ЖУРНАЛ ОБЩЕЙ ХИМИИ, 2019, том 89, № 10, с. 1496-1503
УДК 547.241
СИНТЕЗ ПРОИЗВОДНЫХ
ТЕТРАДЕКАПЕНТАЕНОВОЙ КИСЛОТЫ
© 2019 г. А. О. Колодяжная, О. И. Колодяжный*
Институт биоорганической химии и нефтехимии Национальной академии наук Украины имени В. П. Кухаря,
ул. Мурманская 1, Киев, 02094 Украина
*e-mail: olegkol321@gmail.com
Поступило в Редакцию 4 мая 2019 г.
После доработки 4 мая 2019 г.
Принято к печати 12 мая 2019 г.
Разработан 12-стадийный стереоселективный метод синтеза производных тетрадекапентаеновой кислоты
с использованием фосфорных реагентов. Ключевой стадией синтеза является Z-селективная реакция Вит-
тига между сорбальдегидом и трифенилфосфоний(6-метоксикарбонил)гексанилидом, а также реакция
Рамиреса-Кори-Фукса и перегруппировка Троста-Казмайера. Синтезированный (2E,4E,8Z,10E,12E)-N-
изобутилтетрадека-2,4,6,8,10,12-гексаенамид соответствует природному соединению, имеющему на-
звание γ-Sanshoöl.
Ключевые слова: реакция Виттига, реакция Хорнера-Эммонса, реакция Рамиреса-Кори-Фукса, пере-
группировка Троста-Казмайера, производные тетрадекапентаеновой кислоты
DOI: 10.1134/S0044460X19100032
Производные тетрадекапентаеновой кислоты
Америки. Данное растение встречается также в
представляют собой семейство полиненасыщен-
Австралии, Японии и Китае. В азиатских странах
ных амидов жирных кислот, обнаруженных в раз-
оно известно под названиями «японский (или сы-
личных видах тропических растений, в частности
чуаньский) перец» [1-4]. На сегодняшний день
рода Zanthoxylum bungeanum Maxim, Zanthoxylum
около 50 соединений класса полиненасыщенных
piperitum, Zanthoxylum ailanthoides и др., которые
амидов жирных кислот были выделены из различ-
привлекают внимание многих химиков. Это аро-
ных видов зантоксилума и родственных растений.
матическое дерево или кустарник произрастает
Особенно интересны амиды полиненасыщенных
в теплых регионах Африки, Южной и Северной
жирных кислот, которые проявляют разнообраз-
Схема 1.
CONHR
C(O)NHR
C(O)NHR
D-Sanshoöl
1, J-Sanshoöl
H-Sanshoöl
R = i-Bu, CH2CMe2OH
CONHR
CONHR
E-Sanshoöl
G-Sanshoöl
R = i-Bu, CH2C(OH)Me2, CH2C(OH)2Me.
1496
СИНТЕЗ ПРОИЗВОДНЫХ ТЕТР
АДЕКАПЕНТАЕНОВОЙ КИСЛОТЫ
1497
ную биологическую активность. Они обладают
Арбузова и Аппеля. В работе [8] использовали
антиоксидантной, антигельминтной, выраженной
реакцию Хорнера, протекающую с образованием
антипролиферативной и апоптозной активностью
двойной связи C4=C5 E- и Z-конфигурации [7].
[5, 6]. Особенно интересны представители ряда
Представляет интерес разработка синтеза сое-
природных соединений Sanshoöl, которых извест-
динения 1, который позволил бы получить стере-
но несколько десятков. Было обнаружено, что
охимически чистый продукт, содержащий Z,E-2,4-
γ-Sanshoöl 1 ингибирует ацил-КоА-холестерин-α-
диеновую группу [9, 10]. Для получения такого со-
цилтрансферазу человека. Некоторые представи-
единения, как γ-Sanshoöl, можно использовать сте-
тели ряда Sanshoöl представлены на схеме 1 [7].
реоселективную реакцию Виттига. Нами была по-
Несмотря на интересные фармакологические
ставлена цель получить диеновый фрагмент этого
характеристики, нестабильность соединений этой
соединения с помощью реакции Рамиреса-Кори-
группы (изомеризация, окисление, полимериза-
Фукса [11, 12], в результате которой должна обра-
ция и/или фотодеградация) из-за присутствия со-
зоваться алкиновая группа с последующей ее пе-
пряженной триеновой структуры затрудняет их
регруппировкой в диен [3, 13]. Реакция Рамиреса-
получение синтетическими методами. Особые
Кори-Фукса используется для превращения аль-
трудности представляет достижение высокой се-
дегида в алкин. На первой стадии при взаимодей-
лективности в образовании Z- и E-конфигураций
ствии трифенилфосфина с тетрабромметаном об-
кратных углерод-углеродных связей. Известны
разуется дибромметиленфосфоран, который далее
несколько синтетических подходов к получению
по реакции Виттига с альдегидом превращается
тетрадекапентаеноатов. Наиболее часто на клю-
в 1,1-дибромолефин. На второй стадии реакцией
Фрича-Буттенберга-Вихелля (превращение гало-
чевой стадии синтезов для построения 2,4-ди-
еновой группы используется реакция Хорнера
генэтиленов в ацетилены под действием сильных
оснований) дибромолефин превращали в алкин
или реакция Вудворта-Эммонса [5-10]. Описано
[13]. По сути, реакция Рамиреса-Кори-Фукса
несколько синтетических подходов с использова-
представляет собой реакцию Виттига, объединен-
нием реакции Хорнера для образования двойной
ную с реакцией Фрича-Буттенберга-Вихелля, в
связи C2-C3 [2-9]. Кромби и Фишер [5, 6] исполь-
результате которой 1,1-дибромалкен под действи-
зовали в синтезе тетрадекапентаеноатов МЕМ-
ем бутиллития (или ацетата палладия, трет-бу-
защищенный 4-пентин-1-ол, который был пре-
токсида калия) в толуоле превращается в соответ-
вращен в две стадии в МЕМ-защищенный (Е)-6-
ствующий алкин. Далее в результате обработки
гидроксигекс-2-еналь посредством реакции с эти-
образовавшегося алкина трифенилфосфином по
лортоформиатом в присутствии реагента Гриньяра
методу Троста-Казмайера [14] осуществляется пе-
c последующим стереоселективным восстанов-
регруппировка алкина в 1,3-диен с образованием
лением тройной связи и рН-контролируемой де-
тетрадекапентаеновой кислоты.
