ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2019, Том 55, № 1, с. 67-99
УДК 547.31 + 542.943.5
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ
ОЗОНОЛИЗА АЛКЕНОВ
© 2019 г. Ю. В. Мясоедова*, И. С. Назаров, Г. Ю. Ишмуратов
ФГБУН Уфимский Институт химии РАН (УфИХ РАН),
450054, Россия, Республика Башкортостан, г. Уфа, проспект Октября, 71
*е-mail: legostaevayuv@yandex.ru
Поступила в редакцию 13 июня 2018 г.
После доработки 26 июня 2018 г.
Принята к публикации 2 июля 2018 г.
В обзорной статье систематизированы и описаны данные за последние 10 лет по превращениям перок-
сидных продуктов озонолиза алкенов в различных вариантах (реакциях “расщепления”, под действием
восстановителей и N-содержащих соединений), а также применение этих реакций в направленных
синтезах.
Ключевые слова: озонолиз, пероксидные продукты, N-содержащие соединения, восстановители.
DOI: 10.1134/S0514749219010075
ОГЛАВЛЕНИЕ
ных продуктов разделяют на два типа: протекаю-
щие без изменения достигнутой степени окисле-
1. Введение
75
ния (“реакции расщепления”) и с ее изменением
2. Реакции “расщепления” перекисных продуктов
(реакции окисления и восстановления) [1].
озонолиза
75
3. Превращения перекисных продуктов
2. РЕАКЦИИ “РАСЩЕПЛЕНИЯ” ПЕРЕКИСНЫХ
озонолиза при действии восстановителей
80
ПРОДУКТОВ ОЗОНОЛИЗА
4. N-содержащие соединения в превращениях
перекисных продуктов озонолиза
94
Обычными продуктами расщепления озонидов
являются, соответственно, карбонильные соедине-
1. ВВЕДЕНИЕ
ния и карбоновые кислоты, а направление распада
перекисных соединений существенно зависит от
В настоящее время озонолиз олефинов явля-
природы растворителя и температуры озонирова-
ется хорошо изученным процессом, механизм и
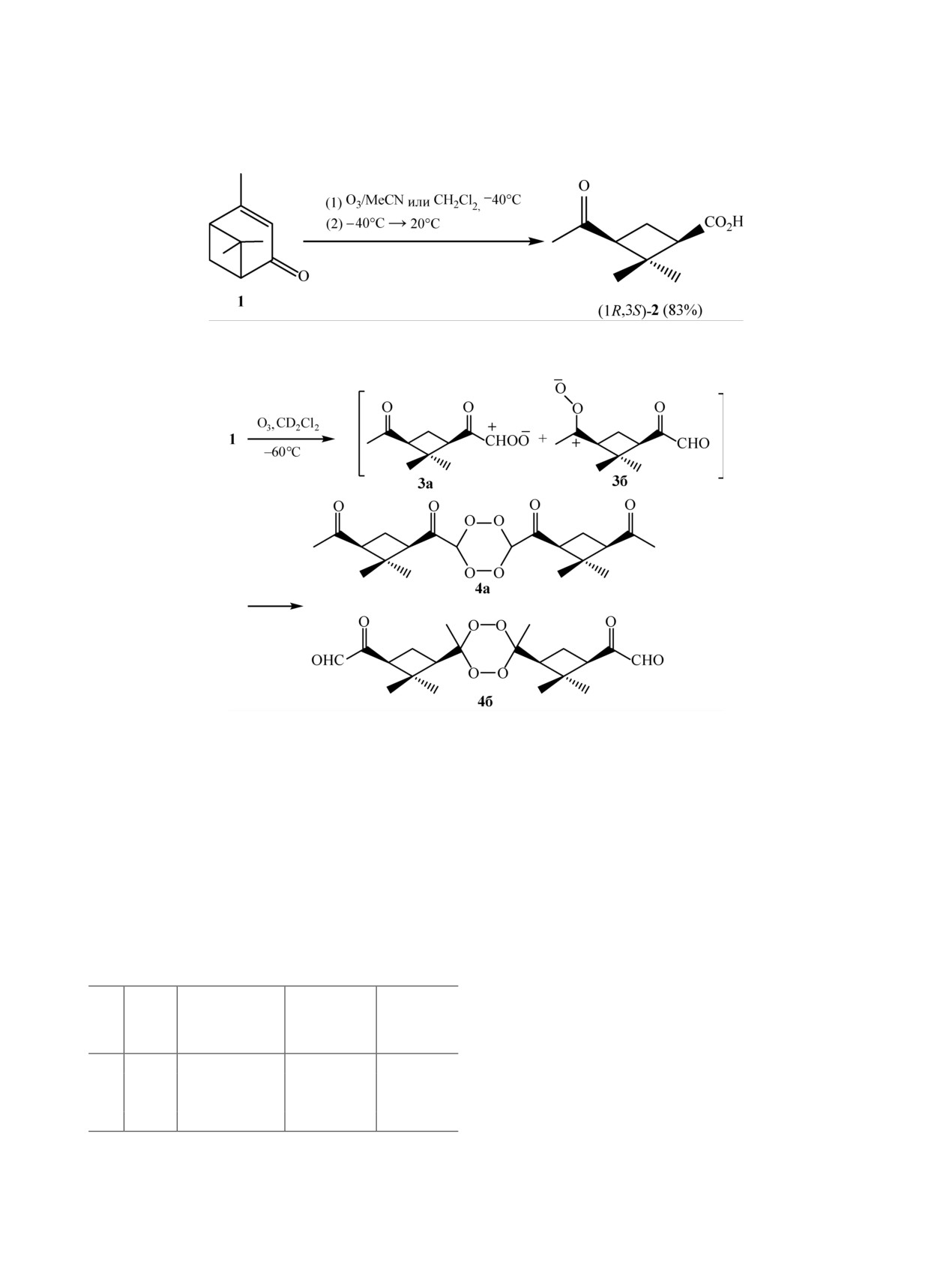
ния. Так, озонолиз вербенона 1 в хлористом ме-
основные закономерности которого приводились
тилене и ацетонитриле при разных температурах
в обзорных статьях [1, 2]. Существенное влияние
(табл. 1) приводит к (1R,3S)-ацетил-2,2-диметил-
на строение неперекисных продуктов оказывают
циклобутанкарбоновой кислоте 2 в качестве ос-
превращения пероксидных продуктов озонолиза,
новного продукта (схема 1) [4].
зависящие от условий проведения реакции (рас-
Анализ ЯМР 1Н и 13С спектров перекисных
творитель, температура), а также используемых
продуктов озонолиза, проведенного при -60°С в
реагентов [2, 3]. При озонировании олефинов
CD2Cl2, свидетельствует о том, что реакция, веро-
достигается степень окисления, промежуточная
ятно, протекает через стадию формирования кар-
между альдегидом или кетоном и карбоновой кис-
бонилоксидов 3а, 3б, которые далее превращаются
лотой, а превращения озонидов и других перекис-
в димерные пероксиды 4а, 4б (схема 2).
67
68
МЯСОЕДОВА и др.
Схема 1.
Схема 2.
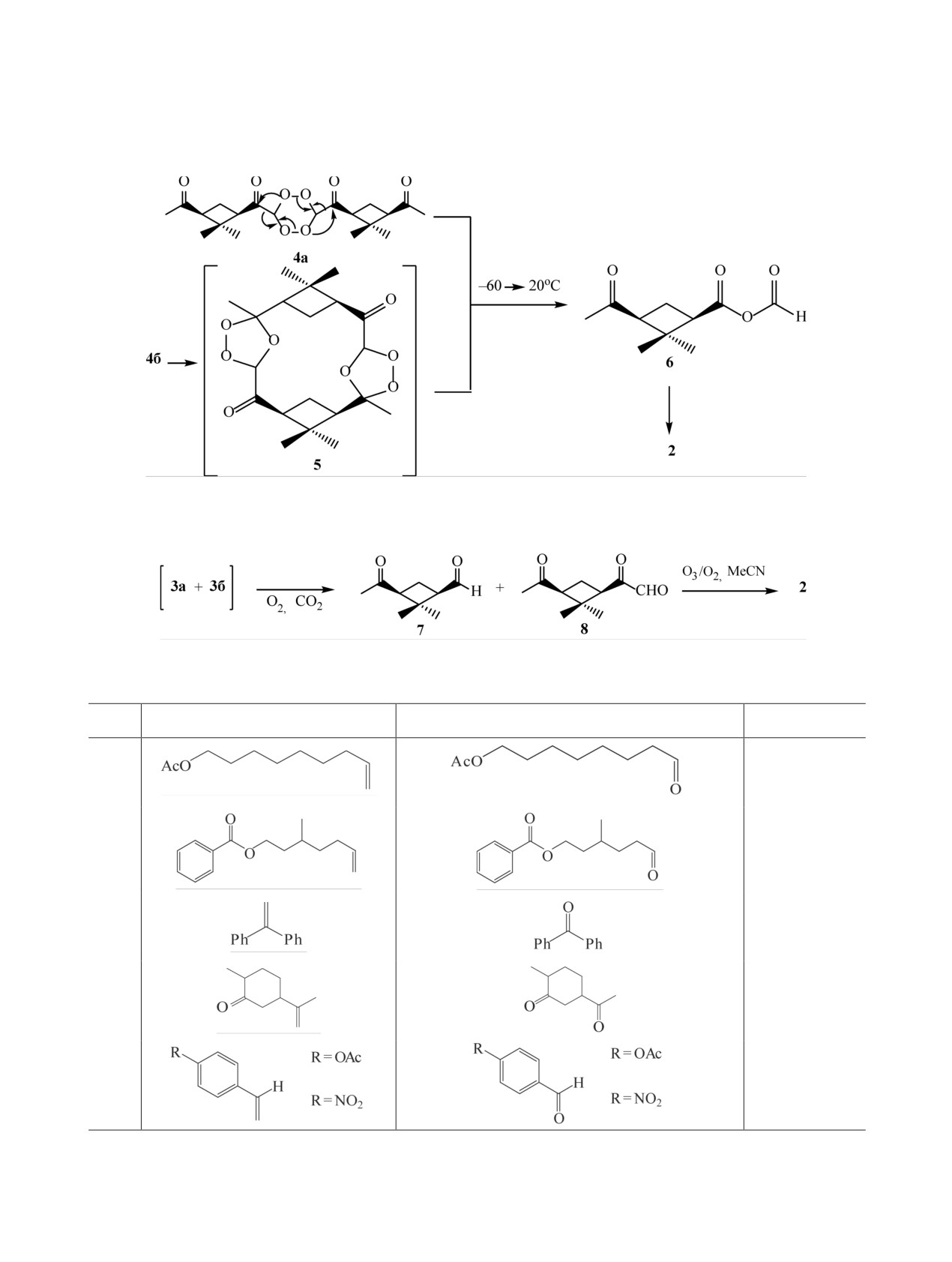
Повышение температуры приводит к перегруп-
тонных растворителях при -20°С протекает, веро-
пировке соединений 4а, 4б (при -40°С наполови-
ятно, по-другому. Альдегидные группы соедине-
ну, а при 0°С - полностью) в смешанный ангидрид
ний 7 и 8, образующихся из цвиттер-ионов 3а, 3б,
6, который, в соответствии со спектральными дан-
окисляются озоном с образованием единственного
ными, образуется из обоих пероксидов. Перегруп-
продукта - кетокислоты 2 (схема 4).
