ЖУРНАЛ ОБЩЕЙ ХИМИИ, 2020, том 90, № 1, с. 31-41
УДК 547.565:544.43:544.18
СОГЛАСОВАННЫЙ ПЕРЕНОС ЭЛЕКТРОНА
И ПРОТОНА В РЕАКЦИИ ГИДРОКСИБЕНЗОЛОВ
С ГИДРАЗИЛЬНЫМ РАДИКАЛОМ В ВОДНЫХ СРЕДАХ
© 2020 г. Н. И. Белаяa,*, А. В. Белыйa, О. М. Заречнаяb,
И. Н. Щербаковc, В. С. Дорошкевичa
a Донецкий национальный университет, ул. Университетская 24, Донецк, 283001 Украина
b Институт физико-органической химии и углехимии имени Л. М. Литвиненко, Донецк, 283114 Украина
c Южный федеральный университет, Ростов-на-Дону, 344006 Россия
*e-mail: nat.iv.belaya@gmail.com
Поступило в Редакцию 12 июня 2019 г.
После доработки 12 июня 2019 г.
Принято к печати 16 июня 2019 г.
Исследованы возможные механизмы переноса электрона в реакции 2,2'-дифенил-1-пикрилгидразила с
рядом природных гидроксибензолов в водных буферных растворах при рН = 2-9 методами спектрофото-
метрии и квантовой химии. Установлено, что в кислых средах перенос электрона осуществляется от мо-
лекулярной формы гидроксибензола к радикалу с последующей потерей протона, а в щелочных средах -
от фенолят-иона на радикал с предшествующей стадией отрыва протона. О реализации указанных ме-
ханизмов свидетельствует зависимость константы скорости реакции от рН среды и наличие корреляции
между свободной энергией Гиббса активации, рассчитанной по уравнению Маркуса и определенной на
основе экспериментальных данных.
Ключевые слова: гидроксибензол, 2,2'-дифенил-1-пикрилгидразил, перенос электрона, энергия реор-
ганизации
DOI: 10.31857/S0044460X20010047
Наиболее актуальным вопросом ингибирования
возрастает [7-9]. В этом случае имеет место со-
радикальных процессов является исследование
пряженный перенос электрона и протона. Если
антиоксидантной активности веществ в водных
потеря протона предшествует лимитирующей
средах [1-3], что связано, во-первых, с имитацией
стадии переноса электрона, то механизм имену-
биологических систем, а во-вторых, с реализацией
ется как SPLET (Sequential Proton Loss-Electron
в полярных растворителях нескольких механиз-
Transfer), и в реакции непосредственно участвует
мов действия антиоксидантов. Так, в неполярных
ионизированная форма антиоксиданта [1, 8-10].
жироподобных средах предполагается механизм
Это характерно для щелочных сред c рН ≥ 8. Если
отрыва атома водорода, который реализуется как
потеря протона следует за медленной стадией пе-
одновременный перенос электрона и протона на
реноса электрона, то имеет место механизм ET-
радикал либо с одной атомной орбитали донора
PT (Electron Transfer-Proton Transfer) (также упо-
(НАТ, Hydrogen Atom Transfer) [4, 5], либо с его
минается как SET-PT или SEPT) [1, 2, 8]. Тогда в
разных атомных орбиталей (РСЕТ, Proton-Coupled
реакции участвует молекулярная форма антиокси-
Electron Transfer ) [6].
данта, что характерно для водных сред с низким
значением рН ≤ 2-3.
В полярных ионизирующих средах (вода, спир-
ты) все значительно сложнее. При увеличении
Исследование механизма SPLET или ET-PT
полярности растворителя скорость радикальной
связано с двумя крайними случаями - либо с силь-
реакции в присутствии антиоксиданта сильно
нокислыми, либо с сильнощелочными средами. В
31
32
БЕЛАЯ и др.
резорцин 2, орцин 3, гидрохинон 4, метиловый
эфир гидрохинона 5, пирогаллол 6, флороглюцин
7 и оксигидрохинон 8.
В смешанном растворителе ДМСО-буфер в
присутствии всех исследуемых фенолов происхо-
дит расходование DPPH▪, которое можно зафик-
сировать в приемлемом временном интервале при
небольшом содержании буфера, до 10 об% (рис. 1).
Установлены частные порядки реакции по DPPH▪
(nDPPH▪) при 10-20-кратном недостатке радикала,
а также общий порядок реакции (n) и константы
t, c
скорости (k), как характеристики антирадикаль-
ной активности (АРА) вещества при эквимольном
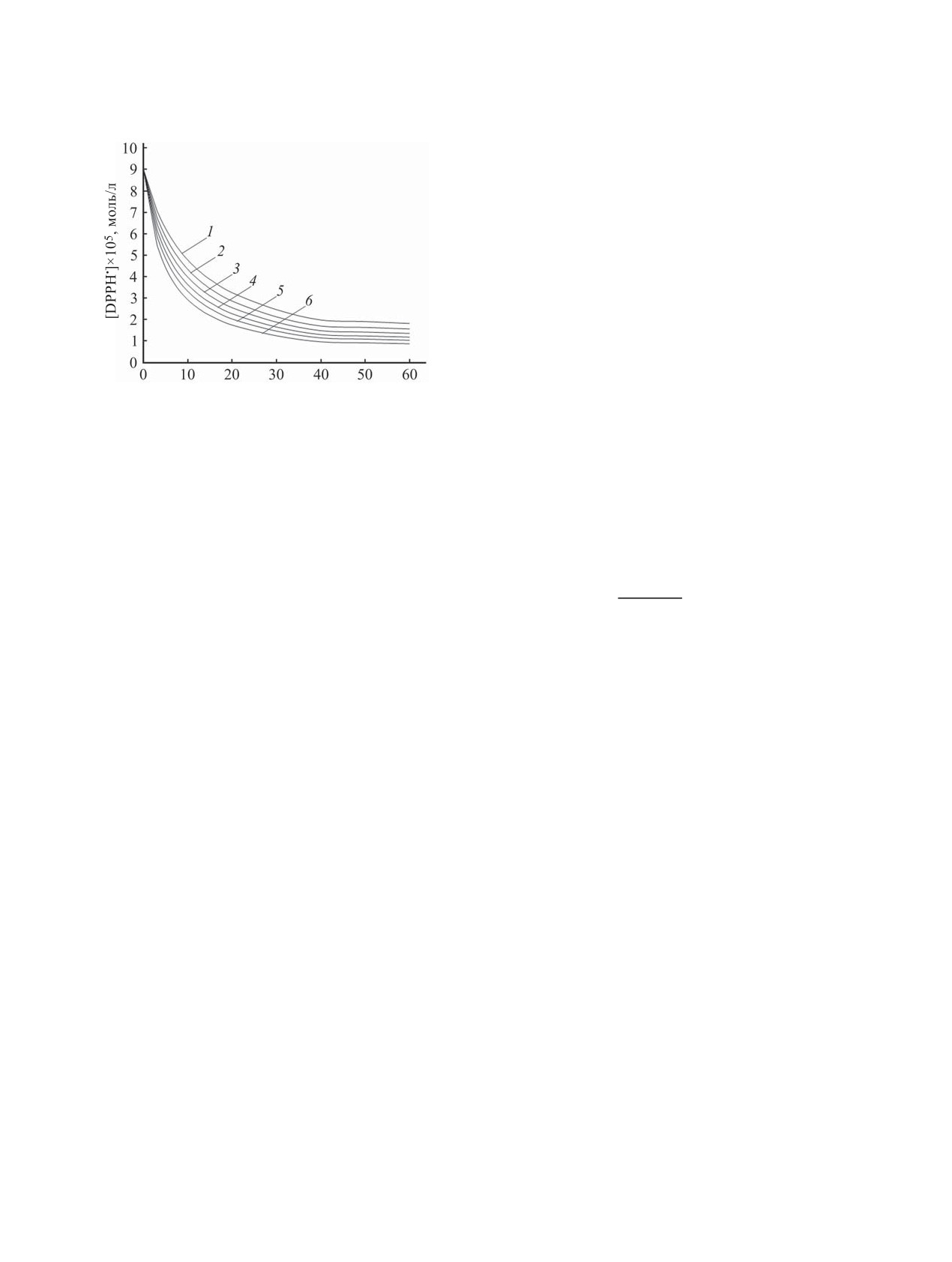
Рис. 1. Кинетические кривые расходования DРРН• в ре-
акции с пирогаллолом (с = 9×10-5 моль/л) при 293±2 K
соотношении фенол-радикал (табл. 1). Для этого
в смеси ДМСО с добавками буфера с рН = 7.35 (об%):
кинетические данные обрабатывали нелинейным
1 - 0, 2 - 2, 3 - 4, 4 - 6, 5 - 8, 6 - 10.
