БИОХИМИЯ, 2023, том 88, вып. 3, с. 508 - 527
УДК 577.113;616.8-056.7
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
Обзор
© 2023 М.И. Шадрина*, П.А. Сломинский
ФГБУ Институт молекулярной генетики
Национального исследовательского центра «Курчатовский институт»,
123182 Москва, Россия; электронная почта: shadrina@img.ras.ru
Поступила в редакцию 02.11.2022
После доработки 25.01.2023
Принята к публикации 25.01.2023
В 2022 году исполняется 25 лет с того момента, когда была идентифицирована первая мутация при
семейной аутосомно-доминантной форме болезни Паркинсона. За эти годы наши представления
о роли генетических факторов в патогенезе семейной и идиопатической форм болезни Паркин-
сона существенно расширились - был выявлен ряд генов семейной формы заболевания, выявлены
ДНК-маркеры повышенного риска развития спорадической формы заболевания. Но, несмотря на
все достигнутые успехи, мы далеки от точной оценки вклада в развитие заболевания как генети-
ческих факторов, так и тем более факторов эпигенетических. В обзоре суммирована накопленная
к настоящему времени информация о генетической архитектуре болезни Паркинсона и сформули-
рованы вопросы, требующие решения и связанные в первую очередь с оценкой эпигенетических
факторов в патогенезе заболевания.
КЛЮЧЕВЫЕ СЛОВА: Болезнь Паркинсона, моногенная форма, спорадическая форма, генетика, анализ
мутаций, полногеномный ассоциативный анализ, эпигенетика, генетический риск.
DOI: 10.31857/S0320972523030107, EDN: QXIEYO
ВВЕДЕНИЕ.
и при учете некоторых особенностей наследо-
БОЛЕЗНЬ ПАРКИНСОНА:
вания доля таких форм может достигать 20%.
КЛИНИЧЕСКАЯ КАРТИНА И ПАТОГЕНЕЗ
В основе развития моторных нарушений
при БП лежит избирательная гибель дофамин-
Болезнь Паркинсона (БП) - нейродегене-
ергических нейронов в черной субстанции
ративное неуклонно прогрессирующее заболе-
(substantia nigra pars compacta) со значитель-
вание, которое характеризуется в первую оче-
ным снижением уровня дофамина в стриату-
редь тетрадой моторных нарушений - таких
ме. Это приводит к нарушению нормального
как тремор (в первую очередь рук), мышечная
функционирования базальных ядер головного
ригидность, брадикенезия и постуральная не-
мозга с нарушением контроля за моторным
устойчивость. По частоте встречаемости среди
поведением. Процессу нейрональной гибели
нейродегенеративных заболеваний БП усту-
в большинстве случаев предшествует появ-
пает только болезни Альцгеймера, и в возрасте
ление так называемых телец Леви и нейритов
более 80 лет симптомы паркинсонизма наблю-
Леви - цитоплазматических включений, со-
даются как минимум у 2% населения в разных
стоящих в первую очередь из фибриллярного
странах мира [1]. Принято выделять семейную
альфа-синуклеина, убиквитина, миелин-ассо-
и спорадическую (или идиопатическую) фор-
циированного белка тау и ряда других белков.
мы БП, причем фенотипически эти формы
Необходимо отметить, что тельца и нейриты
практически не различимы. Семейные формы
Леви выявляют и в других структурах мозга
составляют от 5 до 10% всех случаев заболе-
(дорзальном моторном ядре, таламусе, мин-
вания, но эта оценка может быть занижена, далевидном теле, ядре шва, обонятельных
Принятые сокращения: БП - болезнь Паркинсона; CNV - варианты числа копий ДНК; GWAS - полногеномные
ассоциативные исследования.
* Адресат для корреспонденции.
508
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
509
луковицах, коре больших полушарий), и в свя-
p.Glu46Lys, p.Gly51Asp, p.Ala53Glu, p.Ala53Thr),
зи с этим БП не следует рассматривать как за-
и было показано, что к развитию заболевания
болевание, ограниченное только одним типом
могут приводить мутации с изменением дозы
нейронов и несколькими структурами моз-
гена (дупликации, трипликации, квадрупли-
га [2, 3]. Включения по типу телец Леви най-
кации) без каких-либо нарушений структуры
дены и в периферической нервной системе, в
белка [7]. Частота встречаемости мутаций в
частности, в нейронах подслизистой оболочки
этом гене невелика. Мутации выявлены у при-
кишечника. Вероятно, что эти включения мо-
мерно 0,2% и 1-2% пациентов со спорадиче-
гут образовываться на первом этапе развития
ской и семейной формами заболевания соот-
патологического процесса в различных отде-
ветственно, но они играют роль связующего
лах нервной системы и постепенно распро-
звена между этими формами заболевания и
страняться на другие отделы мозга. Именно
указывают на принципиально важную роль
это лежит в основе неуклонного прогресси-
нарушений структуры и функции альфа-синук-
рования заболевания. Причем на первых его
леина в патогенезе заболевания [8].
стадиях оно не затрагивает дофаминергиче-
Число идентифицированных генов семей-
скую систему черной субстанции и стриатума
ной формы БП неуклонно росло, и к настоя-
с нарушением моторного поведения. На про-
щему времени выявлено более 10 генов, му-
дромальной стадии заболевания наблюдают-
тации в которых однозначно ведут к мендели-
ся нарушения сна и обоняния, дисфункция
рующей БП. В табл. 1 приведено краткое опи-
кишечника и мочеполовой системы, но эти
сание генов c наиболее строго доказанной и не
изменения не специфичны и не могут служить
однократно подтвержденной в различных ис-
критериями диагностики БП. Моторные же
следованиях патогенетической ролью. Но не-
нарушения наблюдаются при гибели значи-
обходимо подчеркнуть, что дискуссия о роли
тельной части дофаминергических нейронов
этих и ряда других генов в этиопатогенезе БП
черной субстанции (не менее 50%) и снижении
продолжается.
уровня дофамина в стриатуме на 70-80% [4].
Один из наиболее ярких примеров такой
На компенсацию снижения уровня дофамина
дискуссии - переоценка роли гена убикви-
в стриатуме направлены основные методы те-
тин карбоксигидролазы UCHL1 (PARK5) (не
рапии заболевания и в первую очередь тера-
включенного нами в табл. 1) в патогенезе БП.
пия предшественником дофамина - леводо-
Впервые мутации в этом гене были выявлены
пой. Кроме этого, на первых этапах лечения
в 1998 г., когда в семье из Германии с позд-
могут использоваться агонисты дофамина или
ним развитием заболевания была выявлена
ингибиторы моноаминоксидазы В или катехол-
косегрегирующая с ним миссенс-мутация
О-метилтрансферазы. На поздних стадиях ис-
p.Ile193Met [9]. Однако с тех пор не удалось
пользуют ряд препаратов, направленных на
выявить ни одного семейного случая БП, свя-
блокирование побочных эффектов леводопы,
занного с мутацией в гене UCHL1 [10, 11].
таких как амантадин при дискинезиях или
В то же время был выявлен ряд полиморф-
апоморфин - при развитии навязчивых со-
ных вариантов гена UCHL1, один из которых
стояний. Но в любом случае терапия является
(миссенс-вариант p.Ser18Tyr) активно изучал-
симптоматической и не направлена на модифи-
ся у спорадических пациентов с БП. Эти ассо-
кацию причин развития нейродегенерации [5].
циативные исследования давали противоречи-
вые результаты, но проведенный метаанализ
выборки размером более 6500 пациентов не
МОНОГЕННЫЕ ФОРМЫ
выявил какой-либо ассоциации этого поли-
БОЛЕЗНИ ПАРКИНСОНА
морфизма с развитием заболевания при ис-
пользовании доминантной, рецессивной и
Впервые точная генетическая природа се-
аддитивной моделей [12]. Необходимо под-
мейной формы БП была показана в 1997 г., ко-
черкнуть, что данный метаанализ был прове-
гда была выявлена миссенс-мутация p.Ala30Thr
ден в первую очередь у пациентов-европеоидов
в гене альфа-синуклеина SNCA в большой не-
и позднее были получены данные о возможной
мецкой семье с аутосомно-доминантным нас-
роли данного полиморфизма в развитии БП в
ледованием БП в четырех поколениях и в
японской популяции [13], но и в этом случае
трех неродственных греческих семьях, где за-
проведенный позднее метаанализ также ис-
болевание наблюдалось в двух и трех поко-
ключил влияние полиморфизма p.Ser18Tyr на
лениях [6]. Позднее в этом гене было иден-
риск развития заболевания у азиатских по-
тифицировано еще несколько патогенетиче-
пуляций [14] как в выборке в целом, так и в
ски значимых миссенс-мутаций (p.Ala30Pro,
селектированных по этничности подвыборках.
БИОХИМИЯ том 88 вып. 3 2023
510
ШАДРИНА, СЛОМИНСКИЙ
БИОХИМИЯ том 88 вып. 3 2023
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
511
Анализ трансгенных мышей с мутациями в
Повышенный риск развития БП выяв-
гене белка убиквитин карбоксигидролазы дал
лен и у гетерозиготных носителей мутаций в
противоречивые результаты. Так, у мышей с
гене GBA - развитие заболевания выявлено
внутригенной делеции в гомологе гена UCHL1
у примерно 10% носителей мутаций, а пене-
наблюдалось снижение уровня моноубикви-
трантность этих мутаций оценивается в 30%
тина с образованием in vivo белковых вклю-
в возрасте 80 лет [23, 24]. Наиболее вероятно,
чений, но без признаков нейродегенерации
что все вызывающие болезнь Гоше мутации
в области черной субстанции [15]. В то же
повышают риск развития БП, причем вероят-
время у трансгенных мышей с мутантным
ность развития паркинсонизма коррелирует с
геном UCHL1 p.193Met в возрасте 20 месяцев
тяжестью клинической картины болезни Гоше,
была выявлена нейродегенерация в области
наблюдаемой у носителей этих мутаций. К на-
черной субстанции с нарушением спонтанной
стоящему времени у больных БП выявлено
двигательной активности [16], которая сопро-
более 130 патогенетически значимых мутаций
вождалась образованием белковых включений
в гене GBA, причем в различных этнических
и нарушением обмена альфа-синуклеина [17].
группах преобладают разные мутации. Так, у
В случае полиморфизма p.Ser18Tyr вариант
евреев-ашкеназов БП связана в основном с му-
18Tyr, в отличие от белка дикого типа, обла-
тациями p.Arg496His, p.Asn370Ser и 84insGC,
дает антиоксидантной активностью как in vitro
а у европеоидов - с мутациями p.Asn370Ser,
в культурах нейрональных клеток, так и in vivo
p.Leu444Pro, p.Arg120Trp, IVS2 + 1G > A,
при введении трансгена в область черной суб-
p.His255Gln,
p.Asp409His,
p.Glu326Lys,
станции [18, 19], что может снижать риск раз-
p.Thr369Мet. В разных популяциях также от-
вития процессов нейродегенерации у носите-
личается частота встречаемости мутантных ва-
лей варианта 18Tyr.
риантов гена GBA, что может влиять на оценку
В итоге совокупность генетических и моле-
кумулятивного риска развития GBA-ассоции-
кулярно-биологических данных не позволяет
рованной БП в разных этнических группах.
однозначно решить вопрос о роли гена UCHL1
Но в любом случае мутации в гене GBA повы-
в патогенезе БП, хотя пока позиция «против»
шают риск развития заболевания с очень дале-
кажется более аргументированной.
кой от 100% пенетрантности.
Второй не включенный в табл. 1 ген - ген
Сниженная пенетрантность наблюдается
глюкоцереброзидазы GBA. Мутации в гене этой
и в случае мутаций в гене киназы 2 с лейцин-
лизосомальной гидролазы были впервые опи-
богатыми повторами (LRRK2 или дардарина).
саны при болезни Гоше - системном заболе-
Ген LRRK2 исторически рассматривается как
вании с нарушением кроветворения и повы-
ген аутосомно-доминантной формы заболе-
шенным риском переломов и разной степенью
вания с пенетрантностью, близкой к 100%.
выраженности неврологических нарушений.
Однако из более чем 100 описанных мута-
В настоящее время в гене GBA описано более
ций в этом гене только для 6 (p.Gly2019Ser,
300 патогенетически значимых мутаций, и в
p.Arg1441Cys/Gly/His, Tyr1699Cys, Ile2020Thr)
данном случае нет никаких сомнений в том,
описана четкая семейная косегрегация заболе-
что мутации в этом гене играют важную роль
вания. Однако и в этом случае пенетрантность
в развитии БП как в гетерозиготном, так и в
мутаций не достигает 100% [25]. Для мутации
гомозиготном или компаундно-гетерозиготном
p.Gly2019Ser она достигает 85% в возрасте 80 лет
состояниях [20-23].
[26], причем ее величина отличается в разных
Но проблема состоит в том, что даже у
этнических группах. Еще более низкой ока-
гомозигот по мутациям в гене глюкоцеребро-
залась пенетрантность для мутаций в кодо-
зидазы GBA симптоматика паркинсонизма раз-
не 1441 [27], причем она отличалась для раз-
вивается только у части носителей мутаций и
ных миссенс-вариантов этой мутации. Точно
независимо от наличия клинического фено-
количественно оценить пенетрантность четы-
типа болезни Гоше. Так, у гомозигот по частой
рех остальных мутаций не удалось в связи с их
и относительно мягкой мутации Asn370Ser (ве-
низкой частотой, но и в этом случае очевидно,
дущей к болезни Гоше типа 1 без выраженных
что она не является полной.
