ЖУРНАЛ ОБЩЕЙ ХИМИИ, 2019, том 89, № 8, с. 1206-1215
УДК 547.558.1
ФУНКЦИОНАЛИЗАЦИЯ
2-ФОСФОРИЛЗАМЕЩЕННЫХ ФЕНОЛОВ
© 2019 г. Ю. И. Рогачёваa, Д. В. Баулинb, *, В. Е. Баулинa, b, А. Ю. Цивадзеb
a Институт физиологически активных веществ Российской академии наук, Черноголовка, Россия
b Институт физической химии и электрохимии имени А. Н. Фрумкина Российской академии наук,
Ленинский пр. 31, Москва, 119991 Россия
*e-mail: ximik1988200811@rambler.ru
Поступило в Редакцию 12 марта 2019 г.
После доработки 12 марта 2019 г.
Принято к печати 14 марта 2019 г.
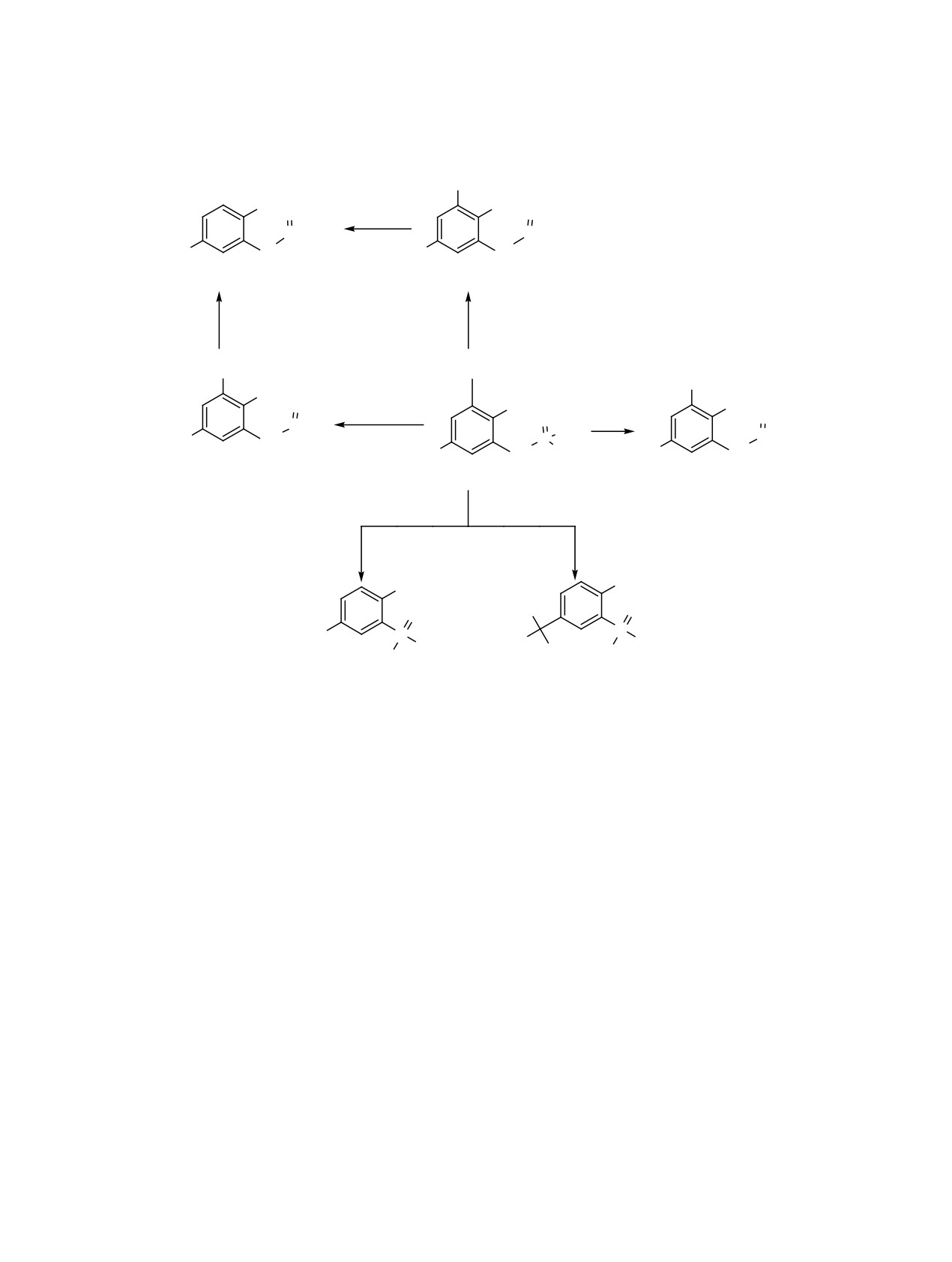
Впервые исследованы реакции нитрования, бромирования, азосочетания, сульфирования и алкилиро-
вания фенольного кольца ряда 2-фосфорилфенолов. С использованием 1,3-фосфат-фосфонатной пе-
регруппировки впервые получены 2,6-дифосфорилфенолы. Изучены особенности алкилирования ок-
сигруппы 2-дифенилфосфорилфенола этиленбром- и этиленхлоргидрином в условиях термического и
микроволнового нагрева.
Ключевые слова: 2-фосфорилзамещенные фенолы, электрофильное замещение, 1,3-фосфат-фосфо-
натная перегруппировка, 2,6-дифосфорилфенолы
DOI: 10.1134/S0044460X19080092
2-Фосфорилзамещенные фенолы и их комплек-
при фосфорильной группе является весьма эффек-
сы с металлами представляют интерес для прове-
тивным подходом к изменению координирующих
дения исследований в области органической, коор-
свойств 2-фосфорилфенолов, что вместе с введе-
динационной, физической и медицинской химии.
нием алкильных и хромофорных заместителей в
В частности, описаны комплексы 2-фосфорилфе-
фенольное кольцо открывает широкие возможно-
нолов c лантанидами (Eu3+, Gd3+, Tb3+, Lu3+), ко-
сти для направленной модификации физико-хи-
торые являются потенциальными компонентами
мических свойств и физиологической активности.
люминесцентных материалов [1, 2]. Установлено,
Если методы получения 2-фосфорилзамещенных
что 2-фосфорилфенолы являются избирательными
фенолов с различными заместителями при фос-
ионофорами по отношению к катиону цезия в при-
форильной группе достаточно хорошо исследова-
сутствии щелочных и щелочноземельных метал-
ны [7-11], то возможности их функционализации
лов [3] и обладают достаточно высокой анальге-
путем введения заместителей в фенольное кольцо
тической и противовоспалительной активностью
практически не изучены.
[4]. 2-Фосфорилфенолы используются для полу-
чения фосфорилсодержащих фталоцианинов
-
В настоящей работе впервые исследованы
перспективных соединений для создания новых
реакции нитрования, бромирования, сульфиро-
функциональных материалов и для применения в
вания, азосочетания и алкилирования феноль-
фотодинамической терапии онкологических забо-
ного кольца 2-фосфорилфенолов 1-4 (схема 1).
леваний [5]. Особый интерес 2-фосфорилфенолы
Восстановлением соответствующих нитро- и диа-
представляют в качестве исходных соединений
зопроизводных впервые синтезирован аминозаме-
для получения фосфорилсодержащих подандов -
щенный 2-фосфорилфенол 17. С использованием
перспективных органических комплексообразую-
1,3-фосфат-фосфонатной перегруппировки впер-
щих соединений [6]. Варьирование заместителей
вые получены 2,6-дифосфорилфенолы 22 и 23.
1206
ФУНКЦИОНАЛИЗАЦИЯ 2-ФОСФОРИЛЗАМЕЩЕННЫХ ФЕНОЛОВ
1207
Схема 1.