методом обобщенного приведенного градиента
[12], реализованного в Solver MS Excel. В качестве
слабоионизирующих растворителях (нейтральные
критерия выбора порядка реакции использовали
и слабокислые среды, спирты), где антиоксидант
параметр S (1), отражающий относительный раз-
диссоциирует незначительно, имеет место комби-
брос вычисленного ряда констант.
нация обоих механизмов [1, 10], поскольку в этом
n
1
S =
6 |ki k|,
(1)
случае в лимитирующей стадии будут участвовать
i=2
k(n
1)
и молекулярная, и ионная формы антиоксиданта.
Механизм SPLET, как правило, связывают с во-
где n - число опытов, соответствующих моментам
-
дными средами и спиртами [1-3], не проводя де-
времени t; ki и
k - константа скорости реакции в
тального анализа влияния рН среды. Если такой
момент времени t и ее среднее значение. Порядок
анализ и проводится, то путем квантово-химиче-
реакции определяли как значение, при котором
ского расчета [3].
относительный разброс S вычисленных по этому
порядку значений констант скорости реакции был
Целью данной работы является исследование
наименьшим. Кинетические кривые, построен-
возможных механизмов реакции ди- и тригидрок-
ные на основе констант реакции второго порядка,
сибензолов (PhOH) с N-центрированным радика-
лом
2,2'-дифенил-1-пикрилгидразилом (DPPH•)
хорошо согласуются с экспериментальными дан-
при разных значениях рН среды с привлечением
ными. Некоторые отклонения наблюдаются при
экспериментальных методов химической кинети-
степени превращения радикала >80%, что обу-
ки и квантовой химии.
словлено влиянием продуктов реакции превраще-
ния гидроксибензолов [13]. В связи с этим расчет
Спектральное определение кинетических
порядков реакции и констант скорости проводили
параметров реакции PhOH с DPPH▪ в буферном
до 50-60%-ной конверсии DPPH•.
растворе при рН = 7.35. Для изучения кинетики
и механизма взаимодействия PhOH с DPPH• при
Полученные данные (табл. 1) показывают, что
293±2 K были использованы диметилсульфоксид,
реакция исследуемых гидроксибензолов с DPPH▪
буферы с рН = 2-9 и их смеси в разных соотноше-
в смеси ДМСО с нейтральным буфером при
ниях. Применение ДМСО необходимо, поскольку
рН = 7.35 имеет первый псевдопорядок по радика-
радикал DPPH• нерастворим в воде, а также для
лу и второй общий порядок. Сравнение значений
снижения скорости исследуемой реакции за счет
k
в смеси ДМСО-буфер показало, что
ДМСО-буфер
подавления диссоциации PhOH. В качестве акцеп-
при добавлении даже небольшого количества бу-
торов радикала были выбраны гидроксибензолы
фера, когда полярность среды изменяется незна-
природного происхождения [11]: пирокатехин 1,
чительно, реакционная способность PhOH увели-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020
СОГЛАСОВАННЫЙ ПЕРЕНОС ЭЛЕКТРОНА И ПРОТОНА
33
Таблица 1. Экспериментальные значения порядков (n) и констант скоростей (k) реакций ди- и тригидроксибензо-
лов с DPPH• в средах с разной кислотностью при 293±2 K
kДМСО,б
kбуфер(pH=2),
kбуфер(pH=7),
kбуфер(pH=9),
kбуфер(pH=9)/
nDPPH▪a
na
л/(моль∙с)
л/(моль∙с)
л/(моль∙с)
л/(моль∙с)
k
буфер(pH=2)
1
0.99
2.01
(3.71±0.14)×102
(3.4±0.14)×102
(2.85±0.09)×103
(1.47±0.05)×104
43
2
1.01
2.03
2.34±0.09
2.43±0.09
69.3±2.7
(2.71±0.09)×102
111
3
0.98
2.00
3.41±0.11
3.54±0.11
(1.67±0.05)×102
(1.31±0.05)×103
370
4
0.99
2.02
(5.52±0.15)×102
(4.74±0.15)×102
(1.16±0.04)×104
(1.66±0.05)×104
35
5
1.02
1.99
(6.22±0.22)×102
(7.01±0.23)×102
(1.34±0.05)×104
(2.93±0.09)×104
42
6
1.00
2.01
(9.8±0.4)×102
(9.9±0.4)×102
(4.13±0.14)×103
(9.5±0.4)×103
10
7
1.01
2.02
1.42±0.05
1.48±0.05
27.5±1.1
54.0±2.2
37
8
1.02
2.00
(7.3±0.3)×102
(9.0±0.3)×102
(5.67±0.15)×103
(1.31±0.05)×104
15
а Порядки реакции определены в смеси ДМСО с 10 об% буферного раствора при рН = 7.35. б Величины kДМСО взяты из работы [6].
чивается. Таким образом, влияние специфической
ченные антиоксиданты проявляют на 1-2 порядка
сольватации, определяемой способностью раство-
большую антирадикальную активность по сравне-
рителя к образованию водородной связи с раство-
нию с ДМСО. На наш взгляд, ускоряющий эффект
ренным веществом, на величину антирадикальной
воды обусловлен усилением диссоциации PhOH, в
активности гидроксибензолов преобладает над не-
результате с DРРН• реагируют как молекулы фено-
специфической сольватацией, связанной с диэлек-
лов (механизм ET-PT), так и фенолят-ионы (меха-
трической проницаемостью среды.
низм SPLET).
Поскольку гидразильный радикал нерастворим
Разделить механизмы ET-PT и SPLET в ней-
в воде, а скорость исследуемой реакции при боль-
тральном буфере крайне сложно. Для исследо-
шом содержании буфера настолько велика, что не
вания индивидуальных механизмов необходимо
позволяет корректно оценить начальные участки
рассмотреть среды с рН, где будут присутствовать
кинетических кривых, величину антирадикальной
преимущественно или ионные, или молекулярные
активности (kбуфер) гидроксибензолов в чистом
формы антиоксиданта.
буфере определяли из линейной зависимости (2)
Методология квантово-химических расче-
величины kДМСО-буфер от объемной доли буфера,
тов. Квантово-химические расчеты проводили с
вводимого в реакционную смесь.