неврологических нарушений) паркинсонизм
Таким образом, выявление в гене LRRK2
развивается только у примерно 9% лиц с таким
мутаций с патогенетически доказанной значи-
генотипом [20]. Компаундные гетерозиготы по
мостью указывает на повышенный риск раз-
этой мутации выявлены у примерно 40% паци-
вития БП. Но точная оценка этого риска на
ентов с болезнью Гоше и паркинсонизмом -
основании только факта выявления мутации
таким образом, суммарно она выявляется
невозможна. Это в целом затрудняет оценку
у 49% пациентов с данным фенотипом.
вклада мутаций в этом гене в риск развития
БИОХИМИЯ том 88 вып. 3 2023
512
ШАДРИНА, СЛОМИНСКИЙ
именно семейных форм БП, и имеющиеся в ли-
и у гетерозиготных носителей мутаций в гене
тературе оценки (согласно которым мутации в
PINK1, но в обоих случаях до настоящего вре-
гене LRRK2 находят у 5% пациентов с семей-
мени не проведено проспективных исследова-
ными формами заболевания и у 1% больных
ний для оценки развития БП у носителей ге-
с идиопатической БП) следует рассматривать
терозиготных мутаций [39]. С другой стороны,
как ориентировочные [28].
не было выявлено статистически достоверных
Крайне интересна ситуация с геном пар-
отличий в частоте гетерозиготных делеций гена
кина PRKN. Диаллельные мутации в этом
паркина при CNV-анализе больших (более
гене с потерей функции в гомозиготном или
2000 человек) выборок пациентов с БП и здо-
компаундном гетерозиготном состоянии ве-
ровых лиц [40].
дут к развитию аутосомно-рецессивной юве-
В целом считается, что моногенные фор-
нильной формы БП, для которой характе-
мы БП наблюдаются у 5-10% пациентов [41].
рен медианный возраст клинического дебюта
При этом основной вклад в развитие соответ-
в 31 год, причем у 16% пациентов первые при-
ственно аутосомно-доминантных и аутосомно-
знаки заболевания наблюдаются в возрасте
рецессивных моногенных форм вносят гены
до 20 лет [29]. В целом считается, что от 10
LRRK2 и PRKN. Необходимо подчеркнуть, что
до 20% пациентов с возрастом дебюта до 40 лет
опубликовано крайне мало работ с широкомас-
являются носителями диаллельных мутаций
штабным анализом большого числа генов. Так,
в гене PRKN, хотя данные по разным популя-
в Бразилии был проведен метаанализ ряда вы-
циям достаточно сильно отличаются [30-32].
полненных в этой стране исследований, охва-
Подавляющую часть этих мутаций составляют
тывающих все основные гены БП. В итоге ока-
возникшие в результате неравного гомологич-
залось, что в не селектированной по семейному
ного кроссинговера большие делеции/дупли-
характеру заболевания выборке самыми часты-
кации, захватывающие несколько экзонов
ми оказались точковые мутации в гене LRRK2
гена PRKN и приводящие к сдвигу рамки счи-
(выявлена у 2,5% пациентов, причем в 2,2% слу-
тывания с образованием нефункционального
чаев найдена мутация p.Gly2019Ser). Мутации
варианта белка паркина. Однако оказалось, что
в гене паркина выявлены у 8,3% пациентов.
такие мутации в гетерозиготном состоянии об-
В Ирландии анализ был ограничен только мута-
наруживаются у пациентов со спорадической
циями в генах PRKN, DJ1, PINK1 и пациентами
формой БП [33-35]. Эти делеции/дуплика-
с ранним возрастом клинического дебюта, и
ции также были выявлены и при популяцион-
были выявлены только мутации в гене PRKN
ных исследованиях здоровых лиц у примерно
у 6,9% обследованных больных. Анализ тех же
5% обследованных [36] - то есть их частота
трех генов был проведен у пациентов с EOPD
превышает популяционную частоту мутаций в
из Центральной Европы (в основном из Поль-
гене дардарина. В связи с этим их можно рас-
ши), мутации в гене PRKN были обнаружены
сматривать как аутосомно-доминантные, при-
у 3,1% пациентов [30, 31, 42]. Часто мутацион-
водящие к форме БП с классическим дебютом
ный скрининг ограничивается только мутацией
в возрасте более 55-60 лет, или как фактор
p.Gly2019Ser - она изучена в большом числе по-
риска развития заболевания (по аналогии с
пуляций мира, и в большинстве из них ее частота
мутациями в гене GBA). Потенциальная пато-
как при семейных, так и спорадических случаях
генетическая роль этих мутаций в гетерозигот-
заболевания не превышает 3-5% [43].
ном состоянии была подтверждена при анализе
Все это говорит о низкой частоте патоге-
функциональной активности нигростриатной
нетически значимых мутаций в мажорных ге-
системы головного мозга использованием
нах семейных форм заболевания, выраженной
флюро-ДОФА [37]. У гетерозиготных носите-
межпопуляционной гетерогенности и необ-
лей мутаций было выявлено снижение уровня
ходимости продолжения работ как по анализу
дофамина в хвостатом ядре и скорлупе, кото-
спектра мутаций при БП с включением в ис-
рое, однако, менее выражено, чем у пациен-
следование более широкой панели генов, так
тов с идиопатической болезнью Паркинсона.
и по поиску новых генов моногенных форм
Такое снижение уровня дофамина вызывает
заболевания [44, 45]. Ряд авторов предлагает
компенсаторную активацию активности в об-
свои варианты панели генов семейных форм
ласти правой дорзальной премоторной коры и
заболевания, но эти панели достаточно силь-
ростральной дополнительной моторной обла-
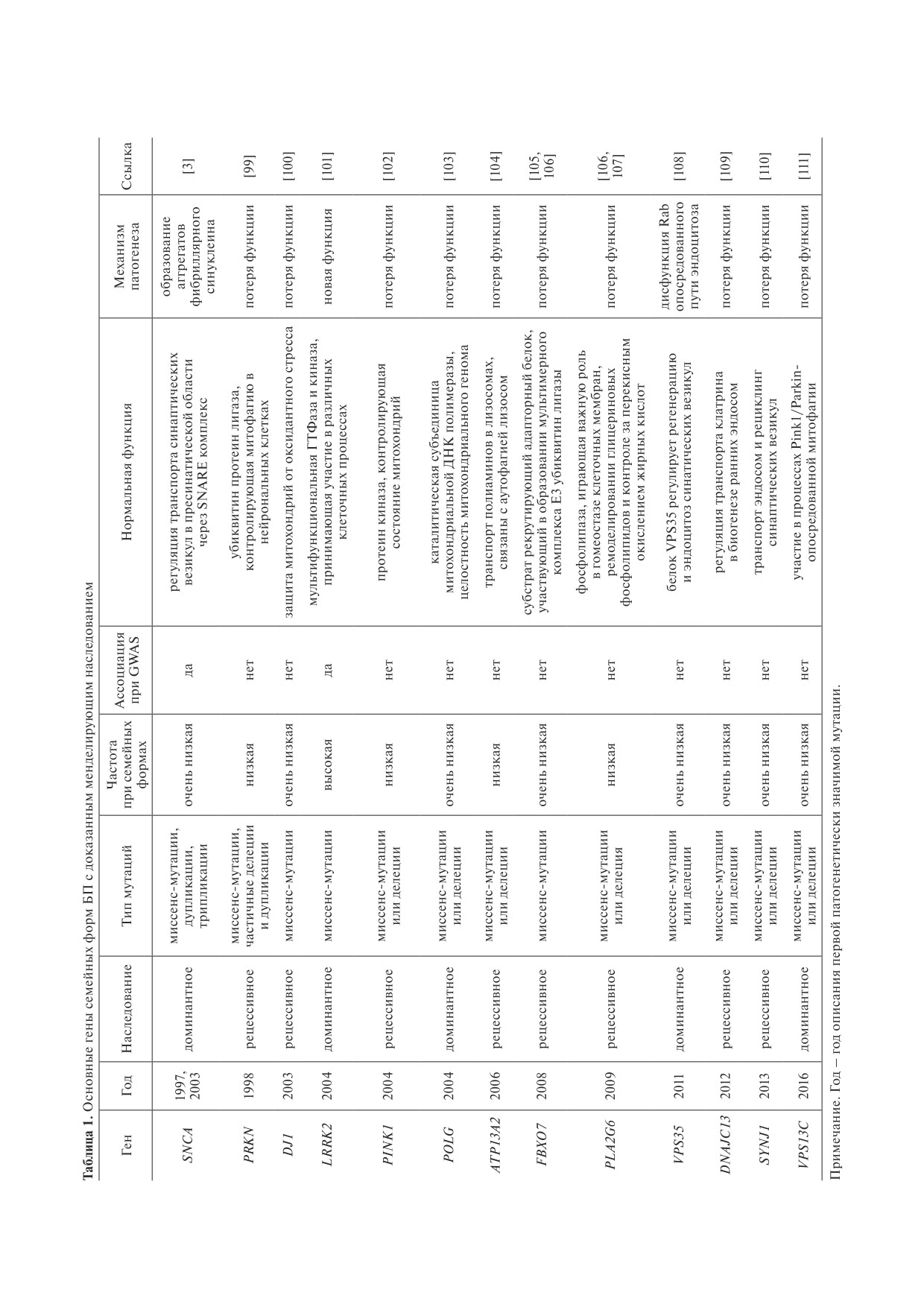
но отличаются (рисунок). В итоге имеющейся
сти [37, 38], что замедляет развитие моторных
в настоящее время информации оказывается
нарушений у гетерозиготных носителей мута-
крайне недостаточно для внедрения в прак-
ций в гене паркина на фоне нарушения обмена
тику алгоритмов генетического тестирования
дофамина. Аналогичная картина наблюдается
риска развития БП. Разработан ряд сильно
БИОХИМИЯ том 88 вып. 3 2023
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
513
Отсутствие полного консенсуса по генам моногенных форм болезни Паркинсона. По периферии в голубых блоках -
предложенные разными авторами гены моногенных форм [7, 41, 47, 48]. В центральном блоке красным выделены гены,
упомянутые в каждом из списков, а зеленым - упомянутые как минимум дважды
отличающихся по числу и набору генов ком-
выборках и с использованием ограниченного
мерческих панелей для такого тестирования,
набора ДНК-маркеров и не дали каких-либо
но ни одна из них не обладает, по мнению экс-
высоко достоверных результатов [49, 50], и, по
пертов, достаточной информативностью без
сути, оказались пробой пера, направленной на
включения в анализ дополнительных данных,
отработку технологии проведения GWAS при
описывающих семейный анамнез и фенотипи-
нейродегенеративных заболеваниях. Такой же
ческие особенности тестируемого. При этом
пробой пера стал и первый метаанализ дан-
минимальная панель из пяти общих для всех
ных GWAS, в рамках которого был проведен
панелей генов (PRKN, LRRK2, SNCA, PINK1,
совместный анализ двух ранее упомянутых
PARK7) по своей эффективности практиче-
работ [51]. Этот анализ позволил выявить три
ски не отличается от максимальной панели
однонуклеотидных ДНК-маркера, ассоцииро-
из 43 генов [46].
ванных с развитием заболевания вне зависи-
мости от используемой стратегии метаанализа,
но ни один из них не достигал уровня полноге-
ПОЛНОГЕНОМНЫЙ АССОЦИАТИВНЫЙ
номой значимости (р < 5⋅10-8). При этом стало
АНАЛИЗ ПРИ ИДИОПАТИЧЕСКОЙ
очевидно, что повышение информативности
БОЛЕЗНИ ПАРКИНСОНА
возможно при значительном увеличении раз-
меров анализируемых выборок (с перехо-
Очевидная важная роль генетических
дом от сотен человек в GWAS-исследованиях
факторов в патогенезе БП и сочетание семей-
2005-2006 гг. к десяткам тысяч - в более позд-
ных и идиопатических форм заболевания при
них работах) и повышении плотности анали-
явном преобладании последних стали осно-
зируемых ДНК-маркеров.
вой для проведения полногеномных ассоциа-
Сочетание технологического развития ме-
тивных исследований или GWAS при идио-
тодологии GWAS (повышение информативно-
патической форме заболевания. Первые такие
сти микрочипов, усовершенствование методов
исследования были проведены в 2005-2006 гг.
статистического анализа) привело к тому, что в
на относительно небольших по численности
настоящее время проведено генотипирование
11
БИОХИМИЯ том 88 вып. 3 2023
514
ШАДРИНА, СЛОМИНСКИЙ
десятков тысяч пациентов с БП и выполнен
очередь, снижает активность лизосомальных
ассоциативный анализ как для самого забо-
ферментов (в том числе GBA). В присутствии
левания, так и для его эндофенотипов (воз-
фибриллярного альфа-синуклеина наблюда-
раст клинического дебюта, характер моторных
ется усиление его фосфорилирования с обра-
нарушений, наличие сопутствующих психо-
зованием нерастворимых включений, в состав
эмоциональных расстройств, скорость про-
которых входит альфа-синуклеин. In vivo у
грессирования заболевания). Всего к настоя-
мышей с недостаточностью ТМЕМ175 наблю-
щему времени, по данным портала «GWAS
далась гибель дофаминергический нейронов
с развитием моторных нарушений
[55-58].
