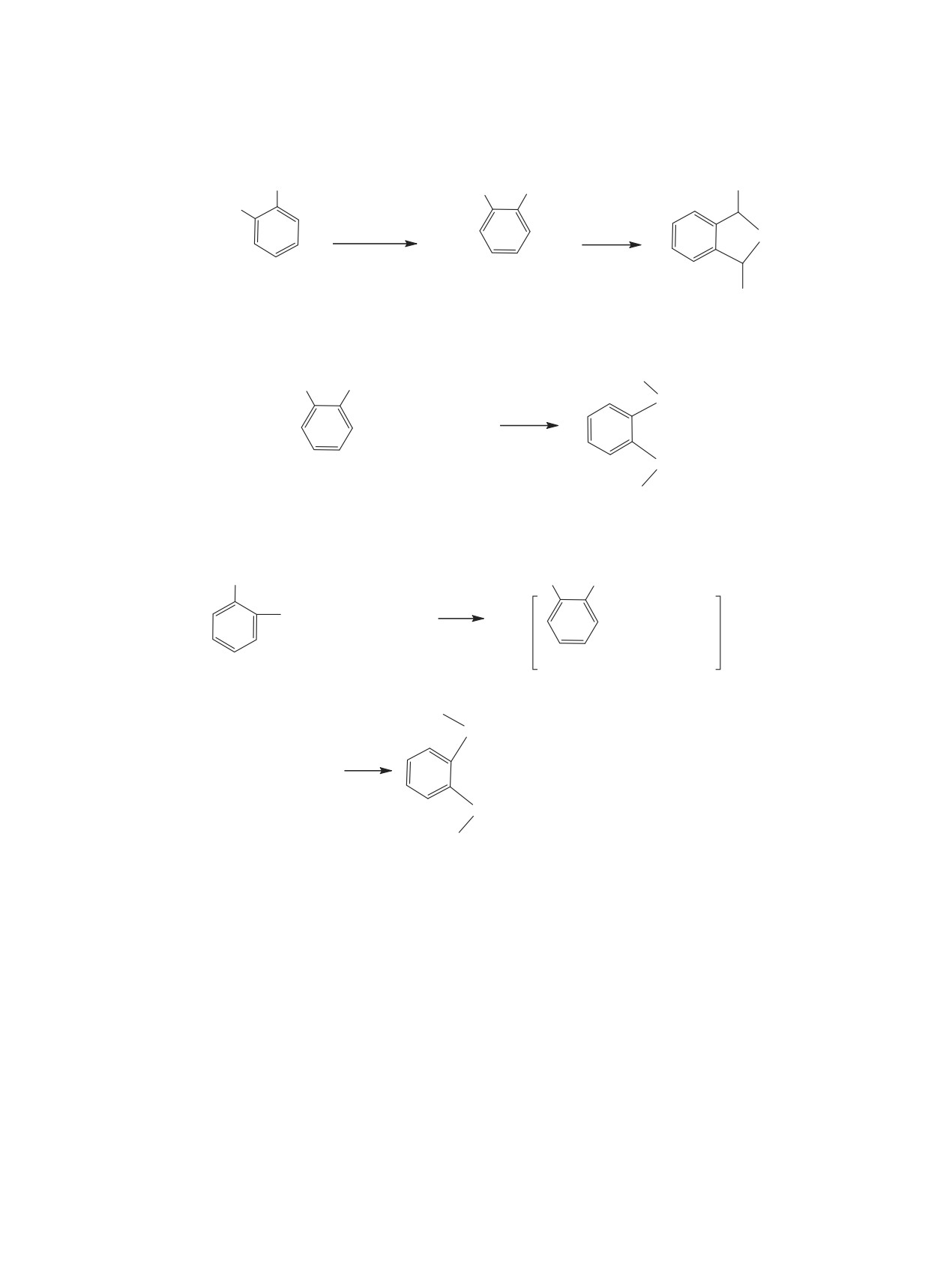ЖУРНАЛ ОБЩЕЙ ХИМИИ, 2019, том 89, № 11, с. 1651-1658
УДК 661.718;661.727;661.2
СИНТЕЗ ФТАЛЕВОГО АЛЬДЕГИДА
И ЕГО ДИАЦЕТАЛЕЙ
© 2019 г. Р. А. Хайруллин, М. Б. Газизов*, Ю. С. Кириллина, С. Ю. Иванова,
О. Д. Хайруллина, К. С. Газизова
Казанский национальный исследовательский технологический университет,
ул. К. Маркса 68, Казань, 420015 Россия
*e-mail: mukattisg@mail.ru
Поступило в Редакцию 22 апреля 2019 г.
После доработки 22 апреля 2019 г.
Принято к печати 25 апреля 2019 г.
Ациклический диацеталь фталевого альдегида без примеси циклического 1,3-дигидро-1,3-диметокси-
бензо[с]фурана образуется при взаимодействии 1,2-бис(дибромметил)бензола с триметилортоформиатом
в соотношении 1:6 при 90°С в присутствии 10 мол% ZnCl2, гидролиз которого приводит к фталевому
альдегиду без выделения HBr. Реакция фталевого альдегида с триметилортомуравьиным эфиром в при-
сутствии трифторуксусной кислоты протекает аномально с образованием его циклического диацеталя.
Проведено фосфорилирование ациклического диацеталя хлорфосфинами и последовательным действием
PCl3 и эфиром кислоты P(III).
Ключевые слова: фталевый альдегид, диацетали фталевого альдегида, 1,3-дигидро-1,3-диметоксибен-
зо[с]фуран, 1,2-бис(дибромметил)бензол, фосфорилирование
DOI: 10.1134/S0044460X19110027
Фталевые альдегиды проявляют высокую бак-
[7], муравьиной [8], фталевой [9], уксусной [8] и
терицидную активность и являются прекрасными
хлороводорода [10], а также гидроксида натрия [7, 10].