ацетализацией. Конденсация Вудворта-Эммонса
и реакция Виттига с (2E,4E)-гекса-2,4-диеналем
Основанный на этой стратегии
12-стадий-
приводит к образованию соответствующего тетра-
ный метод синтеза тетрадекапентаеновой кисло-
декапентаеноата [5-8]. Стратегия этих синтезов
ты представлен на схеме 1. Вначале гексадиенол
заключалась в образовании двойных связей C2=C3,
2 окисляли комплексом пиридина с триоксидом
C4=C5 и C8=C9 посредством стереоселективного
серы по методу Парик-Дёринга [15, 16] с обра-
гидрирования, реакции Вудворта-Эммонса и ре-
зованием сорбинового альдегида 3. Второй ком-
акции Виттига на ключевых стадиях [7, 8]. Авторы
понент реакции - фосфониевую соль 6 - полу-
исследовали образование E-геометрии двойной
чали из 6-бромгексановой кислоты 4 и эфира 5.
связи C8=C9, доступность основных синтетиче-
Фосфониевую соль 6 затем использовали в реак-
ции Виттига с альдегидом 3 (схема 2).
ских блоков и возможность получения модифици-
рованных аналогов. В работе [9] для построения
Наиболее важной задачей синтеза являлось
додекапентаенового скелета этого типа соедине-
достижение высокой степени Z-селективности на
ний использовались реакции Вудворта-Эммонса,
стадии образования соединения 7, имеющего си-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 10 2019
1498
КОЛОДЯЖНАЯ, КОЛОДЯЖНЫЙ
Схема 2.
Py·SO3
OH
O
Me2SO, Et3N
2
3
PPh3
-
Br
CO2H
MeOH Br
CO2Me
CO2Me] Br
[Ph3P+
4
5
6
NaN(SiMe3)2
3
LiAlH4
Ph3P
CO2Me
OH
C6H
6
CO2Me
7
8
Ph3P + CBr4
Py·SO3
Ph3P=CBr2
Br
(1) BuLi
O
Me2SO, Et3N
(2) H2O
Br
H
9
10
11
BuLi
NaOH
Ph3P, ɬɨɥɭɨɥ, '
ClCO2Me
MeOH
PhOH
CO2H
Me
CO2
CO2Me
12
13
14
RNH2
HATU
CONHR
15, 16
R = i-Bu (15), Bu (16).
стему Z,E-кратных связей. С этой целью нам уда-
нии реакции Виттига в отсутствие солей металлов
лось разработать Z-селективную реакцию Виттига
удается существенно повысить Z-селективность
без использования солей металлов. В результате
реакции [17]. При непосредственном взаимодей-
Z-селективной реакции Виттига фосфониевой
ствии фосфониевых солей с альдегидами в при-
соли 6 с сорбиновым альдегидом 3 был полу-
сутствии соответствующих оснований, например
чен метил (6Z,8E,10E)-додека-6,8,10-триеноат
7
бутиллития, образуются металлированные по
(схема 2). Сначала по методу Бестмана из фосфони-
α-углероду фосфониевые соли, которые реагиру-
евой соли 6 реакцией с гексаметилдисилазаном на-
ют с альдегидами по типу Е-селективной реакции
