ЖУРНАЛ ОБЩЕЙ ХИМИИ, 2019, том 89, № 7, с. 1130-1139
УДК 541.64
ДИСПЕРСИОННАЯ ОКИСЛИТЕЛЬНАЯ
ПОЛИМЕРИЗАЦИЯ ПИРРОЛА В ВОДНЫХ РАСТВОРАХ
ПОЛИВИНИЛОВОГО СПИРТА
© 2019 г. Я. О. Межуевa, *, И. В. Плющийa, Ю. В. Коршакa, М. И. Штильманa, И. А. Грицковаb
a Российский химико-технологический университет имени Д. И. Менделеева, Миусская пл. 9, Москва, 125047 Россия
*e-mail: valsorja@mail.ru
b Институт тонких химических технологий имени М. В. Ломоносова,
Российский технологический университет, Москва, Россия
Поступило в Редакцию 13 декабря 2018 г.
После доработки 7 марта 2019 г.
Принято к печати 10 марта 2019 г.
Исследована кинетика окислительной полимеризации пиррола в водном растворе поливинилового
спирта под действием пероксодисульфата аммония. Приведено количественное описание полученных
кинетических данных с учетом образования новой фазы, определены значения энергии активации для
некаталитической и каталитической стадий окислительной полимеризации пиррола. Критически
рассмотрены различные механизмы окислительной полимеризации пиррола и приведено сравнение
вычисленных кинетических параметров с их значениями, описанными в литературе для окислительной
полимеризации пиррола в отсутствии стабилизаторов.
Ключевые слова: полипиррол, окислительная полимеризация, пиррол, поливиниловый спирт, кинетика
DOI: 10.1134/S0044460X19070175
Окислительная полимеризация пиррола является
дается первый порядок реакции по концентрации
основным методом синтеза полипиррола, обладаю-
мономера [9], тогда как при электрохимическом
щего высокой электрической проводимостью, выра-
окислении порядок реакции близок к 0.5 [10], при
женной зависимостью электрической прово-
этом автокаталитического ускорения окисли-
димости от положения протолитического и окисли-
тельной полимеризации пиррола не отмечалось. Об
тельно-восстановительного равновесий, термо-
отсутствии автоускорения также свидетельствуют
стабильностью, а также биосовместимостью [1].
результаты, полученные при исследовании
Совокупность этих свойств определила возмож-
кинетики окислительной полимеризации пиррола в
ность применения полипиррола в молекулярной
воде под действием K3[Fe(CN)6] [11, 12] и FeCl3
электронике, медико-биологических областях и
[13]. При этом в работе
[13] использовали
конструировании сенсорных систем, включая био-
значительные концентрации FeCl3 (>0.1 М.), а
сенсоры [1]. Вместе с тем до настоящего времени
также смесь
1-нафтилсульфокислоты и серной
кинетические аспекты и механизм окислительной
кислоты в качестве источника проивоионов
полимеризации пиррола остаются предметом
допанта, что могло способствовать параллельному
дискуссии, что определяет необходимость проведе-
развитию кислотно-катализируемой олигомериза-
ния дальнейших исследований в этой области.
ции пиррола, которая, как будет показано далее,
подавляет автокаталитическое течение окисли-
Окислительная полимеризация пиррола тради-
тельной полимеризации пиррола.
ционно проводится в среде ацетонитрила или в
воде под действием солей железа(III)
[2-5],
Напротив, в работах [14, 15], в которых была
пероксодисульфатов
[6], а также в условиях
использована низкая концентрация FeCl3, сообщается
электрохимического окисления [7, 8]. Показано,
об автоускорении окислительной полимеризации
что при окислительной полимеризации пиррола в
пиррола и подчинении скорости реакции
среде ацетонитрила под действием FeCl3 наблю-
кинетическому уравнению (1) [15].
1130
ДИСПЕРСИОННАЯ ОКИСЛИТЕЛЬНАЯ ПОЛИМЕРИЗАЦИЯ ПИРРОЛА
1131
d[Py]
Вместе с тем, хотя в работе [15] предпринята
-
= k1[Py][Fe3+] + k2[Py][P],
(1)
dt
попытка объяснения кинетического уравнения (1) в
рамках механизма Плетчера, в действительности
где [Py] - концентрация пиррола, [Fe3+] - концен-
трация FeCl3, [P] - концентрация растущих олиго-
механизмы Плетчера и Диаса не соответствует
этому уравнению. Так, если справедлив механизм
меров пиррола; k1, k2 - константы скоростей реакции.
Плетчера, то уравнение (1) может выполняться,
Подчинение скорости окислительной поли-
если только справедливо равенство (2).
меризации пиррола уравнению (1) предполагает
[P] = λ([Py]0 -[Py]),
(2)
наличие как минимум двух стадий, первая из
которых включает непосредственное окисление
где [Py]0 - начальная концентрация пиррола; [P] -
пиррола, а вторая взаимодействие пиррола и
суммарная концентрация катион-радикалов в
образующихся n-меров пиррола. Механизм первой
системе; λ - константа, не зависящая от концен-
стадии хорошо известен [13-18] и представляет
трации окислителя.
одноэлектронное окисление пиррола с образо-
Вместе с тем вполне понятно, что доля n-меров
ванием соответствующего катион-радикала.
пиррола превращенных в катион-радикалы за счет
Однако предметом обсуждения остается
взаимодействия с окислителем не может не
механизм второй стадии, которая, как полагают,
зависеть от концентрации окислителя. С учетом
непосредственно связана с реализацией роста цепи.
несоответствия кинетических данных механизму
Согласно представлениям Плетчера
[17,
18],
Плетчера, а также его несоответствия квантово-
реализация роста цепи связана с присоединением
химическими расчетам
[19], следует отдать
пиррола к образовавшемуся катион-радикалу
предпочтение механизму Диаса. Однако механизм
пиррола. Результатом присоединения является
Диаса не может объяснить эффект автоускорения
образование катион-радикала димера пиррола,
при окислительной полимеризации пиррола и
который подвержен быстрому окислению до
хорошо согласуется с кинетическими данными,
катиона с последующим депротонированием. В
полученными при проведении полимеризации
итоге
катион-радикал
димера
пиррола
пиррола в среде ацетонитрила, где автоускорение
превращается в димер пиррола, который вновь
не наблюдалось.