дезинфектантами [1]. Различные композиции на
Окислению подвергали: о-ксилол кислородом
их основе широко используются для обеззаражи-
в воде [11], в хлороформе в присутствии перхло-
вания помещений и стерилизации медицинского
рата 10-метил-9-фенил-10-акридиния [12]; нафта-
оборудования, особенно в эндоскопии. Поэтому
лин метапериодатом калия в присутствии аквапен-
является актуальной разработка новых методов
тахлорорутената(III) калия в воде, дихлорметане и
получения фталевых альдегидов из доступного
ацетонитриле [13], броматом калия в присутствии
сырья.
Ru-катализатора в CH2Cl2, H2O и MeCN и с уль-
Известные методы синтеза фталевого альдегида
тразвуковым облучением [14], периодатом натрия
1 основаны на трансформации 1,2-дизамещенных
в CH2Cl2, H2O и MeCN и с ультразвуковым облу-
бензолов с использованием различных типов реак-
чением [15], озоном в метаноле и бутилацетате
ций: гидролиза, окисления, снятия бис(гемдиаце-
[16], иодатом натрия в присутствии катализатора
татной) защиты и восстановления. Первоначально
Ru3(CO)12 в MeCN, H2O и CH2Cl2 [16]; 1,2-ди-
методы синтеза фталевых альдегидов были осно-
(гидроксиметил)бензол диоксидом марганца в
ваны на сернокислотном гидролизе (дымящей или
CH2Cl2 [17], нитратом железа [18], кислородом в
концентрированной серной кислотой) бис(дигало-
присутствии Ru(PPh3)3Cl2 в различных раствори-
генометил)бензолов, которые затем совершенство-
телях [19], перманганатом дипиридинсеребра в
вались способами нейтрализации выделяющегося
бензоле [20], диметилсульфоксидом и оксалилхло-
галогеноводорода [1-6]. Гидролиз ациклических
ридом в CH2Cl2 [21]; 1,2-ди(бромметил)бензол с
диацеталей фталевого альдегида водой осущест-
использованием ди(4-метоксифенил)селеноксида
вляли в присутствии различных кислот: серной
и гидрокарбоната натрия в ацетонитриле [22].
1651
1652
ХАЙРУЛЛИН и др.
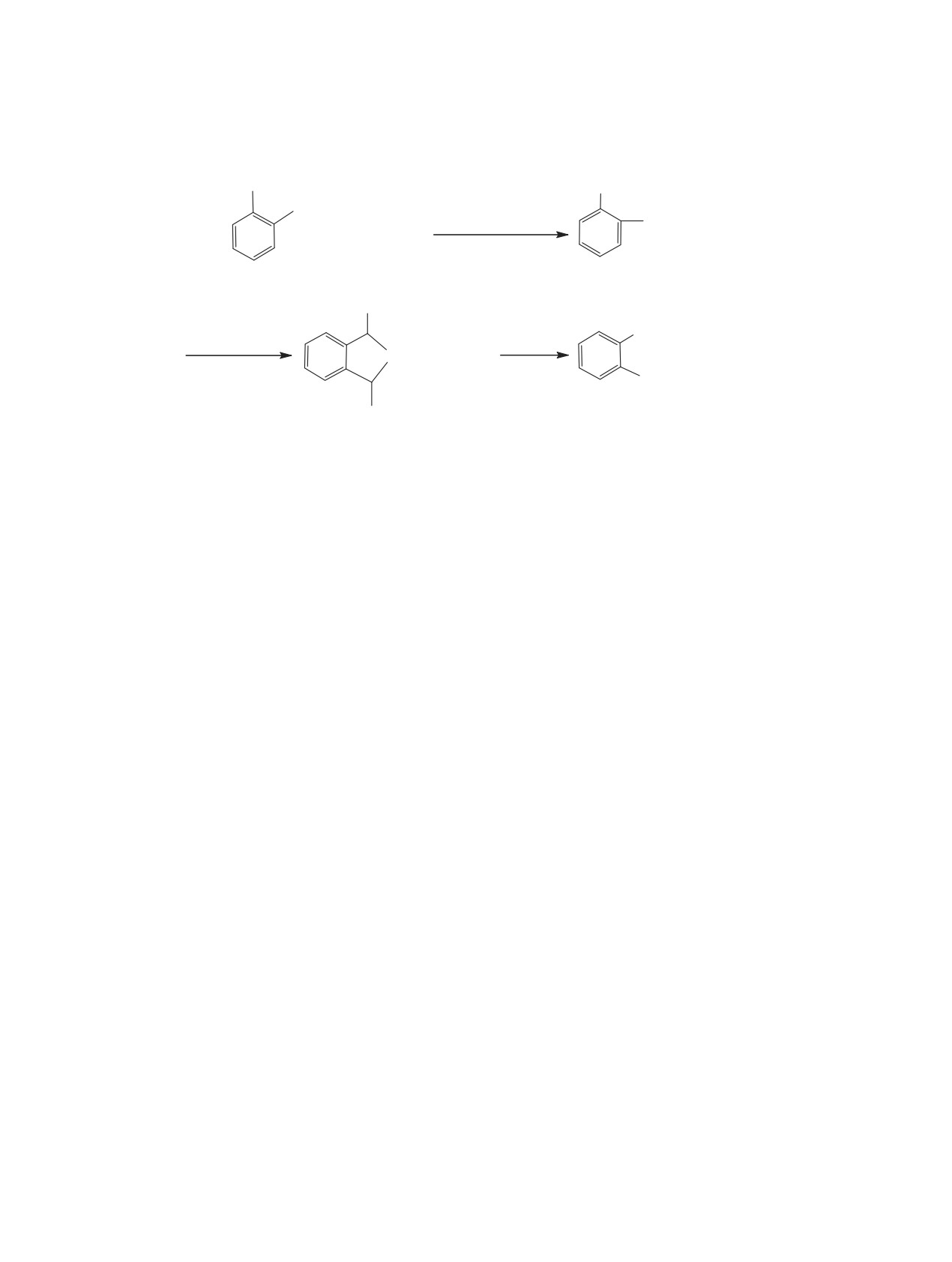
Схема 1.
