БИОХИМИЯ, 2023, том 88, вып. 7, с. 1101 - 1122
УДК 577.2
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ, ВЫЗВАННЫХ
ТЕРАПИЕЙ С ПРИМЕНЕНИЕМ ИНГИБИТОРОВ ТОПОИЗОМЕРАЗ
Обзор
© 2023 Н.А. Ломов1*, В.С. Вьюшков1, М.А. Рубцов1,2
1 Московский государственный университет имени М.В. Ломоносова,
биологический факультет, кафедра молекулярной биологии,
119234 Москва, Россия; электронная почта: lomov@mail.bio.msu.ru
2 Первый МГМУ им. И.М. Сеченова Минздрава России (Сеченовский университет),
Центр индустриальных технологий и предпринимательства, 119435 Москва, Россия
Поступила в редакцию 06.02.2023
После доработки 14.04.2023
Принята к публикации 20.04.2023
Лейкозы - опухолевые заболевания кроветворной ткани. Среди лейкозов выделяют отдельную
группу заболеваний, развитие которых связано с предшествующей химиотерапией - вторичные
лейкозы. Терапия рака ингибиторами ДНК-топоизомераз типа II является одной из самых эффек-
тивных среди химиотерапий. Однако ее побочным эффектом может стать развитие вторичного
лейкоза, характеризующегося хромосомными перестройками с участием генов AML1 или MLL.
Характерный набор рекуррентных перестроек при таком лейкозе отличается от спектра хромо-
сомных перестроек при других неоплазиях. В обзоре рассматриваются факторы, которые влияют
на образование перестроек при обработке клеток ингибиторами ДНК-топоизомераз. К таким фак-
торам в первую очередь относятся подвижность концов разрывов, образование которых предше-
ствует транслокации, и появление новых функций у белков, которые образуются в клетке в резуль-
тате транслокации.
КЛЮЧЕВЫЕ СЛОВА: лейкозы, транслокации, слитые белки, ингибиторы топоизомераз.
DOI: 10.31857/S0320972523070047, EDN: FVBNLW
ВВЕДЕНИЕ
зом являются хромосомные транслокации с
участием гена, ассоциированного с острым
В последние годы благодаря совершенство-
миелоидным лейкозом (Acute myeloid leukemia,
ванию химиотерапевтических подходов про-
AML1), и гена, ассоциированного с лейкозом
должительность жизни пациентов с онколо-
смешанного фенотипа (Mixed lineage leukemia,
гическими заболеваниями заметно возросла.
MLL). Эти гены необходимы для нормального
В то же время увеличение продолжительности
развития клеток крови, поэтому нарушение их
жизни выявляет отдаленные побочные эффек-
функции вследствие транслокации приводит
ты химиотерапии. К числу таких последствий
к опухолевой трансформации кроветворных
относится так называемый вторичный, или
клеток. Считается, что хромосомные пере-
обусловленный терапией, лейкоз. Одной из
стройки возникают из-за ошибочной репара-
частых причин развития вторичного лейкоза
ции двуцепочечных разрывов, вносимых ДНК-
является применение для терапии первичных
топоизомеразой II после ее ингибирования.
опухолей препаратов из класса ингибиторов
В этом обзоре мы рассмотрим молекулярные
ДНК-топоизомераз типа II.
механизмы развития вторичных лейкозов, вы-
Цитогенетической особенностью опухо-
званных терапией ингибиторами ДНК-топо-
левых клеток пациентов со вторичным лейко- изомераз типа II.
Принятые сокращения: ДЦР - двуцепочечный разрыв ДНК; AML1 (или RUNX1) - ген, ассоциированный с острым
миелоидным лейкозом; ETO (или RUNX1T1) - ген, часто перестраивающийся с образованием транслокации t(8;21);
MLL (или KMT2A) - ген, ассоциированный с лейкозом смешанного фенотипа; NHEJ - репарация ДЦР по механизму
негомологичного соединения концов.
* Адресат для корреспонденции.
1101
1
102
ЛОМОВ и др.
ОСТРЫЕ МИЕЛОИДНЫЕ ЛЕЙКОЗЫ,
клетке - активность ферментов во время таких
ВЫЗВАННЫЕ ТЕРАПИЕЙ
процессов, как V(D)J-рекомбинация, переклю-
чение класса антител (CSR) и соматический
Лейкозы и хромосомные транслокации. Лей-
гипермутагенез (SHM) при созревании лим-
козы, или лейкемии, - опухолевые заболева-
фоцитов. С активностью этих ферментов свя-
ния кроветворной ткани, которые развивают-
зывают развитие многих лимфом, например,
ся, когда недифференцированные (бластные)
лимфомы Беркитта, которая сопровождается
клетки теряют способность к образованию
транслокацией t(8;14) между локусом IGH и
зрелых форм и накапливаются в костном мозге
геном MYC [9-11].
и в крови. Считается, что лейкозы возникают в
Другой причиной образования ДЦР в клет-
результате трансформации лишь одной исход-
ках может быть действие цитотоксических аген-
ной клетки - стволовой лейкозной клетки [1].
тов или ионизирующего излучения, что про-
В результате ее деления получается неопласти-
исходит при радио- и химиотерапии опухолей
ческий клон бластных клеток, который вытес-
и аутоиммунных заболеваниях. Накопление
няет нормальные элементы кроветворения из
ДЦР в раковых клетках приводит к останов-
костного мозга и приводит к недостатку зре-
ке деления и развитию в них апоптоза [12].
лых клеток в периферической крови, а ин-
Однако побочным эффектом терапии является
фильтрация лейкозными клетками внутренних
развитие вторичных опухолей. Таким образом,
органов нарушает их работу. Кроме лейкозов,
с появлением различных видов терапии опу-
к злокачественным заболеваниям кроветвор-
холей возник и особый класс заболеваний -
ной системы также относятся лимфомы. При
обусловленные терапией миелоидные неопла-
лимфомах опухолевые клетки поражают лим-
зии (therapy-related myeloid neoplasms, t-MN,
фатическую систему - в первую очередь лим-
или myeloid neoplasms post cytotoxic therapy,
фатические узлы.
MN-pCT), также называемые вторичными
Считается, что к онкогенной трансформа-
неоплазиями.
ции клеток крови и костного мозга приводят
Обусловленные терапией миелоидные нео-
определенные хромосомные транслокации -
плазии составляют 10-20% от всех случаев
обмен участками между разными хромосома-
миелоидных лейкозов. Они развиваются в
ми. Когда в месте контакта двух фрагментов
0,8-6,3% случаев в течение 20 лет после пред-
разных хромосом соединяются фрагменты раз-
шествующей терапии с медианным временем
ных генов - эти гены называют генами-парт-
3-5 лет [13]. Выживаемость при таких вторич-
нерами по транслокации - образуются слитые
ных заболеваниях измеряется месяцами, а не
гены, продукты которых называют слитыми
годами, что делает их одними из самых агрес-
(химерными) белками [2]. Хромосомная пере-
сивных видов рака - они считаются даже более
стройка сама по себе может быть достаточным
опасными, чем аналогичные лейкозы, возни-
событием для развития лейкоза. В пользу это-
кающие de novo [14-18].
го говорят данные высокопроизводительного
Вторичные неоплазии можно разделить на
секвенирования: лейкозы несут очень мало со-
группы в зависимости от терапии, которая им
матических мутаций по сравнению с другими
предшествовала - терапия алкилирующими
видами злокачественных новообразований [3,
агентами, радиотерапия или терапия инги-
4]. Тип хромосомной перестройки в лейкозных
биторами ДНК-топоизомеразы II
[19]. Об-
клетках, в частности то, какие гены являются
условленные алкилирующими агентами и/или
партнерами по транслокации, влияет на разви-
радиотерапией вторичные лейкозы обычно
тие заболевания, прогноз и выбор лечения [5-
возникают через 4-7 лет после терапии и чаще
7]. Определение того, какая именно хромосом-
всего относятся к хроническому лейкозу, тогда
ная транслокация ассоциирована с данным
как обусловленные ингибиторами ДНК-топо-
лейкозом, является первостепенной задачей
изомеразы II вторичные лейкозы развиваются
при постановке диагноза у пациентов [8].
через 1-3 года и относятся к острому мие-
Причины развития лейкозов после терапии
лоидному лейкозу (topoisomerase inhibitor-
опухолей. Хромосомные транслокации возни-
related AML, TI-related AML). Реже вторичные
кают при ошибках репарации двуцепочечных
неоплазии встречаются после терапии анти-
разрывов ДНК (ДЦР), когда концы разных
метаболитами, например метотрексатом [20].
разрывов сшиваются вместе. Поэтому причи-
Примечательно, что обусловленные разной
ной хромосомных транслокаций могут быть
терапией неоплазии характеризуются разными
любые процессы, приводящие к образованию
хромосомными аберрациями. Так, для первой
разрывов в ДНК. Среди естественных причин
группы характерны крупные делеции участ-
возникновения большого количества ДЦР в
ков 5 и 7 хромосом, тогда как для вторичных
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1103
неоплазий, вызванных терапией ингибито-
как в процессе каталитического цикла топо-
рами топоизомераз II, - сбалансированные
изомераза II вносит двуцепочечный разрыв
(реципрокные) транслокации с участием хро-
в ДНК. Ниже пойдет речь о роли топоизомераз
мосом 11 или 21 [13, 21-24]. Терапия ингиби-
в клетке и механизме работы ДНК-топоизо-
торами ДНК-топоизомераз II выделяется в
мераз типа II.
отдельную группу ввиду своего широкого рас-
Топоизомеразы и их активность в клетках.
пространения и вследствие своей эффектив-
К ДНК, представляющей собой двойную спи-
ности.
раль, применимы законы топологии. Важной
проблемой, связанной с топологией ДНК,
является расхождение хромосом после репли-
ТОПОИЗОМЕРАЗНЫЕ ЯДЫ
кации по дочерним клеткам. Так как репли-
КАК ЭФФЕКТИВНОЕ СРЕДСТВО
кация идет полуконсервативно - к каждой
ПРОТИВООПУХОЛЕВОЙ ТЕРАПИИ
цепочке ДНК достраивается комплементарная
ей цепь - то получившиеся хромосомы ока-
В целом, противоопухолевая химиотера-
зываются закручены относительно друг друга
пия стала конвенциальным методом лечения
так же, как были закручены цепочки исходной
только во второй половине ХХ века, до этого
двойной спирали ДНК. Эта ключевая пробле-
момента опухоли лечили хирургически или с
ма полуконсервативной модели репликации
помощью радиотерапии. Экстракт из тканей
была сформулирована еще Фрэнсисом Кри-
Подофилла щитовидного (Podophyllum pelta-
ком и Джеймсоном Уотсоном, а также Максом
tum), или подофиллин, эпизодически приме-
Дельбрюком, который для ее разрешения даже
нялся в медицине против кожных опухолей и
предложил альтернативную модель реплика-
бородавок еще в ХIХ веке [25]. В 1946 г. было
ции - дисперсную модель [31, 32].
показано, что подофиллин препятствует деле-
Проблемы, связанные с топологией ДНК,
нию клеток [26]. Однако попытки применения
решаются ферментами-топоизомеразами с по-
этого экстракта против опухолей были ограни-
мощью временного разрыва цепочки ДНК.
чены его высокой токсичностью [27]. Поэтому
Топоизомеразы типа I вносят одноцепочечный
в 1950-х гг. начались поиски действующей мо-
разрыв, позволяя одной цепочке вращаться во-
лекулы из экстракта Podophyllum. Наилучшее
круг второй, тогда как топоизомеразы типа II
противоопухолевое действие показал бензил-
вносят двуцепочечный разрыв, через который
иден 4′-деметилэпиподофиллин глюкозид, и
может пройти другой фрагмент ДНК [33, 34].
в 1966 г. был синтезирован его синтетический
Для разделения переплетенных хромосом мо-
аналог - этопозид (VP-16), годом позже - те-
жет использоваться только ДНК-топоизомера-
нипозид (VM-26). В 1974 г. завершились докли-
за типа II, что было показано для многих орга-
нические испытания, доказавшие их высокую
низмов [35-38]. Поэтому любым делящимся
эффективность против самых разных видов
клеткам критически необходима работа топо-
опухолей, а с 1983 г. было получено разреше-
изомераз именно типа II.
ние FDA (Food and Drug Administration) США
Задача ДНК-топоизомераз типа II заклю-
на применение этопозида в клинической прак-
чается в контролируемом внесении двуцепо-
тике [25]. На сегодня этопозид и аналогичные
чечных разрывов в ДНК, которые необходимы
по механизму работы вещества находятся на
для расплетения хромосом. Почти все ДНК-
переднем крае терапии самых разных типов
топоизомеразы типа II из разных организмов
опухолей: лейкозов, лимфом, сарком, рака мо-
гомологичны друг другу (их относят к под-
лочной железы, рака легких и других [28, 29].
типу IIA) и работают по одному механиз-
Интересно, что механизм действия это-
му (рис. 1). ДНК-топоизомераза IIA представ-
позида и тенипозида на момент их одобре-
ляет собой гомодимер, мономеры которого
ния еще не был установлен. Было показано,
соединяются друг с другом тремя участками,
что этопозид приводит к фрагментации ДНК
формируя трое «ворот» - N-, ДНК- и C-во-
in vivo, однако он не способен вносить раз-
рота. Ворота последовательно открываются
рывы в очищенную ДНК [30]. Только в 1984 г.
и закрываются, пропуская фрагмент ДНК,
выяснили, что мишенью этопозида являются
называемый T-фрагментом (от
«transport»).
топоизомеразы типа II - ферменты, особенно
ДНК-ворота, расположенные в середине, свя-
востребованные в делящихся клетках, к кото-
зывают другой фрагмент ДНК (G-фрагмент,
рым относятся клетки опухолей. Работа ДНК-
от «gate») и открываются, временно разрывая
топоизомераз типа II, с одной стороны, крайне
его. Когда Т-фрагмент ДНК проходит ДНК-во-
необходима при делении, с другой - заключает
рота и они закрываются, разрыв в G-фрагмен-
в себе смертельную опасность для клетки, так
те лигируется. Образование двуцепочечного
БИОХИМИЯ том 88 вып. 7 2023
1104
ЛОМОВ и др.
разрыва в G-фрагменте происходит за счет двух
реакций переэтерификации: остатки тирозина
каждой из субъединиц топоизомеразы атаку-
ют фосфодиэфирные связи на разных цепях
ДНК, образуя ковалентные связи с 5′-фосфа-
тами нуклеотидов [39-41]. Когда Т-фрагмент
высвобождается через C-ворота, топоизо-
мераза возвращается в исходное состояние и
цикл ее работы завершается. Хотя сама реак-
ция переэтерификации не требует затрат энер-
гии, в ходе каталитического цикла топоизоме-
раза типа IIA гидролизует две молекулы ATP,
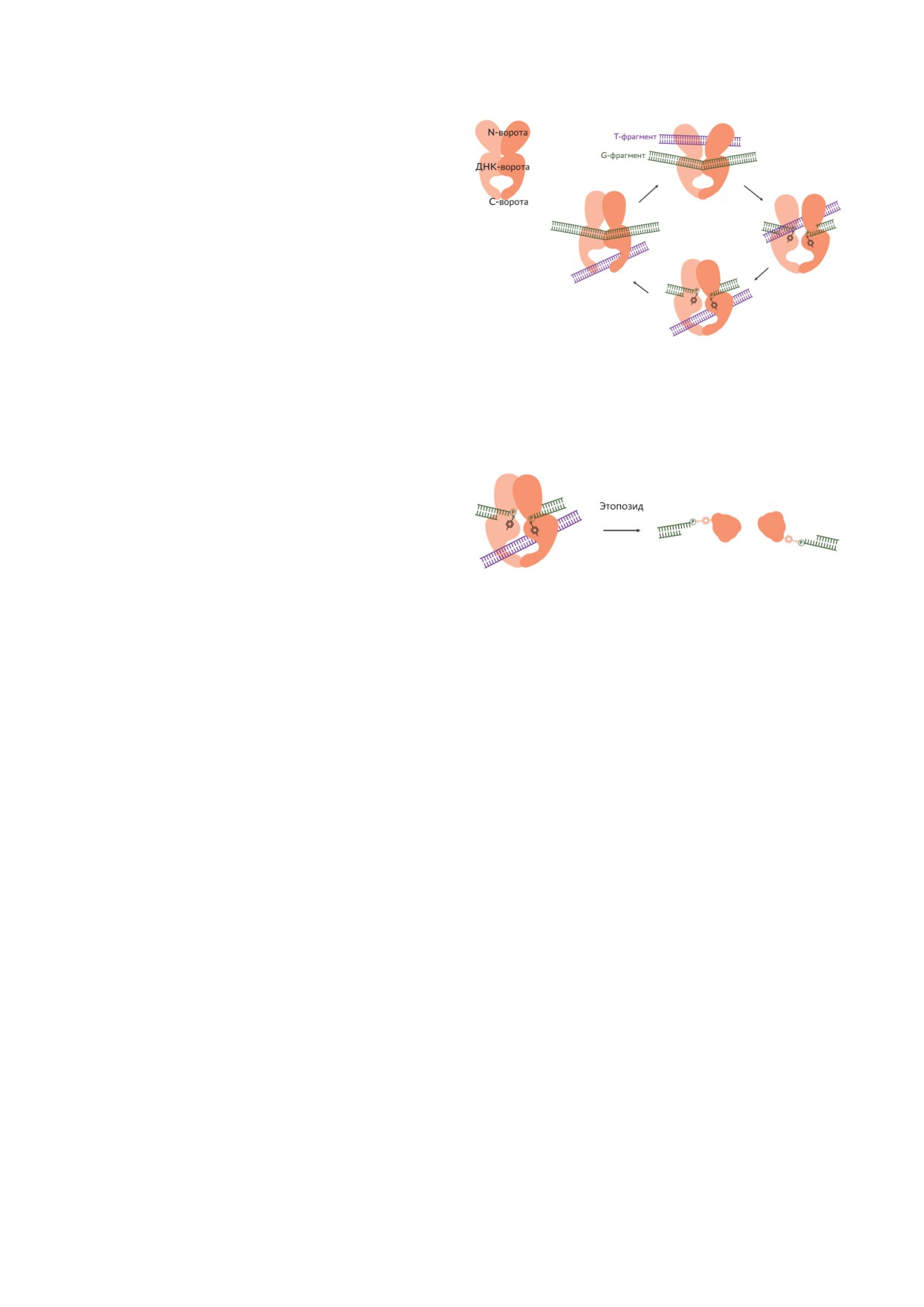
Рис.