окисляется до катион-радикала, взаимодействует с
По всей видимости, механизм Диаса непро-
мономером, превращаясь в катион-радикал
тиворечиво описывает процессы, лежащие в основе
тримера, который теряет электрон и протон,
прироста молекулярной массы n-меров пиррола.
образуя тример, и т. д. [17, 18]. Проведенные
Вместе с тем рост цепи требует постоянного
квантово-химические расчеты, а также анализ
образования катион-радикалов как из n-меров
орбитальной симметрии, выполненные в работе
пиррола, так и из самого пиррола. При этом
[19], показали, что рост цепи должен протекать с
участие в актах рекомбинации двух катион-
высокой энергией активации, что ставит
радикалов n-меров пиррола возможно лишь для
рассматриваемый механизм под сомнение. К
небольших значений n, так как растворимость n-
выводу о невозможности присоединения молекулы
меров пиррола быстро снижается с ростом число n
пиррола к катион-радикалу пиррола, также
[1]. Поэтому вклад процессов роста молекулярной
приводит детальное рассмотрение кинетических
массы за счет рекомбинации двух растворимых n-
закономерностей начальной стадии окислительной
меров пиррола невелик. Таким образом, следует
полимеризации производных пиррола, выполнен-
ожидать, что рост цепи будет обеспечиваться за
ное в работе [20].
счет захвата катион-радикалами n-меров пиррола,
Альтернативной точкой зрения является
находящимися в дисперсной фазе, катион-ради-
механизм Диаса [21], предполагающий рост цепи
калов мономера (пиррола) из объема реакционной
посредством рекомбинации катион-радикалов n-
системы.
меров пиррола с катион-радикалом пиррола. Этот
Здесь возникает некоторое противоречие, связан-
механизм подтверждается кинетическими данными
ное с малой вероятностью описанного реком-
[20] и квантово-химическими расчетами
[19].
бинационного процесса, так как концентрация
Некоторые авторы не исключают возможности
катион-радикалов пиррола должна быть мала.
роста цепи в результате рекомбинации катион-
Однако известно, что окислительная поли-
радикалов n-меров пиррола между собой [15].
меризация пиррола является достаточно быстрым
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 7 2019
1132
МЕЖУЕВ и др.
Схема 1.
2-
S2O8
...-
C+
C+
N
N
N
N
N
N
N
N
H
H
H
H
H
H
H
H
•
−
•
+ CH
N
H
2-
N
S2O8
H
δ-
...-
C+
C+
N
N
N
N
H
H
H
H
δ+
N
H
процессом, приводящим к образованию сшитых
В данной работе исследована кинетика
структур [1].
дисперсионной окислительной полимеризации
пиррола в водных растворах поливинилового
Описанное противоречие можно устранить,
спирта, с использованием которого ранее был
если допустить возможность образования катион-
установлен ряд коллоидно-химических закономер-
радикалов пиррола вблизи агрегатов его n-меров,
ностей этой реакции [22], что позволило выдвинуть
формирующих новую фазу. Для этого достаточно
предположения, объясняющие многие противо-
предположить возможность предварительной
речия, имеющиеся в литературе.
координации пиррола с продуктами его окисли-
тельной полимеризации с последующим окис-
Принимая справедливость механизма Диаса [21]
лением комплекса. В качестве такого комплекса
для окислительной полимеризации пиррола и
можно рассматривать комплекс с переном заряда
дополняя его стадией медленного образования
между пирролом (донор) и окисленными
комплекса с переносом заряда в адсорбционном
участками цепей n-меров пиррола (акцептор),
слое с последующим его быстрым окислением
которые образуются при синтезе полипиррола и
(схема
1), можно дать объяснение эффекту
ответственны за его допирование (схема 1). Ранее
автоускорения при окислительной полимеризации
подобный механизм был предложен без должной
пиррола. Очевидно, что скорость окислительной
аргументации авторами для описания кинетики
полимеризации пиррола должна лимитироваться
окислительной полимеризации пиррола под
скоростью окисления пиррола до катион-радикала,
действием пероксодисульфата аммония [16].
так как стабильность катион-радикалов n-меров
пиррола должна быть значительно выше, а
Как видно из приведенного анализа литературы
скорость их образования больше [1]. Поэтому, в
[9-21], взгляды на механизм окислительной
соответствии со схемой
1, принимается, что
полимеризации пиррола весьма противоречивы,
продукты окислительной полимеризации пиррола
что указывает на необходимость проведения
катализируют окисление пиррола. Причиной
дальнейших исследований с целью объяснения
большей скорости окисления комплекса с переном
имеющихся результатов. Основные сложности,
заряда по сравнению со свободным пирролом,
связаны с необходимостью учета образования
следует считать наличие низколежащих свободных
новой фазы, которую формирует нерастворимый в
орбиталей окисленных фрагментов цепи поли-
реакционной среде полипиррол
[1]. В этом
пиррола, которые также ответственны за высокую
отношении заметный интерес представляет
электрическую проводимость полипиррола. В
исследование кинетики дисперсионной окисли-
целом предполагаемый механизм окислительной
тельной полимеризации пиррола в водных
полимеризации пиррола аналогичен предложен-
растворах полимеров и сравнение с результатами
ному ранее авторами для окислительной поли-
полученными для осадительного режима, которое
меризации анилина, N-этиланилина и фенилен-
позволяет выявить роль образования новой фазы в
диаминов, описанному в работах [23-27]. Поэтому
механизме этой реакции.
