, C. S., Li, F., Kim, J.,
89. Fischer, D. S., Schaar, A. C., and Theis, F. J. (2021)
Ho, S. Y., Matzuk, M. M., Lu, K. H., Wong, S. T. C.,
Learning cell communication from spatial graphs of
and Mok, S. C.
(2019) Systematic identification
cells, bioRxiv, doi: 10.1101/2021.07.11.451750.
of druggable epithelial-stromal crosstalk signaling
90. Van Dam, S., Võsa, U., van der Graaf, A., Franke, L.,
networks in ovarian cancer, J. Natl. Cancer Institute,
and de Magalhães, J. P. (2018) Gene co-expression
111, 272-282, doi: 10.1093/jnci/djy097.
analysis for functional classification and gene-
80. Chua, R. L., Lukassen, S., Trump, S., Hennig, B. P.,
disease predictions, Brief. Bioinform., 19, 575-592,
Wendisch, D., Pott, F., Debnath, O., Thürmann, L.,
doi: 10.1093/bib/bbw139.
Kurth, F., Völker, M. T., Kazmierski, J., Timmer-
91. Rambow, F., Rogiers, A., Marin-Bejar, O., Aibar, S.,
mann, B., Twardziok, S., Schneider, S., Machleidt, F.,
Femel, J., Dewaele, M., Karras, P., Brown, D.,
Müller-Redetzky, H., Maier, M., Krannich, A.,
Chang, Y. H., Debiec-Rychter, M., Adriaens, C.,
Schmidt, S., Balzer, F., et al. (2020) COVID-19 se-
Radaelli, E., Wolter, P., Bechter, O., Dummer, R.,
verity correlates with airway epithelium-immune
Levesque, M., Piris, A., Frederick, D. T., Boland, G.,
cell interactions identified by single-cell analysis,
Flaherty, K. T., et al. (2018) Toward minimal residual
Nat. Biotechnol., 38, 970-979, doi: 10.1038/s41587-
disease-directed therapy in melanoma, Cell, 174, 843-
020-0602-4.
855.e819, doi: 10.1016/j.cell.2018.06.025.
81. Yuan, D., Tao, Y., Chen, G., and Shi, T. (2019)
92. Huynh-Thu, V. A., Irrthum, A., Wehenkel, L., and
Systematic expression analysis of ligand-receptor
Geurts, P. (2010) Inferring regulatory networks from
pairs reveals important cell-to-cell interactions inside
expression data using tree-based methods, PLoS One,
glioma, Cell Commun. Signal., 17, 48, doi: 10.1186/
5, e12776, doi: 10.1371/journal.pone.0012776.
s12964-019-0363-1.
93. Moerman, T., Aibar Santos, S., Bravo González-
82. Rao, V. S., Srinivas, K., Sujini, G. N., and Ku-
Blas, C., Simm, J., Moreau, Y., Aerts, J., and Aerts, S.
mar, G. N. (2014) Protein-protein interaction detec-
(2019) GRNBoost2 and Arboreto: efficient and scal-
tion: methods and analysis, Int. J. Proteomics, 2014,
able inference of gene regulatory networks, Bioinfor-
147648, doi: 10.1155/2014/147648.
matics, 35, 2159-2161, doi: 10.1093/bioinformatics/
83. Wang, Y., Wang, R., Zhang, S., Song, S., Jiang, C.,
bty916.
Han, G., Wang, M., Ajani, J., Futreal, A., and
94. Aibar, S., González-Blas, C. B., Moerman, T., Huynh-
Wang, L. (2019) iTALK: an R package to characterize
Thu, V. A., Imrichova, H., Hulselmans, G., Rambow,
and illustrate intercellular communication, bioRxiv,
F., Marine, J.-C., Geurts, P., and Aerts, J. (2017)
507871, doi: 10.1101/507871.
