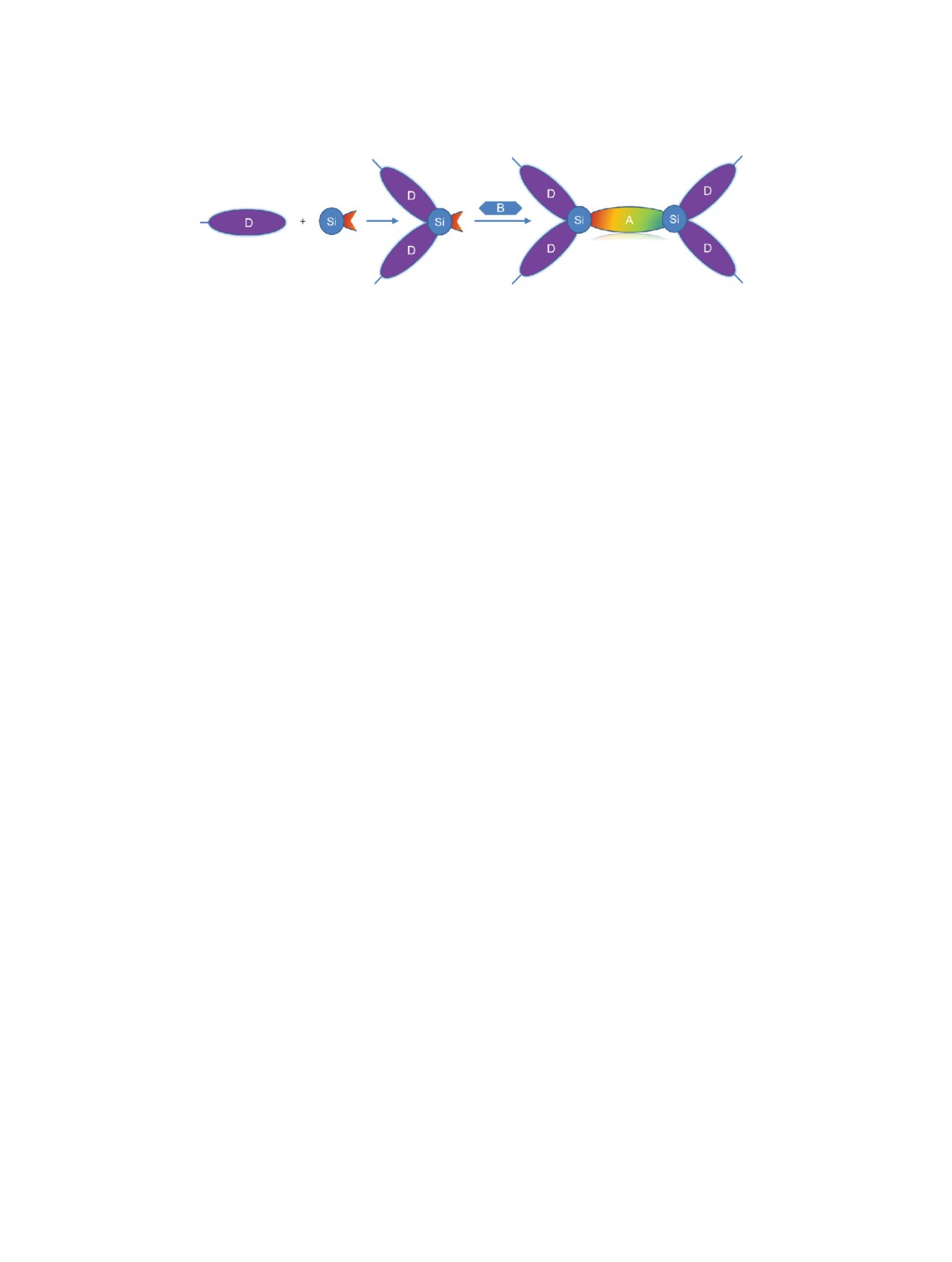ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2019, Том 55, № 1, с. 40-59
УДК 541.64:547(128´1+245)
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ
НАНОСТРУКТУРИРОВАННЫХ
ЛЮМИНОФОРОВ НА ОСНОВЕ ФЕНИЛОКСАЗОЛОВ
© 2019 г. М. С. Скоротецкийa, О. В. Борщевa, Г. В. Черкаевa, С. А. Пономаренкоa, b, *
a ФГБУН Институт синтетических полимерных материалов им. Н.С. Ениколопова РАН,
117393, Россия, г. Москва, ул. Профсоюзная 70
*e-mail: ponomarenko@ispm.ru
b Московский государственный университет имени М.В. Ломоносова, химический факультет,
ГСП-1, 119991, Россия, г. Москва, Ленинские горы 1, стр. 3
Поступила в редакцию 29 октября 2018 г.
После доработки 5 ноября 2018 г.
Принята к публикации 11 ноября 2018 г.
Впервые с использованием реакций Ван Лёссена и катализируемого комплексами палладия прямого С-Н
арилирования оксазолов синтезирован ряд кремнийорганических наноструктурированных люминофоров
(КНЛ), в центре которых находится акцепторный хромофорный фрагмент 1,4-бис(5-фенилоксазо-2-ил)
бензола (POPOP), а на периферии - различные донорные производные п-терфенила и 2,5-дифенилокса-
зола. За счет использования кремнийорганических разветвляющих центров с различной функциональ-
ностью получены КНЛ с разным соотношением донор : акцептор. Выбор данных сопряженных структур
обусловлен их хорошими оптическим характеристиками, благодаря которым полученные КНЛ могут
найти применение в фотонике и оптоэлектронике.
Ключевые слова: прямое C-Н арилирование, реакция Ван-Лессена, фенилоксазол, арилсилан, лю-
минофор, терфенил, POPOP.
DOI: 10.1134/S0514749219010051
ВВЕДЕНИЕ
деле, данная идея не нова и широко использует-
ся в живых организмах, где перенос фотоинду-
Создание “молекулярных антенн”, или как их
цированной энергии необходим для эффективно-
еще называют “светособирающие комплексы”, яв-
го поглощения солнечной энергии и транспорта
ляется чрезвычайно сложной задачей для синтети-
ее к реакционному центру, преобразующему ее
ческой химии. Сам термин “молекулярные антен-
в химический потенциал [4]. На данный момент
ны” был впервые предложен Бальцани (Balzani) и
именно биоорганизмы обладают самой сложной
Леном (Lehn) с соавт. [1-3]. Благодаря разработан-
и совершенной системой переноса энергии по
ным синтетическим подходам и молекулярному
принципу молекулярной антенны. Исследование в
дизайну им удалось получить ряд высокомолеку-
данной области имеет большой потенциал для соз-
лярных дендритных структур на основе комплек-
дания новых способов трансформации и хранения
сов металлов, обладающих внутримолекулярным
солнечной энергии [5-8]. В связи с этим, усилия
переносом энергии. За счет высокой способности
многих исследователей в последнее время были
поглощать свет в видимом диапазоне, полученные
направлены на получение и изучение уникальных
материалы могут найти применение в качестве ан-
оптических свойств люминесцентных разветвлен-
тенн для собирания солнечного света. На самом
ных, сверхразветвленных и дендритных макромо-
40
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
41
R
R
R
R
I
II
III
КНЛ
R
R
R
Общая схема получения КНЛ, где R - солюбилизирующий алкильный заместитель, D и А - донорный и акцепторные фраг-
менты соответственно.
лекул [9-12], организованных по принципу моле-
ные оптические свойства, как поглощение и люми-
кулярной антенны.
несценция, на заданный спектральный диапазон.
Недавно в ИСПМ РАН был разработан новый
Фенилоксазолы и низшие олигофенилены явля-
класс кремнийорганических люминофоров, об-
ются перспективными люминофорами для исполь-
ладающих эффективным внутримолекулярным
зования в структурах КНЛ. На сегодняшний день
переносом энергии по принципу “молекулярной
1,4-бис(5-фенилоксазол-2-ил)бензол
(POPOP),
антенны” [13]. Они представляют собой развет-
п-терфенил и 2,5-дифенилоксазол (PPO), облада-
вленные или дендритные соединения, в которых
ющие высоким квантовым выходом люминесцен-
несколько органических люминофоров двух ти-
ции (Qlum) нашли широкое применение в промыш-
пов ковалентно связаны между собой через атомы
ленности в качестве активаторов (п-терфенил,
кремния, в результате чего они располагаются на
PPO) и сместителя (POPOP) спектров пластмас-
небольшом расстоянии с фиксированным углом
совых сцинтилляторов, лазерной оптике, физике
друг относительно друга. При этом один из них
высоких энергий [15, 16]. Благодаря универсаль-
может служить донором, а другой - акцептором
ной структуре КНЛ, представляется возможным
энергии электронного возбуждения. Кроме того,
соединить в одной молекуле данные люминофо-
в случае подбора люминофоров с определённы-
ры, что позволит получить новые соединения с
ми характеристиками, такие системы обладают
уникальными оптическими характеристиками.
не только эффективным внутримолекулярным пе-
Предполагается, что они будут эффективно погло-
реносом энергии, но и высоким квантовым выхо-
щать свет в ультрафиолетовом диапазоне, соответ-
дом люминесценции. Это позволяет создавать так
ствующему поглощению донорных фрагментов и
называемые “кремнийорганические нанострукту-
обладать люминесценцией с высоким Qlum в более
рированные люминофоры” (КНЛ) [14], к преиму-
длинноволновом диапазоне, соответствующему
ществам которых можно отнести большое сечение
люминесценции POPOP (~420 нм). В связи с этим,
поглощения, огромный псевдостоксовский сдвиг1,
данная работа направлена на разработку условий
позволяющий свести к минимуму эффект самопо-
синтеза КНЛ с центральным акцепторным фраг-
глощения не только в концентрированных раство-
ментом люминофора POPOP и донорными п-тер-
рах таких люминофоров, но и в тонких пленках,
фенильными или PPO фрагментами.
а также дает возможность настраивать такие важ-
--------------
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
1 Стоксовский сдвиг определяется расстоянием между макси-
Синтетические методы получения фенилокса-
мумами поглощения и люминесценции хромофора, однако
в случае КНЛ максимум поглощения не соответствует ко-
золов хорошо известны. С тех пор, как Эмилем
лебательному уровню, с которого происходит испускание
Фишером в 1896 году был впервые получен 2,5-ди-
фотона. Для КНЛ это расстояние фактически определяется
фенилоксазол, описано огромное количество его
разницей между максимумами поглощения донорного и лю-
минесценции акцепторного фрагментов.
производных и методов синтеза [17]. Однако боль-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
42
СКОРОТЕЦКИЙ и др.
Схема 1. Химическая структура оптимальных донорных фрагментов для использования в КНЛ с центральным ак-
цепторным фрагментом POPOP.
шинство из них не подходит для получения крем-
четырьмя периферийными донорными фрагмен-
нийорганических производных, а также других
тами и одним центральным акцепторным (A). В
соединений, неустойчивых в кислой среде. Ранее
этом случае достигается оптимальное сочетание
нами был разработан новый метод получения
простоты синтеза, хороших реакционных выхо-
кремнийорганических производных фенилокса-
дов и желаемых оптических свойств. В то время
зола, включающий постадийное наращивание фе-
как получение монодендрона с тремя донорными
нилоксазольного фрагмента с получением струк-
фрагментами имеет преимущество с точки зрения
туры, идентичной люминофору РОРОР [18, 19]. В
увеличения молярного коэффициента экстинкции,
его основу легла реакция получения 5-замещенных
однако сопряжено с более низкими выходами на
оксазолов из альдегидов по методу Ван-Лёссена и
стадии получения монодендрона и, соответствен-
реакция прямого арилирования, катализируемая
но, усложняет его синтез. На последней стадии
широко применяемым комплексом палладия. Как
полученный функциональный монодендрон после
показано ниже, данный подход оказался не только
нескольких превращений вступает в реакцию с би-
применимым, но и достаточно эффективным для
функциональным прекурсором (B) с образованием
синтеза КНЛ.
целевой структуры КНЛ.
Стратегию синтеза КНЛ на основе фенилок-
п-Терфенил (3Ph), 2',5'-диметил-1,1':4',1''-тер-
сазолов можно разделить на несколько основных
фенил (3Ph-Me2) и PPO (схема 1) обладают при-
этапов (см. рисунок). На первом из них синтези-
влекательным комплексом физико-химических
руют функциональный прекурсор I с солюби-
свойств с точки зрения их использования в каче-
лизирующей группой R, который представляет
стве донорных фрагментов КНЛ: поглощение в
собой уже готовый донорный (D) фрагмент или
УФ диапазоне с большим молярным коэффици-
в дальнейшем будет входить в состав донорного
ентом экстинкции, высокий квантовый выход лю-
фрагмента КНЛ. Для этого удобнее всего исполь-
минесценции, хорошая фотохимическая стабиль-
зовать различные бром- или литийорганические
ность. Диапазоны их поглощения и излучения
производные. Затем синтезируют многофункци-
света хорошо подходят для внутримолекулярного
ональный разветвляющий кремнийорганический
переноса энергии к центральному акцепторному
центр II, который на следующей стадии может ре-
фрагменту, что является необходимым условием
агировать с прекурсором I с образованием функ-
для создания КНЛ по принципу «молекулярной
ционального монодендрона III. В зависимости от
антенны». Основной недостаток 3Ph, в отличие от
количества реакционных групп в разветвляющем
PPO и 3Ph-Me2 - низкая растворимость в большин-
центре, можно получать кремнийорганические
стве органических растворителей, а также в поли-
соединения с различным содержанием донорных
мерной матрице. Поэтому особое внимание при
фрагментов. С точки зрения молекулярного дизай-
молекулярном дизайне было уделено увеличению
на, оптимальной структурой является моноден-
растворимости будущих люминофоров с фрагмен-
дрон с двумя донорными фрагментами, из которо-
тами 3Ph. Для решения этой проблемы в структуре
го на следующей стадии можно получить КНЛ c
целевого КНЛ были использованы разветвленные
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
43
2-этилгексильные заместители, обладающие высо-
Тем не менее, чистое соединение 3 может быть по-
кой солюбилизирующей способностью. В случае с
лучено методом вакуумной сублимации.
