ЖЭТФ, 2019, том 156, вып. 2 (8), стр. 254-261
© 2019
АНАЛИЗ МЕТОДОВ ИССЛЕДОВАНИЯ
КОЛЕБАТЕЛЬНО-ВРАЩАТЕЛЬНОГО СПЕКТРА МОНОМЕРОВ
И ДИМЕРОВ ИЗОТОПОВ ГЕКСАФТОРИДА СЕРЫ
М. Е. Бычков*, Ю. В. Петрушевич, А. Н. Старостин
Государственный научный центр Российской Федерации
«Троицкий институт инновационных и термоядерных исследований»
142190, Троицк, Москва, Россия
Поступила в редакцию 16 февраля 2019 г.,
после переработки 16 февраля 2019 г.
Принята к публикации 18 февраля 2019 г.
Для оценки стабильности системы димеров гексафторида серы, SF6, под воздействием внешнего лазер-
ного излучения с помощью ab initio квантово-химических вычислений рассчитывается колебательно-вра-
щательный спектр мономеров, гомо- и гетеродимеров SF6. На примере простой молекулы CO замечено,
что наиболее распространенный в литературе метод расчетов, основанный на подходе матриц Гессе, не
может быть надежным инструментом при количественных расчетах некоторых молекулярных систем.
В качестве потенциального аналога был опробован алгоритм получения колебательно-вращательного
спектра на основе данных молекулярной динамики.
DOI: 10.1134/S0044451019080042
В работах [1, 2] построен межмолекулярный по-
тенциал взаимодействия, учитывающий дипольные
корреляционные эффекты, на основе которого про-
1. ВВЕДЕНИЕ
веден анализ структуры и колебательно-вращатель-
ного спектра простейших кластерных структур SF6,
Особый научный интерес представляет изуче-
в числе которых димер S(32)F6-S(32)F6. Согласно
ние свойств веществ, образующих многоатомные
этим работам, использование модифицированного
сложные кластеры, в наиболее общих приближени-
потенциала взаимодействия позволяет теоретически
ях. Благодаря доступности высокопроизводитель-
описать расщепление колебательной моды ν3 в клас-
ной вычислительной техники, среди теоретических
терных системах гексафторида серы, наблюдаемое в
подходов большое распространение получили чис-
экспериментах, например, [6,7]. При этом замечено,
ленные методы анализа систем большого числа час-
что один из пиков смещается в область низких час-
тиц. В данной работе основное внимание уделено
тот, в то время как остальные пики, в зависимости
гексафториду серы SF6. В литературе можно встре-
от размера рассматриваемого кластера, сдвигаются
тить большое количество работ [1-9], посвящен-
в область высоких частот. Также можно отметить
ных как теоретическому расчету, так и эксперимен-
работы по схожей тематике [3, 4], в которых изу-
тальному анализу различных характеристик данной
чены кластеры (SF6)N высоких порядков (N > 7).
молекулярной системы. Наибольший интерес пред-
В работах [8, 9] проведен экспериментальный спек-
ставляет анализ колебательно-вращательного спек-
тральный анализ многомолекулярных систем SF6 с
тра кластеров SF6, а именно, полосы частот, соот-
учетом присутствия в образце натуральной примеси
ветствующей колебательной моде ν3 ≈ 947 см-1 мо-
изотопов серы. Колебательно-вращательный спектр
номера SF6 [5], так как данная мода находится в
строится для всего образца в целом, с выделени-
инфракрасном диапазоне и может быть возбуждена
ем частот, соответствующих модам ν3 молекул с
с помощью лазерного излучения в экспериментах по
различным изотопом. Важно, что при проведении
диссоциации.
эксперимента невозможно зафиксировать структу-
ру кластеров, входящих в образец, а полученный
* E-mail: bychkov.m.e@gmail.com
254
ЖЭТФ, том 156, вып. 2 (8), 2019
Анализ методов исследования колебательно-вращательного спектра. . .
спектр имеет вклад как от мономерных молекул,
В общем случае, разложение электронной энер-
так и от димеров, тримеров и т. д. Таким образом,
гии в ряд Тейлора может быть представлено в виде
авторы данной работы ставили перед собой задачу
∑
∂E
1∑
получение расчетного колебательно-вращательного
E(gk) = E0 +
qk +
qjHj,kqk + . . . ,
∂qk
2
спектра свободных димеров SF6 с учетом возможно-
k
j,k
го присутствия в системе наиболее распространен-
где E0
— электронная энергия в некоторой на-
ных изотопов серы.
чальной геометрической конфигурации, (∂E/∂qk) ≡
2. МЕТОД РАСЧЕТОВ
≡ gk — градиент энергии вдоль координаты qk,
Hj,k — матрица Гессе.
Теоретическое построение колебательно-враща-
Если энергия E0 соответствует конфигурации
тельного спектра молекулы может быть выполне-
молекулы в основном состоянии, слагаемое, содер-
но с помощью одного из двух видов методик — че-
жащее градиент, обращается в нуль, а слагаемыми
рез решение стационарного уравнения Шредингера
высших порядков в предположении гармоническо-
или посредством использования данных молекуляр-
го поведения колебаний можно пренебречь. Таким
ной динамики. Под решением стационарного урав-
образом, электронная энергия принимает вид
нения Шредингера понимается анализ нормальных
колебаний молекулы путем построения дискретно-
1∑
E(gk) = E0 +
qjHj,kqk.