70 такого рода ассоциативных исследований,
Также с лизосомальной дисфункцией может
в результате которых обнаружена ассоциация
быть связан ассоциированный с развитием БП
заболевания с более чем 500 однонуклеотид-
ген LAMP3, кодирующий лизосомальный мем-
ными ДНК-маркерами с достоверностью бо-
бранно-ассоциированный белок и влияю-
лее
щий на процессы лизосомальной аутофагии
MONDO_0005180). Однако только для не-
и апоптоза, но его роль в функционировании
скольких генов выявлено по несколько ассо-
нейрональных клеток практически не изуча-
циированных с развитием заболевания ДНК-
лась [59-61]. Крайне мало изучена роль в па-
маркеров, и фактически с учетом числа ассо-
тогенезе БП белка BST1/CD157 - гликопро-
циированных с заболеванием маркеров и сте-
теина из суперсемейства ADP-рибозил циклаз,
пени достоверности ассоциации можно гово-
связываемого в первую очередь с аутоиммун-
рить о шести основных белок-кодирующих
ными, гематологическими и опухолевыми за-
генах, определяющих многофакторный риск
болеваниями. Однако полученные в послед-
развития БП и/или влияющих на клиническое
нее время данные говорят о важной роли этого
течение заболевания (табл. 2).
белка в регуляции обмена окситоцина и раз-
Также два из этих генов, SNCA и LRRK2,
витии нарушений поведения [62-64].
определяют развитие моногенных форм БП,
Подавляющая часть ассоциированных
и они же оказываются наиболее сильно ассо-
с развитием БП ДНК-маркеров оказывает
циированными с идиопатической формой за-
незначительное влияние на риск развития
болевания как по числу ассоциированных
заболевания - для подавляющего большин-
ДНК-маркеров, так и по связанному с ними
ства маркеров относительный риск (OR) по-
относительному риску. C развитием моноген-
вышен или понижен не более чем в 1,5 раза.
ных нейродегенеративных заболеваний свя-
Проведенный в 2019 г. [65] метаанализ всех
заны также мутации в гене ассоциированного
опубликованных к этому времени GWAS-
с микротрубочками тау-белка, но клинические
исследований (7,8 млн (с учетом импьютинга)
проявления этих мутаций связаны в первую
полиморфных ДНК-маркеров, 1,4 млн кон-
очередь с клинической картиной лобно-височ-
трольных образцов ДНК, 37 700 пациентов с
ного слабоумия и эссенциального тремора.
болезнью Паркинсона, 18 600 родственников
Характерные для БП мышечные нарушения
первой степени пациентов с болезнью Пар-
оказываются вторичными по отношению к
кинсона) позволил выявить всего 90 неза-
симптоматике деменции/тремора, и выявля-
висимых и принадлежащих к 78 геномным
емые при полногеномном анализе ассоциа-
областям ассоциированных с развитием забо-
ции отражают генетическую близость широ-
левания ДНК-маркеров. 38 из этих маркеров
кого спектра нейродегенеративных заболева-
ранее описаны не были, что связано в первую
ний [52-54].
очередь с увеличением размера выборок, по-
Роль трех остальных ассоциированных с
зволяющим добиться уровня полногеномной
развитием заболевания генов изучена в на-
значимости для слабо ассоциированных мар-
стоящее время недостаточно, но имеющиеся
керов (с достоверностью около 10-8). Для этих
данные об их функциональной активности
новых ДНК-маркеров была проведена оцен-
позволяют связать их с современной картиной
ка кумулятивного генетического риска (PRS)
этиопатогенеза БП.
развития БП, связанного с полиморфизмом
Ген TMEM175 кодирует трансмембран-
генома, и было показано, что она не превы-
ный лизосомальный белок - ионный канал
шает 36% (если оценка частоты заболевания
для ионов калия и протонов. Моделирование
в популяции у лиц в возрасте 80 лет и старше
недостаточности этого белка в культуре ней-
составляет 2%). При этом необходимо учиты-
рональных клеток in vitro показало, что сни-
вать, что в настоящее время не предложено
жение активности ТМЕМ175 приводит к нару-
общепринятой методологии оценки полило-
шению контроля за рН лизосом. Это, в свою
кусных рисков и, возможно, эта оценка может
БИОХИМИЯ том 88 вып. 3 2023
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
515
Таблица 2. Белок кодирующие гены и ДНК-маркеры, наиболее высоко достоверно ассоциированные с риском развития
идиопатической БП по данным полногеномных ассоциативных исследований
ГЕН
ДНК-маркер
Р
OR
СI
Ссылка
rs11724635
1 × 10-19
1,11
[1,09-1,14]
[112]
rs11724635
9 × 10-18
1,13
[1,1-1,15]
[113]
BST1/CD157
rs11724635
1 × 10-16
1,15
[1,11-1,19]
[114]
rs4266290
8 × 10-11
1,13
[1,08-1,17]
[115]
rs34311866
6 × 10-50
1,23
[1,20-1,27]
[112]
rs34311866
1 × 10-43
1,27
[1,24-1,30]
[113]
TMEM175
rs34311866
6 × 10-11
1,25
[1,17-1,34]
[116]
rs6599388
4 × 10-12
1,16
[1,12-1,20]
[114]
rs356182
5 × 10-123
1,33
[1,30-1,36]
[112]
rs356182
4 × 10-73
1,32
[1,29-1,35]
[113]
SNCA
rs356182
1 × 10-56
1,318
[1,27-1,36]
[115]
rs356219
6 × 10-65
1,29
[1,25-1,33]
[117]
rs12637471
2 × 10-30
1,17
[1,15-1,22]
[112]
rs12637471
2 × 10-21
1,18
[1,15-1,22]
[113]
MCCC1/LAMP3
rs11711441
8 × 10-12
1,19
[1.13-1,25]
[114]
rs10513789
3 × 10-10
1,25
[1,16-1,33]
[118]
rs17649553
1 × 10-68
1,28
[1,25-1,32]
[112]
MAPT
rs17649553
2 × 10-48
1,3
[1,27-1,34]
[113]
rs8070723
7 × 10-12
1,3
[1,19-1,43]
[119]
rs28903073
1 × 10-39
3,12
[2,68-3,62]
[115]
rs34637584
2 × 10-28
9,62
[6,43-14,37]
[118]
LRRK2
rs34778348
3 × 10-21
2,23
[1,89-2,63]
[117]
rs76904798
1 × 10-19
1,15
[1,12-1,19]
[112]
Примечание. Приведены гены, для которых в независимых исследованиях выявлено не менее трех ДНК-маркеров
с р ниже 10-10.
существенно измениться при внедрении в
мов. Дальнейшее продолжение исследований
практику стандартов для расчета PRS [66].
в области широкомасштабного секвенирова-
Таким образом, в настоящее время куму-
ния геномов и экзомов у пациентов позволит
лятивная оценка риска развития БП оказы-
выявить новые локусы и гены, ассоцииро-
вается не очень высокой как для моногенных
ванные с развитием заболевания. При этом
форм, так и для многофакторных вариантов
ключевую роль в такого рода исследованиях
заболевания. Один из возможных вариантов
будет играть интеграция получаемых пер-
связан с недооценкой роли в развитии забо-
вичных данных в большие международные
левания редких мутаций или полиморфиз-
базы данных, объединенных с программами
БИОХИМИЯ том 88 вып. 3 2023
11*
516
ШАДРИНА, СЛОМИНСКИЙ
функциональной аннотации таких редких
генетического риска, основанную на анали-
вариантов. Первый пример такой базы дан-
зе только однонуклеотидных ДНК-маркеров.
ных - база Gene4PD [47], которая интегрирует
С другой стороны, это указывает на важность
результаты всех опубликованных работ по по-
поиска эпигенетических факторов, связанных
иску патогенетически значимых генетических
с развитием заболевания - выявление тех или
вариантов БП и позволяет на одной платфор-
иных изменений эпигенома позволит лучше
ме провести аннотацию выявляемых вариан-
понять вызывающие их генетические причины.
тов с использованием данных по природе и
частоте встречаемости генетического вари-
анта, функциональной аннотации связанных с
ЭПИГЕНЕТИЧЕСКИЕ ФАКТОРЫ
ним белков, транскриптомных и метиломных
В РАЗВИТИИ БОЛЕЗНИ ПАРКИНСОНА
исследований. В итоге все гены на основании
анализа выявленных в них ассоциированных с
В последние годы интерес к роли эпигене-
развитием БП вариантов могут быть отнесены
тических механизмов и факторов в патогенезе
к кандидатным генам заболевания высокой,
нейродегенеративных заболеваний значитель-
средней и низкой достоверности.
но возрос [71, 72]. При этом активно анализи-
Как говорилось выше, в ряде случаев (му-
руются метилирование ДНК, модификация ги-
тации в генах паркина, PINK1, альфа-синук-
стонов, экспрессия различных не кодирующих
леина), кроме точечных мутаций, причиной
белок РНК (микроРНК, малые интерферирую-
заболевания могут быть мутации или поли-
щие РНК, длинные некодирующие РНК). Эпи-
морфизмы типа разных вариантов числа ко-
геном при этом рассматривается не только как
пий ДНК (copy number variation, СNV), при
причина заболевания, но и как возможная те-
которых наблюдается делеция или дуплика-
рапевтическая мишень.
ция фрагментов генома размером более 50 п.н.
Однако при рассмотрении вопросов, свя-
Мутации такого типа могут вносить важный
занных с эпигенетическими механизмами БП,
вклад в общую вариабельность генома [67], и
необходимо учитывать влияние на результаты
включение их в анализ генетического риска
проведенных исследований лимитирующих
многофакторных заболеваний может суще-
факторов.
ственно изменить наши оценки кумулятивного
Первый фактор связан с тем, что подав-
генетического риска. Один из примеров CNV-
ляющая часть эпигенетических работ не свя-
маркера, влияющего на риск развития БП,
зана (по понятным причинам) с изучением
связан с синдромом Х-сцепленной дистонии/
эпигенома черной субстанции и стриатума и
паркинсонизма. Это заболевание вызывается
направлена на анализ периферических тка-
инсерцией сложного ретротранспозона типа
ней, в первую очередь, крови. В некоторых
SINE-VNTR-Alu (SVA) в интрон 32 гена TAF1,
исследованиях был проведен анализ аутопсий-
кодирующего ассоциированный с ТАТА-свя-
ного материала, но в этих случаях изучались
зывающимся белком фактор 1. Эта инсерция
небольшие выборки пациентов, находивших-
приводит к нарушению экспрессии гена TAF1,
ся на поздних стадиях развития заболевания
причем особенно сильно изменяется экспрес-
(четвертая-пятая стадии по Хен-Яру) и при
сия нейронального варианта мРНК. На фе-
наличии различных сопутствующих заболева-
нотип заболевания и уровень мРНК оказы-
ний, по сути, ставших причиной смерти [73,
вает влияние число мономеров (СССТСТ)n в
74]. Также ведется активный анализ эпигенома
VNTR-повторе. Это число варьирует от 34 до 52
у модельных объектов (клеточные линии, в
и влияет как на сравнительную выраженность
том числе индуцированные плюрипотентные
дистонии/паркинсонизма, так и на возраст кли-
клетки и их производные, модельные живот-
нического дебюта заболевания [68-70].
ные с генетическими и токсическими моделя-
Необходимо подчеркнуть, что в этом слу-
ми заболевания), но перенос полученных при
чае мутация не захватывает белок; кодирую-
этом данных на ситуацию у человека требует
щую область гена и ее выявление и подтвер-
осторожности и аккуратности.
ждение патогенетической роли потребовало
Второй фактор связан с длительным бес-
сложного комплекса исследований генома и
симптомным течением заболевания, и, по сути,
транскриптома с привлечением методов ге-
все эпигенетические исследования проводятся
номного редактирования. Это, с одной сторо-
на пациентах с выраженными моторными на-
ны, подчеркивает сложность поиска патогене-
рушениями. В лучшем случае выборки фор-
тически значимых мутаций такого типа и по
мируются из пациентов на первой или ранней
крайне мере частично объясняет полученную в
второй стадии по Хен-Яру с включением в
настоящее время относительно низкую оценку
выборку только пациентов с первично постав-
БИОХИМИЯ том 88 вып. 3 2023
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
517
ленным диагнозом «болезнь Паркинсона» до
употребление кофе, работа с пестицидами и
начала лечения. Это может быть важно, так
тяжелыми металлами [80]. Важно также прово-
как эпигенетические маркеры (например, уро-
дить комплексные исследования с включением
вень экспрессии микроРНК) могут реаги-
в анализ не только профилей метилирования,
ровать на терапевтические воздействия [75].
но также поиска и аннотации дифференци-
В случае выявления эпигенетических измене-
ально экспрессированных генов. Например,
ний возникает вопрос об их причинной связи
в работе Henderson et al. [81] в лимфоцитах
с развитием заболевания - изменения эпи-
периферической крови был выявлен ряд диф-
генома могут быть также следствием пораже-
ференциально метилированных при БП обла-
ния дофаминергических нейронов. Но и такие
стей генома, и были обнаружены связанные с
изменения крайне важны, так как они могут
этими областями генома дифференциально
рассматриваться как маркеры течения пато-
экспрессирующиеся гены, хотя в большинстве
логического процесса. Анализ причинно-след-
случаев дифференциальное метилирование и
ственных связей в этом случае также потребует
дифференциальная экспрессия плохо корре-
анализа различных моделей заболевания, хотя
лировали между собой.