SCENIC: single-cell regulatory network inference and
84. Efremova, M., Vento-Tormo, M., Teichmann, S. A.,
clustering, Nat. Methods, 14, 1083-1086, doi: 10.1038/
and Vento-Tormo, R. (2020) CellPhoneDB: infer-
nmeth.4463.
ring cell-cell communication from combined expres-
95. Langfelder, P., and Horvath, S. (2008) WGCNA:
sion of multi-subunit ligand-receptor complexes,
an R package for weighted correlation network
Nat. Protocols, 15, 1484-1506, doi: 10.1038/s41596-
analysis, BMC Bioinformatics, 9, 1-13, doi: 10.1186/
020-0292-x.
1471-2105-9-559.
85. Cabello-Aguilar, S., Alame, M., Kon-Sun-Tack, F.,
96. Subramanian, A., Tamayo, P., Mootha, V. K.,
Fau, C., Lacroix, M., and Colinge, J.
(2020)
Mukherjee, S., Ebert, B. L., Gillette, M. A., Pau-
SingleCellSignalR: inference of intercellular networks
lovich, A., Pomeroy, S. L., Golub, T. R., Land-
from single-cell transcriptomics, Nucleic Acids Res.,
er, E. S., and Mesirov, J. P. (2005) Gene set en-
48, e55, doi: 10.1093/nar/gkaa183.
richment analysis: a knowledge-based approach for
86. Zhang, Y., Liu, T., Hu, X., Wang, M., Wang, J.,
interpreting genome-wide expression profiles, Proc.
Zou, B., Tan, P., Cui, T., Dou, Y., Ning, L., Huang, Y.,
Natl. Acad. Sci. USA, 102, 15545-15550, doi: 10.1073/
Rao, S., Wang, D., and Zhao, X. (2021) CellCall:
pnas.0506580102.
integrating paired ligand-receptor and transcript-
97. Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A.,
ion factor activities for cell-cell communication,
Wyder, S., Huerta-Cepas, J., Simonovic, M.,
Nucleic Acids Res., 49, 8520-8534, doi: 10.1093/
Doncheva, N. T., Morris, J. H., Bork, P., et al. (2019)
nar/gkab638.
STRING v11: protein-protein association networks
87. Tsuyuzaki, K., Ishii, M., and Nikaido, I.
(2019)
with increased coverage, supporting functional
Uncovering hypergraphs of cell-cell interaction from
discovery in genome-wide experimental datasets,
single cell RNA-sequencing data, bioRxiv, 566182,
Nucleic Acids Res., 47, D607-D613, doi: 10.1093/nar/
doi: 10.1101/566182.
gky1131.
88. Armingol, E., Officer, A., Harismendy, O., and
98. Kim, C. Y., Baek, S., Cha, J., Yang, S., Kim, E.,
Lewis, N. E. (2021) Deciphering cell-cell interac-
Marcotte, E. M., Hart, T., and Lee, I.
(2022)
tions and communication from gene expression,
HumanNet v3: an improved database of human gene
Nat. Rev. Genet., 22, 71-88, doi: 10.1038/s41576-
networks for disease research, Nucleic acids Res., 50,
020-00292-x.
D632-D639, doi: 10.1093/nar/gkab1048.
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
195
99. Xue, Z., Huang, K., Cai, C., Cai, L., Jiang, C.-Y.,
RNA-seq data, Gen. Res., 28, 1217-1227, doi: 10.1101/
Feng, Y., Liu, Z., Zeng, Q., Cheng, L., Sun, Y. E.,
gr.228080.117.
et al. (2013) Genetic programs in human and mouse
110. Serin Harmanci, A., Harmanci, A. O., and Zhou, X.
early embryos revealed by single-cell RNA sequencing,
(2020) CaSpER identifies and visualizes CNV events
Nature, 500, 593-597, doi: 10.1038/nature12364.
by integrative analysis of single-cell or bulk RNA-
100. Wu, H., Chen, S., Yu, J., Li, Y., Zhang, X.-Y.,
equencing data, Nat. Commun., 11, 1-16, doi: 10.1038/
Yang, L., Zhang, H., Jiang, M., Brunicardi, F. C.,
s41467-019-13779-x.