го спектра или с помощью решения колебательно-
2
j,k
вращательного уравнения Шредингера, или с ис-
пользованием матрицы Гессе. Важно отметить, что
Используя полученный электронный потенциал, а
упомянутые ранее теоретические работы [1-4] вы-
также запись для кинетической энергии в виде
полнены как раз с помощью анализа нормальных
колебаний. Использование данных молекулярной
1∑(dqj )2
T =
mj
,
динамики для построения колебательно-вращатель-
2
dt
j
ного спектра менее распространено среди исследова-
телей, так как, зачастую, даже для простых молеку-
где mj — масса ядра j, можно построить 3Nq
урав-
k
лярных систем требуется высокопроизводительная
нений движения
вычислительная система. Однако, как будет пока-
∑
d2qj
зано далее, такой метод, по сравнению с алгорит-
mj
=- Hj,kqk.
мом на основе стационарного уравнения Шрединге-
dt2
k
ра, может обладать рядом важных преимуществ.
Совершая переход к масс-взвешенным декартовым
2.1. Получение колебательно-вращательного
координатам xj = qj
√mj и предполагая периоди-
спектра с помощью матрицы Гессе
ческое поведение координаты во времени, xj (t) =
= x(0) cos ωt, можно получить уравнение для соб-
Расчет колебательно-вращательного спектра в
ственных значений и собственных векторов матри-
стационарном приближении может быть проведен
цы Гессе:
несколькими различными путями: либо построени-
∑
ω2xj =
H′j,kxk,
ем матрицы Гессе, или гессиана, и анализом элемен-
k
тов на ее главной диагонали, либо с помощью реше-
ний колебательно-вращательного уравнения Шре-
где H′j,k — элементы матрицы Гессе в масс-взвешен-
дингера. В большинстве программных комплексов
ных декартовых координатах. Таким образом, для
для квантово-химических расчетов реализован ал-
расчета колебательно-вращательного спектра тре-
горитм с использованием именно гессиана, так как,
буется найти основное состояние молекулы, постро-
формально, поиск полной энергии системы в алго-
ить матрицу Гессе в масс-взвешенных декартовых
ритме Хартри - Фока проводится при каждом цик-
координатах и найти все ее собственные значения,
ле вычислений, в то время как гессиан представля-
отличные от нуля. При этом собственные значе-
ет собой матрицу вторых производных от энергии
ния гессиана являются квадратами колебательно-
по координатам. Следовательно, в процессе цикла
вращательных нормальных частот, а собственные
вычислений рассчитываются все необходимые пере-
функции для каждой моды задают амплитуду или
менные для построения матрицы Гессе, и значитель-
интенсивность колебаний.
ных дополнительных программных усилий не требу-
Данный способ может быть формально исполь-
ется.
зован для любых молекулярных систем, однако
255
М. Е. Бычков, Ю. В. Петрушевич, А. Н. Старостин
ЖЭТФ, том 156, вып. 2 (8), 2019
можно отметить ряд недостатков. Методика постро-
молекулярной динамики. С другой стороны, в слу-
ения матрицы Гессе предполагает расчет энергии ос-
чае динамической задачи все нормальные моды воз-
новного состояния с высокой точностью, так как ма-
буждаются одновременно, а поскольку вклады каж-
лые ошибки в оценке существенно влияют на вид
дой нормальной моды находятся на соответствую-
матрицы вторых производных и, как следствие, на
щей этой моде частоте, характеристики всех актив-
собственные значения и соответствующие им часто-
ных мод можно получить за одно фурье-преобра-
ты. Для ряда молекул, например с атомами тяже-
зование корреляционной функции дипольного мо-
лых металлов, поиск энергии основного состояния
мента. Таким образом, интенсивность колебатель-
с заведомо высокой точностью представляет значи-
но-вращательного спектра соотносится с амплиту-
тельную вычислительную трудность. Кроме того,
дой изменения дипольного момента в автокорреля-
расчет энергии основного состояния проводится в
ционной функции.
приближении нулевой температуры, тогда как мо-
Согласно работам [10, 11], автокорреляционную
лекулы при конечных температурах обладают неко-
функцию можно представить в общем виде:
∫ ∫
торой конформационной динамикой. Наиболее ярко
1
2
〈f(τ)f(t+τ)〉τ =
f (t)e-iωtdt
iωtdω,
это заметно при расчете и анализе белковых струк-
e
2π
тур и некоторых органических молекул. Приближе-
а интенсивность спектральной компоненты может
ние гармонического осциллятора, в свою очередь,
быть тогда записана в форме
также можно считать недостатком метода (как бу-
∫
дет показано в разд. 2.3) даже для простых мо-
A(ω) ∝
〈 μ(τ) μ(t + τ)〉τ e-iωtdt.
лекул учет ангармонических поправок значительно
усложняет вид спектра. Можно отметить, что ис-
Учитывая сказанное выше, можно заметить, что
пользование решений колебательно-вращательного
время цикла вычислений методом на основе мо-
уравнения Шредингера позволяет обойти, как мини-
лекулярной динамики напрямую зависит от скоро-
мум, требование гармонического приближения, од-
сти расчета прямого и обратного фурье-преобразо-
нако решение такого уравнения с ангармоническими
ваний.
поправками для многоатомной системы с большим
Существуют несколько способов получения дан-
числом электронов представляет собой отдельную
ных о временной зависимости дипольного момента
расчетную задачу со своими трудностями.
исходя из результатов цикла молекулярных расче-
тов — с помощью фаз Берри [12] или через схему
максимально локализованных функций Ванье [13].
2.2. Получение колебательно-вращательного
спектра на основе данных молекулярной
Фазы Берри дают величину полного дипольного мо-
мента в моделируемом объеме, что не позволяет эф-
динамики
фективно рассчитывать более одной молекулы за
Построение колебательно-вращательного спект-
один цикл вычислений, в то время как схема функ-
ра с использованием данных расчета на основе зако-
ций Ванье не имеет ограничений на число молекул
нов молекулярной динамики опирается на прибли-
в исследуемом объеме и подходит как для модели-
жение, согласно которому спектральные компонен-
рования отдельно взятой молекулы, так и для ис-
ты получаются фурье-преобразованием автокорре-
следования сложных многосоставных систем — мо-
ляционной функции дипольного момента.