следует еще раз заметить, что перенос дан-
Таким образом, в настоящее время трудно
ных с модельного объекта на человека требует
сделать окончательный вывод о вкладе мети-
осторожности.
лирования ДНК в риск развития БП. Требу-
Первый активно изучаемый при БП эпи-
ется проведение дополнительных широкомас-
генетический фактор - метилирование ДНК.
штабных пролонгированных исследований
Было показано, что при этом заболевании
на больших выборках пациентов, детально
наблюдается глобальное снижение уровня ме-
охарактеризованных как с клинической точ-
тилирования ДНК как в мозге (в том числе в
ки зрения, так и с точки зрения стиля жизни,
стриатуме и черной субстанции), так и в пери-
причем основное внимание должно быть уде-
ферических тканях [76]. Это снижение уровня
лено больным на ранних стадиях заболевания.
метилирования обусловлено в нервной ткани
Аналогичный вывод можно сделать и в отно-
взаимодействием между ДНК-метилтранс-
шении другого активно анализируемого эпи-
феразой 1 (DNMT1) и альфа-синуклеином, в
генетического маркера - уровня экспрессии
результате которого DNMT1 накапливается в
микроРНК.
цитоплазме [77]. Показано также, что DNMT1
Первые работы по анализу изменения экс-
может связываться с геном альфа-синуклеина.
прессии микроРНК в нервной ткани при БП
В интроне 1 этого гена выявлены сайты связы-
были проведены в 2015-2016 гг. и дали весьма
вания, обусловливающие дифференциальное
противоречивые результаты. Спектры диффе-
метилирование ген SNCA. При этом не выяв-
ренциально экспрессирующихся микроРНК
лено какой-либо ассоциации между поли-
практически не перекрывались. Это может
морфными вариантами гена DNMT1 и риском
быть связано как с использованием разных
развития заболевания [78]. Выявлен ряд генов,
отделов мозга (черная субстанция и префрон-
уровень метилирования которых меняется
тальная кора) и применением разных методов
при БП. Например, снижено метилирование
анализа микроРНК (секвенирование, ПЦР
генов транспортера дофамина (DAТ), катехол-
в реальном времени, технология Nanostring),
О-метилтрансферазы (СОМТ), белка прио-
так и с разными клинико-морфологическими
на (PRNP), митохондриальных белков (LARS2,
характеристиками пациентов. При этом необ-
MIR1977 и DDAH2) [79]. Но получаемые при
ходимо отметить, что характеристика изучае-
анализе метилирования результаты плохо вос-
мых лиц во всех публикациях крайне недо-
производимы. Проведенные за последние 10 лет
статочна - например, ни в одной из статей не
полногеномные исследования глобального ме-
приведено информации о стадии заболевания
тилирования в тканях мозга пациентов с БП
и клинической форме БП [73, 82, 83]. Всего к
и здоровых лиц выявили только один ген,
настоящему времени было выявлено 99 диф-
уровень метилирования которого менял-
ференциально экспрессирующихся в области
ся в нескольких работах
- ген цитохро-
черной субстанции микроРНК при БП, при-
ма P450 (СYP2E1) [80]. Низкая воспроизво-
чем для 60 микроРНК уровень экспрессии был
димость может быть, в частности, связана с
повышен, а для 39 - понижен [84]. Данные ра-
необходимостью учета при проведении ана-
боты показали, что спектры микроРНК в мозге
лиза метилирования роли факторов внешней
при БП существенно изменяются и такие изме-
среды, влияющих как на риск развития БП,
нения могут вызывать определенные наруше-
так и на дифференциальное метилирование.
ния в экспрессии генов ряда метаболических
К числу таких факторов относятся курение,
путей, связанных с патогенезом заболевания.
БИОХИМИЯ том 88 вып. 3 2023
518
ШАДРИНА, СЛОМИНСКИЙ
Особый интерес при этом представляют
человека могут влиять на связывание с ними
микроРНК, мишенями которых являются гены
микроРНК и тем самым модулировать экс-
моногенных форм заболевания - такие микро-
прессию гена-мишени. В частности, это было
РНК могут рассматриваться как связующее
продемонстрировано на примере однонуклео-
звено между идиопатическими и семейными
тидного полиморфизма (ОНП) rs10024743 в
формами. Например, у микроРНК miR-7 был
3′-нетранслируемой области гена альфа-сину-
выявлен сайт связывания в 3′-нетранслируе-
клеина в сайте связывания микроРНК miR-34
мой области гена альфа-синуклеина, и было
(редкий аллель по данному ОНП) в системе
показано, что как in vitro, так и in vivo эта микро-
in vitro снижал экспрессию альфа-синуклеина
РНК блокирует трансляцию мРНК альфа-си-
более чем в 2 раза [88]. В 3′-нетранслируемой
нуклеина и снижает уровень этого белка. При
области гена дардарина (LRRK2) был также
блокировании активности микроРНК miR-7
выявлен rs66737902, который ассоциирован
in vivo у мышей наблюдается гибель дофамин-
с риском развития БП и расположен в сайте
ергических нейронов черной субстанции, сни-
связывания микроРНК miR-138-2-3p. Авторы
жение уровня дофамина и накопление в нерв-
считают, что именно нарушения связывания
ной ткани альфа-синуклеина [74]. Однако, в
этой микроРНК с мРНК дардарина приводит
целом, ситуация оказывается более сложной,
к изменению уровня мРНК LRRK2 и развитию
и экспрессия альфа-синуклеина оказывается
патологического процесса [89].
под контролем сразу нескольких микроРНК,
Очень большую роль в формировании ре-
таких как miR-153, miR-203a-3p, miR-203a-3p,
гуляторных сетей микроРНК, возможно, игра-
miR-30b, miR-34b/c, miR-214 и miR-433. Ана-
ют длинные некодирующие РНК (lncRNA) -
логичная ситуация наблюдается в случае дру-
РНК размером более
200 нуклеотидов, не
гих генов семейных форм БП. Так, экспрес-
содержащие открытых рамок считывания и
сия гена паркина регулируется как минимум
не транслирующиеся в белки. Эти РНК могут
четырьмя микроРНК (miR-103a-3p, miR-146a,
считываться со смысловой и антисмысловой
miR-181a, miR-218), гена дардарина - двумя
цепей ДНК и располагаться как в интронах ге-
(miR-205, miR-599), гена белка DJ-1 - двумя
нов, так и в межгенных областях. Точное чис-
(miR-494, miR-4639). В итоге микроРНК мо-
ло таких РНК-транскриптов и кодирующих их
гут быть вовлечены в регуляцию (как позитив-
генов не известно - оно сопоставимо и, воз-
ную, так и негативную) большого количества
можно, превышает число белок-кодирующих
метаболических процессов, так или иначе свя-
генов [90].
занных с развитием БП, таких как митохон-
Описан ряд lncRNA, так или иначе свя-
дриальная дисфункция и оксидантный стресс,
занных с биологическими процессами, ве-
аутофагия, апоптоз, процессы воспаления,
дущими к БП. Так, у мышей с токсическим
экспрессия нейротрофинов [84, 85].
(6-гидроксидофамин) поражением черной
МикроРНК могут оказаться еще и свя-
субстанции lncRNA H19 через связывание мик-
зующим звеном между моногенными фор-
роРНК miR-301b-3p активирует экспрессию
мами заболеваниями, идиопатическими слу-
гена тирозингидроксилазы и сигнальный путь
чаями БП и воздействием факторов внешней
Wnt/бета-катенин и тем самым способствует
среды - в первую очередь таких известных
выживанию дофаминергических нейронов
факторов риска развития болезни Паркин-
области черной субстанции. Снижение уровня
сона, как пестициды [86]. Было показано, что
РНК Н19 вызывает, напротив, усиление про-
пестициды вызывают изменение профиля мик-
цессов нейродегенерации [91]. Ряд lncRNA
роРНК в различных тканях организма, при-
(такие как MALAT1, UCA1, SNGH14) регули-
чем спектр таких изменений во многом пере-
руют процессы обмена альфа-синуклеина и
крывается для разных пестицидов. Например,
формирование его агрегированных форм [92,
ротенон, паракват, органофосфаты и атразин
93]. Важно отметить, что многие из lncRNA
вызывают изменение экспрессии микроРНК
могут оказывать влияние на разные процессы,
miR-34 [87], а как было сказано выше, эта
связанные с развитием нейродегенерации.
микроРНК принимает участие в регуля-
Так, MALAT1, кроме регуляции обмена аль-
ции экспрессии альфа-синуклеина. Другая
фа-синуклеина, принимает участие в модуля-
связь между генетическими факторами и ми-
ции процессов нейровоспаления и активирует
кроРНК может быть обусловлена регуляцией
экспрессию дардарина (LRKK2), тем самым
экспрессии генов микроРНК на уровне тран-
усиливая процессы апоптоза и аутофагии в
скрипции или стабильности транскрипта.
дофаминергических нейронах. Показано, что
Об этом известно крайне мало, но показано,
блокирование экспрессии этой lncRNA может
что отдельные полиморфные сайты в геноме
повышать выживаемость дофаминергических
БИОХИМИЯ том 88 вып. 3 2023
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
519
нейронов [92]. Активность дардарина также
континууме взаимодействующих между собой
может контролировать и другая lncRNA
-
клеточных процессов, приводящих к избира-
NEAT1. Она формирует внутриядерные вклю-
тельной гибели дофаминергических нейронов
чения (параспеклы), в состав которых входит
при нарушении функционирования любого из
ряд клеточных белков, включая дардарин. Это
его звеньев. Эта модель сохраняет свою акту-
снижает уровень активного дардарина в клетке
альность и для идиопатической формы забо-
и тем самым повышает ее устойчивость к ок-
левания
- полногеномный ассоциативный
сидантному стрессу [94].
анализ выявил в качестве основных ассоции-
Эпигенетичесские регуляторные взаимо-
рованных с заболеванием локусов гены SNCA
действия не ограничены тремя выше описан-
и LRKK2. При этом очевидно, что мы пока не
ными механизмами, но остальные варианты
достроили стены до конца и не выявили все
изучены намного хуже. Хотя показана воз-
возможные гены, связанные с семейной и
можная роль в патогенезе БП модификации
спорадической формами заболевания, но ос-
гистонов [95], циклических РНК (circRNA) и
новное уже сделано, и появление нового гена
конкурентных РНК (ceRNA) [96]. Работы по
(генов) не разрушит уже выстроенное здание.
анализу эпигенетического профиля при БП
Но для здания важна взаимная увязан-
должны быть расширены с включением новых
ность его частей, которая в случае организма
моделей заболевания - как клеточных, так и
достигается через эпигенетические взаимодей-
организменных.
ствия, позволяющие обеспечить функциони-
рование всего ансамбля генов при осуществле-
нии ими заданной биологической функции.
ЗАКЛЮЧЕНИЕ
Нами сделаны первые и успешные шаги в этом
направлении, но как раз в эпигенетической
За последние 25 лет удалось выявить ряд
«отделке» здания предстоит сделать еще очень
генетических и эпигенетических факторов,
и очень многое. И это потребует разработки
связанных с патогенезом БП, и на этой осно-
принципиально новых подходов как с точки
ве, по сути, начато строительство своего рода
зрения анализа молекулярных событий, свя-
Кельнского собора или Саграда Фамилия. Осо-
занных с эпигеномом, так и с точки зрения
бенно близок второй вариант - все-таки как
анализа данных с использованием методов
в случае Саграда Фамилия, так и в случае БП
машинного обучения и искусственного интел-
здание еще не построено, но основные кон-
лекта [97, 98].
туры уже видны.
Для БП - это в первую очередь фунда-
Вклад авторов. М.И. Шадрина - написа-
мент, гены основных моногенных форм БП,
ние и редактирование текста; П.А. Сломин-
такие как SNCA, PRKN, LRRK2. Это, по сути,
ский - концепция и написание текста.
закладные камни, на основании которых были
Финансирование. Обзор написан при фи-
предложены первые механизмы этиопатоге-
нансовой поддержке Российского научного
неза заболевания с опорой на формирование
фонда (грант № 20-15-00262, М.И. Шадри-
агрегатов альфа-синуклеина, митохондриаль-
на) и Министерства высшего образования и
ную дисфункцию и нарушение процессов про-
науки РФ (соглашения № 075-15-2021-1357,
теасомной деградации белков. Дальнейшее
П.А. Сломинский).
строительство с выявлением новых генов мо-
Конфликт интересов. Авторы заявляют об
ногенных форм заболевания включило в чис-
отсутствии конфликта интересов.
ло основных патологических механизмов БП
Соблюдение этических норм. Настоящий
лизосомальную дисфункцию и нарушение
обзор не содержит описания каких-либо ис-
процессов везикулярного транспорта. Было
следований с участием людей или животных в
сформулировано представление о едином
качестве объектов.
СПИСОК ЛИТЕРАТУРЫ
1. Bandres-Ciga, S., Ahmed, S., Sabir, M. S.,
Mendez-Del-Barrio, C., Perinan-Tocino, T., Tejera-
Blauwendraat, C., Adarmes-Gomez, A. D., Bernal-
Parrado, C., Vargas-Gonzalez, L., Diez-Fairen, M.,
Bernal, I., Bonilla-Toribio, M., Buiza-Rueda, D.,
Alvarez, I., Tartari, J. P., Buongiorno, M., Aguilar, M.,
Carrillo, F., Carrion-Claro, M., Gomez-Garre, P.,
Gorostidi, A., Bergareche, J. A., Mondragon, E.,
Jesus, S., Labrador-Espinosa, M. A., Macias, D.,
Vinagre-Aragon, A., Croitoru, I., Ruiz-Martinez, J.,
БИОХИМИЯ том 88 вып. 3 2023
520
ШАДРИНА, СЛОМИНСКИЙ
Dols-Icardo, O., Kulisevsky, J., Marin-Lahoz, J.,
Rel. Disord., 18 Suppl
1, S66-S70, doi: 10.1016/
Pagonabarraga, J., Pascual-Sedano, B., Ezquerra, M.,
s1353-8020(11)70022-0.