1. Механизм работы топоизомеразы типа IIA.
которые аллостерически координируют откры-
На схеме показано согласованное изменение конформа-
вание и закрывание ворот во время каталити-
ции фермента, что приводит к последовательному пере-
ческого цикла топоизомеразы [42-44]. Согла-
мещению Т-фрагмента ДНК через G-фрагмент, при этом
сованность в работе ворот крайне важна: если
субъединицы фермента всегда удерживаются вместе.
Для разрыва в G-фрагменте фермент формирует фосфо-
все ворота окажутся одномоментно открыты-
эфирную связь с ДНК через остаток тирозина
ми, это значит, что субъединицы топоизомера-
зы отсоединились друг от друга, и временный
разрыв ДНК превратится в постоянный.
Топоизомеразные яды. На сегодняшний день
изучены различные ингибиторы ДНК-топо-
изомераз типа II (топо2-ингибиторы). По ме-
ханизму действия их принято делить на 2 груп-
пы - ингибиторы каталитического цикла и
Рис. 2. Действие топоизомеразного яда (этопозида) на
топоизомеразные яды (catalytic inhibitors и
ДНК-топоизомеразу II, приводящее к образованию дву-
poisons соответственно). Первые действуют на
цепочечного разрыва ДНК с ковалентно связанной на
конце ДНК субъединицей топоизомеразы - Тор2сс по [48]
любых стадиях каталитического цикла, вклю-
чая связывание и гидролиз ATP, связывание
и разрезание ДНК. Вторая группа - так назы-
(Protein with SprT-like domain at the N terminus)
ваемые топоизомеразные яды, действие кото-
с последующим отщеплением остатка поли-
рых характеризуется повышением в клетке
пептида ДНК-фосфодиэстеразой TDP2 (tyro-
количества ковалентно связанных комплексов
syl DNA phosphodiesterase 2). Нуклеолитиче-
ДНК-фермент (Тор2сс). Большинство ис-
ский путь предполагает отщепление участка
пользуемых на сегодня в терапии опухолей то-
цепочки ДНК, на конце которого ковалентно
по2-ингибиторов относятся к группе топоизо-
пришит белок, с помощью эндонуклеаз систем
меразных ядов [41, 45]. Чаще всего среди них
репарации [48, 49].
используется этопозид, его механизм действия
В любом случае удаление топоизомеразы
подробно изучен. Этопозид связывается с ко-
приводит к образованию в ДНК двуцепочеч-
валентным комплексом топоизомеразы с ДНК
ного разрыва. Множественные ДЦР приводят
и стабилизирует его, препятствуя религирова-
к запуску клеточного ответа на повреждение
нию ДНК [46]. Когда в G-фрагмент ДНК вне-
ДНК, что включает в себя фосфорилирова-
сен разрыв, молекула этопозида нековалентно
ние остатка S139 гистонов Н2A.X, репарацию
связывается с ферментом, что препятствует
ДНК или индукцию апоптоза [50]. Действие
полному закрытию ДНК-ворот и сближению
этопозида можно зарегистрировать по появ-
концов разрыва ДНК (рис. 2).
лению фосфорилированного гистона Н2A.X
Пути удаления топоизомеразы с ДНК. Оста-
(γH2A.X) уже через несколько минут после об-
новленная ДНК-топоизомераза, ковалентно
работки им клеток [51, 52]. Ряд экспериментов
связанная с ДНК, препятствует нормальной
подтверждает ключевую роль ингибирования
транскрипции и репликации, что запускает
ДНК-топоизомеразы II в образовании транс-
процесс ее деградации [47]. В клетках человека
локаций, вызванных соответствующей тера-
существует несколько путей удаления топоизо-
пией. Так, этопозид индуцирует образование
меразы, ковалентно сшитой с ДНК. Их можно
двуцепочечных разрывов в генах AML1, MLL и
разделить на протеолитические и нуклеоли-
их партнеров, а к месту разрыва привлекаются
тический пути. Протеолитические пути пред-
белки репарации [52-56]. Эти разрывы лока-
полагают деградацию белка с помощью про-
лизуются в кластерах точек разрывов, обнару-
теасомы или специальной протеазы SPRTN
женных у пациентов со вторичным лейкозом.
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1105
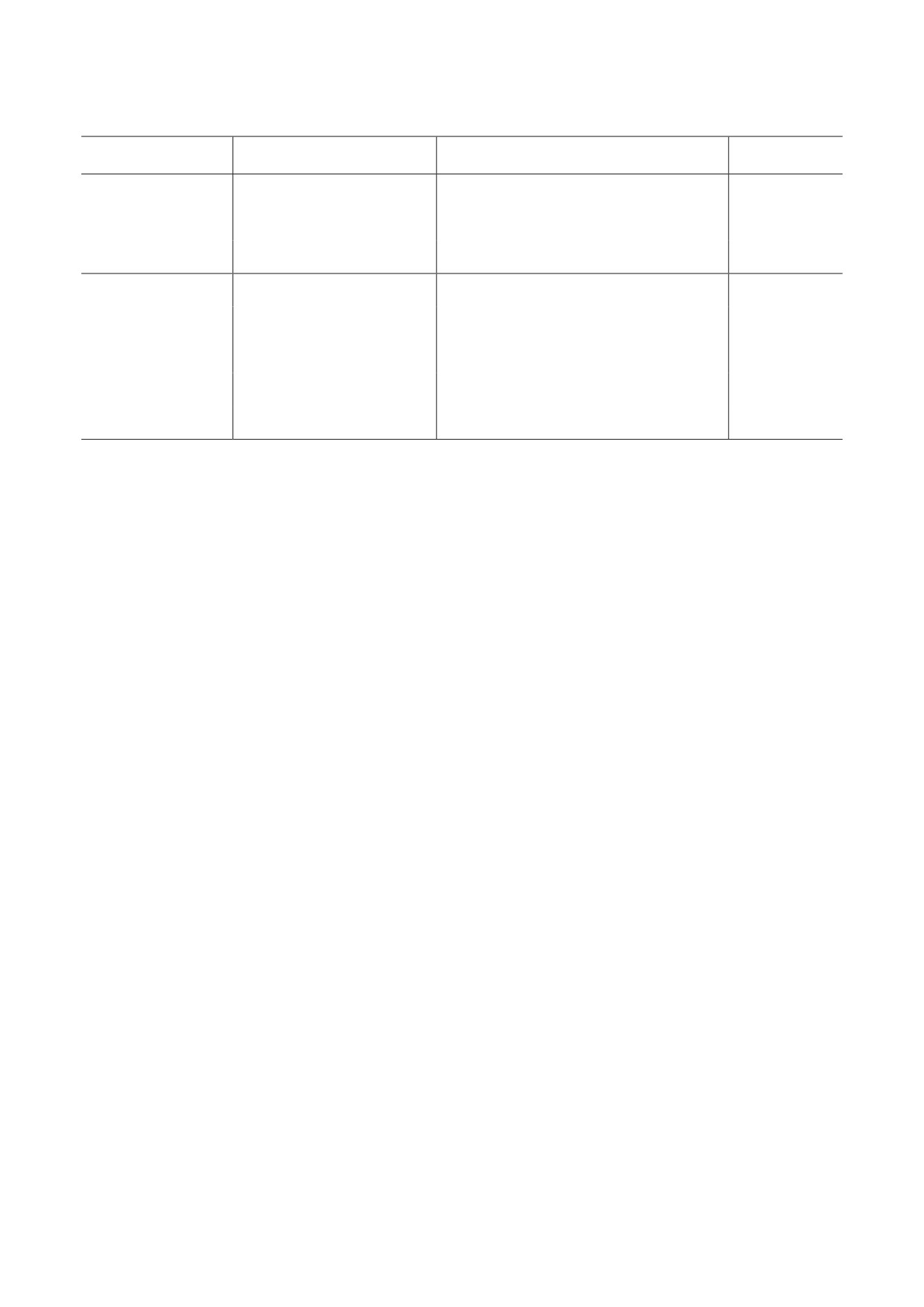
Рекуррентные транслокации, обнаруживаемые при лейкозах, вызванных терапией ингибиторами ДНК-топоизомераз II [57]
Ген
Транслокация
Ген-партнер
Ссылки
t(8;21)(q22;q22)
ETO (RUNX1T1)
[58-62]
AML1 (RUNX1)
t(3;21)(q26.2;q22)
MDS1-EVI1 (MECOM, PRDM3)
[63, 64]
t(1;21)(p36;q21)
PRDM16
[65, 66]
t(9;11)(p22;q23)
AF9 (MLLT3)
[58, 60]
t(4;11)(q21;q23)
AF4 (AFF1, MLLT2)
[67, 68]
MLL (KMT2A)
t(19;11)(q13;q23)
ELL
[60, 69]
t(11;19)(q23;p13.3)
ENL (MLLT1)
[60, 69, 70]
t(11;16)(q23;p13)
CREBBP (CBP)
[71-73]
Примечание. Указаны различные названия генов; полужирным шрифтом выделены те названия, которые приняты за
официальные в современной классификации генов HUGO Gene Nomenclature Committee.
Транслокации, рекуррентные для вторичных
ядра. Чем выше подвижность концов разры-
острых миелоидных лейкозов (ОМЛ), вызван-
вов в ядре, тем меньше на выбор партнера по
ных терапией ингибиторами ДНК-топоизоме-
перестройке будет влиять изначальное распо-
разы II. Вторичные лейкозы, вызванные тера-
ложение перестраивающихся локусов. С одной
пией ингибиторами ДНК-топоизомеразы II,
стороны, различными методами была пока-
характеризуются транслокациями с участием
зана прямая корреляция между частотой фор-
генов AML1 или MLL, лежащих на хромосо-
мирования транслокаций и пространствен-
мах 21 и 11. Причем список перестроек, кото-
ной близостью перестраивающихся локусов
рые раз за разом встречаются при этом типе
[74-77]. С другой стороны, многие исследова-
лейкоза, включает лишь 8 транслокаций (таб-
ния подтверждают повышенную подвижность
лица).
двуцепочечных разрывов в ядре, например,
Представленный набор транслокаций от-
их кластеризацию [78-80]. Кроме того, было
личается ограниченностью: для других опухо-
показано, что вероятность образования кон-
лей характерен гораздо более широкий набор
кретных транслокаций во многом зависит от
хромосомных аберраций, в том числе более
вероятности возникновения разрывов в пере-
широкий набор транслокаций с участием ге-
страивающихся локусах
[75]. Скорее всего,
нов AML1 и MLL. В данном обзоре будут рас-
разные факторы важны в разной степени в
смотрены факторы, обусловливающие воз-
случае той или иной транслокации [57, 81].
никновение транслокаций в случае лейкозов,
Например, в работе по наблюдению за процес-
вызванных терапией ингибиторами ДНК-то-
сом образования транслокаций в живых клет-
поизомераз типа II.
ках было показано, что и изначально близкие,
и далекие локусы формировали транслокации
после внесения в них разрывов, однако чаще
ФАКТОРЫ, ВЛИЯЮЩИЕ
происходили транслокации между близкими
НА ОБРАЗОВАНИЕ
локусами [82].
ХРОМОСОМНЫХ ТРАНСЛОКАЦИЙ
Далее, рассмотрим факторы, которые яв-
ляются определяющими для транслокаций при
Поиск универсальных механизмов образо-
вторичных лейкозах, вызванных терапией то-
вания транслокаций поначалу привел к выяв-
поизомеразными ядами. Во-первых, топоизо-
лению двух основных факторов, определяющих
меразные яды вызывают разрывы в неслучай-
результат перестройки. Первый фактор - вза-
ных участках генома, хотя таких участков и
имное расположение в ядре перестраиваю-
очень много. Работы по секвенированию точек
щихся генов до момента образования в них
разрывов, вызванных обработкой этопозидом,
разрыва. Чем ближе расположены локусы друг к
показывают, что к ним в большей степени чув-
другу в ядре, тем выше вероятность перестрой-
ствительны участки в основаниях хроматино-
ки между ними. Второй фактор - подвижность
вых петель [83, 84]. Также разрывы ДНК, ин-
перестраивающихся генов в пространстве
дуцированные этопозидом, отличаются ходом
5
БИОХИМИЯ том 88 вып. 7 2023
1
106
ЛОМОВ и др.
репарации от прочих ДЦР в клетке - необходи-
В целом, развитие опухоли
- процесс,
мость процессинга концов разрывов приводит
похожий на дарвиновскую эволюцию: среди
к задержке начала их репарации, что способ-
клеток с различными транслокациями отбира-
ствует пространственному разделению концов
ются те, в которых образуются новые функцио-
разрыва. Так, флуоресцентная in situ гибриди-
нальные гены, влияющие на пролиферативные
зация (FISH-анализ) с применением двуцвет-
свойства клетки [91]. Поэтому набор наблюдае-
ной пробы на разрыв (break-apart FISH probe)
мых у пациентов транслокаций будет включать
показывает, что при обработке клеток этопо-
только те транслокации, которые дают клеткам
зидом начало и конец многих аллелей MLL
пролиферативное преимущество. Далее мы
и AML1 расходятся на расстояние, детекти-
рассмотрим, почему транслокации, характер-
руемое с помощью светового микроскопа (не-
ные для вызванных терапией топоизомераз-
сколько сотен нанометров). Такого расхожде-
ными ядами вторичных лейкозов, приводят к
ния концов MLL и AML1 не наблюдается при
раковому перерождению бластных клеток. Для
обработке клеток высокими дозами радиа-
этого рассмотрим строение и функции нор-
ции [57]. Обычно репарация двуцепочечных
мальных белков AML1 и MLL, а также слитых с
разрывов осуществляется по механизму него-
ними белков, которые образуются в результате
мологичного соединения концов (NHEJ). Этот
транслокаций с участием AML1 и MLL.
механизм довольно надежен, и при репарации
концы разрыва сначала быстро соединяются с
помощью комплекса белков и удерживаются
МЕХАНИЗМЫ ОНКОГЕННОСТИ
до момента лигирования ДНК [85-87]. В слу-
ТРАНСЛОКАЦИЙ С УЧАСТИЕМ
чае же индукции разрывов ДНК этопозидом
ГЕНОВ AML1 И MLL
необходимость в процессинге ковалентного
комплекса топоизомеразы с ДНК приводит
Ген AML1. В 1991 г. был идентифицирован
к задержке формирования комплекса белков
точный локус хромосомной перестройки t(8;21)
NHEJ. Это существенно повышает шансы рас-
у пациентов с острым миелоидным лейкозом.
хождения концов разрыва в пространстве ядра
Перестраивающийся ген был, соответственно,
и соединения их с концами другого разрыва с
назван AML1 [92]. Затем ученые выяснили, что
образованием транслокации. Похожая ситуа-
ген AML1 гомологичен гену дрозофилы runt,
ция наблюдается при индукции разрывов эк-
открытому еще в 1980-е гг., и поэтому AML1
топически экспрессируемой в клетках нуклеа-
получил второе название RUNX1, от Runt-re-
зой Cas9 с одновременной обработкой клеток
lated transcription factor [63]. Вскоре было об-
ингибиторами белков системы NHEJ - в этом
наружено, что этот ген часто перестраивается
случае число транслокаций также возрас-
при лейкозах, вызванных терапией ингибито-
тает [88].
рами ДНК-топоизомераз II [93, 94]. В настоя-
В ситуации, когда разрывы дольше оста-
щее время известно, что ген AML1 участвует
ются не сшитыми, их концы демонстрируют
в перестройках не только при ОМЛ, но и при
и большую подвижность в пространстве ядра.
некоторых других видах лейкозов [63, 95].
Так, in vivo наблюдение за фокусами бел-
Изоформы мРНК и белка AML1. Выде-
ка 53BP1, который связывается с разрыва-
ляют три основные изоформы белка: AML1A,
ми ДНК, показывает более высокую подвиж-
AML1B и AML1C. Все они содержат вблизи
ность разрывов, индуцированных этопозидом,
N-конца Runt-домен (runt-homology domain,
по сравнению с разрывами, вызванными ра-
RHD), необходимый для связывания с ДНК и
диацией [89]. К свидетельствам в пользу вы-
для гетеродимеризации AML1 с белком CBFβ
сокой подвижности разрывов можно отнести
(core binding factor β). Гетеродимеризация с
и то, что после обработки клеток этопозидом
CBFβ повышает сродство AML1 к ДНК, хотя
разрывы чаще локализуются вне хромосом-
сам CBFβ не взаимодействует с ДНК [96, 97].
ных территорий, в которых они находятся в
Более длинные изоформы В и С содержат транс-
норме [57, 90]. В связи с повышенной подвиж-
активирующий домен (transactivation domain,
ностью концов разрывов для транслокаций,
TAD) и консервативный участок VWRPY на
происходящих после воздействия этопозида,
C-конце белка, участвующий во взаимодействии
взаимное расположение генов-партнеров в
с некоторыми белками-репрессорами (рис. 3).
ядре уже не имеет определяющего значения.
Нормальная функция AML1. Ген AML1 экс-
Так, 4C-анализ контактов AML1 и MLL не
прессируется в кроветворных клетках на про-
выявляет какого-либо предпочтения для их
тяжении всего онтогенеза [98]. Изучение его
генов-партнеров по транслокациям по сравне-
функции началось с нокаута Aml1 у мышей.