возможно применение кинетического уравнения (3),
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 7 2019
ДИСПЕРСИОННАЯ ОКИСЛИТЕЛЬНАЯ ПОЛИМЕРИЗАЦИЯ ПИРРОЛА
1133
t, с
t, с
Рис. 1. Кинетические кривые для окислительной поли-
Рис. 2. Кинетические кривые для окислительной поли-
меризации пиррола под действием пероксодисульфата
меризации пиррола под действием пероксодисульфата
аммония в водном растворе поливинилового спирта
аммония в водном растворе поливинилового спирта
марки BF-14 в координатах 102ln(102+[M]-1)-t при 298
марки BF-14 в координатах [M]–1ln([M]0 - [M])/[M]-t
(1), 303 (2), 308 (3), 313 K (4).
при 298 (1), 303 (2), 308 (3), 313 K (4).
выведенного впервые с учетом особенностей
концентраций мономера и окислителя, когда оно
гетерогенного течения процесса окислительной
принимает вид (5) [16].
полимеризации в работе [23].
102ln(102 + [M]-1) = 529.8 + kst.
(5)
kc([M]0
- [M])
d[M]
При полимеризации пиррола под действием
-
= ws +
,
(3)
dt
пероксодисульфата аммония в водном растворе
1 + K[M]
поливинилового спирта линейность в координатах
где [M]0, [M] - начальная и текущая концентрация
102ln(102+[M]-1)-t наблюдается в течении первых
s
мономера, [Ox] - концентрация окислителя, ws = k
150 с (рис.
1), затем начинаются отклонения,
[M][Ox] - скорость одноэлектронного окисления
вызванные автоускорением.
пиррола, ks - константа скорости одноэлектронного
окисления пиррола, kc = (3KkM)/dr - константа
Наличие автоускорения, подтверждается сущест-
скорости каталитической стадии, k - константа
вованием линейной зависимости в широком
скорости взаимодействия адсорбированного мономера
диапазоне конверсий пиррола в координатах
с электронодефицитными фрагментами цепей с
[M]–1ln([M]0
-
[M])/[M]-t (рис.
2), которые
образованием комплекса с переносом заряда, d -
отвечают интегральной форме (6) уравнения (4)
плотность полимерного продукта; M - молярная
при выполнении неравенства ks[M][Ox] << kc([M]0 -
масса мономера; r
- средний эквивалентный
[M])/[M]:
радиус частиц; K
- константа адсорбционного
равновесия мономера на поверхности частиц.
[M]–1ln([M]0 - [M])/[M] = kct + C,
(6)
При условии (1 >> K[M]), соответствующему
где С - константа интегрирования.
слабой адсорбции мономера на поверхности
По тангенсам угла наклона зависимостей,
агрегатов его n-меров, уравнение (3) переходит в
приведенных на рис. 1 и 2, возможно определить
уравнение (4), аналогичное уравнению (1).
константы скорости некаталитической и катали-
d[M]
тической стадий для температур 298, 303, 308 и
-
= ks[M][Ox] + kc([M]0 - [M])[M].
(4)
dt
313 K, при которых исследовали кинетику реакции.
Определение констант скоростей одноэлектрон-
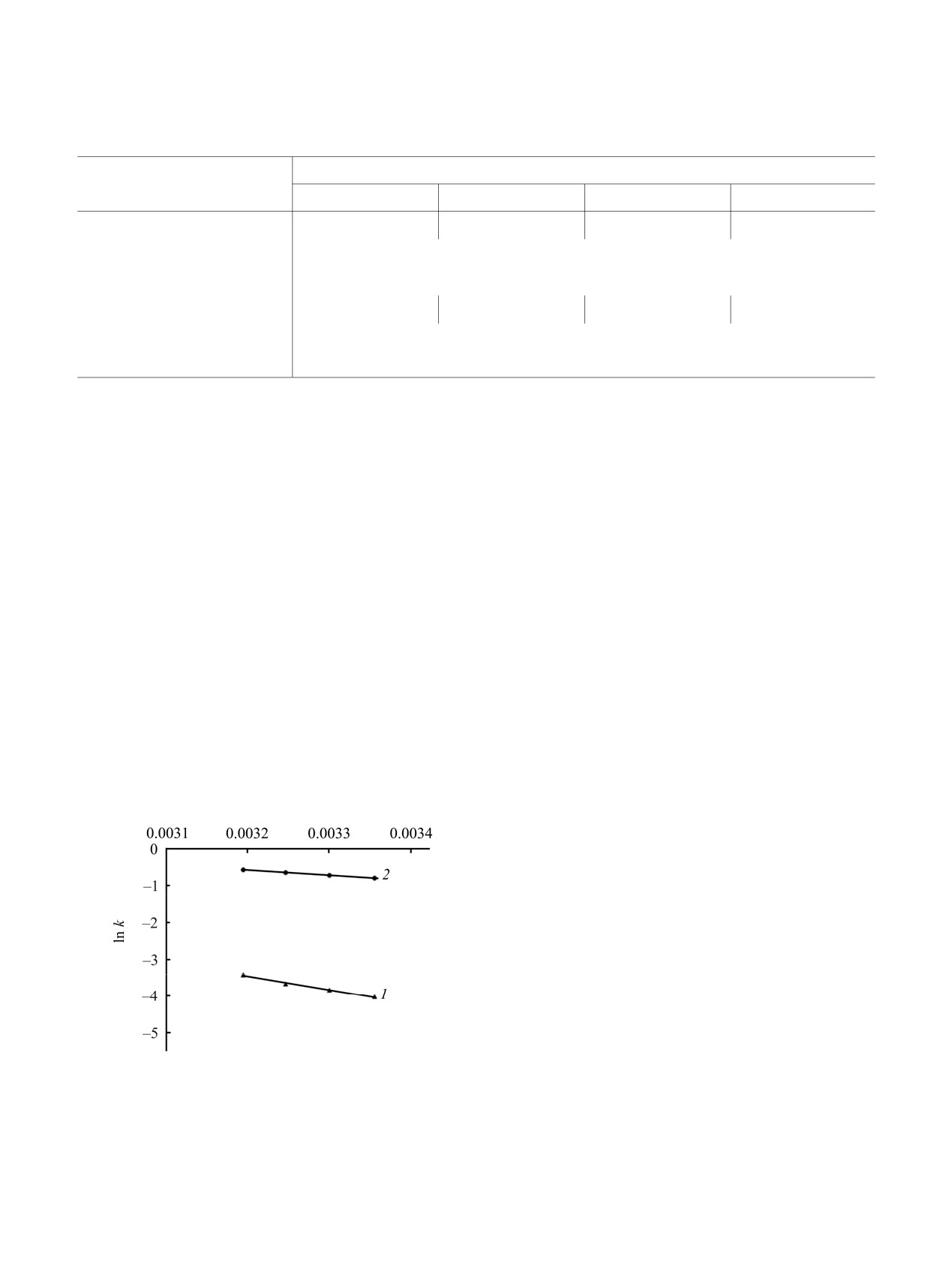
Температурные зависимости констант скоростей
ного окисления возможно после интегрирования
некаталитической и каталитической стадий
уравнения (4) в области низких конверсий пиррола
линейны в аррениусовых координатах и приведены
([M]0
≈
[M]) и подстановки начальных
на рис.
