ЖУРНАЛ ОБЩЕЙ ХИМИИ, 2020, том 90, № 8, с. 1262-1271
УДК 547.661.4;547.639.5;547.1-315
ПОЛИФУНКЦИОНАЛИЗИРОВАННЫЕ ХЕЛАТНЫЕ
ПРОИЗВОДНЫЕ ДИНАФТИЛМЕТАНА
И РЕЗОРЦИНКАЛИКС[4]АРЕНА: СИНТЕЗ,
ВЗАИМОСВЯЗЬ МЕЖДУ СТРУКТУРОЙ ХЕЛАТА
И ЕГО АКЦЕПТОРНЫМИ СПОСОБНОСТЯМИ
© 2020 г. О. С. Серковаa, А. В. Камкинаa, И. Ю. Торопыгинb, В. И. Масленниковаa,*
а Институт биологии и химии, Московский педагогический государственный университет,
ул. Кибальчича 6, Москва, 129164 Россия
b Научно-исследовательский институт биомедицинской химии имени В. Н. Ореховича, Москва, 119121 Россия
*е-mail: him-vim@mail.ru; lvi.maslennikova@mpgu.su
Поступило в Редакцию 17 марта 2020 г.
После доработки 17 марта 2020 г.
Принято к печати 28 марта 2020 г.
Функционализацией метиленбис(нафталин-2-ола/-2,7-диола) и rccc-резорцинкаликс[4]арена получена
серия их производных по группам ОН с 1, 2 и 4 парами хелатных ионофорных заместителей, различа-
ющихся комбинацией электронодонорных атомов (O, S, N, P). С использованием метода жидкостной
экстракции изучено влияние структурных особенностей лиганда на его способность к распознаванию
и связыванию катионов переходных металлов (Cd2+, Ag+, Hg2+).
Ключевые слова: метиленбис(нафталин-2-ол), метиленбис(нафталин-2,7-диол), резорцинкаликс[4]-
арены, фосфорилирование, карбамоилирование
DOI: 10.31857/S0044460X20080144
Полиядерные ароматические соединения с
катионам переходных металлов (Cd2+, Ag+, Hg2+).
хелатными ионофорными функциональными
Для оценки эффективности и селективности свя-
группами на периферии молекулы могут быть
зывания ионов металлов полученными соединени-
использованы в качестве экстрагентов [1, 2], хе-
ями были проведены эксперименты по жидкост-
мосенсоров [3-6], переносчиков ионов [7], пре-
ной экстракции в системе вода-хлороформ
курсоров для конструирования сложных супрамо-
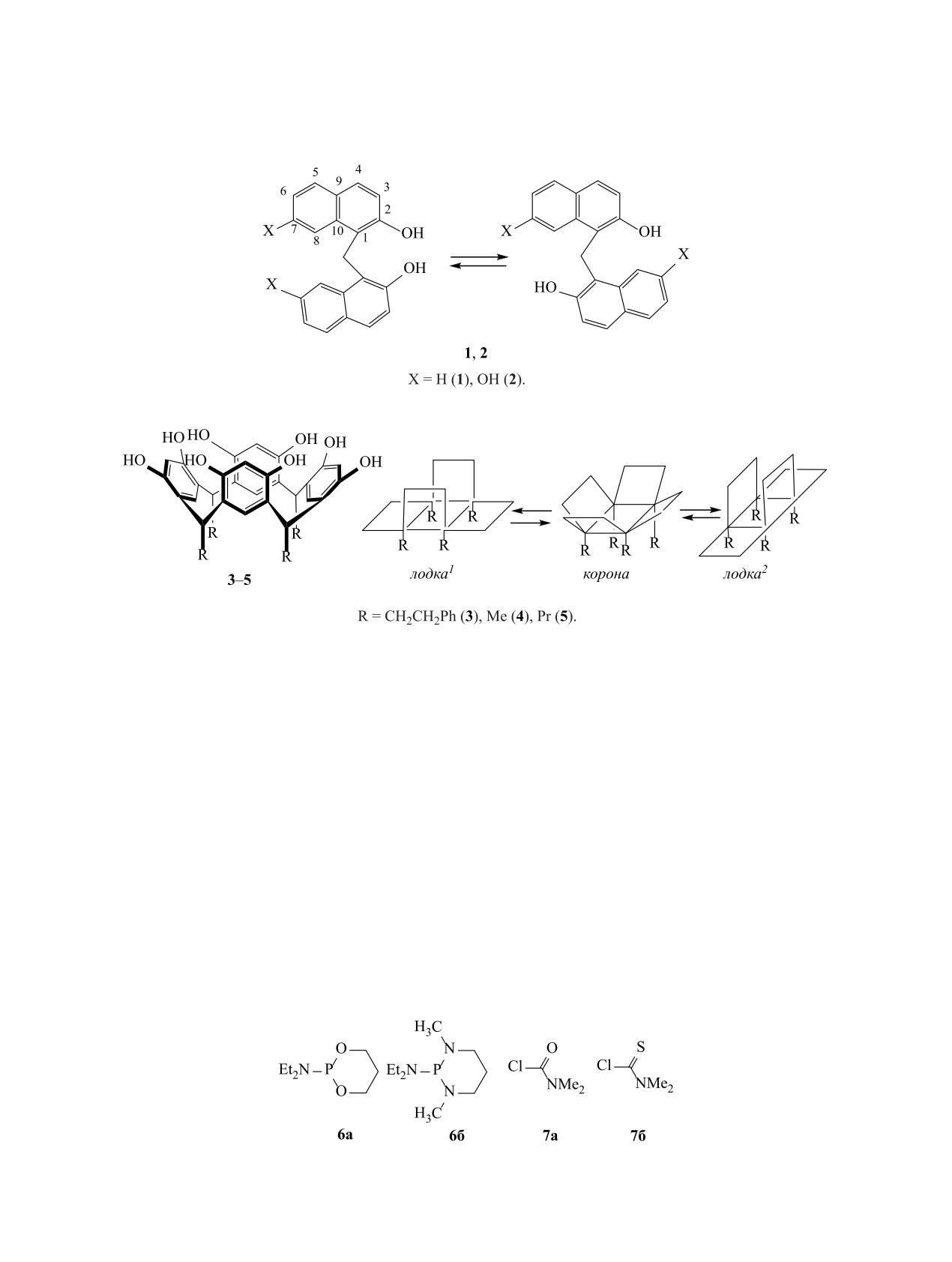
В метиленбис(нафталин-2-оле) 1 и метилен-
лекулярных архитектур [8-9]. Сфера применения
бис(нафталин-2,7-диоле)
2 нафталиновые ядра
веществ такого типа и их эффективность опре-
связаны между собой метиленовым мостиком
деляются природой, количеством, ориентацией
и расположены по отношению друг к другу под
в пространстве ионофорных групп и топологией
углом, величина которого определяется конформа-
молекулы в целом.