нию с другими генами [57].
В этих работах было показано, что основную
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1107
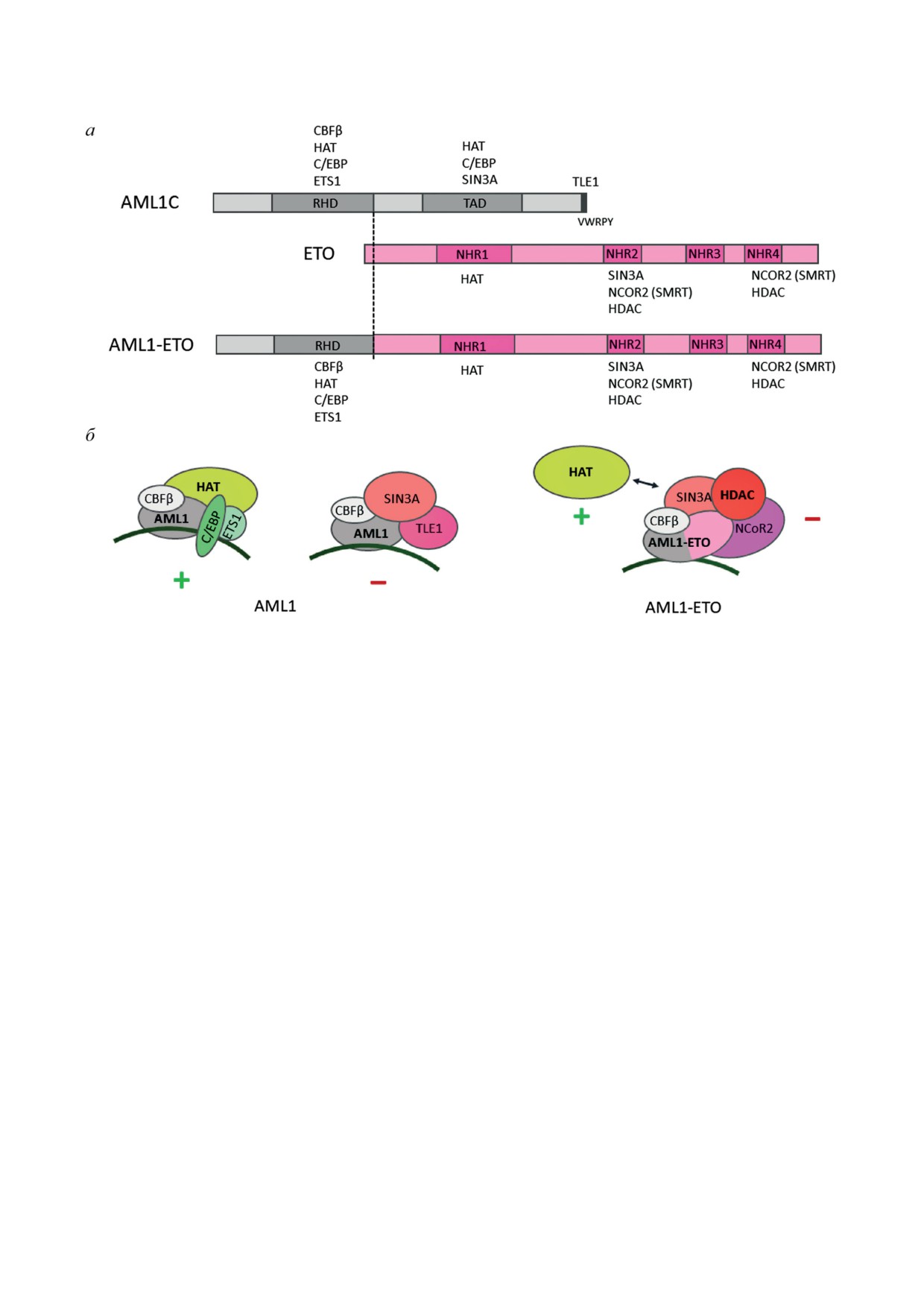
Рис. 3. Белки AML1, ETO и слитый белок AML1-ETO. а - Доменная структура AML1, ETO и слитого белка AML1-
ETO. У AML1: RHD - runt-homology domain, TAD - трансактивирующий домен, на C-конце отмечен пентапеп-
тид VWRPY. У ETO: NHR1-3 - nervy homology regions, MYND - (myeloid, Nervy, and DEAF-1)-домен. Возле доменов
перечислены белки, с которыми они взаимодействуют. б - Комплексы, формируемые на ДНК нормальным AML1
и слитым AML1-ETO. CBFβ, C/EBP, ETS1, TLE1 и SIN3A - белки, взаимодействующие с AML1 или AML1-ETO.
HAT - гистонацетилтрансферазы; HDAC - гистондеацетилазы
свою роль AML1 играет при созревании гемо-
YAP (Yes[-киназа] associated protein), GATA-1
поэтических стволовых клеток в раннем эмбрио-
(названный по узнаваемой им последова-
генезе [99-103]. Дальнейшие опыты, прове-
тельности), ETS1 (Erythroblast Transformation
денные in vitro на клетках гемогенного эндо-
Specific) и другие [105-113]. А в роли рекру-
телия мыши, продемонстрировали направ-
тируемых AML1 репрессоров выступают тран-
ляющую функцию AML1 в гемопоэзе: при
скрипционные факторы TLE1 (Transducin like
дифференцировке этих клеток до гемопоэти-
enhancer of split 1) и SIN3A (гомолог SIN3 дрож-
ческих стволовых клеток AML1 связывается
жей) [114, 115]. Связывание различных белков
с промоторами множества генов, привлекая
зависит от фосфорилирования AML1 и отли-
коактиваторы или корепрессоры. AML1 акти-
чается у разных изоформ [115-117].
вирует гены, участвующие в дифференцировке
Развитие лейкозов при перестройках гена
миелоидных клеток и эритроцитов, а к группе
AML1. Среди партнеров AML1 при рекуррент-
репрессируемых им принадлежат гены, играю-
ных транслокациях, вызванных ингибиторами
щие роль в развитии нервной системы, ске-
топоизомераз, самый частый - ген ETO (Eight
летных мышц и печени [104]. Таким образом,
twenty one), также называемый RUNX1T1 [118].
AML1 является многофункциональным регу-
Оба названия гена отсылают к его транслока-
лятором транскрипции генов, участвующих в
ции t(8;21) с AML1 (RUNX1). Эта хромосомная
дифференцировке гемопоэтических клеток.
транслокация встречается также при других
Для активации генов комплекс AML1/CBFβ
лейкозах и в достаточной мере изучена [119].
способен рекрутировать такие белки, как гис-
При транслокации AML1-ETO в получающем-
тонацетилтрансферазы p300 и CREBBP (cyclic
ся слитом белке присутствует N-конец от белка
AMP response element binding protein), коакти-
AML1, содержащий домен RHD. C-Конец сли-
ватор транскрипции C/EBP (CCAAT-enhancer-
того белка представлен почти полноразмер-
binding protein), транскрипционные факторы
ным белком ETO (рис. 3) [97].
БИОХИМИЯ том 88 вып. 7 2023
5*
1
108
ЛОМОВ и др.
Белок ETO представляет собой транскрип-
для дальнейших преобразований клетки на
ционный репрессор, в структуре которого при-
пути к развитию лейкоза.
сутствуют четыре консервативных участка
Ген MLL. В 1980-х гг. среди пациентов с
NHR (nervy homology regions - гомологичны
острым лейкозом начали выделять подгруппу с
по последовательности белку Nervy дрозо-
крайне неблагоприятным прогнозом. С появ-
филы) [120]. ETO сам по себе не связыва-
лением проточной цитофлуориметрии было
ется с ДНК - к целевому локусу он привле-
обнаружено, что бластные клетки этих агрес-
кается через транскрипционные факторы.
сивных лейкозов часто экспрессируют поверх-
А репрессия осуществляется с помощью ко-
ностные маркеры, свойственные как лимфо-
репрессоров, взаимодействующих с NHR-участ-
идной, так и миелоидной линиям. Иногда во
ками: NCoR1 (nuclear receptor corepressor),
время лечения наблюдалась даже смена набора
SMRT (silencing mediator of retinoid and thy-
этих поверхностных маркеров, и лейкоз, пер-
roid receptors), SIN3A и деацетилаз гисто-
воначально диагностированный как острый
нов [121, 122].
лимфоидный, мог рецидивировать в острый
Ген ETO в норме не экспрессируется в
миелоидный. Соответственно, был введен тер-
кроветворных и лимфоидных органах [123].
мин «лейкоз смешанного фенотипа» [137-139].
Можно заключить, что образование слитого
Вскоре стало ясно, что для этого лейкоза ха-
гена AML1-ETO приводит, во-первых, к появ-
рактерна транслокация локуса 11q23. В 1991 г.
лению практически полноразмерного белка
был идентифицирован ген, который находит-
ETO в клетках, где он не должен присутство-
ся в точке перестройки в клетках таких паци-
вать; а во-вторых, ETO за счет участка AML1
ентов. Соответственно, ген был назван MLL,
привлекается к генам, к которым он не должен
от mixed lineage leukemia (лейкоз смешанного
привлекаться. В итоге репрессируется тран-
фенотипа). Выявленный ген оказался гомо-
скрипция многих генов, экспрессия которых в
логом гена trithorax дрозофилы, кодирующего
норме регулируется транскрипционным фак-
лизинметилтрансферазу, которая активирует
тором AML1 [124-126].
Hox-гены [140, 141]. Ген MLL кодирует лизин-
В реальности ситуация сложнее - экспрес-
метилтрансферазу 2А, и отсюда его другое
сия слитого белка приводит не только к ре-
название - KMT2A (lysine (K)-specific methyl-
прессии многих генов, но и к активации неко-
transferase 2A).
торых других генов с помощью привлекаемых
Хромосомные транслокации с участием
белков-активаторов. Так, было показано, что
гена MLL наблюдаются примерно в 10% слу-
ETO способен привлекать гистонацетилтранс-
чаев острого миелоидного и острого лимфо-
феразы с помощью первого участка NHR [127].
идного лейкозов [142], причем среди лейко-
Вероятно, репрессирующее или активирующее
зов, вызванных терапией топоизомеразными
действие AML1-ETO зависит от сигнальных
ядами, такие транслокации составляют боль-
каскадов, активных в данный момент в клетке,
шую часть [24, 143, 144]. Особенно часто -
и особенностей хроматина в том месте, где на-
до 85% случаев - перестройки MLL встреча-
ходится регулируемый ген [128, 129].
ются при острых лейкозах у детей первого года
Важной особенностью клеток с t(8;21), по-
жизни. Также мутации этого гена обнаружива-
лученных от пациентов, является наличие ин-
ются и при многих других видах рака: в част-
тактного аллеля AML1, что говорит о необхо-
ности, рака толстой кишки, легких, мочевого
димости нормального AML1 для клетки [130].
пузыря, эндометрия и при раке груди [145-148].
По-видимому, интактный AML1 и слитый
Экспрессия перестроившегося MLL, слив-
AML1-ETO конкурируют за связывание с
шегося с одним из своих многочисленных
промоторами генов - нокдаун AML1-ETO в
генов-партнеров, достаточна для запуска со-
этих клетках меняет распределение сайтов свя-
бытий, приводящих к развитию лейкоза, что
зывания нормального AML1 [131, 132].
напрямую показано на мышах [142]. Это объ-
Также стоит отметить, что для развития
ясняется глобальной функцией MLL в клетках,
лейкоза, кроме перестройки AML1-ETO, необ-
где он экспрессируется: комплекс белков, фор-
ходимы дополнительные мутации, по крайней
мирующийся на основе MLL, осуществляет
мере в мышиных моделях [98, 133, 134]. Суще-
эпигенетическую маркировку промоторов тка-
ствуют данные о том, что на развитие лейкоза
неспецифичных генов. Такая маркировка не-
влияет и то, какие изоформы белка получа-
обходима для поддержания паттерна экспрес-
ются в результате альтернативного сплайсинга
сии этих генов в процессе пролиферации и
слитой мРНК [129, 135, 136]. Можно заклю-
дифференциации клеток - другими словами,
чить, что экспрессия слитого белка AML1-
MLL отвечает за «транскрипционную память»
ETO - первый шаг, который создает условия
клеток при их делении [149].
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1109
Ген MLL играет важную роль в эмбрио-
ют привлечение MLL к участкам хроматина
нальном развитии, особенно на ранних ста-
с H3K36me2/3 [167, 168].
диях гемопоэза, хотя и во взрослом возрасте
C-Концевой фрагмент MLL содержит до-
активно экспрессируется в некоторых тканях.
мен TAD (transactivation domain), привлекаю-
Особая роль MLL в эмбриональном развитии
щий гистонацетилтрансферазы H3K27, H3K9
состоит в активации транскрипции HOX-ге-
и H4K16 - p300/CREBBP, MOZ (monocytic
нов и некоторых других генов, связанных с
leukemic zinc-finger protein) и MOF (назван
пролиферацией и регуляцией клеточного ци-
по гомологу из дрозофилы MOF) соответ-
кла [150-154]. Нокаут по Mll приводит к гибели
ственно [169]. На C-конце MLL находится
мышей на ранних стадиях эмбриогенеза из-за
домен SET (Su(var)3-9, Enhancer-of-zeste and
нарушений гемопоэза и дифференциации ге-
Trithorax), который катализирует ди- и три-
мопоэтических стволовых клеток
[155-157].
метилирование H3K4 [170]. Вдобавок C-MLL
Кондиционный нокаут Mll у мышей показыва-
привлекает белки WDR5 (WD repeat-containing
ет, что во взрослом организме экспрессия Mll
protein 5), RbBP5 (Retinoblastoma-binding pro-
необходима клеткам костного мозга [158].
tein 5), ASH2L (Absent, small, or homeotic discs
Структура белка MLL. Структура нормаль-
protein 2 like) и DPY30, и в составе полученно-
ного белка MLL позволяет понять механиз-
го комплекса метилирующая активность SET-
мы его работы в клетке и последствия об-
домена MLL многократно усиливается [145,
разования слитых с MLL белков. MLL - круп-
171-173].
ный многодоменный белок, состоящий из
Важным свойством MLL-комплекса явля-
3969 аминокислот, его молекулярная масса -
ется возможность его переключения между со-
500 кДа (рис. 4). Несколько доменов MLL от-
стояниями транскрипционного активатора и
вечают за взаимодействие с определенными
транскрипционного репрессора. Переключе-
участками хроматина, а также с белками, ра-
ние происходит за счет белка CYP33 (cyclo-
ботающими в комплексе с MLL - это домены
philin 33) - пролин-изомеразы, при связыва-
AT-hook, CxxC, PHD. Каталитический домен
нии с которой происходит цис/транс-изомери-
осуществляет ди- и триметилирование лизина,
зация пролина в составе PHD [174]. В результа-
находящегося в положении 4 полипептидной
те MLL меняет свою структуру так, что вместо
цепи гистона Н3 (H3K4me2/3), что сопряжено
узнавания H3K4me2/3 домен PHD начинает
с активацией транскрипции соответствующих
связывать деацетилазы гистонов, репресси-
генов [145]. После синтеза на рибосоме MLL
рующие транскрипцию [175-177]. Важность
расщепляется протеазой таспазой 1 на два по-
этого механизма подчеркивается тем, что уда-
липептида - N-MLL и C-MLL, которые обра-
ление из MLL участка PHD приводит к опухо-
зуют единый комплекс, взаимодействуя доме-
левой трансформации клетки [178, 179].
нами FYRN и FYRC [159].
Таким образом, белок MLL является регу-
На N-фрагменте MLL находятся все доме-
лятором транскрипции генов, которые несут
ны, отвечающие за связывание транскриби-
метки активного хроматина - определенные
руемой ДНК. AT-hook - участок неспецифи-
гистоновые модификации и неметилирован-
ческого взаимодействия белка с малой борозд-
ные CpG. В зависимости от контекста и ак-
кой ДНК. Домен CxxC обеспечивает уже спе-
тивных сигнальных путей MLL записывает
цифическое связывание MLL с неметилиро-
активирующие модификации с помощью SET-
ванными CpG-динуклеотидами, которые ха-
домена и привлекаемых гистоновых ацетил-
рактерны для активных промоторов [160-162].
трансфераз либо репрессирующие - за счет
Окружающие CxxC-домен участки - pre-CxxC
привлекаемых гистоновых деацетилаз
[149].
и post-CxxC - важны для взаимодействия MLL
Можно заключить, что MLL обеспечивает тран-
с элонгационными факторами РНК-полиме-
скрипционную память при линейной диффе-
разы II PAFc (polymerase associated factor
ренцировке клеток в ходе эмбрионального раз-
complex) [163]. Ближе к середине белка на-
вития и в гемопоэзе.
ходится домен PHD (plant homeodomain),
Развитие лейкозов при перестройках гена
распознающий H3K4me2/3,
- метку про-
MLL. Хромосомные транслокации гена MLL
моторов активных генов [164-166]. Также на
чаще всего происходят с разрывом этого гена в
N-фрагменте MLL находится мотив для связы-
так называемом кластере точек разрыва, BCR
вания адаптерных белков Menin1 и PSIP1 (PC4
(breakpoint cluster region), протяженностью
[positive coactivator 4] And SFRS1 [Serine And
около 10 т.п.н. Этот BCR захватывает интроны
Arginine Rich Splicing Factor
1] Interacting
с 9 по 11. В структуре белка это соответствует
Protein 1; другое название белка - LEDGF/
области домена PHD. В слитых белках присут-
p75), которые дополнительно обеспечива-
ствует N-концевая часть белка MLL, а C-конец
БИОХИМИЯ том 88 вып. 7 2023
1110
ЛОМОВ и др.