3. Вычисленные константы скорости,
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 7 2019
1134
МЕЖУЕВ и др.
Кинетические параметры окислительной полимеризации пиррола под действием пероксодисульфата аммония в
водном растворе поливинилового спирта марки BF-14
Т, K
Параметр
298
303
308
313
ks, л·моль-1·с-1
0.018±0.002
0.022±0.002
0.026±0.003
0.033±0.005
Eas, кДж/моль
31±3
Aas, л·моль-1·с-1
4400±300
kc, л·моль-1·с-1
0.46±0.03
0.49±0.03
0.53±0.03
0.57±0.04
Eac, кДж/моль
11±1
Aac, л·моль-1·с-1
42±3
предэкспоненциальные множители и энергии
активации свидетельствует о значительной упоря-
активации приведены в таблице.
доченности переходного состояния по сравнению с
реагентами, что типично для бимолекулярных
Предполагая, что кинетические параметры Aas и
реакций. Расчет энтальпии активации и энтропии
Eas относятся к элементарной стадии, состоящей в
активации по уравнениям (7) и
(8) для одно-
одноэлектронном переносе с молекулы пиррола на
электронного окисления пиррола пероксоди-
пероксодисульфат-ион, можно вычислить энталь-
сульфатом аммония в отсутствие поливинилового
пию активации и энтропию активации этой стадии
спирта с использованием констант скоростей,
по уравнениям (7) и (8) в соответствии с теорией
приведенных в работе [16], приводит к значениям
активированного комплекса.
81 кДж/моль и
-10 Дж/(моль·K). Уменьшение
ΔH≠ = Eas - RT,
(7)
энтальпии активации стадии одноэлектронного
окисления в присутствии поливинилового спирта
ΔS≠ = Rln(kh/κkBT) + ΔH≠/T.
(8)
свидетельствует о его каталитическом действии,
Энтальпия и энтропия активации для одно-
что, вероятно, связано со стабилизацией электро-
электронного окисления пиррола составляют
фильного переходного состояния нуклеофильными
28 кДж/моль и -183 Дж/(моль·K) соответственно
гидроксильными
группами
поливинилового
при
298 K. Отрицательное значение энтропии
спирта. Об участии звеньев цепей поливинилового
спирта в формировании переходного состояния
также свидетельствует более отрицательное
1/T, K-1
значение энтропии активации для одноэлектрон-
ного окисления пиррола в присутствии поливинил-
ового спирта.
Схожий эффект, связанный с понижением
энергии активации стадии одноэлектронного
окисления пиррола, был отмечен ранее для
окислительной полимеризации пиррола в водных
растворах поли-N-винилпирролидона под дейст-
вием пероксодисульфата аммония [28]. Причем
каталитический эффект, оказываемый поли-N-
винилпирролидоном (12 кДж/моль [28]) для стадии
одноэлектронного окисления пиррола, превосходит
наблюдаемый при использовании поливинилового
Рис. 3. Температурные зависимости констант скоростей
спирта (31 кДж/моль). Это обстоятельство хорошо
реакции ks (1) и kc (2) в аррениусовых координатах для
согласуется с предположением о нуклеофильной
окислительной полимеризации пиррола под действием
пероксодисульфата аммония в водном растворе
стабилизации переходного состояния одноэлек-
поливинилового спирта.
тронного окисления пиррола, так как нуклео-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 7 2019
ДИСПЕРСИОННАЯ ОКИСЛИТЕЛЬНАЯ ПОЛИМЕРИЗАЦИЯ ПИРРОЛА
1135
фильность амидной группы должна превосходить
окисляться с большей скоростью, чем его комплекс
нуклеофильность гидроксильной группы.
с FeCl3. Вероятно, второй порядок по концен-
трации ионов Fe3+ является кажущимся и связан с
Энтальпия и энтропия активации, определенные
частичной дезактивацией FeCl3 вследствие
для одноэлектронного окисления пиррола под
гидролиза, так как при определении порядка
действием хлорида железа(III),
составили
использовались очень низкие концентрации FeCl3.
77.09 кДж/моль и
24.25 Дж/(моль·K) соответ-
ственно. Обращает на себя внимание положи-
В целом автокаталитический характер
тельная величина энтропии активации, определен-
окислительной полимеризации пиррола выражен в
ная в присутствии хлорида железа(III), которая так
значительно меньшей степени, чем для
и не получила в работе [15] объяснения.
