ВВЕДЕНИЕ
Несмотря на стремительное развитие технологии ДНК-секвенирования нового поколения (NGS), позволяющей определять полную нуклеотидную последовательность геномов, ее использование для прочитывания определенных областей генома затруднительно. Это касается исследований, связанных с определением однонуклеотидных полиморфизмов (SNP), анализом перестройки ДНК, а также при изучении подмножеств участков генома, а не всего генома, например кодирующих белок экзонов. Для выполнения таких задач необходимо проводить “целевое обогащение” анализируемой ДНК и создание специализированных библиотек целевых последовательностей для последующего их секвенирования на выбранной платформе NGS.
Удаление той части геномной ДНК, которая не представляет интереса для проводимого исследования, позволяет обеспечить большую глубину покрытия при секвенировании целевых районов генома, увеличить число мишеней, а также значительно сэкономить время и стоимость эксперимента, в том числе за счет упрощения биоинформатического анализа результатов секвенирования.
Методы обогащения мишеней можно разделить на несколько категорий в зависимости от способа захвата целевого участка ДНК. В одних способах обогащения мишеней используют метод гибридизации нуклеиновых кислот. В этом случае денатурированную геномную ДНК “отжигают” с одноцепочечными (оц) ДНК- или РНК-зондами к целевым участкам. Связавшуюся с зондами геномную ДНК выделяют и секвенируют. Другие способы обогащения мишеней основаны на методе ПЦР. Используют мультиплексную ПЦР с десятками тысяч праймеров. Эмульсионная ПЦР позволяет одновременно проводить миллионы цепных реакций и получать миллионы мишеней для секвенирования. В последние годы появились методы обогащения целевых фрагментов ДНК, основанные на использовании РНК-программируемого комплекса CRISPR-Cas9.
СПОСОБЫ ОБОГАЩЕНИЯ ЦЕЛЕВЫХ ФРАГМЕНТОВ ДНК С ПОМОЩЬЮ МЕТОДА МОЛЕКУЛЯРНОЙ ГИБРИДИЗАЦИИ
На первом этапе проводят фрагментацию (физическую или ферментативную) геномной ДНК с последующей ферментативной репарацией концов полученных ДНК-дуплексов и пришивкой к ним специфичных для секвенирующей платформы адаптеров. Адаптеры содержат в своем составе “штрих-код” – уникальную последовательность для каждого образца. После секвенирования биоинформатический анализ позволяет с помощью этого штрих-кода группировать данные, относящиеся к одному образцу. Производители наборов для целевого обогащения ДНК поставляют от 96 до 384 различных адаптеров, содержащих различные штрих-коды. Использование штрих-кодов обеспечивает одновременное секвенирование нескольких образцов и тем самым значительное сокращение временных и финансовых затрат.
Кроме штрих-кода в адаптерах содержатся последовательности праймеров для амплификации. С помощью ПЦР нарабатывается достаточное количество ДНК для последующей стадии гибридизации с одноцепочечными зондами, комплементарными целевым областям генома. Как правило, это ДНК-зонды. Фирма “Agilent” (США) использует в своих наборах SureSelect гибридизационные РНК-зонды. Основное преимущество РНК-зондов – большая стабильность РНК:ДНК-гибридов по сравнению с ДНК:ДНК-дуплексами [1].
Гибридизация на твердой поверхности
М. Zwick с сотрудниками [2] первыми использовали слайды “Affymetrix Chip Design Group” (США) с ковалентно связанными олигонуклеотидами для селекции геномных фрагментов (метод MGS, microarray-based genomic selection).
Протокол MGS включает пять основных этапов (рис. 1):
Рис. 1.
Схема обогащения геномных фрагментов ДНК с помощью слайдов, содержащих олигонуклеотидные зонды (метод MGS).
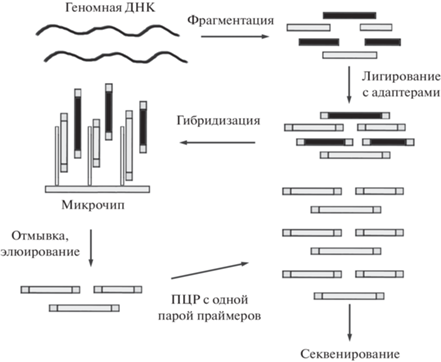
1) расщепление геномной ДНК на случайные фрагменты длиной 300 п.н;
2) репарация концевых фрагментов ДНК с добавлением 3'-выступающих остатков аденина и присоединение уникальных адаптеров с 5'-выступающим тимидином;
3) гибридизация фрагментов ДНК с комплементарными олигонуклеотидами на микрочипе высокой плотности;
4) элюирование фрагментов ДНК, связанных с зондами;
5) амплификация выделенных фрагментов с помощью праймеров, интегрированных в адаптеры.
В методе использовали перекрывающиеся гибридизационные зонды к обеим цепям дуплекса, при этом каждая цепь перекрывалась зондами от 1.5 до 4 раз. На одном слайде содержалось 385 тыс. гибридизационных зондов длиной от 50 до 93 нуклеотидов, позволяющих проводить изотермическую гибридизацию. Зонды покрывали фрагмент генома размером от 4 до 5 млн.п.н. Позднее их число увеличили до 2.1 млн. Эти зонды потенциально могут связаться с 34 млн.п.н. геномной последовательности [3, 4].