CHBr2
CH(OMe)2
CHBr2
CH(OMe)
ZnCl
2
2
+
4 CH(OMe)3
4(HCOOMe + MeBr)
3
2
4
OCH3
CHO
H2O, CF3COOH
H2O, HCl
O
+ 2 MeOH
+ 2 MeOH
'
'
CHO
OCH
3
5
1
Снятие бис(гем-диацетатной) защиты осу-
комнатной температуре сначала образуется цикли-
ществляли нагреванием бис(диацетата) фталевого
ческий диацеталь фталевого альдегида - 1,3-диги-
альдегида с хлоратом поли(4-винилпиридиния)
дро-1,3-диметоксибензо[с]фуран 5, который при
[P(4-VPH)ClO4] в этаноле [23] и гидросульфатом
нагревании с подкисленной водой превращается в
[P(4-VPH)HSO4] в метаноле при ультразвуковoм
альдегид 1 (схема 1).
облучении [24]; выдерживанием бис(диацетата) и
Последняя стадия этой реакционной схемы
хлорида N-сульфонополи-(4-винилпиридиния) в
была реализована отдельно и фталевый альдегид
метаноле [25].
1 был получен с выходом 81%.
Восстановлению подвергали фталевые кис-
Из схемы 1 следует, что диацетали фталевого
лоты и их производные: взаимодействием самой
альдегида 3 и 5 являются промежуточными со-
кислоты с трет-гексил-s-бутоксибораном в ТГФ
единениями, поэтому является актуальной зада-
[26], c LiAlH4 [27]; ее дихлорангидрида с пента-
чей разработка их синтеза. Одним из успешных
координированным гидросиланом [28]; диамида с
методов синтеза ацеталей до сих пор остается
LiAlH4 [29]; динитрила с гидротрис(дибутилами-
реакция легкодоступных органических гем-ди-
но)алюминатом натрия в ТГФ [30].
галогенидов с алкоголятами щелочных металлов
Следует подчеркнуть, что гидролиз легкодо-
[31-37]. Происходит ди(дегалогеналкоксилиро-
ступного 1,2-бис(дибромметил)бензола 2 является
вание) арилдигалогенометанов или бензилиден-
одним из самых старых [1-6], но применяющихся
галогенидов. Часто во взаимодействие вводились
в настоящее время [7-10] способов синтеза фта-
гем-дихлориды [33-36] и значительно реже при-
левого альдегида. Однако данный метод имеет
менялись гем-дибромиды [37-39]. Основными не-
много недостатков, главным из которых является
достатками этого метода являются необходимость
выделение большого количества бромоводорода,
приготовления алкоголята щелочного металла и
корродирующего аппаратуру и ограничивающего
возможность трансформации имеющихся функци-
ее загрузку.
ональных групп под действием сильного основа-
Нами разработан новый способ получения фта-
ния - алкоксид-аниона.
левого альдегида 1 из 1,2-бис(дибромметил)бен-
Мы впервые использовали триалкилортофор-
зола 2, исключающий образование HBr. Сначала
миаты вместо алкоголятов для ди(дегалогеналкок-
тетрабромид
2 под действием триметилорто-
силирования) дигалогенометиларенов [40]. Нами
формиата 4 превращали в ациклический диаце-
был синтезирован ряд ацеталей функционально
таль 3, который при нагревании с подкисленной
замещенных бензальдегидов, в том числе диацета-
водой трансформировался во фталевый альдегид
ли терефталевого альдегида 6, из 1,4-бис(дибром-