PPO и 3Ph-Me2 использовали короткие метильные
Контроль в режиме in situ за степенью завер-
заместители для удобства при расшифровке ЯМР
шенности реакций, а также чистотой конечных и
спектров.
промежуточных соединений здесь и далее прово-
Согласно изложенной выше стратегии синтеза
дили путем анализа на гель-проникающем хрома-
КНЛ, для получения люминофоров с донорными
тографе, оборудованном колонкой, работающей в
3Ph фрагментами первоначально был синтези-
линейном режиме в интервале молекулярных масс
рован растворимый функциональный прекурсор
от 100 до 15000 согласно паспорту (размер пор
4-бром-4''-(2-этилгексил)-1,1':4',1''-терфенил
(3)
500 Å) с диодным матричным детектором. Данная
(схема 2). Для этого взаимодействием 2-этилгек-
система оказалась очень информативной и позво-
силбромида (1) с магнием в диэтиловом эфире
ляет точно отличать даже небольшие молекулы с
разной длиной сопряжения за счет получения од-
получили реактив Гриньяра. Затем полученную
реакционную смесь охлаждали до комнатной тем-
новременно с ГПХ-кривой спектров поглощения
в диапазоне 200-800 нм всех присутствующих на
пературы и прикапывали к раствору 1,4-дибром-
ней пиков. Благодаря этому достигается не только
бензола, взятого в недостатке, и катализатора, под-
оптимальное время прохождения реакции (зача-
держивая температуру ниже 5°С. После окончания
стую в литературе время протекания реакций ме-
реакции кросс-сочетания в условиях Кумады и
таллоорганических реакций существенно завыше-
очистки методом вакуумной дистилляции уда-
но из-за отсутствия такого анализа), но и контроль
лось получить чистое соединение 2 с выходом
высокой чистоты получаемых соединений на ста-
86%. Использование недостатка 1,4-дибромбен-
дии их синтеза и очистки за счет высокой чувстви-
зола относительно алкилбромида 1 обусловлено
тельности данного прибора (чувствительность ди-
как протеканием побочной реакции Вюрца, так и
одной матрицы в сотни раз выше по сравнению с
практически полным отсутствием побочного про-
рефрактометрическим детектором).
цесса замещения второго атома брома в 1,4-ди-
бромбензола на 2-этилгексильной группы. В про-
Синтез
функционального
прекурсора
тивном случае большое количество остаточного
4-бром-2',4'',5'-триметил-1,1':4',1''-терфенила
(6)
1,4-дибромбензола сильно усложняет вакуумную
для получения КНЛ с донорными 3Ph-Me2 фраг-
дистилляцию. Аналогично проводили получе-
ментами проводили аналогично получению сое-
динения 3 (cхема 2). Для этого взаимодействием
ние соединения 3, реакционный выход которого
составил 70%. В данном случае, протекание по-
реактива Гриньяра, полученного из п-бромтолу-
бочной реакции двойного замещения происходит
ола с избытком 1,4-дибром-2,5-диметилбензола
статистически, а близкая растворимость соедине-
был получен 4-бром-2,4',5-триметилбифенил (5).
ния 3 и продукта дизамещенения - 4,4'''-бис(2-э-
После очистки методом вакуумной дистилляции
удалось выделить чистое соединение 5 с выходом
тилгексил)-1,1':4',1'':4'',1'''-кватрофенила - сильно
усложняет очистку. Согласно данным ГПХ, чисто-
63%. Конечный функциональный прекурсор 6 был
та полученного производного терфенила 3 после
синтезирован по реакции Кумады между п-ди-
многократной экстракции из спирта и/или ацетона
бромбензолом и реактивом Гриньяра, полученным
составила 85%. Однако оставшийся нефункцио-
из соединения 5. После перекристаллизации из
этилового спирта было выделено чистое соедине-
нальный кватерфенил 3а в количестве 15% не спо-
ние 6 с выходом 87%.
собен участвовать в дальнейших реакциях и может
быть легко отделен на следующей стадии от моно-
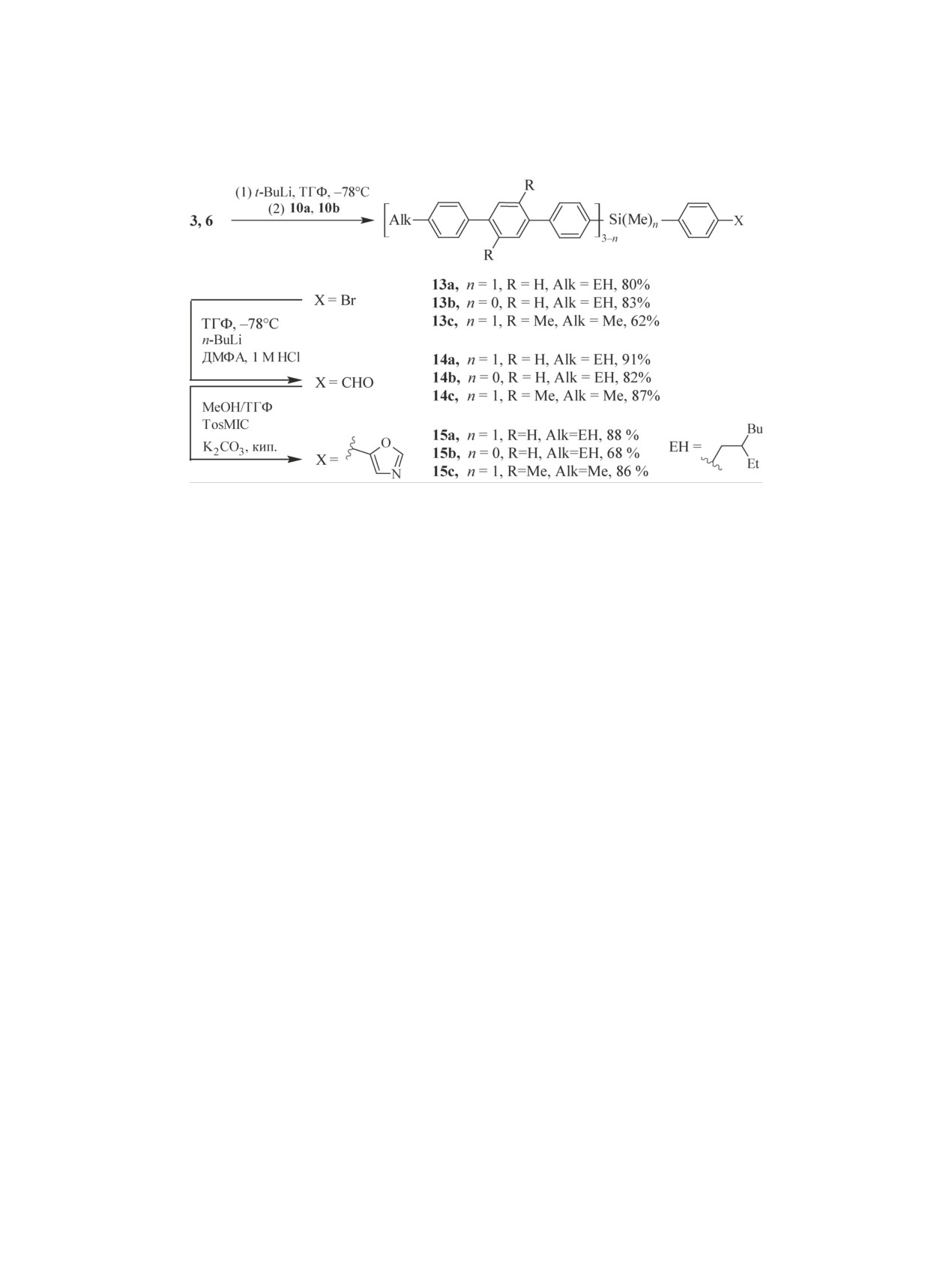
Для объединения функциональных прекурсоров
дендрона 13 (cхема 3), имеющего гораздо лучшую
3 и 6 в структуре целевой кремнийорганической
растворимость в органических растворителях.
молекулярной антенны необходим соответствую-
Поэтому во избежание больших потерь при даль-
щий центр ветвления. В качестве такового были
нейшей экстракции, функциональный прекурсор
выбраны дифункциональный
(4-бромфенил)-
3 был использован без дополнительной очистки.
(метил)дихлорсилан (10a) и трифункциональный
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
44
СКОРОТЕЦКИЙ и др.
Схема
2. Схема синтеза функциональных прекурсоров
3 и
6, а также функциональных разветвляющих
кремнийорганических центров 10a, 10b для КНЛ с донорными 3Ph и 3Ph-Me2 фрагментами.
1
2
3
7
-
9a, 9b
(4-бромфенил)трихлорсилан (10b) (схема 2). Син-
личеств ДМФА. Контроль осуществляли методом
тез этих соединений был осуществлен через
1Н ЯМР спектроскопии, на спектрах которой от-
предварительное получение соответствующих
четливо наблюдалось исчезновение характерных
этоксисиланов:
(4-бромфенил)(метил)диэтокси-
сигналов этокси-групп в районе 1-4 м. д. После
силана
(9a) и
(4-бромфенил)триэтокисилана
вакуумной перегонки было получено соедине-
(9b). Такой подход за счет простоты выделения
ние 10a с выходом 95% и чистотой 98% согласно
этоксисиланов и высоких реакционных выходов
ГЖХ-анализу. Аналогичным образом был получен
оказался более удобным и эффективным, нежели
(4-бромфенил)трихлорсилан (10b). В этом случае
непосредственное получение соответствующих
общий выход за две стадии целевого хлорсилана
хлорсиланов.
чистотой 97% составил 33.5%.
Синтез соединения 9a был осуществлен литии-
Получение функциональных монодендронов с
рованием 1,4-дибромбензола (7) n-BuLi при -78°С
донорными 3Ph и 3Ph-Me2 фрагментами проводи-
в ТГФ с последующей обработкой безводным бро-
ли по специально разработанной трехстадийной
мидом магния для получения реактива Гриньяра,
методике (схема 3). Для этого взаимодействием
который сразу после получения прикапывали к
соединений 3 или 6 с t-BuLi при пониженных тем-
3-кратному избытку метилтриэтоксисилана при
пературах и последующей обработкой соответ-
-15°С. После выделения, упаривания растворите-
ствующим хлорсиланом (10a, 10b) были получены
ля и последующей фракционной дистилляции при
соединения 13a, 13b. После очистки методом коло-
пониженном давлении удалось выделить целевой
ночной хроматографии на силикагеле выход соста-
продукт 9a с выходом 58%. Для получения соот-
вил 621-83%. На следующей стадии литиировани-
ветствующего хлорсилана полученный этокси-
ем соединений 13a, 13b при -78°С с последующей
силан перемешивали при нагревании с избытком
обработкой ДМФА и разрушением полученной
тионилхлорида в присутствии каталитических ко-
соли до альдегида путем подкисления до кислой
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
45
Схема 3. Синтез функциональных монодендронов с донорными 3Ph и 3Ph-Me2 фрагментами.
среды были получены производные бензальдегида
проводили методом колоночной хроматографии
14a, 14b. Выход реакции был определен согласно
на силикагеле в толуоле. Выходы составили 68%
данным 1Н ЯМР-спектроскопии по появлению ха-
и 73%, соответственно. На следующей стадии
рактерного синглета протона альдегидной группы
карбонильную группу защищали диоксановой за-
при 10 м. д.. Далее соединения 14a, 14b без допол-
щитой с использованием неопентингликоля путем
нительной очистки по реакции Ван Лёссена пере-
кипячения в бензоле в присутствии толуолсуль-
водили в 5-замещенные оксазолы 15a-15c. После
фокислоты в качестве катализатора с азеотропной
очистки методом колоночной хроматографии на
отгонкой образующейся воды в процессе реакции.
силикагеле (элюент - толуол) выход чистых сое-
Выбор данного спирта был обусловлен тем, что
динений 15a, 15b составил 68-88%.
более простая диоксалановая защитная группа с
использованием этиленгликоля менее устойчива,
В связи с возможностью оксазольного кольца
что может приводить к ее частичному снятию в
вступать в различные побочные реакции в усло-
виях литиирования, синтез функциональных мо-
процессе очистки на силикагеле.
нодендронов с донорными PPO фрагментами был
Затем из соединений 18a, 18b по реакции пря-
осуществлён по другой методике. Для этого в ка-
мого арилирования с ранее полученными функ-
честве исходных соединений использовали мно-
циональными прекурсорами 20a, 20b по реакции
гофункциональные кремнийорганические центры
Ван Лёссена из соответствующих бензальдегидов
трис(4-бромфенил)метилсилан (16a) или тетра-
были получены соединения 21a-21c с защищен-
кис(4-бромфенил)силан (16b) (схема 4), которые
ной альдегидной группой (схема 5). Их очистке
получали монолитиированием 1,4-дибромбензола
уделяли особое внимание, поскольку наличие не-
и последующим взаимодействием с соответствую-
дозамещенного продукта или исходных субстра-
щим три- или тетрахлорсиланом. Реакция в обоих
тов может повлечь образование на последующих
случаях при точном соблюдении температурных
стадиях побочных продуктов, очистка от которых
режимов по данным аналитической ГПХ проходит
может быть затруднена. Снятие диоксановой за-
практически количественно, что позволило ис-
щиты проводили в водно-ацетоновой смеси в кис-
пользовать полученные соединения 16a-b без до-
лой среде. Контроль осуществляли методом ТСХ
полнительной очистки. Моноитиированием и по-
на силикагеле, а также по появлению характерного
следующим взаимодействием полученных функ-
сигнала альдегидного протона на 1Н ЯМР спектрах
циональных центров с ДМФА были получены со-
при 10 м. д. Поскольку данная реакция является
ответствующие бензальдегиды 17a, 17b. Очистку
равновесной, ни в одном случае не удалось до-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
46
СКОРОТЕЦКИЙ и др.