Аналогичным методом [5] выделяли более 200 тыс. экзонов для секвенирования. В качестве гибридизационных зондов были выбраны олигонуклеотиды к кодирующей последовательности экзона длиной >60 н., перекрывающие последовательность экзона со сдвигом 20 н. Всего использовали в работе 7 слайдов (“Roche NimbleGen”, США), каждый из которых содержал 385 тыс. зондов. В зависимости от применяемого протокола от 55 до 85% выделенных фрагментов ДНК содержали целевые последовательности. Метод позволяет секвенировать до 98% всех целевых экзонов.
Т. Albert и соавт. [6] ввели в схему дополнительную процедуру ‒ амплификацию фрагментов ДНК перед гибридизацией на микрочипе. Секвенирование всего экзома с использованием гибридизации на микрочиповых слайдах доказало свою полезность для выявления редких вариантов генов и мутаций, вызывающих заболевания [7, 8]. Для обогащения 1000 генов из транскриптома хлопка А. Salmon и др. [9] использовали 135 тыс. зондов, содержащихся на слайдах “Roche NimbleGen”.
Nelson с сотрудниками [10] удалось повысить эффективность обогащения целевых мишеней при блокировании побочной гибридизации адаптерных частей зондов с геномной ДНК и дублировании стадии гибридизационного обогащения целевых мишеней.
Для выделения последовательностей генов BRCA1 и TP53 в микрофлюидных кассетах микрочипового ДНК-синтезатора Geniom (“Geniom”, Германия) синтезировали 50-членные олигонуклеотиды [11, 12]. На основе этого синтезатора разработали автоматизированный метод HybSelect для подготовки образцов для высокопроизводительного секвенирования. Метод позволяет параллельно проводить обогащение ≤8 образцов размером от 125 т.п.н. до 1 млн.п.н. Метод был апробирован на анализе 115 генов, ассоциированных с развитием злокачественных опухолей [13].
Несмотря на удовлетворительную производительность, разработанный метод HybSelect не лишен существенных недостатков. Во-первых, для проведения селективного выделения целевых дуплексов требуется много (от 10 до 15 мкг) исходного материала ДНК. Во-вторых, необходимость дорогостоящего специализированного оборудования (станции для проведения гибридизации). В‑третьих, метод хорошо работает при обогащении геномных фрагментов размером ~500 п.н., но значительно менее эффективен в случае коротких экзонов (~120 п.н.) [5]. В-четвертых, HybSelect сложно масштабировать и автоматизировать.
Гибридизация в растворе
Из-за недостатков обогащения целевых фрагментов ДНК на твердой поверхности “Roche NimbleGen” и другие компании перешли на обогащение мишеней в растворе, где гибридизация идет с избытком гибридизационных зондов. Фирма “Roche NimbleGen” прекратила выпуск слайдов для гибридизации геномных фрагментов непосредственно на микрочипе.
Компания “Agilent” была первой, разработавшей коммерческий продукт, использующий гибридизацию олигонуклеотидов с геномными фрагментами ДНК в растворе [14]. В этом методе применяют синтезированные на микрочиповом ДНК-синтезаторе 200-членные олигонуклеотиды, содержащие 170-членную мишеньспецифическую последовательность, фланкированную двумя праймерами для амплификации. Набор синтезированных олигонуклеотидов отщепляют от слайда и подвергают двураундной ПЦР. В первом раунде используют интегрированные в последовательность олигонуклеотидов праймеры, во втором – в мишеньспецифические дуплексы вводят последовательность промотора фага T7. Затем, используя транскрипцию in vitro в присутствии биотин-UTP, получают биотинилированные оцРНК-зонды для выделения целевых геномных фрагментов. Геномную ДНК случайным образом расщепляют на фрагменты длиной ~250 п.н., к которым присоединяют адаптеры. После 12 циклов ПЦР полученные ампликоны гибридизуют с РНК-зондами в растворе. Биотинилированные РНК, содержащие и не содержащие комплементарную геномную ДНК, выделяют из реакционной смеси с помощью магнитных шариков с иммобилизованным стрептавидином. ДНК:РНК-гибриды разрушают и полученную ДНК амплифицируют с помощью ранее введенных в состав фрагментов геномной ДНК адаптеров (рис. 2).
Рис. 2.
Схема обогащения геномных фрагментов ДНК с помощью олигонуклеотидных зондов в растворе, применяемая в наборах Agilent.

Разработанный метод не требует специального оборудования и совместим с различными секвенирующими платформами. Он был апробирован на секвенировании 15 тыс. экзонов (2.5 млн.п.н.) человеческого генома.
К основными достоинствам метода относятся:
1) высокая концентрация оцРНК-зондов, что позволяет повысить эффективность процесса гибридизации с целевыми ДНК-фрагментами;
2) относительно небольшое количество геномной ДНК (0.5‒3.0 мкг) для секвенирования;
3) проведение гибридизации в растворе, что позволяет масштабировать и автоматизировать процесс в отличие от твердофазной гибридизации на слайдах [2];
4) нивелирование различий в гибридизации разных аллелей благодаря использованию протяженных зондов;
5) возможность готовить и характеризовать большие партии РНК-зондов, что позволяет иметь стандартизованный материал для использования в большом числе экспериментов;
6) эффективность при таргетном секвенировании как множества несмежных геномных фрагментов, так и протяженных геномных районов;
7) высокая специфичность – успешно определяют 85‒90% целевых последовательностей.