трия генерировали Р-илид. Таким образом удалось
Виттига или дают смесь E- и Z-алкенов. Например,
получить илид, не закомплексованный с галоге-
в результате реакции фосфониевой соли 6 с сор-
нидом металла, который затем вводили в реакцию
биновым альдегидом в присутствии карбоната це-
Виттига с сорбиновым альдегидом. При проведе-
зия была получена с низким выходом смесь Z- и
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 10 2019
СИНТЕЗ ПРОИЗВОДНЫХ ТЕТР
АДЕКАПЕНТАЕНОВОЙ КИСЛОТЫ
1499
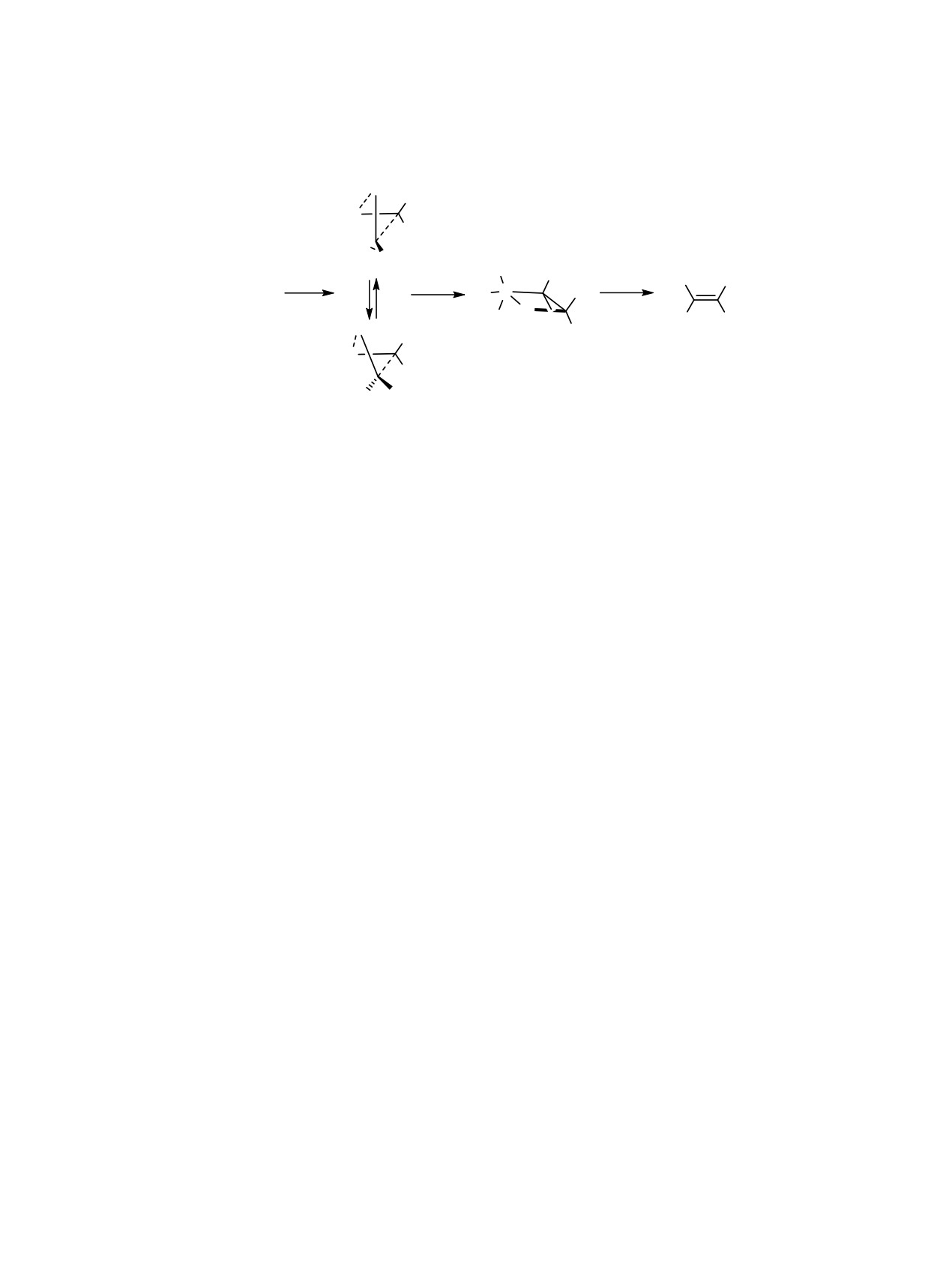
Схема 3.
O
R1
Ph3P
H
Ph3P=CHR1
H R2
Ph
R1
R1
R2
+
Ph
P
R2
O
Ph3PO
Ph
R2CH=O
H
H H
O
H
R1
Ph3P
H
H R2
R1 = (CH2)5CO2Me; R2 = CH3CH=CH-CH=C.
E-стереомеров метил-(8E,10E)-додека-6,8,10-три-
трифенилфосфином и четырехбромистым углеро-
еноата
7
[18]. Z-Стереоселективность реакции
дом для получения дибромолефина 10, который
Виттига объясняется синхронным [2+2]-цикло-
затем превращали в алкиноат 11 [3] под действи-
присоединением С=О группы к илидной связи
ем бутиллития. В результате реакции соединения
Р=С. В результате синхронного [2+2]-циклопри-
11 с бутиллитием и метилхлорформиатом удалось
соединения илида к карбонильной группе образу-
заместить атом водорода концевой СН-алкиновой
ется оксафосфетан, имеющий цис-конфигурацию.
группы метоксикарбонильной группой. На следу-
Этот тип трансформации достигается, если сбли-
ющей стадии синтеза осуществляли перегруппи-
жение реагентов осуществляется в соответствии
ровку алкина 12 в пентаен 13 по методу Троста-
с правилами Вудворда-Хоффмана и приводит к
Казмайера при нагревании алкина в толуоле с три-
минимальному стерическому неблагоприятному
фенилфосфином и фенолом с образованием тетра-
отталкиванию заместителей R1 и R2 (схема 3). На
декапентаената 13 с высокими стереоселективно-
втором этапе реакции происходит син-элиминиро-
стью и выходом. Дальнейший гидролиз эфира 13
вание, которое приводит к образованию олефина
гидроокисью натрия в метаноле приводил к обра-
Z-конфигурации. Таким образом, осуществляется
зованию 2E,4E,8Z,10E,12E-тетрадекапентаеновой
синхронное циклоприсоединение с четырехцен-
кислоты 14 с высокой стереохимической чистотой.
тровым переходным состоянием, в котором сте-
На последней стадии синтеза амидирование кис-
рические факторы и гибридизация атома фосфора
лоты 14 такими первичными аминами, как изобу-
благоприятствуют Z-стереоселективности реак-
тиламин или н-бутиламин, в присутствии реагента
ции [17].