использованием пакета Gaussian 09 [14] в рамках
lnkДМСО-буфер = lnkДМСОwДМСО + lnkбуфер(1 - wДМСО),
(2)
теории функционала плотности (DFT) с гибрид-
где wДМСО, (1 - wДМСО) - доля ДМСО и буфера в
ным функционалом B3LYP, выбор которого обу-
смешанном растворителе.
словлен успешным применении его для соедине-
Значение kбуфер определяли путем экстрапо-
ний фенольного типа [15, 6]. Поиск стабильных
ляции величин kДМСО-буфер на чистый буферный
конформеров гидроксибензолов предварительно
раствор. По полученным в координатах уравне-
проводили методом PM6, используя полученные
ния (2) зависимостям были рассчитаны констан-
структуры в качестве начального приближения
ты скорости реакции kбуфер(pH=7) в чистом ней-
для расчетов на уровне B3LYP/6-311++G(d,p) - для
тральном буфере из углового параметра линейной
систем с закрытыми оболочками (синглетное ос-
регрессии. Вычисленные значения kбуфер(pH=7)
новное состояние) и UB3LYP/6-311++G(d,p) - для
(табл. 1) указывают на то, что в буфере все изу-
систем с открытыми оболочками (дублетное ос-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020
34
БЕЛАЯ и др.
Таблица 2. Значения рKa гидроксибензолов (по первой ступени диссоциации) и распределение их молекулярной и
ионных форм в зависимости от рН среды
[PhO-] ([PhOH]), %
Соединение
Положение ОН группы
pKa
pH = 2
pH = 7.35
pH = 9
1
1
9.34
0 (100)
1.13
(98.87)
31.25
(68.74)
2
1
9.26
1.35
(98.65)
35.01
(64.33)
3
1
9.39
1.02
(98.98)
28.92
(70.66)
4
1
9.68
0.21
(99.79)
17.4
(82.56)
5
1
9.94
0.12
(99.86)
10.29
(89.71)
6
2
8.94
1.93
(97.18)
36.64
(46.27)
7
1
9.13
1.84
(98.16)
42.15
(56.38)
8
2
9.39
0.59
(98.98)
18.88
(70.64)
новное состояние). Геометрия всех структур была
условиям (298 K, 1 атм) в водной среде, если не
оптимизирована по всем независимым перемен-
указано иное.
ным без ограничений по симметрии для водной
Реакции PhOH с DPPH▪ в буферном растворе
среды. Влияние растворителя учитывалось в рам-
при рН = 2. Механизм ET-РТ. Участие в лими-
ках модели поляризуемого континуума РСМ [16].
тирующей стадии ионизированных форм гидрок-
Для построения полости растворенного вещества
сибензолов подтверждается влиянием добавок бу-
задавались радиусы атомных сфер из модели си-
фера с рН = 2 на кинетику исследуемой реакции
лового поля UFF [17]. Характер всех стационар-
в смешанном растворителе ДМСО-буфер (pH =
ных точек определялся расчетом матрицы Гессе.
7). Замена нейтрального буфера на сильнокислый
Частоты нормальных колебаний (в гармоническом
приводит к резкому уменьшению скорости реак-
приближении) и тепловые поправки к свободной
ции (табл. 1) вследствие подавления диссоциации
энергии (с использованием немасштабированных
фенолов.
частот) были рассчитаны теми же методами. Все
Из распределения ионных форм фенолов в за-
полученные результаты относятся к стандартным
висимости от рН среды при 298 K (растворитель –
вода), рассчитанного методом QSPR в программе
1
Marvin 17.21.0. [18], следует (табл. 2, рис. 2), что
4
при рН = 2 в системе присутствуют только моле-
кулярные формы. В этом случае может реализо-
2
вываться ET-PT механизм переноса электрона с
гидроксибензола на радикал с последующей по-
терей протона образующимся катион-радикалом
3
6
(PhOH•+) (3).
5
PhOH + DPPH• ĺ PhOH•+ + DPPH-,
PhOH•+ + H2O ĺ PhO•+ + H3O+,
pH
(3)
DPPH- + H3O+ ĸ DPPH- + H2O.
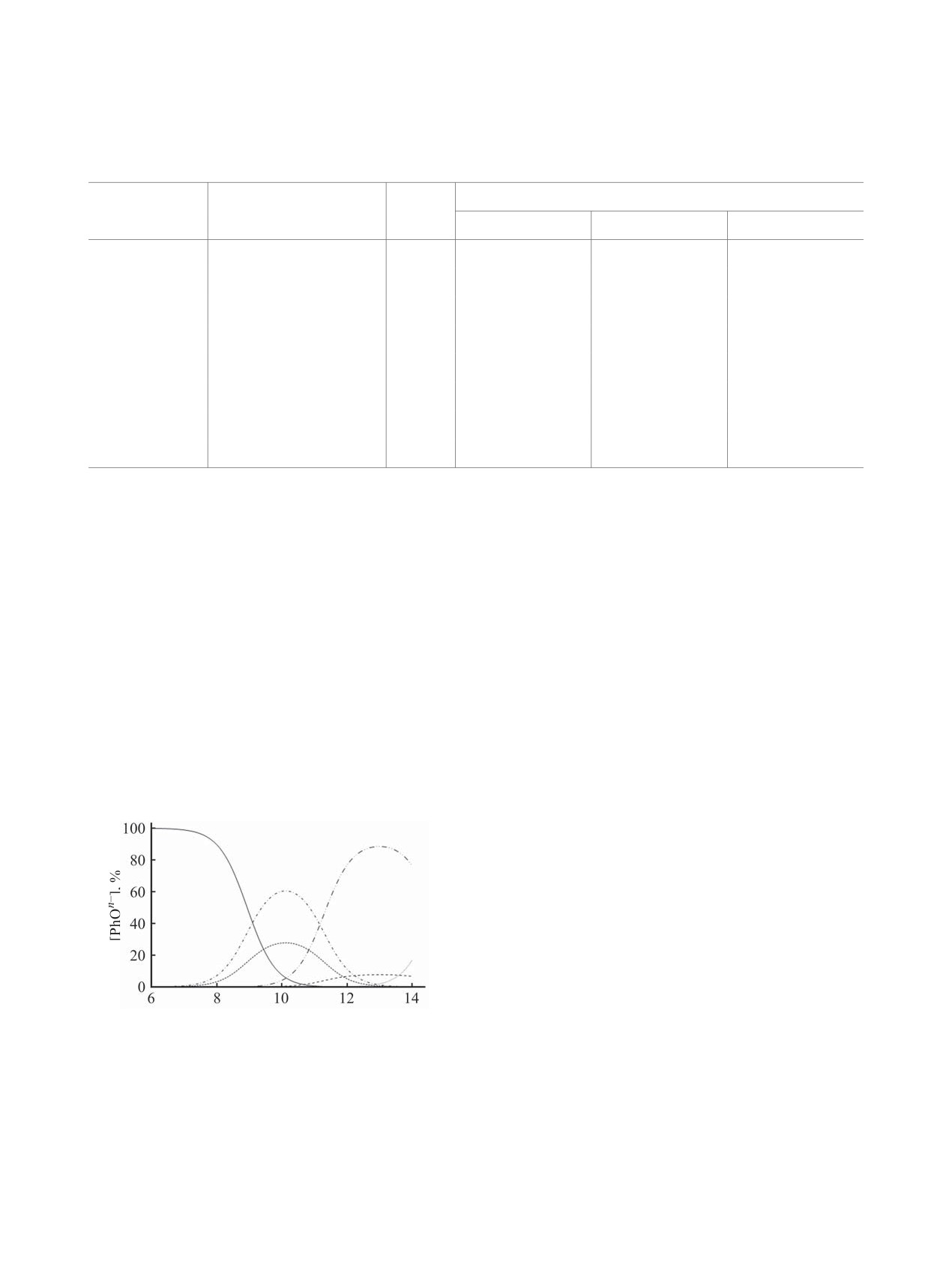
Рис. 2. Распределение доли ионных форм пирогаллола
(PhOН) в зависимости от рН среды (вода, 298 K), рас-
Диссоциация PhOH•+ идет очень быстро, так
считанное методом QSPR в программе Marvin 17.21.