R2
OH
OH
O
O
Fe, HCl
PPh2
PPh2
H2N
(CH2)n
R1
(CH2)n
17-18
5-8
Fe, HCl
HNO3
R2
R2
R2
OH
O
OH
OH
(1) NaH
O
O
Br2
PPh2
R3
(2) +N≡N-Ar
P
PPh2
R1
(CH2)n
R1
(CH2)nR3
R1
(CH2)n
13-16
1-4
9-11
H2SO4 (98%)
t-BuOH, PPA
OH
OH
H3C
O
O
P
NaO3S
P
H
3C
Ph
Ph
CH3
Ph
Ph
19
12
R1 = R2 = H, R3 = Ph, n = 0 (1); R1 = R2 = H, R3 = OEt, n = 0 (2); R1 = R2 = H, R3 = Ph, n = 1 (3); R1 = Et, R2 = H, R3 =
Ph, n = 0 (4); R1 = NO2, R2 = H, n = 0 (5); R1 = R2 = NO2, n = 0 (6); R1 = NO2, R2 = H, n = 1 (7); R1 = Et, R2 = NO2, n = 0
(8); R1 = Br, R2 = H, n = 0 (9); R1 = R2 = Br, n = 1 (10); R1 = Br, R2 = H, n = 1 (11); R1 = N=NPh, R2 = H, n = 0 (13); R1 =
N = NC6H4-4-OMe, R2 = H, n = 0 (14); R1 = N=NPh, R2 = H, n = 1 (15); R1 = Et, R2 = N=NPh, n = 0 (16); n = 0 (17), 1 (18).
Исследованы особенности взаимодействия фос-
вании нитрующей смеси (60%-ной НNO3 и 98%-
форилфенола 1 с этиленбромгидрином в условиях
ной H2SO4) при комнатной температуре основным
термического и микроволнового нагрева.
продуктом нитрования фенола 1 является 4,6-ди-
нитро-2-дифенилфосфорилфенол 6. Нитрование
Электроноакцепторная фосфорильная группа
соединений 1 и 3 раствором азотной кислоты в
в 2-фосфорилфенолах 1, 2 и 4 существенно сни-
ацетонитриле при 60°С дает в качестве основных
жает способность к электрофильному замещению
продуктов мононитросоединения 5 и 7 соответ-
фенольного кольца. В частности, 2-дифенилфос-
ственно, которые легко отделяются от примесей
форилфенол 1 не нитруется разбавленной (30%-
колоночной хроматографией. На основании спек-
ной) азотной кислотой в интервале температур
тров ЯМР 31Р установлено, что избыток азотной
20-60°С. При нитровании фенола 3 в аналогичных
кислоты приводит лишь к незначительному ко-
условиях получено нитросоединение 7 с выходом
личеству динитропроизводного 6. При наличии в
30% (схема 1). Это позволяет полагать, что разде-
исходном 2-фосфорилфеноле заместителя в поло-
ление метиленовым фрагментом фенольного коль-
жении 4 фенольного кольца (соединение 4) нитро-
ца и фосфорильной группы частично нивелирует
вание раствором азотной кислоты в ацетонитриле
ее электронноакцепторной эффект. При использо-
происходит в положение 6 (соединение 8).
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 8 2019
1208
РОГАЧЁВА и др.
Схема 2.
OH
OP(O)Ph2
Ph2P(O)Cl, Py
O
R
P
P
R
R
R
O
1, 2
20, 21
Li
P(O)Ph2
OP(O)Ph2
OH
LDA, ТГФ, -78oC
H+, 20oC
R
O
P
P
R
R
O
R
20a, 21a
22, 23
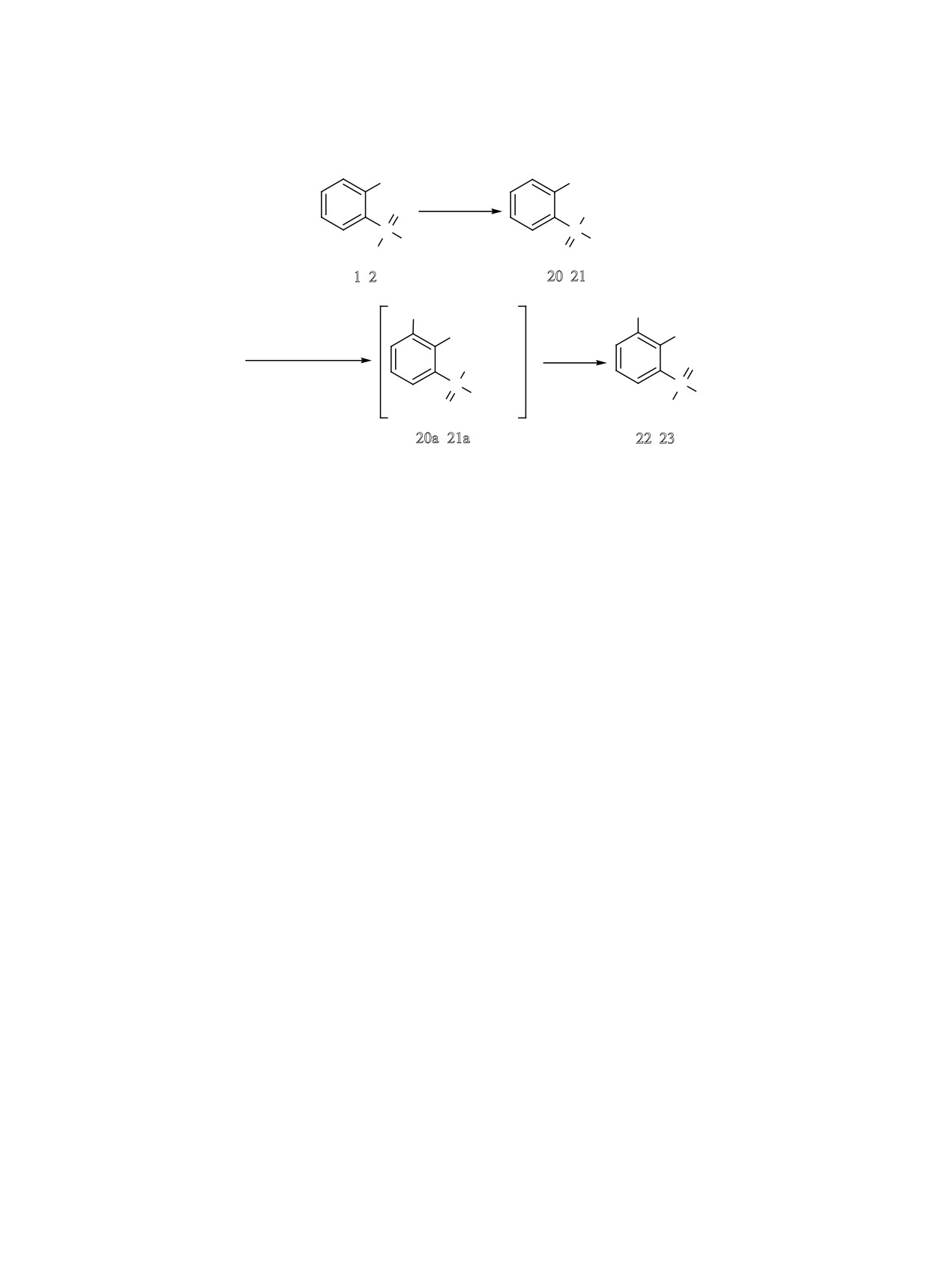
R = Ph (1, 20, 20a, 22), OEt (2, 21, 21a, 23).
Взаимодействием 2-фосфорилированных фе-
использовали эфиры дифенилфосфиновой кис-
нолов 1 и 3 с раствором брома в четыреххлористом
лоты - 2-(дифенилфосфорил)фенилдифенилфос-
углероде в присутствии железа в качестве катали-
финат 20 и 2-(диэтоксифосфорил)фенилдифенил-
затора синтезированы соответствующие 4-бром-
фосфинат 21, которые получены взаимодействием
производные 9 и 11 (схема 1). Избыток брома при-
фенолов 1 и 2 с хлорангидридом дифенилфосфи-
водит к образованию дибромпроизводного 10.