окислительной полимеризации анилина. По всей
видимости, это связано с большей скоростью
По нашему мнению, кажущийся аномальный
некаталитического окисления пиррола, так как он
для бимолекулярных реакций знак энтропии
является более сильным одноэлектронным
активации можно объяснить образованием неустой-
восстановителем, а также значительно более
чивого комплекса ионов Fe3+ с пирролом, который
слабым основанием
[16,
23]. Вероятно, это
далее подвергается мономолекулярному разложе-
является одной из причин, по которой автокатали-
нию (схема
2).
Поэтому одноэлектронное
тическое течение окислительной полимеризации
окисление пиррола ионами Fe3+ следует рассмат-
пиррола может остаться незамеченным при иссле-
ривать не как бимолекулярный элементарный акт,
довании процесса в области низких конверсий
а как последовательность стадий быстрого
мономера. Другая причина, по мнению авторов,
образования комплекса с последующим его
может состоять во введении дефектов в цепь,
мономолекулярным разложением. Как известно,
разрывающих сопряженную систему и препятству-
для мономолекулярных процессов характерны
ющих введению поляронов и биполяронов,
небольшие по величине положительные энтропии
выполняющих функцию акцепторов электронной
активации, что хорошо согласуется с ее величиной,
плотности при образовании комплекса с переносом
определенной в работе [15].
заряда. Так, в работе [14] отмечено наблюдение,
Схема 2.
что окислительная полимеризация пиррола
протекает с эффектом автокатализа только при рН
быстро
медленно
+2
Pyr + Fe3+
Pyr---Fe3+
Pyr+· + Fe
больших 1.3. Вероятно, при меньших значениях рН
происходит протонирование пиррола, приводящее,
В работе [13] также высказано предположение
как известно, к образованию частично деароматизи-
об образовании донорно-акцепторного комплекса
рованного тримера, который способен к
между пирролом и FeCl3 в связи с установленным
окислительной сополимеризации с пирролом [18] с
вторым порядком реакции по концентрации ионов
введением в цепь фрагментов, разрывающих
Fe3+, который был объяснен Льюисом кислотно-
сопряжение по цепи (схема 4). В этом случае,
основным взаимодействием пиррола и ионов Fe3+ с
очевидно, автокатализа наблюдаться не должно
последующим окислением образующегося комплекса
или он может быть в значительной степени
ионами Fe3+ (схема 3).
подавлен вследствие отсутствия или меньшей
концентрации акцепторных фрагментов в цепи.
Схема 3.
Аналогичный процесс олигомеризации пиррола
быстро
Fe+3
может инициироваться и кислотами Льюиса [29], в
Pyr + Fe+3
Pyr---Fe+3медленно Pyr+· + Fe+2 + Fe+3
том числе и FeCl3, если его концентрация
достаточно велика.
Однако результатом образования такого
комплекса (схема 3), должно быть значительное
В то же время кинетические параметры
уменьшение электронной плотности на пирроль-
каталитической стадии (см. таблицу) характе-
ном кольце без введения низколежащих вакансий
ризуют суммарный результат процессов адсорбции
(Fe3+, жесткая кислота Льюиса по Пирсону, и
мономера, образования комплекса с переносом
вакантные d-орбитали имеют высокую энергию),
заряда и его окисления. Поэтому рассмотрение
что должно способствовать увеличению энергии
кинетических параметров каталитической стадии
активации последующего акта окисления под
(kc, Eac, Aac) как соответствующих элементарной
действием Fe3+. Таким образом, пиррол должен
стадии следует признать неправомерным, так же
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 7 2019
1136
МЕЖУЕВ и др.
Схема 4.
N
H+
H
H+
CH+
+HC
N
N
N+
−H+
N
N
N
N
H
H
H
H
H
H
H
-H+
N
H
N
H
Полимерный продукт
N
N
N
H
H
H
как и проведение расчетов активационных
отрицательной и составила -81 кДж/моль [16].
параметров для этой стадии. В частности,
Напротив, при введении в систему поливинилового
константа скорости kc должна быть обратно
спирта энергия активации, как было показано
пропорциональна радиусу частиц дисперсной фазы
выше, положительна и составляет 11 кДж/моль.
в соответствии с уравнением (3) [23]. При этом,
Данный результат вполне понятен, если учесть, что
хотя
размер
частиц
дисперсной
фазы
с ростом температуры в отсутствие стабилизатора
увеличивается с ростом конверсии пиррола при ее
размер образующихся первичных частиц должен
проведении в водном растворе поливинилового
быстро возрастать, что и приводит к снижению kс с
спирта
[22], полученные экспериментальные
ростом температуры, как это следует из уравнения
данные свидетельствуют о том, что константа
(3)
[16]. Этот результат еще раз указывает на
скорости каталитической стадии не зависит от
формальность кинетических параметров катали-
конверсии пиррола. Также о независимости
тической стадии.
константы скорости каталитической стадии от
Еще большее увеличение энергии активации
конверсии пиррола, свидетельствуют результаты
каталитической стадии окислительной полимери-
работ [15, 16], в которых исследовалась кинетика
зации пиррола (31 кДж/моль) наблюдалось при
осадительной окислительной полимеризации
использовании
поли-N-винилпирролидона
с
пиррола без введения стабилизаторов.
молекулярной массой
40000 Да в качестве
По всей видимости, это противоречие является
стабилизатора [28]. Это можно объяснить тем, что
кажущимся. Следует ожидать, что первоначально
электроотрицательность sp2-гибридного атома
образующиеся частицы полипиррола, претер-
кислорода в составе амидной группы поли-N-винил-
певают процессы коагуляции с образованием
пирролидона должна превышать электроотрица-
проницаемых для мономера и окислителя
тельность sp3-гибридного атома кислорода в
агрегатов, что и обеспечивает наблюдаемый рост
составе гидроксильной группы поливинилового
агрегатов, при сохранении почти постоянных
спирта, что отвечает образованию более прочных
размеров первичных частиц. В этом случае
водородных связей с полипирролом при исполь-
константа скорости каталитической стадии не
зовании в качестве стабилизатора поли-N-
должна значительно зависеть от конверсии
винилпирролидона. Таким образом, в присутствии
мономера, что и наблюдается в действительности.