Camara, A., Compta, Y., Fernandez, M., Fernandez-
9.
Leroy, E., Boyer, R., Auburger, G., Leube, B.,
Santiago, R., Munoz, E., Tolosa, E., Valldeoriola, F.,
Ulm, G., Mezey, E., Harta, G., Brownstein, M. J.,
Gonzalez-Aramburu, I., Sanchez Rodriguez, A.,
Jonnalagada, S., Chernova, T., Dehejia, A., Lavedan, C.,
Sierra, M., Menendez-Gonzalez, M., Blazquez, M.,
Gasser, T., Steinbach, P. J., Wilkinson, K. D.,
Garcia, C., Suarez-San Martin, E., Garcia-Ruiz, P.,
and Polymeropoulos, M. H. (1998) The ubiquitin
Martinez-Castrillo, J. C., Vela-Desojo, L., Ruz, C.,
pathway in Parkinson’s disease, Nature, 395, 451-452,
Barrero, F. J., Escamilla-Sevilla, F., Minguez-
doi: 10.1038/26652.
Castellanos, A., Cerdan, D., Tabernero, C., Gomez
10.
Lee, Y. C., and Hsu, S. D. (2017) Familial mutations
Heredia, M. J., Perez Errazquin, F., Romero-Acebal, M.,
and post-translational modifications of UCH-L1 in
Feliz, C., Lopez-Sendon, J. L., Mata, M., Martinez
Parkinson’s disease and neurodegenerative disor-
Torres, I., Kim, J. J., Dalgard, C. L., The American
ders, Curr. Prot. Pept. Sci., 18, 733-745, doi: 10.2174/
Genome Center, Brooks, J., Saez-Atienzar, S.,
1389203717666160217143721.
Gibbs, J. R., Jorda, R., Botia, J. A., Bonet-Ponce, L.,
11.
Wintermeyer, P., Kruger, R., Kuhn, W., Muller, T.,
Morrison, K. E., Clarke, C., Tan, M., Morris, H.,
Woitalla, D., Berg, D., Becker, G., Leroy, E., Poly-
Edsall, C., Hernandez, D., Simon-Sanchez, J., Nalls,
meropoulos, M., Berger, K., Przuntek, H., Schols, L.,
M. A., Scholz, S. W., Jimenez-Escrig, A., Duarte, J.,
Epplen, J. T., and Riess, O. (2000) Mutation analysis
Vives, F., Duran, R., Hoenicka, J., Alvarez, V., Infante, J.,
and association studies of the UCHL1 gene in German
Marti, M. J., Clarimon, J., Lopez de Munain, A.,
Parkinson’s disease patients, Neuroreport, 11, 2079-
Pastor, P., Mir, P., Singleton, A., and International
2082, doi: 10.1097/00001756-200007140-00004.
Parkinson Disease Genomics Consortium (2019) The
12.
Healy, D. G., Abou-Sleiman, P. M., Casas, J. P.,
genetic architecture of Parkinson’s disease in Spain:
Ahmadi, K. R., Lynch, T., Gandhi, S., Muqit, M. M.,
characterizing population-specific risk, differential
Foltynie, T., Barker, R., Bhatia, K. P., Quinn, N. P.,
haplotype structures, and providing etiologic insight,
Lees, A. J., Gibson, J. M., Holton, J. L., Revesz, T.,
Mov. Disord. Offic. J. Mov. Disord. Soc., 34, 1851-1863,
Goldstein, D. B., and Wood, N. W. (2006) UCHL-1
doi: 10.1002/mds.27864.
is not a Parkinson’s disease susceptibility gene, Ann.
2.
Savitt, J. M., Dawson, V. L., and Dawson, T. M.
Neurol., 59, 627-633, doi: 10.1002/ana.20757.
(2006) Diagnosis and treatment of Parkinson disease:
13.
Miyake, Y., Tanaka, K., Fukushima, W., Kiyohara, C.,
molecules to medicine, J. Clin. Invest., 116, 1744-1754,
Sasaki, S., Tsuboi, Y., Yamada, T., Oeda, T., Shi-
doi: 10.1172/JCI29178.
mada, H., Kawamura, N., Sakae, N., Fukuyama, H.,
3.
Vázquez-Vélez, G. E., and Zoghbi, H. Y.
(2021)
Hirota, Y., and Nagai, M. (2012) UCHL1 S18Y variant
Parkinson’s disease genetics and pathophysiolo-
is a risk factor for Parkinson’s disease in Japan, BMC
gy, Annu. Rev. Neurosci., 44, 87-108, doi: 10.1146/
Neurol., 12, 62, doi: 10.1186/1471-2377-12-62.
annurev-neuro-100720-034518.
14.
Sun, S., Zhao, Y., Jin, G., and Kang, H. (2014) Lack
4.
Del Tredici, K., and Braak, H. (2016) Review: sporadic
of association between UCHL1 S18Y gene polymor-
Parkinson’s disease: development and distribution of
phism and Parkinson’s disease in the Asian popu-
α-synuclein pathology, Neuropathol. Appl. Neurobiol.,
lation: a meta-analysis, Neurol. Sci., 35, 1867-1876,
42, 33-50, doi: 10.1111/nan.12298.
doi: 10.1007/s10072-014-1973-4.
5.
Connolly, B. S., and Lang, A. E. (2014) Pharmacolog-
15.
Saigoh, K., Wang, Y. L., Suh, J. G., Yamanishi, T.,
ical treatment of Parkinson disease: a review, JAMA,
Sakai, Y., Kiyosawa, H., Harada, T., Ichihara, N.,
311, 1670-1683, doi: 10.1001/jama.2014.3654.
Wakana, S., Kikuchi, T., and Wada, K.
(1999)
6.
Polymeropoulos, M. H., Lavedan, C., Leroy, E.,
Intragenic deletion in the gene encoding ubiquitin
Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H.,
carboxy-terminal hydrolase in gad mice, Nat. Genet.,
Rubenstein, J., Boyer, R., Stenroos, E. S., Chandra-
23, 47-51, doi: 10.1038/12647.
sekharappa, S., Athanassiadou, A., Papapetro-
16.
Setsuie, R., Wang, Y. L., Mochizuki, H., Osaka, H.,
poulos, T., Johnson, W. G., Lazzarini, A. M.,
Hayakawa, H., Ichihara, N., Li, H., Furuta, A.,
Duvoisin, R. C., Di Iorio, G., Golbe, L. I., and
Sano, Y., Sun, Y. J., Kwon, J., Kabuta, T., Yoshimi,
Nussbaum, R. L. (1997) Mutation in the alpha-
K., Aoki, S., Mizuno, Y., Noda, M., and Wada, K.
synuclein gene identified in families with Parkinson’s
(2007) Dopaminergic neuronal loss in transgenic
disease, Science, 276, 2045-2047, doi: 10.1126/science.
mice expressing the Parkinson’s disease-associated
276.5321.2045.
UCH-L1 I93M mutant, Neurochem. Int., 50, 119-129,
7.
Lunati, A., Lesage, S., and Brice, A. (2018) The genetic
doi: 10.1016/j.neuint.2006.07.015.
landscape of Parkinson’s disease, Rev. Neurolog., 174,
17.
Yasuda, T., Nihira, T., Ren, Y. R., Cao, X. Q., Wada, K.,
628-643, doi: 10.1016/j.neurol.2018.08.004.
Setsuie, R., Kabuta, T., Wada, K., Hattori, N.,
8.
Lesage, S., and Brice, A. (2012) Role of mendelian
Mizuno, Y., and Mochizuki, H. (2009) Effects of
genes in “sporadic” Parkinson’s disease, Parkinson.
UCH-L1 on alpha-synuclein over-expression mouse
БИОХИМИЯ том 88 вып. 3 2023
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
521
model of Parkinson’s disease, J. Neurochem., 108,
28.
Kestenbaum, M., and Alcalay, R. N. (2017) Clinical
932-944, doi: 10.1111/j.1471-4159.2008.05827.x.
features of LRRK2 carriers with Parkinson’s
18.
Kyratzi, E., Pavlaki, M., and Stefanis, L.
(2008)
disease, Adv. Neurobiol., 14, 31-48, doi: 10.1007/
The S18Y polymorphic variant of UCH-L1 confers
978-3-319-49969-7_2.
an antioxidant function to neuronal cells, Hum. Mol.
29.
Guadagnolo, D., Piane, M., Torrisi, M. R., Pizzuti, A.,
Genet., 17, 2160-2171, doi: 10.1093/hmg/ddn115.
and Petrucci, S. (2021) Genotype-phenotype correla-
19.
Xilouri, M., Kyratzi, E., Pitychoutis, P. M.,
tions in monogenic Parkinson’s disease: a review on
Papadopoulou-Daifoti, Z., Perier, C., Vila, M., Mani-
clinical and molecular findings, Front. Neurol., 12,
ati, M., Ulusoy, A., Kirik, D., Park, D. S., Wada, K.,
648588, doi: 10.3389/fneur.2021.648588.
and Stefanis, L.
(2012) Selective neuroprotective
30.
Milanowski, Ł., M., Lindemann, J. A., Hoffman-
effects of the S18Y polymorphic variant of UCH-L1 in
Zacharska, D., Soto-Beasley, A. I., Barcikowska, M.,
the dopaminergic system, Hum. Mol. Genet., 21, 874-
Boczarska-Jedynak, M., Deutschlander, A., Kło-
889, doi: 10.1093/hmg/ddr521.
dowska, G., Dulski, J., Fedoryshyn, L., Friedman, A.,
20.
Riboldi, G. M., and Di Fonzo, A. B. (2019) GBA,
Jamrozik, Z., Janik, P., Karpinsky, K., Koziorowski, D.,
Gaucher disease, and Parkinson’s disease: from
Krygowska-Wajs, A., Jasińska-Myga, B., Opala, G.,
genetic to clinic to new therapeutic approaches, Cells,
Potulska-Chromik, A., Pulyk, A., Rektorova, I.,
8, 364, doi: 10.3390/cells8040364.
Sanotsky, Y., Siuda, J., Sławek, J., Śmiłowska, K.,
21.
Thaler, A., Bregman, N., Gurevich, T., Shiner, T.,
Szczechowski, L., Rudzińska-Bar, M., Walton, R. L.,
Dror, Y., Zmira, O., Gan-Or, Z., Bar-Shira, A.,
Ross, O. A., and Wszolek, Z. K. (2021) Frequency
Gana-Weisz, M., Orr-Urtreger, A., Giladi, N., and
of mutations in PRKN, PINK1, and DJ1 in patients
Mirelman, A. (2018) Parkinson’s disease phenotype
with early-onset Parkinson’s disease from neighboring
is influenced by the severity of the mutations in the
countries in Central Europe, Parkinsonism Rel. Disord.,
GBA gene, Parkinsonism Rel. Disord., 55, 45-49,
86, 48-51, doi: 10.1016/j.parkreldis.2021.03.026.
doi: 10.1016/j.parkreldis.2018.05.009.
31.
Olszewska, D. A., McCarthy, A., Soto-Beasley, A. I.,
22.
Thaler, A., Gurevich, T., Bar Shira, A., Gana Weisz, M.,
Walton, R. L., Ross, O. A., and Lynch, T. (2022)
Ash, E., Shiner, T., Orr-Urtreger, A., Giladi, N., and
PARKIN, PINK1, and DJ1 analysis in early-onset
Mirelman, A. (2017) A “dose” effect of mutations in
Parkinson’s disease in Ireland, Irish J. Med. Sci., 191,
the GBA gene on Parkinson’s disease phenotype,
901-907, doi: 10.1007/s11845-021-02563-w.
Parkinsonism Rel. Disord., 36, 47-51, doi: 10.1016/
32.
Erer, S., Egeli, U., Zarifoglu, M., Tezcan, G.,
j.parkreldis.2016.12.014.
Cecener, G., Tunca, B., Ak, S., Demirdogen, E.,
23.
Gan-Or, Z., Liong, C., and Alcalay, R. N. (2018)
Kenangil, G., Kaleagası, H., Dogu, O., Saka, E., and
GBA-associated Parkinson’s disease and other
Elibol, B. (2016) Mutation analysis of the PARKIN,
synucleinopathies, Curr. Neurol. Neurosci. Rep., 18,
PINK1, DJ1, and SNCA genes in Turkish early-
44, doi: 10.1007/s11910-018-0860-4.
onset Parkinson’s patients and genotype-phenotype
24.
Vieira, S. R. L., and Schapira, A. H. V.
(2022)
correlations, Clin. Neurol. Neurosurg., 148, 147-153,
Glucocerebrosidase mutations and Parkinson’s
doi: 10.1016/j.clineuro.2016.07.005.
disease, J. Neural Transmiss.,
129,
1105-1117,
33.
Shulskaya, M. V., Shadrina, M. I., Fedotova, E. Y.,
doi: 10.1007/s00702-022-02531-3.