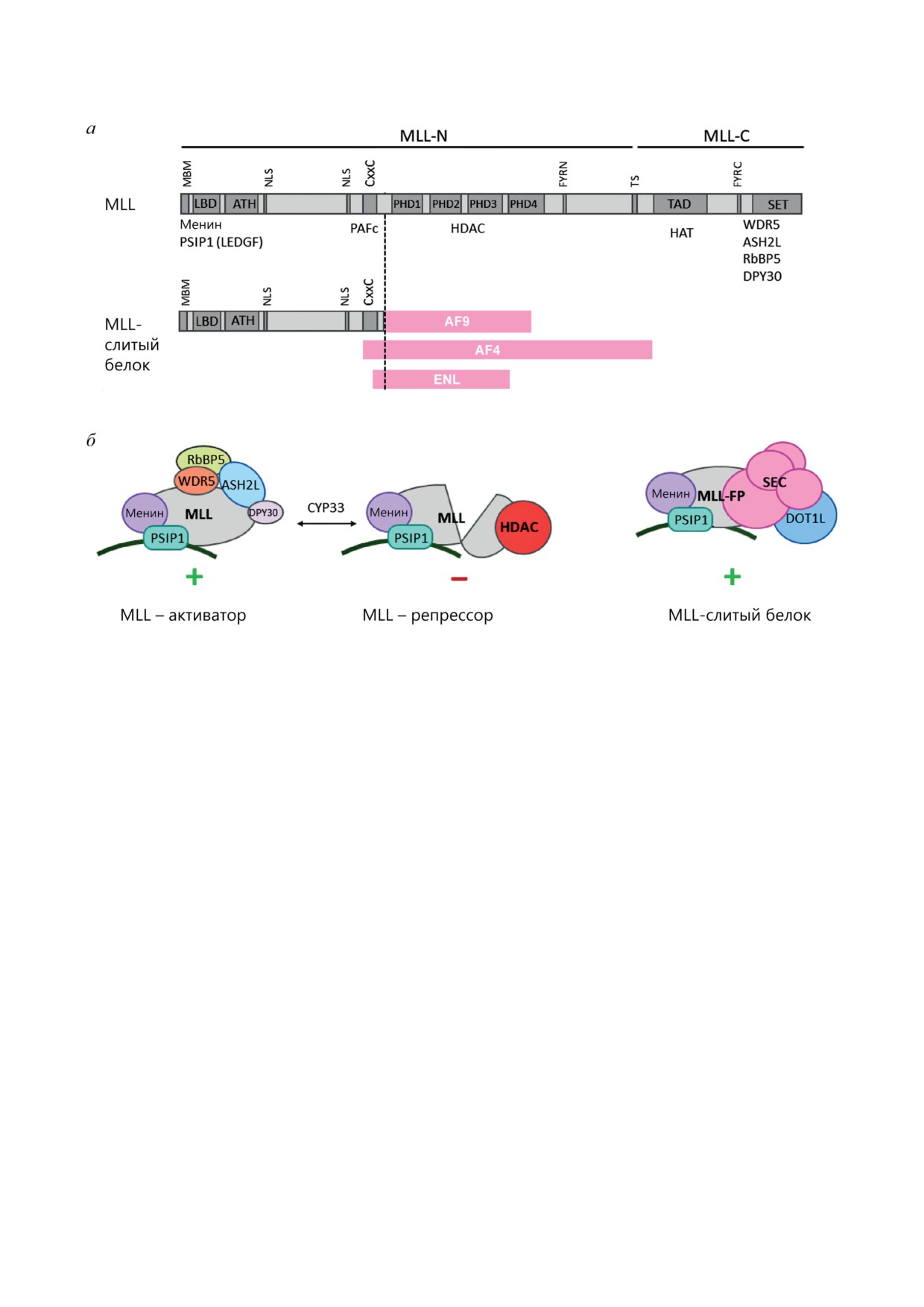
Рис. 4. Белок MLL и слитый с MLL белок. а - Доменная структура MLL и MLL-слитого белка. MLL-N и MLL-C -
фрагменты, образующиеся при расщеплении таспазой; MBM - менин-связывающий мотив; LBD - LEDGF(PSIP1)-
связывающий домен; ATH - участки AT-hook; NLS - сигналы ядерной локализации; CxxC - CxxC-домен и окру-
жающие его участки; PHD1-4 - участки домена PHD; FYRN и FYRC - FY-rich участки N- и C-доменов; TS - сайты
расщепления пре-MLL таспазой; TAD - трансактивирующий домен; SET - SET-домен. Возле доменов перечислены
белки, с которыми они взаимодействуют. б - Комплексы, формируемые нормальным MLL и MLL-слитым белком
(MLL-FP). Менин, PSIP1, DPY30, ASH2L, RbBP5, WDR5, DOT1L - белки, взаимодействующие с MLL и MLL-FP;
HDAC - гистондеацетилазы; SEC - суперэлонгационный комплекс. По [176, 177, 181]
происходит от партнера по транслокации [56,
как раз происходят с этими партнерами, за
176, 180, 182]. Исходя из структуры MLL и
исключением MLL-CREBBP (таблица).
функции его доменов понятно, что образова-
Схожесть транслокаций с участием генов
ние таких слитых белков принципиально нару-
ENL, AF9, AF4, ELL и AF10 объясняется тем,
шает функционирование комплекса на осно-
что они кодируют белки, образующие вме-
ве MLL.
сте с фактором P-TEFb суперэлонгационный
Необычной особенностью транслокаций с
комплекс, необходимый для осуществления
участием гена MLL является то, что существует
эффективной элонгации транскрипции РНК-
более 100 генов-партнеров MLL по транслока-
полимеразой II [185, 186]. И когда MLL ока-
ции [7]. И несмотря на такое разнообразие,
зывается слит с одним из компонентов этого
большинство случаев лейкозов с транслока-
комплекса, то он привлекает к ДНК весь ком-
циями MLL схожи по своим клиническим про-
плекс целиком, а также фактор DOT1L [187,
явлениям [183, 184]. Это можно объяснить,
188]. P-TEFb, входящий в данный комплекс,
во-первых, сходными точками разрыва в сли-
осуществляет фосфорилирование C-концевого
тых генах, и во-вторых, тем, что большинство
домена РНК-полимеразы II, что стимулирует
пациентов с перестройкой MLL несут трансло-
элонгацию транскрипции, а DOT1L осущест-
кации с одним из следующих генов: ENL, AF9,
вляет ди- и триметилирование H3K79 - эта
AF4, ELL и AF10, - тогда как остальные транс-
метка ассоциирована с активным хромати-
локации встречаются крайне редко. В част-
ном [189-190]. Следует также отметить, что
ности, все транслокации MLL, вызванные дей-
при транслокации MLL теряется участок гена,
ствием ингибиторов ДНК-топоизомеразы II,
который кодирует PHD-домен, отвечающий
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1111
за переключение MLL в репрессирующее со-
Общие закономерности развития лейкозов
стояние. В итоге остаток MLL полностью те-
при перестройках AML1 и MLL. Можно поды-
ряет способность к репрессии генов, а слитый
тожить, что гены AML1 и MLL, перестраива-
с ним суперэлонгационный комплекс и фактор
ющиеся в результате терапии ингибиторами
DOT1L напрямую активируют транскрипцию
ДНК-топоизомераз II, оба в норме регулируют
тех генов, с которыми связывается MLL.
транскрипцию в гемопоэтических клетках.
Механизм развития лейкоза при транс-
А образующиеся в результате транслокации сли-
локации MLL включает активацию транскрип-
тые AML1- и MLL-белки приводят к разбалан-
ции генов HoxA-кластера, а также генов, коди-
сировке транскрипции в этих клетках. При
рующих белки сигнальных путей, например,
этом, видимо, не существует единственного
генов малой GTPазы RAC1 и рецепторной
варианта изменения паттерна экспрессии ге-
тирозинкиназы FLT3. Это приводит к опу-
нов - в клоне клеток с транслокацией, скорее
холевой трансформации кроветворных кле-
всего, появляются клетки с разным паттерном
ток. Показано, что слитые MLL-белки сти-
экспрессии. И из этих эпигенетически разно-
мулируют экспрессию гомеобоксных генов
образных клеток постепенно выделяются наи-
HOXA, MEIS1 и PBX, генов-транскрипцион-
более активно делящиеся, которые становятся
ных регуляторов AML1 и MYC и других генов.
лейкозными клетками [196].
Промоторы активированных генов содер-
При развитии опухоли в клетках проис-
жат метки активного хроматина H3K4me3 и
ходят и соматические мутации, которые могут
H3K27ac, а также имеют повышенный уровень
повышать приспособленность опухолевых кло-
H3K79me2 [146, 148, 191].
нов: их возможность сопротивляться иммунной
В случае транслокации MLL-CREBBP ме-
системе и повышенная скорость деления. Од-
ханизм влияния слитого белка на клетку
нако при делении клеток передаются не только
схож - CREBPP является гистонацетилтранс-
мутации, но и эпигенетические метки, причем
феразой, которая навешивает активирую-
накопление эпигенетических изменений про-
щие метки на гистоны, а сам MLL разорван в
исходит гораздо быстрее, чем мутаций [197].
том же месте, что и при описанных выше его
И эффект от изменения уровня экспрессии
транслокациях [133].
гена часто более масштабный, чем от точечной
Также в последнее время накапливается
мутации. Поэтому клетки с транслокациями
все больше данных о том, что для развития
AML1 и MLL, в результате которых наблюда-
лейкоза необходима хотя бы кратковременная
ются масштабные эпигенетические эффекты,
экспрессия слитого гена, полученного в ре-
быстро развиваются в опасные лейкозы.
зультате реципрокной транслокации. Напри-
мер, выдвигается гипотеза, что продукт реци-
прокной транслокации AF4-MLL является
ЗАКЛЮЧЕНИЕ
глобальным активатором транскрипции, так
как C-конец белка MLL вносит активирующие
Вызванные терапией лейкозы - тяжелый
метки неспецифично, в отличие от интактного
побочный эффект терапии первичных опухо-
MLL [192]. Такие модификации могут при-
лей топоизомеразными ядами. «Отравленная»
вести к реактивации участков генома, которые
ДНК-топоизомераза типа II становится при-
уже были инактивированы в процессе диффе-
чиной образования двуцепочечных разрывов
ренцировки клеток. В числе реактивирован-
в геноме, что ведет к гибели клеток первичной
ных могут быть гены, работающие в стволо-
опухоли. Однако в организме существуют и
вых клетках, например, NANOG и OCT4, что
другие активно делящиеся клетки, например -
было показано для клеток с транслокацией
предшественники клеток крови. Поэтому в
t(4;11) [193, 194]. Эксперименты показывают,
ходе терапии топоизомеразными ядами в ге-
что для активации хроматина достаточно экс-
номе таких клеток также возникают разрывы.
прессии AF4-MLL в течение лишь 48 часов,
В частности, разрывы возникают в генах AML1,
при этом изменения в статусе хроматина оста-
MLL и их партнерах по перестройкам. Особый
ются в популяции клеток надолго [195].
характер таких разрывов - наличие на концах
Таким образом, при транслокациях с уча-
разрывов ковалентно связанных субъединиц
стием MLL происходит потеря контроля над
топоизомеразы - осложняет их репарацию.
транскрипцией генов, работающих в первую
Концы разрывов могут отделяться друг от дру-
очередь в незрелых клетках. Активируются ге-
га и встречаться с концами разрывов в генах-
ны, отвечающие за пролиферацию, и теряется
партнерах, в результате чего возникают хромо-
контроль над делением клеток, что приводит к
сомные транслокации. Образующиеся сли-
развитию лейкоза.
тые белки теряют часть функций нормальных
БИОХИМИЯ том 88 вып. 7 2023
1112
ЛОМОВ и др.
белков AML1 и MLL, а также приобретают новые
Несмотря на то что в последние годы было
функции, обусловленные функциональной ак-
получено много экспериментальных подтвер-
тивностью фрагментов (доменов) белков-парт-
ждений такого механизма развития вторич-
неров. В результате в некоторых клетках кост-
ного лейкоза, ряд вопросов еще ожидает
ного мозга нарушается регуляция экспрессии
своего ответа. Среди таких вопросов: какое
генов, в норме регулируемых белками AML1
влияние оказывают различные партнеры по
и MLL, что приводит к усилению экспрессии
перестройкам генов MLL и AML1, а также
генов, стимулирующих пролиферацию клеток
положение точки разрыва внутри этих генов
и к опухолевой трансформации клеток.
на развитие лейкоза, его клиническое про-
Повышенная частота встречаемости транс-
явление и прогноз? Как вещества, применяе-
локаций с участием генов AML1 и MLL вслед-
мые в терапии первичных опухолей совместно
ствие обработки клеток ингибиторами ДНК-
с топоизомеразными ядами, могут влиять на
топоизомераз II (рекуррентный характер таких
возникновение хромосомных транслокаций
перестроек) так же, как и отличие от хромо-
и их спектр? Ответить на эти вопросы помо-
сомных аберраций при других типах вторич-
гут результаты экспериментов на клеточных
ных неоплазий, объясняется целым рядом
моделях, где изучается как действие транс-
факторов, а именно: повышенной частотой
локаций на фенотип клеток [199, 200], так и
возникновения разрывов в этих генах при
сам процесс образования транслокаций [88,
ингибировании ДНК-топоизомеразы и селек-
201]. Будущие исследования на таких моде-
тивным преимуществом, которое дает клеткам
лях могут не только пролить свет на эти во-
экспрессия в них белковых продуктов слитых
просы, но также выявить способы снижения
генов. При этом сложность репарации таких
риска возникновения и развития вторичных
разрывов повышает их подвижность в ядре,
лейкозов.
что позволяет встретиться генам-партнерам,
которые в норме удалены друг от друга.
Вклад авторов. Все авторы участвовали в
Отдельно отметим, что разрывы в ДНК мо-
написании данного обзора.
гут быть индуцированы и не ингибированной
Финансирование. Исследование выполне-
топоизомеразой [49], правда вероятность таких
но при поддержке Министерства науки и выс-
разрывов значительно меньше, чем при инги-
шего образования Российской Федерации
бировании топоизомеразы. Тем не менее для
(соглашение 075-15-2021-1062) и Междисци-
вторичных лейкозов, вызванных предшествую-
плинарной научно-образовательной школы
щей терапией метотрексатом, характерны
Московского университета
«Молекулярные
некоторые транслокации, характерные для лей-
технологии живых систем и синтетическая
козов, вызванных терапией ингибиторами
биология».
ДНК-топоизомеразы
[198]. Влияние мето-
Конфликт интересов. Авторы заявляют об
трексата на образование транслокаций еще
отсутствии конфликта интересов.
предстоит подробно изучить, сейчас имеются
Соблюдение этических норм. Настоящая
предварительные данные, что он повышает
статья не содержит описания каких-либо ис-
вероятность транслокации AML1-ETO в экс-
следований с участием людей или животных в
периментах на клетках [88].
качестве объектов.
СПИСОК ЛИТЕРАТУРЫ
1. Becker, M. W., and Jordan, C. T. (2011) Leukemia
Payne-Turner, D., Vadodaria, B., Boggs, K., Yergeau, D.,
stem cells in 2010: Current understanding and future
Manne, J., Song, G., Edmonson, M., Nagahawatte, P.,
directions, Blood Rev.,
25,
75-81, doi:
10.1016/j.
Wei, L., Cheng, C., Pei, D., Sutton, R., Venn, N. C.,
blre.2010.11.001.
Chetcuti, A., Rush, A., Catchpoole, D., Heldrup, J.,
2. Wang, J. H. (2012) Mechanisms and impacts of
Fioretos, T., Lu, C., Ding, L., Pui, C. H., Shurtleff, S.,
chromosomal translocations in cancers, Front. Med.,
Mullighan, C. G., Mardis, E. R., Wilson, R. K.,
6, 263-274, doi: 10.1007/s11684-012-0215-5.
Gruber, T. A., Zhang, J., and Downing, J. R. (2015)
3. Andersson, A. K., Ma, J., Wang, J., Chen, X., Ged-
The landscape of somatic mutations in infant MLL-
man, A. L., Dang, J., Nakitandwe, J., Holmfeldt, L.,
rearranged acute lymphoblastic leukemias, Nat. Genet.,
Parker, M., Easton, J., Huether, R., Kriwacki, R.,
47, 330-337, doi: 10.1038/ng.3230.
Rusch, M., Wu, G., Li, Y., Mulder, H., Raimondi, S.,
4. Greaves, M. (2015) When one mutation is all it takes, Can-
Pounds, S., Kang, G., Shi, L., Becksfort, J., Gupta, P.,
cer Cell, 27, 433-434, doi: 10.1016/j.ccell.2015.03.016.
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1113
5.
Balgobind, B. V., Raimondi, S. C., Harbott, J.,
Oksenhendler, E., Lipinski, M., and Vassetzky, Y. S.
Zimmermann, M., Alonzo, T. A., Auvrignon, A.,
(2017) HIV Tat induces a prolonged MYC re-
Beverloo, H. B., Chang, M., Creutzig, U., Dworzak,
localization next to IGH in circulating B-cells,
M. N., Forestier, E., Gibson, B., Hasle, H., Harrison,
Leukemia, 31, 2515-2522, doi: 10.1038/leu.2017.106.
C. J., Heerema, N. A., Kaspers, G. J. L., Leszl, A.,
10.