поли-N-винилпирролидона (40000 Да) стерическая
Причем введение стабилизатора оказывает влияние
стабилизация полипиррола должна быть более
не только на подавление процессов коагуляции, но
эффективной, чем в случае использования поли-
и на размер первичных частиц. Это следует из
винилового спирта (45000-50000 Да), способствуя
сравнения энергий активации каталитической
повышению энергии активации каталитической
стадии для окислительной полимеризации пиррола
стадии. Действительно, образование оболочки,
пероксодисульфатом аммония в присутствии
построенной фрагментами гидратированных цепей
поливинилового спирта и его отсутствие. Так, в
водорастворимых полимерных стабилизаторов и
отсутствие поливинилового спирта энергия актива-
оказывающей стерические препятствия коале-
ции каталитической стадии оказалась величиной
сценции, непременно должно создавать экрани-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 7 2019
ДИСПЕРСИОННАЯ ОКИСЛИТЕЛЬНАЯ ПОЛИМЕРИЗАЦИЯ ПИРРОЛА
1137
(а)
(б)
t, с
c(ПВС), оси-моль/л
(в)
(г)
t, с
c(ПВС), оси-моль/л
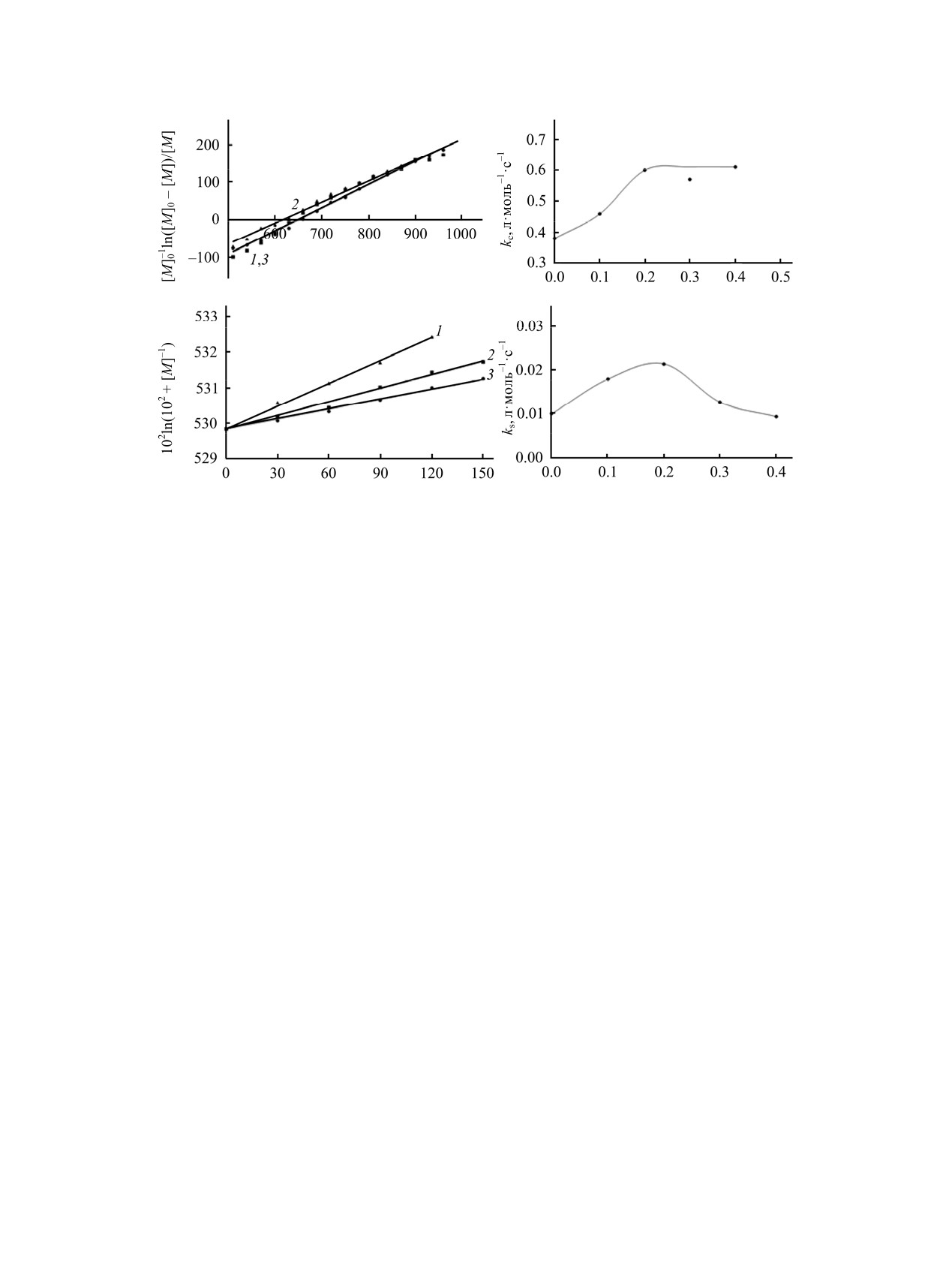
Рис. 4. Кинетические закономерности каталитической (а, б) и некаталитической (в, г) стадий окислительной полимеризации
пиррола в присутствии различных концентраций поливинилового спирта марки BF-14. с = 0.2 (1), 0.3 (2), 0.4 осн-моль/л (3).
Значения констант скоростей в отсутствие поливинилового спирта заимствованы из работы [16].
рование электронодефицитных фрагментов цепей
поливинилового спирта с
84 до
31 кДж/моль.
образующихся электропроводящих полимеров,
Наличие эффекта экранирования пиррола цепями
которые обуславливают автокаталитическое течение
поливинилового спирта согласуется с умень-
процесса. Описанный эффект, по всей видимости,
шением предэкспоненциального множителя с
является общим и отмечался также для окисли-
4.87×1012
[16] до
4425 л·моль-1·с-1. По всей
тельной полимеризации анилина в присутствии
видимости, за столь выраженный эффект
водорастворимых полимерных стабилизаторов [23,
экранирования ответственны
специфические
27, 30].