Abramycheva, N. Y., Limborska, S. A., Illarioshkin, S. N.,
25.
Kluss, J. H., Mamais, A., and Cookson, M. R. (2019)
and Slominsky, P. A. (2017) Second mutation in
LRRK2 links genetic and sporadic Parkinson’s disease,
PARK2 is absent in patients with sporadic Parkinson’s
Biochem. Soc. Transact., 47, 651-661, doi: 10.1042/
disease and heterozygous exonic deletions/dupli-
bst20180462.
cations in parkin gene, Int. J. Neurosci., 127, 781-784,
26.
Lee, A. J., Wang, Y., Alcalay, R. N., Mejia-Santana, H.,
doi: 10.1080/00207454.2016.1255612.
Saunders-Pullman, R., Bressman, S., Corvol, J. C.,
34.
Semenova, E. V., Shadrina, M. I., Slominsky, P. A.,
Brice, A., Lesage, S., Mangone, G., Tolosa, E., Pont-
Ivanova-Smolenskaya, I. A., Bagyeva, G., Illariosh-
Sunyer, C., Vilas, D., Schüle, B., Kausar, F., Foroud, T.,
kin, S. N., and Limborska, S. A. (2012) Analysis of
Berg, D., Brockmann, K., Goldwurm, S., Siri, C.,
PARK2 gene exon rearrangements in Russian patients
Asselta, R., Ruiz-Martinez, J., Mondragón, E.,
with sporadic Parkinson’s disease, Mov. Disord., 27,
Marras, C., Ghate, T., Giladi, N., Mirelman, A., and
139-142, doi: 10.1002/mds.23901.
Marder, K. (2017) Penetrance estimate of LRRK2
35.
Valente, E. M., and Ferraris, A. (2007) Heterozygous
p.G2019S mutation in individuals of non-Ashkenazi
mutations in genes causing parkinsonism: monogenic
Jewish ancestry, Mov. Disord.,
32,
1432-1438,
disorders go complex, Lancet Neurol., 6, 576-578,
doi: 10.1002/mds.27059.
doi: 10.1016/s1474-4422(07)70158-8.
27.
Trinh, J., Guella, I., and Farrer, M. J. (2014) Disease
36.
Brüggemann, N., Mitterer, M., Lanthaler, A. J.,
penetrance of late-onset parkinsonism: a meta-
Djarmati, A., Hagenah, J., Wiegers, K., Winkler, S.,
analysis, JAMA Neurol., 71, 1535-1539, doi: 10.1001/
Pawlack, H., Lohnau, T., Pramstaller, P. P., Klein, C.,
jamaneurol.2014.1909.
and Lohmann, K. (2009) Frequency of heterozygous
БИОХИМИЯ том 88 вып. 3 2023
522
ШАДРИНА, СЛОМИНСКИЙ
Parkin mutations in healthy subjects: need for careful
D’Esposito, M., Simeone, A., Ciullo, M., and
prospective follow-up examination of mutation
Esposito, T. (2021) Identification of sixteen novel
carriers, Parkinsonism Rel. Disord.,
15,
425-429,
candidate genes for late onset Parkinson’s disease,
doi: 10.1016/j.parkreldis.2008.11.014.
Mol. Neurodegener.,
16,
35, doi:
10.1186/s13024-
37.
Pavese, N., Khan, N. L., Scherfler, C., Cohen, L.,
021-00455-2.
Brooks, D. J., Wood, N. W., Bhatia, K. P., Quinn, N. P.,
45.
Shulskaya, M. V., Alieva, A. K., Vlasov, I. N., Zyrin, V. V.,
Lees, A. J., and Piccini, P. (2009) Nigrostriatal
Fedotova, E. Y., Abramycheva, N. Y., Usenko, T. S.,
dysfunction in homozygous and heterozygous parkin
Yakimovsky, A. F., Emelyanov, A. K., Pchelina, S. N.,
gene carriers: an 18F-dopa PET progression study,
Illarioshkin, S. N., Slominsky, P. A., and Shad-
Mov. Disord., 24, 2260-2266, doi: 10.1002/mds.22817.
rina, M. I.
(2018) Whole-exome sequencing in
38.
Van Nuenen, B. F., van Eimeren, T., van der Vegt, J. P.,
searching for new variants associated with the
Buhmann, C., Klein, C., Bloem, B. R., and Siebner, H. R.
development of Parkinson’s disease, Front. Aging
(2009) Mapping preclinical compensation in Par-
Neurosci., 10, 136, doi: 10.3389/fnagi.2018.00136.
kinson’s disease: an imaging genomics approach,
46.
Cook, L., Schulze, J., Verbrugge, J., Beck, J. C.,
Mov. Disord., 24 Suppl
2, S703-S710, doi: 10.1002/
Marder, K. S., Saunders-Pullman, R., Klein, C.,
mds.22635.
Naito, A., and Alcalay, R. N. (2021) The commercial
39.
Marongiu, R., Ferraris, A., Ialongo, T., Michiorri, S.,
genetic testing landscape for Parkinson’s disease,
Soleti, F., Ferrari, F., Elia, A. E., Ghezzi, D.,
Parkinsonism Rel. Disord., 92, 107-111, doi: 10.1016/
Albanese, A., Altavista, M. C., Antonini, A., Barone, P.,
j.parkreldis.2021.10.001.
Brusa, L., Cortelli, P., Martinelli, P., Pellecchia, M. T.,
47.
Li, B., Zhao, G., Zhou, Q., Xie, Y., Wang, Z., Fang, Z.,
Pezzoli, G., Scaglione, C., Stanzione, P., Tinazzi, M.,
Lu, B., Qin, L., Zhao, Y., Zhang, R., Jiang, L., Pan, H.,
Zecchinelli, A., Zeviani, M., Cassetta, E., Gara-
He, Y., Wang, X., Luo, T., Zhang, Y., Wang, Y.,
vaglia, B., Dallapiccola, B., Bentivoglio, A. R., and
Chen, Q., Liu, Z., Guo, J., Tang, B., and Li, J.
Valente, E. M. (2008) PINK1 heterozygous rare
(2021) Gene4PD: a comprehensive genetic database
variants: prevalence, significance and phenotypic
of Parkinson’s disease, Front. Neurosci., 15, 679568,
spectrum, Hum. Mutat.,
29,
565, doi:
10.1002/
doi: 10.3389/fnins.2021.679568.
humu.20719.
48.
Day, J. O., and Mullin, S. (2021) The genetics of
40.
Yu, E., Rudakou, U., Krohn, L., Mufti, K., Rus-
Parkinson’s disease and implications for clinical
key, J. A., Asayesh, F., Estiar, M. A., Spiegelman, D.,
practice, Genes, 12, 1006, doi: 10.3390/genes12071006.
Surface, M., Fahn, S., Waters, C. H., Greenbaum, L.,
49.
Fung, H. C., Scholz, S., Matarin, M., Simón-
Espay, A. J., Dauvilliers, Y., Dupré, N., Rouleau, G. A.,
Sánchez, J., Hernandez, D., Britton, A., Gibbs, J. R.,
Hassin-Baer, S., Fon, E. A., Alcalay, R. N., and Gan-
Langefeld, C., Stiegert, M. L., Schymick, J., Okun, M. S.,
Or, Z. (2021) Analysis of heterozygous PRKN variants
Mandel, R. J., Fernandez, H. H., Foote, K. D.,
and copy-number variations in Parkinson’s disease,
Rodríguez, R. L., Peckham, E., De Vrieze, F. W.,
Mov. Disord., 36, 178-187, doi: 10.1002/mds.28299.
Gwinn-Hardy, K., Hardy, J. A., and Singleton, A.
41.
Jia, F., Fellner, A., and Kumar, K. R. (2022) Mono-
(2006) Genome-wide genotyping in Parkinson’s
genic Parkinson’s disease: genotype, phenotype,
disease and neurologically normal controls: first stage
pathophysiology, and genetic testing, Genes, 13, 471,
analysis and public release of data, Lancet Neurol., 5,
doi: 10.3390/genes13030471.
911-916, doi: 10.1016/s1474-4422(06)70578-6.
42.
Santos-Lobato, B. L., Schumacher-Schuh, A.,
50.
Maraganore, D. M., de Andrade, M., Lesnick, T. G.,
Mata, I. F., Letro, G. H., Braga-Neto, P., Brandão,
Strain, K. J., Farrer, M. J., Rocca, W. A., Pant, P. V.,
P. R. P., Godeiro-Junior, C. O., Coletta, M. V. D.,
Frazer, K. A., Cox, D. R., and Ballinger, D. G. (2005)
Camargos, S. T., Borges, V., Rieder, C. R. M., and
High-resolution whole-genome association study of
Tumas, V. (2021) Genetics of Parkinson’s disease
Parkinson disease, Am. J. Hum. Genet., 77, 685-693,
in Brazil: a systematic review of monogenic forms,
doi: 10.1086/496902.
Arq. Neuropsiquiatr.,
79,
612-623, doi:
10.1590/
51.
Evangelou, E., Maraganore, D. M., and Ioannidis, J. P.
0004-282X-anp-2020-0409.
(2007) Meta-analysis in genome-wide association
43.
Correia Guedes, L., Ferreira, J. J., Rosa, M. M.,
datasets: strategies and application in Parkinson’s
Coelho, M., Bonifati, V., and Sampaio, C. (2010)
disease, PLoS One, 2, e196, doi: 10.1371/journal.
Worldwide frequency of G2019S LRRK2 mutation
pone.0000196.
in Parkinson’s disease: a systematic review, Par-
52.
Leveille, E., Ross, O. A., and Gan-Or, Z. (2021)
kinsonism Rel. Disord., 16, 237-242, doi: 10.1016/
Tau and MAPT genetics in tauopathies and
j.parkreldis.2009.11.004.
synucleinopathies, Parkinsonism Rel. Disord.,
90,
44.
Gialluisi, A., Reccia, M. G., Modugno, N., Nutile, T.,
142-154, doi: 10.1016/j.parkreldis.2021.09.008.
Lombardi, A., Di Giovannantonio, L. G., Pietra-
53.
Clark, L. N., Gao, Y., Wang, G. T., Hernandez, N.,
cupa, S., Ruggiero, D., Scala, S., Gambardella, S.,
Ashley-Koch, A., Jankovic, J., Ottman, R., Leal, S. M.,
Iacoviello, L., Gianfrancesco, F., Acampora, D.,
Rodriguez, S. M. B., and Louis, E. D.
(2022)
БИОХИМИЯ том 88 вып. 3 2023
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
523
Whole genome sequencing identifies candidate
lysosomal membrane permeabilization, Autophagy, 18,
genes for familial essential tremor and reveals
1629-1647, doi: 10.1080/15548627.2021.1995150.
biological pathways implicated in essential tremor
61.
Tanaka, T., Warner, B. M., Odani, T., Ji, Y., Mo, Y. Q.,
aetiology, EBioMedicine, 85, 104290, doi: 10.1016/
Nakamura, H., Jang, S. I., Yin, H., Michael, D. G.,
j.ebiom.2022.104290.
Hirata, N., Suizu, F., Ishigaki, S., Oliveira, F. R.,
54.
Drazich-Taylor, E. H. S., Todd, E., Convery, R.,
Motta, A. C. F., Ribeiro-Silva, A., Rocha, E. M.,
Bocchetta, M., Clarke, M., Warren, J. D., Fox, N. C.,
Atsumi, T., Noguchi, M., and Chiorini, J. A. (2020)
Revesz, T., and Rohrer, J. D. (2023) Q351R MAPT
LAMP3 induces apoptosis and autoantigen release
mutation is associated with a mixed 3R/4R tauo-
in Sjögren’s syndrome patients, Sci. Rep., 10, 15169,
pathy and a slowly progressive cognitive, behav-
doi: 10.1038/s41598-020-71669-5.
ioural and parkinsonian syndrome, J. Neurol.
62.
Chosa, N., and Ishisaki, A.
(2018) Two novel
Neurosurg. Psychiatry,
94,
169-171, doi:
10.1136/
mechanisms for maintenance of stemness in
jnnp-2022-329330.
mesenchymal stem cells: SCRG1/BST1 axis and cell-
55.
Hu, M., Li, P., Wang, C., Feng, X., Geng, Q.,
cell adhesion through N-cadherin, Jpn. Dent. Sci. Rev.,
Chen, W., Marthi, M., Zhang, W., Gao, C., Reid, W.,
54, 37-44, doi: 10.1016/j.jdsr.2017.10.001.
Swanson, J., Du, W., Hume, R. I., and Xu, H. (2022)
63.
Higashida, H., Hashii, M., Tanaka, Y., Matsukawa, S.,
Parkinson’s disease-risk protein TMEM175 is a
Higuchi, Y., Gabata, R., Tsubomoto, M., Seishima, N.,
proton-activated proton channel in lysosomes, Cell,
Teramachi, M., Kamijima, T., Hattori, T., Hori, O.,
185, 2292-2308.e2220, doi: 10.1016/j.cell.2022.05.021.
Tsuji, C., Cherepanov, S. M., Shabalova, A. A.,
56.
Jinn, S., Drolet, R. E., Cramer, P. E., Wong, A. H.,
Gerasimenko, M., Minami, K., Yokoyama, S.,
Toolan, D. M., Gretzula, C. A., Voleti, B., Vassileva, G.,
Munesue, S. I., Harashima, A., Yamamoto, Y.,
Disa, J., Tadin-Strapps, M., and Stone, D. J. (2017)
Salmina, A. B., and Lopatina, O. (2019) CD38,
TMEM175 deficiency impairs lysosomal and mito-
CD157, and RAGE as molecular determinants for
chondrial function and increases α-synuclein
social behavior, Cells, 9, 62, doi: 10.3390/cells9010062.
aggregation, Proc. Natl. Acad. Sci. USA, 114, 2389-
64.