1. Методом ЯМР 1Н нам удалось показать, что при
метил)бензола 7 взаимодействием последнего с
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 11 2019
СИНТЕЗ ФТАЛЕВОГО АЛЬДЕГИДА И ЕГО ДИАЦЕТАЛЕЙ
1653
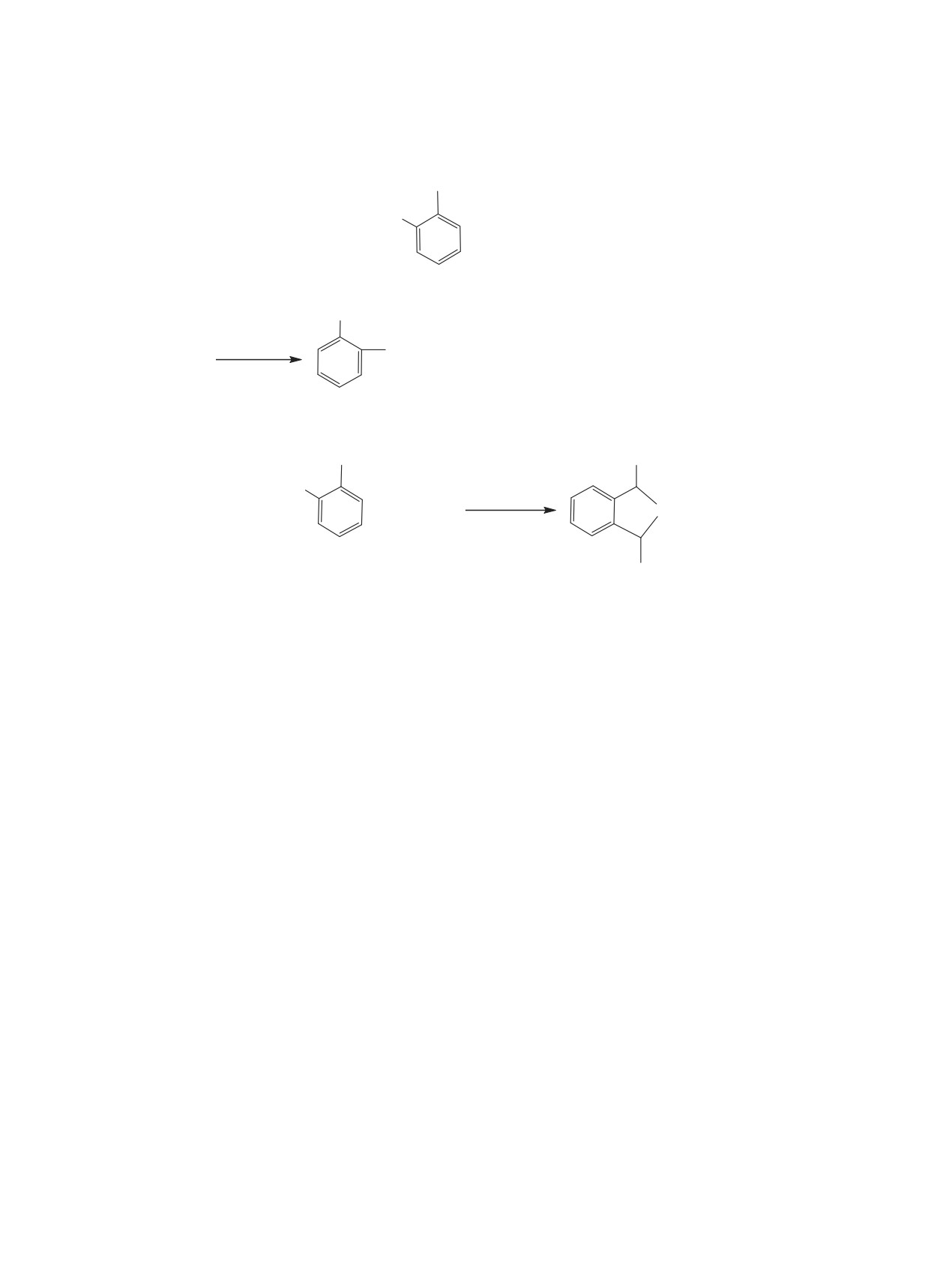
Схема 2.
CHBr2
Br2HC
+ 6CH(OMe)
3
2
4
CH(OMe)2
ZnCl2, 90°ɋ
CH(OMe)2
+ 4HCOOMe + 4MeBr + 2CH(OMe)
3
3
Схема 3.
CHO
OCH3
OHC
CF3COOH
+ CH(OMe)
O
3
OCH3
1
4
5
эфирами ортомуравьиной кислоты 4 при 50°С в те-
образование ациклических диацеталей фталевого
чение 4-5 ч в присутствии 10 мол% хлорида цинка
альдегида 3 с хорошим выходом (схема 2).
в качестве катализатора [41-42].
Таким образом, нами впервые был разрабо-
Соответствующие ациклические диацетали
тан способ получения ациклического диацета-
фталевого альдегида 3 не были описаны в литера-
ля фталевого альдегида 3 без примеси 1,3-диги-
туре, хотя были предприняты попытки их синтеза
дро-1,3-диалкоксибензо[с]фурана 5.
несколькими способами: взаимодействием фтале-
Известно, что диацетали терефталевого альде-
вого альдегида 1 с этанольным раствором триэти-
гида 6 получают взаимодействием самого альде-
лортоформиата (1:1) в присутствии хлорида алю-
гида с ортоэфирами 4 в присутствии кислот [43].
миния в качестве катализатора [43], ацетализацией
Мы впервые установили, что взаимодействие фта-
альдегида 1 метиловым спиртом, используя ката-
левого альдегида 1 с ортоэфирами 4 в присутствии
литические количества TiCl4 и в присутствии NH3
трифторуксусной кислоты в качестве катализатора
или Et3N [38], а также реакцией этого альдегида с
протекает аномально с образованием циклическо-
диметилсульфитом в метаноле [44]. Во всех случа-
го диацеталя 5 с хорошим выходом (схема 3).
ях происходило образование смеси ациклического
Соединение 5 было синтезировано нами так-
диацеталя 3 и 1,3-дигидро-1,3-диалкоксибензо[с]-
же из ациклического диацеталя 3: при взаимодей-
фурана 5.
ствии последнего с PCl3 происходило образование
Мы установили, что, в отличие от 1,4-бис(ди-
соответствующего ди-α-хлорэфира
7, гидролиз
бромметил)бензола 7, в вышеуказанных услови-
которого приводил к циклическому диацеталю 5
ях 1,2-бис(дибромметил)бензол 2 практически не
(схема 4).
вступает во взаимодействие с триметилортофор-
Строение промежуточного соединения 7 было
миатом, происходит распад последнего в метил-
подтверждено данными ЯМР 1Н, а также его ре-
формиат. Однако при 90°С и выше и 3-кратном
акцией с триметилфосфитом, в результате которой
избытке ортоэфира (соотношение 1:6) происходит
был получен дифосфонат 8 (схема 5).
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 11 2019
1654
ХАЙРУЛЛИН и др.
Схема 4.
CH(OMe)2
OCH3
Cl(MeO)HC
CH(OMe)Cl
(MeO)2HC
2PCl3
H2O
O
2MeOPCl2
HCl
OCH
3
3
7
5
Схема 5.
Cl(MeO)HC
CH(OMe)Cl
(MeO)2(O)P
CH(OMe)
+ 2 (MeO)3P
2MeCl
CH(OMe)
(MeO)2(O)P
7
8
Схема 6.