Карпино (HАTU) [19] приводило к образованию
Реакция Виттига в присутствии карбоната це-
амидов 15 и 16 с выходами 30 и 32%.
зия является асинхронным циклоприсоединением
Строение, стереохимическая гомогенность и чи-
по связям P=C и C=O вследствие того, что с со-
стота соединений 15 и 16 подтверждены данными
рбиновым альдегидом реагирует комплекс фос-
спектроскопии ЯМР и ГЖХ-масс-спектрометрии.
фониевой соли с цезием, что благоприятствует
В спектрах ЯМР 1Н и 13С обнаруживаются сигна-
Е-стереоселективности реакции Виттига. В ре-
лы всех групп, входящих в молекулу соединений
зультате образуется смесь Z- и E-алкенов.
15 и 16 соответствующей интенсивности и муль-
На следующем этапе синтеза соединение 7
типлетности. В частности, сигналы протонов крат-
восстанавливали литийалюмогидридом до спир-
ных С=С связей находятся в области 5-6 м. д., а
та 8, который окисляли по методу Парик-Дёринга
сигналы атомов углерода - в области 120-130 м. д.
c образованием альдегида 9 с высоким выходом
В спектрах ЯМР 13С обнаруживается также сигнал
(схема 2). Далее альдегид 9 вводили в реакцию с
группы С=О при ~167 м. д. Спектральные и фи-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 10 2019
1500
КОЛОДЯЖНАЯ, КОЛОДЯЖНЫЙ
зико-химические характеристики амида 15 полно-
нилфосфина в 500 мл хлороформа. Реакционную
стью соответствуют характеристикам природного
смесь кипятили при перемешивании в течение
соединения γ-Sanshoöl, выделенного из растений
3 сут, затем охлаждали до комнатной температуры.
Zanthoxylum piperitum, Zanthoxylum ailanthoides
Растворитель упаривали при пониженном давле-
и др. [20]. Данные хромато-масс-спектрометрии
нии. К остатку добавляли 500 мл метил-трет-бу-
свидетельствуют о высокой чистоте полученных
тилового эфира. Полученную смесь перемешивали
соединений.
1 ч, после чего фильтровали и упаривали. Остаток
сушили в вакууме. Выход 83 г (70%). Спектр ЯМР
Таким образом, разработан стереоселектив-
1Н, δ, м. д.: 1.59 м (2H, CH2), 1.7 м (2H, CH2), 2.4 м
ный 12-стадийный метод синтеза производных
(2H, CH2), 3.6 c (3H, CH3O), 3.857 м (2H, CH2Br),
тетрадекапентаеновой кислоты с использовани-
7.3 м и 7.6 м (15H, C6H5). Спектр ЯМР 13С, δC, м. д.
ем фосфорных реагентов (Z-селективная реак-
(J, Гц): 22.7, 22.3, 22.7, 24.0 д (1JPC = 7.0 Гц), 30.0 д
ция Виттига, реакция Рамиреса-Кори-Фукса,
(3JPC = 8.0 Гц), 33.9, 52.5, 117.8 д (1JPC = 90.0 Гц),
перегруппировка Троста-Казмайера). Ключевой
130, 133 д (JPC = 10.0 Гц), 134.5, 174. Спектр ЯМР
стадией синтеза является Z-селективная реакция
31P: δР 25.0 м. д.
Виттига между синтетически доступными сор-
(2E,4E)-Гекса-2,4-диеналь
(3). К раствору
бальдегидом и (6-этокси-6-оксогексил)трифенил-
20 г (0.2 моль) (2E,4E)-гекса-2,4-диен-1-ола 2 в
фосфонийбромидом.
300 мл ДМСО добавляли 85 мл триэтиламина
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
(0.6 моль), затем при охлаждении и перемеши-
вании небольшими порциями добавляли 97.3 г
В работе использовали реактивы, силикагель,
(0.6 моль) комплекса Py.SO3. Полученную смесь
пластинки ТСХ (Polіgraм SIL G/UV254) фирм
перемешивали при комнатной температуре 3 ч,
«Fluka» и «Мerck».
затем выливали в воду со льдом и экстрагировали
Спектры ЯМР регистрировали на спектроме-
метил-трет-бутиловым эфиром. Объединенные
тре Bruker Avance DRX 400 или 500. В качестве
органические экстракты несколько раз промывали
растворителя использовали CDCl3. Данные ГЖХ-
водой. Растворитель упаривали, остаток перегоня-
МС регистрировали с помощью прибора ВЭЖХ
ли. Выход 3 15 г (84%), т. кип. 60°С (20 мм рт. ст.).
Agilent 1100, оснащенного диодно-матричным и
Спектр ЯМР 1Н, δ, м. д. (J, Гц): 1.89 c (3H, CH3),
масс-селективным детектором Agilent LC/MSD SL.
6.07 м (1H, CH), 6.29 м (1H, CH), 7.02 м (1H, CH),
7.26 м (1H, CH), 9.52 д (1H, CHO, J = 8.0).
Метил-6-бромгексаноат (5). К 300 мл метано-
Метил-(8E,10E)-додека-6,8,10-триеноат
(7).