Созданы коммерческие наборы, основанные на различных вариантах обогащения последовательностей ДНК в растворе: например, Agilent SureSelect (“Agilent”) [15], Illumina TruSeq (“Illumina”, США) [16], Roche NimbleGen SeqCap EZ (“Roche NimbleGen”) [17]. Принципиальная разница между “Agilent” и другими коммерческими наборами заключается в природе гибридизационных зондов: “Agilent” использует 120‒170-членные РНК-пробы, в то время как “Roche NimbleGen” 60‒90-членные, а “Illumina” 95-членные ДНК-зонды [3, 16]. Для процедуры проведения обогащения геномной ДНК в растворе применяются стандартные 96-луночные планшеты и термоциклер, то есть специализированного оборудования в этом случае не требуется.
Модификация метода обогащения целевых фрагментов путем гибридизации олигонуклеотидов с геномными фрагментами ДНК в растворе ‒ транспозонопосредованная фрагментация анализируемой ДНК (рис. 3).
Рис. 3.
Схема использования транспозонопосредованной фрагментации анализируемой ДНК. Комплекс траспозазы с двухцепочечными олигонуклеотидами, содержащими адаптерные участки, связывается с геномной ДНК. Транспозаза расщепляет двухцепочечную геномную ДНК и встраивает олигонуклеотиды на участке разрезания. Полученные молекулы ДНК используют в ПЦР с праймерами, необходимыми для секвенирования.
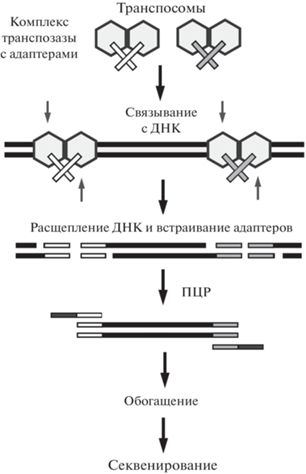
Данная модификация значительно упрощает процесс получения фрагментов анализируемой ДНК, поскольку фрагментация и присоединение адаптеров происходит за одну стадию с применением фермента транспозазы [18, 19]. Транспосомы (комплекс фермента транспозазы с двухцепочечными олигонуклеотидами, содержащими участок связывания с транспозазой и часть последовательности праймера для последующей амплификации) обладают способностью случайным образом связываться с целевыми последовательностями двухцепочечной геномной ДНК. Транспозазы в транспосоме расщепляют ДНК и одновременно встраивают на участке разрезания олигонуклеотиды из комплекса.
Разработанные методы обогащения экзома оказались эффективными для обнаружения мутаций, служащих причиной редких менделевских заболеваний [20‒23], сложных расстройств [24‒27], митохондриальных нарушений [28‒30]. Также их применяли при скрининге потенциальных генетических мутаций, связанных с раковыми заболеваниями [31‒36].
Метод гибридизационного обогащения успешно используют при анализе древней ДНК человека (ancient DNA; аДНК), сохранившейся в человеческих останках. Первым генетическим маркером, проанализированным в палеогенетических исследованиях человека, была митохондриальная ДНК (мтДНК), так как в клетках ее копийность гораздо выше, чем ядерной ДНК. Для гибридизации использовали биотинилированные ДНК или РНК-зонды, направленные на два гипервариабельных сегмента контрольной области мтДНК [37‒43].
Селективное обогащение фрагментов Y-хромосомы аДНК проводили как на твердой поверхности [44], так и в растворе [45].
Carpenter и соавт. [46] разработали метод полногеномного обогащения (whole-genome in-solution capture, WISC) аДНК, используя современную ДНК человека в качестве зондов.
В настоящее время разработаны коммерческие наборы, ориентированные на мтДНК, индивидуальные локусы или целые геномы, такие как myBaitsR3 (“Daicel Arbor Biosciences”, США), которые используют для секвенирования аДНК [47, 48].
Применение смеси олигонуклеотидов, полученных с помощью микрочипового синтезатора, содержащей 962 438 зондов, позволило секвенировать экзоны бурундуков [49]. Из целевых 11 975 экзонов обогатить удалось более 99%. Зонды были рассчитаны на основе известной последовательности генома бурундука Tamias alpinus. Однако эти же зонды были успешно использованы для селективного выделения экзонов родственных видов: Tamias amoenus, Tamias ruficaudus и Tamias striatus.
Аналогичным образом, используя зонды для экзонов генов домашней коровы (Bos taurus), Т. Cosart и др. [50] секвенировали 16 131 экзон быка (Bos indicus) и дикого бизона (Bison bison). Предложенный метод селективного обогащения геномной ДНК применяли для секвенирования ряда хромосом и митоходриального генома западных шимпанзе (Pan troglodytes verus) [51]. R. Tewhey и соавт. [52] для гибридизации использовали перекрывающиеся РНК-зонды длиной 120 нуклеотидов. На мишени размером 3.9 млн.п.н. они картировали 93% мутаций с точностью более 99%. Подход Agilent был успешно применен также при проведении секвенирования мутаций в генах, ответственных за потерю слуха [53].