новой кислоты в присутствии пиридина в качестве
Реакция фенола 1 с избытком трет-бутило-
основания (схема 2).
вого спирта в присутствии полифосфорной кис-
По-видимому, по аналогии с фениловым эфи-
лоты (PPA) дает в качестве основного продукта
ром дифенилфосфиновой кислоты [9] при взаи-
трет-бутильное производное
12. Остальными
модействии эфиров 20 и 21 с диизопропиламидом
продуктами реакции, по-видимому, являются
лития в ТГФ при -78°C на первой стадии обра-
орто-изомер и/или продукт дизамещения.
зуется литийорганические соединения 20а и 21а,
При взаимодействии щелочных суспензий или
которые при повышении температуры перегруп-
растворов 2-фосфорилфенолов 1, 3 и 4 с рядом со-
пировываются в соответствующие фенолы 22 и 23
лей арилдиазония впервые получены соединения
(схема 2). Следует отметить, что в аналогичных
13-16 с хромофорными диазофенильными фраг-
условиях перегруппировка сложного эфира ди-
ментами (схема 1).
фенилфосфиновой кислоты и 2-(дифенилфосфо-
Сульфопроизводное 19 получено взаимодей-
рилметил)фенилфенола не приводит к получению
соответствующего 2,6-дифосфорилфенола - из ре-
ствием 2-фосфорилфенола 1 с избытком серной
акционной смеси выделена и идентифицирована
кислоты при 80°С и выделено в виде натриевой
соли соответствующей сульфокислоты (схема 1).
лишь дифенилфосфиновая кислота.
Известно, что электрофильную 1,3-фосфат-фос-
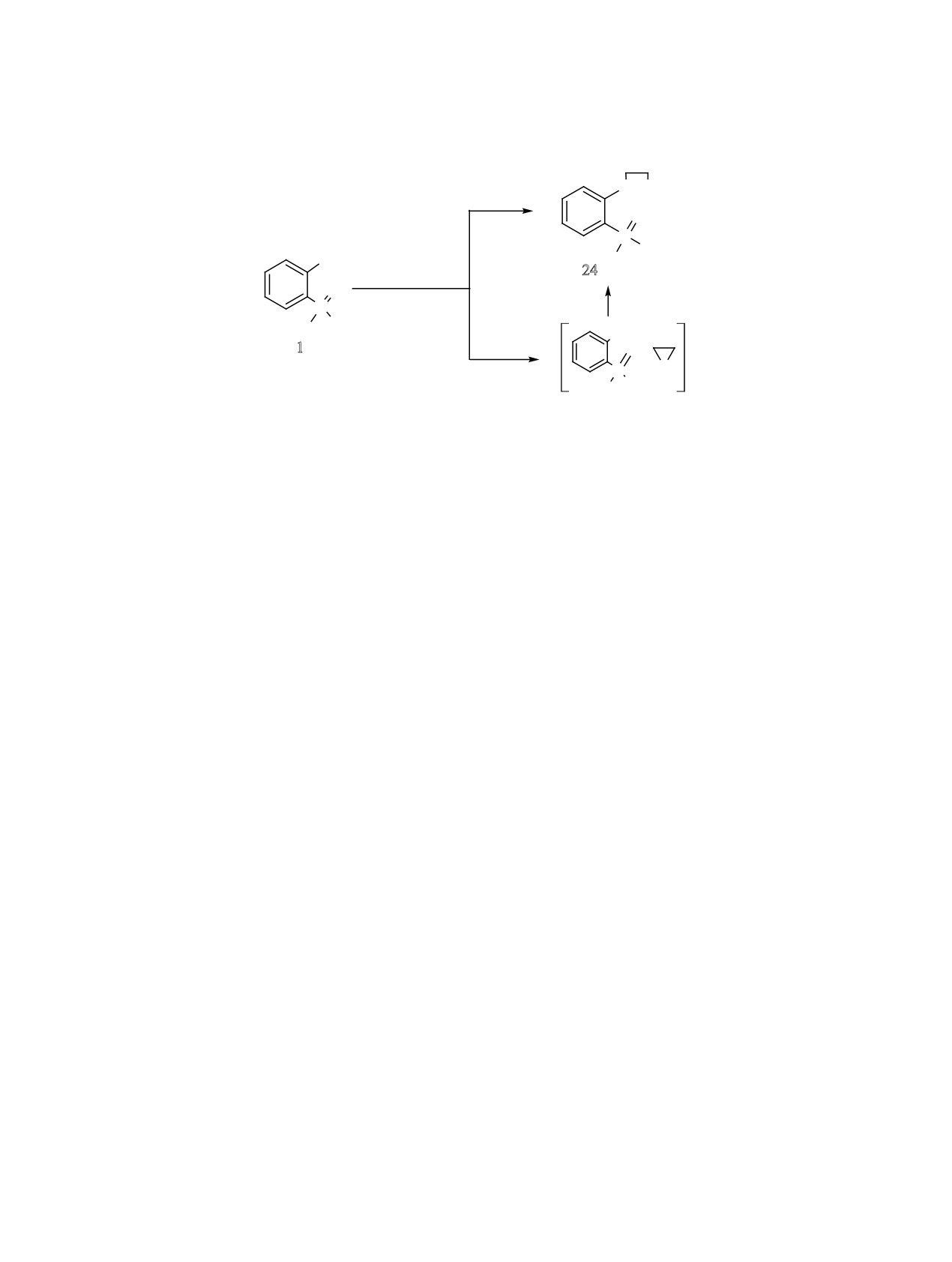
Исследованы особенности взаимодействия фос-
фонатную перегруппировку, включающую разрыв
форилфенола1 с этиленгалогенгидринами(схема3).
O-P связи и образование новой cвязи Ar-P, широко
Так, при взаимодействии натриевого фенолята 1
применяют для получения 2-оксиарилфосфонатов
с этиленбромгидрином в кипящем диоксане при
и бис-2-оксиaрилфосфинатов [12-19]. В настоя-
100°С быстро устанавливается нейтральная среда,
щей работе эта перегруппировка использовалась
и выход соединения 24 составляет 20-25%. Замена
для получения новых 2,6-дифосфорилфенолов 22
этиленбромгидрина на этиленхлоргидрин суще-
и 23 (схема 2). В качестве исходных соединений
ственно не сказывается на выходе соединения 24.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 8 2019
ФУНКЦИОНАЛИЗАЦИЯ 2-ФОСФОРИЛЗАМЕЩЕННЫХ ФЕНОЛОВ
1209
Схема 3.
O
OH
20-25%
O
P
Ph
ONa
Ph
24
Hlg(CH2)2OH
O
100-120oC
94%
P
Ph Ph
OH
1
MW
O
+
P
O
Ph Ph
Hlg = Cl, Br.
Для увеличения выхода 2-дифенилфосфорилфе-
ному замещению фенольного кольца. Наличие
ноксиэтанола 24 в реакционную среду необходимо
метиленового фрагмента между фосфорильной
несколько раз последовательно вводить дополни-
группой и фенольным кольцом уменьшает де-
тельные количества гидроксида натрия и этилен-
зактивирующий эффект фосфорильной группы.
бромгидрина, что существенным образом увели-
Установлено, что электрофильное замещение про-
чивает расход реагентов. По-видимому, в данных
исходит преимущественно в положение 4 феноль-
условиях идет циклизация этиленгалогенгидринов
ного кольца, а при наличии заместителя в этом по-
в низкокипящую окись этилена, которая удаляет-
ложении - в положение 6.