пировка пероксида 4б, возможно, проходит через
Озонолиз алкенов в смеси воды и органиче-
димер 5 (схема 3).
ских растворителей является быстрым, удобным
Согласно полученным результатам, формиро-
и эффективным однореакторным способом син-
вание димерных пероксидов происходит только
теза альдегидов и кетонов, исключающим стадию
при пониженных температурах. Озонолиз в апро-
восстановления образующихся пероксидов
[5].
Предполагается, что добавление воды к карбони-
Таблица 1
локсидам приводит к гем-гидроперокси спиртам,
которые для большинства субстратов разлагаются
Израсходо-
Выход
с выделением альдегида или кетона и H2O2, что
№
Т, °С
Растворитель
ванный
2, %
подтверждается фиксированием в реакционной
О3, моль
смеси стехиометрических количеств Н2О2 по от-
1
-60
СН2Cl2
1.1-1.3
70
ношению к исходному алкену (схема 5).
2
-40
MeCN
1.1-1.3
83
СН2Cl2
1.1-1.3
71
Разработанный метод был проверен на серии
3
0
MeCN
1.5-1.7
57
субстратов (табл. 2). Показано, что в зависимости
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
69
Схема 3.
Схема 4.
-
-
Таблица 2
№
Исходное соединение
Продукт
Выход, %
1
72
2
75
3
54
4
100
5
81
6
100
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
70
МЯСОЕДОВА и др.
Схема 5.
Схема 6.
от исходного алкена альдегиды/кетоны получа-
ной температуре менялось. В течение 14 дней ке-
ются с выходами от средних до количественных
тоны 11а и 11б полностью трансформировались в
[5].
кристаллический лактон 13, который далее исполь-
зовали в синтезе целевого амбраксана 14 (схема 6).
Разработанный способ был применен авторами
[6] в синтезе амбраксана (ambrox®) 14. Окисли-
Обработкой хлористым водородом в метаноле
тельное расщепление смеси изомеров 9 и 10 озо-
пероксиды количественно превращают в метило-
ном в ацетоне, содержащем 5% воды, при -78°С
вые эфиры соответствующих карбоновых кислот
дало смесь соединений 11а, 11б и 12 в соотноше-
[2]. Авторами [7] было обнаружено, что в услови-
нии 3 : 1, которое при выдерживании при комнат-
ях частичного озонолиза (S)-(-)-лимонена 15 в си-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
71
Схема 7.
или
-PrOH
Схема 8.
стеме циклогексан-метанол при 2-4°С образуются
зующихся перекисных продуктов. Такой подход
озониды 16а, 11б в виде смеси (2 : 3) диастереоме-
был использован для получения метиловых эфи-
ров, дальнейшая обработка которых метанольным
ров бензил- 21 и фенокси- 24 -уксусных кислот из
раствором хлороводорода приводит к циклизации
бензил- 20 и фенил- 23 -аллиловых эфиров [10].
промежуточных продуктов и образованию сме-
Установлено, что низкотемпературный (-65°С)
си (4 : 1) двух соединений, основной из которых
озонолиз субстратов 20 и 23 в присутствии NaOH
идентифицирован как сложный эфир 17, а минор-
приводит с высоким выходом к эфирам 21 и 24,
ный - как соответствующий альдегид 18 [8]. Заме-
соответственно. Побочными продуктами являют-
на MeOH на i-PrOH приводит к тем же озонидам
ся соответствующие алкоксиуксусные альдегиды
16а, 16б, однако при обработке HCl в i-PrOH с вы-
22 и 25 (схема 8).
ходом 50% образуется изопропиловый эфир 19 [9]
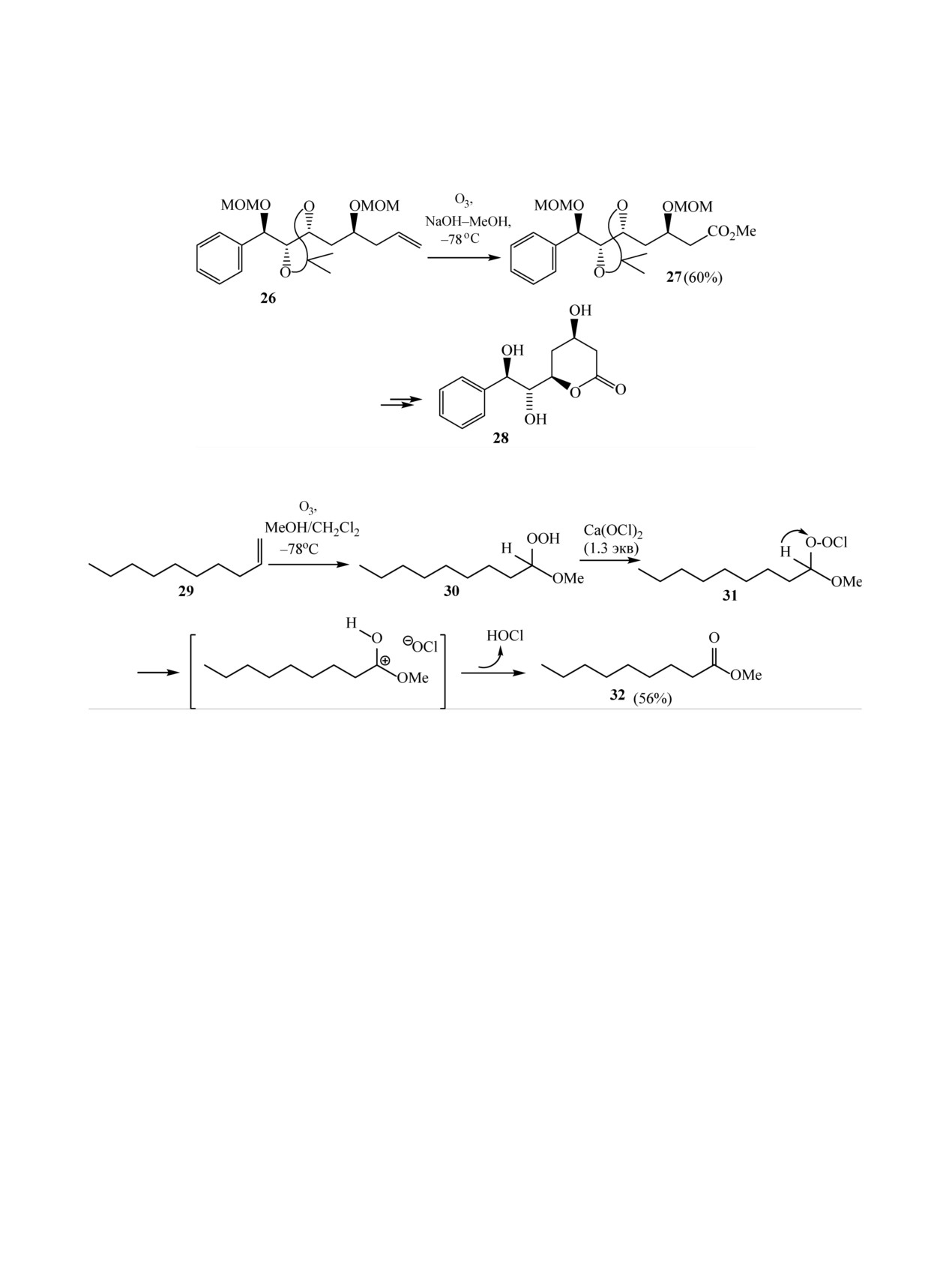
Озонированием МОМ-эфира 26 в МеОН при
(схема 7).
-78°С в присутствии NaOH получают соответ-
Озонированием алкенов в смеси метанола,
ствующий сложный эфир 27, что является одной из
хлористого метилена и гидроксида натрия по-
ключевых стадий стереоселективного синтеза при-
лучают соответствующие сложные эфиры без
родного стириллактона лейокарпина (leiocarpin) С
дополнительной обработки промежуточно обра-
28 (схема 9) [11].
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
72
МЯСОЕДОВА и др.
Схема 9.
Схема 10.
Гипохлориты эффективно дегидратируют ги-
сидных продуктов озонолиза чаще применяются
дропероксиацетали, давая соответствующие слож-
восстановители.
ные эфиры [12]. Реакция, которая выполняется со
Одним из наиболее часто используемых для
стехиометрическим количеством Ca(OCl)2, вклю-
получения карбонильных соединений восстано-
чает, по-видимому, гетеролитическое расщепление
вителем является диметилсульфид [2]. Взаимо-
первичного хлоропероксида 31. Данные реагенты
действие пероксидов с диметилсульфидом хорошо
могут быть применены в озонолизе алкенов, что
изучено, применение этого реагента в реакциях
позволяет осуществлять удобный однореакторный
“озонолиза - восстановления” показано на боль-
синтез сложных эфиров (схема 10).
шом количестве примеров, поэтому данный реа-
гент широко используется в направленном органи-
ПРЕВРАЩЕНИЯ ПЕРЕКИСНЫХ ПРОДУКТОВ
ческом синтезе [14-26].
ОЗОНОЛИЗА ПОД ДЕЙСТВИЕМ
Представлен новый подход к замещенным 2-ок-
ВОССТАНОВИТЕЛЕЙ
со-1,3-пропандиолам из аддуктов Морита-Бэй-
лис-Хиллмана (МБХ) [27]. В данной стратегии
Превращения перекисных продуктов озонолиза
замещенные
2-оксо-1,3-пропандиолы получают
под действием окислителей или восстановителей
озонолизом аллильных ацетатов 36-38, получен-
широко используются как в препаративном орга-
ных из соответствующих диолов 33-35, при -72°С
ническом синтезе, так и в промышленной техноло-
в метаноле. Последующее восстановление проме-
гии [13]. В последние годы для разрушения перок-
жуточных пероксидов диметилсульфидом ведет к
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
73
Схема 11.