С помощью гибридизации в растворе с ДНК-олигонуклеотидами были получены для секвенирования образцы ретровирусов, интегрированные в геном человека [54].
Добавление коммерческого препарата C0t1 DNA при проведении гибридизации, как на поверхности, так и в растворе, увеличивает ее специфичность [14, 55]. C0t1 DNA состоит из коротких фрагментов (50–300 п.н.) плацентарной ДНК человека, обогащенной повторяющимися последовательностями. При гибридизации на слайде или в растворе C0t1 DNA обычно добавляют соответственно в 5- и 20-кратном избытке по отношению к геномной ДНК.
СПОСОБЫ ОБОГАЩЕНИЯ ЦЕЛЕВЫХ ФРАГМЕНТОВ ДНК, ОСНОВАННЫЕ НА ИСПОЛЬЗОВАНИИ МЕТОДА МОЛЕКУЛЯРНОЙ ИНВЕРСИИ
Разработанные М. Nilsson с соавт. [56] инверсионные зонды в сочетании с мультиплексной ПЦР нашли широкое применение при детектировании точечных мутаций. Так, М. Akhras и др. [57] использовали их для обогащения мишеней NGS. Схема применения таких зондов (Connector Inversion Probe; CIPer) приведена на рис. 4. Разработанный метод был успешно апробирован авторами при генотипировании вируса папилломы человека (HPV), ассоциированных с онкологическими заболеваниями вирусов, скрининге мутаций антибиотикорезистентности в геноме гонококка (Neisseria gonorrhoeae).
Рис. 4.
Схема обогащения целевых фрагментов ДНК с использованием инверсионных зондов CIPer. а – Структура CIPe-r. AS и ES – 20-звенные олигонуклеотиды, гомологичные целевым фрагментам геномной ДНК; R – сайт рестрикции; U1 и U2 – универсальные праймеры для амплификации инвертированного зонда. б – Циклизация CIPer путем заполнения бреши ДНК-полимеразой и лигирования. в – Обогащение кольцевой ДНК за счет деградации линейной формы ДНК экзонуклеазами I и III. г – Линеаризация кольцевой формы ДНК путем расщепления сайта рестрикции. д – Амплификация полученной линейной формы ДНК с праймерами U1 и U2.
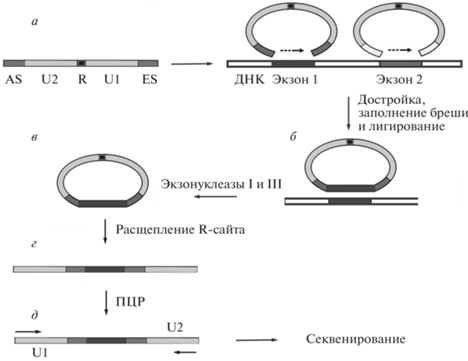
Метод коммерциализирован под названием HaloPlex (“Halo Genomics”, Швеция). В настоящее время набор производит компания “Agilent Technologies”. Их использование позволяет выделять 21 тыс. генов общим размером в 37 млн.п.н. Зонды такого типа получили общее название “молекулярные инверсионные пробы” (molecular inversion probes, MIP).
Способ обогащения целевой ДНК с использованием MIP был применен для исследования большой группы пациентов с дистонией на наличие патогенных и редких вариантов гена ATM [58], для анализа мутаций гена GNAS, вызывающих внутримышечную миксому [59].
Применение MIP в совокупности с таргетным РНК-секвенированием нового поколения позволило авторам создать независимую от гистологии молекулярную диагностику опухолей головного мозга [60]. Аналогичный подход использовали для анализа профилей экспрессии генов в случае почечно-клеточного рака [61].
Применение MIP для амплификации вариабельных областей V1, V2, V3, V6 и V7 гена 16S рРНК и последующее NGS полученных ампликонов позволило создать метод диагностики бактериальных возбудителей [62]. MIP оказались эффективными при создании автоматизированного процесса анализа мутаций генов BRCA1 и BRCA2 с целью диагностики рака молочной железы [63]. Использование MIP при секвенировании гена SCN1A позволило создать метод диагностики родительского мозаицизма при синдроме Даве [64]. Аналогично был проведен анализ мозаичности гена PIK3CA при нарушении развития [65].
Y. Wada и соавт. [66], используя MIP, выявляли в генах семейства PPAR/RXR мутации, ассоциированные с развитием шизофрении. NGS с использованием MIP применяли при анализе мутаций генов ионных каналов при диабетической периферической нейропатии [67] и для анализа мутаций в генах QRDR [68], определяющих устойчивость к ципрофлоксацину в бактериях, вызывающих сибирскую язву, чуму и туляремию. Разработана методика NGS с применением MIP, предназначенная для исследований в области фармакогеномики [69].
По сравнению с другими способами обогащения мишеней методы на основе MIP имеют ряд преимуществ. Они высокоспецифичны к мишеням, работают с мизерным количеством (≤5 пг) геномной ДНК и не требуют этапов предварительной обработки, таких как фрагментация ДНК. Кроме того, при использовании для NGS ограниченного числа MIP можно применять для их синтеза традиционные колоночные и планшетные синтезаторы.