Ortolan, E., Augeri, S., Fissolo, G., Musso, I., and
2394, doi: 10.1073/pnas.1616332114.
Funaro, A. (2019) CD157: From immunoregulatory
57.
Krohn, L., Öztürk, T. N., Vanderperre, B., Ouled
protein to potential therapeutic target, Immunol. Lett.,
Amar Bencheikh, B., Ruskey, J. A., Laurent, S. B.,
205, 59-64, doi: 10.1016/j.imlet.2018.06.007.
Spiegelman, D., Postuma, R. B., Arnulf, I.,
65.
Blauwendraat, C., Nalls, M. A., and Singleton, A. B.
Hu, M. T. M., Dauvilliers, Y., Högl, B., Stefani, A.,
(2020) The genetic architecture of Parkinson’s
Monaca, C. C., Plazzi, G., Antelmi, E., Ferini-
disease, Lancet Neurol., 19, 170-178, doi: 10.1016/
Strambi, L., Heidbreder, A., Rudakou, U., Cochen
S1474-4422(19)30287-X.
De Cock, V., Young, P., Wolf, P., Oliva, P., Zhang, X. K.,
66.
Wand, H., Lambert, S. A., Tamburro, C., Iacocca, M. A.,
Greenbaum, L., Liong, C., Gagnon, J. F., Desautels, A.,
O’Sullivan, J. W., Sillari, C., Kullo, I. J., Rowley, R.,
Hassin-Baer, S., Montplaisir, J. Y., Dupré, N.,
Dron, J. S., Brockman, D., Venner, E., McCarthy, M. I.,
Rouleau, G. A., Fon, E. A., Trempe, J. F., Lam-
Antoniou, A. C., Easton, D. F., Hegele, R. A.,
oureux, G., Alcalay, R. N., and Gan-Or, Z. (2020)
Khera, A. V., Chatterjee, N., Kooperberg, C., Ed-
Genetic, structural, and functional evidence Link
wards, K., Vlessis, K., Kinnear, K., Danesh, J. N.,
TMEM175 to synucleinopathies, Ann. Neurol., 87,
Parkinson, H., Ramos, E. M., Roberts, M. C.,
139-153, doi: 10.1002/ana.25629.
Ormond, K. E., Khoury, M. J., Janssens, A., Goddard,
58.
Wie, J., Liu, Z., Song, H., Tropea, T. F., Yang, L.,
K. A. B., Kraft, P., MacArthur, J. A. L., Inouye, M.,
Wang, H., Liang, Y., Cang, C., Aranda, K., Loh-
and Wojcik, G. L. (2021) Improving reporting stan-
mann, J., Yang, J., Lu, B., Chen-Plotkin, A. S.,
dards for polygenic scores in risk prediction studies,
Luk, K. C., and Ren, D. (2021) A growth-factor-
Nature, 591, 211-219, doi: 10.1038/s41586-021-03243-6.
activated lysosomal K+ channel regulates Parkinson’s
67.
Pang, A. W., MacDonald, J. R., Pinto, D., Wei, J.,
pathology, Nature, 591, 431-437, doi: 10.1038/s41586-
Rafiq, M. A., Conrad, D. F., Park, H., Hurles, M. E.,
021-03185-z.
Lee, C., Venter, J. C., Kirkness, E. F., Levy, S., Feuk, L.,
59.
Lunding, L. P., Krause, D., Stichtenoth, G., Stamme, C.,
and Scherer, S. W. (2010) Towards a comprehensive
Lauterbach, N., Hegermann, J., Ochs, M., Schuster, B.,
structural variation map of an individual human
Sedlacek, R., Saftig, P., Schwudke, D., Wegmann, M.,
genome, Genome Biol.,
11, R52, doi:
10.1186/
and Damme, M. (2021) LAMP3 deficiency affects
gb-2010-11-5-r52.
surfactant homeostasis in mice, PLoS Genet., 17,
68.
Ng, A. R., Jamora, R. D. G., and Rosales, R. L.
e1009619, doi: 10.1371/journal.pgen.1009619.
(2021) X-linked dystonia Parkinsonism: crossing
60.
Tanaka, T., Warner, B. M., Michael, D. G.,
a new threshold, J. Neural Transm., 128, 567-573,
Nakamura, H., Odani, T., Yin, H., Atsumi, T.,
doi: 10.1007/s00702-021-02324-0.
Noguchi, M., and Chiorini, J. A. (2022) LAMP3
69.
Di Lazzaro, G., Magrinelli, F., Estevez-Fraga, C.,
inhibits autophagy and contributes to cell death by
Valente, E. M., Pisani, A., and Bhatia, K. P.
БИОХИМИЯ том 88 вып. 3 2023
524
ШАДРИНА, СЛОМИНСКИЙ
(2021) X-linked Parkinsonism: phenotypic and
80.
Schaffner, S. L., and Kobor, M. S. (2022) DNA meth-
genetic heterogeneity, Mov. Disord., 36, 1511-1525,
ylation as a mediator of genetic and environmental
doi: 10.1002/mds.28565.
influences on Parkinson’s disease susceptibility: im-
70.
Bragg, D. C., Sharma, N., and Ozelius, L. J. (2019)
pacts of alpha-Synuclein, physical activity, and pes-
X-Linked Dystonia-Parkinsonism: recent advances,
ticide exposure on the epigenome, Front. Genet., 13,
Curr. Opin. Neurol.,
32,
604-609, doi:
10.1097/
971298, doi: 10.3389/fgene.2022.971298.
wco.0000000000000708.
81.
Henderson, A. R., Wang, Q., Meechoovet, B., Sin-
71.
Marques, S. C., Oliveira, C. R., Pereira, C. M.,
iard, A. L., Naymik, M., De Both, M., Huentel-
and Outeiro, T. F. (2011) Epigenetics in neuro-
man, M. J., Caselli, R. J., Driver-Dunckley, E., and
degeneration: a new layer of complexity, Prog. Neuro-
Dunckley, T. (2021) DNA methylation and expression
psychopharmacol. Biol. Psychiatry.,
35,
348-355,
profiles of whole blood in Parkinson’s disease, Front.
doi: 10.1016/j.pnpbp.2010.08.008.
Genet., 12, 640266, doi: 10.3389/fgene.2021.640266.
72.
Mohd Murshid, N., Aminullah Lubis, F., and
82.
Hoss, A. G., Labadorf, A., Beach, T. G., La-
Makpol, S.
(2022) Epigenetic changes and its
tourelle, J. C., and Myers, R. H. (2016) MicroRNA
intervention in age-related neurodegenerative diseases,
profiles in Parkinson’s disease prefrontal cortex, Front.
Cell. Mol. Neurobiol., 42, 577-595, doi: 10.1007/
Aging Neurosci., 8, 36, doi: 10.3389/fnagi.2016.00036.
s10571-020-00979-z.
83.
Nair, V. D., and Ge, Y. (2016) Alterations of miRNAs
73.
Briggs, C. E., Wang, Y., Kong, B., Woo, T. U.,
reveal a dysregulated molecular regulatory network in
Iyer, L. K., and Sonntag, K. C. (2015) Midbrain do-
Parkinson’s disease striatum, Neurosci. Lett., 629, 99-
pamine neurons in Parkinson’s disease exhibit a dys-
104, doi: 10.1016/j.neulet.2016.06.061.
regulated miRNA and target-gene network, Brain Res.,
84.
Santos-Lobato, B. L., Vidal, A. F., and Ribeiro-
1618, 111-121, doi: 10.1016/j.brainres.2015.05.021.
Dos-Santos, Â. (2021) Regulatory miRNA-mRNA
74.
McMillan, K. J., Murray, T. K., Bengoa-Vergniory, N.,
networks in Parkinson’s disease, Cells,
10,
1410,
Cordero-Llana, O., Cooper, J., Buckley, A., Wade-
doi: 10.3390/cells10061410.
Martins, R., Uney, J. B., O’Neill, M. J., Wong, L. F.,
85.
Saghazadeh, A., and Rezaei, N. (2022) MicroRNA
and Caldwell, M. A. (2017) Loss of microRNA-7
machinery in Parkinson’s disease: a platform for
regulation leads to alpha-synuclein accumulation and
neurodegenerative diseases, Exp. Rev. Neurother., 22,
dopaminergic neuronal loss in vivo, Mol. Ther., 25,
427-453, doi: 10.1586/14737175.2015.1114886.
2404-2414, doi: 10.1016/j.ymthe.2017.08.017.
86.
Ahmed, H., Abushouk, A. I., Gabr, M., Negida, A.,
75.
Alieva, A., Filatova, E. V., Karabanov, A. V.,
and Abdel-Daim, M. M. (2017) Parkinson’s disease
Illarioshkin, S. N., Limborska, S. A., Shadrina, M. I.,
and pesticides: a meta-analysis of disease connection
and Slominsky, P. A. (2015) miRNA expression is high-
and genetic alterations, Biomed. Pharmacother., 90,
ly sensitive to a drug therapy in Parkinson’s disease,
638-649, doi: 10.1016/j.biopha.2017.03.100.
Parkinsonism Rel. Disord., 21, 72-74, doi: 10.1016/
87.
Aloizou, A. M., Siokas, V., Sapouni, E. M., Sita, N.,
j.parkreldis.2014.10.018.
Liampas, I., Brotis, A. G., Rakitskii, V. N., Bury-
76.
Wang, C., Chen, L., Zhang, M., Yang, Y., and
kina, T. I., Aschner, M., Bogdanos, D. P., Tsatsa-
Wong, G. (2020) PDmethDB: A curated Parkinson’s
kis, A., Hadjigeorgiou, G. M., and Dardiotis, E. (2020)
disease associated methylation information database,
Parkinson’s disease and pesticides: are microRNAs
Computat. Struct. Biotechnol. J.,
18,
3745-3749,
the missing link? Sci. Total Environ., 744, 140591,
doi: 10.1016/j.csbj.2020.11.015.
doi: 10.1016/j.scitotenv.2020.140591.
77.
Desplats, P., Spencer, B., Coffee, E., Patel, P.,
88.
Kabaria, S., Choi, D. C., Chaudhuri, A. D.,
Michael, S., Patrick, C., Adame, A., Rockenstein, E.,
Mouradian, M. M., and Junn, E. (2015) Inhibition
and Masliah, E. (2011) Alpha-synuclein sequesters
of miR-34b and miR-34c enhances α-synuclein
Dnmt1 from the nucleus: a novel mechanism for
expression in Parkinson’s disease, FEBS Lett., 589,
epigenetic alterations in Lewy body diseases, J. Biol.
319-325, doi: 10.1016/j.febslet.2014.12.014.
Chem., 286, 9031-9037, doi: 10.1074/jbc.C110.212589.
89.
Cardo, L. F., Coto, E., Ribacoba, R., Mata, I. F.,
78.
Pezzi, J. C., de Bem, C. M., da Rocha, T. J.,
Moris, G., Menéndez, M., and Alvarez, V. (2014)
Schumacher-Schuh, A. F., Chaves, M. L., Rieder, C. R.,
The screening of the 3′-UTR sequence of LRRK2
Hutz, M. H., Fiegenbaum, M., and Camozzato, A. L.
identified an association between the rs66737902
(2017) Association between DNA methyltransferase
polymorphism and Parkinson’s disease, J. Hum.
gene polymorphism and Parkinson’s disease, Neurosci.
Genet., 59, 346-348, doi: 10.1038/jhg.2014.26.
Lett., 639, 146-150, doi: 10.1016/j.neulet.2016.12.058.
90.
Nadhan, R., Isidoro, C., Song, Y. S., and
79.
Angelopoulou, E., Paudel, Y. N., Papageorgiou, S. G.,
Dhanasekaran, D. N. (2022) Signaling by LncRNAs:
and Piperi, C. (2022) Environmental impact on the
structure, cellular homeostasis, and disease pathology,
epigenetic mechanisms underlying Parkinson’s disease
Cells, 11, 2517, doi: 10.3390/cells11162517.
pathogenesis: a narrative review, Brain Sci., 12, 175,
91.
Jiang, J., Piao, X., Hu, S., Gao, J., and Bao, M. (2020)
doi: 10.3390/brainsci12020175.
LncRNA H19 diminishes dopaminergic neuron loss by
БИОХИМИЯ том 88 вып. 3 2023
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
525
mediating microRNA-301b-3p in Parkinson’s disease
103. Cohen, B. H., Chinnery, P. F., and Copeland, W. C.
via the HPRT1-mediated Wnt/β-catenin signaling
(1993) POLG-related disorders, in GeneReviews
pathway, Aging,
12,
8820-8836, doi:
10.18632/
(Adam, M. P., Everman, D. B., Mirzaa, G. M.,
aging.102877.
Pagon, R. A., Wallace, S. E., Bean, L. J. H.,
92. Abrishamdar, M., Jalali, M. S., and Rashno, M. (2022)
Gripp, K. W., and Amemiya, A., eds) University of
MALAT1 lncRNA and Parkinson’s disease: the role
Washington, Seattle.
in the pathophysiology and significance for diagnostic
104. Van Veen, S., Martin, S., Van den Haute, C., Benoy, V.,
and therapeutic approaches, Mol. Neurobiol.,
59,
Lyons, J., Vanhoutte, R., Kahler, J. P., Decuypere, J. P.,
5253-5262, doi: 10.1007/s12035-022-02899-z.