Схема 12.
2-оксо-1,3-пропандиолам 39-41 (схема 11) с выхо-
тилсульфидом [28, 29]. При обычном озонирова-
дами от 80 до 91%.
нии озоном в смеси с кислородом могут окислять-
Циклические 42 и ациклические 43 1,4-дие-
ся некоторые субстраты, например, легко окисля-
ны, полученные в результате Со-катализируемых
ющиеся на воздухе производные 1,4-циклогекса-
реакций Дильса-Альдера алкинов с 1,3-диенами
диена. Вот почему авторы [28] абсорбировали О3
и 1,4-гидровинилирования алкенов и 1,3-диенов,
на крупном силикагеле при низкой температуре,
превращают в дикарбонильныe соединения
44
десорбцию О3 проводили повышением темпера-
озонолизом с дальнейшим восстановлением диме-
туры и десорбцией азотом несодержащего О2 озо-
Таблица 3
№
R
Ацилирование, %a
Озонолиз, %a
1
H
36, 85
39, 91
2
OMe
37, 90
40, 80
3
NO2
38, 90
41, 82
a Выход на выделенный и очищенный продукт.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
74
МЯСОЕДОВА и др.
Схема 13.
РМАС
РМОС
на. Для того чтобы охарактеризовать 1,3-дикарбо-
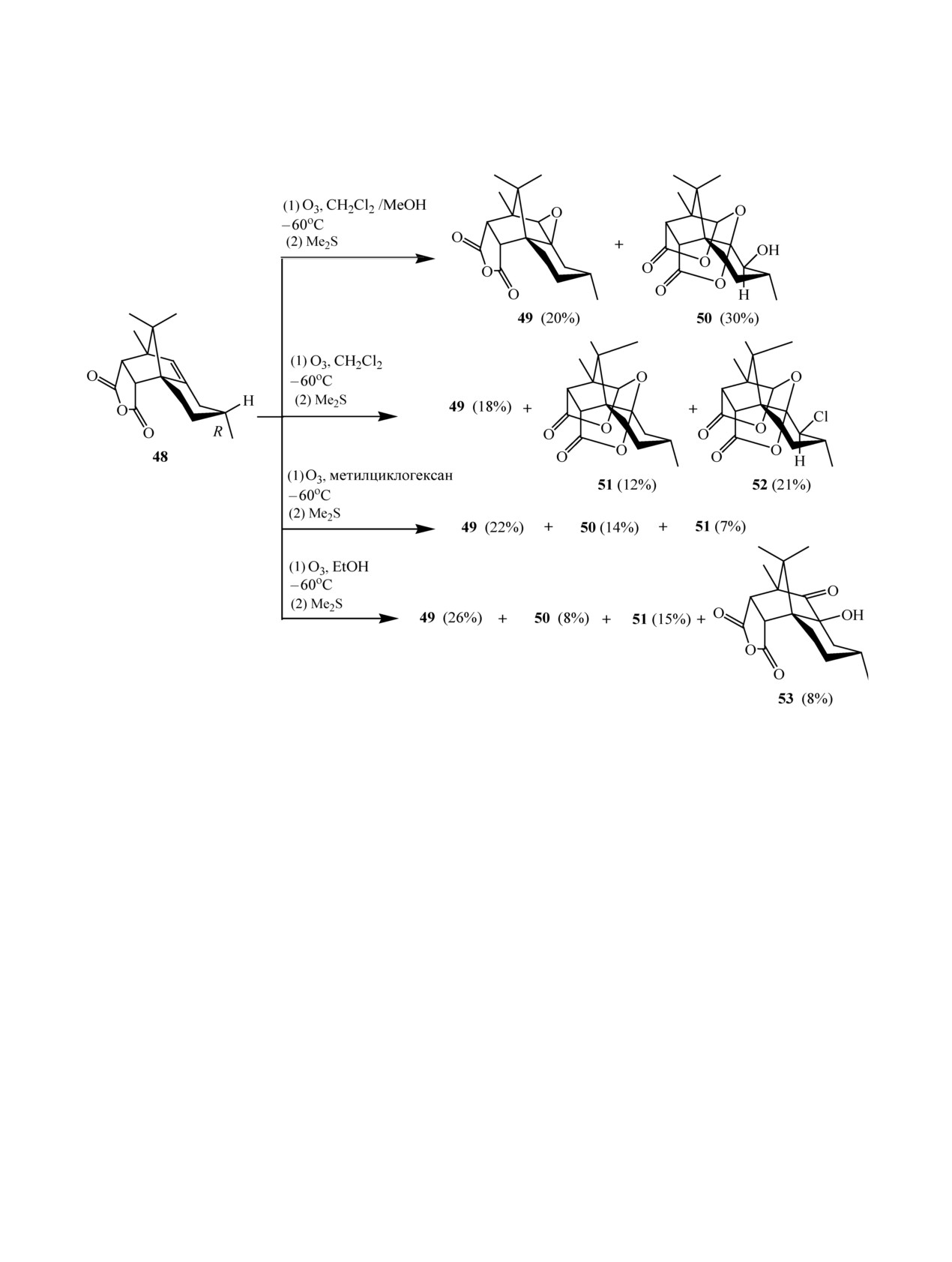
Авторами статьей [31, 32] показано, что в за-
нильные производные 44, они были превращены
висимости от используемого растворителя озоно-
в соответствующие фенилпиразолы 45 реакцией с
лиз ангидрида 48 проходит по-разному. Окисле-
фенилгидразином (схема 12).
ние озоном субстрата 48 в смеси CH2Cl2-МеОН
приводит к эпоксиду 49 и гидрокси-бис-лактону
Озонолиз с последующим восстановлением
50, а в CH2Cl2 - к трем продуктам: эпоксиду 49,
может успешно применяться для получения про-
бис-лактону 51, и, неожиданно, к хлоро-бис-лак-
изводных поликарбонатов, содержащих альде-
тону 52 - производному гидрокси-бис-лактона
гидные фрагменты - потенциальных платформ в
синтезе соединений с различными свойствами и
50. Структура лактонов 51 и 52 доказана с помо-
щью РСА [32]. Низкая реакционная способность
видами активности [30]. Так, альдегид-замещен-
наблюдалась также при использовании в качестве
ный поли(5-метил-5-оксоэтилоксикарбонил-1,3-ди-
растворителя метилциклогексана. Применение
оксан-2-он) (PMOC), полученный in situ озоноли-
EtOH привело к полиоксигенированным соеди-
зом аллил-функционализированного полимерного
предшественника (PMAC) с последующей обра-
нениям: эпоксиду 49 в качестве основного про-
дукта, бис-лактонам 50 и 51, а также кетону 53,
боткой Me2S, обрабатывали в присутствии ацетата
образующемуся в результате перегруппировки
натрия гидрофобным O-бензил- или гидрофильным
эпоксида 49 (схема 14).
O-(карбоксиметил)- гидроксиламинными реагента-
ми. В результате, первый из полученных полиме-
В современной органической химии также ак-
ров 46 растворим в большинстве органических
туально применение диметилсульфида при восста-
растворителей, а 47 - в воде и метаноле (схема 13).
новлении продуктов озонолиза полициклических
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
75
Схема 14.
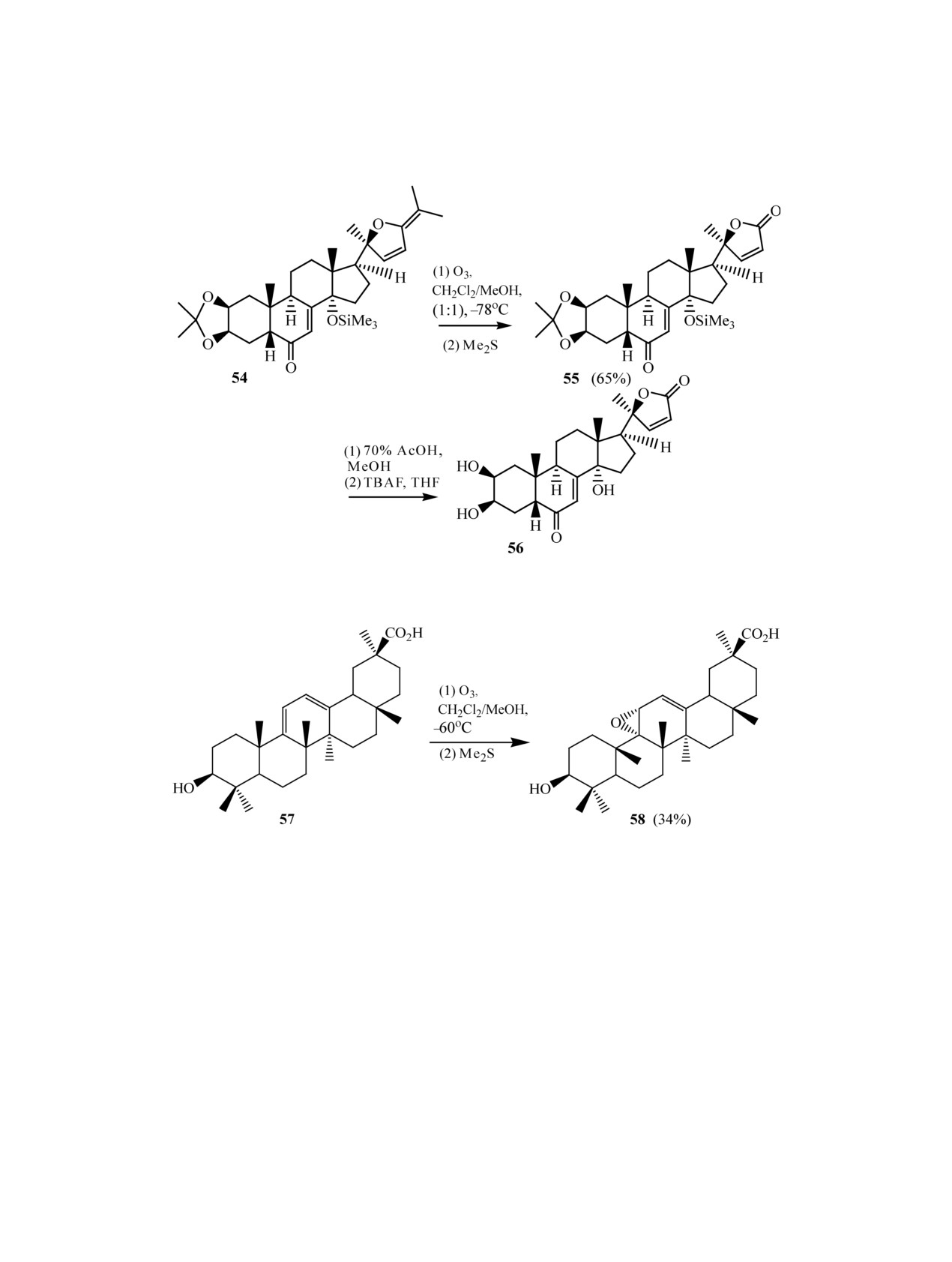
субстратов, например, стероидной природы. Так,
Несмотря на то, что Me2S является наиболее
авторами [33] впервые реализован синтез природ-
широко применяемым реагентом для восстанов-
ного фитоэкдистероида сидистерона (sidisterone)
ления промежуточных пероксидов, он имеет недо-
56, включающий восстановительный озонолиз эк-
статки: сильно летуч и имеет неприятный запах,
зо-циклической двойной связи дигидрофураново-
поэтому в качестве восстанавливающего агента
го производного 54, приводящий к целевому γ-лак-
иногда используют тиомочевину. Авторами [36]
тону 55 с хорошим выходом (схема 15). При этом
показано, что озонолиз непредельного кетона 59
продуктов окисления эндо-циклических двойных
приводит обычно к озониду 60 в виде смеси ди-
связей не наблюдалось.