Для создания тысяч MIP, захватывающих большое число целей одновременно, обычно используют микрочиповые синтезаторы. При получении одноцепочечных олигонуклеотидов с помощью микрочиповых синтезаторов необходимо провести ряд последовательных стадий, таких как расщепление эндо- или экзонуклеазами, очистку полученных продуктов в геле и амплификацию. D. Bang с сотрудниками [70] разработали значительно более простой и эффективный протокол применения MIP из синтезированных на микрочиповых синтезаторах олигонуклеотидов ‒ с использованием только одного фермента и двухцепочечных MIP-зондов. Метод был успешно апробирован на 11 510 дуплексных MIP, предназначенных для выделения 3554 экзонов 228 генов в образце геномной ДНК.
СПОСОБЫ ОБОГАЩЕНИЯ ЦЕЛЕВЫХ ФРАГМЕНТОВ ДНК С ПОМОЩЬЮ ПЦР
Моноплексная ПЦР
ПЦР с момента появления в 1980 годах предназначалась для наработки целевого фрагмента ДНК с помощью праймеров для амплификации [71]. Именно поэтому технология ПЦР была применена для NGS. В моноплексной ПЦР в одной пробирке с помощью одной пары праймеров нарабатывают только один ДНК-дуплекс. После амплификации всех целевых фрагментов их объединяют в эквимолярных количествах для создания библиотеки для секвенирования. Получение ампликонов с помощью моноплексной ПЦР может быть очень трудоемким процессом, поэтому эта стадия была автоматизирована. Например, для упрощения процедуры получения ампликонов компания “Fluidigm” (Канада) разработала микрофлюидное устройство Access Array system, позволяющее автоматизировать процесс получения целевых ампликонов. Одновременно можно получить 480 различных ампликонов [72].
Эмульсионная технология RainDance (“Bio-Rad”, США) [73], использующая моноплексную ПЦР, значительно более производительна по сравнению с описанной выше. В ней генерируют миллионы капель двух типов: один содержит геномную ДНК (1 молекула геномной ДНК, дезоксинуклеозидтрифосфаты, ДНК-полимераза в буфере), а другой только молекулы одной пары праймеров. Затем эти типы капель смешивают в соотношении 1 : 1. Примерно 1.5 млн капель ПЦР собирают в одну пробирку для ПЦР объемом 0.2 мл, амплифицируют в стандартном термоциклере с последующим разрушением эмульсии с высвобождения ампликонов в раствор. После очистки смесь секвенируют.
Технологии как RainDance, так и Fluidigm совместимы с большинством коммерческих секвенаторов и требуют только несколько нанограммов исходного материала для получения ампликонов для секвенирования. Однако технология RainDance позволяет получать значительно больше ампликонов одновременно: до 20 тыс. на образец по сравнению с 480 для Fluidigm. В продуктах компании “Bio-Rad” есть панели ампликонов, связанных с мутациями в генах, ассоциированных с онкологическими заболеваниями и аутизмом.
Мультиплексная ПЦР
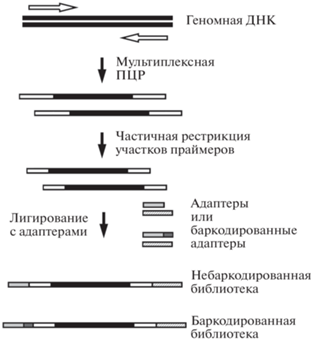
Мультиплексная ПЦР ‒ реакция, в которой несколько пар праймеров одновременно генерируют несколько ампликонов из одного и того же исходного материала. До недавнего времени использование мультиплексной ПЦР было ограничено ‒ из-за высокого уровня неспецифической амплификации. Однако в последние годы несколько компаний разработали алгоритмы, позволяющие значительно повысить селективность мультиплексной ПЦР.
Компания “Illumina” разработала вариант мультиплексной ПЦР, включающий следующие стадии:
1) гибридизацию олигонуклеотидов к их комплементарным последовательностям в интересующей области;
2) достраивание одного из пары олигонуклеотидов;
3) сшивание достроенного фрагмента со вторым олигонуклеотидом;
4) ПЦР-амплификацию полученного фрагмента ДНК с использованием универсальных праймеров, содержащих индексные последовательности.
Разработанный подход позволяет создавать разнообразные панели, в том числе и нестандартные. Пользовательские панели могут быть созданы для 1536 мишеней в геномах человека, мыши, крысы и коровы. Сконструированы панели TruSeq Amplicon Cancer Panel (“Illumina”), нацеленные на гены, связанные с развитием рака. Одна из них создана для секвенирования 212 ампликонов 48 ассоциированных со злокачественными опухолями генов. TruSight Tumor Panel (“Illumina”) разработана для получения 174 ампликонов, покрывающих сайты мутаций в 26 ассоциированных с развитием рака генах [74].
Компания “Thermo Fisher Scientific” (США) разработали технологию Ion AmpliSeq, позволяющую проводить до 24 тыс. амплификаций в одной пробирке (рис. 5).
В протоколе этого метода предусмотрено использование всего 1 нг ДНК и РНК. Технология AmpliSeq получила широкое распространение во всем мире. Так, ее использовали в исследованиях злокачественных опухолей [75], наследственных заболеваний [76] и бактерий [77].