CH(OMe)2
Cl(MeO)HC
CH(OMe)Cl
+ 2R2PCl
CH(OMe)2
+ 2 R2POMe
3
9
7
11
R2(O)P
CH(OMe)
+ 2 MeCl
CH(OMe)
R2(O)P
10
R = Et (ɚ), Ph (ɛ).
Диацеталь 3 реагирует также со вторичны-
го альдегида без выделения HBr превращением
ми хлорфосфинами 9 с образованием третич-
1,2-бис(дибромметил)бензола в ациклический
диацеталь под действием триметилортоформиата
ных дифосфиноксидов 10. Очевидно, что реак-
с последующим гидролизом последнего до альде-
ция протекает через промежуточное образование
гида. Предложен новый метод синтеза ацикличе-
ди-α-хлорэфира 7 и О-метилфосфинитов 11, кото-
ского диацеталя фталевого альдегида без примеси
рые, взаимодействуя между собой, образуют ди-
циклического диацеталя - 1,3-дигидро-1,3-диме-
фосфиноксиды 10 (схема 6).
токсибензо[с]фурана - взаимодействием 1,2-бис-
В заключение следует отметить, что нами
(дибромметил)бензола с триметилортоформиа-
разработан новый способ получения фталево-
том в соотношении 1:6 при 90°С в присутствии
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 11 2019
СИНТЕЗ ФТАЛЕВОГО АЛЬДЕГИДА И ЕГО ДИАЦЕТАЛЕЙ
1655
10 мол% ZnCl2. Также предложены два спосо-
Бис(диметиловый)ацеталь фталевого аль-
ба получения циклического диацеталя фталевого
дегида (3). Смесь 12.00 г (0.028 моль) 1,2-бис-
альдегида: реакцией самого альдегида с триметил-
(дибромметил)бензола 2, 17.83 г (0.168 моль)
ортоформиатом в присутствии трифторуксусной
триметилортоформиата 4 и 0.57 г (0.0042 моль)
кислоты и последовательным взаимодействием
хлорида цинка нагревали при 90°C в течение
диацеталя с PCl3 и гидролизом промежуточно об-
5 ч. Реакционную массу охлаждали, обрабатыва-
разующегося ди-α-хлорэфира; реализовано фос-
ли 50 мл изооктана и фильтровали. Изооктан уда-
форилирование диацеталя вторичными хлорфос-
ляли в вакууме, остаток перегоняли. Выход 5.25 г
финами и последовательным действием PCl3 и
(85%), бесцветная жидкость, т. кип.
65-66°С
эфиром кислоты P(III).
(0.3 мм рт. ст.). Спектр ЯМР 1Н (CDCl3), δ, м. д.:
3.29 с (12Н, OMe), 5.67 с (2Н, СНO2), 7.30 д. д
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
= 3.4, 6.4 Гц). Спектр
и 7.57 д. д (4Н, С6H4, JHH
Спектры ЯМР 1Н и 13С снимали на при-
ЯМР 13С (CDCl3), δС, м. д.: 47.76 (4С, OMe), 95.38
борах Tesla BS-567A (100 МГц) и AVANCE
(2С, СН), 121.55 (2С, м-CHAr), 122.91 (2С, о-CHAr),
400WB (400.13 и 100.61 МГц). Спектры ЯМР
130.49 (2С, 2CAr).
31Р регистрировали на приборе AVANCE 400WB
1,2-Ди[(хлор)метоксиметил]бензол
(7).
К
(161.98 МГц), химические сдвиги ядер фосфора
6.04 г (0.044 моль) PCl3 добавляли по каплям рас-
указаны относительно 85%-ной H3PO4.
твор 2.54 г (0.011 моль) бис(диметилового)ацеталя
В работе использовали коммерческие о-ксилол,
фталевого альдегида 3 в 5 мл СCl4 при темпера-
бром, триметилортоформиат, трихлорид фосфора.
туре 5°C. Перемешивали реакционную массу в те-
1,2-Бис(дибромметил)бензол 2 синтезировали по
чение 1 ч при этой же температуре. Соединение
методике [1]. Растворители очищали и абсолюти-
термически лабильно и было идентифицировано
зировали, как описано в работе [45].
в неочищенном виде после удаления растворите-
Фталевый альдегид (1). а. Смесь 12.00 г
ля и легколетучих продуктов в глубоком вакууме
(0.028 моль)
1,2-бис(дибромметил)бензола
2,
(0.05 мм рт. ст.) на холоду. Получали 2.30 г (89%) не-
17.83 г (0.168 моль) триметилортоформиата 4 и
очищенного 1,2-бис[(хлор)метоксиметил]бензола
0.57 г (0.0042 моль) хлорида цинка нагревали при
7. Спектр ЯМР 1Н (CDCl3), δ, м. д.: 3.86 с (6Н,
90°C в течение 5 ч. Реакционную массу охлажда-
ОМе), 66.92 c (2H, CHCl), 7.53 м и 7.76 м (4Н,
ли, обрабатывали 50 мл изооктана и фильтрова-
С6Н4).
ли. К раствору добавляли 5 г (0.278 моль) воды и
1,3-Дигидро-1,3-диметоксибензо[с]фуран (5).