Wang, C., and Wu, S. (2018) Single-cell transcriptome
111. Gao, R., Bai, S., Henderson, Y. C., Lin, Y.,
analyses reveal molecular signals to intrinsic and
Schalck, A., Yan, Y., Kumar, T., Hu, M., Sei, E.,
acquired paclitaxel resistance in esophageal squamous
Davis, A., et al. (2021) Delineating copy number and
cancer cells, Cancer Lett., 420, 156-167, doi: 10.1016/
clonal substructure in human tumors from single-cell
j.canlet.2018.01.059.
transcriptomes, Nat. Biotechnol., 39, 599-608, doi:
101. Lu, J., Chen, Y., Zhang, X., Guo, J., Xu, K., and
10.1038/s41587-020-00795-2.
Li, L. (2022) A novel prognostic model based on
112. Menyailo, M. E., Zainullina, V. R., Tashireva, L. A.,
single-cell RNA sequencing data for hepatocellular
Zolotareva, S. Y., Gerashchenko, T. S., Alifanov, V. V.,
carcinoma, Cancer Cell Int., 22, 1-12, doi: 10.1186/
Savelieva, O. E., Grigoryeva, E. S., Tarabanovska-
s12935-022-02469-2.
ya, N. A., Popova, N. O., Khozyainova, A. A.,
102. Lee, W.-P., and Tzou, W.-S. (2009) Computational
Choinzonov, E. L., Cherdyntseva, N. V., Perelmu-
methods for discovering gene networks from expression
ter, V. M., and Denisov, E. V. (2022) Heterogeneity of
data, Brief. Bioinform., 10, 408-423, doi: 10.1093/
circulating epithelial cells in breast cancer at single-cell
bib/bbp028.
resolution: identifying tumor and hybrid cells, bioRxiv,
103. Matsumoto, H., Kiryu, H., Furusawa, C., Ko, M. S.,
doi: 10.1101/2021.11.24.469962.
Ko, S. B., Gouda, N., Hayashi, T., and Nikai-
113. Müller, S., Liu, S. J., Di Lullo, E., Malatesta, M.,
do, I. (2017) SCODE: an efficient regulatory net-
Pollen, A. A., Nowakowski, T. J., Kohanbash, G.,
work inference algorithm from single-cell RNA-Seq
Aghi, M., Kriegstein, A. R., Lim, D. A., and Diaz, A.
during differentiation, Bioinformatics, 33, 2314-2321,
(2016) Single-cell sequencing maps gene expression
doi: 10.1093/bioinformatics/btx194.
to mutational phylogenies in PDGF-and EGF-driven
104. Pös, O., Radvanszky, J., Buglyó, G., Pös, Z., Rus-
gliomas, Mol. Syst. Biol., 12, 889, doi: 10.15252/
nakova, D., Nagy, B., and Szemes, T.
(2021)
msb.20166969.
DNA copy number variation: Main character-
114. McKenna, A., Hanna, M., Banks, E., Sivachenko, A.,
istics, evolutionary significance, and pathologi-
Cibulskis, K., Kernytsky, A., Garimella, K., Altshul-
cal aspects, Biomed. J., 44, 548-559, doi: 10.1016/
er, D., Gabriel, S., Daly, M., and DePristo, M. A.
j.bj.2021.02.003.
(2010) The Genome Analysis Toolkit: a MapReduce
105. Lye, Z. N., and Purugganan, M. D. (2019) Copy
framework for analyzing next-generation DNA se-
number variation in domestication, Trends Plant Sci.,
quencing data, Gen. Res., 20, 1297-1303, doi: 10.1101/
24, 352-365, doi: 10.1016/j.tplants.2019.01.003.
gr.107524.110.
106. Zhao, Y., Carter, R., Natarajan, S., Varn, F. S., Comp-
115. Wu, T. D., Reeder, J., Lawrence, M., Becker, G., and
ton, D. A., Gawad, C., Cheng, C., and Godek, K. M.