Grande, B. M., Gerhard, D. S., Jiang, A., Griner,
Litvinko, N., Lo Nigro, L., Morimoto, A., Perot, C.,
N. B., Abramson, J. S., Alexander, T. B., Allen, H.,
Pieters, R., Reinhardt, D., Rubnitz, J. E., Smith,
Ayers, L. W., Bethony, J. M., Bhatia, K., Bowen, J.,
F. O., Stary, J., Stasevich, I., Strehl, S., Taga, T.,
Casper, C., Choi, J. K., Culibrk, L., Davidsen, T. M.,
Tomizawa, D., Webb, D., Zemanova, Z., Zwaan,
Dyer, M. A., Gastier-Foster, J. M., Gesuwan, P.,
C. M., and Van Den Heuvel-Eibrink, M. M. (2009)
Greiner, T. C., Gross, T. G., Hanf, B., Harris, N. L.,
Novel prognostic subgroups in childhood
11q23/
He, Y., Irvin, J. D., Jaffe, E. S., Jones, S. J. M.,
MLL-rearranged acute myeloid leukemia: results of
Kerchan, P., Knoetze, N., Leal, F. E., Lichtenberg,
an international retrospective study, Blood, 114, 2489-
T. M., Ma, Y., Martin, J. P., Martin, M. R., Mbulaiteye,
2496, doi: 10.1182/blood-2009-04-215152.
S. M., Mullighan, C. G., Mungall, A. J., Namirembe, C.,
6.
Döhner, H., Estey, E., Grimwade, D., Amadori, S.,
Novik, K., Noy, A., Ogwang, M. D., Omoding, A.,
Appelbaum, F. R., Büchner, T., Dombret, H., Ebert,
Orem, J., Reynolds, S. J., Rushton, C. K., Sandlund,
B. L., Fenaux, P., Larson, R. A., Levine, R. L., Lo-
J. T., Schmitz, R., Taylor, C., Wilson, W. H., Wright,
Coco, F., Naoe, T., Niederwieser, D., Ossenkoppele,
G. W., Zhao, E. Y., Marra, M. A., Morin, R. D.,
G. J., Sanz, M., Sierra, J., Tallman, M. S., Tien, H. F.,
and Staudt, L. M. (2019) Genome-wide discovery of
Wei, A. H., Löwenberg, B., and Bloomfield, C. D.
somatic coding and noncoding mutations in pediatric
(2017) Diagnosis and management of AML in adults:
endemic and sporadic Burkitt lymphoma, Blood, 133,
2017 ELN recommendations from an international
1313-1324, doi: 10.1182/blood-2018-09-871418.
expert panel, Blood,
129,
424-447, doi:
10.1182/
11.
Lieber, M. R. (2016) Mechanisms of human lymphoid
blood-2016-08-733196.
chromosomal translocations, Nat. Rev. Cancer, 16,
7.
Meyer, C., Burmeister, T., Gröger, D., Tsaur, G.,
387-398, doi: 10.1038/nrc.2016.40.
Fechina, L., Renneville, A., Sutton, R., Venn, N. C.,
12.
Roos, W. P., and Kaina, B. (2006) DNA damage-
Emerenciano, M., Pombo-De-Oliveira, M. S., Barbieri
induced cell death by apoptosis, Trends Mol. Med., 12,
Blunck, C., Almeida Lopes, B., Zuna, J., Trka, J.,
440-450, doi: 10.1016/j.molmed.2006.07.007.
Ballerini, P., Lapillonne, H., De Braekeleer, M.,
13.
Bhatia, S. (2013) Therapy-related myelodysplasia and
Cazzaniga, G., Corral Abascal, L., Van Der Velden,
acute myeloid leukemia, Semin. Oncol., 40, 666-675,
V. H. J., Delabesse, E., Park, T. S., Oh, S. H., Silva,
doi: 10.1053/j.seminoncol.2013.09.013.
M. L. M., Lund-Aho, T., Juvonen, V., Moore, A. S.,
14.
Schoch, C., Kern, W., Schnittger, S., Hiddemann, W.,
Heidenreich, O., Vormoor, J., Zerkalenkova, E.,
and Haferlach, T. (2004) Karyotype is an independent
Olshanskaya, Y., Bueno, C., Menendez, P., Teigler-
prognostic parameter in therapy-related acute myeloid
Schlegel, A., Zur Stadt, U., Lentes, J., Göhring, G.,
leukemia (t-AML): an analysis of
93 patients
Kustanovich, A., Aleinikova, O., Schäfer, B. W.,
with t-AML in comparison to 1091 patients with
Kubetzko, S., Madsen, H. O., Gruhn, B., Duarte, X.,
de novo AML, Leukemia, 18, 120-125, doi: 10.1038/
Gameiro, P., Lippert, E., Bidet, A., Cayuela, J. M.,
sj.leu.2403187.
Clappier, E., Alonso, C. N., Zwaan, C. M., Van Den
15.
Borthakur, G., and Estey, E. E. (2007) Therapy-related
Heuvel-Eibrink, M. M., Izraeli, S., Trakhtenbrot, L.,
acute myelogenous leukemia and myelodysplastic
Archer, P., Hancock, J., Möricke, A., Alten, J.,
syndrome, Curr. Oncol. Rep., 9, 373-377, doi: 10.1007/
Schrappe, M., Stanulla, M., Strehl, S., Attarbaschi, A.,
s11912-007-0050-z.
Dworzak, M., Haas, O. A., Panzer-Grümayer, R.,
16.
Godley, L. A., and Larson, R. A. (2008) Therapy-
Sedék, L., Szczepa, T., Caye, A., Suarez, L., Cavé, H.,
related myeloid leukemia, Semin. Oncol., 35, 418-429,
and Marschalek, R. (2018) The MLL recombinome
doi: 10.1053/j.seminoncol.2008.04.012.
of acute leukemias in 2017, Leukemia, 32, 273-284,
17.
Østgård, L. S. G., Medeiros, B. C., Sengeløv, H.,
doi: 10.1038/leu.2017.213.
Nørgaard, M., Andersen, M. K., Dufva, I., Friis, L. S.,
8.
Lomov, N., Zerkalenkova, E., Lebedeva, S., Viush-
Kjeldsen, E., Marcher, C. W., Preiss, B., Severinsen, M.,
kov, V., and Rubtsov, M. A. (2021) Cytogenetic
and Nørgaard, J. M. (2015) Epidemiology and clinical
and molecular genetic methods for chromosomal
significance of secondary and therapy-related acute
translocations detection with reference to the KMT2A/
myeloid leukemia: A national population-based cohort
MLL gene, Crit. Rev. Clin. Lab. Sci., 58, 180-206,
study, J. Clin. Oncol., 33, 3641-3649, doi: 10.1200/
doi: 10.1080/10408363.2020.1844135.
JCO.2014.60.0890.
9.
Germini, D., Tsfasman, T., Klibi, M., El-Amine, R.,
18.
Tiruneh, T., Enawgaw, B., and Shiferaw, E. (2020)
Pichugin, A., Iarovaia, O. V., Bilhou-Nabera, C.,
Genetic pathway in the pathogenesis of therapy-
Subra, F., Bou Saada, Y., Sukhanova, A., Boutboul, D.,
related myeloid neoplasms: a literature review, Oncol.
Raphaël, M., Wiels, J., Razin, S. V., Bury-Moné, S.,
Ther., 8, 45-57, doi: 10.1007/s40487-020-00111-7.
БИОХИМИЯ том 88 вып. 7 2023
1114
ЛОМОВ и др.
19.
Pedersen-Bjergaard, J., Andersen, M. K., Christian-
ethylidene-beta-D-glucopyranoside) (etoposide) cy-
sen, D. H., and Nerlov, C. (2002) Genetic pathways
totoxicity, Cancer Res., 43, 120-124.
in therapy-related myelodysplasia and acute myeloid
31.
Watson, J. D., and Crick, F. H. C. (1953) Genetical
leukemia, Blood, 99, 1909-1912, doi: 10.1182/blood.
implications of the structure of deoxyribonucleic acid,
V99.6.1909.
Nature, 171, 964-967, doi: 10.1038/171964b0.
20.
Czader, M., and Orazi, A. (2009) Therapy-related
32.
Delbrück, M. (1954) On the replication of desoxy-
myeloid neoplasms, Am. J. Clin. Pathol., 132, 410-425,
ribonucleic acid (DNA), Proc. Natl. Acad. Sci. USA,
doi: 10.1309/AJCPD85MCOHHCOMQ.
40, 783-788, doi: 10.1073/pnas.40.9.783.
21.
Kayser, S., Döhner, K., Krauter, J., Köhne, C.-H.,
33.
McKie, S. J., Neuman, K. C., and Maxwell, A. (2021)
Horst, H. A., Held, G., von Lilienfeld-Toal, M.,
DNA topoisomerases: Advances in understanding
Wilhelm, S., Kündgen, A., Götze, K., Rummel, M.,
of cellular roles and multi-protein complexes via
Nachbaur, D., Schlegelberger, B., Göhring, G.,
structure-function analysis, BioEssays, 43, e2000286,
Späth, D., Morlok, C., Zucknick, M., Ganser, A.,
doi: 10.1002/bies.202000286.
Döhner, H., Schlenk, R. F., and German-Austrian
34.
Champoux, J. J. (2001) DNA topoisomerases: struc-
AMLSG (2011) The impact of therapy-related acute
ture, function, and mechanism, Annu. Rev. Biochem.,
myeloid leukemia (AML) on outcome in 2853 adult
70, 369-413, doi: 10.1146/annurev.biochem.70.1.369.
patients with newly diagnosed AML, Blood, 117, 2137-
35.
Bliska, J. B., and Cozzarelli, N. R. (1987) Use of site-
2145, doi: 10.1182/blood-2010-08-301713.
specific recombination as a probe of DNA structure
22.
McNerney, M. E., Godley, L. A., and Le Beau, M. M.
and metabolism in vivo, J. Mol. Biol., 194, 205-218,
(2017) Therapy-related myeloid neoplasms: When
doi: 10.1016/0022-2836(87)90369-X.
genetics and environment collide, Nat. Rev. Cancer,
36.
Zechiedrich, E. L., and Cozzarelli, N. R. (1995) Roles
17, 513-527, doi: 10.1038/nrc.2017.60.
of topoisomerase IV and DNA gyrase in DNA unlink-
23.
Ratain, M. J., and Rowley, J. D. (1992) Therapy-
ing during replication in Escherichia coli, Genes Dev.,
related acute myeloid leukemia secondary to inhibitors
9, 2859-2869, doi: 10.1101/gad.9.22.2859.
of topoisomerase II: From the bedside to the target
37.
Yang, L., Wold, M. S., Li, J. J., Kelly, T. J., and Liu,
genes, Ann. Oncol., 3, 107-111, doi: 10.1093/oxford-
L. F. (1987) Roles of DNA topoisomerases in simian
journals.annonc.a058121.
virus 40 DNA replication in vitro, Proc. Natl. Acad. Sci.
24.
Mauritzson, N., Albin, M., Rylander, L., Billström, R.,
USA, 84, 950-954, doi: 10.1073/pnas.84.4.950.
Ahlgren, T., Mikoczy, Z., Björk, J., Strömberg, U.,
38.
Buchenau, P., Saumweber, H., and Arndt-Jovin, D. J.
Nilsson, P., Mitelman, F., Hagmar, L., and Johans-
(1993) Consequences of topoisomerase II inhibition in
son, B. (2002) Pooled analysis of clinical and cyto-
early embryogenesis of Drosophila revealed by in vivo
genetic features in treatment-related and de novo
confocal laser scanning microscopy, J. Cell Sci., 104
adult acute myeloid leukemia and myelodysplastic
(Pt 4), 1175-1185, doi: 10.1242/jcs.104.4.1175.
syndromes based on a consecutive series of 761 patients
39.
Liu, L. F., Rowe, T. C., Yang, L., Tewey, K. M., and
analyzed 1976-1993 and on 5098 unselected cases
Chen, G. L. (1983) Cleavage of DNA by mammalian
reported in the literature 1974-2001, Leukemia, 16,
DNA topoisomerase II, J. Biol. Chem., 258, 15365-
2366-2378, doi: 10.1038/sj.leu.2402713.
15370, doi: 10.1016/S0021-9258(17)43815-4.
25.
Hande, K. R. (1998) Etoposide: four decades of devel-
40.
Schmidt, B. H., Osheroff, N., and Berger, J. M.
opment of a topoisomerase ii inhibitor, Eur. J. Cancer,
(2012) Structure of a topoisomerase II-DNA-nucle-
34, 1514-1521, doi: 10.1016/S0959-8049(98)00228-7.
otide complex reveals a new control mechanism for
26.
King, L. S., and Sullivan, M. (1946) The similarity of
ATPase activity, Nat. Struct. Mol. Biol., 19, 1147-1154,
the effect of podophyllin and colchicine and their use
doi: 10.1038/nsmb.2388.
in the treatment of condylomata acuminata, Science,
41.
Nitiss, J. L. (2009) DNA topoisomerase II and its
104, 244-245, doi: 10.1126/science.104.2698.244.
growing repertoire of biological functions, Nat. Rev.
27.
Greenspan, E. M. (1950) Effect of alpha-peltatin,
Cancer, 9, 327-337, doi: 10.1038/nrc2608.
beta-peltatin, and podophyllotoxin on lymphomas and
42.
Bates, A. D., Berger, J. M., and Maxwell, A.
other transplanted tumors, J. Natl. Cancer Inst., 10,
(2011) The ancestral role of ATP hydrolysis in
1295-1333.
type II topoisomerases: prevention of DNA dou-
28.
Baldwin, E. L., and Osheroff, N. (2005) Etoposide, to-
ble-strand breaks, Nucleic Acids Res., 39, 6327-6339,
poisomerase II and cancer, Curr. Med. Chem. Antican-
doi: 10.1093/nar/gkr258.
cer Agents, 5, 363-372, doi: 10.2174/1568011054222364.
43.
Bush, N. G., Evans-roberts, K., and Maxwell, A.
29.
Burden, D. A., and Osheroff, N. (1998) Mechanism
(2015) Macromolecules DNA topoisomerases, EcoSal
of action of eukaryotic topoisomerase II and drugs
Plus, 6, 1-34, doi: 10.1128/ecosalplus.ESP-0010-2014.
targeted to the enzyme, Biochim. Biophys. Acta, 1400,
44.
Bax, B. D., Murshudov, G., Maxwell, A., and
139-154, doi: 10.1016/S0167-4781(98)00132-8.
Germe, T. (2019) DNA topoisomerase inhibitors:
30.
Wozniak, A. J., and Ross, W. E. (1983) DNA damage as
trapping a DNA-cleaving machine in motion, J. Mol.
a basis for 4’-demethylepipodophyllotoxin-9-(4,6-O-
Biol., 431, 3427-3449, doi: 10.1016/j.jmb.2019.07.008.
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1115
45.
Vann, K. R., Oviatt, A. A., and Osheroff, N. (2021)
Austin, C. A. (2012) Model for MLL translocations
Topoisomerase II poisons: converting essential en-
in therapy-related leukemia involving topoisomerase
zymes into molecular scissors, Biochemistry, 60, 1630-
IIβ-mediated DNA strand breaks and gene proximity,
1641, doi: 10.1021/acs.biochem.1c00240.
Proc. Natl. Acad. Sci. USA,
109,
8989-8994,
46.
Robinson, M. J., and Osheroff, N. (1991) Effects of
doi: 10.1073/pnas.1204406109.
antineoplastic drugs on the post-strand-passage DNA
57.
Lomov, N. A., Viushkov, V. S., Ulianov, S. V.,
cleavage/religation equilibrium of topoisomerase II,
Gavrilov, A. A., Alexeyevsky, D. A., Artemov, A. V.,
Biochemistry, 30, 1807-1813, doi: 10.1021/bi00221a012.
Razin, S. V., and Rubtsov, M. A. (2022) Recurrent
47.
Yan, H., Tammaro, M., and Liao, S. (2016) Collision
translocations in topoisomerase inhibitor-related leu-
of trapped topoisomerase 2 with transcription and
kemia are determined by the features of DNA breaks
replication: generation and repair of DNA double-
rather than by the proximity of the translocating genes,
strand breaks with 5′ adducts, Genes (Basel), 7, 32,
Int. J. Mol. Sci., 23, 9824, doi: 10.3390/ijms23179824.
doi: 10.3390/genes7070032.
58.
Rowley, J. D., and Olney, H. J. (2002) International
48.
Riccio, A. A., Schellenberg, M. J., and Williams,
Workshop on the relationship of prior therapy to bal-
R. S. (2020) Molecular mechanisms of topoisomerase
anced chromosome aberrations in therapy-related
2 DNA-protein crosslink resolution, Cell. Mol. Life
myelodysplastic syndromes and acute leukemia: over-
Sci., 77, 81-91, doi: 10.1007/s00018-019-03367-z.
view report, Genes Chromosomes Cancer, 33, 331-345,
49.
Swan, R. L., Cowell, I. G., and Austin, C. A. (2022)
doi: 10.1002/gcc.10040.
Mechanisms to repair stalled topoisomerase II-DNA
59.
Pedersen-Bjergaard, J., Andersen, M. K., Andersen,
covalent complexes, Mol. Pharmacol., 101, 24-32,
M. T., and Christiansen, D. H. (2008) Genetics of
doi: 10.1124/molpharm.121.000374.
therapy-related myelodysplasia and acute myeloid
50.
Tomicic, M. T., and Kaina, B. (2013) Topoisomerase
leukemia, Leukemia,
22,
240-248, doi:
10.1038/
degradation, DSB repair, p53 and IAPs in cancer
sj.leu.2405078.
cell resistance to camptothecin-like topoisomerase I
60.
Imamura, T., Taga, T., Takagi, M., Kawasaki, H.,
inhibitors, Biochim. Biophys. Acta Rev. Cancer, 1835,
Koh, K., Taki, T., Adachi, S., Manabe, A., and
11-27, doi: 10.1016/j.bbcan.2012.09.002.
Ishida, Y. (2018) Nationwide survey of therapy-related
51.
Montecucco, A., and Biamonti, G. (2007) Cellular
leukemia in childhood in Japan, Int. J. Hematol., 108,
response to etoposide treatment, Cancer Lett., 252,
91-97, doi: 10.1007/s12185-018-2439-x.
9-18, doi: 10.1016/j.canlet.2006.11.005.
61.