1 - PhOН, 2 - PhO- (2-ОН), 3 - PhO- (1-ОН), 4 - PhO2-
как его кислотность много выше, чем исходной
(1,3-ОН), 5 - PhO2- (1,2-ОН), 6 - PhO3- (1,2,3-ОН).
молекулы PhOH.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020
СОГЛАСОВАННЫЙ ПЕРЕНОС ЭЛЕКТРОНА И ПРОТОНА
35
Механизм НАТ с участием молекулярной фор-
Мы применили подобный подход к реакциям
мы (4) для данных реакций не рассматривался,
ди- и тригидроксибензолов с DРРН• в водных сре-
поскольку вероятность его протекания в полярных
дах. На основе полученных данных рассчитывали
средах крайне мала, что подтверждается как лите-
изменение энергии Гиббса (ΔG0ET-PT) реакции (3)
ратурными данными [1-3, 7-10], так и проведен-
и (ΔEET-PT) в соответствии с выражениями (7) и
ными раннее исследованиями [6].
(8).
PhOH + DPPH• → PhO• + DPPH - H.
(4)
ΔG0ET- PT = (GPhOH•+ + GDPPH-) - (GPhOH + GDPPH•), (7)
Теоретическую идентификацию того или иного
ΔEET-PT = (EPhOH•+ + EDPPH-) - (EPhOH + EDPPH•). (8)
механизма реакции переноса электрона проводили
Здесь GPhOH, GPhOH•+, GDPPH-, GDPPH• - энергия
с применением упрощенной формы классической
Гиббса молекулы гидроксибензола и его катион-ра-
теории Маркуса, которая, несмотря на иногда су-
дикала, аниона и радикала DРРН• соответственно;
щественные количественные расхождения, обе-
EPhOH, EPhOH•+ - полная электронная энергия моле-
спечивает хорошее качественное согласие с экспе-
кулы гидроксибензола и его катион-радикала, рас-
риментом для множества реакций [10, 19].
считанного из геометрии PhOH, EDPPH•, EDPPH- -
Уравнение Маркуса, базирующееся на теории
общая электронная энергия радикала и его аниона,
переходного состояния [20], позволяет связать ки-
рассчитанного из геометрии DРРН•.
нетический параметр реакции переноса электрона -
По величине рассчитанной энергии Гиббса
свободную энергию активации ΔG≠ с термодина-
видно (табл. 3), что реакция (3) эндоэргична
мическим - стандартной свободной энергией ре-
(ΔG0ET-PT > 0). Несмотря на то, что теоретические
акции ΔG° (5).
свободные энергии активации ΔG≠ET-PT завышены
по сравнению с экспериментальными значениями
O
'G
z
(5)
'G
(1 +
)2,
ΔG≠exp(pH=2) для кислых сред на 25-78 кДж/моль,
4
O
они хорошо коррелируют между собой.
где λ - энергия реорганизации, необходимая для
предварительной частичной перестройки среды и
ΔG≠ET-PT = -(95±11) + (3.25±0.17) ΔG≠exp(pH=2),
(9)
ядерной конфигурации реагентов, которая созда-
r2 = 0.982, F = 352, p < 0.00000, Sest = 3.32.
ет условия для последующего быстрого перехода
Здесь n - число опытов; ~r - коэффициент корре-
электрона от донора к акцептору. Для оценки λ
ляции; ~r2 - коэффициент детерминации; F - крите-
часто используется метод приближенного расче-
рий Фишера; p - уровень значимости, при котором
та, основанный на разнице энергий начальных и
может быть принята нуль-гипотеза (о равенстве
конечных состояний электронного переноса (так
нулю истинного углового коэффициента уравне-
называемый четырехточечный метод [19]).
ния регрессии); Sest - стандартноее отклонение.
λ ≈ ΔE - ΔG0,
(6)
Регрессионный анализ проводили в программе
где ∆E - разность полных электронных энергий
Statistica Demo 6.0.
реагентов и продуктов для вертикального пере-
Зависимость ΔG≠ET-PT от ΔG≠exp(pH=9) имеет наи-
носа электрона. Соответствующие энергии E рас-
более низкий коэффициент корреляции (r = 0.928)
считываются без термических и колебательных
для щелочных сред, что логично, поскольку при
поправок для 0 K, так как относятся к нестацио-
рН = 9 доля активных ионных форм в системе
нарным состояниям.
велика (табл. 2), а значит антирадикальная актив-
В работе [10] описанная приближенная схема
ность будет определяться более быстрым механиз-
расчета была успешно реализована для большого
мом SPLET.
набора реакций антиоксидантов со свободными
Наличие лучшей линейной зависимости в кис-
радикалами, затем теоретические значения ΔG≠
лых средах свидетельствует о возможности реа-
сравнивали с ΔG≠exp, рассчитанными на основе
лизации механизма ЕТ-РТ. Завышенные по срав-
экспериментальных констант скоростей (k) реак-
нению с экспериментальными значения энергии
ции с использованием формализма теории пере-
активации связаны, по-видимому, с участием в
ходного состояния.
реакции переноса электрона не индивидуальной
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020
36
БЕЛАЯ и др.
Таблица 3. Расчетные (ΔG≠) и экспериментальные (ΔG≠exp) величины свободных энергий активации, энергий реор-
ганизации (λ) и свободных энергий (∆G°) реакций (3) и (13)
G°ET-PT,
λET-PT,
ΔG≠ET-PT,
ΔG°SPLET,
λSPLET,
ΔG≠SPLET,
ΔG≠exp(pH=2),
ΔG≠exp(pH=7),
ΔG≠exp(pH=9),
кДж/моль
кДж/моль
кДж/моль
кДж/моль
кДж/моль
кДж/моль
кДж/моль
кДж/моль
кДж/моль
1
97.4
88.8
97.6
-63.1
74.3
0.42
58.5
53.3
49.2
2
124.8
70.1
135.4
-26.4
58.7
4.45
70.8
62.5
59.1
3
120.6
68.2
130.7
-30.2
56.9
3.14
69.9
60.3
55.2
4
93.8
71.6
95.5
-60.4
77.5
0.93
57.7
49.8
48.9
5
84.2
72.2
84.7
-64.1
72.9
0.27
56.8
49.4
47.5
6
88.7
99.0
88.9
-38.7
58.9
1.73
55.9
52.4
50.3
7
128.1
74.8
137.6
-20.6
59.1
6.26
72.0
64.8
63.1
8
82.2
70.9
82.6
-78.2
92.6
0.56
56.1
51.6
49.5
молекулы гидроксибензола, как предполагается в
так и в качестве его акцептора. Исходя из расчетных
большинстве работ [1, 2, 10], а комплекса с водо-
энтальпий образования донорных (ΔH0PhOH···H
=
2O
родной связью PhOH···H2O (10) [4].