ся из реакционной смеси. Проведение синтеза в
Исследовано взаимодействие 2-(дифенилфос-
закрытом сосуде (запаянной ампуле) при соотно-
форил)фенилдифенилфосфината и
2-(диэтокси-
шении реагентов 1:1 и температуре 100-120°С в
фосфорилфенил)дифенилфосфината с ди(изо-
течение 12 ч повышает выход целевого соедине-
пропил)амидом лития как вариант внутримоле-
ния 24 до 94%. В данном случае образующаяся
кулярного электрофильного замещения, в резуль-
окись этилена остается в реакционной среде и
тате получены новые 2,6-дифосфорилфенолы
практически количественно взаимодействует со
с препаративными выходами. Установлено, что
свободным фенолом. Проведение данной реакции
О-алкилирование в 2-фосфорилфенолах этиленга-
в герметично закрытой лабораторной системе с
логенгидринами протекает как прямое замещение
фокусированным микроволновым нагревом со-
галогена фенолят-ионом, так и через образование
кращает время процесса до 10 мин. При мощно-
циклической окиси этилена, с последующим рас-
сти 30 Вт и максимальном значении температуры
крытием цикла путем взаимодействия с феноль-
120°С выход соединения 24 составил 91%. При
ной ОН-группой.
этом использование избытка этиленбромгидрина
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
приводит к образованию смолообразных неиден-
тифицированных соединений.
Спектры ЯМР 1H и 31Р записаны на спектроме-
Строение и состав всех полученных соедине-
тре Bruker СХР-200. Температуры плавления изме-
ний установлены методами спектроскопии ЯМР и
рены на приборе Boetius PHMK 05. Для проведе-
элементного анализа (см. таблицу).
ния синтезов с применением микроволнового на-
Изучение ряда реакций электрофильного заме-
грева использовали установку с фокусированным
щения дифенилфосфорил-, дифенилфосфорилме-
микроволновым нагревом (Focused MicrowaveTM
тил- и диэтоксифосфорилфенолов показало, что
Synthesis) типа DISCOVER (CEM Corp., США, мак-
электроноакцепторная фосфорильная группа су-
симальная мощность - 300 Вт, частота - 2450 МГц),
щественно снижает способность к электрофиль-
снабженную датчиками давления и температуры.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 8 2019
1210
РОГАЧЁВА и др.
Данные элементного анализа 2-фосфорилфенолов 4-24
Найдено, %
Вычислено, %
№
Брутто-формула
С
Н
Р
С
Н
Р
4
74.32, 74.65
5.56, 6.03
9.43, 9.68
C20H19O2P
74.52
5.94
9.61
5
63.58, 63.94
4.10, 4.21
9.06, 9.14
C18H14NO4P
63.72
4.16
9.13
6
56.17, 56.31
3.27, 3.45
8.01, 8.11
C18H13N2O6P
56.26
3.41
8.06
7
64.51, 65.62
4.52, 4.61
8.58, 8.82
C19H16NO4P
64.59
4.56
8.77
8
65.27, 65.43
4.89, 4.97
8.39, 8.47
C20H18NO4P
65.39
4.94
8.43
9
57.88, 57.94
3.74, 3.86
7.91, 8.31
C18H14BrO2P
57.93
3.78
8.30
10
48.86, 48.98
3.14, 3.33
6.63, 6.68
C19H15Br2O2P
48.96
3.24
6.65
11
58.89, 58.95
4.13, 4.18
7.98, 8.02
C19H16BrO2P
58.94
4.16
8.00
12
75.37, 75.43
6.57, 6.63
8.78, 8.86
C22H23O2P
75.41
6.62
8.84
13
72.28, 71.41
4.79, 4.86
7.66, 7.89
C24H19 N2O2P
72.35
4.81
7.77
14
70.06, 70.10
4.88, 4.96
7.21, 7.25
C25H21N2O3P
70.09
4.94
7.23
15
72.77, 72.84
5.10, 5.16
7.49, 7.53
C25H21N2O2P
72.81
5.13
7.51
16
73.19, 73.25
5.39, 5.46
7.23, 7.28
C26H23N2O2P
73.23
5.44
7.26
17
69.88, 69.93
5.18, 5.23
9.99, 10.04
C18H16NO2P
69.90
5.21
10.01
18
70.53, 70.61
5.59, 5.63
9.53, 9.59
C19H18NO2P
70.58
5.61
9.58
19
54.51, 54.57
3.53, 3.59
7.79, 7.85
C18H14NaO5PS
54.55
3.56
7.82
20
72.86, 72.93
4.83, 4.90
12.50, 12.56
C30H24O3P2
72.87
4.89
12.53
21
61.20, 61.70
5.58, 5.66
14.37, 14.41
C22H24O5P2
61.40
5.62
14.39
22
72.86, 72.90
4.87, 4.93
12.49, 12.55
C30H24O3P2
72.87
4.89
12.53
23
61.00, 60.90
5.51, 5.67
14.00, 14.49
C22H24O5P2
61.40
5.60
14.40
24
70.07, 70.09
5.55, 5.78
8.92, 8.99
C20H19O3P
70.08
5.70
9.20
Контроль за протеканием реакций и степенью чи-
2-Дифенилфосфорил-4-этилфенол (4) полу-
стоты синтезируемых соединений осуществляли
чен аналогично соединению 1 [4] из 28.0 мл 1.96 н.
методом тонкослойной хроматографии на пласти-
раствора n-BuLi в гексане, 9.3 г (56.0 ммоль) ме-
нах Silufol. Проявление хроматограмм осуществля-
токсиметилового эфира 4-этилфенола в 90 мл ТГФ
ли парами иода. Для колоночной хроматографии
и 11.8 г (50.0 ммоль) хлорангидрида дифенил-
фосфиновой кислоты. Выход 12.60 г (78%), т. пл.
использовали силикагель марки L (100-160 мкм).
198-200°С (этанол). Спектр ЯМР 1Н (СDCl3), δ,
Эксперименты с использованием литийоргани-
м. д.: 1.15 т (3Н, Ar-CH2CH3, 3JHH = 7.3 Гц), 2.45 к
ческих соединений проводили в атмосфере сухого
(2Н, Ar-CH2CH3, 3JHH = 7.3 Гц), 6.77 д (1Н, Ar-Н,
аргона.
3JHP = 8.3 Гц), 6.95 д. д (4JHH = 8.3 Гц, 1Н, Ar-Н),
Синтез соединений 1-3 описан нами ранее в ра-
7.30 д (1Н, Ar-Н, 3JHH = 10.0 Гц), 7.60 м (6Н, Ph-Н),
ботах [4, 7, 20] соответственно.
7.75 м (4Н, Ph-Н), 11.00 с (1Н, ArOH). Спектр
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 8 2019
ФУНКЦИОНАЛИЗАЦИЯ 2-ФОСФОРИЛЗАМЕЩЕННЫХ ФЕНОЛОВ
1211
ЯМР 13С (CDCl3), δC, м. д.: 16.40 (CH3CH2), 28.28
2-Дифенилфосфорил-4-этил-6-нитрофенол
(CH3CH2), 118.90 д (Ar, JCP = 8.2 Гц), 129.00 д (Ph,
(8) получали аналогично соединению 5 из 1.10 г
JCP = 12.5 Гц), 130.80 д (Ar, JCP = 10.0 Гц), 132.35
(3.40 ммоль)
2-дифенилфосфорил-4-этилфенола
д. д (Ar, JCP = 52.8 Гц), 132.50 д (Ph, JCP = 10.8 Гц),
4 и 0.23 мл HNO3 (d = 1.5 г/мл) в 10 мл ацетони-
132.80 д (Ph, JCP = 2.5 Гц), 136.00 (Ar), 164.00 (Ar).
трила. Выход 0.76 г (64%), т. пл. 229-231°С (эта-
Спектр ЯМР 31Р (СDCl3): δP 40.7 м. д.