ционным состоянием молекулы в растворе [10, 11],
Нами рассматриваются пути синтеза произво-
в свою очередь зависящим от природы растворите-
дных метиленбис(нафталин-2-ола/-2,7-диола) и
ля и заместителей в положениях 2,2ʹ и 7,7ʹ нафта-
rccc-резорцинкаликс[4]арена. В молекулах синте-
линовых циклов. Структура и цис/транс-конфор-
зированных соединений находятся 1, 2 и 4 пары
мационные переходы молекул 1, 2 представлены
хелатных ионофорных групп, различающихся
на схеме 1.
комбинацией электронодонорных атомов (O, S, N,
P), эти структурные особенности лигандов влияют
Резорцинкаликс[4]арены
3-5
обладают
на их акцепторные способности по отношению к
all-cis-конфигурацией заместителей R в мети-
1262
ПОЛИФУНКЦИОНАЛИЗИРОВАННЫЕ ХЕЛАТНЫЕ ПРОИЗВОДНЫЕ ДИНАФТИЛМЕТАНА
1263
Схема 1.
Схема 2.
леновых мостиках и находятся в конформации
водит к увеличению количества молекул, находя-
лодка. Для данной конфигурации характерна ин-
щихся в конформации лодка.
терковерсия лодка-корона-лодка (схема 2) [12-
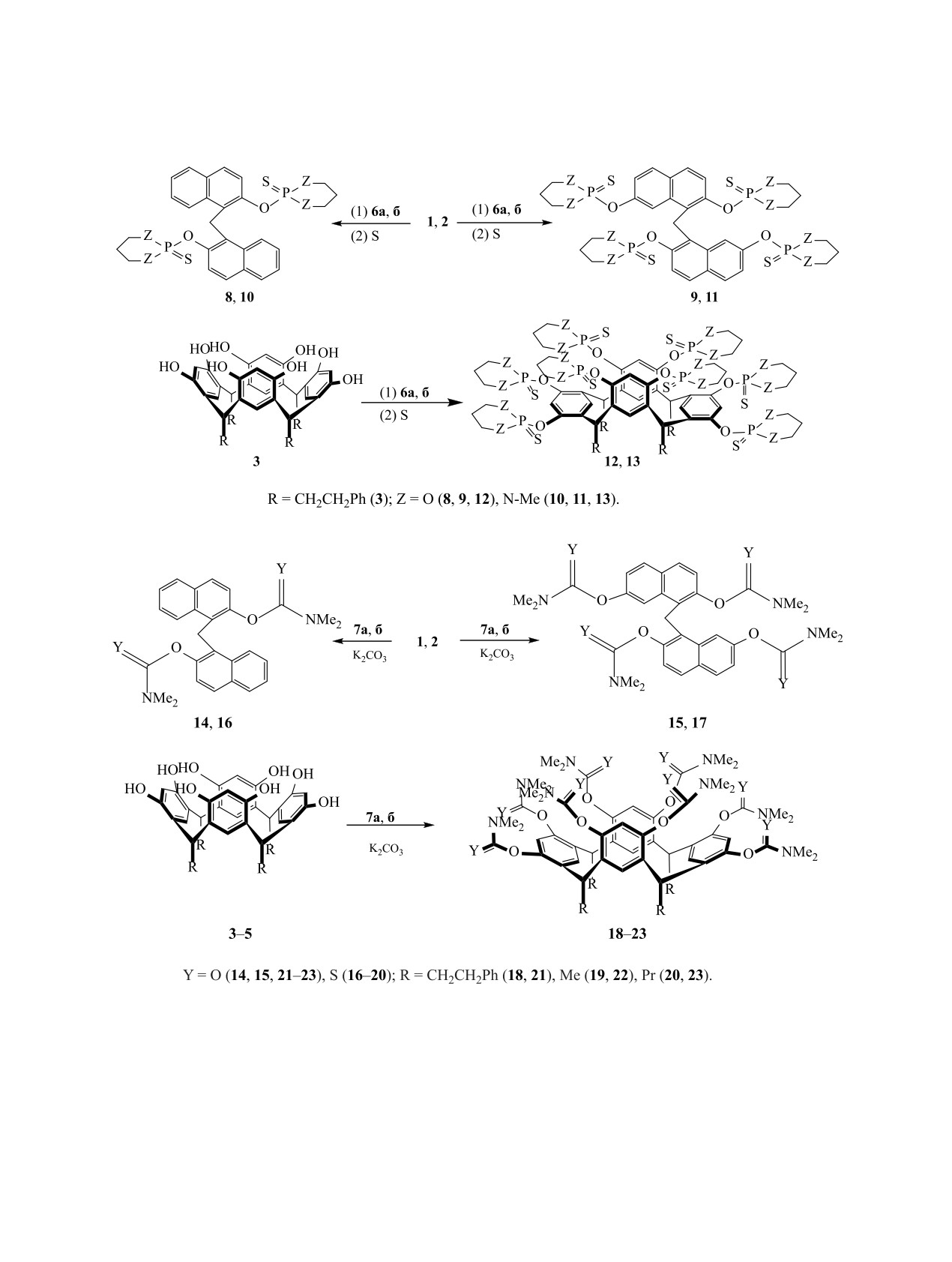
С целью введения в соединения 1-5 ионофорных
14], при этом изменяется размер полости и моле-
групп различного типа в качестве реагентов были
кула подстраивается к ионам различного диаметра.
использованы
N,N-диэтил-1,3,2-диоксафосфи-
Интенсивность интерконверсии и угол отклонения
нан-2-амин 6a, 1,3-диметил-N,N-диэтил-1,3,2-диа-
бензольных ядер от плоскости макроцикла зависят
зафосфинан-2-амин 6б, диметилкарбамоил- и ди-
от объема и природы вводимых в резорцинарен
метилкарбамотиоилхлориды 7a, б (схема 3).
функциональных групп, от заместителей R и ис-
При функционализации гидроксипроизводных
пользуемого растворителя.