Схема 4. Схема синтеза разветвляющих кремнийорганических центров для монодендронов с донорными PPO
фрагментами.
Схема 5. Синтез функциональных монодендронов с донорными PPO фрагментами.
19,
20a,
20b,
биться полного снятия защитной группы. Однако
22a-22c получали 5-замещенные производные ок-
не вступающая в реакцию на следующей стадии
сазола 23a-23c. После очистки методом колоноч-
диоксановая группа позволяет выделить непроре-
ной хроматографии на силикагеле в смеси толуол-
агировавшие соединения 21a-21c в чистом виде.
этилацетат, были получены целевые функциональ-
Затем по реакции Ван-Лёссена из бензальдегидов
ные монодендроны с выходом 75-97%.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
47
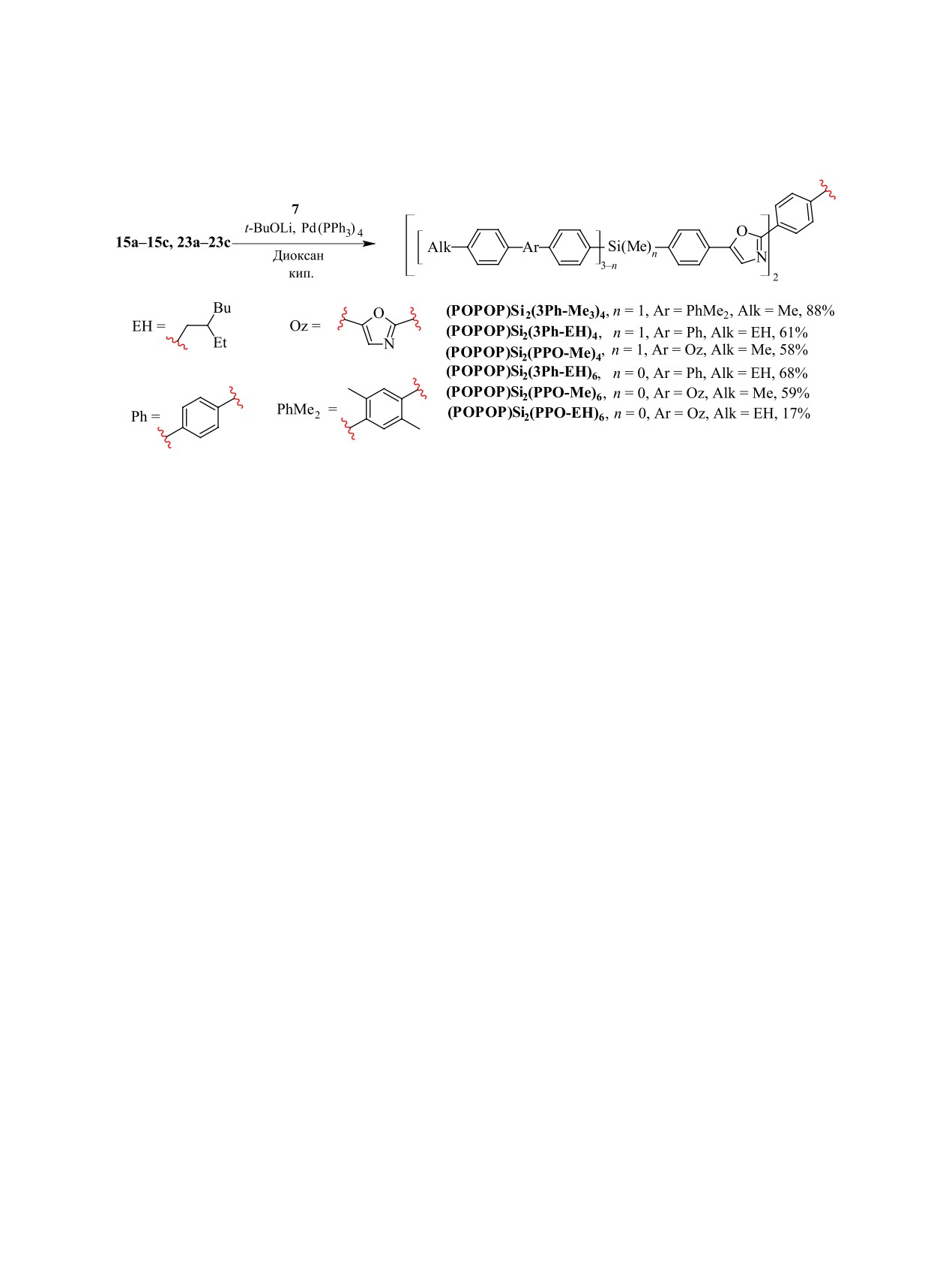
Схема 6. Получение КНЛ на основе фенилоксазолов с различными донорными фрагментами.
Таким образом, были успешно получены не-
Все полученные КНЛ представляют собой ин-
симметричные функциональные монодендроны
дивидуальные соединения разветвленной струк-
15а-15с и 23а-12с с оксазольной группой в фо-
туры, состоящие из двух типов хромофорных
кальной точке, которые на последней стадии вво-
фрагментов, соединенных через атомы кремния.
дили в реакцию кросс-сочетания с 1,4-дибром-
В центральной части в качестве акцептора нахо-
бензолом в условиях прямого С-Н арилирования
дится фрагмент люминофора POPOP. В качестве
(cхема 6).
донорных фрагментов используются различные
После
тщательной
очистки
путем
производные 3Ph, 3Ph-Me2 и PPO с метильными
и 2-этилгексильными солюбилизирующими груп-
перекристаллизации из диоксана для соединений
с донорными 3Ph фрагментами и классической
пами. Следует отметить, что в результате были
получены КНЛ с одинаковыми хромофорными
хроматографии на силикагеле для соединений с
донорными 3Ph-Me2 и PPO фрагментами удалось
фрагментами, но различным соотношением до-
нор-акцептор, составившим 4 : 1 - для соедине-
получить искомые разветвленные КНЛ с выходами,
варьирующемся в пределах 58-88% для соединений
ний (POPOP)Si2(3Ph-EH)4 и (POPOP)Si2(PPO-
Me)4, - или 6 : 1 - для соединений и (POPOP)
(POPOP)Si2(3Ph-Me3)4,
(POPOP)Si2(3Ph-EH)4,
Si2(3Ph-EH)6 и (POPOP)Si2(PPO-Me)6 (схема 7).
(POPOP)Si2(PPO-Me)4, (POPOP)Si2(PPO-EH)6,
(POPOP)Si2(PPO-Me)6 и 17% для (POPOP)Si2(3Ph-
EH)6 (cхема 7). Низкий выход для последнего
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
соединения объясняется сложной очисткой,
2.1 Реагенты, растворители и материалы
которую не удалось оптимизировать, в то время как
реакционный выход составил не менее 86%. Особое
2.5 М и 1.6 M растворы н-бутиллития (n-BuLi)
поведение продемонстрировал (POPOP)Si2(PPO-
в гексане,
1.6 М раствор трет-бутиллития
Me)6. В отличие от других соединений, хорошо
(t-BuLi) в пентане, тионилхлорид, (п-бромфенил)
растворимых в таких органических растворителях
триметилсилан, толуолсульфонилметил изоциа-
как толуол, ТГФ, хлороформ, он обладает заметной
нид (TosMIC), безводный карбонат калия,
растворимостью (>5 % масс.) только в хлороформе.
п-метилбензальдегид, 1,2-дибромэтан, 4,4'-дибром-
В то время как его растворимость в ТГФ составила
бифенил, 1-бром-2-этил-гексан, п-дибромбен-зол
лишь 1.9 г/л и еще меньше в толуоле. Очистку
(Acros organics), а так же тетракис(трифенил-
в данном случае удалось осуществить методом
фосфин)палладий(0) (Pd(PPh3)4), магний,
экстракции сначала из толуола, а потом хлороформа.
1,4-дибром-2,5-диметилбензол, [1,1'-бис(дифенил-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
48
СКОРОТЕЦКИЙ и др.
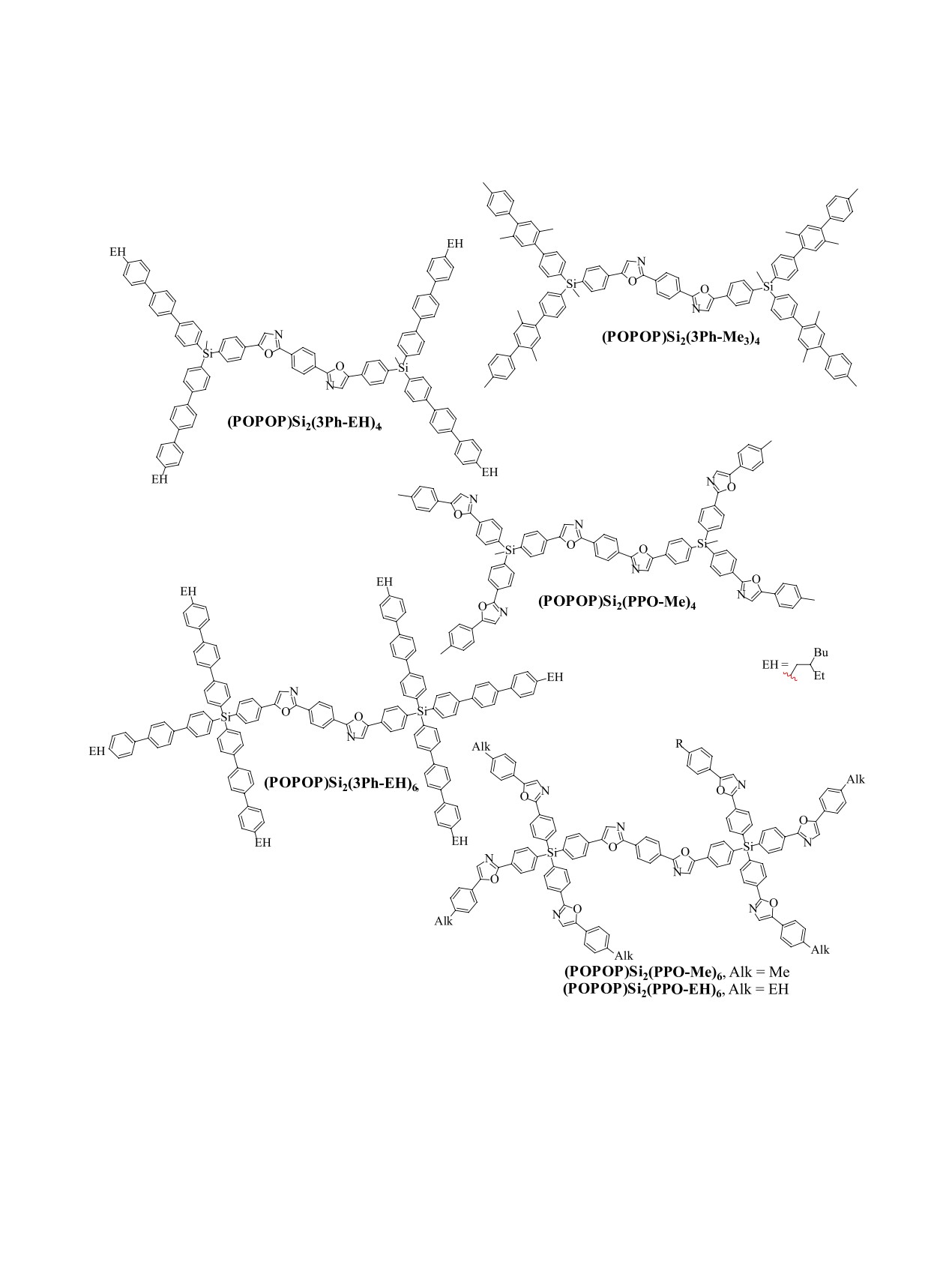
Схема 7. Структурные формулы полученных КНЛ.
фосфино)ферроцен] дихлорпалладий(II) (Pd(dppf)
трет-Бутилат лития получали взаимодействием
Cl2), неопентилгликоль (Sigma-Aldrich), были
2.5 M раствора n-BuLi с трет-бутанолом в абс.
использованы без дополнительной очистки.
ТГФ с последующим упариванием летучих
Этоксисиланы (ООО “Пента”) отчищали дистил-
компонентов. ТГФ, диоксан и другие растворители
ляцией непосредственно перед использованием.
абсолютизировали согласно хорошо известным
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
49
методикам и хранили над гранулами цеолита с
ра CE1106 (Италия) с погрешностью 0.3-0.5%.
размером пор 3 Å. Синтез соединений 5, 6 и 13с
Сжигание осуществляли в склянке Шенингера с
описан ранее [14].
использованием щелочного раствора пероксида
водорода в качестве сорбента. Для определения
Для препаративной колоночной хроматографии
кремния применяли спектрофотометрический
использовали силикагель зернистостью 40-60 мкм
анализ.
с размерами пор 60 Å (“Merck”). Для тонкослой-
ной хроматографии применяли пластинки Sorbfil
2.3 Методики получения соединений
(“Сорбполимер”) с нанесённым слоем люминофо-
ра.
1-Бром-4-(2-этилгексил)бензол (2). К суспен-
Все реакции, если особо не оговорено, прово-
зии 9,8 г (0,403 моль) магния в 50 мл эфира при-
дили в атмосфере аргона и в абсолютизированных
капали раствор 60 мл (0,336 моль) 1-бром-2-этил-
растворителях.