Brauer, M. J. (2016) GMAP and GSNAP for Genomic
(2019) Single-cell RNA sequencing reveals the impact
Sequence Alignment: Enhancements to Speed,
of chromosomal instability on glioblastoma cancer
Accuracy, and Functionality, in Statistical Genomics,
stem cells, BMC Med. Genom., 12, 1-16, doi: 10.1186/
Springer, pp. 283-334.
s12920-019-0532-5.
116. Li, H., Handsaker, B., Wysoker, A., Fennell, T.,
107. Zhou, B., Ho, S. S., Zhang, X., Pattni, R., Harak-
Ruan, J., Homer, N., Marth, G., Abecasis, G., and
singh, R. R., and Urban, A. E. (2018) Whole-genome
Durbin, R.
(2009) The sequence alignment/map
sequencing analysis of CNV using low-coverage and
format and SAMtools, Bioinformatics, 25, 2078-2079,
paired-end strategies is efficient and outperforms
doi: 10.1093/bioinformatics/btp352.
array-based CNV analysis, J. Med. Genet., 55, 735-
117. Liu, F., Zhang, Y., Zhang, L., Li, Z., Fang, Q.,
743, doi: 10.1136/jmedgenet-2018-105272.
Gao, R., and Zhang, Z. (2019) Systematic comparative
108. Shao, X., Lv, N., Liao, J., Long, J., Xue, R., Ai, N.,
analysis of single-nucleotide variant detection methods
Xu, D., and Fan, X. (2019) Copy number variation
from single-cell RNA sequencing data, Gen. Biol., 20,
is highly correlated with differential gene expression:
1-15, doi: 10.1186/s13059-019-1863-4.
a pan-cancer study, BMC Med. Genet., 20, 1-14,
118. Ludwig, L. S., Lareau, C. A., Ulirsch, J. C., Chris-
doi: 10.1186/s12881-019-0909-5.
tian, E., Muus, C., Li, L. H., Pelka, K., Ge, W.,
109. Fan, J., Lee, H.-O., Lee, S., Ryu, D.-E., Lee, S.,
Oren, Y., Brack, A., et al. (2019) Lineage tracing in
Xue, C., Kim, S. J., Kim, K., Barkas, N., Park, P. J.,
humans enabled by mitochondrial mutations and
et al. (2018) Linking transcriptional and genetic tumor
single-cell genomics, Cell, 176, 1325-1339. e1322,
heterogeneity through allele analysis of single-cell
doi: 10.1016/j.cell.2019.01.022.
БИОХИМИЯ том 88 вып. 2 2023
196
ХОЗЯИНОВА и др.
119. Wilson, G. W., Derouet, M., Darling, G. E., and
130. Shlyueva, D., Stampfel, G., and Stark, A.
(2014)
Yeung, J. C. (2021) scSNV: accurate dscRNA-seq SNV
Transcriptional enhancers: from properties to genome-
co-expression analysis using duplicate tag collapsing,
wide predictions, Nat. Rev. Genet., 15, 272-286, doi:
Gen. Biol., 22, 1-27, doi: 10.1186/s13059-021-02364-5.
10.1038/nrg3682.
120. Yao, Y., and Dai, W. (2014) Genomic instability
131. Wray, G. A. (2007) The evolutionary significance of
and cancer, J. Carcinog. Mutagen., 5, 1000163, doi:
cis-regulatory mutations, Nat. Rev. Genet., 8, 206-216,
10.4172/2157-2518.1000165.
doi: 10.1038/nrg2063.
121. Fu, Y., Zhang, F., Zhang, X., Yin, J., Du, M.,
132. Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang,
Jiang, M., Liu, L., Li, J., Huang, Y., and Wang, J.
H. Y., and Greenleaf, W. J. (2013) Transposition of
(2019) High-throughput single-cell whole-genome
native chromatin for multimodal regulatory analysis
amplification through centrifugal emulsification and
and personal epigenomics, Nat. Methods, 10, 1213,
eMDA, Commun. Biol., 2, 1-10, doi: 10.1038/s42003-
doi: 10.1038/nmeth.2688.