–15.8 кДж/моль) и акцепторных (ΔH0PhOH···H
=
2O
–7 кДж/моль) Н-комплексов гидрохинона, более
PhOH + H2O →← PhOH···H2O,
устойчивыми являются донорные комплексы
PhOH···H2O + DPPH• → PhOH•+···H2O + DPPH-,
(рис. 3a), что подтверждается литературными дан-
PhOH•+···H2O →← PhO• + H3O+.
(10)
ными [22, 23]. Поэтому дальнейший расчет про-
Мы оценили возможность реализации меха-
водилb для структур, в которых донором протона
низма ЕТ-РТ с участием в лимитирующей стадии
является фенол.
(10) комплекса PhOH···H2O, используя комбини-
О наличии межмолекулярной водородной свя-
рованный подход, улучшающий точность расчета
зи PhOH···H2O свидетельствует [24] ряд характер-
[21], в котором сочетаются явный учет гидратации
ных признаков (табл. 4): межмолекулярная связь
фенолов одной молекулой воды с континуальной
r(Н∙∙∙О) значительно короче суммы ван-дер-ва-
моделью РСМ.
альсовых радиусов, составляющих для H и O
Гидроксибензол относительно молекулы воды
0.12 и 0.14 нм соответственно; в Н-комплексе дли-
может выступать как в качестве донора протона,
на фенольной O-H связи r(О-Н) увеличивается по
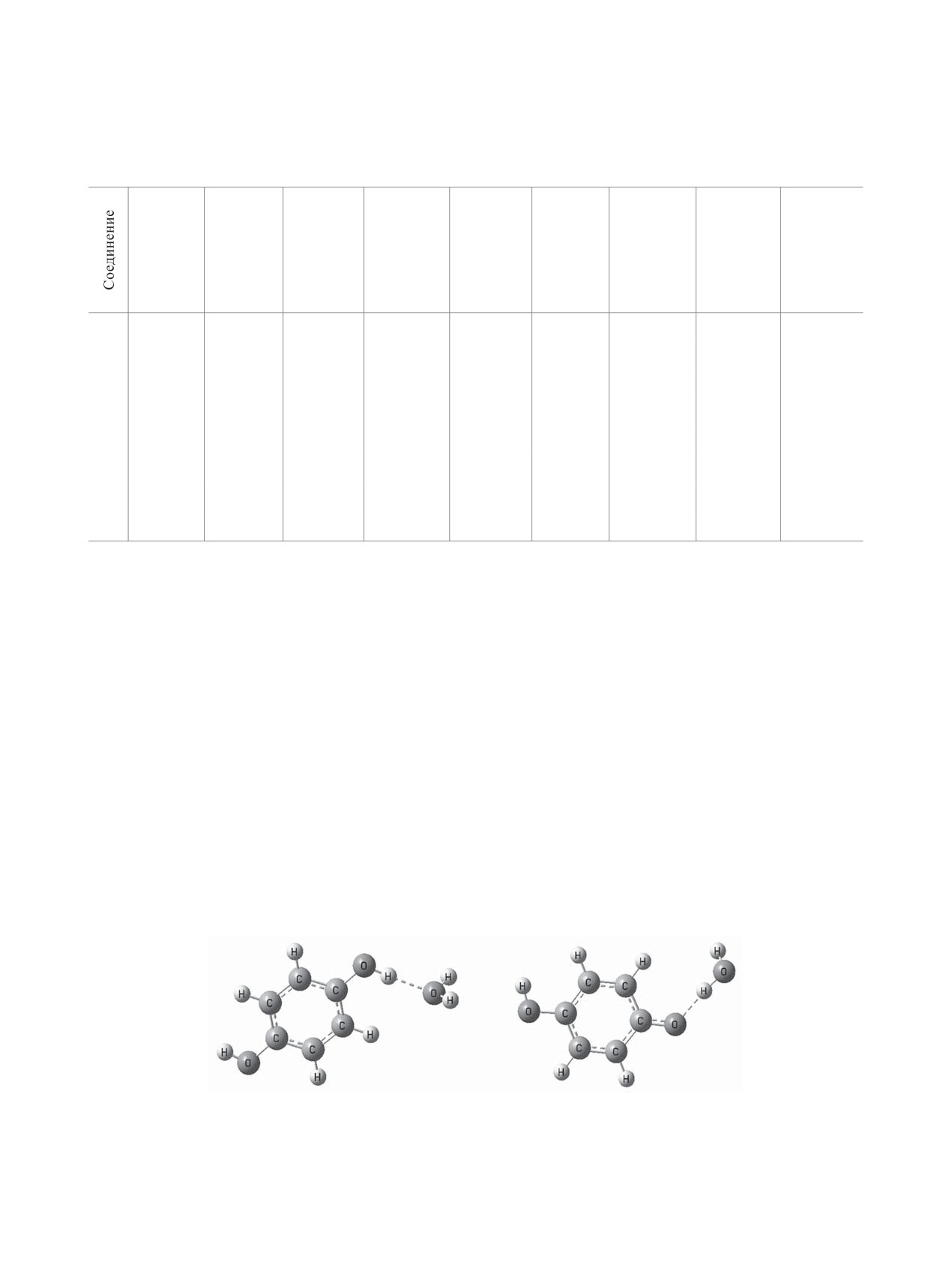
ɚ
ɛ
Рис. 3. Структура донорного межмолекулярного (а) и акцепторного ион-молекулярного (б) Н-комплекса молекулы и аниона
гидрохинона с водой (растворитель - вода) по данным метода B3LYP/6-311++G(d,p), PCM.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020
СОГЛАСОВАННЫЙ ПЕРЕНОС ЭЛЕКТРОНА И ПРОТОНА
37
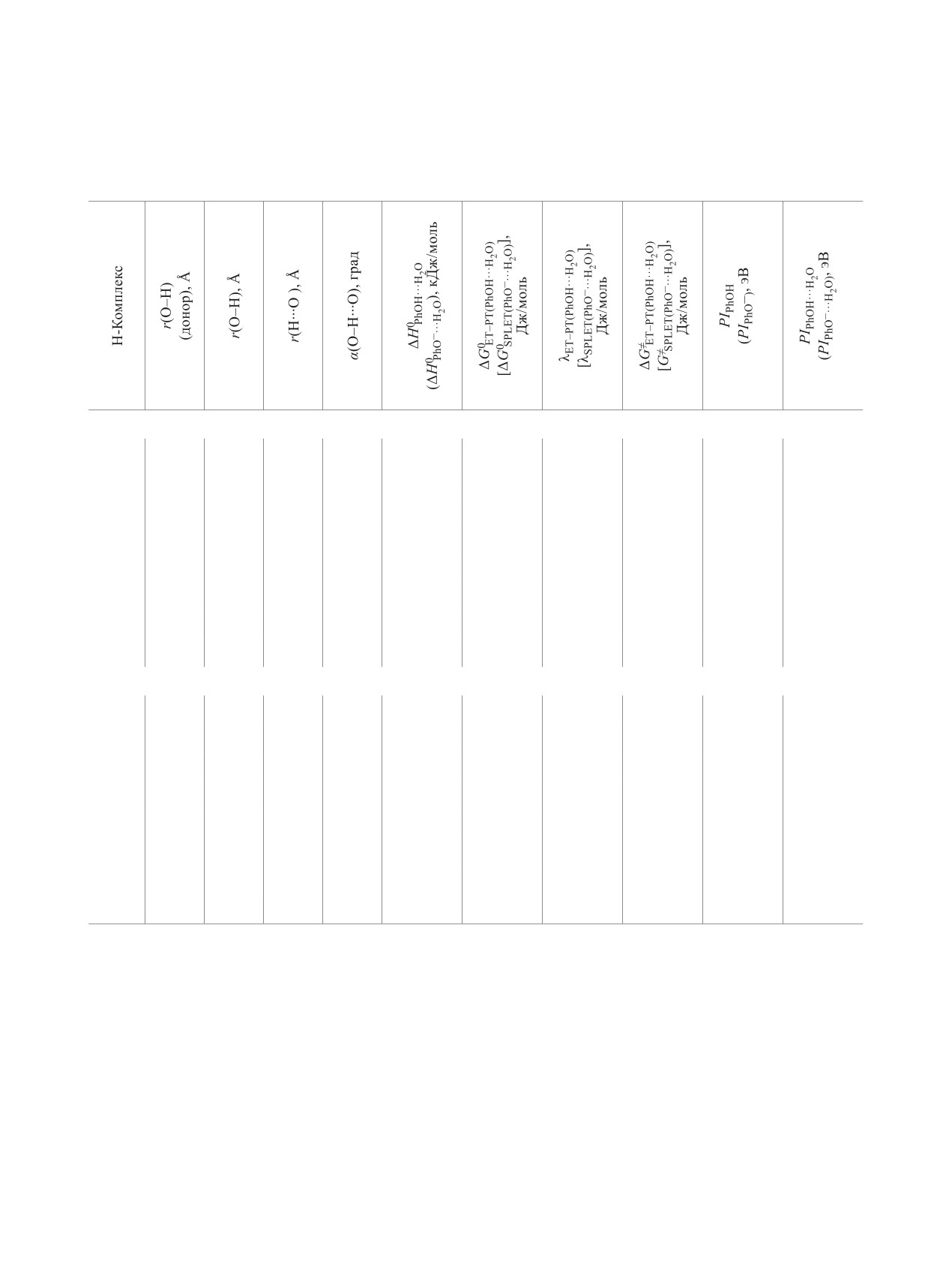
Таблица 4. Параметры геометрических комплексов PhOH···H2O и PhO-···H2O, энтальпии их образования (∆Н°),
потенциалы ионизации (PI), а также энергии реорганизации (λ), свободные энергии активации (ΔG≠) и свободные
энергии (∆G°) реакций (10) и (17)
PhOH···H2O
1
0.969
0.983
1.748
178
-19.1
81.9
87.6
82.0
5.87
5.73
2
0.972
0.981
1.782
177
-16.6
112.0
68.2
119.0
6.08
5.89
3
0.972
0.980
1.786
171
-16.0
103.7
71.3
107.4
6.03
5.81
4
0.971
0.979
1.794
177
-15.8
78.5
71.2
78.6
5.74
5.53
5
0.971
0.979
1.796
177
-15.8
67.1
74.5
67.3
5.65
5.45
6
0.970
0.984
1.744
178
-19.4
68.1
106.7
71.6
5.72
5.50
7
0.972
0.981
1.778
177
-17.2
112.6
73.7
117.8
6.10
5.92
8
0.970
0.983
1.742
178
-19.5
64.0
76.4
64.5
5.61
5.44
PhO-···H2O
1
0.964
1.014
1.562
169
-52.1
-28.4
73.0
6.8
4.29
4.48
2
1.006
1.626
175
-31.8
-9.9
67.9
12.4
4.51
4.72
3
1.007
1.623
174
-32.2
-15.5
68.0
10.1
4.48
4.65
4
1.010
1.608
175
-33.9
-44.5
72.8
5.7
4.15
4.36
5
1.007
1.619
174
-32.0
-44.0
92.0
5.8
4.11
4.31
6
1.024
1.529
168
-36.9
-24.0
79.0
7.1
4.28
4.55
7
1.005
1.630
175
-31.2
-4.8
68.5
14.8
4.56
4.77
8
1.012
1.571
169
-50.5
-32.8
73.5
5.6
4.25
4.43
сравнению с изолированным гидроксибензолом;
Экспериментальные и теоретические иссле-
угол водородной связи α(О-Н∙∙∙О) близок к 180°
дования [25-27] свидетельствуют о том, что при
(171-178°); энтальпия образовании комплексов
наличии водородных связей между растворителем
ΔrH0PhOH···H2O составляет 15-20 кДж/моль, что ха-
и растворенным веществом электронодонорные
рактерно для сильных водородных связей (11).