гексана (1) в 500 мл ТГФ. Затем смесь перемеши-
2.2 Физико-химические методы исследования
вали при кипении в течение 1.5 ч. Во второй колбе
растворили 65 г (0.276 моль) 1,4-дибромбензола и
Спектры ЯМР 1Н регистрировали на спектро-
0.58 г (2 ммоль) катализатора Pd(dppf)Cl2 в 100 мл
метре “Bruker AC-250” (250 МГц). Спектры ЯМР
эфира и охладили реакционный сосуд до темпера-
13C и 29Si регистрировали на спектрометре “Bruker
туры 0°С, после чего прикапали свежеполученный
AVII-300” (75,5 МГц и 59,6 МГц соответствен-
реактив Гриньяра из первой колбы, не поднимая
но). В качестве стандарта использовали остаточ-
температуру выше 5°С. Затем охлаждение убра-
ный сигнал растворителя. Спектры обрабатыва-
ли и перемешивали реакционную смесь в течение
ли на компьютере с использованием программы
20 ч. После окончания реакции смесь вылили в
“ACDLabs”.
400 мл ледяной воды и 200 мл диэтилового эфира.
Масс-спектры (MALDI) регистрировали на
Полученную смесь промыли дистиллированной
приборе Autoflex II Bruker (FWHM 18000), снаб-
водой (3 раза по 300 мл), осушили над сульфа-
женным азотным лазером (рабочая длинна волны
том натрия и растворитель упарили на роторном
337 нм) и времяпролетным детектором в режиме
испарителе при пониженном давлении. Продукт
отражения. Конечный спектр является суммар-
очищали вакуумной перегонкой (128-130°C/6.3 ×
ным 300 спектров, полученных в различных об-
10-4 Бар). Выход составил 63.6 г (86% от теории)
ластях образца. 2,5-дигидроксибензойная кислота
продукта 98%-ной чистоты (по данным ГЖХ).
(Acros, 99%) и α-циано-4-гидроксикоричная кис-
Спектр ЯМР 1H (J, Гц, CDCl3) δ, м. д.: 0.87 м (6Н),
лота (Acros, 99%) были использованы в качестве
1.26 м (8Н), 1.54 м (1Н), 2.49 д (J = 7.0, 2Н), 7.02 м
матрицы.
(J = 8.5, 2Н), 7.38 м (J = 8.2, 2Н). Спектр ЯМР 13С
ГПХ-анализ осуществляли на приборе
(CDCl3) δ, м. д.: 10.73; 14.12; 22.99; 26.28; 28.77;
“Shimadzu” (Япония), детекторы - рефрактометр
32.19; 39.46; 40.99; 119.19; 130.91; 131.08; 140.81.
RID-10A и диодная матрица SPD-M10AVP, колон-
MALDI-MS: найдено m/z 268.19 [M]+; вычислено
ка - “Phenomenex” (США) 7.8 × 300 мм, заполнен-
268.08.
ная сорбентом “Phenogel” с размерами пор 500 Å
4-Бромо-4''-(2-этилгексил)-1,1':4',1''-терфе-
(температура термостатривания 40°С), элюент -
нил (3). Синтез проводили по методике, аналогич-
ТГФ со скоростью потока 1 мл мин-1 .
ной получению соединения 2 из 1.68 г (70 ммоль)
Газовую хроматографию осуществляли на
магния, 20 г (66.8 ммоль) 1-бром-4-(2-этилгексил)
хроматографе марки “Хроматэк - Кристалл 5000.2”
бензола (2), 41.7 г (133.7 ммоль) 4,4'-дибромбифе-
(Россия). Детектор по теплопроводности, длина
нила и 0.2 г (0.3 ммоль) Pd(dppf)Cl2. Продукт очи-
колонок - 2 м, диаметр - 3 мм. Газ-носитель -
щали многократной экстракцией из спирта и/или
гелий, скорость 30 мл мин-1, неподвижная фаза
ацетона. Выход составил 23.1 г (70% от теории) с
SE-30 (5%), нанесенная на “Chromaton-N-AW”
содержанием основного продукта не менее 85%
Элементный анализ С, Н, N, проводили с ис-
(по данным ГПХ). Полученный продукт использо-
пользованием CHN автоматического анализато-
вали без дополнительной очистки в последующем
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
50
СКОРОТЕЦКИЙ и др.
синтезе. Чистое соединение 3 для анализа было
в гексане,
3.36 г
(0,14 моль) магния,
11,76 мл
выделено методом вакуумной сублимации. Спектр
(0.136 моль) 1,2-дибромэтана и 125 мл (0.76 моль)
ЯМР 1H (J, Гц, CDCl3) δ, м. д.: 0.91 м (6Н), 1.24-
тетраэтоксисилана. После упаривания раствори-
1.44 м (8Н), 1.62 м (J = 5.5, 1Н), 2.60 д (J = 6.7,
теля и других летучих компонентов полученную
2Н), 7.26 м (J = 8.6, 2Н), 7.49-7.71 м (10Н). Спектр
реакционную смесь очищали методом вакуумной
ЯМР 13С (CDCl3) δ, м. д.: 10.81; 14.16; 23.07; 25.45;
дистилляции. Выход чистого для анализа соеди-
28.88; 32.38; 39.77; 41.09; 76.70; 77.01; 77.33;
нения 9b составил 18.67 г (46% от теории) (92-
121.51; 126.70; 127.20; 127.00; 128.60; 129.70.
97°C/3,7 × 10-4 Бар; лит. данные 104 ± 1°С/0.64-
131.90; 137.70; 138.50; 139.70; 140.50; 141.40.
0.65 мБар [20]). Спектр ЯМР 1Н (J, Гц, CDCl3) δ,
Найдено, %: С 74.30; Н 7.08; Br 18.74. Вычислено
м. д.: 1.24 т (J = 7.33, 9H), 3.36 м (J = 7.33, 6H),
для C26H29Br, %: С 74.10; Н 6.94; Br 18.96.
7.54 с (4H).
(4-Бромфенил)(метил)диэтоксисилан
(9a).
(4-Бромфенил)(метил)дихлорсилан (10a). К
К раствору
35 г
(0.148 моль) п-дибромбензола
10 г (34.6 ммоль) соединения 9a и 100 мкл ДМФА в
в 650 мл абс. ТГФ при -78°С прикапали 60 мл
качестве катализатора при 60°С прикапали 13.6 мл
(0.148 моль) 2.5 M раствора n-BuLi в гексане, не
(0.188 моль) тионилхлорида. Протекание реакции
подымая температуру выше -70 °С. Реакционную
контролировали по исчезновению характерных
смесь перемешивали при этой температуре в те-
пиков этокси-групп на 1Н ЯМР спектре. Чистый
чение 1 ч. Во второй колбе к суспензии 3.92 г
продукт был получен вакуумной перегонкой (55-
(0.1632 моль) магния в 20 мл эфира прикапали
57°C/3 ×
10-4 Бар; лит. данные
57°С/0.12 мБар
раствор 13.7 мл (0.159 моль) дибромэтана в 90 мл
[21]). Выход соединения 10a с чистотой 98% (со-
гласно ГЖХ-анализу) составил 8.87 г (95% от тео-
эфира. После этого смесь перемешивали с на-
рии). Спектр ЯМР 1H (J, Гц, CDCl3) δ, м. д.: 1.02 с
гревом до кипения в течение 30 мин и охладили
(3Н), 7.59 м (4Н).
до комнатной температуры. Прилили весь зара-
нее приготовленный эфирный раствор комплекса
(4-Бромфенил)трихлорсилан
(10b). Синтез
бромида магния с эфиром к содержимому первой
проводили по методике, аналогичной получению
колбы. После этого реакционную смесь переме-
соединения 10а из 10 г (31.3 ммоль) соединения
шивали в течение 30 минут и дали самопроизволь-
9b, 100 мкл ДМФА и 13.6 мл (0,188 моль) тио-
но нагреться до комнатной температуры (20°С).
нилхлорида. Чистый продукт был получен вакуум-
Полученный реактив Гриньяра 8 прикапали к
ной перегонкой (62-64°C/4.2 × 10-4 Бар). Выход
раствору 158 г (0.89 моль) метилтриэтоксисилана
соединения 10b с чистотой 97% (согласно ГЖХ-
в 100 мл ТГФ при температуре не выше 5°С, и
анализу) составил 8.8 г (91% от теории). Спектр
затем перемешивали при комнатной температуре.
ЯМР 1Н (CDCl3) δ, м. д.: 7.67 с (4H).
По окончании реакции к реакционной смеси до-
(4-Бромфенил)(бис[4''-(2-этилгексил)-
бавили 800 мл дистиллированной воды и экстра-
1,1':4',1''-терфенил-4-ил])метилсилан
(13a).
гировали дважды диэтиловым эфиром (800 мл).
К раствору 12,79 г (25.8 ммоль) соединения 3 в
Объединенные органические вытяжки промыли
500 мл ТГФ прикапали 30.7 мл (51.6 ммоль) 1.7 M
водой до нейтральной среды, осушили над Na2SO4
раствора t-BuLi в пентане, поддерживая темпе-
и упарили летучие компоненты на роторном ис-
ратуру не выше -25°С. После этого реакционной
парителе. После вакуумной дистилляции выход
смеси дали самопроизвольно нагреться до комнат-
продукта 9a чистотой 97% составил 24.12 г (58%
ной температуры, затем снова охладили до -78°С
от теории) (71-78°C/3-4 × 10-4 Бар). Спектр ЯМР
и одной порцией добавили 3.137 г (11.6 ммоль)
1H (J, Гц, CDCl3) δ, м. д.: 0.34 с (3Н), 1.23 т (6Н),
хлорсилана 10a. Контроль реакции осуществля-
3.79 м (J = 7.33, 4Н), 7.51 с (4Н).
ли методом ГПХ и тонкослойной хроматографии.
(4-Бромфенил)триэтоксисилан (9b). Синтез
Через 40 мин убрали охлаждающую баню и смеси
проводили по методике, аналогичной получению
самопроизвольно позволяли нагреться до комнат-
соединения 9а из 30 г (0.127 моль) п-дибромбен-
ной температуры. Полученный раствор вылили
зола, 51 мл (0.127 моль) 2.5 M раствора n-BuLi
в 500 мл ледяной воды и экстрагировали дважды
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
51
диэтиловым эфиром (400 мл). Объединенный ор-
77.04; 77.35; 126.47; 126.66; 126.69; 127.37; 127.46;
ганический слой промыли водой до нейтральной
128.77; 129.7; 133.7; 135.8; 135.9; 137.00; 137.80;
реакции и сушили над Na2SO4. После упаривания
139.00; 140.50; 141.30; 142.10; 145.00; 193.00.
растворителя и вакуумирования при 1 мБар по-
Найдено, %: С 86.45; Н 8.15; Si 3.24; вычислено
лучили 11.49 г. После очистки методом класси-
для C60H66OSi, %: С 86.69; Н 8.00; O 1.92; Si 3.38.
ческой колоночной хроматографии на силикагеле
5-(4-{Метил[бис(4''-(2-этилгексил)-1,1':4',1''-
( элюент - смесь гексан-толуол 5 : 1) выделили
терфенил-4-ил)]силил}фенил)-1,3-оксазол
8.0 г (80% от теории) чистого для анализа моно-
(15a). К раствору 2.58 г (3.1 ммоль) соединения
дендрона 13a. Спектр ЯМР 1H (J, Гц, CDCl3) δ,
14a и 0.636 г (3.2 ммоль) TosMIC в смеси 40 мл ме-
м. д.: 0.90-0.95 м (15Н), 1.28-1.40 м (16Н), 1.64 м
танола и 15 мл ТГФ добавили 0.858 г (6.2 ммоль)
(J = 5.9, 2Н), 2.61 м (J = 6.6, 4Н), 7.26 м (J = 8.1,
карбоната калия. Смесь перемешивали при кипя-
4Н), 7.47 м (J = 8.1, 2Н), 7.55-7.62 м (6Н), 7.65 м
чении в течение 2 ч, контролируя прохождение
(J = 8.1, 4Н), 7.89 м (J = 8.1, 4Н), 7.71 с (8Н). 13С
реакции по ТСХ. Полученный раствор вылили
ЯМР (CDCl3), δ, м. д.: -3.29; 10.84; 14.18; 21.48;
в 100 мл ледяной воды и экстрагировали дваж-
23.08; 25.48; 28.90; 32.41; 39.80; 41.12; 76.71;
ды диэтиловым эфиром (200 мл). Объединенные
77.03; 77.35; 124.55; 125.32; 126.57; 126.69; 127.35;
органические вытяжки промыли водой до ней-
127.46; 128.24; 129.10; 129.70; 131.20; 134.20;
тральной реакции и осушили над Na2SO4. После
135.00; 135.80; 137.00; 137.80; 137.90; 139.40;
упаривания растворителя и очисткой методом
140.40; 141.30; 141.90. Найдено, %: С 80.07; Н
колоночной хроматографии на силикагеле (элю-
7.65; Br 9.00; Si 2.98; вычислено для C59H65BrSi,
ент - толуол) выделили 2.38 г чистого для ана-
%: С 80.33; Н 7.43; Br 9.06; Si 3.18.
лиза соединения 15a (88% от теории). Спектр
4-{Метил[бис(4''-(2-этилгексил)-1,1':4',1''-
ЯМР 1H (J, Гц, CDCl3) δ, м. д.: 0.92-0.97 м (12Н),
терфенил-4-ил)]силил}бензальдегид
(14a). К
0.98 с (3Н), 1.29-1.42 м (16Н), 1.66 м (J = 5.9, 2Н),
раствору 1.05 г (1.2 ммоль) монодендрона 13a в
2.63 д (J = 6.6, 4Н), 7.28 м (J = 8.1, 4Н), 7.45 с
20 мл ТГФ при -78°С прикапали 0.9 мл (1.4 ммоль)
(1Н), 7.59 м (J = 8.1, 4Н), 7.67-7.75 м (20Н), 7.97 с
2.5 M раствора n-BuLi в гексане, поддерживая
(1Н). 13С ЯМР (CDCl3), δ, м. д.: -3.29; 10.83; 14.18;
температуру не выше -70°С. Затем реакционную
23.08; 25.48; 28.9; 32.40; 39.80; 41.11; 76.72; 77.04;
смесь перемешивали в течение часа при этой тем-
77.35; 122.09; 123.8; 126.57; 126.68; 127.35; 127.46;
пературе и добавили 0.18 мл (2,3 ммоль) ДМФА.