На основе технологии AmpliSeq разработаны наборы для исследования либо определенных генов (например, Ion AmpliSeq TP53 Panel, Ion A-mpliSeq BRCA1 и BRCA2 Panel), либо заболеваний (например, муковисцидоза, деменции, рака толстой кишки и легкого). Создан набор для полноэкзомного секвенирования, содержащий 294 тыс. пар праймеров.
СПОСОБЫ ОБОГАЩЕНИЯ ЦЕЛЕВЫХ ФРАГМЕНТОВ ДНК С ПОМОЩЬЮ СИСТЕМЫ CRISPR-Cas9
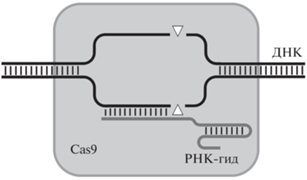
В последние годы стали появляться методы обогащения целевых фрагментов ДНК, основанные на использовании РНК-программируемого комплекса CRISPR-Cas9 [78, 79]. Системы CRISPR-Cas обеспечивают бактериям приобретенный иммунитет к вирусам. CRISPR-ассоциированный белок Cas9 ‒ это эндонуклеаза, которая использует направляющую последовательность (РНК-гид), чтобы сформировать комплекс с ДНК-мишенями, что позволяет Cas9 произвести сайтспецифичный двухцепочечный разрыв в ДНК (рис. 6). Эта система направленно действует на любую последовательность геномной ДНК.
Рис. 6.
Схема комплекса РНК-гид:Cas9 с ДНК-мишенью. Треугольниками показаны места сайтспецифичного разрезания ДНК.
Истощение фоновых ДНК
Прежде всего, система CRISPR-Cas была использована для обогащения целевых ДНК путем истощения нежелательных. Метод обогащения получил название DASH (Depletion of Abundant Sequences by Hybridization) [80]. На первом этапе конструируют библиотеку фрагментов ДНК для секвенирования, содержащую в частности праймеры для ее амплификации. Разрушение ненужных для секвенирования последовательностей в полученной библиотеке проводят с помощью РНК-гидов, направленных на неинформативные последовательности. После амплификации дуплексов, содержащих в своем составе оба амплификационных праймера, библиотеку секвенируют. Используя этот метод, J. DeRisi с сотрудниками [80] удалось снизить содержание митохондриальной рРНК в клетках HeLa на два порядка и обогатить содержание целевых последовательностей патогенов в образцах пациентов. Модифицированный метод DASH был применен S. Bae с сотрудниками [81] для разработки сверхчувствительного способа обнаружения циркулирующих опухолевых ДНК.
R. Stevens и др. [82] разработали метод выделения длинных фрагментов ДНК (10‒36 т.п.н.) с помощью система CRISPR-Cas. После расщепления исходной ДНК эндонуклеазой Cas9 оба конца целевого фрагмента ДНК остаются связанными с ферментом. Обработка реакционной смеси экзонуклеазами приводит к разрушению фоновых последовательностей, но не затрагивает защищенные Cas9 целевые последовательности. Разработанный метод позволяет обогатить мишени в 30‒600 раз.
Применение гель-электрофореза, аффинной хроматографии и гибридизации
В нижеприведенных методах ДНК разрезают с использованием Cas9 и целевые фрагменты выделяют электрофорезом в агарозном геле. Такие методики позволяют выделять очень большие неповрежденные участки генома. Bennett-Baker & Mueller [83] с помощью пульсирующего электрофореза выделили геномные фрагменты размером 2 млн.п.н. со стократным обогащением. Т. Gabrieli с соавт. [84] усовершенствовали метод и апробировали его на секвенировании бактериального фрагмента длиной 200 т.п.н. с помощью нанопорового секвенатора. В рассмотренном методе используют приборы для импульсного гель-электрофореза, а также ДНК с чрезвычайно высокой молекулярной массой, что доступно далеко не во всех лабораториях. Однако есть другие методы, которые обходят эти проблемы.
D. Nachmanson с соавт. [85] разрезали геномную ДНК на фрагменты длиной ~500 п.н. с помощью набора РНК-гидов и выделяли целевые фрагменты гель-электрофорезом либо экстракцией геномной ДНК. Таким способом удалось добиться обогащения в 49 тыс. раз. J. Lee и др. [86] гидролизовали целевую последовательность размером 13 т.п.н. на фрагменты, размеры которых были оптимальны для создания библиотеки для секвенатора Illumina. Целевые фрагменты выделяли с помощью гель-электрофореза либо конъюгированных со стрептавидином шариков. В последнем случае РНК-гиды были биотинилированы.
Идея обогащения целевых ДНК путем выделения комплекса РНК-гид:Cas9:ДНК реализована в нескольких патентах. Например, авторы работ [87, 88] выделяли комплекс, включающий биотинилированный фермент Cas9, с помощью стрептавидинсодержащих шариков. D. Bang и др. [89] использовали для этой цели нуклеазу Cas9, содержащую полигистидиновый тег, и металл-хелатную хроматографию. Х. Xu и соавт. [90] выделяли комплекс путем гибридизации удлиненного РНК-гида с комплементарным олигонуклеотидом, иммобилизованным на твердой поверхности. А. Aalipour и др. [91] использовали каталитически неактивный белок Cas9 с полигистидиновым тегом для выделения целевых ДНК и аллельспецифическую количественную ПЦР для выявления редких мутаций ДНК. Разработанный метод позволяет выявить одну такую мутацию на фоне 1000 аллелей дикого типа.