астереомеров с выходом 86%, восстановление ко-
В случае использования диметилсульфида для
торого in situ тиомочевиной дает дикарбонильное
восстановления пероксидных продуктов озоно-
соединение 61 с выходом 70% (схема 17).
лиза стерически затрудненных двойных связей
Другим эффективным восстановителем пере-
возможно образование не только кетонов и аль-
кисных продуктов озонолиза до карбонильных
дегидов [34]. Так, озонолиз кислоты 57 в CH2Cl2-
соединений является трифенилфосфин. Следует
MeOH при -60°С с последующим действием Me2S
отметить, что восстановление PPh3 проходит, в
приводит к сложной смеси продуктов окисления,
отличие от Me2S, быстро и практически без об-
из которой хроматографически выделен в качестве
разования аномальных продуктов [2], поэтому он
основного продукта 9α,11α-эпоксид 58 с выходом
широко используется в направленных синтезах
34% (схема 16) [35].
[37-50]. Так, PPh3 был применен авторами [51]
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
76
МЯСОЕДОВА и др.
Схема 15.
Схема 16.
при восстановлении продуктов озонолиза мети-
сенный на твердую подложку (полистирол-связан-
леновых производных 62а-62д в синтезе поли-
ный PPh2 (PS-PPh2)), легко удаляемый обычным
карбонильных соединений
63а-63д (таблица 4).
фильтрованием. Были подобраны оптимальные
Условия реакции: 1. O3, CH2Cl2, -78°С; 2. PPh3
условия восстановления (2 экв. PS-PPh2, пере-
(2.0 экв).
мешивание 24 ч, метод Б), приводящие к полной
Для термически неустойчивых формилуксус-
конверсии в альдегиды 65а-65е (схема 18), в том
числе в формилуксусный амид 65е, без разложе-
ных эфиров использование стандартной методи-
ния продуктов реакции (табл. 5) [52].
ки восстановления (1.5 экв. PPh3, перемешивание
16 ч, метод А) не является оптимальным, т.к. пред-
Для демонстрации синтетических возможно-
полагает очистку перегонкой, приводящую к сни-
стей разработанного метода был получен тиазо-
жению выходов, особенно у соединений с высокой
лидин 68, являющийся интермедиатом для 6-не-
температурой кипения. Для исключения стадии
замещенного β-лактама 69 - пенама, как пред-
очистки был применен трифенилфосфин, нане-
ставителя β-лактамного семейства антибиотиков.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
77
Таблица 4
№
Исходное соединение
Продукт
Выход, %
O
Me
Me
1
91
O
O
62а
63а
Me O
Me
2
97
O
O
62б
63б
O
O
O
N
Me
N
Me
3
86
O
O
O
O
62в
63в
O
O
O
Me
Me
Me
Me
4
75
O
O
O
62г
63г
O
5
69
O
62д
63д
Ди-трет-бутилгидромуконовый эфир
66 пре-
го пенам 69 легко получают селективным снятием
вратили в альдегид с использованием метода Б,
трет-бутиловой защиты с последующей лактами-
отфильтровали и реакцией с метиловым эфиром
зацией (схема 19) [52].
пенициламина 67 перевели с высокими выходом
Полистирол-связанный PPh2 был применен
и стереоселективностью в триазол 68, из которо-
также в синтезе лактама 70 (схема 20) [53].
Схема 17.
Схема 18.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
78
МЯСОЕДОВА и др.
Таблица 5
Выход (%),
Выход (%),
Опыт
R
Условия восстановления
метод А
метод Б
А
MeO
Метод Б: 2.0 экв. PS-PPh2, -60°С → ком. темп., 24 ч
–
90
Б
EtO
Метод А: 1.5 экв. PPh3, -60°С→ ком темп., 16 ч, или метод Б
65
95
В
i-PrO
Методы А или Б
63
Колич.
Г
t-BuO
Методы А или Б
46
Колич.
Д
BnO
Методы А или Б
24
Колич.
Е
i-PrNH
Метод Б
-
Колич.
Одним из стандартных и широко применямых
Озонолитическое расщепление винилиденовой
методов получения спиртов является восстано-
группы замещенного циклопентана 71 в присут-
вительное расщепление перекисных продуктов
ствии в качестве индикатора Судан III с после-
озонолиза алкенов комплексными гидридами ще-
дующим селективным восстановлением NaBH4
лочных металлов, например, боргидридом натрия
в EtOH привели к диолу 72, выделенному в виде
[54-57].
смеси (3.4 : 1) диастереомеров (схема 21) [58].
Схема 19.
Схема 20.
Схема 21.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
79
Схема 22.
Схема 23.
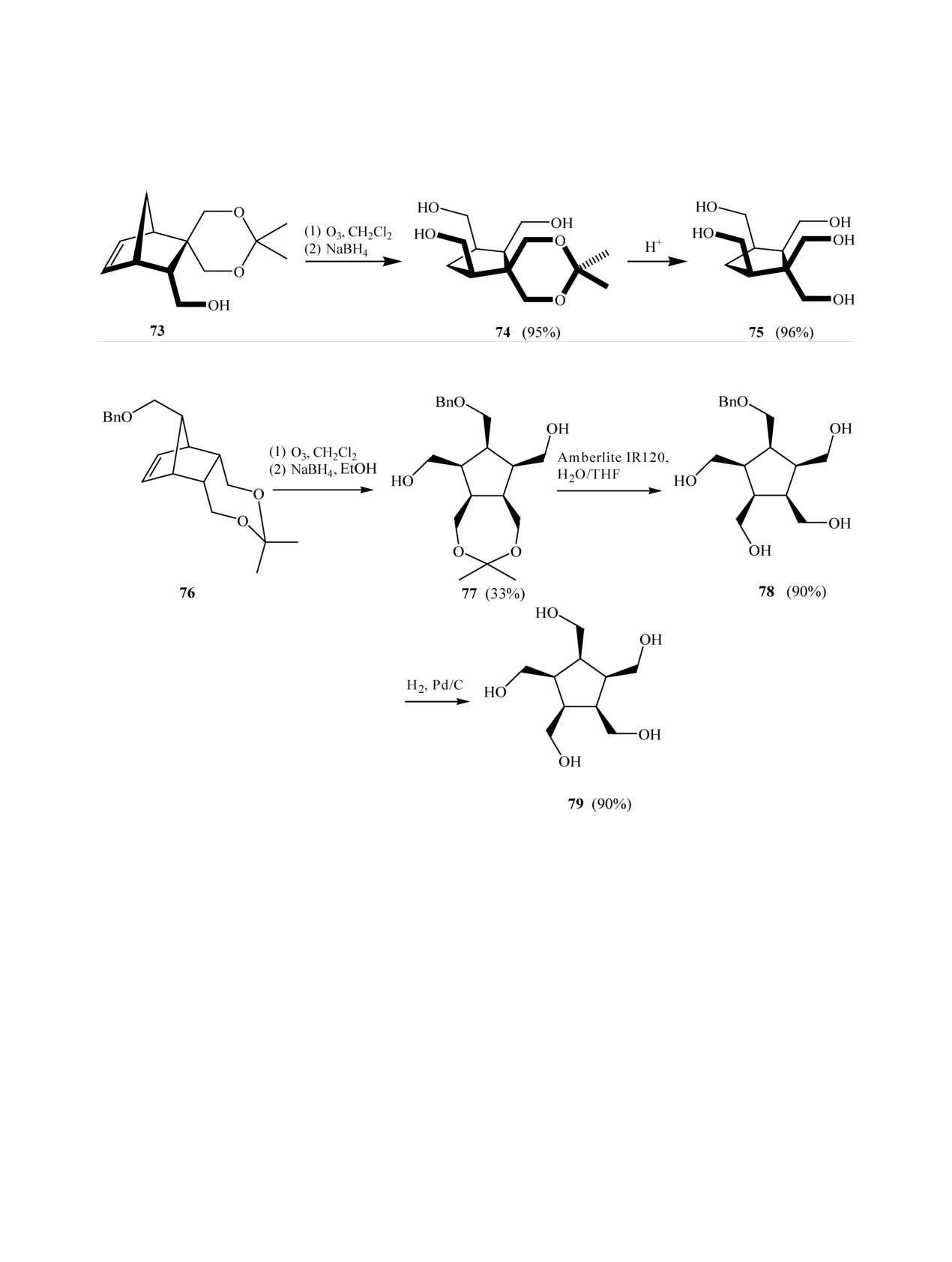
Одной из стадий синтеза сферических полио-
цис-1,2,3,4,5-пентакис(гидроксиметил)циклопен-
лов является озонолиз спиро-соединения 73 при
тан 79 с 90% выходом (схема 23) [60].
-60°С в присутствии Судан III. После обработ-
Восстановительная обработка перекисных
ки этанольным раствором NaBH4 было получено
продуктов озонолиза в CH2Cl2 при
-60°С
1,3-диоксановое производное пентаола 74 с коли-
ангидрида 48 избытком NaBH4 дает в качестве
чественным выходом, переведенное в целевое сое-
основных продуктов дигидроксилактон
80 и
динение 75 снятием защиты (схема 22) [59].