лекулярных кластеров, молекул в растворителях и
Интегральный коэффициент Aν ИК-поглощения
т. д.
в решении колебательно-вращательного уравнения
В схеме Ванье каждой паре электронов припи-
Шредингера представим в виде
сывается некоторый центр Ванье rj , так что полный
)2
дипольный момент выражается в виде
1
Naπ
( ∂μ
Aν =
,
∑
∑
4πε0
3c2
∂xk
μ = -2e rj + e ZiRi,
j
i
где ε0 — диэлектрическая постоянная, Na — посто-
янная Авогадро, c — скорость света, μ — дипольный
где Zi и Ri — соответственно заряд и положение яд-
момент молекулы, xk — масс-взвешенная декарто-
ра. В общем случае выбор способа задания положе-
ва координата вдоль нормальной моды k. Производ-
ний rj и Ri влияет на полный дипольный момент, од-
ная от дипольного момента по координатам долж-
нако поскольку для вычислений необходимы только
на быть рассчитана в основном состоянии, что де-
временные производные, данное влияние не имеет
лает невозможным такой расчет при использовании
принципиального значения.
256
ЖЭТФ, том 156, вып. 2 (8), 2019
Анализ методов исследования колебательно-вращательного спектра. . .
I, отн. ед.
0.25
1.0
2141.78
2186.57
0.20
0.15
0.8
0.10
0.6
0.05
2160.1
0
1800 1900 2000 2100 2200 2300 2400 2500 2600
0.4
, см-1
0.2
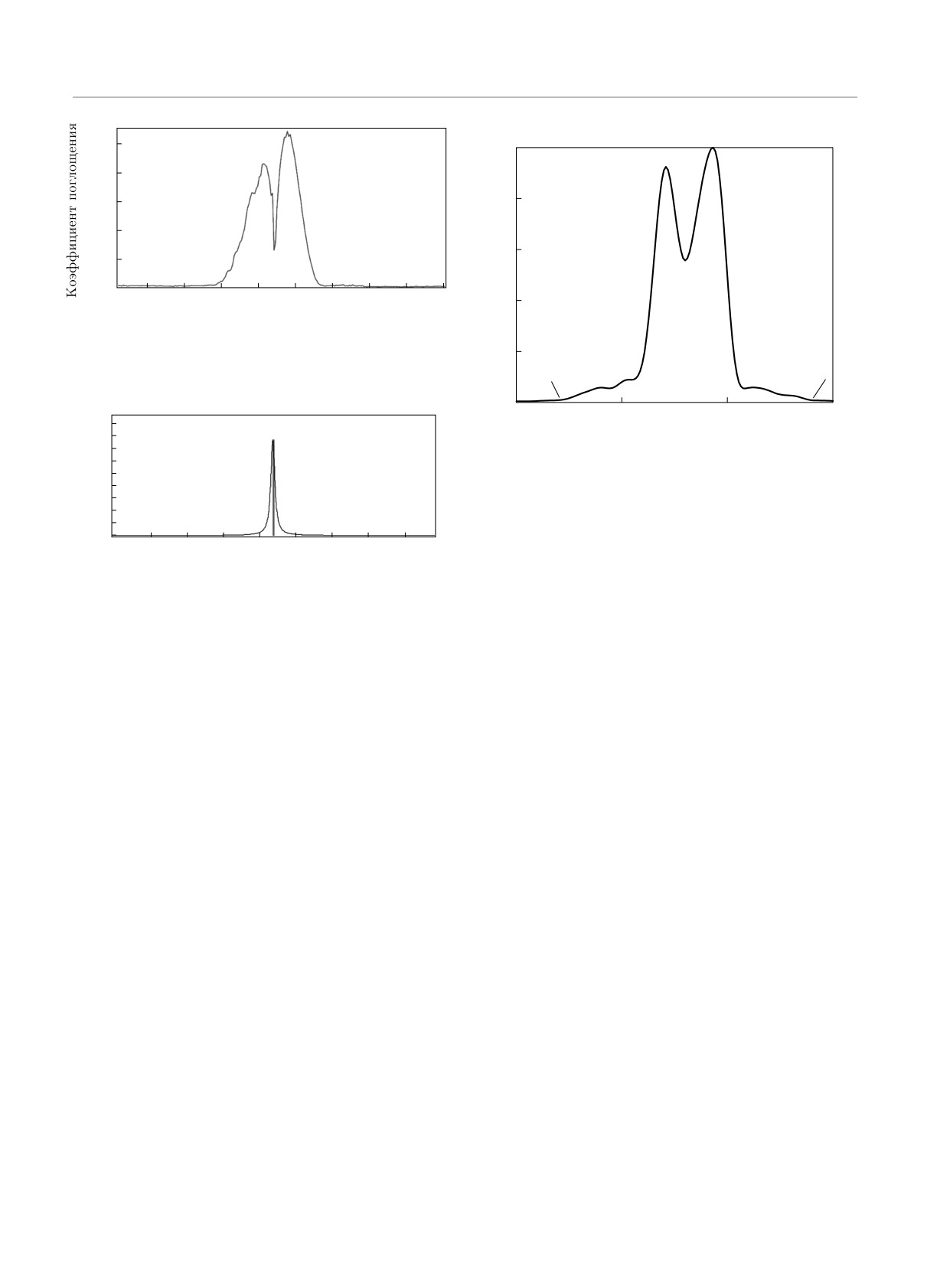
Рис.