ла при охлаждении и перемешивании по каплям
а. К раствору 73 г (0.16 моль) (6-этокси-6-ок-
добавляли 30 мл (0.384 моль) хлористого тионила,
согексил)трифенилфосфонийбромида
6 и
15 г
затем к полученной смеси по каплям при переме-
(0.16 моль) (2E,4E)-гекса-2,4-диеналя 3 в 1000 мл
шивании добавляли 50 г (0.256 моль) 6-бромгекса-
хлористого метилена добавляли 200 г (0.62 моль)
новой кислоты 4. Реакционную смесь кипятили 3 ч
карбоната цезия. Полученную смесь кипятили
при перемешивании. После удаления растворите-
24 ч, затем добавляли целит и перемешивали еще
ля получили вязкую жидкость, которую использо-
10 мин при комнатной температуре. Смесь филь-
вали дальше без дополнительной очистки. Выход
тровали, затем упаривали. К остатку добавляли
53.5 г (100%). Спектр ЯМР 1Н), δ, м. д.: 1.45 м (2H,
100 мл гексана для высаживания трифенилфос-
CH2), 1.64 м (2H, CH2), 1.77 м (1H, CH2), 1.86 м
финоксида. Осадок отфильтровывали, фильтрат
(1H, CH2), 2.31 м (2H, CH2), 3.39 (1H, CH2Br), 3.52
упаривали. Остаток хроматографировали (этила-
м (1H, CH2Br), 3.65 c (3H, МeO).
цетат-гексан, 10:90). Выход 7 г (21%), желтоватая
(6-Meтокси-6-оксогексил)трифенилфос-
жидкость, смесь Z- и E-стереомеров в соотноше-
фонийбромид (6). К раствору 53.5 г (0.26 моль)
нии 3:1 [18].
метил-6-бромгексаноата 5 в 500 мл хлорофор-
б. Из 73 г (0.16 моль) (6-этокси-6-оксогексил)-
ма прибавляли раствор 67.1 г (0.26 моль) трифе-
трифенилфосфонийбромида 6 в 500 мл бензола и
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 10 2019
СИНТЕЗ ПРОИЗВОДНЫХ ТЕТР
АДЕКАПЕНТАЕНОВОЙ КИСЛОТЫ
1501
0.2 моль гексаметилсилазана лития генерирова-
метилен-этилацетат-гексан, 20:5:75). Выход 4 г
ли соответствующий илид по методу Бестманна
(80%). Спектр ЯМР 1Н, δ, м. д. (J, Гц): 1.64 м (2H,
[21, 22]. К реакционной смеси прибавляли 15 г
СH2), 1.77 м (3H, СH3), 2.22 м (2H, СH2), 2.30 м
(0.16 моль) альдегида 3. Смесь перемешивали в
(2H, СН2), 5.40 д. т (1H, CH=C, 3JHH = 11.0, 3JHH =
течение ночи при комнатной температуре, после
8.0), 5.73 д. к (1H, CH=C, 3JHH = 14.0, 3JHH = 8.0),
чего фильтровали и упаривали. К остатку добавля-
6.05 м (3H, CH=C), 6.31 м (1H, CH=C), 9.76 с (1H,
ли 100 мл гексана для высаживания трифенилфос-
CHO). Спектр ЯМР 13С, δC, м. д.: 18.5, 22.0, 27.5,
финоксида. Осадок отфильтровывали, фильтрат
28.2, 43.7, 126.0, 127.6, 131.0, 131.8, 132.2, 132.6,
упаривали. Остаток хроматографировали (этила-
202.4. Найдено, %: C 81.05; H 10.38. C12H18O.
цетат-гексан, 10:90). Выход 22 г (65%), желтова-
Вычислено, %: C 80.85; H 10.18.
тая жидкость. Спектр ЯМР 1Н, δ, м. д.: 1.42 м (2H,
(9E,11E)-1,1-Дибромтридека-1,7,9,11-тетраен
CH2), 1.65 (2H, CH2), 1.77 м (3H, CH3), 2.21 м (2H,
(10). К раствору 17.8 г (0.054 моль) четырехбро-
CH2), 2.32 м (2H, CH2), 3.67 с (3H, CH3), 5.64 м
мистого углерода в 100 мл хлористого метилена
(1H, CH), 5.72 м (1H, CH), 6.02 м (2H, CH + CH),
при охлаждении до -70°С по каплям добавляли
6.19 м (1H, CH), 6.35 м (1H, CH), 6.46 м (1H, CH).
раствор 26.23 г (0.1 моль) трифенилфосфина в
Спектр ЯМР 13С, δC, м. д.: 18.0, 18.4, 24.5, 28.0,
100 мл хлористого метилена. Смесь перемешива-
29.1, 34.0, 60.0, 126.0, 128.6, 130.7, 131.2, 132.0,
ли 30 мин, затем повышали температуру до 0°С
133.0, 173.9. Найдено, %: C 75.6; H 9.8. C13H20O2.
и добавляли раствор 4 г (0.022 моль) (8E,10E)-
Вычислено, %: C 74.96; H 9.68.
додека-6,8,10-триеналя 9 в 15 мл хлористого ме-
(8E,10E)-Додека-6,8,10-триен-1-ол (8). К су-
тилена. Полученную смесь перемешивали еще 1 ч
спензии 2.55 г (0.067 моль) литийалюмогидрида в
при комнатной температуре, затем добавляли гек-
100 мл ТГФ при перемешивании и охлаждении до
сан для высаживания трифенилфосфиноксида.
0°С по каплям прибавляли раствор 7 г (0.034 моль)
Раствор декантировали с осадка. Осадок раство-
метилдодека-6,8,10-триеноата 7 в 100 мл ТГФ.