динафтилметана 1, 2 и резорцинкаликс[4]арена
Каликc[4]арены 3-5 с незамещенными гидрок-
3 фосфинанами 6a, б был использован однореак-
сильными группами вследствие высокой скорости
торный процесс: первоначально в диоксане про-
интерконверсии в основном находятся в конфор-
водили фосфорилирование соединений 1-3, за-
мации корона. При тотальной функционализации
тем для окисления образовавшихся производных
гидроксильных групп с увеличением объема заме-
трехвалентного фосфора добавляли раствор серы
стителей интерконверсия замедляется, и это при-
в бензоле и выдерживали реакционные смеси 12-
Схема 3.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 8 2020
1264
СЕРКОВА и др.
Схема 4.
Схема 5.
24 ч (схема 4). Ацилирование соединений 1-5 ди-
соединений 9-13 полностью соответствуют приве-
метилкарбамоил- и диметилкарбамотиоилхлори-
денным формулам. В спектре ЯМР 31Р соединения
дами 7а, б проводили при кипячении в ацетоне в
10 наблюдается синглетный сигнал в характерной
присутствии карбоната калия (схема 5).
для диамидо(тио)фосфатов области (табл. 1), а в
Бистиофосфат
8 был охарактеризован ра-
спектрах ЯМР 1Н, 13С - по одному набору сигна-
нее [10]. Данные элементного анализа, ИК и
лов всех видов протонов и углеродных ядер, что
масс-спектров выделенных с хорошими выходами
указывает на симметричность молекулы и эквива-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 8 2020
ПОЛИФУНКЦИОНАЛИЗИРОВАННЫЕ ХЕЛАТНЫЕ ПРОИЗВОДНЫЕ ДИНАФТИЛМЕТАНА
1265
Таблица 1. Выходы, данные ЯМР 31Р, ИК и масс-спектров соединений 9-15
№ соединения
Выход, %
δР, м. д.
ν, cм-1
m/z
9
80
56.46, 56.54
880.2 (P=S)
875 [M]+
10
71
74.44
874.7 (P=S)
624 [M]+
11
61
74.81, 75.27
872.6 (P=S)
978 [M]+
12
81
55.39, 56.38
880.7 (P=S)
2017 [M + Na]+
13
61
72.05, 72.67
871.7 (P=S)
2190 [M- CH3]+
14
79
-
1703.3 (С=О)
442 [M]+
15
84
-
1711.3 (С=О)
615 [M]+
Таблица 2. Степень экстракции (E, %) пикратов металлов соединениями 8-13
Количество электронодонорных
E, %
Соединение
Электронодорная группа
групп
Ag+
Hg2+
Cd2+
8
2
-
7.2
10.8
10
4
45.0
15.0
18.8
12
8
13.1
7.2
4.8
9
2
40.1
89.0
12.9
11
4
65.6
93.4
18.8
13
8
51.4
57.2
3.6
лентность циклических фрагментов. В спектрах
7,7ʹ нафталиновых колец. В ИК спектрах обоих
ЯМР 31Р соединений 9, 11 регистрировали по 2 син-
соединений присутствовали интенсивные полосы
глетных сигнала с равными интегральными интен-
карбонильных групп (табл. 1). Данные элементно-
сивностями (табл. 1), а в спектрах ЯМР 1Н, 13С -
го анализа и масс-спектрометрии соответствовали
удвоение сигналов протонов и углеродных ядер
присутствию в соединениях 14, 15 двух и четырех
фрагментов фосфинана, попарно локализованных
карбамидных групп соответственно. Карбамотиоа-
в положениях 2,2ʹ и 7,7ʹ нафталиновых колец. В
ты 16-20, а также карбаматы 21-23 охарактеризова-
спектрах ЯМР 31Р соединений 12, 13 фиксировали
ны ранее [15, 16].
по 2 синглетных сигнала с равными интегральны-
Для оценки рецепторной активности соедине-
ми интенсивностями (табл. 1), в спектрах ЯМР 1Н,
ний 8-23 использовали пикратный метод жидкост-
13С - двойной набор сигналов протонов и углерод-
ной экстракции катионов переходных металлов
ных ядер фрагментов фосфинана и бензольных
(Cd2+, Ag+, Hg2+) из водной фазы в хлороформ.
ядер макроциклического остова, что полностью
Остаточное количество металла в водной фазе
соответствует спектральным характеристикам
определяли методом УФ спектроскопии, измеряя
перфосфорилированного резорцинкаликс[4]арена
значения оптической плотности водной фазы до и
в конформации заторможенная лодка [12-14].
после экстракции.
Соединения 14, 15 получены с выходами 79,
Изучение экстракционной способности про-
84% соответственно (табл. 1). В спектрах ЯМР 1Н,
изводных динафтилметана и резорцинкаликс[4]-
13С дикарбамата 14 фиксировали по одному на-
арена 8-13 показало, что лиганды 8, 10, 12 (соот-
бору сигналов Н и С атомов карбамидных групп
ветственно с 2, 4 и 8 диоксафосфинановыми за-
и динафтилметанового остова, в аналогичных
местителями) проявляют низкую активность по
спектрах тетракарбамата 15 наблюдалось удвое-
отношению к исследованным катионам (табл. 2).
ние сигналов протонов и углеродных атомов ме-
Наибольшую эффективность экстракции (EAg+
тильных и карбонильных групп карбамидных
45%) в этом ряду продемонстрировало соедине-
фрагментов, находящихся в положениях 2,2ʹ и
ние 10, обладающее в трансоидной конформации
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 8 2020
1266
СЕРКОВА и др.
Таблица 3. Степень экстракции (E, %) пикратов металлов соединениями 14-23
Степень экстракции (E, %)
№
Заместитель
Ag+
Hg2+
Cd2+
14
-
17.5
-
15
-
10.4
10.6
16
-
4.0
-
17
45.9
42.8
5.8
18
78.0
93.4
66.4
19
65.0
90.5
70.0
20
63.5
87.0
66.4
21
8.2
7.5
91.0
22
1.0
1.0
81.0
23
10.7
-
72.6
двумя равноценными хелатными парами (схема 1).
электронодонорных атомов, приведены в табл. 3.