лактон 81 (схема 24) [31].
Озонирование непредельного ацетонида 76 с
Обработка в аналогичных условиях диэфира 82
последующим восстановлением NaBH4 дало диол
не привела, как предполагалось, к диолу, но были
77 с выходом 33%, который затем гидролизовали в
получены лактоны 83 и 84 с общим выходом 10%
(схема 25) [60].
монозащищенный пентаол 78 с 90% выходом, сня-
тием бензильной защиты в котором гидрогеноли-
Механизм образования лактонов 83 и 84 авто-
зом на Pd-катализаторе получен цис,цис,цис,цис,-
ры [60] объясняют по аналогии с восстановлением
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
80
МЯСОЕДОВА и др.
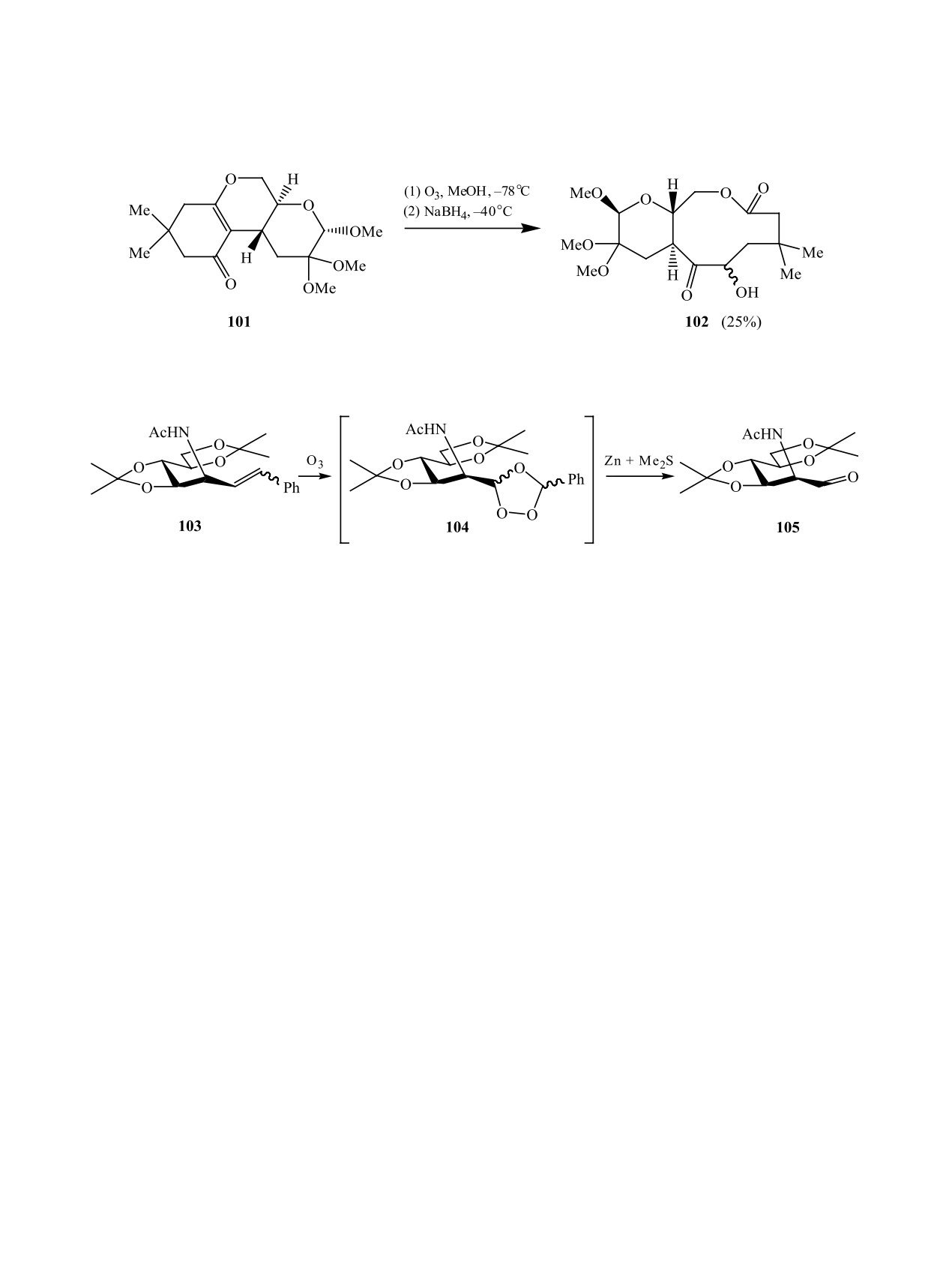
Схема 24.
Схема 25.
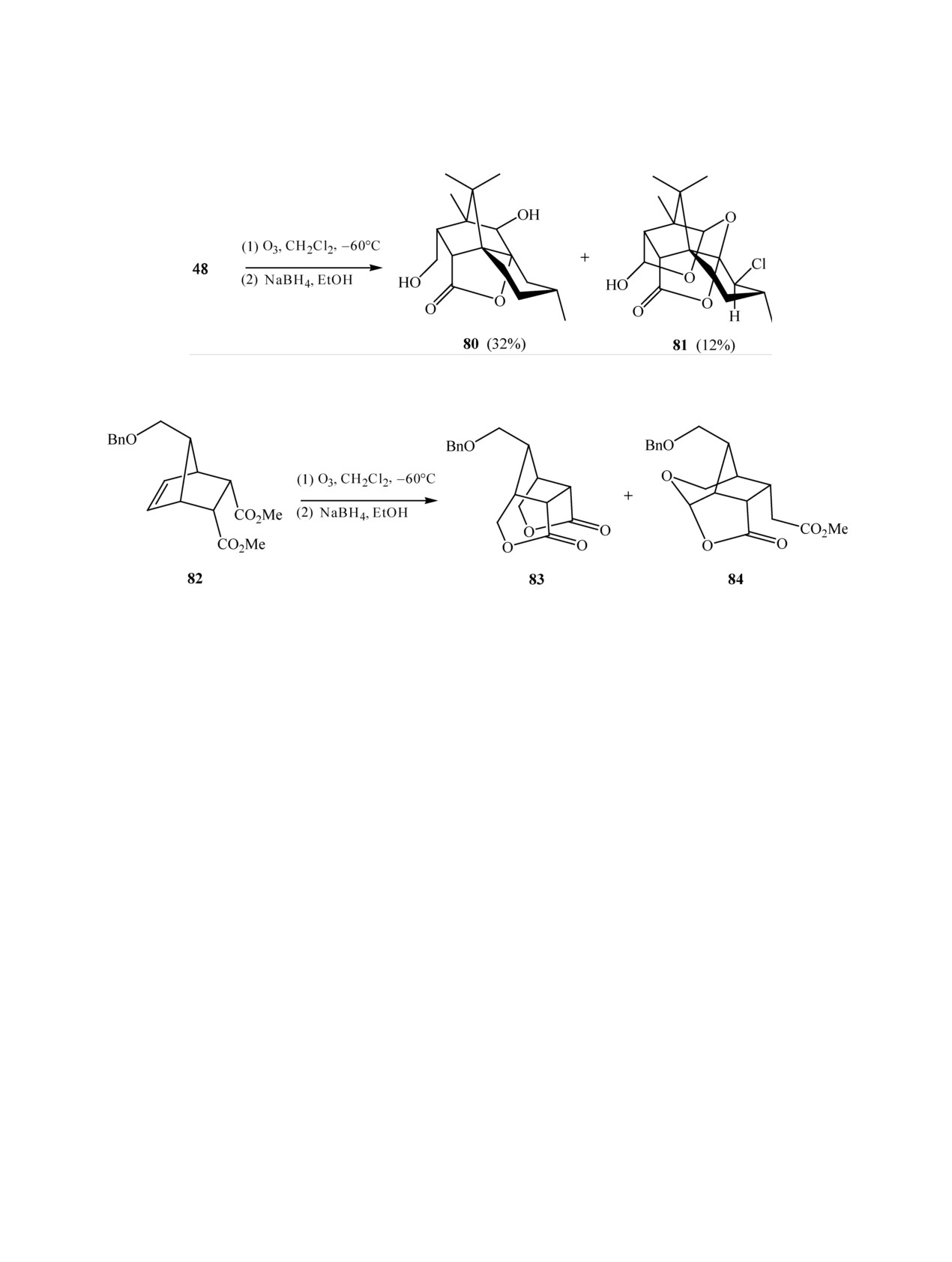
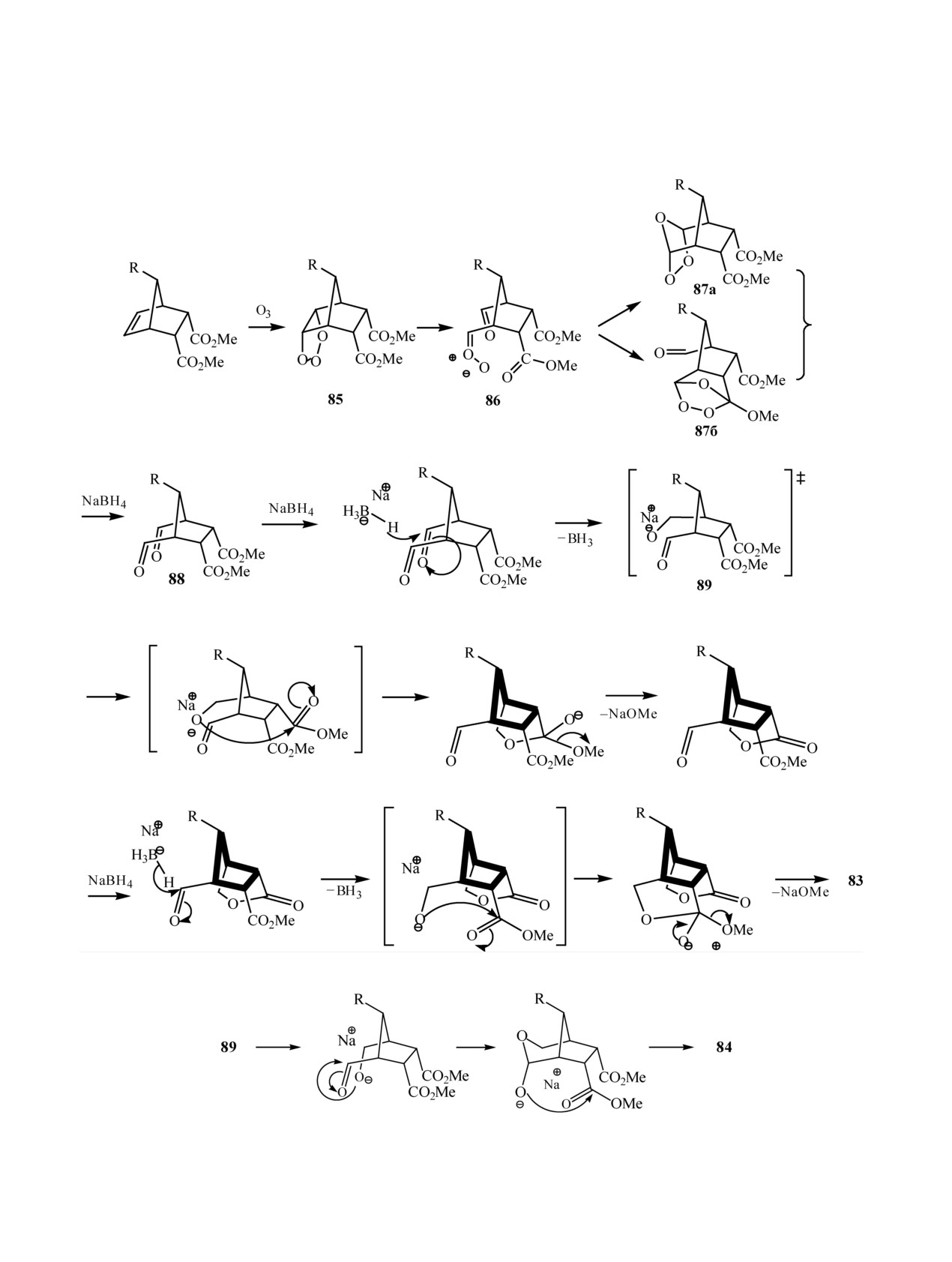
NaBH4 продуктов озонолиза других напряженных
был получен тетразамещенный циклопентанон 92
молекул. В соответствии с механизмом Криге, по-
(схема 27) [61].