128.24; 128.64; 129.05; 129.70; 134.37; 135.80;
Убрали охлаждающую баню и дали смеси само-
135.90; 137.20; 137.80; 139.00; 140.00; 141.00;
произвольно нагреться до комнатной температуры.
141.90; 150.60; 150.60; 151.50; 151.50. Найдено,
Получившуюся соль подкислили 1 М раствором
%: С 85.69; Н 7.68; N 1.61; Si 3.31; вычислено для
НСl до кислой среды, вылили в 100 мл ледяной
C62H67NOSi, %: С 85.57; Н 7.76; N 1.61; Si 3.23.
воды и экстрагировали дважды диэтиловым эфи-
2,2'-Бензол-1,4-диилбис[5-(4-{бис[4''-(2-
ром (150 мл). Объединенные органические слои
этилгексил)-1,1':4',1''-терфенил-4-ил](метил)
промыли водой до нейтральной реакции и высу-
силил}фенил)-1,3-оксазол]
((POPOP)Si2(3Ph-
шили над Na2SO4. После упаривания растворителя
EH)4). Дегазированный раствор 1.5 г (1.63 ммоль)
и очистки методом колоночной хроматографии на
соединения 15a, 0.327 г (4.1 ммоль) трет-бути-
силикагеле (элюент - толуол) выделили 0.9 г (91%
лата лития, 47 мг (0.04 ммоль) Pd(PPh3)4 и 0.193 г
от теории) чистого для анализа соединения 14a.
(0.8 ммоль) 1,4-дибромбензола в 20 мл абс. диок-
Спектр ЯМР 1H (J, Гц, CDCl3) δ, м. д.: 0.92-0.97 м
сана кипятили 1.5 ч до полного исчезновения про-
(12Н), 0.97 (c, 3Н), 1.30-1.43 м (16Н), 1.66 м (J =
дукта моноприсоединения - контроль за прохожде-
5.9, 2Н), 2.63 м (J = 6.6, 4Н), 7.28 м (J = 8.1, 4Н),
нием реакции осуществляли методом ТСХ и ГПХ.
7.60 д (J = 8.1, 4Н), 7.67 м (J = 8.1, 4Н), 7.69-7.75 м
По завершении реакции смесь вылили в 200 мл
(12Н), 7.82 м (J = 8.1, 2Н), 7.92 м (J = 8.1, 2Н),
дистиллированной воды и экстрагировали дважды
10.1 с (1Н). 13С ЯМР (CDCl3) δ, м. д.: -3.40; 10.84;
диэтиловым эфиром. Объединенные органические
14.18; 23.08; 25.48; 28.90; 32.41; 39.80; 41.12; 76.72;
слои промали водой до нейтральной реакции и
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
52
СКОРОТЕЦКИЙ и др.
сушили над Na2SO4. После упаривания раствори-
данным 1Н ЯМР-спектроскопии реакция прошла
теля, реакционную смесь пропустили через слой
на 83%. Использовали полученное соединение
силикагеля (элюент - смесь толуол-этилацетат 20 :
14b в последующем синтезе без дополнительной
1) для удаления следов катализатора. Очистку про-
очистки. Спектр ЯМР 1H (J, Гц, CDCl3) δ, м. д.:
водили методом перекристаллизации из диоксана,
0.87-0.96 м (18Н), 1.24-1.40 м (24Н), 1.63 м (J =
что позволило выделить 0.91 г чистого для анали-
5.9, 3Н), 2.60 д (J = 6.7, 6Н), 7.26 м (J = 7.9, 6Н),
за соединения (POPOP)Si2(3Ph-EH)4 (61% от тео-
7.58 м (J = 8.2, 6Н), 7.67-7.79 м (25Н), 7.93 м (3Н),
рии). Спектр ЯМР 1H (J, Гц, CDCl3) δ, м. д.: 0.90-
10.11 с (1Н).
0.96 м (24Н), 0.99 с (6Н), 1.29-1.42 м (32Н), 1.66 м
5-(4-{Трис[4''-(2-этилгексил)-1,1':4',1''-
(J = 5.9, 4Н), 2.62 м (J = 6.6, 8Н), 7.28 м (J = 8.8,
терфенил-4-ил]силил}фенил)-1,3-оксазол (15b).
8Н), 7.56-7.61 м (10Н), 7.69-7.75 м (36Н), 7.81 м
Синтез проводили аналогично методике полу-
(J = 8.1, 4Н), 8.27 с (4Н). 13С ЯМР (CDCl3) δ, м. д.:
чения соединения 15a из 5.27 г (4.6 ммоль) бен-
-3.27; 10.83; 14.17; 23.07; 25.47; 28.89; 32.40; 39.79;
зальдегида 14b, 0.93 г (4.8 ммоль) TosMIC и 1.25 г
41.11; 76.71; 77.03; 77.35; 123.66; 124.46; 126.58;
(9.1 ммоль) карбоната калия. Реакционную смесь
126.68; 126.72; 127.35; 127.46; 128.68; 128.79;
перемешивали при кипячении в течение 2 ч, кон-
129.70; 134.40; 135.80; 136.00; 137.20; 138.00;
тролируя прохождение реакции по ТСХ. После
139.40; 140.40; 141.30; 141.90; 151.70; 160.70.
очистки методом колоночной хроматографии на
Найдено, %: С, 85.81; H 7.63; N 1.45; Si 2.93; вы-
силикагеле в толуоле выделили 3.71 г чистого для
числено для C130H136N2O2Si2, %: С 86.04; H 7.55;
анализа соединения 15b (68% от теории). Спектр
N 1.54; Si 3.10. MALDI-TOF m/z найдено 1814.69
ЯМР 1H (J, Гц, CDCl3) δ, м. д.: 0.87-0.95 м (18Н),
[M]+, рассчитано - 1813.01.
1.26-1.40 (м, 24Н), 1.63 м (J = 5.9, 3Н), 2.59 д (J =
(4-Бромфенил){трис[4''-(2-этилгек-
6.7, 6Н), 7.26 м (J = 7.94, 6Н), 7.44 с (1Н), 7.57 м (J =
сил)-1,1':4',1''-терфенил-4-ил]}силан
(13b).
8.2, 6Н), 7.69-7.78 м (28Н), 7.97 с (1Н). 13С ЯМР
Синтез проводили аналогично методике получе-
(CDCl3) δ, м. д.: 10.78; 14.15; 23.04; 25.4; 28.84;
ния соединения 13a из 24 мл (38.4 ммоль) 1.6 M
32.33; 39.74; 41.06; 122.14; 123.75; 126.57; 126.65;
раствора т-BuLi в пентане, 9.32 г (18.83 ммоль)
127.33; 127.44; 128.77; 129.67; 132.47; 135.28;
соединения 3 и 1.65 г (5.7 ммоль) соединения 10b.
136.89; 136.98; 137.73; 139.27; 140.39; 141.26;
После очистки методом колоночной хроматогра-
142.04. 29Si ЯМР (CDCl3) δ, м. д.: -14.39. Найдено,
фии на силикагеле (элюент - смесь гексан-толуол
%: С 87.44; H 7.87; N 1.14; Si 2.53; вычислено для
5 : 1) выделили 5.73 г (83% от теории) чистого для
C87H93NOSi, %: С 87.31; Н 7.83; N 1.17; Si 2.35.
анализа монодендрона 13b. Спектр ЯМР 1H (J, Гц,
2,2'-Бензол-1,4-диил-бис[5-(4-{трис[4''-(2-
CDCl3) δ, м. д.: 0.87-0.95 м (18Н), 1.25-1.38 м
этилгексил)-1,1':4',1''-терфенил-4-ил]силил}
(24Н), 1.63 м (J = 5.9, 3Н), 2.59 д (J = 6.7, 6Н),
фенил)-1,3-оксазол]
((POPOP)Si2(3Ph-EH)6).
7.25 д (J = 7.6, 6Н), 7.55-7.61 м (10Н), 7.72 м (24Н).
Синтез проводили аналогично методике получе-
13С ЯМР (CDCl3), δ, м. д.: 0.75; 14.11; 23.01; 25.38;
ния соединения (POPOP)Si2(3Ph-EH)4 из 3.9 г
28.82; 32.31; 39.71; 41.03; 124.84; 126.54; 126.62;
(3.19 ммоль) соединения 15b, 0.64 г (7.98 ммоль)
127.3;
127.41;
129.64;
131.18;
132.27;
133.23;
трет-бутилата лития, 62 мг (0.05 ммоль) Pd(PPh3)4
136.81;
137.7;
137.93;
139.22;
140.37;
141.23;
и
0.376 г
(1.59 ммоль)
1,4-дибромбензола.
142.04. 29Si ЯМР (CDCl3) δ, м. д.: -14.16. Найдено,
Реакционную смесь кипятили 2 ч. После упарива-
%: С 83.63; Н 7.71; Br 6.41; Si 2.22; вычислено для
ния растворителя, реакционную смесь пропусти-
C84H91BrSi, %: С 83.48; Н 7.59; Br 6.61; Si 2.32.
ли через слой силикагеля (элюент - смесь толуол-
4-{Трис[4''-(2-этилгексил)-1,1':4',1''-терфе-
этилацетат 20 : 1) от следов катализатора. Очистку
нил-4-ил]силил} бензальдегид
(14b). Синтез
проводили методом перекристаллизации из диок-
проводили аналогично методике получения со-
сана, что позволило выделить 2.41 г чистого для
единения 14a из 5.7 г (4.8 ммоль) монодендрона
анализа соединения (POPOP)Si2(3Ph-EH)6 (68%
13b, 3.45 мл (5,52 ммоль) 1,6 M раствора n-BuLi
от теории). Спектр ЯМР 1H (J, Гц, CDCl3) δ, м. д.:
в гексане и 0.85 мл (10 ммоль) ДМФА. Согласно
0.86-0.95 м (36Н), 1.25-1.40 м (48Н), 1.63 м (J =
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
53
5.9, 6Н), 2.59 д (J = 6.4, 12Н), 7.26 м (J = 7.9, 12Н),
растворителя и очистки методом колоночной хро-
7.55-7.61 м (14Н), 7.69-7.84 м (56Н), 8.25 с (4Н).
матографии на силикагеле (элюент - смесь толу-
13С ЯМР (CDCl3) δ, м. д.: 10.83; 14.05; 23.04; 25.66;
ол-этилацетат 20 : 1) выделили 2.7 г чистого для
28.98; 32.57; 39.93; 41.18; 123.78; 126.63; 126.70;
анализа соединения (POPOP)Si2(3Ph-Me3)4 (71%
126.80; 127.37; 127.48; 128.95; 129.69; 132.69;
от теории). Спектр ЯМР 1H (J MГц, CDCl3), δ, м. д.:
135.5;
136.96;
137.11;
137.90;
139.42;
140.54;
0.98 с (6Н), 2.31 с (12Н), 2.33 с (12Н), 2.43 с (12Н),
141.30; 142.19; 151.82; 160.82. 29Si ЯМР (CDCl3)
7.19 м (J = 5.8, 8H), 7.23-7.31 м (16H), 7.43 д (J =
δ, м. д.: -14.47. Найдено, %: С 87.93; H 7.92;
7.9, 8H), 7.56 с (2H), 7.65 м (J = 7.9, 8H), 7.74 м (J =
N 1.16; Si 2.14; вычислено для C180H188N2O2Si2,
7.9, 4H), 7.80 м (J = 8.2, 4H), 8.25 с (4H). 13С ЯМР
%: С 87.61; H 7.68; N 1.14; Si 2.28.
(CDCl3), δ, м. д.: -3.23; 19.97; 20.00; 21.18; 123.61;
124.38; 126.69; 128.58; 128.76; 128.80; 128.85;
4-{Метил[бис(2',4'',5'-триметил-1,1':4',1''-
129.09; 131.82; 131.97; 132.54; 132.69; 133.77;
терфенил-4-ил)]силил}бензальдегид (14с). Син-
тез проводили аналогично методике получения
135.04; 135.97; 136.41; 137.39; 138.67; 140.37;
140.91; 142.97; 151.75; 160.61. 29Si ЯМР (CDCl3),
соединения 14a из 4 г (5.4 ммоль) соединения 13c,
δ, м. д.: -10.96. Найдено, %: С 85.84; H 6.21;
3.9 мл (6.2 ммоль) 1.6 М раствор n-BuLi в гексане и
N 1.70; Si 3.42; вычислено для C110H96N2O2Si2, %:
0.84 мл (10 ммоль) ДМФА. По данным ЯМР спек-
С 86.12; H 6.31; N 1.83; Si 3.66.
троскопии реакция прошла на 87%. Соединение
14с использовали без дополнительной очистки
Трис(4-бромфенил)(метил)силан (16a). Син-
в последующем синтезе. Спектр ЯМР 1H (J, Гц,
тез и выделение продукта проводили согласно
ДМСО-d), δ, м. д.: 0.96 с (3Н), 2.23 с (6Н), 2.27 с
методике, описанной в работе [22], из 34.62 г
(6Н), 2.38 с (6Н), 7.1 м (J = 7.9, 4H), 7.22 с (8H),
(0.146 моль) п-дибромбензола, 59 мл (0.146 моль)
7.39 м (J = 7.9, 4H), 7.59 м (J = 8.2, 4H), 7.78 м (J =
2.5 M раствора н-BuLi в гексане и
5.74 мл
7.9, 2H), 7.93 м (J = 7.9, 2H), 10.05 с (1H).