Изменение структуры ионофорных групп неза-
Как и следовало ожидать исходя из теории ЖМКО
висимо от их количества и от топологии полици-
Пирсона, содержащие в связывающих сайтах С=О
клической платформы приводит к повышению
группу ди- и тетракарбамоилпроизводные 14, 15
экстракционной способности соединений 9, 11, 13
проявили низкую акцепторную способность по от-
с диазафосфинановыми заместителями по сравне-
ношению ко всем исследованным катионам (Cd2+,
нию с с диоксафосфинановыми производными 8,
Ag+, Hg2+). При замене карбамоильных групп на
10, 12. Эффективность извлечения из водной фазы
карбамотиоильные эффективность тетрафункцио-
катионов Ag+ повышается в ряду соединений 9 <
нализированного производного 17 увеличилась до
13 < 11, а катионов Нg2+ - в ряду соединений 13 <
46% (Ag+) и 43% (Нg2+) экстракции (табл. 3). В то
9 < 11 (табл. 2). Максимальные акцепторные спо-
же время эффективность экстракции дифункцио-
собности и в этом случае проявляет производное
нализированного производного 16 снизилась до
динафтилметана 11 с двумя хелатными группами
4%, что можно объяснить трансоидной конформа-
в молекуле, способное извлекать ионы Ag+ на 65,
цией ароматического остова молекулы, в которой
а ионы Нg2+ на 93%. Высокую экстракционную
карбамотиоильные группы удалены друг от друга
активность (E 89%) по отношению к катионам
(схема 1).
Нg2+ продемонстрировало и соединение 9, име-
В отличие от соединений 16, 17 окта(карбамо-
ющее только одну пару хелатных заместителей.
тиоил)резорцинкаликс[4]арены
18-20, содержа-
Относительно низкие степени экстракции выше
щие в составе связывающих сайтов мягкие атомы
указанных катионов производным резорцинка-
серы, продемонстрировали высокую эффектив-
ликс[4]арена 13 с 8 электронодонорными фраг-
ность экстракции катионов переходных металлов
ментами (табл. 2) можно объяснить затруднением
(табл. 3), что полностью согласуется с принципом
интерконверсии (схема 2) объемными диазафос-
ЖМКО Пирсона. Степень экстракции катионов
финановыми заместителями, фиксирующими его
Hg2+ повышалась в ряду соединений 20 < 19 < 18 и
молекулы в конформации заторможенная лодка
для соединения 18 достигала 93%. Эффективность
с разобщенными в пространстве ионофорными
извлечения катионов ртути подтверждали при по-
группами (схема 2).
мощи атомно-адсорбционной спектроскопии. При
Экстракционные характеристики серии карба-
соотношении соединение 18-металл 1:2 доля Hg2+
матов и карбамотиоатов 14-23, в которых присут-
в образце снижалась со 100 до 0.39 мкг/л. Степень
ствуют ациклические ионофорные группы с отли-
экстракции катионов Ag+ и Cd2+ соединениями
чающимися от описанных выше композициями
18-20 была несколько ниже и варьировалась от 63
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 8 2020
ПОЛИФУНКЦИОНАЛИЗИРОВАННЫЕ ХЕЛАТНЫЕ ПРОИЗВОДНЫЕ ДИНАФТИЛМЕТАНА
1267
до 78% (табл. 3), что обусловлено различной ком-
в узнавании, связывании и извлечении из водной
плементарностью исследуемых ионов к полости
среды в органическую катионов Cd2+.
резорцинкаликс[4]арена.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В отличие от соединений 14-15 октакарбамоил-
Все эксперименты проводили в безводных, не
производные резорцинкаликс[4]арена 21-23 про-
содержащих следов кислорода растворителях в
демонстрировали крайне высокую селективность
атмосфере аргона. Спектры ЯМР 1Н, 13С и 31Р (с
и эффективность по отношению к ионам Сd2+
использованием остаточных сигналов раствори-
(табл. 3), степень экстракции которых из водной
фазы в органическую повышалась в ряду соеди-
телей в качестве внутреннего стандарта и 85%-
нений 23 < 22 < 21 и для октакарбамоилрезорцин-
ной Н3РО4 в качестве внешнего стандарта) для
каликс[4]арена 21 достигала 91%, тогда как для
всех соединений записывали на спектрометре Jeol
остальных рассмотренных катионов (Hg2+, Ag+)
ECX-400 (рабочая частота для ядер 13С 100.5 МГц,
не превышала 10%. Эта особенность карбамоили-
для ядер 31Р - 161.8 МГц). Для точного отнесения
рованных резорцинкаликс[4]аренов 21-23 проти-
сигналов соединений 9-15 использовали Н-Н го-
воречит теории ЖМКО Пирсона, так как катионы
моядерный двойной резонанс и 1H-13C 2D-корре-
кадмия относятся к мягким кислотам. По-види-
ляцию. Масс-спектры регистрировали на приборе
мому, столь высокая селективность и эффектив-
Bruker Ultraflex TOF/TOF (Bruker Daltonics GmbH),
ность связывания обусловлены комплементарно-
матрица - антрацен-1,8,9-триол. Элементный ана-
стью полости лигандов именно этому катиону, и
лиз проводили на CHN анализаторе Thermo Flash
инкапсулирование Сd2+ внутри полости резор-
EA112. ИК спектры регистрировали на спектроме-
цинарена происходит не столько за счет влияния
тре NICOLETE 380 Thermo в режиме отражения в
донорных атомов заместителей, сколько за счет
диапазоне 4000-500 см-1 на стекле ZnSe.
сильного катион-π-взаимодействия. Аргументом
Метиленбис(нафталин-2-ол)
1,
метилен-
в пользу данного предположения может служить
бис(нафталин-2,7-диол)
2, резорцинкаликс[4]
максимальная эффективность связывания Сd2+ со-
арены 3-5, N,N-диэтил-1,3,2-диокса(диаза)фосфи-
единением 21 с фенетильными заместителями в
нан-2-амины 6a, б синтезировали по известным ме-
метиленовых мостиках, способствующими стаби-
тодикам [11, 17-20].
лизации конформационного состояния молекулы,
Общая методика синтеза соединений 8-11.
близкого к короне (схема 2). За счет стекинг-вза-
Смесь 0.1 ммоль соединения 1 (2) и 0.3 (0.6) ммоль
имодействия арильных групп образуется полость
N,N-диэтил-1,3,2-диокса(диаза)фосфинан-2-амина
определенного размера, закрытая с одной стороны
6a, б в 2 мл диоксана выдерживали 48 ч при ком-
фенильными группами.
натной температуре. Затем к реакционной смеси
Таким образом, синтезированы новые серии
добавляли раствор 0.4 (0.8) ммоль серы в 2 мл бен-
олигофункционализированных rccc-резорцинка-
зола и перемешивали 12 ч при 50-55°С (6а) или
ликс[4]аренов и динафтилметанов с заданными
24 ч при 20-25°С (6б). Для выделения продукта
количеством и ориентацией хелатных ионофорных
реакции реакционную смесь упаривали на 2/3 и
групп, закрепленных на полициклической матри-
приливали 10 мл гексана. Осадок отфильтровыва-
це. Продемонстрирована зависимость способно-
ли, промывали 2 мл гексана, и сушили в вакууме
сти синтезированных соединений к распознава-
(15 мм рт. ст.) при 75°С.