нол). Спектр ЯМР 1Н (СDCl3), δ, м. д.: 1.24 т (3Н,
СН3СН2-Ar, 3JHH = 7.8 Гц), 2.67 к (2Н, СН3СН2-Ar,
2-Дифенилфосфорил-4-нитрофенол
(5).
К
2JHH = 7.4 Гц), 7.50 м (6Н, Ph-Н), 7.77 м (4Н, Ph-Н),
раствору 1.00 г (3.40 ммоль) фенола 1 в 10 мл аце-
8.11 с (1Н, Ar-Н), 8.17 д (1Н, Ar-Н, 3JHP = 13.1 Гц),
тонитрила добавляли 0.23 мл HNO3 (d = 1.5 г/мл).
11.20 с (1Н, ArOH). Спектр ЯМР 31Р (СDCl3): δP
Смесь перешивали 0.5 ч при 60°С, затем выли-
28.36 м. д.
вали в стакан со льдом. Осадок отфильтровали и
очищали колоночной хроматографией, элюент -
2-Дифенилфосфорил-4-бромфенол
(9).
К
СHCl3. Выход 0.58 г (50%), т. пл. 243-245°С (эта-
суспензии 2.00 г (6.80 ммоль) 2-дифенилфосфо-
нол). Спектр ЯМР 1Н (ДМСО-d6-CCl4), δ, м. д.:
рилфенола 1 и 0.10 г (0.18 ммоль) железного по-
7.05 м (1Н, Ar-Н), 7.56 м (4Н, Ph-Н), 7.67 м (6Н,
рошка в 15 мл безводного СCl4 добавляли 1.20 г
Ph-Н), 8.30 д (1Н, Ar-Н, 2JHH = 4.0 Гц), 8.70 д (1Н,
(6.80 ммоль) брома. Смесь перемешивали 3 ч при
Ar-Н, 3JHP = 6.0 Гц), 11.80 с (1Н, ArОН). Спектр
50°С и еще 2 ч при 25°С, затем выливали в 100 мл
ЯМР 13С (CDCl3), δC, м. д.: 119.90 д (Ar, JCP =
воды и экстрагировали СHCl3 (3×30 мл). Экстракт
7.2 Гц), 128.80 д (Ar, JCP = 11.6 Гц), 129.60 д (Ph,
сушили Na2SO4, затем удаляли растворитель в ва-
JCP = 13.8 Гц), 130.16 (Ar), 132.40 д (Ph, JCP =
кууме. К остатку добавляли 25 мл эфира, осадок
11.6 Гц), 133.80 д (Ph, JCP = 2.2), 164.00 (Ar).
отфильтровывали и сушили. Выход 1.06 г (42%),
Спектр ЯМР 31Р (СDCl3): δP 40.96 м. д.
т. пл. 218-220°С (бензол). Спектр ЯМР 1Н (СDCl3),
δ, м. д.: 7.08 д (1Н, Ar-Н, 3JHP = 8.4 Гц), 7.56 м (12H,
2-Дифенилфосфорил-4,6-динитрофенол
(6).
Ar-H + Ph-H), 10.40 с (1Н, ArOH). Спектр ЯМР 31Р
Смесь 1.00 г (3.40 ммоль) соединения 1, 0.35 г
(СDCl3): δP 38.85 м. д.
(3.4 ммоль) 60%-ной НNO3 (d = 1.37 г/мл) и 10 мл
98%-ной H2SO4 перемешивали 4 ч при 25°С, затем
2-Дифенилфосфорилметил-4,6-дибромфенол
выливали в 100 мл холодной воды. Осадок отфиль-
(10) получали аналогично из 2.00 г (6.80 ммоль)
тровывали, промывали на фильтре водой и суши-
соединения 1, 0.02 г (0.36 ммоль) железного по-
ли в вакуумном эксикаторе над P2O5. Выход 0.90 г
рошка и 2.40 г (13.60 ммоль) брома в 25 мл СCl4.
(69%), т. пл. 158-160°С (ДМФА). Спектр ЯМР 1Н
Выход 3.13 г (51%), т. пл. 178-180°С (этанол).
(СDCl3), δ, м. д.: 7.74 м (10Н, Ph-H), 8.70 д (1Н,
Спектр ЯМР 1Н (СDCl3), δ, м. д.: 3.74 д [2Н,
Ar-Н, 3JHP = 9.3 Гц), 9.10 c (1Н, Ar-Н), 12.60 с (1Н,
СН2Р(О), 2JHH = 13.1 Гц], 6.90 с (1Н, Ar-Н), 7.60 м
Ar-ОН). Спектр ЯМР 13С (CDCl3), δC, м. д.: 125.92
(10Н, 1Н, Ar-Н + Ph-Н), 9.00 с (1Н, ArOH). Спектр
(Ar), 127.55 д (Ar, JCP = 109.5 Гц), 129.52 д (Ph,
ЯМР 31Р (СDCl3): δP 39.92 м. д.
JCP = 12.1 Гц), 132.40 д (Ph, JCP = 11.2 Гц), 133.90
2-Дифенилфосфорилметил-4-бромфенол
д (Ph, JCP = 2.6), 134.60 д (JCP = 10.77, Ar), 161.30
(11) получали аналогично из 2.10 г (6.80 ммоль)
(Ar). Спектр ЯМР 31Р (СDCl3): δP 33.45 м. д.
2-дифенилметилфосфорилфенола 3, 0.02 г (0.36
2-Дифенилфосфорилметил-4-нитрофенол
ммоль) железного порошка и 1.20 г (6.80 ммоль)
брома в 15 мл СCl4. Выход 1.16 г (44%), т. пл. 194-
(7) получали аналогично соединению 5 из 1.05 г
(3.40 ммоль) 2-дифенилфосфорилметилфенола 3 и
195°С (изопропанол). Спектр ЯМР 1Н (СDCl3), δ,
м. д.: 3.64 д [2Н, СН2Р(О), 2JHP = 12.2 Гц], 6.65 д
0.23 мл HNO3 (d = 1.5 г/мл) в 10 мл ацетонитри-
ла. Выход 0.67 г (58%), т. пл. 179-181°С (этанол).
(1Н, Ar-Н, 3JHH = 5.6 Гц), 7.02 д (1Н, Ar-Н, 3JHH =
7.4 Гц), 7.27 с (1Н, Ar-Н), 7.47 м (6Н, Ph-Н), 7.75
Спектр ЯМР 1Н (ДМСО-d6), δ, м. д.: 3.87 д (2Н, Ar-
м (4Н, Ph-Н), 9.74 с (1Н, ArOH). Спектр ЯМР 31Р
Н, 2JHH = 13.8 Гц), 6.80 д (1Н, Ar-Н, 3JHH = 7.5 Гц),
(СDCl3): δP 33.64 м. д.
7.50 м (6Н, Ph-Н), 7.80 м (4Н, Ph-Н + 1Н, Ar-Н),
8.10 м (1Н, Ar-Н), 10.94 с (1Н, ArOH). Спектр ЯМР
2-Дифенилфосфорил-4-трет-бутилфенол
31Р (ДМСО-d6): δP 33.24 м. д.
(12). Смесь 5.00 г (17.00 ммоль) фосфорилфенола
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 8 2019
1212
РОГАЧЁВА и др.
1, 6.00 г полифосфорной кислоты и 10 мл t-BuOH
ной суспензии гидрида натрия в вазелиновом масле
перемешивали 8 ч при 80°С, затем разбавляли
и 5.30 г (50.00 ммоль) фенилдиазония. Выход 2.49 г
100 мл холодной воды и экстрагировали СHCl3
(48%), т. пл.