019-0401-y.
133. Wu, S. J., Furlan, S. N., Mihalas, A. B., Kaya-
122. Schnepp, P. M., Chen, M., Keller, E. T., and Zhou, X.
Okur, H. S., Feroze, A. H., Emerson, S. N., Zheng, Y.,
(2019) SNV identification from single-cell RNA se-
Carson, K., Cimino, P. J., and Keene, C. D. (2021)
quencing data, Hum. Mol. Genet., 28, 3569-3583,
Single-cell CUT&Tag analysis of chromatin mod-
doi: 10.1093/hmg/ddz207.
ifications in differentiation and tumor progression,
123. Ramazzotti, D., Angaroni, F., Maspero, D., Ascola-
Nat. Biotechnol., 39, 819-824, doi: 10.1038/s41587-
ni, G., Castiglioni, I., Piazza, R., Antoniotti, M., and
021-00865-z.
Graudenzi, A. (2022) Variant calling from scRNA-
134. Zhou, W., Ji, Z., Fang, W., and Ji, H. (2019) Global
seq data allows the assessment of cellular identity in
prediction of chromatin accessibility using small-cell-
patient-derived cell lines, Nat. Commun., 13, 1-3,
number and single-cell RNA-seq, Nucleic Acids Res.,
doi: 10.1038/s41467-022-30230-w.
47, e121-e121, doi: 10.1093/nar/gkz716.
124. Zhou, Z., Xu, B., Minn, A., and Zhang, N. R. (2020)
135. Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S.
DENDRO: genetic heterogeneity profiling and sub-
(2015) The MEME suite, Nucleic Acids Res., 43,
clone detection by single-cell RNA sequencing, Ge-
W39-W49, doi: 10.1093/nar/gkv416.
nome Biol., 21, 1-15, doi: 10.1186/s13059-019-1922-x.
136. Schep, A. N., Wu, B., Buenrostro, J. D., and Green-
125. McCarthy, D. J., Rostom, R., Huang, Y., Kunz,
leaf, W. J. (2017) chromVAR: inferring transcrip-
D. J., Danecek, P., Bonder, M. J., Hagai, T., Lyu, R.,
tion-factor-associated accessibility from single-cell
Wang, W., Gaffney, D. J., Simons, B. D., Stegle, O.,
epigenomic data, Nat. Methods, 14, 975-978, doi:
and Teichmann, S. A. (2020) Cardelino: computational
10.1038/nmeth.4401.
integration of somatic clonal substructure and single-
137. Moody, J., Kouno, T., Suzuki, A., Shibayama, Y.,
cell transcriptomes, Nat. Methods,
17,
414-421,
Terao, C., Chang, J.-C., López-Redondo, F.,
doi: 10.1038/s41592-020-0766-3.
Yip, C. W., Ando, Y., Yamamoto, K., Carninci, P.,
126. Ciccolella, S., Ricketts, C., Soto Gomez, M., Patter-
Shin, J. W., and Hon, C.-C. (2021) Profiling of
son, M., Silverbush, D., Bonizzoni, P., Hajirasouli-
transcribed cis-regulatory elements in single cells,
ha, I., and Della Vedova, G. (2020) Inferring cancer
bioRxiv, doi: 10.1101/2021.04.04.438388
progression from Single-Cell Sequencing while al-
138. Pliner, H. A., Packer, J. S., McFaline-Figueroa, J. L.,
lowing mutation losses, Bioinformatics, 37, 326-333,
Cusanovich, D. A., Daza, R. M., Aghamirzaie, D.,
doi: 10.1093/bioinformatics/btaa722.
Srivatsan, S., Qiu, X., Jackson, D., and Minkina, A.
127. Mehrabadi, F. R., Marie, K. L., Pérez-Guijarro, E.,
(2018) Cicero predicts cis-regulatory DNA interac-
Malikić, S., Azer, E. S., Yang, H. H., Kızılkale, C.,
tions from single-cell chromatin accessibility data,
Gruen, C., Robinson, W., Liu, H., et al.