Gustafson, S. A., Lin, P., Chen, S. S., Chen, L.,
52.
Kantidze, O. L., and Razin, S. V. (2007) Chemo-
Abruzzo, L. V., Luthra, R., Medeiros, L. J., and
therapy-related secondary leukemias: a role for
Wang, S. A. (2009) Therapy-related acute myeloid
DNA repair by error-prone non-homologous end
leukemia with t(8;21) (q22;q22) shares many features
joining in topoisomerase II - induced chromosom-
with de novo acute myeloid leukemia with t(8;21)
al rearrangements, Gene, 391, 76-79, doi: 10.1016/
(q22;q22) but does not have a favorable outcome,
j.gene.2006.12.006.
Am. J. Clin. Pathol., 131, 647-655, doi: 10.1309/
53.
Smith, K. A., Cowell, I. G., Zhang, Y., Sondka, Z.,
AJCP5ETHDXO6NCGZ.
and Austin, C. A. (2014) The role of topoisomerase II
62.
Slovak, M. L., Bedell, V., Popplewell, L., Arber, D. A.,
beta on breakage and proximity of RUNX1 to partner
Schoch, C., and Slater, R. (2002) 21q22 balanced
alleles RUNX1T1 and EVI1, Genes Chromosomes
chromosome aberrations in therapy-related hema-
Cancer, 53, 117-128, doi: 10.1002/gcc.22124.
topoietic disorders: report from an international
54.
Zhang, Y., Strissel, P., Strick, R., Chen, J., Nucifora, G.,
workshop, Genes Chromosomes Cancer, 33, 379-394,
Le Beau, M. M., Larson, R. A., and Rowley, J. D.
doi: 10.1002/gcc.10042.
(2002) Genomic DNA breakpoints in AML1/RUNX1
63.
Sood, R., Kamikubo, Y., and Liu, P. (2017) Role of
and ETO cluster with topoisomerase II DNA cleav-
RUNX1 in hematological malignancies, Blood, 129,
age and DNase I hypersensitive sites in t(8;21) leu-
2070-2082, doi: 10.1182/blood-2016-10-687830.
kemia, Proc. Natl. Acad. Sci. USA, 99, 3070-3075,
64.
Tanaka, K., Oshikawa, G., Akiyama, H., Ishida, S.,
doi: 10.1073/pnas.042702899.
Nagao, T., Yamamoto, M., and Miura, O. (2017)
55.
Strissel, P. L., Strick, R., Tomek, R. J., Roe, B. A.,
Acute myeloid leukemia with t(3;21)(q26.2;q22) de-
Rowley, J. D., and Zeleznik-Le, N. J. (2000) DNA
veloping following low-dose methotrexate therapy
structural properties of AF9 are similar to MLL
for rheumatoid arthritis and expressing two AML1/
and could act as recombination hot spots resulting
MDS1/EVI1 fusion proteins: A case report, Oncol.
in MLL/AF9 translocations and leukemogenesis,
Lett., 14, 97-102, doi: 10.3892/ol.2017.6151.
Hum. Mol. Genet.,
9,
1671-1679, doi:
10.1093/
65.
Sato, Y., Izumi, T., Kanamori, H., Davis, E. M.,
hmg/9.11.1671.
Miura, Y., Larson, R. A., Le Beau, M. M., Ozawa, K.,
56.
Cowell, I. G., Sondka, Z., Smith, K., Lee, K. C.,
and Rowley, J. D. (2002) t(1;3)(p36;p21) is a recurring
Manville, C. M., Sidorczuk-Lesthuruge, M., Rance,
therapy-related translocation, Genes Chromosomes
H. A., Padget, K., Jackson, G. H., Adachi, N., and
Cancer, 34, 186-192, doi: 10.1002/gcc.10055.
БИОХИМИЯ том 88 вып. 7 2023
1116
ЛОМОВ и др.
66.
Duhoux, F. P., Ameye, G., Montano-Almendras,
strand breaks in mammalian cells, Nat. Cell Biol., 9,
C. P., Bahloula, K., Mozziconacci, M. J., Laibe, S.,
675-682, doi: 10.1038/ncb1591.
Wlodarska, I., Michaux, L., Talmant, P., Richebourg, S.,
75.
Zhang, Y., McCord, R. P., Ho, Y.-J., Lajoie, B. R.,
Lippert, E., Speleman, F., Herens, C., Struski, S.,
Hildebrand, D. G., Simon, A. C., Becker, M. S., Alt,
Raynaud, S., Auger, N., Nadal, N., Rack, K.,
F. W., and Dekker, J. (2012) Chromosomal trans-
Mugneret, F., Tigaud, I., Lafage, M., Taviaux, S.,
locations are guided by the spatial organization
Roche-Lestienne, C., Latinne, D., Libouton, J. M.,
of the genome, Cell, 148, 908-921, doi: 10.1016/
Demoulin, J. B., and Poirel, H. A. (2012) PRDM16
j.cell.2012.02.002.
(1p36) translocations define a distinct entity of
76.
Engreitz, J. M., Agarwala, V., and Mirny, L. A. (2012)
myeloid malignancies with poor prognosis but may
Three-dimensional genome architecture influences
also occur in lymphoid malignancies, Br. J. Haematol.,
partner selection for chromosomal translocations in
156, 76-88, doi: 10.1111/j.1365-2141.2011.08918.x.
human disease, PLoS One, 7, e44196, doi: 10.1371/
67.
Andersen, M. K., Christiansen, D. H., Jensen, B. A.,
journal.pone.0044196.
Ernst, P., Hauge, G., and Pedersen-Bjergaard, J.
77.
Sathitruangsak, C., Righolt, C. H., Klewes, L., Tung
(2001) Therapy-related acute lymphoblastic leukae-
Chang, D., Kotb, R., and Mai, S. (2017) Distinct and
mia with MLL rearrangements following DNA to-
shared three-dimensional chromosome organization
poisomerase II inhibitors, an increasing problem: re-
patterns in lymphocytes, monoclonal gammopathy
port on two new cases and review of the literature since
of undetermined significance and multiple myeloma,
1992, Br. J. Haematol., 114, 539-543, doi: 10.1046/
Int. J. Cancer, 140, 400-410, doi: 10.1002/ijc.30461.
j.1365-2141.2001.03000.x.
78.
Falk, M., Lukasova, E., Gabrielova, B., Ondrej, V.,
68.
Cowell, I. G., and Austin, C. A. (2012) Mechanism of
and Kozubek, S. (2007) Chromatin dynamics during
generation of therapy related leukemia in response to
DSB repair, Biochim. Biophys. Acta, 1773, 1534-1545,
anti-topoisomerase II agents, J. Environ. Res. Public
doi: 10.1016/j.bbamcr.2007.07.002.
Health, 9, 2075-2091, doi: 10.3390/ijerph9062075.
79.
Aymard, F., Aguirrebengoa, M., Guillou, E., Javierre,
69.
Moorman, A. V., Hagemeijer, A., Charrin, C., Rieder,
B. M., Bugler, B., Arnould, C., Rocher, V., Iacovoni,
H., and Secker-Walker, L. M. (1998) Clinical profile
J. S., Biernacka, A., Skrzypczak, M., Ginalski, K.,
of 53 patients, Leukemia, 12, 805-810, doi: 10.1038/
Rowicka, M., Fraser, P., and Legube, G.
(2017)
sj.leu.2401016.
Genome-wide mapping of long-range contacts unveils
70.
Meyer, C., Burmeister, T., Strehl, S., Schneider, B.,
clustering of DNA double-strand breaks at damaged
Hubert, D., Zach, O., Haas, O., Klingebiel, T.,
active genes, Nat. Struct. Mol. Biol., 24, 353-361,
Dingermann, T., and Marschalek, R. (2007) Spliced
doi: 10.1038/nsmb.3387.
MLL fusions: a novel mechanism to generate func-
80.
Wang, H., Nakamura, M., Abbott, T. R., Zhao, D.,
tional chimeric MLL-MLLT1 transcripts in t(11;19)
Luo, K., Yu, C., Nguyen, C. M., Lo, A., Daley,
(q23;p13.3) leukemia, Leukemia,
21,
588-590,
T. P., La Russa, M., Liu, Y., and Qi, L. S. (2019)
doi: 10.1038/sj.leu.2404542.
CRISPR-mediated live imaging of genome editing and
71.
Lavau, C., Du, C., Thirman, M., and Zeleznik-Le, N.
transcription, Science, 365, 1301-1305, doi: 10.1126/
(2000) Chromatin-related properties of CBP fused to
science.aax7852.
MLL generate a myelodysplastic-like syndrome that
81.
Ramsden, D. A., and Nussenzweig, A. (2021) Mecha-
evolves into myeloid leukemia, EMBO J., 19, 4655-
nisms driving chromosomal translocations: lost in time
4664, doi: 10.1093/emboj/19.17.4655.
and space, Oncogene, 40, 4263-4270, doi: 10.1038/
72.
Yoo, B. J., Nam, M. H., Sung, H. J., Lim, C. S.,
s41388-021-01856-9.
Lee, C. K., Cho, Y. J., Lee, K. N., and Yoon, S. Y.
82.
Roukos, V., Voss, T. C., Schmidt, C. K., Lee, S.,
(2011) A case of therapy-related acute lymphoblastic
Wangsa, D., and Misteli, T. (2013) Spatial dynamics
leukemia with t(11;19) (q23;p13.3) and MLL/MLLT1
of chromosome translocations in living cells, Science,
gene rearrangement, Korean J. Lab. Med., 31, 13-17,
341, 660-664, doi: 10.1126/science.1237150.
doi: 10.3343/kjlm.2011.31.1.13.
83.
Kantidze, O. L., and Razin, S. V. (2009) Chroma-
73.
Xie, W., Tang, G., Wang, E., Kim, Y., Cloe, A.,
tin loops, illegitimate recombination, and genome
Shen, Q., Zhou, Y., Garcia-Manero, G., Loghavi, S.,
evolution, BioEssays,
31,
278-286, doi:
10.1002/
Hu, A. Y., Wang, S., Bueso-Ramos, C. E., Kantarjian,
bies.200800165.
H. M., Medeiros, L. J., and Hu, S. (2020) t(11;16)
84.
Canela, A., Maman, Y., Huang, S. N., Wutz, G.,
(q23;p13)/KMT2A-CREBBP in hematologic malig-
Tang, W., Zagnoli-Vieira, G., Callen, E., Wong, N.,
nancies: presumptive evidence of myelodysplasia or
Day, A., Peters, J. M., Caldecott, K. W., Pommier, Y.,
therapy-related neoplasm? Ann. Hematol., 99, 487-
and Nussenzweig, A. (2019) Topoisomerase II-in-
500, doi: 10.1007/s00277-020-03909-7.
duced chromosome breakage and translocation is de-
74.
Soutoglou, E., Dorn, J. F., Sengupta, K., Jasin, M.,
termined by chromosome architecture and transcrip-
Nussenzweig, A., Ried, T., Danuser, G., and
tional activity, Mol. Cell, 75, 252-266.e8, doi: 10.1016/
Misteli, T. (2007) Positional stability of single double-
j.molcel.2019.04.030.
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1117
85.
Graham, T. G. W., Walter, J. C., and Loparo, J. J.
96. Tahirov, T. H., and Bushweller, J. (2017) Structure
(2016) Two-stage synapsis of DNA ends during non-
and biophysics of CBFβ/RUNX and its translo-
homologous end joining, Mol. Cell, 61, 850-858,
cation products, Adv. Exp. Med. Biol., 962, 21-31,
doi: 10.1016/j.molcel.2016.02.010.
doi: 10.1007/978-981-10-3233-2_2.
86.
Chaplin, A. K., Hardwick, S. W., Stavridi, A. K., Buehl,
97. Lam, K., and Zhang, D.-E. (2012) RUNX1 and
C. J., Goff, N. J., Ropars, V., Liang, S., De Oliveira,
RUNX1-ETO: roles in hematopoiesis and leukemo-
T. M., Chirgadze, D. Y., Meek, K., Charbonnier, J. B.,
genesis, Front. Biosci. Landmark Ed., 17, 1120-1139,
and Blundell, T. L. (2021) Cryo-EM of NHEJ super-
doi: 10.2741/3977.
complexes provides insights into DNA repair, Mol. Cell,
98. Ichikawa, M., Yoshimi, A., Nakagawa, M., Nishi-
81, 3400-3409.e3, doi: 10.1016/j.molcel.2021.07.005.
moto, N., Watanabe-Okochi, N., and Kurokawa, M.
87.
DeFazio, L. G., Stansel, R. M., Griffith, J. D., and
(2013) A role for RUNX1 in hematopoiesis and
Chu, G. (2002) Synapsis of DNA ends by DNA-de-
myeloid leukemia, Int. J. Hematol., 97, 726-734,
pendent protein kinase, EMBO J., 21, 3192-3200,
doi: 10.1007/s12185-013-1347-3.
doi: 10.1093/emboj/cdf299.
99. Chen, M. J., Yokomizo, T., Zeigler, B. M., Dzierzak, E.,
88.
Shmakova, A., Lomov, N., Viushkov, V., Tsfasman, T.,
and Speck, N. A. (2009) Runx1 is required for the
Kozhevnikova, Y., Sokolova, D., Pokrovsky, V., Syr-
endothelial to haematopoietic cell transition but
kina, M., Germini, D., Rubtsov, M., and Vassetzky, Y.
not thereafter, Nature, 457, 887-891, doi: 10.1038/
(2023) Cell models with inducible oncogenic translo-
nature07619.
cations allow to evaluate the potential of drugs to favor
100. Ottersbach, K. (2019) Endothelial-to-haematopoietic
secondary translocations, Cancer Commun., 43, 154-
transition: An update on the process of making blood,
158, doi: 10.1002/cac2.12370.
Biochem. Soc. Trans., 47, 591-601, doi: 10.1042/
89.
Krawczyk, P. M., Borovski, T., Stap, J., Cijsouw, T.,
BST20180320.
ten Cate, R., Medema, J. P., Kanaar, R., Franken,
101. Wang, Q., Stacy, T., Binder, M., Marin-Padilla, M.,
N. A. P., and Aten, J. A. (2012) Chromatin mobility is
Sharpe, A. H., and Speck, N. A. (1996) Disruption
increased at sites of DNA double-strand breaks, J. Cell
of the Cbfa2 gene causes necrosis and hemorrhaging
Sci., 125, 2127-2133, doi: 10.1242/jcs.089847.
in the central nervous system and blocks definitive
90.
Glukhov, S. I., Rubtsov, M. A., Alexeyevsky, D. A.,
hematopoiesis, Proc. Natl. Acad. Sci. USA, 93, 3444-
Alexeevski, A. V., Razin, S. V., and Iarovaia, O. V.
3449, doi: 10.1073/pnas.93.8.3444.
(2013) The broken MLL gene is frequently located
102. Growney, J. D., Shigematsu, H., Li, Z., Lee, B. H.,
outside the inherent chromosome territory in human
Adelsperger, J., Rowan, R., Curley, D. P., Kutok,
lymphoid cells treated with DNA topoisomerase II
J. L., Akashi, K., Williams, I. R., Speck, N. A., and
poison etoposide, PLoS One, 8, e75871, doi: 10.1371/
Gilliland, D. G. (2005) Loss of Runx1 perturbs adult
journal.pone.0075871.
hematopoiesis and is associated with a myeloprolifer-
91.
Nowell, P. C. (1976) The clonal evolution of tumor
ative phenotype, Blood, 106, 494-504, doi: 10.1182/
cell populations, Science, 194, 23-28, doi: 10.1126/
blood-2004-08-3280.
science.959840.
103. Ichikawa, M., Goyama, S., Asai, T., Kawazu, M.,
92.
Miyoshi, H., Shimizu, K., Kozu, T., Maseki, N.,
Nakagawa, M., Takeshita, M., Chiba, S., Ogawa, S.,
Kaneko, Y., and Ohki, M. (1991) (8;21) breakpoints
and Kurokawa, M. (2008) AML1/Runx1 negative-
on chromosome 21 in acute myeloid leukemia are
ly regulates quiescent hematopoietic stem cells in
clustered within a limited region of a single gene,
adult hematopoIesis, J. Immunol., 180, 4402-4408,
AML1, Proc. Natl. Acad. Sci. USA, 8, 10431-10434,
doi: 10.4049/jimmunol.180.7.4402.
doi: 10.1073/pnas.88.23.10431.
104. Tanaka, Y., Joshi, A., Wilson, N. K., Kinston, S.,
93.
Roulston, D., Espinosa, R., Nucifora, G., Larson,
Nishikawa, S., and Göttgens, B. (2012) The transcrip-
R. A., Le Beau, M. M., and Rowley, J. D. (1998)
tional programme controlled by Runx1 during early
CBFA2(AML1) Translocations with novel partner
embryonic blood development, Dev. Biol., 366, 404-
chromosomes in myeloid leukemias: association with
419, doi: 10.1016/j.ydbio.2012.03.024.
prior therapy, Blood, 92, 2879-2885, doi: 10.1182/
105. Yamagata, T., Maki, K., and Mitani, K. (2005) Runx1/
blood.V92.8.2879.
AML1 in normal and abnormal hematopoiesis, Int. J.
94.
Pedersen-Bjergaard, J., and Rowley, J. D.
(1994)
Hematol., 82, 1-8, doi: 10.1532/IJH97.05075.
The balanced and the unbalanced chromosome aber-
106. Elagib, K. E., Racke, F. K., Mogass, M., Khetawat, R.,
rations of acute myeloid leukemia may develop in dif-
Delehanty, L. L., and Goldfarb, A. N. (2003) RUNX1
ferent ways and may contribute differently to malignant
and GATA-1 coexpression and cooperation in mega-
transformation, Blood, 83, 2780-2786, doi: 10.1182/
karyocytic differentiation, Blood,
101,
4333-4341,
blood.V83.10.2780.2780.
doi: 10.1182/blood-2002-09-2708.