213-215°С (этанол). Спектр ЯМР
(3×30 мл). Экстракт промывали водой (3×30 мл),
1Н (СDCl3), δ, м. д.: 3.85 д [2Н, СН2Р(О), 2JHH =
затем 10%-ным раствором NaHCO3 (5×30 мл) и
11.8 Гц], 7.09 д (1Н, Аr-Н, 3JHH = 8.6 Гц), 7.40-7.65
снова водой (3×30 мл). После удаления раство-
м (10Н, Ph-Н), 7.73-7.84 м (7Н, Ar), 11.70 c (1Н,
рителя остаток хроматографировали, элюент -
АrOH). Спектр ЯМР 31Р (СDCl3): δP 39.70 м. д.
СHCl3. Выход 4.40 г (70%), т. пл. 213-215°С (эта-
2-Дифенилфосфорил-4-этил-6-(фенилдиа-
нол) (т. пл. 213-215°С [19]). Спектр ЯМР 1Н
зенил)фенол (16) получали аналогично из 4.00 г
(СDCl3), δ, м. д.: 1.15 c [9Н, Ar-С(СН3)3], 6.85-7.80
(12.40 ммоль) фенола 4, 0.54 г (12.40 ммоль) 55%-
м (13Н, Ar-Н + Ph-Н), 10.50 с (1H, ArОН). Спектр
ной суспензии гидрида натрия в вазелиновом масле
ЯМР 31Р (СDCl3): δP 40.10 м. д.
и 5.30 г (50.00 ммоль) фенилдиазония. Выход 1.19 г
2-Дифенилфосфорил-4-фенилдиазенил-
(48%), т. пл. 141-143°С (этанол). Спектр ЯМР 1Н
фенол (13). К суспензии 3.60 г (12.4 ммоль) фено-
(СDCl3), δ, м. д.: 1.30 т (3Н, СН3СН2Ar, 3JHH =
ла 1 в 30 мл безводного диоксана добавляли 0.54 г
7.6 Гц), 2.76 м (2Н, СН3СН2Ar), 7.42-7.98 м (17Н,
(12.40 ммоль) 55%-ной суспензии гидрида натрия
Ph-H + Ar-H), 13.53 с (1Н, ArOH). Спектр ЯМР 31Р
в вазелиновом масле. Смесь кипятили 0.5 ч, затем
(СDCl3): δP 28.34 м. д.
охлаждали до 0°С и по каплям при температуре
2-Дифенилфосфорил-4-аминофенол (17). а.
2-5°С добавляли к раствору фенилдиазония, по-
К 1.00 г (2.94 ммоль) 2-дифенилфосфорил-4-ни-
лученного из 4.70 г (50.00 ммоль) анилина, 3.50 г
трофенола 5 добавляли 0.50 г (8.92 ммоль) же-
(50.00 ммоль) NaNO2 и 20 мл 35%-ной HCl. Смесь
лезных стружек в 10 мл HCl. Смесь кипятили 2 ч,
перемешивали 2 ч при комнатной температуре,
затем охлаждали. Осадок отфильтровывали,
затем отфильтровывали осадок, промыли разбав-
промывали холодной водой. Остаток растворяли
ленной HCl (1:3, 3×30 мл) и сушили. Выход 2.27 г
в 20 мл СНСl3. Полученный раствор промывали
(46%), т. пл. 193-195°С (этанол). Спектр ЯМР 1Н
разбавленным (1:1) раствором HCl (2×15 мл), затем
(СDCl3), δ, м. д.: 7.08 м (1Н, Аr-Н), 7.55 м (7Н, Рh-Н),
водой (2×15 мл) и упаривали в вакууме. Остаток
7.76 м (6Н, Ph-Н), 7.82 м (2Н, Рh-Н), 8.05 д (1Н, Аr-
хроматографировали на колонке с силикагелем,
Н, 3JHH = 7.1 Гц), 8.25 м (1Н, Аr-Н, 3JHP = 11.0 Гц),
элюент - CHCl3. Выход 0.64 г (64%), т. пл. 196-
11.25 c (1Н, АrОН). Спектр Спектр ЯМР 31Р
197°С (изопропанол). Спектр ЯМР 1Н (ДМСО-d6),
(СDCl3): δP 41.10 м. д.
δ, м. д.: 4.76 с (2Н, Ar-NH2), 6.70 м (3Н, Ar-H), 7.58
2-Дифенилфосфорил-4-(4-метоксифенил-
м (10Н, Ph-H), 9.59 с (1Н, ArOH). Спектр ЯМР 31Р
диазенил)фенол (14) получали аналогично из
(ДМСО-d6): 30.41 м. д.
3.60 г
(12.40 ммоль) соединения
3,
0.54 г
б. Суспензию 1.00 г (2.51 ммоль) 2-дифенил-
(12.40 ммоль) 55%-ной суспензии гидрида натрия
фосфорил-4-фенилдиазенилфенола 13 и 0.50 г
в вазелиновом масле и 6.75 г (50.00 ммоль) 4-ме-
(8.92 ммоль) железных стружек в 10 мл HCl
токсифенилдиазония. Выход 2.39 г (45%), т. пл.
кипятили 2 ч, затем выливали в 50 мл холодной
259-260°С (этанол). Спектр ЯМР 1Н (ДМСО-d6-
воды. Смесь экстрагировали СHCl3 (3×25 мл).
CCl4), δ, м. д.: 2.40 с (3Н, СН3ОPh), 7.00 м (1Н,
Органический слой промывали насыщенным рас-
Ar-Н), 7.10 д (2Н, СН3ОAr, 2JHH = 6.9 Гц), 7.55 м
твором NaHCO3 (3×25 мл), водой (2×25 мл), затем
(6Н, Ph-H), 7.73 м (4Н, Ph-H), 7.81 д (2Н, СН3ОAr,
упаривали в вакууме. Остаток сушили в вакуум-
3JHH = 7.1 Гц), 7.95 д (1Н, Ar-Н, 3JHH = 5.8 Гц), 8.20
ном эксикаторе над P2O5. Выход 0.73 г (73%), т. пл.
д (1Н, Ar-Н, 3JHH = 5.8 Гц), 12.50 c (1Н, ArОН).
196-198°С (изопропанол). Спектр ЯМР получен-
Спектр ЯМР 31Р (ДМСО-d6-CCl4): δP 40.87 м. д.
ного соединения идентичен спектру ЯМР соеди-
нения 17, полученному по методу а.
2-Дифенилфосфорилметил-4-(фенилдиа-
зенил)фенол (15) получали аналогично из 3.82 г
2-Дифенилфосфорилметил-4-аминофенол
(12.40 ммоль) фенола 3, 0.54 г (12.40 ммоль) 55%-
(18) Получили аналогично из 1.00 г (3.53 ммоль)
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 8 2019
ФУНКЦИОНАЛИЗАЦИЯ 2-ФОСФОРИЛЗАМЕЩЕННЫХ ФЕНОЛОВ
1213
2-дифенилфосфорилметил-4-нитрофенола
7 и
2,6-Бис(дифенилфосфорил)фенол (22). К рас-
0.50 г (8.92 ммоль) железных стружек в 10 мл HCl.
твору 1.02 г (10.00 ммоль) диизопропиламина в
Выход 0.71 г (70%), т. пл. 170-172°С (изопропа-
35 мл безводного ТГФ при температуре -75±5°С
нол). Спектр ЯМР 1Н (СDCl3), δ, м. д.: 3.80 д [2Н,
при перемешивании по каплям добавляли 10.00 мл
СН2Р(О), 4JHP = 12.4 Гц], 6.11 д (1Н, Ar-Н, 2JHH =
1.0 н. раствора n-BuLi в гексане. Смесь переме-
18.0 Гц), 6.46 м (1Н, Ar-Н), 6.86 м (1Н, Ar-Н), 7.72
шивали 0.5 ч. К полученному раствору ди(изо-
м (10Н, Ph-H), 10.70 с (1Н, ArОН). Спектр ЯМР 31Р
пропил)амида лития при температуре -75±5°С
(СDCl3): δP 38.96 м. д.