сле первоначального присоединения молекулы О3
Описан новый метод синтеза тетрагидрофу-
образуется примозононид 85, который затем пре-
рановых колец, включающий озонолиз диенов,
вращается в альдегидокарбонилоксид 86. Послед-
содержащих свободные гидроксильные группы в
ний подвергается внутримолекулярному 1,3-ди-
γ-положении. При изучении процесса озонолиза
полярному присоединению как по альдегидной,
сопряженных двойных связей в диене 93 неболь-
так и сложноэфирной группам, приводя, соответ-
шим избытком О3 при разных температурах [62]
ственно, к озонидам 87а и 87б, восстановление ко-
показано, что при -70°С основным продуктом
торых NaBH4 дает промежуточный диальдегид 88.
после боргидридного восстановления был диол
Затем боргидрид натрия реагирует с одной из аль-
95- продукт расщепления С12-С13 двойной связи.
дегидных групп с образованием на первой стадии
Озонирование при 0°С привело к соединению 94
натриевого алкоголята 89. Последний может ата-
(схема 28), содержащему на 2 углеродных атома
ковать как сложноэфирную группу, приводя к об-
меньше, чем в исходном диене, из-за разрыва свя-
разованию лактонных фрагментов соединения 83,
зи С13-С14 (схема 29).
так и альдегидную, образуя продукт 84 (схема 26).
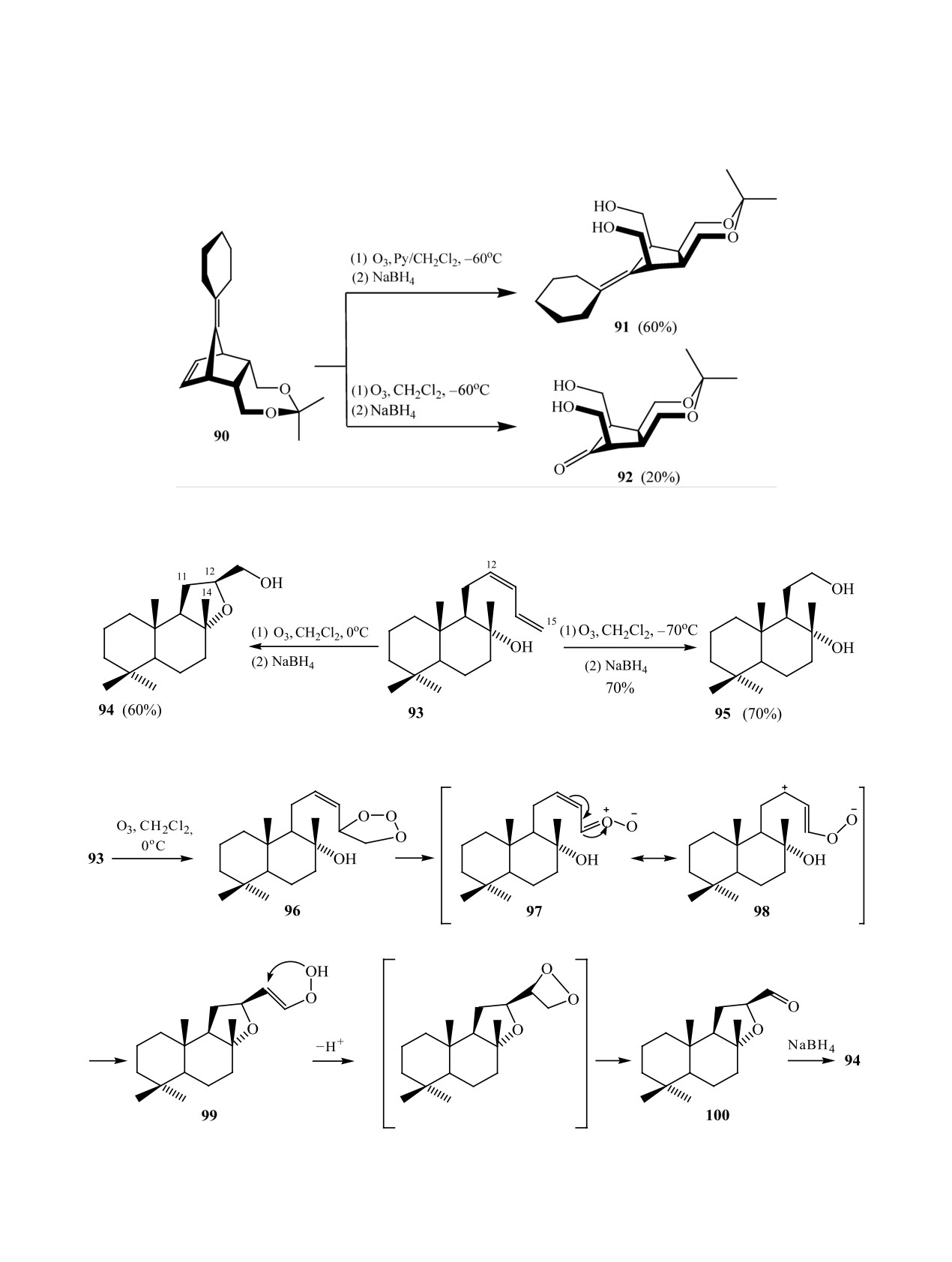
Образование спирта 94 авторы [62] объясня-
Показано, что при исчерпывающем озониро-
ют через карбонилоксид Криге 97, стабилизация
вании диенового ацетонида 90 с неравноценными
которого ведет к перераспределению частичного
двойными связями при -60°С в CH2Cl2 в присут-
положительного заряда на атоме С12 соедине-
ствии пиридина реагировала только двойная связь
ния 98, последующая внутримолекулярная атака
норборненового фрагмента, и после обработки
НО-группы в котором приводит к соответствую-
этанольным раствором NaBH4 был получен диол
щему гетероциклу 99. Поскольку стабильность
91 с выходом 60%. Озонолизом 90 при той же тем-
образующегося винилового гидропероксида край-
пературе без пиридина после обработки NaBH4
не мала, он перегруппировывается в альдегид 100,
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
81
Схема 26.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
82
МЯСОЕДОВА и др.
Схема 27.
Схема 28.
Схема 29.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
83
Схема 30.
Схема 31.
возможно, через напряженный четырехчленный
смесью мелкодисперсного Zn и Me2S за 24 ч коли-
циклический пероксид, который далее восстанав-
чественно без образования побочных продуктов и
ливается NaBH4 в спирт 94 (схема 29).
эпимеризации (схема 31) [65].
Озонолиз эндо-циклических двойных связей в
Применив Zn в условиях реакции Клемменсена
конденсированных системах с последующей обра-
(схема 32), авторами [66] впервые было выполне-
боткой промежуточных пероксидов NaBH4 пред-
но однореакторное восстановительное расщепле-
лагается авторами [63] как один из методов синте-
ние экзо-олефинов последовательными реакция-
за среднециклических лактонов. Так, из триметил-
ми озонолиза и восстановления по Клемменсену,
кеталя 101 получен стабильный замещенный дека-
результаты которого приведены в таблице 6. На-
нолид 102 в виде смеси (2 : 1) двух диастеромеров
пример, в случае алкена 106 был достигнут выход
(схема 30).
72%, в отличие от ранее описанного 3-хстадийно-
Из неорганических восстановителей перекис-
го синтеза, приводящего к целевому лактаму 107 с
ных продуктов озонолиза олефинов до карбо-
общим выходом 49% [67].
нильных соединений часто применяют цинковую
Для демонстрации эффективности предложен-
пыль в уксусной кислоте [2, 3, 64], но иногда пе-
ного метода авторами [68] было показано, что
роксиды оказываются достаточно устойчивыми к
полученный из алкена 106 стандартным спосо-
ее действию. Так, восстановительная обработка
бом с использованием Me2S кетон 108 достаточно
промежуточного озонида 104 (продукта озонолиза
сложно дезоксигенируется (схема 33): обработка
бис-диоксолана 103), приводящая к альдегиду 105,
такими системами как тиоацеталь - никель Ренея,
оказалась затруднена. При использовании стан-
NH2NHTs - NaBH4, NH2NHTs - NaBH3CN снижа-
дартных методик восстановления (Zn, PPh3, Me2S)
целевой альдегид не был получен с удовлетво-
ет выходы или разрушает продукты (табл. 7). В
рительными выходами, кроме того, часто наблю-
связи с этим, разработанный однореакторный ме-
далась эпимеризация атома углерода при ацета-
тод “озонолиз - восстановление по Клеменсену”
мидной группе. Тем не менее, условия обработки
является более эффективным для применения в
были оптимизированы: озонид 104 восстановили
направленном синтезе природных соединений.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
84
МЯСОЕДОВА и др.