(2021)
Mol. Cell, 71, 858-871.e858, doi: 10.1016/j.molcel.
Profiles of expressed mutations in single cells reveal
2018.06.044.
subclonal expansion patterns and therapeutic
139. Oczko-Wojciechowska, M., Pfeifer, A., Jarzab, M.,
impact of intratumor heterogeneity, bioRxiv, doi:
Swierniak, M., Rusinek, D., Tyszkiewicz, T.,
10.1101/2021.03.26.437185.
Kowalska, M., Chmielik, E., Zembala-Nozynska, E.,
128. Ziegenhain, C., Vieth, B., Parekh, S., Reinius, B.,
Czarniecka, A., et al. (2020) Impact of the tumor
Guillaumet-Adkins, A., Smets, M., Leonhardt, H.,
microenvironment on the gene expression profile in
Heyn, H., Hellmann, I., and Enard, W.
(2017)
papillary thyroid cancer, Pathobiology, 87, 143-154,
Comparative analysis of single-cell RNA sequencing
doi: 10.1159/000507223.
methods, Mol. Cell, 65, 631-643.e634, doi: 10.1016/
140. Pape, J., Magdeldin, T., Stamati, K., Nyga, A.,
j.molcel.2017.01.023.
Loizidou, M., Emberton, M., and Cheema, U.
129. Kim, T.-K., and Shiekhattar, R. (2015) Architectural
(2020) Cancer-associated fibroblasts mediate can-
and functional commonalities between enhancers
cer progression and remodel the tumouroid stro-
and promoters, Cell, 162, 948-959, doi: 10.1016/
ma, Br. J. Cancer, 123, 1178-1190, doi: 10.1038/
j.cell.2015.08.008.
s41416-020-0973-9.
БИОХИМИЯ том 88 вып. 2 2023
ВОЗМОЖНОСТИ КОМПЛЕКСНОГО АНАЛИЗА ДАННЫХ
197
141. Liu, J., Li, P., Wang, L., Li, M., Ge, Z., Noordam, L.,
146. Moses, L., and Pachter, L. (2022) Museum of spatial
Lieshout, R., Verstegen, M. M., Ma, B., and Su, J.
transcriptomics, Nat. Methods,
19,
534-546, doi:
(2021) Cancer-associated fibroblasts provide a
10.1038/s41592-022-01409-2.
stromal niche for liver cancer organoids that confers
147. Hahaut, V., Pavlinic, D., Carbone, W., Schuierer, S.,
trophic effects and therapy resistance, Cell. Mol.
Balmer, P., Quinodoz, M., Renner, M., Roma, G.,
Gastroenterol. Hepatol., 11, 407-431, doi: 10.1016/
Cowan, C. S., and Picelli, S. (2022) Fast and highly
j.jcmgh.2020.09.003.
sensitive full-length single-cell RNA sequencing
142. Moriel, N., Senel, E., Friedman, N., Rajewsky, N.,
using FLASH-seq, Nat. Biotechnol., 40, 1447-1451,
Karaiskos, N., and Nitzan, M. (2021) NovoSpaRc:
doi: 10.1038/s41587-022-01312-3.
flexible spatial reconstruction of single-cell gene
148. Cao, J., Cusanovich, D. A., Ramani, V., Aghamir-
expression with optimal transport, Nat. Protocols, 16,
zaie, D., Pliner, H. A., Hill, A. J., Daza, R. M.,
4177-4200, doi: 10.1038/s41596-021-00573-7.
McFaline-Figueroa, J. L., Packer, J. S., Christian-
143. Ren, X., Zhong, G., Zhang, Q., Zhang, L., Sun, Y.,
sen, L., et al. (2018) Joint profiling of chromatin ac-
and Zhang, Z. (2020) Reconstruction of cell spatial
cessibility and gene expression in thousands of single
organization from single-cell RNA sequencing data
cells, Science, 361, 1380-1385, doi: 10.1126/science.
based on ligand-receptor mediated self-assembly, Cell
aau0730.