и перемешивании по каплям добавляли раствор
4.94 г (10.00 ммоль) соединения 20 в 35 мл без-
2-Дифенилфосфорил-4-оксибензолсуль-
водного ТГФ. Реакционную смесь перемешивали
фонат натрия (19). Смесь 1.00 г (3.40 ммоль)
45 мин при температуре -75±5°С, затем подни-
2-дифенилфосфорилфенола 1 и 15 мл 98%-ной
мали температуру до комнатной и перемешивали
H2SO4 перемешивали 4 ч при 80°С, затем выли-
еще 2 ч. После удаления растворителя к остатку
вали в 300 мл насыщенного раствора NaCl. Через
добавляли 15 мл разбавленной HCl (1:3), смесь
24 ч отфильтровывали осадок, который затем
экстрагировали СHCl3 (3×15 мл). Органический
кристаллизовали из насыщенного раствора NaCl.
слой промывали водой, сушили Na2SO4 и упарили
Выход 0.85 г (63%,) т. разл. ≥ 230°С. Спектр ЯМР
в вакууме. Остаток хроматографировали, элюент -
1Н (D2О), δ, м. д.: 6.84-7.44 м (11Н), 7.76 м (2Н),
СHCl3-этанол (1:0.35). Выход 3.29 г (66%), т. пл.
11.20 с (1Н, ArОН). Спектр ЯМР 31Р (D2О): 35.08.
), δ,
202-205°С (гексан). Спектр ЯМР 1Н (СDCl3
2-(Дифенилфосфорил)фенилдифенилфос-
м. д.: 7.24 м (3Н, Ar-Н), 7.38-7.50 м (12Н, Ph-Н),
финат (20). К раствору 6.40 г (21.74 ммоль) 2-ди-
7.76-7.80 м (9Н, Ph-Н), 12.20 с (1Н, ArOH). Спектр
фенилфосфорилфенола 1 и 1.69 г (21.74 ммоль)
ЯМР 31Р (СDCl3), δP, м. д.: 32.11, 34.37.
безводного пиридина в 50 мл безводного бензола
2-Диэтоксифосфорил-6-дифенилфосфорил-
при перемешивании при комнатной температуре
фенол
(23) получали аналогично из
1.09 г
добавляли 5.12 г (21.74 ммоль) хлорангидрида ди-
(10.00 ммоль) диизопропиламина, 11.67 мл 1.0 н.
фенилфосфиновой кислоты. Реакционную массу
раствора n-BuLi в гексане и 5.30 г (10.00 ммоль)
кипятили 2 ч, затем вылили в стакан с 100 мл воды.
соединения 21. Выход 4.35 г (88%), т. пл. 80-82°С
Смесь экстрагировали бензолом (3×30 мл), про-
(гептан). Спектр ЯМР 1Н (СDCl3), δ, м. д.: 1.37 т (6Н,
мывали разбавленной HCl (1:3), затем водой (3×
СН3CH2О, 3JHH = 7.5 Гц), 3.95 м (4Н, СН3CH2О),
30 мл), сушили Na2SO4, затем упаривали в вакууме.
7.25 м (1Н, Ar-H), 7.00-7.95 м (13Н, Ph-H + ArH-H),
Остаток кристаллизовали из смеси бензол-
10.00 с (1Н, ArOH). Спектр ЯМР 31Р (СDCl3), δP,
гексан. Выход 7.23 г (69%), т. пл. 175-177°С (бен-
м. д.: 19.56, 32.55.
зол). Спектр ЯМР 1Н (СDCl3), δ, м. д.: 7.01 м (1Н,
2-Дифенилфосфорилфеноксиэтанол (24). а.
Ar-Н), 7.23-7.56 м (13Н, Ar-H + Ph-H), 7.63-7.90 м
К раствору 8.40 г (30.00 ммоль) 2-дифенилфос-
(10Н, Ar-H + Ph-H). Спектр ЯМР 31Р (СDCl3), δP,
форилфенола 1 в 50 мл абсолютного спирта при-
м. д.: 28.72, 32.25.
бавляли 1.30 г (30.00 ммоль) NaH в виде 55%-ной
2-(Диэтоксифосфорил)фенилдифенил-
суспензии в вазелиновом масле. Реакционную
фосфинат (21) получали аналогично из 5.00 г
смесь кипятили 20 мин, затем упаривали в ва-
(21.74 ммоль)
2-диэтоксифосфорилфенола
4,
кууме. К остатку добавляли 50 мл абсолютного
1.71 г (21.73 ммоль) безводного пиридина и 5.12 г
эфира. Осадок отфильтровывали и сушили в ва-
(21.73
ммоль) хлорангидрида дифенилфос-
кууме (1 мм рт. ст., 80°С, 6 ч). К полученному
финовой кислоты в 50 мл абсолютного бензола.
феноляту добавляли 4.30 г (35.00 ммоль) свеже-
Выход 5.70 г (63%), т. пл. 65-67°С (гексан).
перегнанного этиленбромгидрина в 50 мл без-
Спектр ЯМР 1Н (CDCl3), δ, м. д.: 1.26 т (6Н,
водного диоксана и кипятили 1 ч. При этом рН
СН3CH2, 3JHH = 7.5 Гц), 4.16 м (4Н, СН3CH2),
среды составлял 7. По данным ЯМР 31Р и ТСХ,
7.20 м (1Н, Ar-Н), 7.10-8.32 м (13Н, Ar-H + Ph-H).
конверсия исходного фенола не превышала 20%.
Спектр ЯМР 31Р (СDCl3), δP, м. д.: 20.33, 33.03.
Реакционную смесь охлаждали до 25°С, добавля-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 8 2019
1214
РОГАЧЁВА и др.
ли 1.30 г (15.00 ммоль) 55%-ой суспензии NaH в
ФОНДОВАЯ ПОДДЕРЖКА
вазелиновом масле. Смесь нагревали до кипения
Работа выполнена при частичной финансовой
и перемешивали 20 мин, затем добавляли 4.30 г
поддержке Российского фонда фундаментальных
(35.00 ммоль) этиленбромгидрина и кипятили 1 ч.
исследований (грант № 19-03-00262) в рамках го-
В реакционной среде рН = 7 и конверсия исходно-
сударственного задания. Института физиологиче-
го фенола не превышала 40%. Процедуру добав-
ски активных веществ РАН.
ления NaH и этиленбромгидрина повторяли еще
2 раза, затем удаляли растворитель в вакууме. К
КОНФЛИКТ ИНТЕРЕСОВ
остатку добавляли 15 мл ацетона, смесь кипятили
15 мин, затем отфильтровывали осадок исходного
Авторы заявляют об отсутствии конфликта
соединения 1, который промыли горячим ацето-
интересов.
ном (2×15 мл) и сушили 5 ч при 100°С (m = 2.01 г,
СПИСОК ЛИТЕРАТУРЫ
т. пл. 232-233°С). Фильтрат упаривали до объе-
ма 10 мл, охлаждали до 0°С и отфильтровывали
1.
Shuvaev S., Kotova O., Utochnikova V., Vaschenko A.,
осадок. Выход 7.71 г (76%), т. пл. 154-155°С (аце-
Puntus L., Baulin V., Kuzmina N., Tzivadze A. // Inorg.
Chem. Commun. 2012. Vol. 20. P. 73. doi 10.1016/j.
тон). Спектр ЯМР 1Н (СDCl3), δ, м. д.: 3.56 т (2Н,
inoche.2012.02.020
СН2ОН, 3JHH = 7.4 Гц), 4.02 т (2Н, СН2ОАr, 3JHH =
2.
huvaev S., Bushmarinov I.S., Sinev I., Dmitrienko A.O.,
7.4 Гц), 6.92 м (2Н, Ar-Н), 7.40-7.61м (6Н, Ph-Н),
Lyssenko K.A., Baulin V., Grünert W., Tsivadze A.Yu.,
7.68-7.80 м (6Н, Ar-H + Ph-H), 5.04 с (1Н, ArOH).