3 капли HCl, и полученную смесь нагревали (80°С)
2 ч с одновременной отгонкой образующегося ме-
а. К раствору 2.3 г (0.0098 моль) 1,2-ди[(хлор)-
тилового спирта. Органический слой отделяли и
метоксиметил]бензола 7 в 5 мл CCl4 добавляли
сушили MgSO4. Растворитель удаляли и кристал-
2 мл (0.11 моль) воды при комнатной температу-
лический остаток перекристаллизовывали из пе-
ре, затем перемешивали 0.5 ч при этой же тем-
тролейного эфира. Выход 2.97 г (79%), т. пл. 58°С
пературе. Отделяли водный слой, органический
(т. пл. 56.5-58°С [46]). Спектр ЯМР 1Н (ацетон-d6),
слой сушили K2CO3. Удаляли растворитель в
δ, м. д.: 7.85 д. д и 8.01 д. д (4Н, С6Н4, JHH = 3.2,
вакууме, остаток перегоняли. Получали 1.27 г
5.2 Гц), 10.51 с (2Н, CHО). Спектр ЯМР 13С (аце-
(72%) соединения 5 в виде смеси цис/транс-и-
тон-d6), δС, м. д.: 130.72 (2С, о-CHAr), 133.68 (2C,
зомеров в соотношении 65:35, т. кип. 62-63°С
м-CHAr), 136.65 (2С, ССНО), 192.00 (2С, СНО).
(0.2 мм рт. ст.). Спектр ЯМР 1Н (CDCl3), δ, м. д.:
б. К раствору
1.8 г
(0.01 моль)
1,3-диги-
3.32 с (6Н, ОМе, транс-изомер), 3.35 с (6Н, ОМе,
дро-1,3-диметоксибензо[с]фурана 5 в 10 мл CCl4
цис-изомер), 5.99 с (2Н, СНОМе, цис-изомер),
добавляли 3 мл воды, 3-4 капли HCl и кипятили
6.24 с (2Н, СНОМе, транс-изомер), 7.37 м и
полученную смесь с одновременной отгонкой об-
7.43 м (4Н, CHAr). Спектр ЯМР 13С (CDCl3), δС,
разующегося метилового спирта. Остаток сушили
м. д.: 53.28 (2С, ОМе, транс-изомер), 55.45 (2С,
в вакууме и перекристаллизовывали из петролей-
ОМе, цис-изомер), 105.25 (2С, СНО, цис-изомер),
ного эфира. Выход 1.08 г (81%).
106.39 (2С, СНО, транс-изомер), 122.92, 129.47 и
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 11 2019
1656
ХАЙРУЛЛИН и др.
138.85 (6Н, С6Н4, транс-изомер), 122.97, 129.36 и
1,2-Бензолбис[(метоксиметил)диэтилфосфи-
138.68 (6Н, С6Н4, цис-изомер).
ноксид (10б). К 2.48 г (0.02 моль) диэтилхлорфос-
фина при перемешивании и охлаждении (10°С)
б. К смеси 1.0 г (0.0075 моль) фталевого альде-
добавляли по каплям 2.26 г (0.01 моль) соединения
гида 4 и 3.18 г (0.03 моль) триметилортомуравьи-
3. Полученную кристаллическую массу перекри-
ного эфира 6 добавляли 2 капли трифторуксусной
сталлизовывали из изооктана. Выход 2.77 г (74%),
кислоты. Температура реакционной смеси повы-
т. пл. 102-104°С. Спектр ЯМР 1Н (CDCl3), δ,
шалась до 42°С. Через 24 ч удаляли в вакууме из-
м. д.: 0.91 т, 0.96 т и 1.29 т (12Н, CH2CH3, JHH = 7.6,
быток ортоэфира и другие легколетучие соедине-
3JPH = 6.8 Гц), 1.47-1.62 м и 1.84-1.93 м (8Н,
ния, остаток перегоняли. Выход 1.17 г (85%).
РСН2), 5.86 д (2Н, CHР, 2JРH = 12.0 Гц), 7.29 д.
д (2Н, м-CHAr, JHH = 5.2, 3.6 Гц), 7.42 д. д (2Н,
1,2-Бензолбис[(метоксиметил)диметокси-
о-CHAr, 3JРH = 5.2, JHH = 3.6 Гц). Спектр ЯМР 31Р
фосфонат] (8). К раствору 3.7 г (0.157 моль)
(CDCl3): δР 53.99 м. д. Найдено, %: С 57.63; Н 8.52;
бис[(хлор)метоксиметил]бензола
7, полученно-
Р 16.48. С18Н32О4Р2. Вычислено, %: С 57.75; Н
го из
3.87 г
1,2-бис(диметоксиметил)бензола,
8.62; Р 16.55.
в 10 мл безводного бензола в токе осушенного
аргона добавляли раствор 4.1 г (0.034 моль) три-
ФОНДОВАЯ ПОДДЕРЖКА
метилфосфита в 5 мл бензола. Реакционную мас-
су перемешивали при 20°С в течение 1 ч, затем
Работа выполнена при финансовой под-
нагревали (60°С) в течение 2 ч и вакуумировали
держке Министерства образования и науки
при 0.02 мм рт. ст. 1 ч (60°С). Выход 5.2 г (82%),
РФ в рамках базовой части госзадания (проект
вязкая жидкость. Спектр ЯМР 1Н (CDCl3), δ, м. д.:
№ 4.5348.2017/8.9).
3.31 с (6Н, ОМе), 3.53 д и 3.71 д (12Н, РОМе, JНH =
КОНФЛИКТ ИНТЕРЕСОВ
10.4 Гц), 5.12 д (2Н, СНР, 2JРH = 12.8 Гц), 7.28 д. д
(2Н, м-CHAr, JНH = 5.2, 3.6 Гц), 7.50 д (2Н, о-CHAr,
Авторы заявляют об отсутствии конфликта
JНH = 3.6 Гц). Спектр ЯМР 13С (CDCl3), δС, м. д.:
интересов.
53.11 и 53.93 (2С, ОМе), 58.38 д (4С, РОСН3, 2JРС =
6.3 Гц), 75.20 д (2С, СН, 1JРС = 166.5 Гц), 127.13,
СПИСОК ЛИТЕРАТУРЫ
128.29 и 133.57 (6С, С6Н4). Найдено, %: С 44.21; Н
1. Снелл Дж., Вайсбергер А. Синтез органических пре-
6.62; Р 15.98. С14Н24О8Р2. Вычислено, %: С 43.99;
паратов. III. Сб. 3. М.: ИЛ, 1952. С. 397.