(или электроноакцепторные) свойства сольва-
ΔH0PhOH···H2O = (HPhOH···H2O) - (HPhOH + HH2O),
(11)
тированной частицы изменяются по сравнению
где HPhOH, HH2O, HPhOH···H2O - энтальпии гидрок-
с изолированной формой, что находит объясне-
сибензола, воды и их комплекса.
ние как с точки зрения электростатических, так
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020
38
БЕЛАЯ и др.
и межорбитальных взаимодействий. Так, мо-
фенолят-ионов достигает 42% (табл. 2), превы-
лекулы воды, действующие при ассоциации с
шают константы в кислых средах на 1-3 порядка
растворенным веществом как доноры Н-связи,
(табл. 1).
уменьшают электронодонорные свойства обра-
Для лимитирующей стадии (13) механизма
зовавшегося Н-комплекса, тогда как молекулы
SPLET были рассчитаны значения G≠SPLET по фор-
воды, действующие как акцепторы H-связи, их
муле (6) с использованием ΔG0SPLET и ΔESPLET в со-
увеличивают. Количественной мерой электроно-
ответствии с выражениями (14) и (15).
донорных свойств Н-комплексов, ответственных
ΔG0SPLET = (GPhO• + GDPPH-) - (GPhO- + GDPPH•),
(14)
за их антирадикальную активность, является по-
ΔESPLET = (EPhO• + EDPPH-) - (EPhO- + EDPPH•).
(15)
тенциал ионизации. Первые адиабатические по-
Здесь GPhO-, GPhO•, GDPPH-, GDPPH• - свободная
тенциалы ионизации (PIPhOH···H2O) вычисляли как
энергия Гиббса фенолят-иона гидроксибензола и
разницу энтальпий гидратированных катион-ра-
его радикала, аниона и радикала DРРН• соответ-
дикалов (HPhOH···H2O•+), оптимизированных в их
ственно; EPhO-, EPhO• - общая электронная энергия
основном состоянии, и энтальпий соответствую-
фенолят-иона гидроксибензола и его радикала,
щих нейтральных комплексов (HPhOH···H2O) (12).
рассчитанного из геометрии PhO; EDPPH•, EDPPH- -
Аналогичным образом рассчитывали потенциал
общая электронная энергия радикала и его аниона,
ионизации для индивидуальных фенолов (PIPhOH).
рассчитанного из геометрии DРРН•.
PIPhOH···H
= HPhOH···H
(12)
2O
2O•+ - HPhOH···H2O+.
В случае механизма SPLET энергия реорганиза-
Как следует из приведенных данных (табл. 4),
ции (λSPLET) для всех изученных PhOH меньше по
потенциал ионизации комплексов PhOH···H2O
сравнению с λET-PT (табл. 3). Реакция (13) экзоэр-
меньше, чем у изолированной молекулы PhOH.
гична, а ее свободная энергия активации ΔG≠SPLET
Образование Н-связи даже с одной молекулой
мала, что характеризует электронный перенос от
воды понижает потенциал ионизации фенола и
фенолят-иона на радикал как наиболее вероятный
тем самым способствует более быстрому межмо-
механизм действия гидроксибензолов в щелочных
лекулярному переносу электрона на радикал с
средах. Высокая корреляция величин ΔG≠SPLET и
формированием заряженных систем - гидратиро-
ΔG≠exp(pH=9) наблюдается при повышении рН сре-
ванного катион-радикала фенола PhOH•+···H2O
ды (16). Для аналогичной зависимости в кислых
и аниона DPPH-, стабилизируемых полярной
средах этот показатель значительно ниже и состав-
водной средой. Действительно, вычисленная сво-
ляет 0.913.