нию и связыванию катионов переходных металлов
от структурных особенностей лиганда. Выявлено,
2,2ʹ-[1,1ʹ-Метиленди(нафталин-2-илокси)]-
что окта(карбамотиоил)резорцинкаликс[4]арены
бис(1,3,2-диоксафосфинан-2-сульфид) (8). Вы-
проявляют активность по отношению ко всем ука-
ход 89%, белый порошок, т. пл. 244-246°С [10].
занным катионам (Cd2+, Ag+, Hg2+); динафтилме-
Спектр ЯМР 1Н (СDCl3), δ, м. д.: 1.58 д. д (2Н, СН2,
тановые лиганды, содержащие диамидофосфат-
4JРН = 2.3, 2JНН = 14.6 Гц), 2.24-2.34 м (2Н, СН2),
ные заместители, являются высокоэффективными
4.21-4.28 уш. м (8Н, ОСН2), 4.86 с (2Н, CН2), 7.40-
экстрагентами катионов Hg2+; октакарбамоилиро-
7.46 м (6H, H3,6,7), 7.74 д (2H, H8, 3JНН = 9.2 Гц),
ванные резорцинкаликс[4]арены демонстрируют
7.83 д (2H, H4, 3JНН = 9.6 Гц), 8.12 д (2H, H5, 3JНН =
крайне высокую эффективность и селективность
9.6 Гц). Спектр ЯМР 31Р (CDCl3): δР 56.73 м. д.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 8 2020
1268
СЕРКОВА и др.
2,2ʹ,2ʹʹ,2ʹʹʹ-{1,1ʹ-Метиленбис[(нафталин-
д (2H, CH2, 3JHH = 13.3 Гц), 1.73 д (2H, CH2, 3JHH =
2,7-диил)бис(окси)]}тетра(1,3,2-диоксафосфи-
13.7 Гц), 1.89-2.09 уш. м (2H, CH2), 2.12-2.22 уш.
нан-2-сульфид) (9). Выход 80%, белый порошок,
м (2H, CH2), 2.55 д (6H, NCH3, 3JРH = 13.3 Гц), 2.67
т. пл. 142-144°С. ИК спектр, ν, см–1: 880.2 с (P=S).
д (6H, NCH3, 3JРH = 13.7 Гц), 2.74-2.82 уш. м (8Н,
Спектр ЯМР 1Н (СDCl3), δ, м. д.: 1.63 д (2Н, СН2,
NCH2), 2.93 д (6H, NCH3, 3JРH = 12.4 Гц), 2.95 д
2JНН = 14.6 Гц), 1.78 д. д (2Н, СН2, 4JРН = 2.3, 2JНН =
(6H, NCH3, 3JРH = 10.8 Гц), 3.08-3.20 уш. м (8H,
14.6 Гц), 2.22-2.39 м (4Н, СН2), 4.10-4.18 м (4Н,
NCH2), 3.39-3.45 м (8H, NCH2), 3.58-3.64 м (8H,
ОСН2), 4.24-4.37 м (8Н, ОСН2), 4.47-4.53 м (4Н,
NCH2), 5.02 с (2H, CH2), 7.32 д (2H, H3., 3JHH =
ОСН2), 4.86 с (2Н, CН2), 7.37-7.42 м (4H, H3,6),
8.2 Гц), 7.53-7.57 м (4H, H4,6), 7.64 уш. д (4H, H5,8,
7.75 д (2H, H4, 3JНН = 9.2 Гц), 7.83 д (2H, H5, 3JНН =
3JHH = 8.7 Гц). Спектр ЯМР 13С (CDCl3), δС, м. д.:
8.7 Гц), 7.86 с (2H, H8). Спектр ЯМР 13С (CDCl3),
23.78 (СH2), 24.08 (СН2), 24.90 (CH2), 37.67 д
δС, м. д.: 24.90 (СН2), 25.79 д (СН2, 3JСР = 7.6 Гц),
(NCH3, 2JРС = 35.6 Гц), 37.98 д (NCH3, 2JРС =
25.98 д (СН2, 3JСР = 7.7 Гц), 69.00 д (ОСН2, 3JСР =
34.5 Гц), 49.97 д (NCH2, 2JРС = 41.2 Гц), 50.37 д
(NCH2, 2JРС = 45.0 Гц), 115.28 д (С3, 3JРС = 5.8 Гц),
9.6 Гц), 69.04 д (ОСН2, 3JСР = 8.6 Гц), 114.34 д
119.98 (С8), 120.25 (С6), 126.45 д (С1, 3JРС = 5.8 Гц),
(С8, 3JСР = 5.9 Гц), 119.83 (С6,3), 125.1 д (С1, 3JСР =
127.72 (С4/5), 128.64 (С10), 129.60 (С4/5), 134.47 (С9),
7.7 Гц), 128.22 (С4), 129.16 (С9), 130.67 (С5), 134.12
151.17 д (С2, 2JРС = 9.6 Гц,), 154.19 д (С7, 2JРС =
(С10), 147.75 д (C2, 2JСР = 6.7 Гц), 149.40 д (C7,
9.6 Гц). Спектр ЯМР 31Р (CDCl3), δР, м. д.: 75.27,
2JСР = 7.7 Гц). Спектр ЯМР 31Р (CDCl3), δР, м. д.:
74.81. Масс-спектр (MALDI), m/z: 978 [M+] (вы-
56.46, 56.54. Масс-спектр (MALDI), m/z: 875 [M+]
числено для C41H60N8O4P4S4: 980). Найдено, %: С
(вычислено для C33H36O12P4S4: 876). Найдено, %:
50.17; H 6.20; N 11.29. C41H60N8O4P4S4. Вычисле-
С 45.23; H 4.15 C33H36O12P4S4. Вычислено, %: С
но, %: С 50.19; Н 6.16; N 11.42.
45.21; Н 4.14.
Октакис-2-{2,8,14,20-тетра(2-фенилэтил)-
2,2ʹ-[1,1ʹ-Метиленбис(нафталин-2-илокси)]-
каликс[4]арен-4,6,10,12,16,18,22,24-октаил(ок-
бис(1,3-диметил-1,3,2-диазафосфинан-2-суль-
таокси)}октакис(1,3,2-диоксафосфинан-2-суль-
фид) (10). Выход 71%, белый порошок, т. пл.