Н 6.33; Р 16.20.
2. Weygand F., Vogelbach K., Zimmermann K. //
Chem. Ber. 1947. Vol. 80. P. 391. doi 10.1002/
1,2-Бензолбис[(метоксиметил)дифенилфос-
финоксид] (10а). К раствору 2.5 г (0.011 моль) ди-
cber.19470800504
3. Ruggli P., Brandt F. // Helv. Chim. Acta. 1944. Vol. 27.
фенилхлорфосфина в 10 мл изооктана при переме-
P. 274. doi 10.1002/hlca.19440270132
шивании добавляли по каплям 1.28 г (0.0055 моль)
4. Ruggli P., Mathez M. // Helv. Chim. Acta. 1946. Vol. 29.
соединения 3. Температура реакционной смеси
P. 1235. doi 10.1002/hlca.19460290536
поднималась до 27°С. Смесь нагревали при темпе-
5. Zhu P.C., Roberts C.G., Murrieta Y.T. Pat. US
ратуре 45-50°С 2.5 ч, затем оставляли на сутки при
2005/171201 (2004).
комнатной температуре. Кристаллы отфильтро-
6. Wawzonek S., Karll R. E. // J. Am. Chem. Soc. 1948.
вывали и сушили. Выход 2.45 г (79%), т. пл. 183-
Vol. 70. P. 1666. doi 10.1021/Ja01184a517
184°С. Спектр ЯМР 1Н (ацетон-d6), δ, м. д.: 3.46 с
7. Karlheinz G., Klaus P., Rudolf H. Pat. US 2006/293542
(6Н, ОCH3), 6.48 д (2Н, CHР, 2JРH = 10.8 Гц), 6.82
(2005).
д. д и 6.90 д. д (4Н, С6Н4, JHH = 5.6, 3.6 Гц), 7.97 д.
д и 7.66 д. д (8Н, о-CHAr, 3JРH = 11.0, JHH = 7.2 Гц),
8. Giselbrecht K., Hillisch W. Pat. US 2006/199870
7.37 т (4Н, п-CHAr, JHH = 6.0 Гц), 7.48-7.59 м (8Н,
(2005).
м-CHAr). Спектр ЯМР 31Р (CDCl3): δP 31.67 м. д.
9. Brewer B.N., Mead K.T., Pittman C.U., Lu K.,
Найдено, %: С 72.27; Н 5.51; Р 10.81. С34Н32О4Р2.
Zhu P.C. // J. Heterocycl. Chem. 2006. Vol. 43. P. 361.
Вычислено, %: С 72.08; Н 5.69; Р 10.93.
doi 10.1002/Jhet.5570430216
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 11 2019
СИНТЕЗ ФТАЛЕВОГО АЛЬДЕГИДА И ЕГО ДИАЦЕТАЛЕЙ
1657
10. Zhu P.C., Brewer B.N., Lu K. Pat. US 2007/4808 (2006).
P. 625. doi 10.1002/cber.19510840712
11. Kayan B., Oezen R., Gizir A.M., Kus N.S. // Org.
30. Cha J.S., Jeoung M.K., Kim J.M., Kwon O.O., Lee J.C. //
Prep. Proc. Int. 2005. Vol. 37. P. 83. doi 10.1080/
Org. Prep. Proc. Int. 1994. Vol. 26. P. 583. doi
00304940509355405
10.1080/00304949409458063
12. Ohkubo K., Suga K., Morikawa K., Fukuzumi S. / J.
31. Smith M.B., March J. March’s Advanced Organic
Am. Chem. Soc. 2003. Vol. 125. P. 12850. doi 10.1021/
Chemistry: Reactions, Mechanisms, and Structure. New
Ja036645r
York: John Wiley and Sons, 2013. 2047 p.
13. Shoair A. // J. Mol. Liq. 2015. Vol. 206. P. 68. doi
32. Яновская Л.А., Юфит С.С., Кучеров В.Ф. Химия
10.1016/J.molliq.2015.01.023
ацеталей. М.: Наука, 1975. 273 с.
14. Khorshidi A. // Chinese J. Catal. 2016. Vol. 37. P. 153.
33. Monroe E., Hand C. // J. Am. Chem. Soc. 1950. Vol. 72.
doi 10.1016/S1872-2067(15)61001-4
P. 5345. doi 10.1021/Ja01167a605
15. Tabatabaeian K., Zanjanchi M.A., Mahmoodi N.O.,
34. Moffett R.B. // Org. Synth. Cоll. 1963. Vol. 4. P. 427.
Eftekhari T. // RSC Adv. 2015. Vol. 5. P. 101013. doi
35. Sсhank K. // Chem. Ber. 1967. Vol. 100. P. 2292. doi
10.1039/C5RA18179H
10.1002/cber.19671000725.
16. Reintjens R.W.E., Broxterman Q.B., Kotthaus M.,
36. Cavallini G. // J. Med. Chem. 1964. Vol. 7. P. 255. doi
Poechlauer P. Pat. WO 2007/134847 (2006).
10.1021/Jm00333a003
17. Endo K., Takahashi H., Aihara M. // Heterocycles. 1996.
37. Kober E., Grundmann C. // J. Am. Chem. Soc. 1958.
Vol. 42. P. 589. doi 10.3987/COM-95-S48
Vol. 80. P. 5547. doi 10.1021/Ja01553a058.