Kuzmina N. // E. J. Inorg. Chem. 2013. Vol. 2013. N 27.
Спектр ЯМР 31Р (СDCl3): δP 26.43 м. д.
P. 4823. doi 10.1002/ejic.201300540
б. Смесь 3.20 г (10.00 ммоль) предварительно
3.
Иванова И.С., Баулин В.E., Пятова Е.Н., Илюхин А.Б,
полученного из соединения 1 фенолята натрия,
Галкина Е.Н., Якушевa И.А., Дороватовский П.В.,
1.50 г (12.00 ммоль) свежеперегнанного этилен-
Цивадзе А.Ю. // ЖОХ. 2018. Т. 88. № 9. С. 1524. doi
бромгидрина и 5 мл безводного диоксана выдер-
10.1134/S0044460X18090172
живали 12 ч в запаянной стеклянной ампуле при
4.
Цветков Е.Н., Сюндюкова В.Х., Баулин В.Е. // ЖОХ.
100°С. После охлаждения реакционную смесь рас-
1987. Т. 57. Вып. 11. С. 2456.
творяли в 30 мл СHCl3, добавляли 50 мл воды и
5.
Калашникова И.П., Баулин Д.В., Баулин В.Е.,
подкисляли конц. HCl до рН = 1. Отделяли органи-
Цивадзе А.Ю. // ЖОХ. 2018. Т. 88. № 9. С. 1510.
ческий слой, промывали водой (2×20 мл), сушили
doi 10.1134/S0044460X18090159; Kalashnikova I.P.,
Na2SO4 и упаривали в вакууме. Остаток растворя-
Baulin D.V., Baulin V. E., Tsivadze A. Yu. // Russ. J.
Gen. Chem. 2018. Vol. 88. N 9. P.1853. doi 10.1134/
ли в 7 мл ацетона, охлаждали до 0°С и отфильтро-
S1070363218090153
вывали осадок. Выход 3.20 г (94%).
6.
Баулин В.Е. Автореф. дис. … докт. хим. наук.
в. Смесь 3.20 г (10.00 ммоль) предварительно
Черноголовка, 2012. 40 с.
полученного из соединения 1 фенолята натрия,
7.
Баулин В.Е., Калашникова И.П., Вихарев Ю.Б.,
1.50 г (12.00 ммоль) этиленбромгидрина и 5 мл
Вихарева Е.А., Баулин Д.В., Цивадзе А.Ю.
//
безводного диоксана в специальном закрытом со-
ЖОХ. 2018. Т. 88. № 9. С. 1438. doi 10.1134/
суде помещали в лабораторную систему с фоку-
S0044460X18090044; Baulin V.E., Kalashnikova I.P.,
сированным микроволновым нагревом, который
Vikharev Yu.B., Vikhareva E.A., Baulin D.V., Tsivad-
осуществляли 0.6 ч при мощности 30 Вт. При этом
ze A.Yu. // Russ. J. Gen. Chem. 2018. Vol. 88. N 9.
максимальное значение температуры достигало
P. 1786. doi 10.1134/S1070363218090049
120°С. После охлаждения смесь растворяли в 25 мл
8.
Rauchfuss T.B. // Inorg. Chem. 1977. Vol. 16. N 11.
СHCl3, добавляли 50 мл воды и подкисляли конц.
Р. 2966. doi 10.1021/ic50177a062
HCl до рН = 1. Органический слой отделяли, про-
9.
Melvin L.S. // Tetrahedron Lett. 1981. Vol. 22. N 35. P.
мывали водой (2×25мл), сушили Na2SO4 и упари-
3375. doi 10.1016/S0040-4039(01)81909-2
ли в вакууме. Остаток растворяли в 7 мл ацетона,
10.
Dashan L., Trippett S. // Tetrahedron Lett. 1983. Vol.
охлаждали до 0°С и отфильтровывали осадок.
24. P. 2033. doi 10.1016/S0040-4039(00)81838-9
Выход 2.95 г (91%).
11.
Dhawan B., Redmore D. // Synth. Commun. 1985.Vol. 15.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 8 2019
ФУНКЦИОНАЛИЗАЦИЯ 2-ФОСФОРИЛЗАМЕЩЕННЫХ ФЕНОЛОВ
1215
N 5. P. 411. doi 10.1080/00397918508063819
17. Miller J. // J. Org. Chem. 1987. Vol. 52. N 2. P. 322. doi
10.1021/jo00378a044
12.
Onys’ko P., Suvalova E., Chudakova T., Sinitsa A. //
Нeteroatom Chem. 1993. Vol. 4. N 4. P. 361. doi
18. Mills R.J, Horvath R.F., Sibi M.P., Snieckus V. //
10.1002/hc.520040408
Tetrahedron Lett. 1985. Vol. 26. N 9. P. 1145. doi
10.1016/S0040-4039(00)98418-1
13.
Consiglio G., Failla S., Finocchiaro P., Siracusa V. //
Phosphorus, Sulfur, Silicon, Relat. Elem.
2002.
19. Евреинов В.И., Баулин В.Е., Вострокнутова З.Н.,
Сафронова З.В., Крашакова И.Б., Сюндюкова В.Х.,
Vol. 177. N 11. P. 2561. doi 10.1080/10426500214564
Цветков Е.Н. // Изв. АН СССР. Сер. хим. 1991.
14.
Consiglio G., Failla S., Finocchiaro P., Siracusa V. //
№ 9. С. 1992.
Phosphorus, Sulfur, Silicon, Relat. Elem. 2008. Vol. 134.
20. Евреинов В.И., Баулин В.Е., Вострокнутова З.Н.,
N 1. P. 413. doi 10.1080/10426509808545482
Бондаренко Н.А., Сюндюкова В.Х., Цветков Е.Н. //
15.
Minaeva L., Patrikeeva L., Kabachnik M., Beletskaya I.,
Изв. АН СССР. Сер. хим. 1989. № 9. С. 1990; Evrei-
Orlinson B., Novakov I. // Нeteroatom Chem. 2011.
nov V.I., Baulin V.E., Vostroknutova Z.N., Bondaren-
Vol. 22. N 1. P. 55. doi 10.1002/hc.20656
ko N.A., Syundyukova V.Kh., Tsvetkov E.N. // Bull.
16.
Häbich D., Effenberger F. // Synthesis. 1978. Vol. 10.
Acad. Sci. USSR. Div. Chim. Sci. 1989. Vol. 38.
P. 755. doi 10.1055/s-1978-24879
Р. 1828.
Functionalization of 2-Phosphoryl-Substituted Phenols
Yu. I. Rogachevaa, D. V. Baulinb, *, V. E. Baulina, b, and A. Yu. Tsivadzeb
a Institute of Physiologically Active Substances of the Russian Academy of Sciences, Chernogolovka, Russia
b A.N. Frumkin Institute of Physical Chemistry and Electrochemistry of the Russian Academy of Sciences,
Leninskii pr. 31, Moscow, 119991 Russia
*e-mail: ximik1988200811@rambler.ru
Received March 12, 2019; revised March 12, 2019; accepted March 14, 2019
The nitration, bromination, azocoupling, sulfonation and alkylation reactions of the phenolic ring of some
of 2-phosphorylphenols were investigated for the first time. 2,6-Diphosphorylphenols were synthesized by
1,3-phosphate-phosphonate rearrangement. The some aspects of the alkylation reaction of hydroxyl group of
2-diphenylphosphorylphenol with 2-bromoethanol and 2-cloroethanol under microwave heating were inves-
tigated.
Keywords: 2-phosphoryl-substituted phenols, electrophilic substitution, 1,3-phosphate-phosphonate rear-
rangement, 2,6-diphosphorylphenols
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 89 № 8 2019