взаимодействия между молекулами пиррола и
цепями поливинилового спирта посредством
Примечательно, что увеличение концентрации
образования водородных связей.
поливинилового спирта в реакционной системе
приводит к увеличению константы скорости
Таким образом, исследована кинетика окисли-
каталитической стадии до определенного значения,
тельной полимеризации пиррола в водном растворе
а далее не оказывает значительного влияния, что
поливинилового спирта под действием пероксоди-
связано с насыщением поверхности частиц
сульфата аммония. Установлено, что реакция
дисперсной фазы стабилизатором (рис. 4а, б). При
является автокаталитической и дано коли-
этом константа скорости одноэлектронного
чественное
описание
экспериментальных
окисления пиррола проходит через максимум
кинетических кривых. Проведено сопоставление
(рис. 4в, г), что, вероятно, является результатом
полученных результатов с данными по кинетике
соотношения
нуклеофильной
стабилизации
окислительной полимеризации пиррола под
электрофильного переходного состояния одно-
действием солей железа(III) и пероксодисульфата
электронного переноса и периферической
аммония, описанными в литературе.
сольватации (экранирования) молекул пиррола
цепями поливинилового спирта. Наличие
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
нуклеофильной стабилизации подтверждается
уменьшением энергии активации одноэлектрон-
Пиррол (extra pure, Acros Organics) и
ного окисления пиррола в присутствии
пероксодисульфат
аммония
марки
ЧДА
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 7 2019
1138
МЕЖУЕВ и др.
использовали без дополнительной очистки. В
Shashwati Sen, Patil V.B. // Soft Nanosci. Lett. 2011.
качестве стабилизатора использовали поливини-
Vol. 1. P. 6. doi 10.4236/snl.2011.11002
ловый спирт марки BF-14 (CCP, Тайвань) со
6. Ayad M.M. // J. Polym. Sci. (A). 1994. Vol. 32. N 1.
степенью полимеризации 1100-1200 и степенью
P. 9. doi 10.1002/pola.1994.080320102
гидролиза >98.5%.
7. Khati K., Joshi I., Bisht A., Zaidi M.G.H. // IOSR J.
Appl. Chem. 2016. Vol. 9. N 1. P. 42. doi 10.9790/5736-
Методика эксперимента. В герметичном
09114248
толстостенном реакторе в 70 мл дистиллированной
8. Heinze J. // Synth. Met. 1991. Vol. 43. P. 2805. doi
воды растворяли навеску поливинилового спирта
10.1016/0379-6779(91)91183-B
BF-14 при 353 K и интенсивном перемешивании.
9. Cavallaro S., Colligiani A., Cum G. // J. Therm. Anal.
Приготовленный раствор охлаждали до комнатной
1992. Vol. 38. N 12. P. 2649. doi 10.1007/bf01979741
температуры и растворяли 0.067 г (10-3 моль)
10. Otero T.F., Angulo E. // J. Appl. Electrochem. 1992.
пиррола. Также в 30 мл дистиллированной воды
Vol. 22. N 4. P. 369. doi 10.1007/bf01092691
растворяли
0.456 г
(2×10-3 моль) пероксоди-
11. Schlenoff J.B., Fong Y., Shen C. // Macromolecules.
сульфата аммония.
Приготовленные растворы
1991. Vol.
24. N
25. P.
6791. doi
10.1021/
предварительно термостатировали при установлен-
ma00025a039
ной температуре, а затем смешивали. Определение
12. Fong Y., Chen C., Schlenoff J.B.// MRS Proceedings.
текущей концентрации пиррола проводили
1992. Vol. 247. P. 693. doi 10.1557/proc-247-693
потенциометрическим методом в соответствии с
13. Planche M.F., Thiéblemont J.C., Mazars N., Bidan G. //
принципом, описанным в работе
[23], с
J. Appl. Polym. Sci. 1994. Vol. 52. N 13. P. 1867. doi
использованием рН-метра ИПЛ-311.
10.1002/app.1994.070521304
14. Bjorklund R.B. // J. Chem. Soc. Faraday Trans. 1. 1987.
Реакцию проводили при температурах 298, 303,
Vol. 83. N 5. P. 1507. doi 10.1039/f19878301507
308 и 313 K при концентрации поливинилового
15. Tan Y., Ghandi K. // Synth. Met. 2013. Vol. 175. P. 183.
спирта BF-14
4.4 г/л. Также кинетические
doi 10.1016/j.synthmet.2013.05.014
измерения проводили при 298 K и различных
16. Межуев Я.О., Коршак Ю.В., Штильман М.И.,
концентрациях поливинилового спирта BF-14:
Пискарева А.И. // Вестн. ВГУ. Сер. химия, биология,
0.88, 1.32 и 1.76 г/л.
фармация. 2012. № 2. С. 42.
17. Asavapiriyanont S., Chandler G.K., Gunawardena G.A.,
ФОНДОВАЯ ПОДДЕРЖКА
Pletcher D. // J. Electroanal. Chem. 1984. Vol. 177.
P. 229. doi 10.1016/0022-0728(84)80225-9
Работа выполнена при финансовой поддержке
18. Sabouraud G., Sadki S., Brodie N., Sabouraud G. //
Российского химико-технологического университета
Chem. Soc. Rev. 2000. Vol. 29. N 5. P. 283. doi
им. Д.И. Менделеева (проект № 006-2018).
10.1039/a807124a
19. Takakubo M. // J. Electroanal. Chem. Int. Electrochem.
КОНФЛИКТ ИНТЕРЕСОВ
1989. Vol. 258. N 2. P. 303. doi 10.1016/0022-0728(89)
85116-2
Авторы заявляют об отсутствии конфликта
20. Andrieux C.P., Audebert P., Hapiot P., Saveant J.M. //
интересов.