Другой способ применения системы CRISPR-Cas для обогащения целевых последовательностей ‒ FLASH (Finding Low-Abundance Sequences by Hybridization) ‒ связан с подготовкой библиотек для секвенирования. В этом методе исходную геномную ДНК подвергают дефосфорилированию и разрезают с использованием технологии CRISPR-Cas. Продукты расщепления, содержащие концевые фосфатные группы на целевых фрагментах, способны присоединять адаптеры для секвенирования. После амплификации происходит значительное обогащение библиотеки целевыми последовательностями ДНК. Так, при анализе маркеров устойчивости к антибиотикам удалось достичь 5000-кратного обогащения библиотеки анализируемыми ДНК [92]. Если применять методы секвенирования, позволяющие “прочитывать” протяженные участки ДНК, необходимости в амплификации ДНК нет. Так, N. Hafford-Tear и соавт. [93], используя комплекс CRISPR-Cas и метод одномолекулярного секвенирования в реальном времени (PacBio SMRT), проанализировали мутации, ассоциированные с эндотелиальной дистрофией роговицы (дистрофией Фукса). Варианты метода FLASH получили распространение в нанопоровом секвенировании [94‒96].
Систему CRISPR/Cas9 также используют для фрагментации ДНК с целью генерации фрагментов одинаковой длины. В наиболее широко используемом методе – обработке утразвуком – генерируются фрагменты ДНК произвольного размера, что может приводить к проблемам при секвенировании, связанным с низким уровнем “покрытия”, неравномерным “покрытием” и ложными мутациям. Подход, названный CRISPR-DS, успешно использован для секвенирования экзонных областей TP53 [85]. Целевые фрагменты ДНК для секвенирования могут быть выделены с помощью простого фракционирования по молекулярному весу, что приводит к обогащению примерно в 49 тыс. раз. Метод позволяет снизить на один–два порядка массу исследуемой ДНК.
На основе системы CRISPR-Cas быстро растет разнообразие методов обогащения целевых последовательностей ДНК, однако до сих пор ни один из них не достиг эффективности таковых с применением гибридизации нуклеиновых кислот.
ПРИМЕРЫ ИСПОЛЬЗОВАНИЯ ТАРГЕТНОГО СЕКВЕНИРОВАНИЯ В МЕДИЦИНСКОЙ ДИАГНОСТИКЕ
Методы NGS быстро развиваются и стремительно внедряются в практическую медицину. Многие заболевания человека имеют генетическую основу. Некоторые из них обусловлены отсутствием или дисфункцией определенного белка из-за мутаций в кодирующем гене. Так обстоит дело с заболеваниями менделевской наследственности, такими как болезнь Хантингтона, талассемия и около тысячи других наследственных редких заболеваний [97]. Все больше генетических вариантов и полиморфизмов идентифицируют в качестве факторов риска орфанных заболеваний [98]. Часто развитие злокачественных новообразований тоже опосредовано генетически: мутацией одного или нескольких генов, которые либо повышают риск злокачественной трансформации клеток (например, мутации зародышевой линии), либо способствуют развитию опухоли (онкогены), либо нарушают клеточные механизмы, контролирующие пролиферацию клеток (гены-супрессоры), как это происходит при соматических мутациях [99]. Таргетное или полноэкзомное секвенирование генома пациента позволяют поставить диагноз и выработать адекватную тактику лечения.
Использование в медицинской диагностике NGS не ограничено генетическими заболеваниями. Высокопроизводительное секвенирование применяют и в исследованиях инфекционных заболеваний человека [100]. Эту технологию эффективно используют для выявления малярийных плазмодиев (Plasmodium spp.) и возбудителей микозов. Так, для обнаружения генома Plasmodium falciparum на фоне геномной ДНК человека применяли метод гибридизационного обогащения в растворе [101, 102]. Предложенный подход был распространен на анализ Plasmodium vivax [103] и Candida albicans [104]. Предварительное обогащение генома бактерий с последующим секвенированием может быть очень полезным в клинической практике. А. Brown с соавт. [105] использовали NGS для определения устойчивости Mycobacterium tuberculosis H37Rv к противомикробным препаратам.
Применив для полногеномного обогащения РНК-зонды в растворе, М. Christiansen и др. [106] описали быстрый способ обнаружения и анализа генома Chlamydia trachomatis, превышающий на порядок по чувствительности другие известные методы. Аналогичным образом предлагают проводить мониторинг некультивируемого менингококка Neisseria meningitides в клинических образцах [107].
В вирусологических исследованиях технология NGS находит применение при выявлении лекарственной устойчивости патогенов, разработке новых лекарственных средств и вакцин [108].
Первым технологию целевого обогащения вирусных геномов из клинических образцов использовали D. Depledge с соавт. [109]. С помощью 120-членных РНК-зондов авторы провели обогащение геномов вирусов семейства Herpesviridae: варицелла-зостер, Эпштейна‒Барр и герпесвируса, ассоциированного с саркомой Капоши. Полноразмерные геномы этих вирусов были реконструированы и использованы в работах по исследованию структуры и разнообразию вирусной популяции. Применяя предложенный подход, многие авторы исследовали геномы вируса Эпштейна‒Барр [110], вируса Зика [111], норовируса [112], вируса Ласса [113], вируса гепатита С [114], герпесвируса-7 человека [115], вирусов простого герпеса-1 и -2 [116].