95.
De Braekeleer, E., Ferec, C., and De Braekeleer, M.
107. Kim, W. Y., Sieweke, M., Ogawa, E., Wee, H. J.,
(2009) RUNX1 translocations in malignant hemopa-
Englmeier, U., Graf, T., and Yoshiaki, I. (1999) Mutual
thies, Anticancer Res., 29, 1031-1038.
activation of Ets-1 and AML1 DNA binding by direct
БИОХИМИЯ том 88 вып. 7 2023
1118
ЛОМОВ и др.
interaction of their autoinhibitory domains, EMBO J.,
118. Huret, J. L., Ahmad, M., Arsaban, M., Bernheim, A.,
18, 1609-1620, doi: 10.1093/emboj/18.6.1609.
Cigna, J., Desangles, F., Guignard, J. C., Jacquemot-
108. Zhang, D. E., Hetherington, C. J., Meyers, S.,
Perbal, M. C., Labarussias, M., Leberre, V., Malo, A.,
Rhoades, K. L., Larson, C. J., Chen, H. M., Hiebert,
Morel-Pair, C., Mossafa, H., Potier, J. C., Texier, G.,
S. W., and Tenen, D. G. (1996) CCAAT enhancer-
Vigui, F., Wan-Senon, S. Y. C., Zasadzinski, A., and
binding protein (C/EBP) and AML1 (CBF alpha2)
Dessen, P. (2013) Atlas of genetics and cytogenetics in
synergistically activate the macrophage colony-
oncology and haematology in 2013, Nucleic Acids Res.,
stimulating factor receptor promoter, Mol. Cell. Biol.,
41, D920-D924, doi: 10.1093/nar/gks1082.
16, 1231-1240, doi: 10.1128/MCB.16.3.1231.
119. Peterson, L. F., and Zhang, D. E. (2004) The 8;21
109. Kitabayashi, I., Yokoyama, A., Shimizu, K., and
translocation in leukemogenesis, Oncogene, 23, 4255-
Ohki, M. (1998) Interaction and functional cooper-
4262, doi: 10.1038/sj.onc.1207727.
ation of the leukemia- associated factors AML1 and
120. Davis, J. N., McGhee, L., and Meyers, S. (2003)
p300 in myeloid cell differentiation, EMBO J., 17,
The ETO (MTG8) gene family, Gene, 303, 1-10,
2994-3004, doi: 10.1093/emboj/17.11.2994.
doi: 10.1016/S0378-1119(02)01172-1.
110. Yagi, R., Chen, L. F., Shigesada, K., Murakami, Y.,
121. Hiebert, S. W., Reed-Inderbitzin, E. F., Amann, J.,
and Ito, Y. (1999) A WW domain-containing Yes-
Irvin, B., Durst, K., and Linggi, B. (2003) The t(8;21)
associated protein (YAP) is a novel transcriptional
fusion protein contacts co-repressors and histone
co-activator, EMBO J., 18, 2551-2562, doi: 10.1093/
deacetylases to repress the transcription of the p14ARF
emboj/18.9.2551.
tumor suppressor, Blood Cells Mol. Dis., 30, 177-183,
111. Segrelles, C., Paramio, J. M., and Lorz, C. (2018)
doi: 10.1016/S1079-9796(03)00021-4.
The transcriptional co-activator YAP: A new player
122. Hayashi, Y., Harada, Y., and Harada, H.
(2022)
in head and neck cancer, Oral Oncol., 86, 25-32,
Myeloid neoplasms and clonal hematopoiesis from
doi: 10.1016/j.oraloncology.2018.08.020.
the RUNX1 perspective, Leukemia, 36, 1203-1214,
112. Riddell, A., McBride, M., Braun, T., Nicklin, S. A.,
doi: 10.1038/s41375-022-01548-7.
Cameron, E., Loughrey, C. M., and Martin, T. P.
123. Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C.,
(2020) RUNX1: an emerging therapeutic target for
Oksvold, P., Mardinoglu, A., Sivertsson, Å., Kampf, C.,
cardiovascular disease, Cardiovasc. Res., 116, 1410-
Sjöstedt, E., Asplund, A., Olsson, I. M., Edlund, K.,
1423, doi: 10.1093/cvr/cvaa034.
Lundberg, E., Navani, S., Szigyarto, C. A. K.,
113. Kellaway, S. G., Coleman, D. J. L., Cockerill,
Odeberg, J., Djureinovic, D., Takanen, J. O., Hober, S.,
P. N., Raghavan, M., and Bonifer, C. (2022) Molecu-
Alm, T., Edqvist, P. H., Berling, H., Tegel, H.,
lar basis of hematological disease caused by inherited
Mulder, J., Rockberg, J., Nilsson, P., Schwenk, J. M.,
or acquired RUNX1 mutations, Exp. Hematol., 111,
Hamsten, M., Von Feilitzen, K., Forsberg, M.,
1-12, doi: 10.1016/j.exphem.2022.03.009.
Persson, L., Johansson, F., Zwahlen, M., Von
114. Lutterbach, B., Westendorf, J. J., Linggi, B., Isaac, S.,
Heijne, G., Nielsen, J., and Pontén, F. (2015) Tissue-
Seto, E., and Hiebert, S. W. (2000) A mechanism of
based map of the human proteome, Science, 347,
repression by acute myeloid leukemia-1, the target of
1260419, doi: 10.1126/science.1260419.
multiple chromosomal translocations in acute leukemia,
124. Perry, C., Eldor, A., and Soreq, H. (2002) Runx1/
J. Biol. Chem., 275, 651-656, doi: 10.1074/jbc.275.1.651.
AML1 in leukemia: disrupted association with diverse
115. Imai, Y., Kurokawa, M., Yamaguchi, Y., Izutsu, K.,
protein partners, Leuk. Res., 26, 221-228, doi: 10.1016/
Nitta, E., Mitani, K., Satake, M., Noda, T., Ito, Y.,
S0145-2126(01)00128-X.
and Hirai, H. (2004) The corepressor mSin3A regu-
125. Gelmetti, V., Zhang, J., Fanelli, M., Minucci, S.,
lates phosphorylation-induced activation, intranuclear
Pelicci, P. G., and Lazar, M. A. (1998) Aberrant
location, and stability of AML1, Mol. Cell. Biol., 24,
recruitment of the nuclear receptor corepressor-
1033-1043, doi: 10.1128/MCB.24.3.1033-1043.2004.
histone deacetylase complex by the acute myeloid
116. Seo, W., Tanaka, H., Miyamoto, C., Levanon, D.,
leukemia fusion partner ETO, Mol. Cell. Biol., 18,
Groner, Y., and Taniuchi, I. (2012) Roles of VWRPY
7185-7191, doi: 10.1128/MCB.18.12.7185.
motif-mediated gene repression by Runx proteins
126. Lutterbach, B., Westendorf, J. J., Linggi, B., Patten, A.,
during T-cell development, Immunol. Cell Biol., 90,
Moniwa, M., Davie, J. R., Huynh, K. D., Bardwell,
827-830, doi: 10.1038/icb.2012.6.
V. J., Lavinsky, R. M., Rosenfeld, M. G., Glass, C.,
117. Alkadi, H., McKellar, D., Zhen, T., Karpova, T.,
Seto, E., and Hiebert, S. W. (1998) ETO, a target of
Garrett, L. J., Gao, Y., Alsadhan, A. A., Kwon, E. M.,
t(8;21) in acute leukemia, interacts with the N-CoR
Greene, J. J., Ball, D. A., Cheng, L., Sorrell, A. D.,
and mSin3 corepressors, Mol. Cell. Biol., 18, 7176-
and Liu, P. P. (2018) The VWRPY domain is essential
7184, doi: 10.1128/MCB.18.12.7176.
for RUNX1 function in hematopoietic progenitor
127. Wang, L., Gural, A., Sun, X. J., Zhao, X., Perna, F.,
cell maturation and megakaryocyte differentiation,
Huang, G., Hatlen, M. A., Vu, L., Liu, F., Xu, H.,
Blood,
132 (Supplement
1),
1319, doi:
10.1182/
Asai, T., Xu, H., Deblasio, T., Menendez, S.,
blood-2018-99-113400.
Voza, F., Jiang, Y., Cole, P. A., Zhang, J., Melnick, A.,
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1119
Roeder, R. G., and Nimer, S. D. (2011) The leukemo-
137. Slany, R. K. (2009) The molecular biology of mixed
genicity of AML1-ETO is dependent on site-specific
lineage leukemia, Haematologica,
94,
984-993,
lysine acetylation, Science, 333, 765-769, doi: 10.1126/
doi: 10.3324/haematol.2008.002436.
science.1201662.
138. Mirro, J., Zipf, T. F., Pui, C. H., Kitchingman, G.,
128. Otálora-Otálora, B. A., Henríquez, B., López-Kleine, L.,
Williams, D., Melvin, S., Murphy, S. B., and Stass, S.
and Rojas, A. (2019) RUNX family: oncogenes or
(1985) Acute mixed lineage leukemia: Clinicopatho-
tumor suppressors (review), Oncol. Rep., 42, 3-19,
logic correlations and prognostic significance, Blood,
doi: 10.3892/or.2019.7149.
66, 1115-1123, doi: 10.1182/blood.V66.5.1115.1115.
129. Rejeski, K., Duque-Afonso, J., and Lübbert, M.
139. Mirro, J., Kitchingman, G. R., Williams, D. L.,
(2021) AML1/ETO and its function as a regula-
Murphy, S. B., Zipf, T. F., and Stass, S. A. (1986)
tor of gene transcription via epigenetic mechanisms,
Mixed lineage leukemia: the implications for
Oncogene, 40, 5665-5676, doi: 10.1038/s41388-021-
hematopoietic differentiation, Blood, 68, 597-599,
01952-w.
doi: 10.1182/blood.V68.2.597.597.
130. Ben-Ami, O., Friedman, D., Leshkowitz, D., Golden-
140. Ziemin-Van Der Poel, S., Mccabe, N. R., Gill, H. J.,
berg, D., Orlovsky, K., Pencovich, N., Lotem, J.,
Espinosa, R., Patel, Y., Harden, A., Rubinelli, P.,
Tanay, A., and Groner, Y.
(2013) Addiction of
Smith, S. D., Lebeau, M. M., Rowley, I. D., and
t(8;21) and inv(16) acute myeloid leukemia to na-
Diaz, M. O. (1991) Identification of a gene, MLL, that
tive RUNX1, Cell Rep., 4, 1131-1143, doi: 10.1016/
spans the breakpoint in 11q23 translocations associated
j.celrep.2013.08.020.
with human leukemias, Proc. Natl. Acad. Sci. USA, 88,
131. Van der Kouwe, E., and Staber, P. B. (2019) RUNX1-
10735-10739, doi: 10.1073/pnas.88.23.10735.
ETO: Attacking the epigenome for genomic instable
141. Djabali, M., Selleri, L., Parry, P., Bower, M., Young,
Leukemia, Int. J. Mol. Sci., 20, 350, doi: 10.3390/
B. D., and Evans, G. A. (1992) A trithorax-like gene
ijms20020350.
is interrupted by chromosome 11q23 translocations in
132. Ptasinska, A., Assi, S. A., Mannari, D., James, S. R.,
acute leukaemias, Nat. Genet., 2, 113-118, doi: 10.1038/
Williamson, D., Dunne, J., Hoogenkamp, M., Wu, M.,
ng1092-113.
Care, M., McNeill, H., Cauchy, P., Cullen, M., Tooze,
142. Marschalek, R. (2011) Mechanisms of leukemogenesis
R. M., Tenen, D. G., Young, B. D., Cockerill, P. N.,
by MLL fusion proteins, Br. J. Haematol., 152, 141-
Westhead, D. R., Heidenreich, O., and Bonifer, C.
154, doi: 10.1111/j.1365-2141.2010.08459.x.
(2012) Depletion of RUNX1/ETO in t(8;21) AML
143. Super, H. J. G., McCabe, N. R., Thirman, M. J.,
cells leads to genome-wide changes in chromatin
Larson, R. A., Le Beau, M. M., Pedersen-Bjergaard, J.,
structure and transcription factor binding, Leukemia,
Philip, P., Diaz, M. O., and Rowley, J. D. (1993)
26, 1829-1841, doi: 10.1038/leu.2012.49.
Rearrangements of the MLL gene in therapy-re-
133. Yuan, Y., Zhou, L., Miyamoto, T., Iwasaki, H.,
lated acute myeloid leukemia in patients previous-
Harakawa, N., Hetherington, C. J., Burel, S. A.,
ly treated with agents targeting DNA-topoisomer-
Lagasse, E., Weissman, I. L., Akashi, K., and Zhang,
ase II, Blood, 82, 3705-3711, doi: 10.1182/blood.V82.
D.-E. (2001) AML1-ETO expression is directly in-
12.3705.3705.
volved in the development of acute myeloid leuke-
144. Felix, C. A. (1998) Secondary leukemias induced by
mia in the presence of additional mutations, Proc.
topoisomerase-targeted drugs, Biochim. Biophys. Acta,
Natl. Acad. Sci. USA, 98, 10398-10403, doi: 10.1073/
1400, 233-255, doi: 10.1016/S0167-4781(98)00139-0.
pnas.171321298.
145. Rao, R. C., and Dou, Y. (2015) Hijacked in cancer:
134. Higuchi, M., O’Brien, D., Kumaravelu, P., Lenny, N.,
The KMT2 (MLL) family of methyltransferases, Nat.
Yeoh, E. J., and Downing, J. R. (2002) Expression of a
Rev. Cancer, 15, 334-346, doi: 10.1038/nrc3929.
conditional AML1-ETO oncogene bypasses embryon-
146. Borkhardt, A., Wuchter, C., Viehmann, S., Pils, S.,
ic lethality and establishes a murine model of human
Teigler-Schlegel, A., Stanulla, M., Zimmermann, M.,
t(8;21) acute myeloid leukemia, Cancer Cell, 1, 63-74,
Ludwig, W. D., Janka-Schaub, G., Schrappe, M.,
doi: 10.1016/S1535-6108(02)00016-8.
and Harbott, J. (2002) Infant acute lymphoblastic leu-
135. Kozu, T., Fukuyama, T., Yamami, T., Akagi, K.,
kemia - combined cytogenetic, immonophenotypi-
and Kaneko, Y. (2005) MYND-less splice variants of
cal and molecular analysis of 77 cases, Leukemia, 16,
AML1-MTG8 (RUNX1=CBFA2T1) are expressed in
1685-1690, doi: 10.1038/sj.leu.2402595.
leukemia with t(8;21), Genes Chromosomes Cancer, 43,
147. Pieters, R., Schrappe, M., De Lorenzo, P., Hann, I.,
45-53, doi: 10.1002/gcc.20165.
De Rossi, G., Felice, M., Hovi, L., LeBlanc, T.,
136. Yan, M., Kanbe, E., Peterson, L. F., Boyapati, A.,
Szczepanski, T., Ferster, A., Janka, G., Rubnitz, J.,
Miao, Y., Wang, Y., Chen, I. M., Chen, Z., Rowley,
Silverman, L., Stary, J., Campbell, M., Li, C. K.,
J. D., Willman, C. L., and Zhang, D. E. (2006) A pre-
Mann, G., Suppiah, R., Biondi, A., Vora, A., and
viously unidentified alternatively spliced isoform of
Valsecchi, M. G. (2007) A treatment protocol for
t(8;21) transcript promotes leukemogenesis, Nat.
infants younger than 1 year with acute lymphoblastic
Med., 12, 945-949, doi: 10.1038/nm1443.
leukaemia (Interfant-99): an observational study and
БИОХИМИЯ том 88 вып. 7 2023
1120
ЛОМОВ и др.
a multicentre randomised trial, Lancet, 370, 240-250,
159. García-Alai, M. M., Allen, M. D., Joerger, A. C., and
doi: 10.1016/S0140-6736(07)61126-X.
Bycroft, M. (2010) The structure of the FYR domain
148. Creutzig, U., Zimmermann, M., Dworzak, M.,
of transforming growth factor beta regulator 1, Protein
Bourquin, J.-P., von Neuhoff, C., Sander, A., Stary, J.,
Sci., 19, 1432-1438, doi: 10.1002/pro.404.
and Reinhardt, D.
(2012) Additional prognostic
160. Birke, M., Schreiner, S., García-Cuéllar, M.-P.,
impact of specific sites of extramedullary leukemia in
Mahr, K., Titgemeyer, F., and Slany, R. K. (2002)
pediatric AML with MLL-rearrangements but not in
The MT domain of the proto-oncoprotein MLL
core-binding factor (CBF) AML, Blood, 120, 2621,
binds to CpG-containing DNA and discriminates
doi: 10.1182/blood.V120.21.2621.2621.
against methylation, Nucleic Acids Res., 30, 958-965,
149. Marschalek, R. (2016) Systematic classification of
doi: 10.1093/nar/30.4.958.
mixed-lineage leukemia fusion partners predicts ad-
161. Allen, M. D., Grummitt, C. G., Hilcenko, C., Min,
ditional cancer pathways, Ann. Lab. Med., 36, 85-100,
S. Y., Tonkin, L. M., Johnson, C. M., Freund,
doi: 10.3343/alm.2016.36.2.85.
S. M., Bycroft, M., and Warren, A. J.
(2006)
150. Liu, H., Cheng, E. H. Y., and Hsieh, J. J. D. (2009)
Solution structure of the nonmethyl-CpG-binding
MLL fusions: pathways to leukemia, Cancer Biol.
CXXC domain of the leukaemia-associated MLL
Ther., 8, 1204-1211, doi: 10.4161/cbt.8.13.8924.
histone methyltransferase, EMBO J., 25, 4503-4512,
151. Guenther, M. G., Jenner, R. G., Chevalier, B.,
doi: 10.1038/sj.emboj.7601340.