фид) (12). Смесь 0.12 ммоль резорцинкаликс[4]-
221-222°С. ИК спектр, ν, см-1: 874.7 c (P=S).
арена 3 и 1.2 ммоль N,N-диэтил-1,3,2-диоксафос-
Спектр ЯМР 1Н (СDCl3), δ, м. д.: 1.76 д (2Н, CH2,
финан-2-амина 6a в 1.5 мл диоксана выдержива-
3JHH = 12.4 Гц), 2.23 д (2Н, CH2, 3JHH = 12.4 Гц),
ли 24 ч при 85-90°С. Затем к реакционной смеси
2.98 д (12Н, NCH3, 3JHH = 13.8 Гц), 3.13-3.21 уш.
добавляли раствор 1.8 ммоль серы в 2 мл бензола
м (4Н, NCH2), 3.53-3.62 уш. м (4Н, NCH2), 5.01 c
и выдерживали 12 ч при 50-55°С. Для выделения
(2H, CH2), 7.23 д ( 4H, H3,6, 3JHH = 5.0 Гц), 7.57 д
продукта реакции реакционную смесь упаривали
(2H, H7, 3JHH = 9.1 Гц), 7.63-7.67 м (4H, H4,8), 8.14
на 2/3 и приливали 10 мл гексана. Осадок отфиль-
д (2H, H5, 3JHH = 7.8 Гц). Спектр ЯМР 13С (CDCl3),
тровывали, промывали 2 мл гексана и сушили в
δС, м. д.: 23.83 (CH2), 23.87 (СH2), 38.05 д ( NCH3,
вакууме (1 мм рт. ст.) при 75°С. Выход 81%, белый
2JРС = 3.8 Гц,), 50.48 (NCH2), 120.48 д (С3, 3JРС =
порошок, т. разл. 240-243°С. ИК спектр, ν, см-1:
2.9 Гц), 124.54 (С6/7), 125.01 (С8), 126.16 (С6/7),
880.7 c (P=S). Спектр ЯМР 1Н (СDCl3), δ, м. д.:
127.18 д (С1, 2JРС = 5.75 Гц), 128.04 (С4/5), 128.38
1.90 уш. с (8H, CH2), 2.31 уш. с (16H, CH2CH2Ph),
(С4/5), 131.24 (С10), 133.77 (С9), 146.95 д (С2, 2JРС =
2.74 уш. с (8Н, CH2), 3.32 уш. с (32Н, ОCH2), 4.77
12.4 Гц,). Спектр ЯМР 31Р (CDCl3): δР 74.44 м. д.
т (4H, CH, 3JHH = 7.3 Гц), 6.34 с (2Н, Н3), 7.11 м
Масс-спектр (MALDI), m/z: 624 [M+] (вычислено
(20Н, Н-Ph), 7.51 с (2Н, Н3), 7.64 с (2Н, Н5), 8.07 с
для C31H38N4O2P2S2: 624). Найдено, %: С 59.59;
(2Н, Н5). Спектр ЯМР 13С (CDCl3), δС, м. д.: 25.99
H 6.15; N 9.00. C31H38N4O2P2S2. Вычислено, %: С
(СH2), 34.53 (СН2СН2Ph), 36.64 (СН2СН2Ph), 42.26
59.60; Н 6.13; N 8.97.
(С1), 69.30 д (OCH2, 2JРС = 8.6 Гц), 111.79 уш. с
2,2ʹ,2ʹʹ,2ʹʹʹ-{1,1ʹ-Метиленбис[(нафталин-
(С3), 125.92 (СН2СН2Ph), 126.90 уш. с (C2), 128.45
2,7-диил)диокси]}тетра(1,3-диметил-1,3,2-диа-
(СН2СН2Ph), 128.54 (СН2СН2Ph), 132.85 уш. с (С5),
зафосфинан- 2-сульфид) (11). Выход 61%, белый
141.48 (СН2СН2Ph), 147.64 уш. с (С4), 148.03 уш. с
порошок, т. пл. 134-135°С. ИК спектр, ν, см-1:
(С4). Спектр ЯМР 31Р (CDCl3), δР, м. д.: 56.38,
872.6 c (P=S). Спектр ЯМР 1Н (СDCl3), δ, м. д.: 1.58
55.39. Масс-спектр (MALDI), m/z: 2017 [M + Na+]
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 8 2020
ПОЛИФУНКЦИОНАЛИЗИРОВАННЫЕ ХЕЛАТНЫЕ ПРОИЗВОДНЫЕ ДИНАФТИЛМЕТАНА
1269
(вычислено для C84H96O24P8S8Na: 2015). Найдено,
приливали 3.0 (6.0) ммоль диметилкарбамоилхло-
%: С 50.55; H 4.90. C84H96O24P8S8. Вычислено, %:
рида 7a. Реакционную смесь кипятили при пере-
С 50.60; Н 4.85.
мешивании 6 ч, затем охлаждали и удаляли аце-
тон. К остатку добавляли 15 мл 5%-ной серной
Октакис(1,3-диметил)-октакис-2-
кислоты. Образовавшийся осадок отфильтровыва-
{2,8,14,20-тетра(2-фенилэтил)каликс[4]-
ли, промывали 60 мл воды и 20 мл гексана, затем
арен-4,6,10,12,16,18,22,24-октаил(октаокси)}-
сушили 5 ч при 80°С (1мм рт. ст.).
октакис(1,3,2-диазафосфинан-2-сульфид)
(13).
Смесь 0.12 ммоль резорцинкаликс[4]арена 3 и
[1,1ʹ-Метиленбис(нафталин-2-ил)]бис(диме-
1.2 ммоль
1,3-диметил-N,N-диэтил-1,3,2-диаза-
тилкарбамат) (14). Выход 79%, светло-коричне-
фосфинан-2-амина 6б в 1.5 мл диоксана выдержи-
вый порошок, т. пл. 138-140°С. ИК спектр, ν, см-1:
вали 48 ч при комнатной температуре. Затем к реак-
с 1703 (С=О). Спектр ЯМР 1Н (CDCl3), δ, м. д.: 2.31
ционной смеси добавляли раствор 1.8 ммоль серы
с (6Н, NCH3), 2.73 с (6Н, NCH3), 4.77 с (2Н, СН2),
в 2 мл бензола и перемешивали 24 ч при 20-25°С.
7.14 д (2Н, H3, 3JНН = 8.7 Гц), 7.40-7.47 м (4Н, H6,7),
Для выделения продукта реакции реакционную
7.72 д (2Н, Н4, 3JНН = 8.7 Гц), 7.82 д. д (2Н, Н8, 4JНН =
смесь упаривали на 2/3 и приливали 10 мл гексана.