ряли в хлористом метилене, затем добавляли гек-
Полученную смесь перемешивали 1 ч при комнат-
сан с последующей декантацией. Объединенные
ной температуре, затем добавляли воду. Осадок
экстракты упаривали в вакууме, остаток хромато-
отфильтровывали, фильтрат упаривали. Остаток
графировали (хлористый метилен-гексан, 20:80).
хроматографировали (хлористый метилен-этила-
Выход 3.6 г (48%). Спектр ЯМР 1Н, δ, м. д. (J, Гц):
цетат-гексан, 20:5:75). Выход 5 г (82%), желтая
1.24 д. д (2H, CH2, 3JНН = 4.0, 3JНН = 4.0), 1.536
жидкость. Спектр ЯМР 1Н, δ, м. д. (J, Гц): 1.4 м
м (2H, CH2), 1.75 д (3H, CH3, 3JНН = 8.0), 2.07 м
(4H, CH2), 1.5 м (4H, CH2), 1.77 м (3H, CH3), 2.2 м
(2H, СН2), 2.2 м (2H, СН2), 5.38 м (1H, CH=C),
(2H, CH2), 3.64 т (2H, CH2O, J = 8.0), 5.39 м (1H,
5.70 м (1H, CH=C), 6.08 м (2H, CH=C), 6,37 м (1H,
CH), 5.69 м (2H, CH), 6.00 м (1H, CH), 6.12 м (1H,
CH=C). Найдено, %: C 46.28; H 5.25; Br 48.21. C13
CH), 6.34 м (1H, CH).
H18 Br2. Вычислено, %: C 46.74; H 5.43. Br 47.83.
(8E,10E)-Додека-6,8,10-триеналь (9). К раство-
Метилтетрадека-8,10,12-триен-2-иноат
(12).
ру 5.0 г (0.028 моль) (8E,10E)-додека-6,8,10-триен-
К раствору 3.6 г (0.0108 моль) (9E,11E)- 1,1-дибро-
1-ола 8 в 100 мл абсолютного диметилсульфокси-
мотридека-1,7,9,11-тетраена 10 в 100 мл ТГФ при
да добавляли 11.5 мл (0.083 моль) триэтиламина,
перемешивании и охлаждении до -78°С прибав-
затем при охлаждении и перемешивании неболь-
ляли 9.5 мл 2.5 М. раствора бутиллития в гексане
шими порциями добавляли 13.25 г (0.083 моль)
(0.024 моль). Реакционную смесь перемешивали
комплекса Py.SO3. Полученную смесь переме-
1 ч при -78°С, затем 1 ч при комнатной темпера-
шивали при комнатной температуре 3 ч, затем
туре. После добавления 0.65 мл (8.4 ммоль) ме-
выливали в воду со льдом и экстрагировали ме-
тилхлорформиата при -78°С смесь постепенно
тил-трет-бутиловым эфиром. Объединенные
нагревали до комнатной температуры и переме-
экстракты трижды промывали водой и насыщен-
шивали 16 ч, затем обрабатывали насыщенным
ным раствором хлористого натрия. Полученный
раствором хлористого аммония и экстрагировали
раствор концентрировали при пониженном дав-
диэтиловым эфиром (3×50 мл). Объединенные
лении, остаток хроматографировали (хлористый
экстракты сушили Na2SO4 затем упаривали при
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 10 2019
1502
КОЛОДЯЖНАЯ, КОЛОДЯЖНЫЙ
пониженном давлении. Остаток хроматографиро-
126.7, 127.2, 130.7, 131.3, 1321, 139.0, 148.4, 170.4.
вали (этилацетат-гексан, 5:95). Выход 2.2 г (83%).
(2E,4E,8Z,10E,12E)-N-Изобутилтетрадека-
Спектр ЯМР 1Н, δ, м. д.: 1.24 м (2H, CH2), 1.42 м
2,4,6,8,10,12-гексаенамид (γ-Sanshoöl)
(15). К
(2H,CH2), 1.54 м (2H, CH2), 1.75c (3H, CH), 2.07 м
раствору 2E,4E,8Z,10E,12E-тетрадекапентаеновой
(2H, CH2), 2.2 м (2H, CH2), 5.38 м (1H, CH), 5.70 м
кислоты 0.28 г (0.00128 моль) в смеси 2 мл аце-
(1H, CH), 6.08 м (1H, CH), 6.37 м (1H, CH). Спектр
тонитрила и 1 мл CHCl3 при перемешивании в
ЯМР 13С, δC, м. д.: 17. 8, 18.5, 27.5, 27.9, 29.8, 52.3,
атмосфере азота при комнатной температуре до-
72.8, 89.9, 128.6, 129.3, 130.2, 131.2, 132.2, 154.2.