Схема 32.
При действии солей металлов пероксиды мо-
то-заместителя, по-видимому, направляет разло-
гут быть фрагментированы по С-С связи: при-
жение промежуточных соединений с образовани-
менялись обработка гексагидратом хлорида же-
ем α,β-ненасыщенного кетона 111.
леза, нитратом железа, нагревание в присутствии
При изучении разложения перекисных продук-
Cu(OAc)2·H2O, смесью солей железа и меди [2, 3].
тов озонолиза (+)-β- 112 и (+)-α- 116 пиненов в ме-
Так, (R)-(+)-6-метилциклогекс-2-ен-1-он 111 был
таноле солями Fe(III) было установлено, что при
получен озонолизом
(2R,5R)-(+)-транс-дигидро-
обработке FeCl3·6H2O как при кипячении, так и
карвона 109 в метаноле при -30°С, приводящим
при комнатной температуре из β-пинена 112 полу-
к метоксигидропероксиду 110, с последующим
чается нопинон 113, но в смеси с пара-изопропил-
восстановлением при комнатной температуре сме-
фенолом 114 либо хлоркетоном 115. При исполь-
сью моногидрата ацетата меди (II) и гептагидрата
зовании Fe(NO3)3·9H2O терпен 112 количественно
сульфата железа (II) [69] (схема 34). Наличие ке-
превращается в нопинон 113 (схема 35) [70].
Таблица 6
№
Алкен
Продукт
Растворитель, время
Выход %
1
i-PrOH/CH2Cl2, 0.5 ч
87
2
MeOH/ CH2Cl2,
75
1 ч
3
i-PrOH/CH2Cl2, 0.75 ч
72
4
MeOH/ CH2Cl2, 1 ч
86
Таблица 7
№
Условия
Выход 107a, %
1
(1) HSCH2CH2SH, TiCl4, -15°С; (2) Ni Ренея, EtOH, Δ
35
2
(1) NH2NHTs, MeOH, Δ; 2. NaBH4, MeOH, Δ
Разложение продукта
3
(1) NH2NHTs, MeOH, Δ; 2. NaBH3CN, TsOH, THF, Δ
60
4
Активированный Zn, HCl, Et2O, 0°С
80, низкая воспроизводимость
5
Zn, TMSCl-H2O (5 : 2), THF, 5°С
86
a Общий выход выделенного продукта.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
85
Схема 33.
Схема 34.
Схема 35.
Схема 36.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
86
МЯСОЕДОВА и др.
Схема 37.
Разложение перекисных продуктов озонолиза
тиловому эфиру 20-гидрокси-25,26,27-тринорэк-
(+)-α-пинена 116 под действием FeCl3·6H2O при
дизон-23-карбоновой кислоты 122 (схема 38).
комнатной температуре либо кипячении приводит
Авторами [73] исследованы озонолитические
к смеси ацеталя 117 и сложного эфира 118 в соот-
превращения (R)-4-ментен-3-она 123 в CH2Cl2 или
ношении 5 : 3 (схема 36) [70].
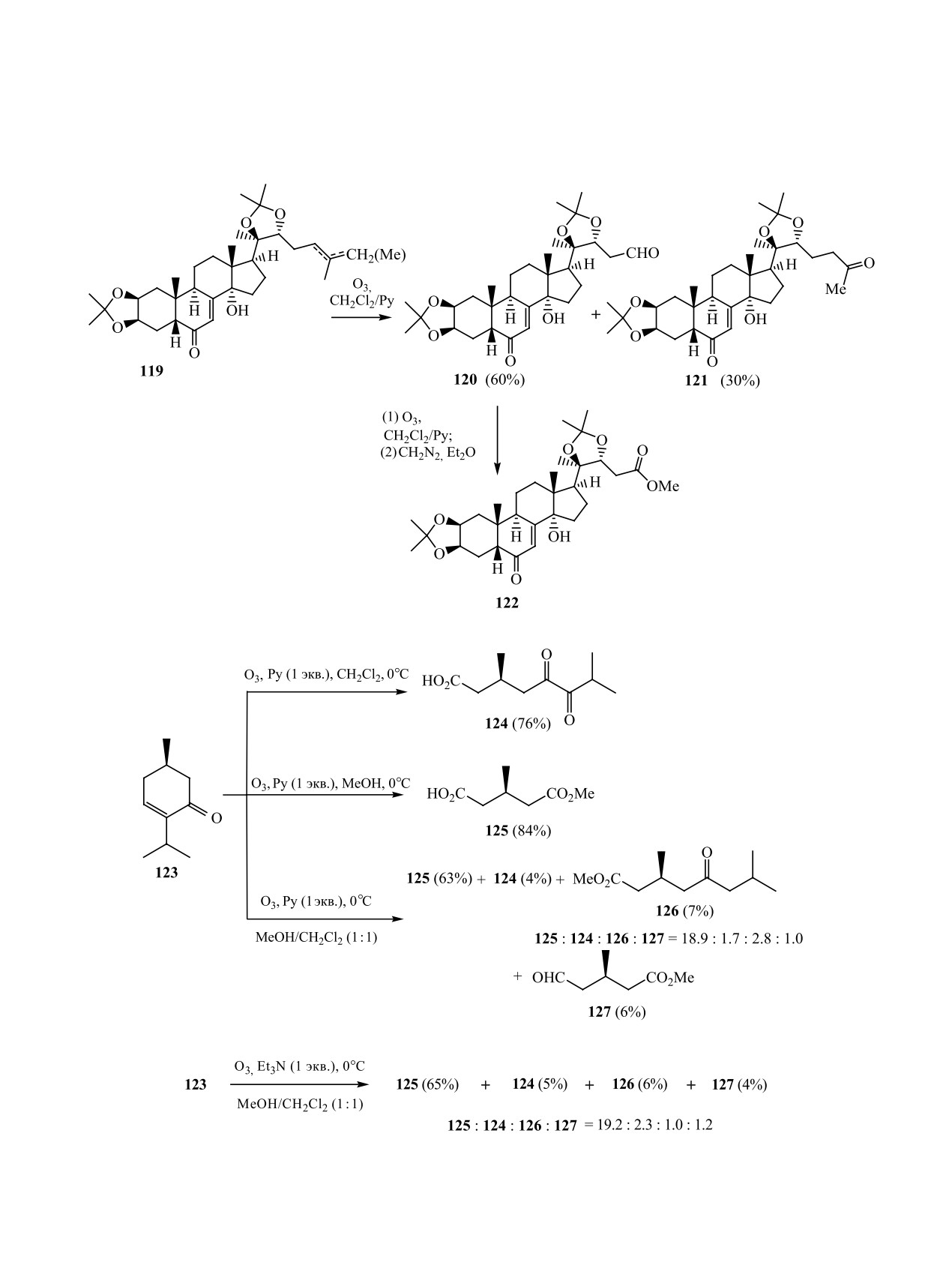
MeOH и их смеси в присутствии Py и Et3N. Пока-
В 2012 г. авторы [71] предложили использова-
зано, что озонолиз енона 123 в CH2Cl2 с добавкой
ние нового реагента - дитионита натрия, позволя-
Py приводит с выходом 76% к дикетокислоте 124.
ющего быстро превращать алкены в соответству-
При замене CH2Cl2 на MeOH и сохранении осталь-
ющие функционализированные производные с
ных параметров с выходом 84% получен мономе-
высокими выходами и без необходимости допол-
тиловый эфир (3R)-метилглутаровой кислоты 125.
нительной хроматографической очистки. Реакция
Применение в качестве растворителя в реакции
проходит за 60-260 мин при использовании от 1 до
озонолиза смеси (1 : 1) CH2Cl2 и MeOH снизило
1.8 экв. Na2S2O4 (схема 37).
выход эфирокислоты 125 до 63%. При этом в каче-
стве минорных продуктов были зарегистрированы
N-СОДЕРЖАЩИЕ СОЕДИНЕНИЯ
дикетокислота 124, кетоэфир 126 и альдегидоэфир
В ПРЕВРАЩЕНИЯХ ПЕРЕКИСНЫХ
127 (схема 39).
ПРОДУКТОВ ОЗОНОЛИЗА
Аналогичные результаты были получены при
замене Py на Et3N (схема 40) [73].