2.3, 3JНН = 7.3 Гц), 8.19 д (2Н, Н5, 3JНН = 8.2 Гц).
Осадок отфильтровывали, промывали 2 мл гексана
Спектр ЯМР 13С (CDCl3), δС, м. д.: 24.09 (СН2),
и сушили в вакууме (1 мм рт. ст.) при 75°С. Выход
35.62 (NCH3), 36.44 (NCH3), 122.56 (С3), 124.29
61%, белый порошок, т. пл. 277-278°С. ИК спектр,
(С8), 125.01 (С6/7), 126.21 (С6/7), 127.76 (С4), 127.83
ν, см-1: 871.7 c (P=S). Спектр ЯМР 1Н (СDCl3), δ,
(С1), 128.68 (С5), 132.06 (С10), 133.27(С9), 147.30
м. д.: 1.33 д (4H, CH2, 3JHH = 13.3 Гц), 1.50 д (4H,
(С2), 154.46 (С=О). Масс-спектр (MALDI), m/z: 442
CH2, 3JHH = 13.7 Гц), 1.94-1.99 уш. м (4H, CH2),
[М]+ (вычислено для C27H26N2O4: 442). Найдено,
2.08-2.16 уш. м (4H, CH2), 2.22-2.28 уш. м (4H,
%: C 73.26; H 5.95; N 6.34. C27H26N2O4 Вычислено,
CH2CH2Ph), 2.55-2.67 уш. м (12H, CH2CH2Ph),
%: C 73.28; H 5.92; N 6.33.
2.81 д (12H, NCH3, 3JРH = 13.8 Гц), 2.87 д (12H,
[1,1ʹ-Метиленбис(нафталин-2,7-диил)]тетра-
NCH3, 3JPH = 14.2 Гц), 2.93 д (12H, NCH3, 3JPH =
(диметилкарбамат) (15). Выход 84%, светло-ко-
13.8 Гц), 2.94 д (12H, NCH3, 3JPH = 14.2 Гц), 2.98-
ричневый порошок, т. пл 213-214°С. ИК спектр,
3.05 уш. м (8Н, NCH2), 3.28-3.35 уш. м (4Н, NCH2),
ν, см-1: c 1711.3 (С=О). Спектр ЯМР 1Н (CDCl3), δ,
3.37-3.43 уш. м (4H, NCH2), 4.83 т (4H, CH, 3JHH =
м. д.: 2.13 с (6Н, NCH3), 2.67 с (6Н, NCH3), 3.10 с
6.4 Гц), 6.44 с (2Н, Н3), 6.87-6.95 м (20Н, Н-Ph),
(6Н, NCH3), 3.12 с (6Н, NCH3), 4.64 с (2Н, СН2), 7.06
7.52 с (2Н, Н3), 7.85 с (2Н, Н5), 8.07 с (2Н, Н5).
д (2Н, H3, 3JНН = 8.7 Гц,), 7.29 д. д (2Н, H6, 4JНН =
Спектр ЯМР 13С (CDCl3), δС, м. д.: 22.29 (СH2),
2.3, 3JНН = 9.2 Гц), 7.9 д (2Н, Н4, 3JНН = 8.7 Гц),
22.32 (СН2), 23.25 (СН2), 23.26 (СН2), 34.51 (СН2С-
7.82 д (2Н, Н5, 3JНН = 8.7 Гц), 7.89 д (2Н, Н8, 3JНН =
Н2Ph), 36.06 (СН2СН2Ph), 37.43(С1), 37.84 (NCH3),
2.2 Гц). Спектр ЯМР 13С (CDCl3), δС, м. д.: 24.51
38.08 (NCH3), 38.15 ( NCH3), 38.18 (NCH3), 49.65
(СН2), 35.36 (NCH3), 36.27 (NCH3), 36.64 (NCH3),
д (NCH2, 2JРС = 61.0 Гц), 50.33 д (NCH2, 2JРС =
36.80 (NCH3), 115.24 (С3), 121.01 (С8), 122.24 (С6),
63.9 Гц,), 111.18 (С3), 112.04 (С3), 125.57 (СН2С-
127.21 (С1), 127.40 (С4/5), 129.61 (С10), 129.67 (С4/5),
Н2Ph), 125.69 (С2), 125.90 д (С5, 2JРС = 10.53 Гц),
134.01 (С9), 148.03 (С2), 150.08 (С7), 154.19 (С=О),
128.01 (СН2СН2Ph), 128.13 (СН2СН2Ph), 128.99
154.99 (С=О). Масс-спектр (MALDI), m/z: 615 [М]+
(С2), 131.13 д (С5, 2JРС = 7.7 Гц), 141.31 (СН2С-
(вычислено для C33H36N4O8: 616). Найдено, %: C
Н2Ph), 147.60 д (С4, 2JРС = 9.6 Гц), 149.20 д (С4, 2JРС=
64.30; H 5.90; N 9.11. C33H36N4O8 Вычислено, %: C
10.5 Гц). Спектр ЯМР 31Р (CDCl3), δР, м. д.: 72.67,
64.27; H 5.88; N 9.09.
72.05. Масс-спектр (MALDI), m/z: 2190 [M - CH3]+
Изучение экстракционной способности сое-
(вычислено для C99H141N16O8P8S8: 2187). Найдено,
динений 8-23. Пикраты металлов получали сли-
%: С 54.39; H 6.50; N 9.92. C100H144N16O8P8S8. Вы-
ванием водных растворов пикриновой кислоты и
числено, %: С 54.53; Н 6.59; N 10.17.
растворов предварительно оттитрованных нитра-
Общая методика синтеза соединений 14, 15.
тов тяжелых металлов. Водные растворы пикратов
К суспензии 1.0 ммоль динафтилметана 1 (2) и
(5 мл), содержащие избыток металла, и растворы
3.0 (6.0) ммоль карбоната калия в 10 мл ацетона
исследуемых соединений в СНСl3 (5 мл) переме-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 8 2020
1270
СЕРКОВА и др.
шивали 60 мин при комнатной температуре и вы-
C4CS00211C
держивали 15 мин для разделения фаз. Начальные
7. Gokel G.W., Negin S. // Adv. Drug Del. Rev. 2012.
концентрации соединений 8-23 [L] в органиче-
Vol. 64. P. 784. doi 10.1016/j.addr.2012.01.011
ской фазе, а солей металлов и пикрата в водной
8. Raynal М., Ballester P., Vidal-Ferrana A., van Leeu-
wen P.W.N.M. // Chem. Soc. Rev. 2014. Vol. 43. P. 1734.