Т. Wylie и др. [117] создали гибридизационную панель ViroCap, предназначенную для обогащения нуклеиновой кислоты ДНК- и РНК-содержащих вирусов 34 семейств, которые инфицируют позвоночных и включают 190 вирусных родов и 337 видов. Панель была создана на основе анализа 1 × 109 геномных последовательностей, а ее применение позволило увеличить число прочтений вирусных геномов в 296‒674 раз. Разработанная панель может быть востребована как в фундаментальных, так и в прикладных исследованиях.
Т. Briese и др. [118] разработали платформу VirCapSeq-VERT для секвенирования вирома. В систему входит около 2 млн зондов, которые охватывают геномы вирусов, поражающих позвоночных, включая человека. Список всех родов вирусов, которые инфицируют позвоночных, был составлен из списка основных видов Международного комитета по таксономии вирусов (International Committee on Taxonomy of Viruses). Всего для выбора зондов было использовано 342 438 белоккодирующих вирусных последовательностей.
Библиотека биотинилированных олигонуклеотидов была синтезирована на основе массивов данных NimbleGen (NimbleGen Systems Inc., США) и использована для захвата в растворе вирусных нуклеиновых кислот. Использование VirCapSeq-VERT привело к 100–10 000-кратному увеличению числа прочтений вирусного генома в образцах крови и гомогенатов тканей по сравнению с секвенированием Illumina. Для технологии VirCapSeq-VERT предел обнаружения сравним со специфической ПЦР в реальном времени в сыворотке, крови и экстрактах тканей. Кроме того, этот метод позволяет идентифицировать новые вирусы, геномы которых примерно на 40% отличаются от известных вирусов, использованных для разработки библиотеки зондов.
В 2018 году был разработан аналогичный подход ‒ ViroFind [119]. Библиотека ViroFind включает 165 433 вирусных зонда, которые охватывают геномы 535 отобранных ДНК- и РНК-вирусов, инфицирующих человека или способных вызывать зооноз. ViroFind использовали для обнаружения и анализа всех вирусных популяций в головном мозге пяти пациентов с прогрессирующей многоочаговой лейкоэнцефалопатией. По сравнению с прямым глубоким секвенированием, с помощью ViroFind удалось обогатить вирусные последовательности, присутствующие в клинических образцах, до 127 раз. Предложенный подход был распространен на анализ ретровирусных геномов (вирус Т-клеточного лейкоза типа 1 человека и вирус иммунодефицита типа 1 человека), интегрированных в геном человека [54]. При использовании вирусспецифичных зондов обогащение геномных последовательностей вирусов в сотни и тысячи раз превосходило этот показатель при прямом секвенировании.
ЗАКЛЮЧЕНИЕ
Наиболее эффективными методами таргетного обогащения в настоящее время считаются основанные на гибридизации в растворе, мультиплексной ПЦР и молекулярной инверсии. Разработаны эффективные компьютерные программы, позволяющие проводить подбор олигонуклеотидов для таргетного и полногеномного секвенирования. Ряд фирм производит наборы для полноэкзомного обогащения, которые при этом постоянно совершенствуются. Эти методы основаны на гибридизации в растворе. Проведены сравнительные анализы специфичности и производительности таких наборов [120‒123].
В целом, все платформы работают хорошо, но между ними выявляют и небольшие различия. Так, РНК-зонды образуют более прочные комплексы с мишенью по сравнению с ДНК-зондами. При сверхглубоком секвенировании РНК-зонды, направленные на обе цепи мишени, работают во всех случаях лучше, чем РНК-зонды направленные только на одну цепь мишени. ДНК-зонды более эффективны для анализа GC-богатых участков, а РНК-зонды – для AT-областей.
С развитием методов таргетного и полноэкзомного секвенирования появляются новые методы медицинской диагностики. Для развития высокоэффективных современных методов диагностики, основанных на использовании NGS, требуется не только инструментальная база (секвенаторы ДНК), но и современная индустрия для получения массива синтетических олигонуклеотидов (сотен тысяч и миллионов праймеров для амплификации ДНК и зондов для гибридизации). Традиционные колоночные и планшетные синтезаторы ДНК малопригодны для получения таких массивов олигонуклеотидов. Для такого рода задач необходимы микрочиповые синтезаторы ДНК. Экспорт высокопроизводительных зарубежных синтезаторов в нашу страну был запрещен с момента их создания. Учитывая исключительную важность таких приборов для современной фундаментальной и прикладной науки, с целью создания макета микрочипового синтезатора олигонуклеотидов Сибирским отделением Российской академии наук был организован консорциум, в который вошли следующие институты: Институт химической биологии и фундаментальной медицины, Институт физики полупроводников, Институт органической химии, Институт автоматики и электрометрии. В настоящее время эта работа успешно завершена [124, 125]. Разработанный макет позволяет синтезировать до 12 тыс. олигонуклеотидов в одном эксперименте. Полученный прибор открывает возможность проведения исследований с использованием таргетного секвенирования и развивать синтетическую биологию в нашей стране на мировом уровне.
Написание обзора не потребовало специального финансирования.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.