1. Колебательно-вращательный спектр молекулы
CO [5]
2042.02
2280.22
0
I, отн. ед.
2000
2100
2200
2300
2177.14
1.6
, см-1
1.2
Рис. 3. Расчетный колебательно-вращательный спектр по-
0.8
глощения молекулы CO по данным метода молекулярной
динамики
0.4
0
0
500
1000 1500 2000 2500 3000 3500 4000
ного комплекса для квантово-химических расчетов
Firefly [14]. Температурная зависимость спектра за-
, см-1
дается через лоренцево уширение.
Рис. 2. Колебательно-вращательный спектр поглощения
Анализ элементов матрицы Гессе, т. е. нормаль-
молекулы CO, полученный с помощью матрицы Гессе
ных колебаний, в стационарном случае не позволя-
ет получить экспериментальную зависимость спект-
ра, так как расщепление пика вызвано ангармониз-
Важно отметить, что при вычислениях с помо-
мом колебаний двухатомной молекулы CO. Нельзя
щью молекулярной динамики, в отличие от мето-
при этом не отметить, что в абсолютных значениях
да матриц Гессе, не делается никаких существен-
метод матриц Гессе дает оценку частотной полосы,
ных приближений: расчет может происходить при
близкую к экспериментальным показателям, хотя и
любой конечной температуре; не требуется высокой
не воспроизводит вид спектра.
точности оценки энергии, так как в основном состо-
На рис. 3 представлен расчетный колебательно-
янии вычисляется только конфигурация начальных
вращательный спектр, полученный при помощи вы-
молекулярных орбиталей; не вводится никаких при-
числения автокорреляционной функции дипольного
ближений на вид осцилляций, так что корреляци-
момента на основе данных молекулярной динами-
онная функция дипольного момента автоматически
ки. Можно отметить явное качественное совпадение
содержит ангармонические поправки.
вида спектра с экспериментальной зависимостью на
рис. 1, в то время как абсолютные значения при этом
2.3. Сравнение методов на примере
также оказываются близкими, хотя и не совсем сов-
молекулы CO
падают с экспериментальными — спектральные пи-
ки на частотах ω ≈ 2120 см-1 и ω ≈ 2178 см-1 [5], а в
На примере простой молекулы CO можно явно
расчетах — при ω ≈ 2141 см-1 и ω ≈ 2186 см-1. От-
продемонстрировать относительные достоинства
носительная ошибка вычислений, однако, в самом
метода получения колебательно-вращательного
грубом случае не превышает 0.1 %.
спектра с помощью данных молекулярной дина-
мики. На рис. 1 представлена экспериментальная
спектральная зависимость коэффициента поглоще-
3. РЕЗУЛЬТАТЫ РАСЧЕТОВ
ния по данным из [5].
На рис. 2 приведен расчетный колебательно-вра-
С помощью алгоритма, описанного в разд. 2.2,
щательный спектр, полученный с помощью постро-
были рассчитаны колебательно-вращательные спек-
ения матрицы Гессе с использованием программ-
тры для мономеров и основных димеров гексафто-
257
5
ЖЭТФ, вып. 2 (8)
М. Е. Бычков, Ю. В. Петрушевич, А. Н. Старостин
ЖЭТФ, том 156, вып. 2 (8), 2019
рида серы. Для получения данных молекулярной
динамики использовался программный комплекс
I, отн. ед.
для квантово-химических ab initio расчетов с от-
1.0
947.77
крытым кодом CP2K [15-18]. Все вычисления про-
а
водились методами молекулярной динамики [19-
0.8
21] в виртуальном термостате
[22] с валентно-
расщепленным базисом DZVP (double-zeta-valen-
S 32)F(6
ce-polarised) [23-26], а в качестве потенциала взаи-
0.6
модействия DFT [27-30] цикла использовался функ-
ционал LDA-семейства PADE [31]. Для всех молеку-
0.4
лярных систем высчитывалась траектория из 50 ты-
сяч шагов, которая с помощью комплекса TRAVIS
0.2
595.55 606.75
[32] затем пересчитывалась в колебательно-враща-
тельный спектр.