(0.0489 моль) трихлорметилсилана. Выход сое-
5-(4-{Метил[бис(2',4'',5'-триметил-1,1':4',1''-
динения 16a 97%-ной чистоты (по данным ГПХ)
составил 24.1 г (96% от теории). Полученный про-
терфенил-4-ил)]силил}фенил)-1,3-оксазол
дукт использовали без дополнительной очистки
(15c). Синтез проводили аналогично методике
в последующем синтезе. Спектр ЯМР 1H (J, Гц,
получения соединения 15a из 3.7 г (5.3 ммоль) со-
CDCl3), δ, м. д.: 0.81 с (3Н), 7.33 м (J = 7.9, 6H),
единения 14c, 1.09 г (5.6 ммоль) TosMIC и 1.48 г
(10.7 ммоль) карбоната калия. Смесь перемеши-
7.52 м (J = 8.2, 6H).
вали при кипячении в течение 2 ч. После очистки
Тетракис(4-бромфенил)силан (16b). Синтез и
методом колоночной хроматографии на силикаге-
выделение продукта проводили согласно методике,
ле (элюент толуол) выделили 3.39 г чистого для
описанной в работе [23], из 36.18 г (0.1563 моль)
анализа соединения 15c (86% от теории). Спектр
п-дибромбензола, 60 мл (0.15 моль) 2.5 M раствора
ЯМР 1H (J, Гц, ДМСО-d), δ, м. д.: 0.94 с (3Н), 2.23
n-BuLi в гексане и 4,32 мл (0,0376 моль) трихлор-
с (6Н), 2.27 с (6Н), 2.38 с (6Н), 7.09 м (J = 8.8, 4H),
метилсилана. Выход соединения 16b 97%-ной чи-
7.22 с (8H), 7.39 м (J = 7.9, 4H), 7.58-7.67 м (7H),
стоты (по данным ГПХ) составил 22,5 г (90% от
7.75 м (J = 8.2, 2H), 8.36 с (1H).
теории). Полученный продукт использовали без
дополнительной очистки в последующем синтезе.
2,2'-Бензол-1,4-диилбис[5-(4-{метил-[бис-
Спектр ЯМР 1H (J, Гц, CDCl3), δ, м. д.: 7.36 м (J =
(2',4'',5'-триметил-1,1':4',1''-терфенил-4-ил)]
8.5, 8H), 7.54 м (J = 8.5, 8H).
силил}фенил)-1,3-оксазол]
((POPOP)Si2(3Ph-
Me3)4). Синтез проводили аналогично методике
4-[Бис(4-бромфенил)(метил)силил]бензаль-
получения соединения (POPOP)Si2(3Ph-EH)4 из
дегид (17a). Синтез проводили аналогично мето-
3.8 г (5.2 ммоль) соединения 15c, 1.04 г (13 ммоль)
дике получения соединения 14a из 5 г (9.8 ммоль)
трет-бутилата лития,
60 мг
(0.05 ммоль)
соединения 16a, 6.42 мл 1.6 M раствора n-BuLi
Pd(PPh3)4 и 0.585 г (2.47 ммоль) 1,4-дибромбензо-
в гексане и 1,51 мл (19.6 ммоль) ДМФА. После
ла. Реакционную смесь кипятили в течение 4 ч до
упаривания растворителя и очистки методом ко-
полного завершения реакции. После упаривания
лоночной хроматографии на силикагеле (элю-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
54
СКОРОТЕЦКИЙ и др.
ент - толуол) выделили 3.06 г (68% от теории)
дили аналогично методике получения соедине-
чистого для анализа соединения 16a.Спектр ЯМР
ния 18a из 7.33 г (12.2 ммоль) соединения 17b,
1H (J, Гц, CDCl3), δ, м. д.: 0.86 с (3Н), 7.33 м (4H,
11.68 г (48.7 ммоль) неопентилгликоля и 0.46 г
J = 8.2), 7.53 м (4H, J = 8.2), 7.64 м (4H, J = 7.9),
(2.4 ммоль) п-толуолсульфокислоты. Реакционную
7.86 м (4H, J = 7.9), 10.05 с (1 Н). 13С ЯМР (CDCl3),
смесь кипятили в течение 12 ч. Выход составил
δ, м. д.: -3.73; 125.05; 128.82; 131.37; 133.24; 135.61;
8.28 г (98% от теории) соединения 18b 99%-ной
чистоты (согласно ГПХ). Полученный продукт ис-
136.63; 137.04; 143.44; 192.43. 29Si ЯМР (CDCl3), δ,
м. д.: -9.95. Найдено, %: С 51.98; H 3.59; Br 34.42;
пользовали без дополнительной очистки в после-
дующем синтезе. Спектр ЯМР 1H (J, Гц, CDCl3),
Si 6.11; вычислено для C20H16Br2OSi, %: С 52.19;
δ, м. д.: 0.72 с (3 Н), 1.31 с (3 Н), 3.73 д (2H, J =
H 3.50; Br 34.72; Si 6.10.
10.7), 3.79 (c, 2H, J = 11.3), 5.43 с (1Н), 7.35 м (6H,
4-[Трис(4-бромфенил)силил]бензальдегид
J = 8.2), 7.49-7.57 м (10H). 13С ЯМР (CDCl3), δ,
(17b). Синтез проводили аналогично методике по-
м. д.: 21.83; 22.98; 30.23; 77.66; 101.40; 125.17;
лучения соединения 14a из 11,6 г (17,8 ммоль) со-
125.91; 131.28; 131.87; 133.16; 136.23; 137.66;
единения 16b, 11,68 мл (18,7 ммоль) 1,6 М n-BuLi
140.31. 29Si ЯМР (CDCl3), δ, м. д.: -13.80. Найдено,
и 2,07 мл (35,6 ммоль) ДМФА. После очистки ме-
%: С 52.74; H 4.00; Br 34.49; Si 4.25; вычислено
тодом колоночной хроматографии на силикагеле
для C30H27Br3O2Si, %: С 52.42; H 3.96; Br 34.88;
(элюент - толуол) удалось получить 7.8 г чисто-
Si 4.09.
го для анализа соединения 17b (73% от теории).
4-(2-Этилгексил)бензальдегид
(19). Синтез
Спектр ЯМР 1H (J, Гц, CDCl3), δ, м. д.: 7.36 м (6H,
проводили аналогично методике получения сое-
J = 8.2), 7.56 м (6H, J =8.5), 7.68 м (2H, J = 8.2), 7,89
динения 14a из 25.5 г (94.7 ммоль) соединения 2,
м (2H, J = 8.2 Гц), 10.07 с (1Н).
39.8 мл (99.5 ммоль) 2.5 М раствора n-BuLi в гек-
Бис(4-бромфенил)[4-(5,5-диметил-1,3-диок-
сане и 8 мл (0.1 моль) ДМФА. Очистку проводили
сан-2-ил)фенил]метилсилан (18a). Раствор 3.95 г
методом колоночной хроматографии на силика-
(8.58 ммоль) соединения 17a, 3.577 г (34.3 ммоль)
геле (элюент - толуол), что позволило выделить
неопентилгликоля и 0.32 г (1.7 ммоль) п-толуо-
27.31 г (74 % от теории) чистого для анализа сое-
лсульфокислоты в бензоле кипятили с насадкой
динения 19. Спектр ЯМР 1H (J, Гц, CDCl3), δ, м. д.:
Дина-Старка для азеотропной отгонки воды. По
0.80-0.97 м (6H), 1.17-1.36 м (8Н), 1,62 м (J = 6.4,
завершении реакции через 17 ч добавили 1 мл
1Н), 2.61 д (2Н, J = 7.3), 7.32 м (2H, J = 8.2), 7.81 м
триэтиламина, смесь вылили в 200 мл дистилли-
(2H, J = 7.9), 9.98 с (1Н).
рованной воды и экстрагировали дважды диэти-
5-[4-(2-Этилгексил)фенил]-1,3-оксазол
ловым эфиром (150 мл). Объединенные органиче-
(20a). Синтез проводили аналогично методике
ские слои промыли водой до нейтральной реакции
получения соединения 15a из 20.5 г (94.0 ммоль)
и осушили над Na2SO4. После упаривания раство-
соединения 19, 19.27 г (98.7 ммоль) TosMIC и
рителя выделили 4.32 г (93% от теории) соедине-
25.99 г (0.188 моль) безводного карбоната калия.
ния 18a 99%-ной чистоты (согласно ГПХ). Спектр
Реакционную смесь перемешивали при кипячении
ЯМР 1H (J, Гц, CDCl3), δ, м. д.: 0.79 с (3Н), 0.81 с
в течение 1.5 ч. После чистки методом колоночной
(3Н), 1.30 с (3Н), 3.66 д (2H, J = 10.4), 3.79 д (2H, J =
хроматографии на силикагеле (элюент - толуол)
11.3), 5.41 с (1Н), 7.31 м (4H, J = 8.5), 7.46-7.55 м
выделили 19.03 г чистого для анализа соедине-
(8H). 13С ЯМР (CDCl3), δ, м. д.: -3.55; 21.85; 22.99;
ния 20a (79% от теории). Спектр ЯМР 1H (J, Гц,
30.23; 77.64; 101.45; 124.62; 125.74; 131.10; 134.26;
CDCl3), δ, м. д.: 0.89 т (6H, J = 7.32), 1.23-1.36 м
135.19; 135.54; 136.71; 139.93. 29Si ЯМР (CDCl3), δ,
(8Н), 1.58 (м 1Н, J = 5.4), 2.56 д (2Н, J = 7.0), 7.22 м
м. д.: -10.25. Найдено, %: С 54.85; H 4.69; Br 29.06;
(2H, J = 8.2), 7.32 с (1Н), 7.57 м (2H, J = 8.2), 7.91 с
Si 5.25; вычислено для C25H26Br2O2Si, %: С 54.96;
(1Н). 13С ЯМР (CDCl3), δ, м. д.: 10.74. 14.10. 22.98.
H 4.80; Br 29.25; Si 5.14.
25.36. 28.77. 32.25. 39.89. 41.02. 120.80. 124.17.
Трис(4-бромфенил)[4-(5,5-диметил-1,3-ди-
125.13. 129.69. 142.75. 150.10. 151.75. Найдено
оксан-2-ил)фенил]силан
(18b). Синтез прово-
(%): С 79.51; H 9.15; N 5.29; вычислено (%) для
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
55
C17H23NO: С 79.33; H 9.01; N 5.44; O, 6.22.
18b, 6.24 г (39,2 ммоль) соединения 20b, 6.27 г
(78,5 ммоль) трет-бутилата лития и
453 мг
5-(4-Метилфенил)-1,3-оксазол (20b). Синтез
(0.39 ммоль) Pd(PPh3)4. После очистки методом
проводили согласно методике, описанной в рабо-
колоночной хроматографии на силикагеле (элюент
те [24], из 5 г (41.6 ммоль) п-метилбензальдегида,
- смесь толуол-этилацетат 20 : 1) выход составил
8.53 г (43.7 ммоль) TosMIC, 11.5 г (83.2 ммоль)
10.08 г чистого для анализа соединения 21b (92%
безводного карбоната калия. После очистки ме-
от теории). Спектр ЯМР 1H (J, Гц, CDCl3), δ,
тодом колоночной хроматографии на силикагеле
м. д.: 0.83 с (3Н), 1.32 с (3Н), 2.41 с (9Н), 3.69 д
(элюент - смесь толуол-этилацетат 20 : 1) выде-
(2H, J = 11.0), 3.81(д, 2H, J = 10.7), 5.47 с (1.Н),
лили 6.01 г (91% от теории) чистого для анализа
7.27 м (6H, J = 8.2), 7.44 с (3Н), 7.59-7.67 м (10H),
соединения 20b. Спектр ЯМР 1H (J, Гц, CDCl3), δ,
7.71 м (6H, J = 8.2), 8.14 м (6H, J = 8.2). 13С ЯМР
м. д.: 2.39 с (3Н), 7.24 м (J = 7.9, 2Н), 7.31 с (1Н),
(CDCl3), δ, м. д.: 21.35; 21.84; 23.01; 30.25; 77.67;
7.55 м (J = 8.2, 2Н), 7.90 с (1Н); 13С (CDCl3), δ,
101.49; 122.97; 124.17; 125.10; 125.52; 125.93;
м. д.: 21.33. 120.81. 124.32. 129.57. 138.66. 150.11.
128.17; 128.72; 128.98; 129.59; 133.51; 135.88;
151.69. Найдено, %: С 75.39; H 5.59; N 8.84; вы-
136.46; 136.71; 138.58; 140.24; 151.68;
160.51.
числено для С10H9NO, %: С 75.45; H 5.70; N 8.80.
29Si ЯМР (CDCl3), δ, м. д.: -14.30. Найдено, %:
5,5'-({[4-(5,5-Диметил-1,3-диоксан-2-ил)
С 77.96; H 5.68; N 4.45 Si 2.98; вычислено для
фенил](метил)силандиил}дибензол-4,1-ди-
C60H51N3O5Si, %: С 78.15; H 5.57; N 4.56; Si 3.05.
ил)бис[2-(4-метилфенил)-1,3-оксазол]
(21a).