Gelders, G., Lambie, E., Zielich, J., Swinnen, J. V.,
93. Taghizadeh, E., Gheibihayat, S. M., Taheri, F.,
Annaert, W., Agostinis, P., Ghesquière, B., Verhelst, S.,
Afshani, S. M., Farahani, N., and Saberi, A. (2021)
Baekelandt, V., Eggermont, J., and Vangheluwe, P.
LncRNAs as putative biomarkers and therapeutic
(2020) ATP13A2 deficiency disrupts lysosomal poly-
targets for Parkinson’s disease, Neurol. Sci., 42, 4007-
amine export, Nature, 578, 419-424, doi: 10.1038/
4015, doi: 10.1007/s10072-021-05408-7.
s41586-020-1968-7.
94. Simchovitz, A., Hanan, M., Niederhoffer, N.,
105. Joseph, S., Schulz, J. B., and Stegmüller, J. (2018) Mech-
Madrer, N., Yayon, N., Bennett, E. R., Green-
anistic contributions of FBXO7 to Parkinson’s disease,
berg, D. S., Kadener, S., and Soreq, H. (2019) NEAT1
J. Neurochem., 144, 118-127, doi: 10.1111/jnc.14253.
is overexpressed in Parkinson’s disease substantia
106. Zhou, Z. D., Lee, J. C. T., and Tan, E. K. (2018)
nigra and confers drug-inducible neuroprotection
Pathophysiological mechanisms linking F-box only
from oxidative stress, FASEB J., 33, 11223-11234,
protein 7 (FBXO7) and Parkinson’s disease (PD),
doi: 10.1096/fj.201900830R.
Mutat. Res. Rev. Mutat. Res., 778, 72-78, doi: 10.1016/
95. Li, Y., Gu, Z., Lin, S., Chen, L., Dzreyan, V., Eid, M.,
j.mrrev.2018.10.001.
Demyanenko, S., and He, B. (2022) Histone deacety-
107. Lin, G., Lee, P. T., Chen, K., Mao, D., Tan, K. L.,
lases as epigenetic targets for treating Parkinson’s
Zuo, Z., Lin, W. W., Wang, L., and Bellen, H. J.
disease, Brain Sci., 12, 672, doi: 10.3390/brains-
(2018) Phospholipase PLA2G6, a Parkinsonism-
ci12050672.
associated gene, affects Vps26 and Vps35, retromer
96. Zhang, H., Yao, L., Zheng, Z., Koc, S., and Lu, G.
function, and ceramide levels, similar to α-synuclein
(2022) The role of non-coding RNAs in the patho-
gain, Cell Metab., 28, 605-618.e606, doi: 10.1016/
genesis of Parkinson’s disease: recent advance-
j.cmet.2018.05.019.
ment, Pharmaceuticals,
15,
811, doi:
10.3390/
108. Sassone, J., Reale, C., Dati, G., Regoni, M.,
ph15070811.
Pellecchia, M. T., and Garavaglia, B. (2021) The role
97. Helmy, M., Eldaydamony, E., Mekky, N., Elmogy, M.,
of VPS35 in the pathobiology of Parkinson’s disease,
and Soliman, H. (2022) Predicting Parkinson’s disease
Cell. Mol. Neurobiol.,
41,
199-227, doi:
10.1007/
related genes based on PyFeat and gradient boosted
s10571-020-00849-8.
decision tree, Sci. Rep., 12, 10004, doi: 10.1038/
109. Roosen, D. A., Blauwendraat, C., Cookson, M. R., and
s41598-022-14127-8.
Lewis, P. A. (2019) DNAJC proteins and pathways to
98. Farrow, S. L., Schierding, W., Gokuladhas, S.,
parkinsonism, FEBS J., 286, 3080-3094, doi: 10.1111/
Golovina, E., Fadason, T., Cooper, A. A., and
febs.14936.
O’Sullivan, J. M. (2022) Establishing gene regulatory
110. Choudhry, H., Aggarwal, M., and Pan, P. Y. (2021)
networks from Parkinson’s disease risk loci, Brain,
Mini-review: synaptojanin 1 and its implications in
145, 2422-2435, doi: 10.1093/brain/awac022.
membrane trafficking, Neurosci. Lett., 765, 136288,
99. Quinn, P. M. J., Moreira, P. I., Ambrósio, A. F.,
doi: 10.1016/j.neulet.2021.136288.
and Alves, C. H. (2020) PINK1/PARKIN signalling
111. Monfrini, E., Spagnolo, F., Canesi, M., Seresini, A.,
in neurodegeneration and neuroinflammation,
Rini, A., Passarella, B., Percetti, M., Seia, M., Gold-
Acta Neuropathol. Commun., 8, 189, doi: 10.1186/
wurm, S., Cereda, V., Comi, G. P., Pezzoli, G., and
s40478-020-01062-w.
Di Fonzo, A. (2022) VPS13C-associated Parkinson’s
100. Dolgacheva, L. P., Berezhnov, A. V., Fedotova, E. I.,
disease: two novel cases and review of the literature,
Zinchenko, V. P., and Abramov, A. Y. (2019) Role of
Parkinsonism Rel. Disord., 94, 37-39, doi: 10.1016/
DJ-1 in the mechanism of pathogenesis of Parkinson’s
j.parkreldis.2021.11.031.
disease, J. Bioenerg. Biomembr.,
51,
175-188,
112. Chang, D., Nalls, M. A., Hallgrímsdóttir, I. B.,
doi: 10.1007/s10863-019-09798-4.
Hunkapiller, J., van der Brug, M., Cai, F., Kerch-
101. Usmani, A., Shavarebi, F., and Hiniker, A. (2021) The
ner, G. A., Ayalon, G., Bingol, B., Sheng, M., Hinds, D.,
cell biology of LRRK2 in Parkinson’s disease, Mol.
Behrens, T. W., Singleton, A. B., Bhangale, T. R.,
Cell. Biol., 41, 1-16, doi: 10.1128/mcb.00660-20.
and Graham, R. R.
(2017) A meta-analysis of
102. Tanaka, K.
(2020) The PINK1-Parkin axis: an
genome-wide association studies identifies 17 new
overview, Neurosci. Res., 159, 9-15, doi: 10.1016/
Parkinson’s disease risk loci, Nat. Genet., 49, 1511-
j.neures.2020.01.006.
1516, doi: 10.1038/ng.3955.
БИОХИМИЯ том 88 вып. 3 2023
526
ШАДРИНА, СЛОМИНСКИЙ
113. Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B.,
Do, C. B., Eriksson, N., Factor, S. A., Farrer, M. J.,
Hernandez, D. G., Saad,M., DeStefano, A. L., Kara, E.,
Foroud, T., Gasser, T., Hamza, T., Hardy, J. A.,
Bras, J., Sharma, M., Schulte, C., Keller, M. F.,
Heutink, P., Hill-Burns, E. M., Klein, C., La-
Arepalli, S., Letson, C., Edsall, C., Stefansson, H.,
tourelle, J. C., Maraganore, D. M., Martin, E. R.,
Liu, X., Pliner, H., Lee, J. H., Cheng, R., Ikram, M. A.,
Martinez, M., Myers, R. H., Nalls, M. A., Pan-
Ioannidis, J. P., Hadjigeorgiou, G. M., Bis, J. C.,
kratz, N., Payami, H., Satake, W., Scott, W. K.,
Martinez, M., Perlmutter, J. S., Goate, A., Marder, K.,
Sharma, M., Singleton, A. B., Stefansson, K., Toda, T.,
Fiske, B., Sutherland, M., Xiromerisiou, G.,
Tung, J. Y., Vance, J., Wood, N. W., Zabetian, C. P.,
Myers, R. H., Clark, L. N., Stefansson, K.,
Young, P., Tanzi, R. E., Khoury, M. J., Zipp, F.,
Hardy, J. A., Heutink, P., Chen, H., Wood, N. W.,
Lehrach, H., Ioannidis, J. P., and Bertram, L. (2012)
Houlden, H., Payami, H., Brice, A., Scott, W. K.,
Comprehensive research synopsis and systematic
Gasser, T., Bertram, L., Eriksson, N., Foroud, T., and
meta-analyses in Parkinson’s disease genetics:
Singleton, A. B. (2014) Large-scale meta-analysis of
the PDGene database, PLoS Genet., 8, e1002548,
genome-wide association data identifies six new risk
doi: 10.1371/journal.pgen.1002548.
loci for Parkinson’s disease, Nat. Genet., 46, 989-993,
118. Do, C. B., Tung, J. Y., Dorfman, E., Kiefer, A. K.,
doi: 10.1038/ng.3043.
Drabant, E. M., Francke, U., Mountain, J. L.,
114. Nalls, M. A., Plagnol, V., Hernandez, D. G.,
Goldman, S. M., Tanner, C. M., Langston, J. W.,
Sharma, M., Sheerin, U. M., Saad, M., Simón-
Wojcicki, A., and Eriksson, N. (2011) Web-based
Sánchez, J., Schulte, C., Lesage, S., Sveinbjörns-
genome-wide association study identifies two
dóttir, S., Stefánsson, K., Martinez, M., Hardy, J.,
novel loci and a substantial genetic component for
Heutink, P., Brice, A., Gasser, T., Singleton, A. B.,
Parkinson’s disease, PLoS Genet.,
7, e1002141,
and Wood, N. W. (2011) Imputation of sequence
doi: 10.1371/journal.pgen.1002141.
variants for identification of genetic risks for
119. Spencer, C. C., Plagnol, V., Strange, A., Gardner, M.,
Parkinson’s disease: a meta-analysis of genome-wide
Paisan-Ruiz, C., Band, G., Barker, R. A., Bellenguez, C.,
association studies, Lancet, 377, 641-649, doi: 10.1016/
Bhatia, K., Blackburn, H., Blackwell, J. M., Bramon, E.,
s0140-6736(10)62345-8.
Brown, M. A., Brown, M. A., Burn, D., Casas, J. P.,
115. Pickrell, J. K., Berisa, T., Liu, J. Z., Ségurel, L.,
Chinnery, P. F., Clarke, C. E., Corvin, A., Crad-
Tung, J. Y., and Hinds, D. A. (2016) Detection and
dock, N., Deloukas, P., Edkins, S., Evans, J.,
interpretation of shared genetic influences on
42
Freeman, C., Gray, E., Hardy, J., Hudson, G.,
human traits, Nat. Genet., 48, 709-717, doi: 10.1038/
Hunt, S., Jankowski, J., Langford, C., Lees, A. J.,
ng.3570.
Markus, H. S., Mathew, C. G., McCarthy, M. I.,
116. Rodrigo, L. M., and Nyholt, D. R. (2021) Imputation
Morrison, K. E., Palmer, C. N., Pearson, J. P.,
and reanalysis of ExomeChip Data identifies
Peltonen, L., Pirinen, M., Plomin, R., Potter, S.,
novel, conditional and joint genetic effects on
Rautanen, A., Sawcer, S. J., Su, Z., Trem-
Parkinson’s disease risk, Genes, 12, 689, doi: 10.3390/
bath, R. C., Viswanathan, A. C., Williams, N. W.,
genes12050689.
Morris, H. R., Donnelly, P., and Wood, N. W.
117. Lill, C. M., Roehr, J. T., McQueen, M. B.,
(2011) Dissection of the genetics of Parkinson’s
Kavvoura, F. K., Bagade, S., Schjeide, B. M.,
disease identifies an additional association
5′ of
Schjeide, L. M., Meissner, E., Zauft, U., Allen, N. C.,
SNCA and multiple associated haplotypes at 17q21,
Liu, T., Schilling, M., Anderson, K. J., Beecham, G.,
Hum. Mol. Genet.,
20,
345-353, doi:
10.1093/
Berg, D., Biernacka, J. M., Brice, A., DeStefano, A. L.,
hmg/ddq469.
GENETIC ARCHITECTURE OF PARKINSON’S DISEASE
Review
M. I. Shadrina* and P. A. Slominsky
Institute of Molecular Genetics of National Research Centre “Kurchatov Institute”,
123182 Moscow, Russia; e-mail: shadrina@img.ras.ru
Year 2022 marks the 25th anniversary of the first mutation in familial autosomal dominant Parkinson’s dis-
ease. Over the years, our understanding of the role of genetics in the pathogenesis of familial and idiopathic
БИОХИМИЯ том 88 вып. 3 2023
ГЕНЕТИЧЕСКАЯ АРХИТЕКТУРА БОЛЕЗНИ ПАРКИНСОНА
527
forms of Parkinson’s disease has expanded significantly - a number of genes for the familial form of the
disease have been identified, and DNA markers of an increased risk of developing a sporadic form of the
disease have been identified. But, despite all the successes achieved, we are far from an accurate assessment
of the contribution to the development of the disease as a whole, both genetic factors and (and even more
so) epigenetic factors. The review summarizes the information accumulated to date on the genetic architec-
ture of Parkinson’s disease and formulates issues that need to be addressed and are primarily related to the
assessment of epigenetic factors in the pathogenesis of the disease.
Keywords: Parkinson’s disease, monogenic form, sporadic form, genetics, mutation analysis, genome-wide association
analysis, epigenetics, genetic risk
БИОХИМИЯ том 88 вып. 3 2023