623.04
0
600
700
800
900
1000
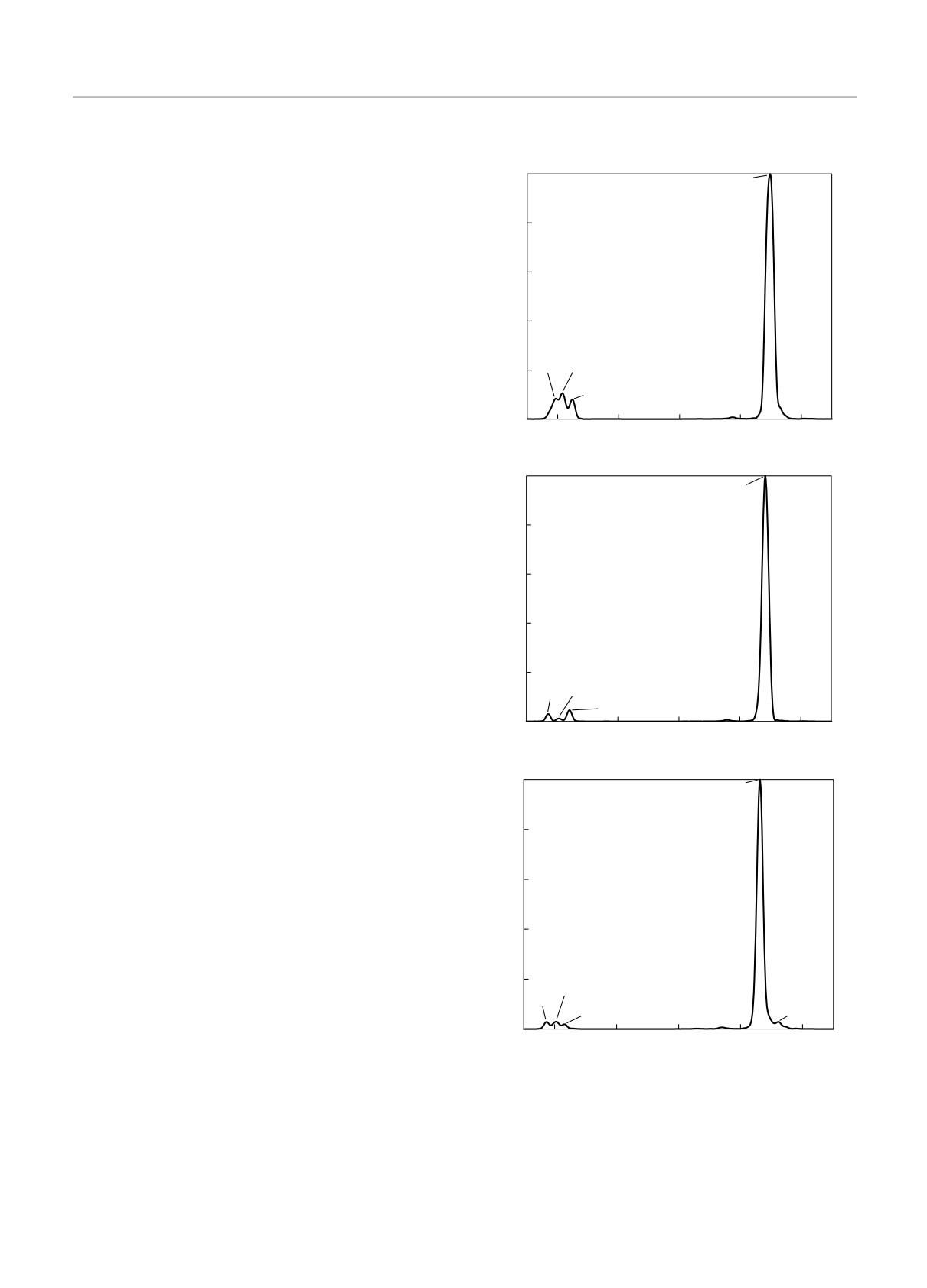
3.1. Мономеры
, см-1
I, отн. ед.
На рис. 4 представлены расчетные спектры для
1.0
941.66
мономеров SF6. Необходимо отметить, что, несмот-
ря на повышение требуемой точности расчетов, вы-
б
0.8
бор более богатого базиса или иного функционала,
не удается приблизиться к экспериментальным дан-
ным в абсолютных показателях с помощью реше-
0.6
S 33)F(6
ний, встречающихся в литературе. В то же время
отклонение вычислений с базисом DZVP и функ-
0.4
ционалом PADE от экспериментальных данных со-
ставляет всего около 0.05 %, так как расчетная час-
0.2
тота пика канала ν3 для самого распространенно-
584.36601.66
го мономера S(32)F6 оказывается равной 899 см-1
621.00
при табличном значении в 947 см-1 [5]. Учиты-
0
600
700
800
900
1000
вая, что цикл вычислений качественно воспроизво-
, см-1
дит экспериментальную зависимость с некоторым
I, отн. ед.
постоянным сдвигом (разность частот малого пи-
1.0
931.48
ка и канала ν3 составляет около 333 см-1 в экс-
перименте и 338 см-1 в расчетах для мономера
в
0.8
S(32)F6), мы решили, учитывая свойства фурье-пре-
образования, отнормировать расчетный спектр от-
носительно экспериментальных значений. Таким об-
0.6
разом, все дальнейшие графики были построены
S 34)F(
6
с учетом поправочного сдвига в фиолетовую об-
0.4
ласть на 48 см-1 и при спектральном разрешении
1.02 см-1.
Важно отметить, что расчет методом построе-
0.2
ния матриц Гессе с помощью аналогичных числен-
586.39601.66
615.91
959.98
ных инструментов для системы гексафторида серы
0
приводит к похожему сдвигу в область низких час-
600
700
800
900
1000
тот расчетного спектра. Однако, в отличие от ал-
, см-1
горитма молекулярной динамики, качественно ко-
Рис. 4. Колебательно-вращательные спектры поглощения
лебательно-вращательный спектр воспроизводится
мономеров S(32)F6 (а), S(33)F6 (б) и S(34)F6 (в)
с существенными ошибками — разность частот ма-
лого пика и канала ν3 составляет около 270 см-1 и
заметно отличается от экспериментальных данных.
258
ЖЭТФ, том 156, вып. 2 (8), 2019
Анализ методов исследования колебательно-вращательного спектра. . .