2,2',2''-({[4-(5,5-Диметил-1,3-диоксан-2-
Раствор 4.14 г (7.5 ммоль) соединения 18a, 2.65 г
ил)фенил]силантриил}трибензол-4,1-диил)
(16.68 ммоль) соединения 20b, 3.33 г (41.7 ммоль)
трис{5-[4-(2-этилгексил)фенил]-1,3-оксазол}
трет-бутилата лития и
192 мг
(0.16 ммоль)
(21c). Синтез проводили аналогично методике
Pd(PPh3)4 в 100 мл диоксана перемешивали при
получения соединения 21a из 2.5 г (3.6 ммоль)
кипячении в течение двух часов. По завершении
соединения 18b, 3.37 г (13.1 ммоль) соединения
реакции смесь вылили в 200 мл дистиллирован-
20a, 2.62 г (32.7 ммоль) трет-бутилата лития и
ной воды и экстрагировали дважды диэтиловым
150 мг (0.13 ммоль) Pd(PPh3)4. После очистки
эфиром. Объединенные органические вытяжки
методом колоночной хроматографии на силикагеле
промыли водой до нейтральной реакции и осуши-
(элюент - смесь толуол-этилацетат 20 : 1) выход
ли над Na2SO4. После упаривания растворителя и
составил 3.78 г чистого для анализа соединения
очистки методом колоночной хроматографии на
силикагеле (элюент - смесь толуол-этилацетат
21c (85% от теории). Спектр ЯМР 1H ЯМР (J, Гц,
CDCl3), δ, м. д.: 0.82 с (3Н), 0.89 м (18H), 1.24-
20 : 1) получили 4.36 г (82% от теории) чисто-
1.37 м (27Н), 1.61 м (3Н), 2.57 д (6Н, J = 7.0), 3.69 д
го для анализа соединения 21a. Спектр ЯМР 1H
(2H, J = 11.0), 3.81 д (2Н, J = 11.3), 5.46 с (1Н),
(J Гц, CDCl3), δ, м. д.: 0.82 с (3Н), 0.90 с (3Н), 1,31
7.24 м (6H, J = 8.2), 7.44 с (3Н), 7.60-7.74 м (16H),
с (3Н), 2.41 с (6Н), 3.70 д (2H, J = 10.4), 3.79 д (2H,
J = 11.0), 5.43 с (1Н), 7.26 м (4H, J = 8.5), 7.42 с
8.14 м (6H, J = 8.5). 13С ЯМР (CDCl3), δ, м. д.:
10.75; 14.09; 21.85; 22.98; 23.02; 25.40; 28.79; 30.25;
(2Н), 7.56 с (4Н), 7.63 м (8H, J = 7.9), 8.08 м (4H,
32.29; 39.96; 41.05; 77.70; 101.53; 123.00; 124.08;
J = 8.2). 13С ЯМР (CDCl3), δ, м. д.: -3.54; 21.36;
125.28; 125.54; 125.95; 128.75; 129.73; 133.54;
21.85; 23.00; 30.23; 77.64; 101.51; 122.82; 124.16;
135.93; 136.46; 136.73; 140.28; 142.72; 151.79;
125.10; 125.41; 125.75; 128.29; 129.59; 135.34;
135.64; 135.78; 138.33; 138.56; 139.87; 151.58;
160.56. 29Si ЯМР (CDCl3), δ, м. д.: -14.32. Найдено:
С 80.03; H 7.84; N 3.39; Si 2.18; вычислено для
160.63. 29Si ЯМР (CDCl3), δ, м. д.: -10.58. Найдено,
C81H93N3O5Si: С 79.96; H 7.70; N 3.45; Si 2.31.
%: С 76.63; H 6.15; N 4.10; Si 3.87; вычислено для
C45H42N2O4Si, %: С 76.89; H 6.02; N 3.99; Si 4.00.
4-[Метил(бис{4-[2-(4-метилфенил)-1,3-окса-
зол-5-ил]фенил})силил] бензальдегид
(22a).
2,2',2''-({[4-(5,5-Диметил-1,3-диоксан-2-ил)
Раствор 5.23 г (7.44 ммоль) соединения 21a в
фенил]силантриил}трибензол-4,1-диил)трис[5-
70 мл ацетона с 10 мл 1 М раствора HCl кипяти-
(4-метилфенил)-1,3-оксазол]
(21b).
Синтез
ли в течение 27 ч до полного снятия защитной
проводили аналогично методике получения
группы. Контроль осуществляли методом ТСХ.
соединения 21a из 8,17 г (11.9 ммоль) соединения
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
56
СКОРОТЕЦКИЙ и др.
По завершении реакции смесь вылили в 200 мл
Гц, CDCl3), δ, м. д.: 0.85-0.93 м (18H), 1.24-1.33 м
дистиллированной воды и экстрагировали дваж-
(24Н), 1.61 м (3Н), 2.57 д (6Н, J = 7.0), 7.24 м (6H,
ды диэтиловым эфиром. Объединенные органи-
J = 8.5), 7.45 с (3Н), 7.61-7.74 м (12H), 7.82 м (2H,
ческие вытяжки промыли водой до нейтральной
J = 7.9), 7.95 д (2H, J = 8.2), 8.18 м (6H, J = 8.5),
реакции и осушили над Na2SO4. После упарива-
10.11 с (1Н). 13С ЯМР (CDCl3), δ, м. д.: 10.75; 14.08;
ния растворителя и очистки методом колоночной
22.98; 25.39; 28.79; 32.28; 39.96; 41.05; 123.04;
хроматографии на силикагеле (элюент - смесь то-
124.09; 125.20; 125.73; 128.90; 129.09; 129.20;
луол-этилацетат 3 : 1) выделили 4.12 г (90% от те-
129.76; 134.87; 136.62; 136.69; 136.84; 137.30;
ории) чистого для анализа соединения 22a. Спектр
141.43; 142.83; 151.91; 160.36; 192.39. 29Si ЯМР
ЯМР 1H (J, Гц, CDCl3), δ, м. д.: 0.97 с (3Н), 2.41
(CDCl3): δ = -14.28. Найдено, %: С 80.55; H 7.55;
с (6Н), 7.26 м (4H, J = 7.9), 7.43 с (2Н), 7.61-7.66
N 3.64; Si 2.48; вычислено для C76H83N3O4Si, %:
м (8H), 7.73 м (2H, J = 7.9), 7.90 м (2H, J = 7.9),
С 80.74; H 7.40; N 3.72; Si 2.48.
8.13 м (4H, J = 8.2), 10.07 с (1Н). 13С ЯМР (CDCl3),
5,5'-({Метил[4-(1,3-оксазол-5-ил)фенил]
δ, м. д.: -3.75; 21.35; 122.90; 124.16; 125.05; 125.58;
силандиил}дибензол-4,1-диил)бис[2-(4-ме-
128.68; 128.82; 129.60; 135.57; 135.73; 137.01;
тилфенил)-1,3-оксазол] (23a). К раствору 4.5 г
137.13; 138.64; 143.73; 151.69; 160.41;
192.47.
(7.3 ммоль) соединения 22a и 1.5 г (7.6 ммоль)
29Si ЯМР (CDCl3), δ, м. д.: -10.31. Найдено, %:
TosMIC в 150 мл метанола и 10 мл ТГФ добавили
С 78.16; H 5.38; N 4.27; Si 4.31; вычислено для
2.02 г (14.6 ммоль) карбоната калия. Смесь переме-
C40H32N2O3Si, %: С 77.89; H 5.23; N 4.54; Si 4.55.
шивали при кипячении в течение 2 ч, контролируя
4-(Трис{4-[5-(4-метилфенил)-1,3-оксазол-
прохождение реакции по ТСХ. Полученный рас-
2-ил]фенил}силил)бензальдегид
(22b). Синтез
твор вылили в 200 мл ледяной воды и дважды экс-
проводили аналогично методике получения соеди-
трагировали диэтиловым эфиром. Объединенные
нения 22a из 10.05 г (10.13 ммоль) соединения 21b
органические слои промыли водой до нейтральной
и 20.3 мл 1М раствора HCl. После очистки методом
реакции и осушили над Na2SO4. После упарива-
колоночной хроматографии на силикагеле (элюент
ния растворителя и очисткой методом колоночной
- смесь толуол-этилацетат 3 : 1) выход чистого
хроматографии на силикагеле (элюент - смесь то-
для анализа соединения 22b составил 7.53 г (89%
луол-этилацетат 3 : 1) получили 4.4 г (92% от тео-
от теории). Спектр ЯМР 1H (J, Гц, CDCl3), δ, м. д.:
рии) чистого для анализа соединения 23a. Спектр
2.41 с (9Н), 7.27 м (6H, J = 7.9), 7.44 с (3Н), 7.63 м
ЯМР 1H (J, Гц, CDCl3), δ, м. д.: 0.95 с (3Н), 2.41 с
(6H, J = 8.2), 7.71 м (6H, J = 8.2), 7.82 м (2H, J =
(6Н), 7.26 м (4H, J = 7.9), 7.43 с (3Н), 7.61-7.71 м
7.9), 7.95 м (2H, J = 8.2), 8.17 м (6H, J = 8.5), 10.11
(12H), 7.95 с (1H), 8.12 м (4H, J = 8.2). 13С ЯМР
с (1Н). Спектр ЯМР 13С ЯМР (CDCl3), δ, м. д.:
(CDCl3), δ, м. д.: -3.65; 21.35; 122.15; 122.81;
21.35; 76.57; 76.99; 77.42; 123.04; 124.22; 125.08;
123.78; 124.16; 125.06; 125.52; 128.44; 128.82;
125.74; 128.90; 129.10; 129.63; 134.87; 136.68;
129.60; 135.59; 135.76; 135.93; 137.92; 138.62;
136.84; 137.30; 138.70; 141.43; 151.84; 160.35;
150.67; 151.27; 151.65; 160.52. 29Si ЯМР (CDCl3), δ,
192.39. 29Si ЯМР (CDCl3), δ, м. д.: -14.28. Найдено,
м. д.: -10.58. Найдено, %: С 76.74; H 5.21; N 6.25;
%: С 79.17; H 5.05; N 5.20; Si 3.28; вычислено для
Si 4.08; вычислено для C42H33N3O3Si, %: С 76.92;
C55H41N3O4Si, %: С 79.02; H 4.94; N 5.03; Si 3.36.
H 5.07; N 6.41; Si 4.28.
4-[Трис(4-{5-[4-(2-этилгексил)фенил]-1,3-
2,2',2''-({[4-(1,3-Оксазол-5-ил)фенил]силан-
оксазол-2-ил}фенил)силил] бнзальдегид (22c).
триил}трибензол-4,1-диил)трис[5-(4-метилфе-
Синтез проводили аналогично методике получе-
нил)-1,3-оксазол] (23b). Синтез проводили анало-
ния соединения 22a из 3.38 г (2.78 ммоль) соеди-
гично методике получения соединения 23a из 8.4 г
нения 21c и 10 мл 1 М раствора HCl. После очист-
(10.0 ммоль) соединения 22b, 2.05 г (10.5 ммоль)
ки методом колоночной хроматографии на силика-
TosMIC и 2.77 г (20.1 ммоль) карбоната калия.
геле (элюент - смесь толуол-этилацетат 3 : 1) вы-
После очистки методом колоночной хроматогра-
ход для анализа чистого соединения 22c составил
фии на силикагеле (элюент - смесь толуол-эти-
2.79 г (89% от теории). Спектр ЯМР 1H ЯМР (J,
лацетат 3 : 1) выход чистого для анализа соедине-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
57
ния 23b составил 6.55 г (75% от теории). Спектр
Si2(PPO-Me)4. Спектр ЯМР 1H (J, Гц, CDCl3), δ,
ЯМР 1H (J, Гц, CDCl3), δ, м. д.: 2.41 с (9Н), 7.27 м
м. д.: 0.97 с (6Н), 2.40 с (12Н), 7.26 м (8H, J = 8.5),
(6H, J = 7.6), 7.44 с (4Н), 7.61-7.76 м (16H), 7.97
7.43 с (5Н), 7.56 с (2Н), 7.61-7.69 м (20H), 7.78
с (6H), 7.71 м (6H, J = 8.2). Спектр ЯМР 13С ЯМР
м (4H, J = 8.2), 8.13 м (8H, J = 8.2), 8.24 с (4H).
(CDCl3), δ, м. д.: 21.35; 122.39; 122.99; 123.90;
13С ЯМР (CDCl3), δ, м. д.: -3.51; 21.42; 123.04;
124.17; 125.06; 125.62; 128.87; 129.18; 129.61;
123.79; 124.26; 124.64; 125.23; 125.61; 126.75;
133.63; 135.52; 136.67; 136.86; 138.64; 150.76;
128.67; 128.79; 128.98; 129.67; 135.68; 135.90;
151.17; 151.74; 160.42. 29Si ЯМР (CDCl3): δ -14.34.
136.08; 137.97; 138.66; 151.57; 151.74; 160.64;
Найдено, %: С 78.12; H 4.92; N 6.10; Si 3.04; вы-
160.73. 29Si ЯМР (CDCl3), δ, м. д.: -10.57. Найдено,
числено для C57H42N4O4Si, %: С 78.24; H 4.84;
%: С 77.83; H 5.10; N 5.83; Si 4.44; вычислено для
N 6.40; Si 3.21.
C90H68N6O6Si2, %: С 78.01; H 4.95; N 6.06; Si 4.05.
2,2',2''-({[4-(1,3-Оксазол-5-ил)фенил]силан-
2,2',2'',2''',2'''',2'''''-[Бензол-1,4-диилбис(1,3-
триил}трибензол-4,1-диил)трис{5-[4-(2-этил-
оксазол-2,5-биилбензол-4,1-диилсилантетраил-
гексил)фенил]-1,3-оксазол} (23c). Синтез прово-
трибензол-4,1-диил)]гексакис[5-(4-метилфе-
дили аналогично методике получения соединения
нил)-1,3-оксазол]
((POPOP)Si2(PPO-Me)6).
23a из 3.37 г (2,98 ммоль) соединения 22c, 0.611 г
Син-тез проводили аналогично методике полу-
(3.1 ммоль) TosMIC и 0.824 г (5.96 ммоль) карбо-
чения соединения (POPOP)Si2(3Ph-EH)4 из 6.3 г
ната калия. После очистки методом колоночной
(7.2 ммоль) соединения 23b, 1.44 г (18.0 ммоль)
хроматографии на силикагеле (элюент - смесь то-
трет-бутилата лития,
83 мг
(0.07 ммоль)
луол-этилацетат 3 : 1) выход чистого для анализа
Pd(PPh3)4 и 0.809 г (3.4 ммоль) 1,4-дибромбензо-
соединения 23c составил 3.38 г (97% от теории).