Res., 30, 763-778, doi: 10.1038/s41422-020-0353-2.
149. Chen, S., Lake, B. B., and Zhang, K. (2019) High-
144. Chen, L., and Flies, D. B. (2013) Molecular mecha-
throughput sequencing of the transcriptome and
nisms of T cell co-stimulation and co-inhibition, Nat.
chromatin accessibility in the same cell, Nat. Bio-
Rev. Immunol., 13, 227-242, doi: 10.1038/nri3405.
technol.,
37,
1452-1457, doi:
10.1038/s41587-019-
145. Ramilowski, J. A., Goldberg, T., Harshbarger, J.,
0290-0.
Kloppmann, E., Lizio, M., Satagopam, V. P., Itoh, M.,
150. Zachariadis, V., Cheng, H., Andrews, N., and
Kawaji, H., Carninci, P., Rost, B., and Forrest, A. R.
Enge, M. (2020) A highly scalable method for joint
(2015) A draft network of ligand-receptor-mediated
whole-genome sequencing and gene-expression pro-
multicellular signalling in human, Nat. Commun., 6,
filing of single cells, Mol Cell, 80, 541-553.e545,
7866, doi: 10.1038/ncomms8866.
doi: 10.1016/j.molcel.2020.09.025.
OPPORTUNITIES OF COMPLEX ANALYSIS
IN SINGLE-CELL RNA SEQUENCING
Review
A. A. Khozyainova1*, A. A. Valyaeva2, M. S. Arbatsky2, S. V. Isaev3,4,
P. S. Iamshchikov1,5, E. V. Volchkov6, M. S. Sabirov7, V. R. Zainullina1, V. I. Chechekhin2,
R. S. Vorobev1, M. E. Menyailo1, P. A. Tyurin-Kuzmin2, and E. V. Denisov1
1 Cancer Research Institute Tomsk NRMC,
634050 Tomsk, Russia; e-mail: khozyainova@onco.tnimc.ru
2 Lomonosov Moscow State University, 119991 Moscow, Russia
3 Research Institute of Personalized Medicine,
National Center for Personalized Medicine of Endocrine Diseases,
The National Medical Research Center for Endocrinology, 117036 Moscow, Russia
4 Phystech School of Biological and Medical Physics,
Moscow Institute of Physics and Technology (National Research University),
115184 Dolgoprudny, Russia
5 National Research Tomsk State University, 634050 Tomsk, Russia
6 Dmitry Rogachev National Research Center of Pediatric Hematology, Oncology and Immunology,
117198 Moscow, Russia
7 Koltzov Institute of Developmental Biology, 119334 Moscow, Russia
Single-cell RNA sequencing (scRNA-seq) is a revolutionary tool for studying the physiology of normal
and pathologically altered tissues. This approach provides information about the molecular features
(gene expression, mutations, chromatin accessibility, etc.) of cells, opens up the possibility to analyze cell
differentiation trajectories/phylogeny and cell-cell interactions and allows discovering new cell types and
previously unexplored processes. From a clinical point of view, scRNA-seq allows a deeper and more detailed
analysis of the molecular mechanisms of various diseases and serves as the basis for the development of new
БИОХИМИЯ том 88 вып. 2 2023
198
ХОЗЯИНОВА и др.
preventive, diagnostic and therapeutic solutions. This review describes the different approaches to analysis of
scRNA-seq data, reviews the strengths and weaknesses of bioinformatic tools, provides recommendations and
examples of their successful use and suggests potential directions for improvement. It also emphasizes the need
to create new, including multi-omics, protocols for the preparation of DNA/RNA libraries of single cells in
order to obtain a more complete and systematic understanding of each cell.
Keywords: single-cell RNA sequencing, cell cycle, clustering, differential expression, cell type, trajectory inference,
cell-cell interaction, gene regulatory network, copy number variation, single nucleotide variant, phylogenetics,
epigenomics, spatial transcriptomics
БИОХИМИЯ том 88 вып. 2 2023