Таблица. Расстояния Δν3 между пиками для раз-
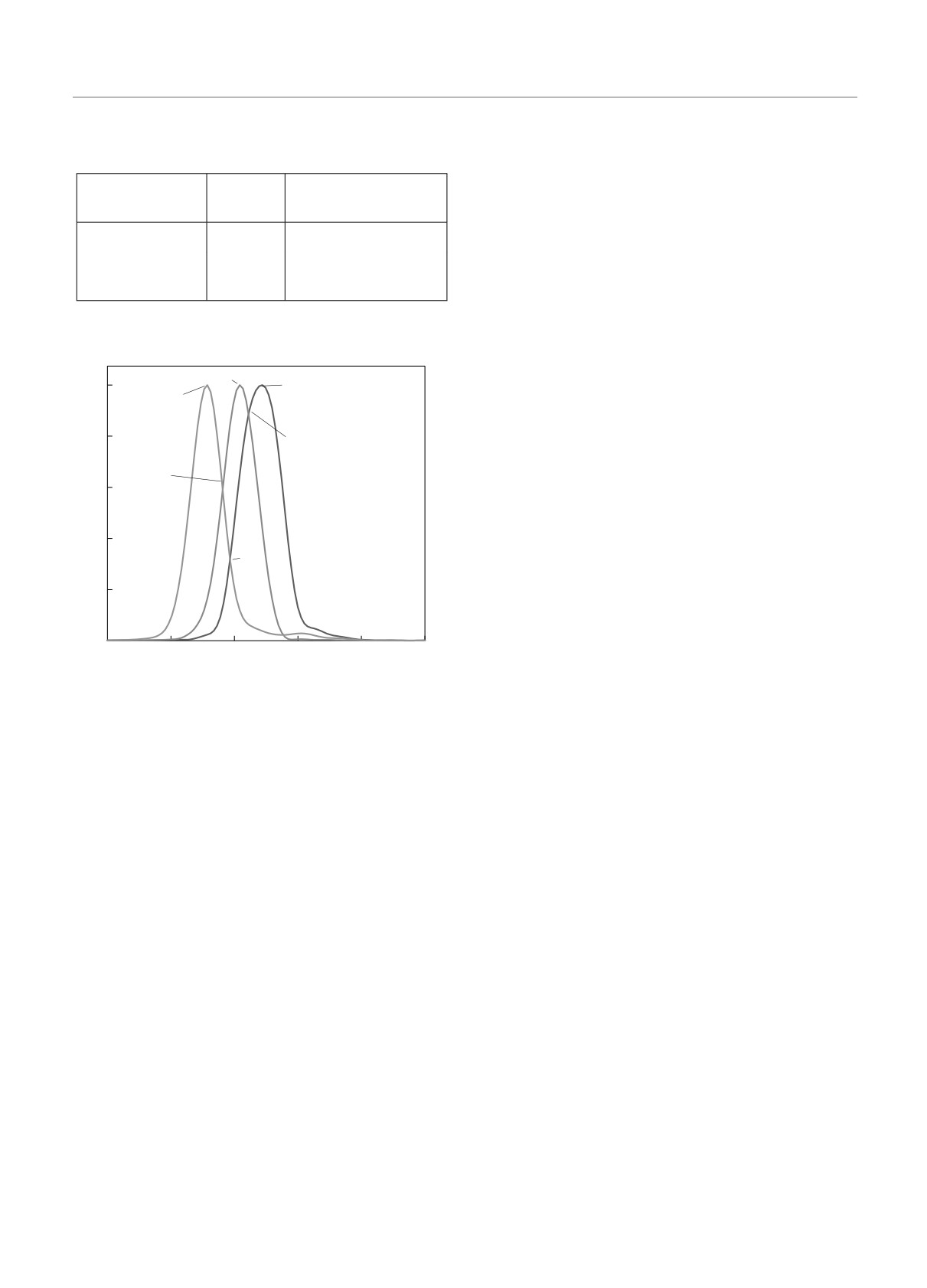
На рис. 5 построен график влияния изотопи-
личных мономеров
ческого эффекта на колебательно-вращательный
,
спектр мономеров гексафторида серы для канала ν3
Расчет, Эксперимент [8, 9],
а расстояние между пиками для различных мономе-
см-1
см-1
ров с изотопами серы представлено в таблице.
Таким образом, все расчетные данные для моно-
Δν3(S(32)-S(33))
6.11
7
меров, полученные с помощью методов молекуляр-
Δν3(S(32)-S(34))
16.29
17
ной динамики с учетом сдвига, воспроизводят экс-
Δν3(S(33)-S(34))
10.18
10
периментальные значения.
3.2. Димеры
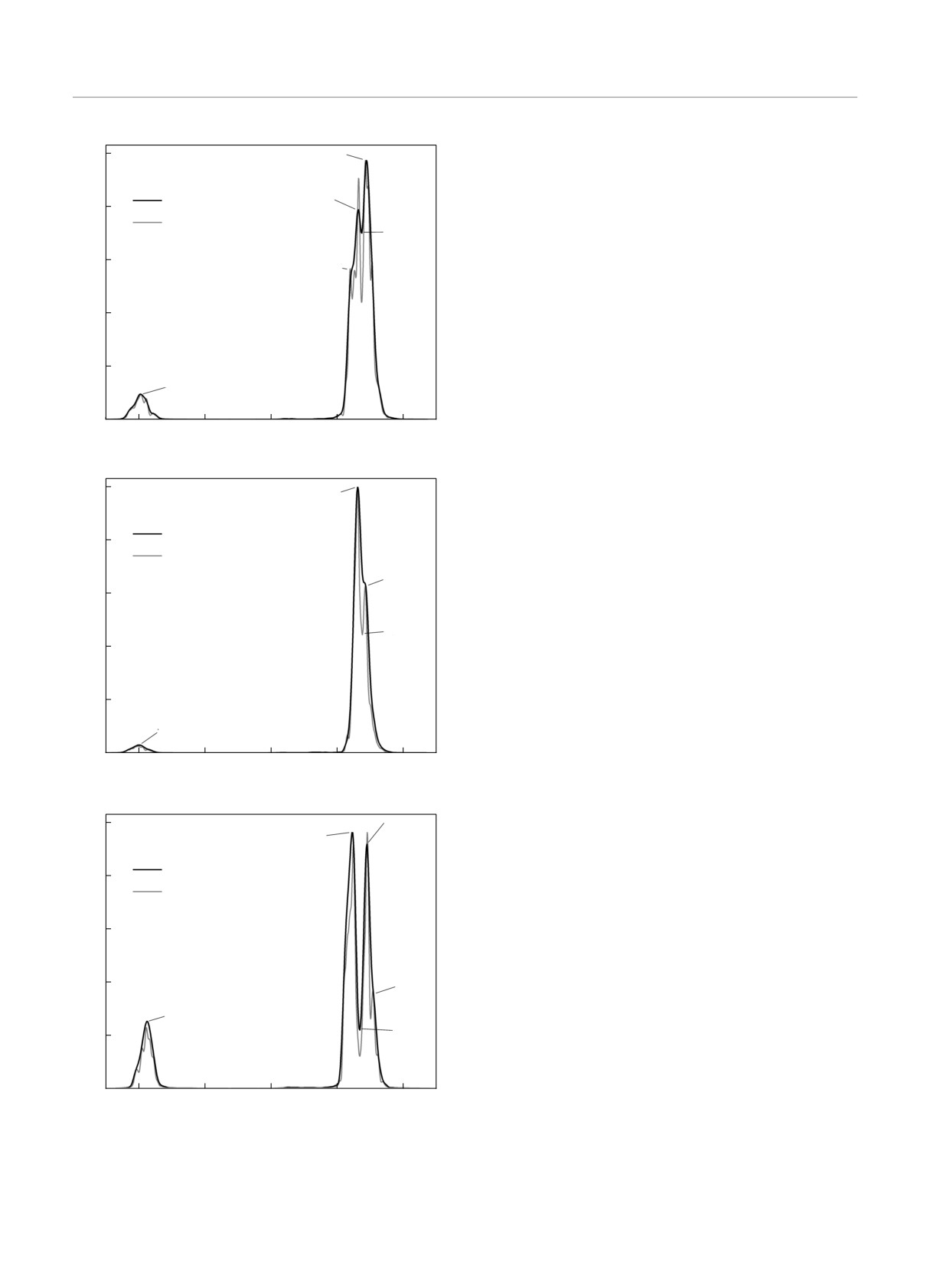
I, отн. ед.
Для димеров гексафторида серы, SF6, также
941.66
1.0
947.77
были проведены расчеты согласно разд. 2.2. Учи-
931.48
тывая распространенность изотопов серы, был
рассчитан колебательно-вращательный спектр
0.8
944.71
гомодимера S(32)F6-S(32)F6 и двух гетеродимеров,
S(32)F6-S(33)F6 и S(32)F6-S(34)F6. На рис. 6 представ-
936.57
0.6
лены расчетные спектры при двух спектральных
разрешениях, 1.02 см-1 и 0.51 см-1.
Можно заметить, что димеризация существен-
0.4
но не изменяет вида спектра — аналогично слу-
938
.60
чаю моделирования мономеров, малый спектраль-
0.2
ный пик соответствует различным колебательным
1
2
3
модам атомов фтора и не меняет своего положе-
ния при изменении массы центрального атома се-
0
900
920
940
960
980
1000
ры, оставаясь в пределах частот от 604 до 607 см-1,
, см-1
в то время как больший пик соответствует различ-
ным колебательным модам центральных атомов се-
Рис. 5. Влияние массы атома S на канал ν3 колебатель-
ры. Как и предсказывается теорией [1,2], канал ν3 в
но-вращательного спектра поглощения мономера SF6: кри-
кластерных молекулярных структурах SF6 расщеп-
вая 1 — S(32); 2 — S(33); 3 — S(34). Спектральное разреше-
ляется на две полосы. Наиболее явно такое расщеп-
ние 1.02 см-1