J. Phys. Chem. 1991. Vol. 95. N 24. P. 10158. doi
10.1021/j100177a096
СПИСОК ЛИТЕРАТУРЫ
21. Genies E.M., Bidan G., Diaz A.F. // J. Electroanal.
Chem. Int. Electrochem. 1983. Vol. 149. N 1-2. P. 101.
1. Wallace G.G., Teasdale P.R., Spinks G.M., Kane-
doi 10.1016/s0022-0728(83)80561-0
Maguire L.A. Conductive electroactive polymers:
intelligent polymer systems. Florida: CRC Press, 2008.
22. Меньшикова А.Ю., Шабсельс Б.М., Евсеева Т.Г. //
264 p.
ЖПХ. 2003. Т. 76. № 5. С. 851; Men’shikova A.Y.,
Shabsel’s B.M., Evseeva T.G. // Russ. J. Appl. Chem.
2. Pron A., Kucharski Z., Budrowski C., Zagorska M.,
2003. Vol.
76. N
5.
Р.
822. doi
10.1023/
Krichene S., Suwalski J., Dehe G., Lefrant S. // J. Chem.
A:1026094211161
Phys. 1985. Vol. 83. P. 5923. doi 10.1063/1.449624
23. Межуев Я.О., Коршак Ю.В., Штильман М.И.,
3. Armes S.P. // Synth. Met. 1987. Vol. 20. P. 365. doi
Соловьева И.В. // ЖОХ. 2014. T. 84. № 12. C. 2029;
10.1016/0379-6779(87)90833-2
Mezhuev Ya.O., Korshak Yu.V., Shtilman M.I.,
4. Machida S., Miyata S., Techagumpuch A. // Synth. Met.
Solovyova I.V. // Russ. J. Gen. Chem. 2014. Vol. 84.
1989. Vol. 31. P. 311. doi 10.1016/0379-6779(89)90798-4
N 12. P. 2445. doi 10.1134/S1070363214120160
5. Chougule M.A., Pawar S.G., Godse P.R., Mulik R.N.,
24. Межуев Я.О., Коршак Ю.В., Штильман М.И. //
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 7 2019
ДИСПЕРСИОННАЯ ОКИСЛИТЕЛЬНАЯ ПОЛИМЕРИЗАЦИЯ ПИРРОЛА
1139
Хим. физика. Т. 34. № 3. С. 76; Mezhuev Ya.O.,
27. Межуев Я.О., Коршак Ю.В., Штильман М.И. // Усп.
Korshak Yu.V., Shtilman M.I. // Russ. J. Phys. Chem.
хим. 2017. T.
86.
№ 12. C. 1271; Mezhuev Ya.O.,
(B).
2015. Vol.
9. N
2. P.
306. doi
10.1134/
Korshak Yu.V., Shtilman M.I. // Russ. Chem. Rev. 2017.
S1990793115020098
Vol. 86(12). P. 1271. doi 10.1070/RCR4755
28. Межуев Я.О., Коршак Ю.В., Штильман М.И.,
25. Страхов И.С., Межуев Я.О., Коршак Ю.В.,
Пискарева А.И. // Вестн. ВГУ. Сер. химия, биология,
Штильман М.И. // ЖОХ. 2016. Т. 86. № 12. С. 2050;
фармация. 2013. № 1. С. 24.
Strakhov I.S., Mezhuev Ya.O., Korshak Yu.V., Shtil-
29. Can M., Özaslan H., Işıldak Ö., Pekmez N.Ö., Yıldız A. //
man M.I. // Russ. J. Gen. Chem. 2016. Vol. 86. N 12.
Polymer. 2004. Vol. 45. N 20. P. 7011. doi 10.1016/
P. 2682. doi 10.1134/S1070363216120185
j.polymer.2004.08.003
26. Межуев Я.О., Коршак Ю.В., Штильман М.И.,
30. Межуев Я.О., Коршак Ю.В., Штильман М.И. //
Похил С.Э., Страхов И.С. // ЖОХ. 2015. T. 85. № 6.
ЖОХ. 2016. Т. 86. № 11. С. 1878; Mezhuev Ya.O.,
C. 1017; Mezhuev Ya.O., Korshak Yu.V., Shtilman M.I.,
Korshak Yu.V., Shtil’man M.I. // Russ. J. Gen. Chem.
Pokhil S.E., Strakhov I.S. // Russ. J. Gen. Chem. 2015.
2016. Vol.
86. N
11. P.
2520. doi
10.1134/
Vol. 85. P. 1482. doi 10.1134/S1070363215060213
S1070363216110190
Dispersion Oxidative Polymerization of Pyrrole
in Aqueous Solutions of Polyvinyl Alcohol
Ya. O. Mezhuyeva, *, I. V. Plyushchiia, Yu. V. Korshaka, M. I. Shtil’mana, and I. A. Gritskovab
a D.I. Mendeleev University of Chemical Technology, Miusskaya pl. 9, Moscow, 125047 Russia
* e-mail: valsorja@mail.ru
b M.V. Lomonosov Institute of Fine Chemical Technologies, Russian University of Technology, Moscow, Russia
Received December 13, 2018; revised March 7, 2019; accepted March 10, 2019
The kinetics of the oxidative polymerization of pyrrole in an aqueous solution of polyvinyl alcohol under the
action of ammonium peroxodisulfate was studied. A quantitative description of the obtained kinetic data with
regard to the formation of a new phase was given, the values of activation energy for the non-catalytic and
catalytic stages of the oxidative polymerization of pyrrole were determined. Various mechanisms of the
oxidative polymerization of pyrrole were critically examined and the comparison of the calculated kinetic
parameters with their values described in the literature for the oxidative polymerization of pyrrole in the absence
of stabilizers was given.
Keywords: polypyrrole, oxidative polymerization, pyrrole, polyvinyl alcohol, kinetics
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 7 2019