18. Namboodiri V.V., Polshettiwar V., Varma R.S. //
38. Clerici A., Pastori N., Porta O. // Tetrahedron. 1998.
Tetrahedron Lett. 2007. Vol. 48. P. 8839. doi 10.1016/
Vol. 54. P. 15679. doi 10.1016/S0040-4020(98)00982-X
J.tetlet.2007.10.068
39. Hamada N., Kazahaya K., Shimizu H., Sato T. // Synlett.
19. Takezawa E., Sakaguchi S., Ishii Y. // Org. Lett. 1999.
2004. P. 1074. doi 10.1055/s-2004-820038
Vol. 1. P. 713. doi 10.1021/ol990117w
40. Газизов М.Б., Газизов К.М., Каримова Р.Ф., Пудо-
20. Firouzabadi H., Vessal B., Naderi M. // Tetrahedron
вик М.А., Садыкова А.И., Синяшин О.Г. // Докл. АН.
Lett. 1982. Vol. 23. P. 1847. doi 10.1016/S0040-
2001. Т. 381. С. 207; Gazizov M.B., Gazizov K.M.,
4039(00)86758-1
Pudovik M.A., Mukhamadiev A.A., Karimova R.F.,
21. Li S., Zhou L., Song Z., Bao F., Kanno K., Takahashi T. //
Sadykova A.I., Sinyashin O.G. // Doklady Chem. 2001.
Heterocycles. 2007. Vol. 73. P. 519. doi 10.3987/COM-
Vol. 381. P. 321. doi 10.1023/A:1012928708045
07-S(U)28
41. Газизов М.Б., Иванова С.Ю., Башкирцева Н.Ю., Хай-
22. Ariyoshi K., Aso Y., Otsubo T., Ogura F. // Chem. Lett.
руллина О.Д., Хайруллин Р.А., Газизова О.В. // Изв.
1984. Vol. 13. P. 891. doi 10.1246/cl.1984.891
АН. Сер. хим. 2017. С. 1230; Gazizov M.B., Ivano-
23. Khaligh N.G. // Chinese J. Catal. 2014. Vol. 35. P. 329.
va S.Yu., Bashkirtseva N.Yu., Khairullina O.D.,
doi 10.1016/S1872-2067(12)60750-5
Khairullin R.A., Gazizova O.V. // Russ. Chem. Bull.
24. Khaligh N.G., Shirini F. // Ultrasonics Sonochemistry.
2017. Vol. 66. P. 1230. doi 10.1007/s11172-017-1877-6
2013. Vol. 20. P. 19. doi 10.1016/J.ultsonch.2012.07.016
42. Газизов М.Б., Иванова С.Ю., Ибрагимов Ш.Н.,
25. Shirini F., Jolodar O.G. // J. Mol. Catal. (A). 2012.
Газизова К.С., Хайруллин Р.А., Медведева К.А. //
Vol. 356. P. 61. doi 10.1016/J.molcata.2012.01.002
ЖОХ. 2016. Т. 86. С. 1570; Gazizov M.B., Ivano-
26. Cha J.S., Chang S.W., Mi Kim J., Kwon O.O., Lee J.C. //
va S.Yu., Ibragimov Sh.N., Gazizova K.S., Khairul-
Org. Prep. Proc. Int. 1997. Vol. 29. P. 665. doi
lin R.A., Medvedeva K.A. // Russ. J. Gen. Chem. 2016.
10.1080/00304949709355246
Vol. 86. P. 2132. doi 10.1134/S1070363216090279
27. Weygand F., Eberhardt G., Linden H., Schäfer F.,
43. Powell M.R., Rexford D.R. // J. Org. Chem. 1953.
Eigen I. // Angew. Chem. 1953. Vol. 65. P. 525. doi
Vol. 18. P. 810. doi 10.1021/Jo50013a006
10.1002/ange.19530652102
44. Schmitz Е. // Chem. Ber. 1958. Vol. 91. P. 410. doi
28. Corriu R.J.P., Lanneau G.F., Perrot M. // Tetrahedron
10.1002/cber.19580910228
Lett. 1988. Vol. 29. P. 1271. doi 10.1016/S0040-
45. Гордон А. Спутник Химика. М.: Мир, 1976. 541с.
4039(00)80274-9
46. McDonald R.S., Martin E.V. // Canad. J. Chem. 1979.
29. Weygand F., Tietjen D. // Chem. Ber. 1951. Vol. 84.
Vol. 57. P. 506. doi 10.1139/v79-084
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 11 2019
1658
ХАЙРУЛЛИН и др.
Synthesis of Phthalic Aldehyde and Its Diacetals
R. A. Khairullin, M. B. Gazizov*, Yu. S. Kirillina,
S. Yu. Ivanova, O. D. Khairullina, and K. S. Gazizova
Kazan National Research Technological University, ul. K. Marksa 68, Kazan, 420015 Russia
* e-mail: mukattisg@mail.ru
Received April 22, 2019; revised April 22, 2019; accepted April 25, 2019
Acyclic phthalaldehyde diacetal without cyclic 1,3-dihydro-1,3-dimethoxybenzo[c]furan impurity is formed
by the reaction of 1,2-bis(dibromomethyl)benzene with trimethylorthoformate in the ratio of 1:6 at 90°C in the
presence of 10 mol% of ZnCl2. Hydrolysis of phthalaldehyde diacetal leads to the formation of phthalaldehyde
without HBr evolution. The reaction of phthalaldehyde with trimethyl orthomoformate in the presence
of trifluoroacetic acid proceeds abnormally with the formation of its cyclic diacetal. Acyclic diacetal is
phosphorylated by chlorophosphines and the sequential action of PCl3 and P(III) acid ester.
Keywords: phthalaldehyde, phthalaldehyde diacetals, 1,3-dihydro-1,3-dimethoxybenzo[c]furan, 1,2-bis(dibro-
momethyl)benzene, phosphorylation
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 11 2019