ление наблюдается на рис. 6в: одна полоса сдвигает-
ся в сторону низких частот относительно канала ν3
свободного мономера, а другая — в область высоких
Сдвиг спектра, при этом, не только не улучшает ре-
частот.
зультаты, но и идейно противоречит методу постро-
Можно отметить, что спектральный пик при бо-
ения матриц Гессе, основанному на точном расчете
лее высоких частотах соответствует колебательным
энергии основного состояния системы.
модам свободного мономера S(32)F6, связанных с ос-
Результаты вычислений методом молекулярной
цилляциями центрального атома серы, в то время
динамики достаточно хорошо согласуются с доступ-
как пик при более низких частотах соответствует
ными экспериментальными данными. Так, напри-
как модам свободного мономера с изотопным ато-
мер, для мономера S(33)F6 расчетное положение пи-
мом серы, так и различным модам смешанных ос-
ка канала ν3 соответствует ω = 942 см-1, тогда
цилляций центральных атомов.
как в работе [8] получено значение около 940 см-1,
что фактически совпадает с расчетным значением,
представленным на рис. 4б. В то же время для моно-
4. ЗАКЛЮЧЕНИЕ
мера S(34)F6 рассчитанное значение положения пи-
ка канала ν3, равное ω = 931 см-1 и приведенное на
Метод построения колебательно-вращательного
рис. 4в, также хорошо согласуется с данными рабо-
спектра с использованием матрицы Гессе, наибо-
ты [8], где представлено значение 930 см-1. Кроме
лее часто встречающийся в литературе, как вид-
того, в работе [9] приводится значение 930 см-1, что
но из пробного расчета для простой молекулы CO,
также соответствует проведенным вычислениям.
не всегда может воспроизвести экспериментальную
259
5*
М. Е. Бычков, Ю. В. Петрушевич, А. Н. Старостин
ЖЭТФ, том 156, вып. 2 (8), 2019
I, отн. ед.
спектральную зависимость. В качестве альтернати-
1.0
953.36
вы может быть использован расчет с помощью мо-
а
лекулярной динамики, основанный на вычислении
-1
автокорреляционной функции дипольного момента.