бавляли 0.2 г (0.002 моль) изобутиламина, 0.35 мл
(2E,4E,8Z,10E,12E)-Метилтетрадекапен-
(0.0025 моль) триэтиламина и 0.62 г (0.0019 моль)
таноат (13). К раствору 2.2 г (0.001 моль) метилте-
HАTU. Смесь перемешивали 30 мин, затем разбав-
традека-8,10,12-триен-2-иноата 12 в 30 мл толуола
ляли 50 мл диэтилового эфира и промывали после-
добавляли 2.48 г (0.01 моль) трифенилфосфина и
довательно насыщенным раствором хлорида на-
0.89 г (5.0 ммоль) фенола. Смесь перемешивали при
трия, 1 н. раствором HCl, водой и 5%-ным раство-
50°С в атмосфере азота в течение 16 ч, затем кон-
ром NaHCO3. Органический слой сушили Na2SO4,
центрировали при пониженном давлении. Остаток
фильтровали и упаривали в вакууме. Выход 0.15 г
хроматографировали (хлористый метилен-гексан,
(43%), бесцветное твердое вещество [20]. Спектр
10:90, затем этилацетат-гексан, 10:90). Выход 2.2 г
ЯМР 1Н, δ, м. д. (J, Гц): 0.93 д (6H, CH3, 3JHH =
(100%), желтоватое масло [18]. Спектр ЯМР 1Н, δ,
7.0 Гц), 1.77 д (3H, CH3, 3JHH = 7.0), 1.7 м (1H, СН),
м. д.: 1.8 м (3Н, CH3), 2.05 м (2Н, CH2), 3.75 c (3H,
2.25 т (2H, СН2, 3JНН = 7.0), 2.30 т (2H, CH2, 3JНН =
OCH3), 5.4 м (1H, CH), 5.7 м (1H, CH), 5.8 м (1H,
7.0), 3.15 м (2H, СН2), 5.34 д. т (1H, CH=C, 3JНН =
CH), 6.2 м (1H, CH), 6.3 м (1H, CH), 6.4 м (1H, CH),
10.0, 3JНН = 8.0), 5.47 д. т (1H, CH=C, 3JHH 14.0,
6.5 м (1H, CH), 6.7 м (1H, CH). Спектр ЯМР 13С,
3JHH = 7.0), 6.45 д (1H, CH=C, 3JHH = 15.0), 6.06-
δC, м. д.: 18.4, 27.0, 33.1, 51.5, 119.2, 125.4, 128.9,
6.13 м (5H, CH=C), 6.29-6.33 м (1H, CH=C), 7.16
129.7, 129.8, 130.2, 131.9, 133.6, 143.7, 145.2, 167.8.
д. д (1H, CH=C, 3JHH = 15.0, 3JHH = 11.0). Спектр
Найдено, %: 78.03; H 9.12. C16H22O2. Вычислено,
ЯМР 13С, δC, м. д.: 18.43, 20.2, 27.2, 28.7, 33.0,
%: 78.01; H 9.00.
47.0, 122.4, 125.4, 128.9, 129.5, 130.0, 130.1, 132.0,
2E,4E,8Z,10E,12E-Тетрадекапентаеновая
133.4, 141.0, 141.8, 166.5. Масс-спектр, m/z: 274.4
кислота (14). К раствору 2.2 г (9.5 ммоль) метил-
[М + H]+.
тетрадекапентаноата в 30 мл метанола добавляли
(2E,4E,8Z,10E,12E)-N-Бутилтетрадека-
1.9 г (0.047 моль) гидроокиси натрия при пере-
2,4,6,8,10,12-гексаенамид (16) получали анало-
мешивании. Смесь перемешивали при комнатной
гично. Выход 50%. Спектр ЯМР 1Н, δ, м. д. (J, Гц):
температуре в течение 2 ч. Метанол упаривали при
0.84 т (3H, CH3, 3JHH = 7.0), 1.3 м (4H, CH2), 1.84 к
пониженном давлении, остаток растворяли в 30 мл
(3H, CH3, 3JHH = 7.0), 2.1 м (2H, СН2, 3JHH = 7.0),
воды. Продукт реакции трижды экстрагировали
2.30 т (2H, СН2, 3JHH = 7.0), 3.15 т (2H, CH2N, 3JНН =
диэтиловым эфиром, водный слой подкисляли 1 н.
7.0), 5.34 д. т (1H, СH=С, 3JНН = 10, 3JНН = 7.5), 5.47
раствором HCl до pH = 1. Объединенные экстрак-
д. т (1H, CH=C, 3JНН = 14.0, 3JHH = 7.0), 6.45 д (1H,
ты промывали насыщенным раствором хлористо-
CH=C, 3JНН = 15.0), 6.06-6.13 м (5H, СН=С), 6.29-
го натрия, сушили Na2SO4 и концентрировали при
6.33 м (1H, CH=C), 7.16 д. д (1H, CH=C, 3JНН =
пониженном давлении. Остаток перекристаллизо-
15.0, 3JНН = 11.0). Спектр ЯМР 13С, δC, м. д.: 14.23,
вывали из смеси CHCl3-гексан. Получили стерео-
18.5, 20.2, 28.7, 31.5, 32.2, 40.0, 43.0, 122.2, 125.2,
химически чистую кислоту 14, при нагревании не-
129.0, 129.9, 130.5, 130.9, 132.0, 133.6, 141.2, 142.0,
устойчива. Выход 0.55 г (25%). Спектр ЯМР 1Н, δ,
м. д. (J, Гц): 1.78 д (3H, CH3, 3JНН = 7.0 Гц), 2.30 м
167.0. Масс-спектр, m/z: 274.4 [М + H]+.
(4H, CH2), 5.34 м (1H, HC=C), 5.74 м (2H, СН=С),
5.77 м (1H, CH=C), 6.08 т (1H, СН=С, 3JНН = 11.0),
КОНФЛИКТ ИНТЕРЕСОВ
6.20 м (4H, СН=С), 6.30 м (1H, СН=С), 7.33 д. д
(1H, СН=С, 3JНН = 16.0, 4JНН = 10). Спектр ЯМР
Авторы заявляют об отсутствии конфликта
13С, δC, м. д.: 18.5, 27.1, 32.2, 120.8, 124.4, 125.9,
интересов.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 10 2019
СИНТЕЗ ПРОИЗВОДНЫХ ТЕТР
АДЕКАПЕНТАЕНОВОЙ КИСЛОТЫ
1503
СПИСОК ЛИТЕРАТУРЫ
11. Corey E.J., Fuchs P.L. // Tetrahedron Lett. 1972. Vol. 13.
P. 3769. doi10.1016/S0040-4039(01)94157-7
1. Bader M., Stark T.D., Dawid C., Lösch S., Hofmann T. //
12. Heravi M.M., Asadi S., Nazari N., Lashkariani B.M. //
J. Agric. Food Chem. 2014. Vol. 62. N 12. P. 2479. doi
Curr. Org. Chem. 2015. Vol. 19. N 22. P. 2196. doi 10.