бодная энергия активации ΔG≠ET-PT(PhOH···H2O) для
ΔG≠SPLET = (47.2±0.4) + (2.55±0.14)ΔG≠exp(pH=9),
(16)
Н-комплексов снижается по сравнению с ΔG≠ET-PT
r2 = 0.981, F = 310, p < 0.00000, Sest = 0.84.
на 15-20 кДж/моль и приближается к эксперимен-
Значительное отклонение ΔG≠SPLET от экспери-
тальным значениям ΔG≠exp(pH=2) в кислых средах
ментальных значений, как и в предыдущем слу-
(табл. 3, 4). Относительная погрешность расчета
чае, связано с использованием в расчетах неги-
снижается на 23-33%.
дратированных фенолят-ионов. Если считать, что
Реакции PhOH с DPPH▪ в буферном растворе
в лимитирующей стадии (17) принимает участие
при рН = 9. Механизм SPLET. Подтверждением
ион-молекулярный акцепторный Н-комплекс ги-
реализации механизма SPLET (13), чувствитель-
дроксибензол-вода (рис. 3б), потенциал иониза-
ного к изменению рН среды, является резкий рост
ции которого выше, чем у соответствующего ин-
констант скоростей реакции в щелочной среде за
дивидуального фенолят-иона, то полученные зна-
счет роста концентрации ионов и изменения их ка-
чения ΔG≠SPLET(PhO-···H2O) для Н-комплексов стано-
чественного состава (рис. 3).
вятся ближе к экспериментальным ΔG≠exp(pH=9) по
PhO-H + H2O →← PhO- + H3O+,
абсолютному значению, а их корреляция остается
PhO- + DPPH• → PhO• + DPPH-,
прежней.
DPPH- + H3O+ →← DPPH-H + H2O.
(13)
PhO-···H2O + DPPH• → PhO•···H2O + DPPH-.
(17)
Константы kбуфер(pH=9), определенные по урав-
Обращает на себя внимание величина
нению (1) в щелочном буфере, где доля активных
ΔG≠exp(pH=7), определенная в нейтральном буфере.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020
СОГЛАСОВАННЫЙ ПЕРЕНОС ЭЛЕКТРОНА И ПРОТОНА
39
Ее корреляция с ΔG≠SPLET и ΔG≠ET-PT имеет при-
Реакции PhOH с DPPH• проводили в ДМСО и
мерно одинаковые коэффициенты ~r, равные 0.952
его смеси с буферами, из которых предварительно
и 0.961 соответственно. По-видимому, в нейтраль-
удалялся кислород путем барботирования аргоном
ных средах, где присутствуют и молекулярная и
в течение 15-20 мин, что позволило исключить воз-
ионная формы антиоксиданта (табл. 2), будут ре-
можные реакции гидроксибензолов и продуктов
ализовываться оба механизма, на что указывают
их превращения с участием кислорода. Кинетику
и другие исследования механизмов действия фе-
реакции изучали методом спектрофотометрии
нольных антиоксидантов [10].
на приборе Specord S300 UV-VIS (Германия) при
293±2 K в интервале начальных концентраций
Таким образом, на основании анализа зависи-
реагирующих веществ 10-3-10-5 моль/л [31, 32].
мости константы скорости реакции PhOH с DPPH•
Раствор PhOH в буфере смешивали с раствором
от рН среды и расчета свободной энергии Гиббса
DPPH• в ДМСО непосредственно в кювете и из-
активации по уравнению Маркуса предложены
меряли значение оптической плотности каждые
возможные механизмы переноса электрона с фе-
0.5 с. Концентрацию DPPH• рассчитывали по оп-
нольного антиоксиданта на радикал. В кислых
тической плотности через молярный коэффициент
средах электронный перенос осуществляется от
экстинкции, равный εДМСО = 1.2×104 л/(моль·см).
межмолекулярного Н-комплекса PhOH···H2O на
DPPH• с последующей потерей протона, а в ще-
КОНФЛИКТ ИНТЕРЕСОВ
лочных средах - от фенолят-иона на радикал с
предшествующей стадией отрыва протона. В ней-
Авторы заявляют об отсутствии конфликта
тральных средах, вероятнее всего, реализуются
интересов.
два пути одновременно. Идентификация указан-
СПИСОК ЛИТЕРАТУРЫ
ных механизмов в водных средах в зависимости от
рН среды позволит подбирать дескрипторы стро-
1. Galano A., Mazzone G., Alvarez-Diduk R., Marino T.,
ения антиоксиданта, по величине которых можно
Alvarez-Idaboy J.R., Russo N. // Annu. Rev. Food Sci.
прогнозировать его антирадикальную и биологи-
Technol. 2016. Vol. 7. P. 335. doi 10.1146/annurev-
ческую активность.
food-041715-033206
2. Milenković D., Yorović J., Jeremić S., Marković J.M.D.,
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Avdović E.H., Marković Z. // J. Chem. 2017. Vol. 2017.
В работе использовали свободный стабильный
P. 1. doi 10.1155/2017/5936239
радикал DPPH• и диметилсульфоксид (Merck).
3. Amić A., Marković Z., Klein E., Marković J.M.D.,
Раствор радикала в ДМСО имеет интенсивный
Milenković D. // Food Chem. 2017. Vol. 246. P. 481.
фиолетовый цвет с максимумом поглощения при
doi 10.1016/j.foodchem.2017.11.100
длине волны 520 нм. При хранении DPPH• в ДМСО
4. Litwinienko G., Ingold K.U. // Acc. Chem. Res. 2007.
в темноте на протяжении 72 ч интенсивность мак-
Vol. 40. N 3. P. 222. doi 10.1021/ar0682029
симума поглощения его спектра остается неизмен-
5. Foti M.C., Daquino C., Mackie I.D., DiLabio G.A.,
ной. ДМСО очищали по известной методике [28].
Ingold K.U. // J. Org. Chem. 2008. Vol. 73. P. 9270. doi
Гидроксибензолы марки ХЧ подвергали много-
10.1021/jo8016555
кратной перекристаллизации из этилового спирта
6. Белая Н.И, Белый А.В., Заречная О.М., Щерба-
и сушили при 40°С в атмосфере азота, после чего
ков И.Н., Дорошкевич В.С. // ЖОХ. 2018. Т. 88. №
сублимировали в вакууме. Оксигидрохинон синте-
7. С. 1057; Belaya N.I., Belyi A.V., Zarechnaya O.M.,
зировали по известной методике [29].
Scherbakov I.N., Doroshkevich V.S. // Russ. J. Gen.
Для задания рН водных растворов использова-
Chem. 2018. Vol. 88. N 7. P. 1351. doi 10.1134/
ли солянокислую (рН = 2), фосфатную (рН = 7.35)
S1070363218070010
и борно-щелочную (рН = 9) буферные системы,
7. Волков В.А., Мисин В.М. // Кинетика и катализ. 2015.
приготовленные по методике [30, 28]. Точные зна-
Т. 56. № 1. С. 48; Volkov V.A., Misin V.M. // Kinetics
чения рН приготовленных буферных растворов
and Catalysis. 2015. Vol. 56. N 1. P. 43. doi 10.1134/
контролировали с помощью иономера И-160МИ.
S0023158415010139
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020
40
БЕЛАЯ и др.