1.02 см
941.66
0.8
В отличие от алгоритма построения матриц Гессе,
0.51 см-1
946.75
использование молекулярной динамики не требует
повышенной точности расчета энергии системы и
0.6
929.44
свободно от ряда достаточно сильных приближе-
ний — гармоничности колебаний и нулевой темпе-
0.4
ратуры — однако требует существенных вычисли-
тельных мощностей.
Данный алгоритм был опробован на хорошо изу-
0.2
ченной молекулярной системе гексафторида серы,
604.71
SF6. Удалось показать, что, несмотря на наличие по-
правочного сдвига, расчетные характеристики мо-
0
600
700
800
900
1000
номерных изотопных структур SF6 совпадают с
, см-1
экспериментальными. Анализ гомо- и гетеродиме-
I, отн. ед.
ров SF6 с изотопными атомами серы подтверждает
1.0
937.59
б
предсказываемое теорией и экспериментом расщеп-
ление полосы ν3.
-1
1.02 см
0.8
0.51 см-1
949.80
ЛИТЕРАТУРА
0.6
1.
T. A. Beu and K. Takeuchi, J. Chem. Phys. 103, 6394
944.20
(1995).
0.4
2.
T. A. Beu, Z. Phys. D 31, 95 (1994).
3.
A. Boutin, J. B. Maillet, and A. H. Fuchs, J. Chem.
0.2
Phys. 99, 9944 (1993).
607.77
4.
A. Boutin, B. Rousseau, and A. H. Fuchs, Chem.
0
600
700
800
900
1000
Phys. Lett. 218, 122 (1993).
, см-1
I, отн. ед.
5.
NIST Chemistry WebBook, ed. by P. J. Linstrom and
W. G. Mallard, NIST Standard Reference Database
1.0
950.82
в
927.41
Number 69, National Institute of Standards and
Technology (2018).
-1
1.02 см
0.8
6.
F. Huisken and M. Stemmler, Chem. Phys. 132, 351
0.51 см-1
(1989).
0.6
7.
B. Heijmen, A. Bizzarri, S. Stolte, and J. Reuss,
Chem. Phys. 132, 331 (1989).
961.00
0.4
8.
Т. Д. Коломийцева, В. А. Кондауров, Е. В. Седел-
кова, Д. Н. Щепкин, Опт. и спектр. 92, 512 (2002).
604.71
938.60
0.2
9.
C. Brodbeck, J. Rossi, H. Strapelias, and J.-P. Bou-
anich, Chem. Phys. 54, 1 (1980).
0
10.
M. Thomas, M. Brehm, R. Fligg et al., Phys. Chem.
600
700
800
900
1000
Chem. Phys. 15, 6608 (2013).
, см-1
11.
M. Thomas, M. Brehmb, and B. Kirchner, Phys.
Рис.
6. (В
цвете онлайн)
Колебательно-вращательные
Chem. Chem. Phys. 17, 3207 (2015).
спектры поглощения гомодимеров S(32)F6-S(32)F6 (а) и ге-
теродимеров S(32)F6-S(33)F6 (б), S(32)F6-S(34)F6 (в)
260
ЖЭТФ, том 156, вып. 2 (8), 2019
Анализ методов исследования колебательно-вращательного спектра. . .
12. R. D. King-Smith and D. Vanderbilt, Phys. Rev.
21. J. VandeVondele and J. Hutter, J. Chem. Phys. 118,
B 47, 1651 (1993).
4365 (2003).
22. S. Nose, J. Chem. Phys. 81, 511 (1984).
13. N. Marzari and D. Vanderbilt, Phys. Rev. B 56,
12847 (1997).
23. J. VandeVondele and J. Hutter, J. Chem. Phys. 127,
114105 (2007).
chem.msu.su/graw/firefly/index.html (2017).
24. M. Krack, Theor. Chem. Accounts 114 (1-3), 145
(2005).
15. J. Hutter, M. Iannuzzi, F. Schiffmann, and J. Van-
deVondele, CP2K: Atomistic Simulations of Con-
25. C. Hartwigsen, S. Goedecker, and J. Hutter, Phys.
densed Matter Systems, Wiley Interdisciplinary Re-
Rev. B 58, 3641 (1998).
views-Computational Molecular Science, 4 (1), 15-25
26. S. Goedecker, M. Teter, and J. Hutter, Phys. Rev.
(2014).
B 54, 1703 (1996).
16. J. VandeVondele, M. Krack, F. Mohamed et al., J.
27. S. Grimme, S. Ehrlich, and L. Goerigk, J. Comp.
Computer Phys. Commun. 167(2), 103 (2005).
Chem. 32, 1456 (2011).
17. O. Schuett, P. Messmer, J. Hutter, and J. Vande-
28. S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, J.
Vondele, GPU-Accelerated Sparse Matrix-Matrix
Chem. Phys. 132, 154104 (2010).
Multiplication for Linear Scaling Density Functional
Theory, Electronic Structure Calculations on
29. M. Frigo and S. G. Johnson, Proc. IEEE 93, 216
Graphics Processing Units, John Wiley & Sons, New
(2005).
York (2016), pp. 173-190.
30. G. Lippert, J. Hutter, and M. Parrinello, Mol. Phys.
92, 477 (1997).
18. U. Borstnik, J. VandeVondele, V. Weber, and J. Hut-
ter, Parallel Computing 40 (5-6), 47 (2014).
31. J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev.
Lett. 77, 3865 (1996).
19. S. Nose, Mol. Phys. 52, 255 (1984).
32. M. Brehm and B. Kirchner, J. Chem. Inf. Model. 51,
20. J. Kolafa, J. Comp. Chem. 25, 335 (2004).
2007 (2011).
261