фазе варьировали в следующих пределах [L]0 = 2×
doi: 10.1039/C3CS60037H
10-4 моль/л, [MOH]0 = 2∙10-4 моль/л, [HPic]0 =
9. Deraedt C., Astruc D. // Coord. Chem. Rev. 2016.
2.5×10-4 моль/л соответственно. Оптические плот-
Vol. 324. P. 106. doi 10.1016/j.ccr.2016.07.007
ности водной фазы до и после экстракции (А0 и Аi
10. Баталова Т.А., Расадкина Е.Н., Васянина Л.К.,
соответственно) определяли методом УФ спектро-
Бельский В.К., Нифантьев Э.Е. // ЖОХ. 1997. Т. 67.
скопии на приборе SHIMADZU PharmaSpec UV
С. 1497; Batalova T.A., Rasadkina E.N., Vasyani-
1700 в диапазоне длин волн от 200 до 900 нм.
na L.K., Belsky V.K., Nifantyev E.E. // Russ. J. Gen.
Степень экстракции (Е, %) вычисляли по соот-
Chem. 1997. Vol. 67. P. 1406.
ношению (1).
11. Maslennikova V.I., Sotova T. Yu., Vasyanina L.K.,
Lyssenko K.A., Antipin M.Yu., Adamson S.O., Demen-
tyev A.I., Habicher W.D., Nifantyev E.E. // Tetrahedron.
2007. Vol. 63. P. 4162. doi 10.1016/j.tet.2007.02.095
ФОНДОВАЯ ПОДДЕРЖКА
12. Hogberg, A.G.S. // J. Org. Chem 1980. Vol. 45. P. 4498.
Работа выполнена при финансовой поддержке
doi 10.1021/jo01310a046
Российского фонда фундаментальных исследова-
13. Hogberg, A.G.S. // J. Am. Chem. Soc 1980. Vol. 102.
ний (проект 18-03-00347a).
P. 6046. doi 10.1021/ja00539a012
14. Maslennikova V.I., Serkova O.S., Gruner M., Goutal S.,
КОНФЛИКТ ИНТЕРЕСОВ
Bauer I., Habicher W.D., Lyssenko K.A., Antipin M.Y.,
Nifantyev E.E. // Eur. J. Org. Chem. 2004. P. 4884. doi
Авторы заявляют об отсутствии конфликта ин-
10.1002/ejoc.200400363
тересов.
15. Maslennikova V.I., Serkova O.S., Shelenkova L.V.,
СПИСОК ЛИТЕРАТУРЫ
Vasyanina L.K., Tarasenko D.V., Nifantiev E.E. //
Tetrahedron Lett. 2012. Vol. 53. N 7. P. 886. doi
1. Leoncini A., Huskens J., Verboom W. // Chem. Soc. Rev.
10.1016/j.tetlet.2011.12.034
2017. Vol. 46. P. 7229. doi 10.1039/C7CS00574A
16. Burikhina A.V, Serkova O.S., Tarasenko D.V., Levi-
2. Konczyk J., Nowik-Zajac A., Kozlowski C. A. //
Separ. Sci. Technol. 2016. Vol. 51. P. 2394. doi
na I.I., Gorlova A.V., Maslennikova V.I. // Arkivoc.
10.1080/01496395.2016.1209219
2016. Vol. 3. P. 325. doi 10.3998/ark.5550190.p009.492
3. Pinalli R., Pedrini A., Dalcanale E. // Chem. Soc. Rev.
17. Wolff W. // Chem. Ber. 1893. Vol. 26. P. 85. doi 10.1002/
2018. Vol. 47. P. 7006. doi 10.1039/C8CS00271A
cber.18930260118
4. Burilov V.A., Mironova D.A., Ibragimova R.R.,
18. Tucker J.A., Knobler C.B., Trueblood K.N., Cram D.J. //
Solovieva S.E., Konig B., Antipin I.S. // RSC Adv. 2015.
J. Am. Chem. Soc. 1989. Vol. 111. P. 3688. doi 10.1021/
Vol. 5. P. 101177. doi 10.1039/C5RA18294H
ja00192a028
5. Collin S., Giraud N., Dumont E., Reinaud O. // Org.
19. Нифантьев Э.Е., Сорокина С.Ф., Борисенко А.А. //
Chem. Front. 2019. Vol. 6. P. 1627. doi 10.1039/
ЖОХ. 1985. Т. 55. C. 1665.
C9QO00263D
20. Нифантьев Э.Е., Завалишина А.И., Сорокина С.Ф.,
6. Rebilly J.-N., Colasson B., Bistri O., Over D., Reinaud O. //
Борисенко А.А., Смирнова Е.И., Курочкин В.В., Мо-
Chem. Soc. Rev. 2015. Vol. 44. P. 467. doi 10.1039/
исеева Л.И. // ЖОХ. 1979. Т. 49. С. 64.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 8 2020
ПОЛИФУНКЦИОНАЛИЗИРОВАННЫЕ ХЕЛАТНЫЕ ПРОИЗВОДНЫЕ ДИНАФТИЛМЕТАНА
1271
Polyfunctional Chelate Derivatives of Dinaphthylmethane
and Resorcincalix[4]arene: Synthesis, Relationship
between Chelate Structure and Its Acceptor Ability
O. S. Serkovaa, A. V. Kamkinaa, I. Yu. Toropyginb, and V. I. Maslennikovaa,*
a Institute of Biology and Chemistry, Moscow State Pedagogical University, Moscow, 129164 Russia
b V.N. Orekhovich Research Institute of Biomedical Chemistry, Moscow, 119121 Russia
*e-mail: him-vim@mail.ru; vi.maslennikova@mpgu.su
Received March 17, 2020; revised March 17, 2020; accepted March 28, 2020
Functionalization of methylenebis(naphthalene-2-ol/-2,7-diol) and rccc-resorcincalix[4]arene gave a series
of their derivatives by OH groups with 1, 2, and 4 pairs of chelate ionophore substituents differing in the
combination of electron-donating atoms (O, S , N, P). Using the method of liquid extraction, the effect of the
structural features of the ligand on its ability to recognize and bind transition metal cations (Cd2+, Ag+, Hg2+)
was studied.
Keywords: methylenebis(naphthalene-2-ol), methylenebis(naphthalene-2,7-diol), resorcincalix[4]arenes,
phosphorylation, carbamoylation
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 90 № 8 2020