8. Mazzone G., Russo N., Toscano M. // Comput.
17. Rappe A.K., Casewit C.J., Colwell K.S., Goddard W.A.,
Theoret. Chem. 2016. Vol. 1077. P. 39. doi 10.1016/j.
Skiff W.M. // J. Am. Chem. Soc. 1992. Vol. 114. N 25.
comptc.2015.10.011
P. 10024. doi 10.1021/ja00051a040
9. Musialik M., Litwinienko G. // Org. Lett. 2005. Vol. 7.
N 22. P. 4951. doi 10.1021/ol051962j
com
10. Galano A., Alvarez-Idaboy J.R. // J. Comput. Chem.
19. Nelsen S.F., Weaver M.N., Luo Y. // J. Phys. Chem. (A).
2013. Vol. 34. P. 2430. doi 10.1002/jcc.23409
2006 Vol. 110. N. 41. P. 11665. doi 10.1021/jp064406v
11. Vermerris W., Nicolson R. Phenolic Compound
20. Marcus R.A. // J. Chem. Phys. 1965. Vol. 43. N 2.
Biochemistry. Dodrecht: Springer, 2006. 276 p.
P. 679. doi 10.1063/1.1696792
12. Сухарев А.Г., Тимохов А.В., Федоров В.В. Курс ме-
21. Skyner R.E., McDonagh J.L., Groom C.R., van Mourika T.,
тодов оптимизации. М.: ФИЗМАТЛИТ, 2005. 368 с.
Mitchell J.B.O. // Phys. Chem. Chem. Phys. 2015.
13. Белая Н.И., Белый А.В., Заречная О.М., Щерба-
Vol. 17. Р. 6174. doi 10.1039/c5cp00288e
ков И.Н., Помещенко А.И., Горбань О.А. // Кинетика
22. Ahn D.-S., Jeon I.-S., Jang S.-H., Park S.-W., Lee S.,
и катализ. 2019. Т. 60. № 1. С. 118; Belaya N.I.,
Cheong W. // Bull. Korean Chem. Soc. 2003. Vol. 24.
Belyi A.V., Zarechnaya O.M., Shcherbakov I.N.,
N 6. P. 695. doi 10.5012/bkcs.2003.24.6.695
Pomeshchenko A.I., Gorban’ O.A. // Kinetics and
23. Yamabe S., Yamazaki S. // Int. J. Quantum Chem. 2017.
Catalysis. 2019. Vol. 60. N 1. Р. 28. doi 10.1134/
Vol. 118. N 6. P. 1. doi 10.1002/qua.25510
S0023158419010014
24. Arunan E., Desiraju G.R., Klein R.A., Sadlej J., Scheiner S.,
14. Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E.,
Alkorta I., Clary D.C., Crabtree R.H., Dannenberg J.J.,
Robb M.A., Cheeseman J.R., Scalmani G., Barone V.,
Hobza P., Kjaergaard H.G., Legon A.C., Mennucci B.,
Mennucci B., Petersson G.A., Nakatsuji H., Caricato M.,
Nesbitt D.J. // Pure Appl. Chem. 2011. Vol. 83. N 8.
Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G.,
P. 1637. doi 10.1351/PAC-REC-10-01-02
Sonnenberg J.L., Hada M., Ehara M., Toyota K.,
25. Самуилов А.Я., Самуилов Я.Д. // Бутлеровск. сообщ.
Fukuda R., Hasegawa J., Ishida M., Nakajima T.,
2011. Т. 28. № 19. С. 1.
Honda Y., Kitao O., Nakai H., Vreven T., Montgo-
mery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M.,
26. Tentscher P.R., Seidel R., Winter B., Guerard J.J.,
Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N.,
Arey J.S. // J. Phys. Chem. (B). 2015. Vol. 119. N 1.
Keith T., Kobayashi R., Normand J., Raghavachari K.,
P. 238. doi 10.1021/jp508053m
Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M.,
27. Ghosh D., Roy A., Seidel R., Winter B., Bradforth S.,
Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B.,
Krylov A.I. // J. Phys. Chem. (B). 2012. Vol. 116. N 24.
Bakken V., Adamo C., Jaramillo J., Gomperts R.,
P. 7269. doi org/10.1021/jp301925k
Stratmann R.E., Yazyev O., Austin A.J., Cammi R.,
28. Armarego W.L.F., Chai C.L.L. Purification of Laboratory
Pomelli C., Ochterski J.W., Martin R.L., Morokuma K.,
Chemicals. Burlington: Elsevier Science, 2003. 608 p.
Zakrzewski V.G., Voth G.A., Salvador P., Dannen-
29. Препаративная органическая химия / Под ред.
berg J.J., Dapprich S., Daniels A.D., Farkas O.,
Н.С. Вульфсона. М.: ГХИ, 1959. 889 с.
Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J.
Gaussian 09, Revision B.01 Gaussian, Inc., Wallingford
30. Рабинович В.А., Хавин З.Я. Краткий химический
CT, 2010.
справочник. Л.: Химия, 1991. 432 с.
15. Weinberg D.R., Gagliardi C.J., Hull J.F., Murphy C.F.,
31. Mendoza-Wilson A.M., Santacruz-Ortega H., Balan-
Kent C.A., Westlake B., Paul A., Ess D.H., McCaf-
drán-Quintana R.R. // J. Mol. Struct. 2011. Vol. 995.
ferty G.D., Meyer T.J. // Chem Rev. 2007. Vol. 107.
P. 134. doi 10.1016/j.molstruc.2011.04.004
N 11. P. 5004. doi 10.1021/cr0500030
32. Petrović Z.D., Đorović J., Simijonović D., Petrović V.P.,
16. Tomasi J., Mennucci B., Cammi R. // Chem. Rev. 2005.
Marković Z. // RSC Adv. 2015. Vol. 5. P. 24094. doi
Vol. 105. N 8. P. 2999. doi 10.1021/cr9904009
10.1039/C5RA02134K
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020
СОГЛАСОВАННЫЙ ПЕРЕНОС ЭЛЕКТРОНА И ПРОТОНА
41
Concurred Transfer of Electron and Proton in the Reaction
of Hydroxybenzenes with a Hydrazyl Radical in Aqueous Media
N. I. Belayaa,*, A. V. Belyia, O. M. Zarechnayab, I. N. Shcherbakovc, and V. S. Doroshkevicha
a Donetsk National University, ul. Universitetskaya 24, Donetsk, 283001 Ukraine
b L. M. Litvinenko Institute of Physical and Organic Chemistry and Coal Chemistry, Donetsk, 283114 Ukraine
c Southern Federal University, Rostov-on-Don, 344006 Russia
*e-mail: nat.iv.belaya@gmail.com
Received June 12, 2019; revised June 12, 2019; accepted June 16, 2019
Plausible mechanisms of electron transfer in the reaction of 2,2'-diphenyl-1-picrylhydrazyl with a number
of natural hydroxybenzenes in aqueous buffer solutions at pH = 2-9 were studied using spectrophotometry
and quantum chemistry methods. In acidic media electron transfer is carried out from the molecular form of
hydroxybenzene to the radical with subsequent loss of the proton, and in alkaline media from the phenolate
ion to the radical with the previous stage of proton detachment. The implementation of these mechanisms
is evidenced by the dependence of the reaction rate constant on the pH of the medium and the presence of a
correlation between the Gibbs free activation energy calculated according to the Marcus equation and determined
on the basis of experimental data.
Keywords: hydroxybenzene, 2,2'-diphenyl-1-picrylhydrazyl, electron transfer, reorganization energy
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 1 2020