ла. Реакционную смесь кипятили 1,5 ч до полно-
Спектр ЯМР 1H ЯМР (J, Гц, CDCl3), δ, м. д.: 0.83-
го завершения реакции. После очисткиметодом
0.95 м (18H), 1.22-1.36 м (24 Н), 1.60 м (3Н), 2.57 д
экстракции из толуола и хлороформа выделили
(6Н, J = 7.0), 7.24 м (6H, J = 8.2), 7.45 с (3Н), 7.46 с
5.6 г (59% от теории) чистого для анализа со-
(1Н), 7.61-7.79 м (16H), 7.97 с (1Н), 8.17 м (6H, J =
единения (экстракт из хлороформа) (POPOP)
Si2(PPO-Me)6. Спектр ЯМР 1H (J, Гц, CDCl3), δ,
8.2). 13С ЯМР (CDCl3), δ, м. д.: 10.75; 14.07; 22.97;
25.42; 28.79; 32.30; 39.97; 41.05; 122.41; 122.94;
м. д.: 2.40 с (18Н), 7.27 м (12H, J = 7.9), 7.44 (с,
123.94; 124.09; 125.22; 125.67; 128.87; 129.22;
6Н), 7.59-7.85 м (34H), 8.17 м (12H, J = 8.2), 8.25 с
(4H). Спектр ЯМР 13С ЯМР (CDCl3), δ, м. д.: 21.35;
129.75; 133.68; 135.60; 136.70; 136.88; 142.82;
151.88; 160.46. 29Si ЯМР (CDCl3), δ, м. д.: -14.34.
123.00; 123.81; 124.17; 124.80; 125.06; 125.63;
126.70; 128.67; 128.88; 129.21; 129.60; 133.62;
Найдено, %: С 79.81; H 7.24; N 4.94; Si 2.27; вы-
135.53; 136.69; 136.90; 138.63; 151.36; 151.74;
числено для C78H84N4O4Si, %: С 80.10; H 7.24;
N 4.79; Si 2.40.
160.42v 160.72. 29Si ЯМР (CDCl3), δ, м. д.: -14.33.
Найдено, %: С 78.80; H 4.68; N 5.97; Si 3.24; вы-
5,5',5'',5'''-{Бензол-1,4-диилбис[1,3-оксазол-
числено для C120H86N8O8Si2, %: С 79.01; H 4.75;
2,5-диилбензол-4,1-диил(метилсилантриил)
N 6.14; Si 3.08.
дибензол-4,1-диил]}тетракис[2-(4-метилфе-
нил)-1,3-оксазол] (POPOP)Si2(PPO-Me)4). Синтез
2,2',2'',2''',2'''',2'''''-[Бензол-1,4-диилбис(1,3-
проводили аналогично методике получения соеди-
оксазол-2,5-биилбензол-4,1-диилсилантетра-
нения (POPOP)Si2(3Ph-EH)4 из 3.25 г (4.95 ммоль)
илтрибензол-4,1-диил)]гексакис{5-[4-(2-этилгек-
соединения 23a, 0.99 г (12.3 ммоль) трет-бутила-
сил)фенил]-1,3-оксазол} [(POPOP)Si2(PPO-EH)6].
та лития, 57 мг (0.05 ммоль) Pd(PPh3)4 и 0.557 г
Синтез проводили аналогично методике получе-
(2.36 ммоль)
1,4-дибромбензола. Реакционную
ния соединения (POPOP)Si2(3Ph-EH)4 из 2.6 г
смесь кипятили в течение 60 мин до полного завер-
(2.22 ммоль) соединения 23с, 0.44 г (5.55 ммоль)
шения реакции. После очистки методом колоноч-
трет-бутилата лития, 25 мг (0.02 ммоль) Pd(PPh3)4
ной хроматографии на силикагеле (элюент - смесь
и
0.249 г
(1.05 ммоль)
1,4-дибромбензола.
толуол-этилацетат 3 : 1) выделили 2.3 г (71% от
Реакционную смесь кипятили 1.5 ч до полного за-
теории) чистого для анализа соединения (POPOP)
вершения реакции. После очистки методом клас-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
58
СКОРОТЕЦКИЙ и др.
сической хроматографии на силикагеле (элюент -
нений доказаны с использованием комплекса со-
смесь толуол-этилацетат 3 : 1) выделили 0.45 г
временных физико-химических методов анализа,
(17% от теории) чистого для анализа соединения
включающих гель-проникающую хроматографию,
(POPOP)Si2(PPO-EH)6. Спектр ЯМР 1H ЯМР (J,
ядерный магнитный резонанс на ядрах 1Н-, 13С- и
Гц, CDCl3), δ, м. д.: 0.84-0.93 м (36H), 1.23-1.37 м
29Si, элементный анализ и масс-спектроскопию
(49Н), 1.61 м (6Н), 2.57 д (12Н, J = 7.0), 7.24 м
с лазерной десорбцией в присутствии матрицы с
(12H, J = 8.2), 7.45 с (6Н), 7.59 с (2Н), 7.64 м (12H,
времяпролетной детекцией (MALDI-TOF). За счет
J = 8.2), 7.71-7.85 м (20H), 7.95 м (12H, J = 8.2),
использования ГПХ с чувствительным матричным
8.25 с (4Н). 13С ЯМР (CDCl3), δ, м. д.: 10.73; 14.08;
фотодетектором достигается контроль высокой чи-
22.96; 25.36; 28.76; 32.25; 39.93; 41.02; 123.01;
стоты синтезированных соединений. Полученные
123.82; 124.05; 125.21; 125.63; 126.72; 128.70;
КНЛ могут найти применение в устройствах и ма-
128.89; 129.22; 129.73; 133.63; 135.54; 136.69;
териалах для органической фотоники, где необхо-
димо большое сечение поглощения в УФ диапазо-
136.91; 142.75; 151.39; 151.82; 160.44;
160.75.
не (270-330 нм) и яркая люминесценция в районе
29Si ЯМР (CDCl3), δ, м. д.: -14.33. Найдено, %:
420 нм [25, 26].
С 80.40; H 7.26; N 4.45; Si 2.20; вычислено для
C162H170N8O8Si2,
%: С 80.63; H 7.10; N 4.64;
ФОНДОВАЯ ПОДДЕРЖКА
Si 2.33.
Работа выполнена при финансовой поддерж-
ВЫВОДЫ
ке Министерства науки и высшего образования
Российской Федерации (госзадание, ведущая на-
Таким образом, в данной работе был успешно
учная школа НШ-5698.2018.3), Программы фун-
синтезирован ряд новых кремнийорганических
даментальных исследований Президиума РАН
наноструктурированных люминофоров с одина-
№ 32 “Наноструктуры: физика, химия, биоло-
ковым центральным акцепторным фрагментом,
гия, основы технологий”, и Российского Фонда
идентичным люминофору POPOP, и разными до-
Фундаментальных Исследований (грант № 16-03-
норными фрагментами (п-терфениил или PPO) на
01118).
периферии. Для этого были использована после-
довательность реакций взаимодействия хлорсила-
КОНФЛИКТ ИНТЕРЕСОВ
нов с литий- или магнийорганическими прекурсо-
рами, синтез оксазолов по Ван Лёссену и катали-
Авторы заявляют об отсутствии конфликта ин-
зируемые комплексом палладия реакции кросс-со-
тересов.
четания, характеризующиеся высокой селектив-
ностью и хорошими реакционными выходами.
СПИСОК ЛИТЕРАТУРЫ
Следует отметить, что в данной работе впервые
для синтеза КНЛ описано использование реак-
1. Balzani V., Campagna S., Denti G., Juris A., Serroni S.,
ции прямого С-Н арилирования. Выбор вышеопи-
Venturi M. Sol. Energy Mater. Sol. Cells. 1995, 38, 159.
санных сопряженных структур для синтеза КНЛ
2
Alpha B., Ballardini R., Balzani V., Lehn J.M.,
обусловлен сочетанием уникальных оптических
Perathoner S., Sabbatini N. Photochem. Photobiol.
1990, 52, 299.
характеристик и подходящим спектральным диа-
пазоном для эффективного внутримолекулярного
3. Jullien L., Canceill J., Valeur B., Bardez E., Lefèvre J.-
переноса фотоиндуцированной энергии по прин-
P., Lehn J.-M., Marchi-Artzner V., Pansu R. J. Am.
Chem. Soc. 1996, 118, 5432.
ципу “молекулярной антенны”. Для соединений
с п-терфенильными и 2,5-дифенилоксазольными
4. Croce R., van Amerongen H. Nat. Chem. Biol. 2014,
10, 492.
донорными фрагментами были получены олиго-
меры с различным соотношением донор-акцептор.
5. Trofymchuk K., Reisch A., Didier P., Fras F., Gilliot P.,
Молекулярное строение и химическая структура
Mely Y., Klymchenko A.S. Nat. Photon. 2017, 11, 657.
полученных промежуточных и конечных соеди-
6. Arrigo A., La Ganga G, Nastasi F., Serroni S.,
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
СИНТЕЗ КРЕМНИЙОРГАНИЧЕСКИХ НАНОСТРУКТУРИРОВАННЫХ ЛЮМИНОФОРОВ
59
Santoro A., Santoni M.-P., Galletta M., Campagna S.,
troscopy. John Wiley & Sons Inc., 2003.
Puntoriero F. C. R. Chimie. 2017, 20(3), 209.
18. Skorotetcky М.S., Borshchev O.V., Surin N.M.,
7.
Frischmann P.D., Mahata K., Wȕrthner F. Chem. Soc.
Odarchenko Y., Pisarev S.A., Peregudova S.M.,
Rev. 2012, 42(4),1847.
Törnroos K.W., Chernyshov D., Ivanov D.A.,
8.
He Z., Ishizuka T., Jiang D. Polym. J. 2007, 39(9), 889.
Ponomarenko S.A. Dyes Pigm. 2017, 141, 128.
9.
Ziessel R., Harriman A. Chem. Commun. 2011, 47, 611.
19. Skorotetcky M.S., Surin N.M., Borshchev O.V.,
Ponomarenko S.A. Mendeleev Commun. 2017, 27,
10.
Adronov A., Fréchet M.J. Chem. Commun. 2000, 18,
377.
1701.
11.
Ensslen P., Wagenknecht H.-A. Acc. Chem. Res. 2015,
20. Starikova T.Y., Surin N.M., Borshchev O.V.,
48(10), 2724.
Pisarev S.A., Svidchenko E.A., Fedorov Y.V.,
Ponomarenko S.A. J. Mater. Chem. C. 2016, 4, 4699.
12.
Romano F., Yu Y., Korgel B.A., Bergamini G., Ceroni P.
Top Curr. Chem. (Z) 2016, 374, 53.
21. Пономаренко С.А., Борщёв О.В., Лупоносов Ю.Н.,
Музафаров А.М., Сурин Н.М. Пат. RU 2396290C1.
13.
Luponosov Y.N., Ponomarenko S.A., Surin N.M.,
Borshchev O.V., Shumilkina E.A., Muzafarov A.M.
22. Wang D., Niu Y., Wang Y., Han J., Feng S. J. Organomet.
Chem. Mater. 2009, 21(3), 447.
Chem. 2010, 695(21), 2329.
14.
Ponomarenko S.A., Surin N.M., Borshchev O.V.,
23. Yu H., Shen C., Tian M., Qu J., Wang Z. Macromolecules,
Luponosov Y.N., Akimov D.Y., Alexandrov I.S.,
2012, 45(12), 5140.
Burenkov A.A., Kovalenko A.G., Stekhanov V.N.,
24. Hachiya H., Hirano K., Satoh T., Miura M. Angew.
Kleymyuk E.A., Gritsenko O.T., Cherkaev G.V.,
Chem. Int. Ed. 2010, 49, 2202.
Kechek’yan A.S., Serenko O.A., Muzafarov A.M. Sci.
25. Ponomarenko S.A., Surin N.M., Borshchev O.V.,
Rep. 2014, 4, 6549.
Skorotetcky M.S., Muzafarov A.M. Proc. of SPIE,
15.
Гринев Б.В., Сенчишин В.Г. Пластмассовые сцин-
2015, 9545, 954509.
тилляторы. X.: Акта, 2003, 324 с.
26. Ponomarenko S.A., Borshchev O.V., Surin N.M.,
16.
Kumari S., Sahare P.D. Adv. Porous Mater. 2013, 1, 114.
Skorotetcky M.S., Kleymyuk E.A., Starikova T.Yu.,
17.
Palmer D. C. Oxazoles:synthesis, reactions, and spec-
Tereshenko A.S. Proc. of SPIE, 2017, 10344, 103440N.
Synthesis of Nanostructured Organosilicon Luminophores
Based on Phenyloxazoles
M. S. Skorotetskiia, O. V. Borshcheva, G. V. Cherkaeva, and S. A. Ponomarenkoa, b, *
a Enikolopov Institute of Synthetic Polymeric Materials RAS (ISPM RAS),
117393, Russia, Moscow, yl. Profsoyuznaya 70
*e-mail: ponomarenko@ispm.ru
b Lomonosov Moscow State University, Faculty of Chemistry, 119991, Russia, Moscow, GSP-1, Leninskie gory 1, str. 3
Received Octobber 29, 2018
Revised November 5, 2018
Accepted November 11, 2018
A number of nanostructured organosilicon luminophores (NOLs) in the center of which there was the acceptor
chromophore fragment 1,4-bis(5-phenyloxazo-2-yl)benzene (POPOP) and on the periphery, various donor derivatives
of p-terphenyl and 2,5-diphenyloxazole, were synthesized using Van Loessen’s reactions and the direct C-H arylation
catalyzed by palladium complexes. Due to the use of organosilicon branching centers with different functionalities,
NOLs with different donor : acceptor ratios were obtained. The choice of these conjugated structures is due to their
good optical characteristics, thus, the resulting NOLs can be used in photonics and optoelectronics.
Keywords: arylation, Van Leussen reaction, phenylozazole, arylsilane, luminophore, terphenyl, POPOP
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019