10.1021/jf500399w
2174/1385272819666150619174010
2. Wang Y., Li C.-H., Luo B., Sun Y.N., Kim Y.H., Wei A.-Z.,
13. Rezaei H., Yamanoi S., Chemla F., Normant J.F. // Org.
Gao J.-M. // Molecules. 2016. Vol. 21. N 10. P.1416.
Lett. 2000.Vol. 4. N 2. P. 419. doi 10.1021/ol991117z
doi10.3390/molecules21101416
14. Trost B.M., Kazmaier U. // J. Am. Chem. Soc. 1992.
3. Chrum J.J., Cullen D.J., Bowman L., Toy P.H. // Nat.
Vol. 114. N 20. P. 7933. doi 10.1021/ja00046a062
Prod. Rep. 2018. Vol. 35. N 1. P. 54. doi 10.1039/
15. Tidwell T.T. // Org. React. 1990. Vol. 39. P. 297. doi
c7np00044h
10.1002/0471264180.or039.03
4. You Y., Zhou M., Lu H., Shirima G.G., Cheng Y., Liu X. //
Food Sci. Biotechnol. 2015. Vol. 24. N 6. P. 2169. doi
16. Parikh J.R., Doering W.V.E. // J. Am. Chem. Soc. 1967.
10.1007/s10068-015-0289-3
Vol. 89. N 21. P. 5505. Doi 10.1021/ja00997a067
5. Crombie L., Fisher D. // Tetrahedron Lett. 1985. Vol. 26.
17. Kolodiazhnyi O.I. Phosphorus Ylides. Chemistry and
N 20. P. 2481. doi10.1016/S0040-4039(00)94859-7
Application in Organic Synthesis. Weinheim; New
York; Chichester: J. Wiley-VCH, 1999. 565 p.
6. Crombie L., Fisher D. // Tetrahedron Lett. 1985, Vol. 26.
N 20, 2477. doi10.1016/S0040-4039(00)94858-5
18. Xia X., Toy P.H. // Synlett. 2014. Vol. 25. N 19. P. 2787.
doi 10.1055/s-0034-1379215
7. Jang K.H., Chang Y.H., Kim D.-D., Oh K.-B., Oh U.,
Shin J. // Arch. Pharm. Res. 2008. Vol. 31. N 5. P. 569.
19. Carpino L.A. // J. Am. Chem. Soc. 1993. Vol. 115. N 10.
doi 10.1007/s12272-001-1194-5
P. 4397. doi 10.1021/ja00063a082
8. Aoki K., Igarashi Y., Nishimura H., Morishita I., Usui K. //
20. Yasuda I., Takeya K., Itokawa H. // Chem. Pharm. Bull.
Tetrahedron Lett. 2012. Vol. 53. N 45. P. 6000. doi
1981. Vol. 2. N 6. P. 1791. doi 10.1248/cpb.29.1791
10.1016/j.tetlet.2012.08.135
21. Cushman M., Casimiro-Garcia A., Williamson K.,
9. Mugnaini C., Corelli F. // Synthesis 2016. Vol. 48. N 13.
Rice W.G. // Bioorg. Med. Chem. Lett. 1998. Vol. 8. N
P. 2085. doi 10.1055/s-0035-1561580
2. P. 195. doi 10.1016/S0960-894X(97)10214-1
10. Kolodiazhna A., Kolodiazhnyi O. // Phosphorus,
22. Bestmann H.J., Stransky W., Vostrowsky O. // Chem.
Sulfur, Silicon, Relat. Elem. 2018. Vol. 193. N 11. doi
Ber. 1976. Bd 109. N 5. S. 1694 doi 10.1002/
10.1080/10426507.2018.1514404
cber.19761090513
Synthesis of Tetradecapentaenoic Acid Derivatives
A. O. Kolodyazhnaya and O. I. Kolodyazhny*
V.P. Kukhar Institute of Bioorganic Chemistry and Petrochemistry of the National Academy of Sciences of Ukraine,
ul. Murmanskaya 1, Kiev, 02094 Ukraine
*e-mail: olegkol321@gmail.com
Received May 4, 2019; revised May 4, 2019; accepted May 12, 2019
A 12-stage stereoselective method for the synthesis of tetradecapentaenoic acid derivatives using phosphoric
reagents was developed. The key step in the synthesis is the Z-selective Wittig reaction between sorbaldehyde
and triphenylphosphonium (6-methoxycarbonyl)hexanilide, as well as the Ramirez-Corey- Fuchs reaction and
the Trost-Kazmaier rearrangement. The synthesized (2E,4E,8Z,10E,12E)-N-isobutyltetradeca-2,4,6,8,10,12-
hexaenamide corresponds to a natural compound called γ-Sanshoöl.
Keywords: Wittig reaction, Horner-Emmons reaction, Ramirez-Corey-Fuchs reaction, Trost-Kazmaier
rearrangement, tetradecapentaenoic acid derivatives
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 10 2019