Nakamura, T., Croce, C. M., Canaani, E., and Young,
162. Risner, L. E., Kuntimaddi, A., Lokken, A. A., Achille,
R. A. (2005) Global and Hox-specific roles for the
N. J., Birch, N. W., Schoenfelt, K., Bushweller, J. H.,
MLL1 methyltransferase, Proc. Natl. Acad. Sci. USA,
and Zeleznik-Le, N. J. (2013) Functional specificity
102, 8603-8608, doi: 10.1073/pnas.0503072102.
of CpG DNA-binding CXXC domains in mixed
152. Ayton, P. M., and Cleary, M. L. (2003) Transformation
lineage leukemia, J. Biol. Chem., 288, 29901-29910,
of myeloid progenitors by MLL oncoproteins is
doi: 10.1074/jbc.M113.474858.
dependent on Hoxa7 and Hoxa9, Genes Dev., 17,
163. Muntean, A. G., Tan, J., Sitwala, K., Huang, Y.,
2298-2307, doi: 10.1101/gad.1111603.
Bronstein, J., Connelly, J. A., Basrur, V., Elenitoba-
153. Wang, P., Lin, C., Smith, E. R., Guo, H., Sanderson,
Johnson, K. S. J., and Hess, J. L. (2010) The PAF
B. W., Wu, M., Gogol, M., Alexander, T., Seidel, C.,
complex synergizes with MLL fusion proteins at HOX
Wiedemann, L. M., Ge, K., Krumlauf, R., and
loci to promote leukemogenesis, Cancer Cell, 17,
Shilatifard, A. (2009) Global analysis of H3K4 meth-
609-621, doi: 10.1016/j.ccr.2010.04.012.
ylation defines MLL family member targets and
164. Wang, Z., Song, J., Milne, T. A., Wang, G. G.,
points to a role for MLL1-mediated H3K4 methyl-
Li, H., Allis, C. D., and Patel, D. J. (2010) Pro isom-
ation in the regulation of transcriptional initiation by
erization in MLL1 PHD3-bromo cassette connects
RNA polymerase II, Mol. Cell. Biol., 29, 6074-6085,
H3K4me readout to CyP33 and HDAC-mediated
doi: 10.1128/MCB.00924-09.
repression, Cell, 141, 1183-1194, doi: 10.1016/j.cell.
154. Artinger, E. L., Mishra, B. P., Zaffuto, K. M., Li,
2010.05.016.
B. E., Chung, E. K. Y., Moore, A. W., Chen, Y.,
165. Chang, P. Y., Hom, R. A., Musselman, C. A., Zhu, L.,
Cheng, C., and Ernst, P. (2013) An MLL-dependent
Kuo, A., Gozani, O., Kutateladze, T. G., and Cleary,
network sustains hematopoiesis, Proc. Natl. Acad. Sci.
M. L. (2010) Binding of the MLL PHD3 finger to
USA, 110, 12000-12005, doi: 10.1073/pnas.1301278110.
histone H3K4me3 is required for MLL-dependent
155. Yu, B. D., Hess, J. L., Horning, S. E., Brown, G. A. J.,
gene transcription, J. Mol. Biol.,
400,
137-144,
and Korsmeyer, S. J. (1995) Altered Hox expression
doi: 10.1016/j.jmb.2010.05.005.
and segmental identity in Mll-mutant mice, Nature,
166. Roloff, T. C., Ropers, H. H., and Nuber, U. A.
378, 505-508, doi: 10.1038/378505a0.
(2003) Comparative study of methyl-CpG-binding
156. Yagi, H., Deguchi, K., Aono, A., Tani, Y., Kishimoto, T.,
domain proteins, BMC Genomics, 4, 1, doi: 10.1186/
and Komori, T. (1998) Growth disturbance in fetal
1471-2164-4-1.
liver hematopoiesis of Mll-mutant mice, Blood, 92,
167. Yokoyama, A., and Cleary, M. L. (2008) Menin
108-117, doi: 10.1182/blood.V92.1.108.413k11_108_117.
critically links MLL protein with LEDGF on cancer-
157. Ernst, P., Fisher, J. K., Avery, W., Wade, S., Foy, D.,
associated target genes, Cancer Cell,
14,
36-46,
and Korsmeyer, S. J. (2004) Definitive hematopoiesis
doi: 10.1016/j.ccr.2008.05.003.
requires the mixed-lineage leukemia gene, Dev. Cell, 6,
168. Hughes, C. M., Rozenblatt-Rosen, O., Milne, T. A.,
437-443, doi: 10.1016/S1534-5807(04)00061-9.
Copeland, T. D., Levine, S. S., Lee, J. C., Hayes, D. N.,
158. McMahon, K. A., Hiew, S. Y. L., Hadjur, S., Veiga-
Shanmugam, K. S., Bhattacharjee, A., Biondi, C. A.,
Fernandes, H., Menzel, U., Price, A. J., Kioussis, D.,
Kay, G. F., Hayward, N. K., Hess, J. L., and Meyer-
Williams, O., and Brady, H. J. M. (2007) Mll has a
son, M. (2004) Menin associates with a trithorax fam-
critical role in fetal and adult hematopoietic stem cell
ily histone methyltransferase complex and with the
self-renewal, Cell Stem Cell, 1, 338-345, doi: 10.1016/
Hoxc8 locus, Mol. Cell, 13, 587-597, doi: 10.1016/
j.stem.2007.07.002.
S1097-2765(04)00081-4.
БИОХИМИЯ том 88 вып. 7 2023
МЕХАНИЗМЫ РАЗВИТИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ
1121
169. Slany, R. K. (2016) The molecular mechanics of
181. Han, Q. L., Zhang, X. L., Ren, P. X., Mei, L. H.,
mixed lineage leukemia, Oncogene, 35, 5215-5223,
Lin, W. H., Wang, L., Cao, Y., Li, K., and Bai, F.
doi: 10.1038/onc.2016.30.
(2022) Discovery, evaluation and mechanism study
170. Del Rizzo, P. A., and Trievel, R. C. (2011) Substrate and
of WDR5-targeted small molecular inhibitors for
product specificities of SET domain methyltransferases,
neuroblastoma, Acta Pharmacol. Sin., 44, 877-887,
Epigenetics, 6, 1059-1067, doi: 10.4161/epi.6.9.16069.
doi: 10.1038/s41401-022-00999-z.
171. Dou, Y., Milne, T. A., Ruthenburg, A. J., Lee, S., Lee,
182. Wright, R. L., and Vaughan, A. T. M. (2014) A sys-
J. W., Verdine, G. L., Allis, C. D., and Roeder, R. G.
tematic description of MLL fusion gene formation,
(2006) Regulation of MLL1 H3K4 methyltransferase
Crit. Rev. Oncol. Hematol., 91, 283-291, doi: 10.1016/
activity by its core components, Nat. Struct. Mol. Biol.,
j.critrevonc.2014.03.004.
13, 713-719, doi: 10.1038/nsmb1128.
183. Ross, M. E., Zhou, X., Song, G., Shurtleff, S. A.,
172. Patel, A., Dharmarajan, V., Vought, V. E., and
Girtman, K., Williams, W. K., Liu, H. C., Mahfouz, R.,
Cosgrove, M. S. (2009) On the mechanism of multiple
Raimondi, S. C., Lenny, N., Patel, A., and Downing,
lysine methylation by the human mixed lineage leuke-
J. R. (2003) Classification of pediatric acute lympho-
mia protein-1 (MLL1) core complex, J. Biol. Chem.,
blastic leukemia by gene expression profiling, Blood,
284, 24242-24256, doi: 10.1074/jbc.M109.014498.
102, 2951-2959, doi: 10.1182/blood-2003-01-0338.
173. Li, Y., Han, J., Zhang, Y., Cao, F., Liu, Z., Li, S.,
184. Ferrando, A. A., Armstrong, S. A., Neuberg, D. S.,
Wu, J., Hu, C., Wang, Y., Shuai, J., Chen, J., Cao, L.,
Sallan, S. E., Silverman, L. B., Korsmeyer, S. J.,
Li, D., Shi, P., Tian, C., Zhang, J., Dou, Y., Li, G.,
and Look, A. T. (2003) Gene expression signatures
Chen, Y., and Lei, M. (2016) Structural basis for
in MLL-rearranged T-lineage and B-precursor acute
activity regulation of MLL family methyltransferases,
leukemias: dominance of HOX dysregulation, Blood,
Nature, 530, 447-452, doi: 10.1038/nature16952.
102, 262-268, doi: 10.1182/blood-2002-10-3221.
174. Lloyd, N. R., and Wuttke, D. S. (2021) Cyp33 binds
185. Lin, C., Smith, E. R., Takahashi, H., Lai, K. C.,
AU-rich RNA motifs via an extended interface that
Martin-Brown, S., Florens, L., Washburn, M. P.,
competitively disrupts the gene repressive Cyp33-
Conaway, J. W., Conaway, R. C., and Shilatifard, A.
MLL1 interaction in vitro, PLoS One, 16, 1-18,
(2010) AFF4, a component of the ELL/P-TEFb elon-
doi: 10.1371/journal.pone.0237956.
gation complex and a shared subunit of MLL chime-
175. Fair, K., Anderson, M., Bulanova, E., Mi, H.,
ras, can link transcription elongation to leukemia, Mol.
Tropschug, M., and Diaz, M. O. (2001) Protein inter-
Cell, 37, 429-437, doi: 10.1016/j.molcel.2010.01.026.
actions of the MLL PHD fingers modulate MLL tar-
186. Yokoyama, A., Lin, M., Naresh, A., Kitabayashi, I.,
get gene regulation in human cells, Mol. Cell. Biol., 21,
and Cleary, M. L. (2010) A higher-order complex
3589-3597, doi: 10.1128/MCB.21.10.3589-3597.2001.
containing AF4 and ENL family proteins with P-TEFb
176. Xia, Z. B., Anderson, M., Diaz, M. O., and Zeleznik-
facilitates oncogenic and physiologic MLL-dependent
Le, N. J. (2003) MLL repression domain interacts with
transcription, Cancer Cell, 17, 198-212, doi: 10.1016/
histone deacetylases, the polycomb group proteins
j.ccr.2009.12.040.
HPC2 and BMI-1, and the corepressor C-terminal-
187. Bitoun, E., Oliver, P. L., and Davies, K. E. (2007)
binding protein, Proc. Natl. Acad. Sci. USA, 100, 8342-
The mixed-lineage leukemia fusion partner AF4 stimu-
8347, doi: 10.1073/pnas.1436338100.
lates RNA polymerase II transcriptional elongation and
177. Grow, E. J., and Wysocka, J. (2010) Flipping MLL1’s
mediates coordinated chromatin remodeling, Hum.
switch one proline at a time, Cell, 141, 1108-1110,
Mol. Genet., 16, 92-106, doi: 10.1093/hmg/ddl444.
doi: 10.1016/j.cell.2010.06.013.
188. Mueller, D., García-Cuéllar, M. P., Bach, C., Buhl, S.,
178. Sha, L., Ayoub, A., Cho, U. S., and Dou, Y. (2020)
Maethner, E., and Slany, R. K. (2009) Misguided
Insights on the regulation of the MLL/SET1 family
transcriptional elongation causes mixed lineage leu-
histone methyltransferases, Biochim. Biophys. Acta
kemia, PLoS Biol., 7, e1000249, doi: 10.1371/journal.
Gene Regul. Mech.,
1863,
194561, doi:
10.1016/
pbio.1000249.
j.bbagrm.2020.194561.
189. Nguyen, A. T., and Zhang, Y. (2011) The diverse
179. Chen, J., Santillan, D. A., Koonce, M., Wei, W.,
functions of Dot1 and H3K79 methylation, Genes
Luo, R., Thirman, M. J., Zeleznik-Le, N. J., and
Dev., 25, 1345-1358, doi: 10.1101/gad.2057811.
Diaz, M. O. (2008) Loss of MLL PHD finger 3 is
190. Godfrey, L., Crump, N. T., Thorne, R., Lau, I. J.,
necessary for MLL-ENL-induced hematopoietic
Repapi, E., Dimou, D., Smith, A. L., Harman, J. R.,
stem cell immortalization, Cancer Res., 68, 6199-6207,
Telenius, J. M., Oudelaar, A. M., Downes, D. J.,
doi: 10.1158/0008-5472.CAN-07-6514.
Vyas, P., Hughes, J. R., and Milne, T. A. (2019)
180. Chan, A. K. N., and Chen, C. W. (2019) Rewiring the
DOT1L inhibition reveals a distinct subset of en-
epigenetic networks in MLL-rearranged leukemias:
hancers dependent on H3K79 methylation, Nat. Com-
Epigenetic dysregulation and pharmacological inter-
mun., 10, 2803, doi: 10.1038/s41467-019-10844-3.
ventions, Front. Cell Dev. Biol., 7, 1-15, doi: 10.3389/
191. Prange, K. H. M., Mandoli, A., Kuznetsova, T.,
fcell.2019.00081.
Wang, S. Y., Sotoca, A. M., Marneth, A. E., Van Der
6
БИОХИМИЯ том 88 вып. 7 2023
1122
ЛОМОВ и др.
Reijden, B. A., Stunnenberg, H. G., and Martens,
197. Greaves, M., and Maley, C. C. (2012) Clonal evolution
J. H. A. (2017) MLL-AF9 and MLL-AF4 oncofusion
in cancer, Nature,
481,
306-313, doi:
10.1038/
proteins bind a distinct enhancer repertoire and target
nature10762.
the RUNX1 program in 11q23 acute myeloid leukemia,
198. Anoun, S., Lamchahab, M., Qachouh, M.,
Oncogene, 36, 3346-3356, doi: 10.1038/onc.2016.488.
Benchekroun, S., and Quessar, A. (2013) A case
192. Marschalek, R.
(2020) The reciprocal world of
of acute myeloid leukemia caused by low dose
MLL fusions: A personal view, Biochim. Biophys.
methotrexate used to treat a rheumatoid arthritis
Acta Gene Regul. Mech., 1863, 194547, doi: 10.1016/
patient, Iran. J. Blood Cancer, 5, 25-28.
j.bbagrm.2020.194547.
199. Maddalo, D., Manchado, E., Concepcion, C. P.,
193. Gaussmann, A., Wenger, T., Eberle, I., Bursen, A.,
Bonetti, C., Vidigal, J. A., Han, Y. C., Ogrodowski, P.,
Bracharz, S., Herr, I., Dingermann, T., and
Crippa, A., Rekhtman, N., Stanchina, E. De, Lowe,
Marschalek, R. (2007) Combined effects of the two
S. W., and Ventura, A. (2014) In vivo engineering
reciprocal t(4;11) fusion proteins MLL·AF4 and
of oncogenic chromosomal rearrangements with
AF4·MLL confer resistance to apoptosis, cell cycling
the CRISPR/Cas9 system, Nature, 516, 423-428,
capacity and growth transformation, Oncogene, 26,
doi: 10.1038/nature13902.
3352-3363, doi: 10.1038/sj.onc.1210125.
200. Torres, R., Martin, M. C., Garcia, A., Cigudosa,
194. Eberle, I., Pless, B., Braun, M., Dingermann, T., and
J. C., Ramirez, J. C., and Rodriguez-Perales, S. (2014)
Marschalek, R. (2010) Transcriptional properties of
Engineering human tumour-associated chromosomal
human NANOG1 and NANOG2 in acute leukemic
translocations with the RNA-guided CRISPR-
cells, Nucleic Acids Res., 38, 5384-5395, doi: 10.1093/
Cas9 system, Nat. Commun., 5, 1-8, doi: 10.1038/
nar/gkq307.
ncomms4964.
195. Wilhelm, A., and Marschalek, R. (2021) The role of
201. Sall, F. B., Shmakova, A., Karpukhina, A., Tsfasman,
reciprocal fusions in MLL-r acute leukemia: studying
T., Lomov, N., Canoy, R. J., Boutboul, D.,
the chromosomal translocation t(4;11), Oncogene, 40,
Oksenhendler, E., Toure, A. O., Lipinski, M., Wiels,
6093-6102, doi: 10.1038/s41388-021-02001-2.
J., Germini, D., and Vassetzky, Y. (2023) Epstein-
196. Ferrando, A. A., and López-Otín, C. (2017) Clonal
Barr virus reactivation induces MYC-IGH spatial
evolution in leukemia, Nat. Med., 23, 1135-1145,
proximity and t(8;14) in B cells, J. Med. Virol., 95,
doi: 10.1038/nm.4410.
e28633, doi: 10.1002/jmv.28633.
MECHANISMS FOR THE DEVELOPMENT OF THERAPY-RELATED
LEUKAEMIA CAUSED BY TOPOISOMERASE INHIBITORS
Review
N. A. Lomov1*, V. S. Viushkov1, and M. A. Rubtsov1,2
1 Department of Molecular Biology, Faculty of Biology, Lomonosov Moscow State University,
119234 Moscow, Russia; e-mail: lomov@mail.bio.msu.ru
2 Department of Biochemistry, Center for Industrial Technologies and Entrepreneurship
I. M. Sechenov First Moscow State Medical University (Sechenov University), 119435 Moscow, Russia
Leukemia is a blood cancer that originates in the blood and bone marrow. Therapy-related leukemia is a
leukemia associated with prior chemotherapy. Cancer therapy with DNA-topoisomerase II inhibitors is one
of the most effective among chemotherapies. However, its side effect can be the development of secondary
leukemia, characterized by chromosomal rearrangements involving the AML1 or MLL gene. The set of re-
current translocations in such leukemia differs from the set of chromosomal rearrangements in other neo-
plasia. We review the factors that drive the translocations upon treatment of cells with DNA-topoisomerase
inhibitors. Such factors primarily include the mobility of double-strand DNA ends prior to translocation
and the gain of functions of the fusion proteins that are formed in the cell as a result of translocation.
Keywords: leukemia, translocations; fusion proteins, DNA topoisomerase II inhibitors
БИОХИМИЯ том 88 вып. 7 2023