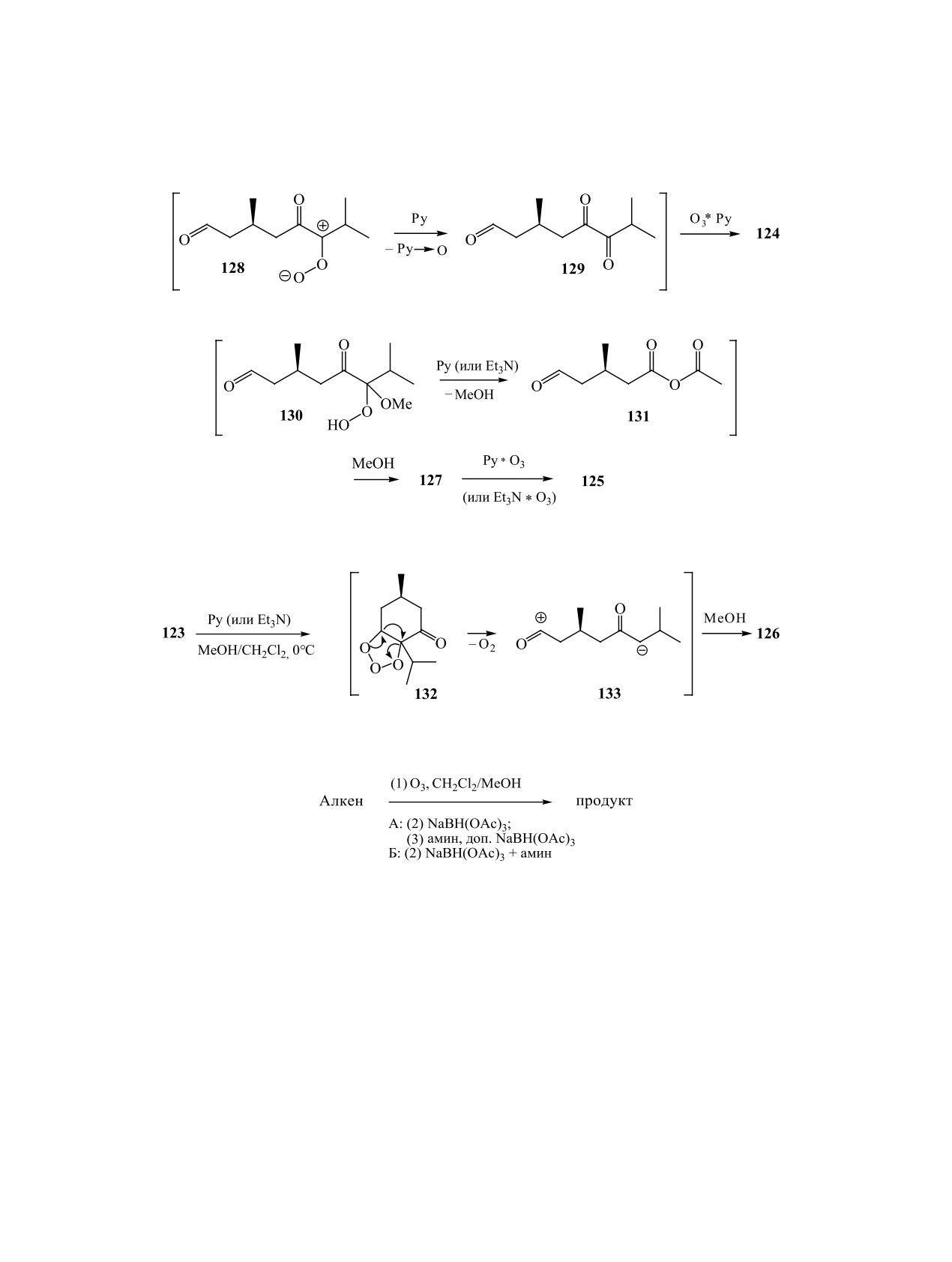
Применение азотсодержащих органических со-
Образование дикетокислоты 124 из пероксида
единений в превращениях пероксидных продуктов
128 авторы [73] объясняют следующими вероят-
озонолиза представлено тетрацианэтиленом, пи-
ными превращениями: на первой стадии пиридин
ридином, аммиаком, третичными аминами, ами-
выступает как восстановитель карбонилоксида
но-N-оксидами, производными гидроксиламина и
128 до альдегидодикетона 129, на второй - обра-
гидразина [2].
зует комплекс с озоном. Этот комплекс, в свою
Карбонильные соединения получаются в одну
очередь, является известным эффективным окис-
стадию при озонировании в присутствии соедине-
лителем альдегидов до соответствующих кислот
ний - акцепторов пероксидного кислорода. Прак-
(схема 41) [74].
тический интерес представляет «неперекисный»
Альдегидоэфир 127 и монометиловый эфир
восстановительный озонолиз, осуществляемый в
(3R)-метилглутаровой кислоты 125 образуются по
присутствии пиридина либо третичных алифати-
вероятной схеме 42, при этом на первой стадии пи-
ческих аминов. Так, озонолиз смеси (2 : 1) ∆24- и
∆25- изомеров 119 в CH2Cl2 в присутствии Py при-
ридин (или триэтиламин), вероятно, катализируют
водит к альдегиду 120 и кетону 121, которые были
превращение гидроперекиси 130 в ангидрид 131.
выделены после колоночной хроматографии на
Образование кетоэфира
126 обусловлено,
силикагеле с выходами 60 и 30%, соответственно
по-видимому, возможностью метанолиза проме-
[72]. Окисление озоном полученного альдегида
жуточного ацилкатиона 133 - продукта перегруп-
120 в Ру с последующей обработкой реакционной
пировки стерически напряженного молозонида
смеси раствором диазометана в Et2O привело к ме-
132 (схема 43).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
87
Схема 38.
Схема 39.
Схема 40.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
88
МЯСОЕДОВА и др.
Схема 41.
Схема 42.
Схема 43.
Схема 44.
Авторами [75] предлагается однореакторный
(схема 45). Озонолиз 134 с последующей обра-
метод превращения алкенов в амины последова-
боткой образующегося гидропероксиацеталя фе-
нилгидразином и триацетоксиборгидридом на-
тельными реакциями озонолиза и восстановитель-
трия приводят к фенилгидразону 135, добавление
ного аминирования (схема 44). Озонирование ал-
NaBH3CN к которому дает фенилгидразид 136,
кенов осуществляют в смеси МеОН-CH2Cl2, далее
быстро окисляющийся на воздухе до диазена 137
реакционную массу обрабатывают последователь-
с выходом 57%.
но триацетоксиборгидридом натрия (3 экв) и ами-
Озонолизом
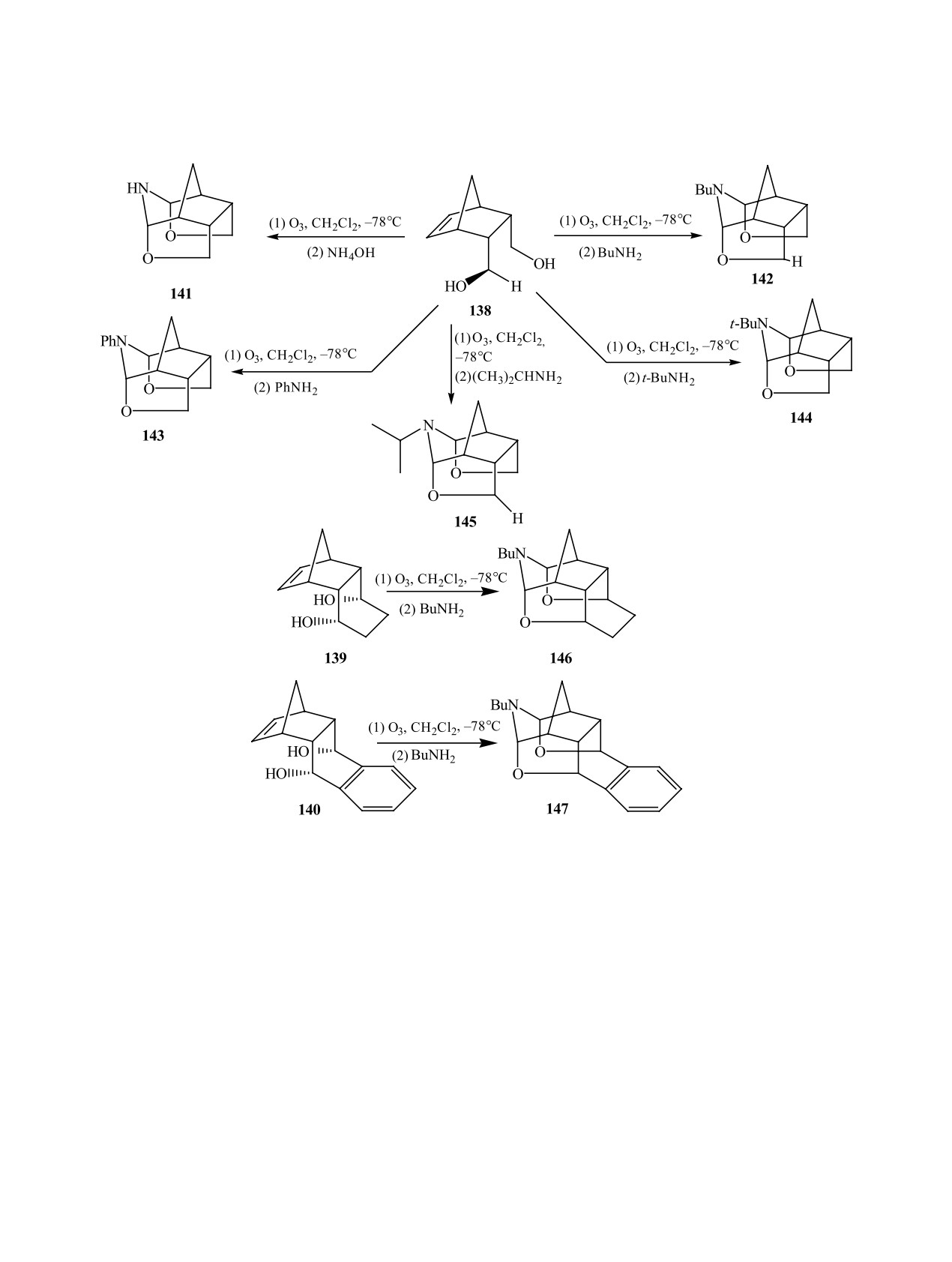
2,3-бис-эндо-диолов
138-140 в
ном (1-2 экв) (метод А) либо смесью NaBH(OAc)3
CH2Cl2 при -78°С с последующей добавкой ами-
и амина (метод Б) (табл. 8).
нов cинтезированы ацетальные производные -
Подобную процедуру авторы [75] применили
аза-клетки 141-147 с хорошими выходами (60-
для синтеза гидразонов, гидразинов и диазенов
75%) (схема 46) [76].
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
89
Схема 45.
Механизм реакции включает 1,3-диполярное
лишь несколькими примерами, причем все реак-
циклоприсоединение молекулы озона к двойной
ции были выполнены в MeOH [2]. Впоследствии
связи в 138, приводящее к примозониду 148. Раз-
было исследовано применение гидрохлорида ги-
рушение 1,2,3-триоксолана 148 дает карбонилок-
дроксиламина как восстановителя пероксидных
сид 149, который в результате внутримолекуляр-
продуктов озонолиза алкенов различного стро-
ного нуклеофильного присоединения НО-группы
ения и происхождения в изопропиловом спирте
к карбонилоксидной и альдегидной превращает-
[77], смеси АсОН-CH2Cl2 [78], тетрагидрофуране
ся в лактологидропероксид 150. Протонирование
[79]. Кроме того изучалось влияние добавки воды
НО- либо НОО-групп соединения 150 с последую-
в качестве сорастворителя [80, 81]. При этом были
щим отщеплением Н2О или Н2О2 приводит к оксо-
получены функционализированные производные,
ниевым ионам 151а и 151б. Нуклеофильное при-
содержащие сложноэфирную, нитрильную и ке-
соединение молекулы амина к оксониевым ионам
то-группы. Причем нитрильные производные 154
151а, 151б с последующей дегидратацией ведет к
и 156 впервые были зафиксированы при озонолизе
аза-клеткам 141-145, вероятно, через интермедиа-
циклооктена 153 и касторового масла 155 в мета-
ты 152а, 152б и 152в, 152г (схема 47).
ноле (схема 48) [82].
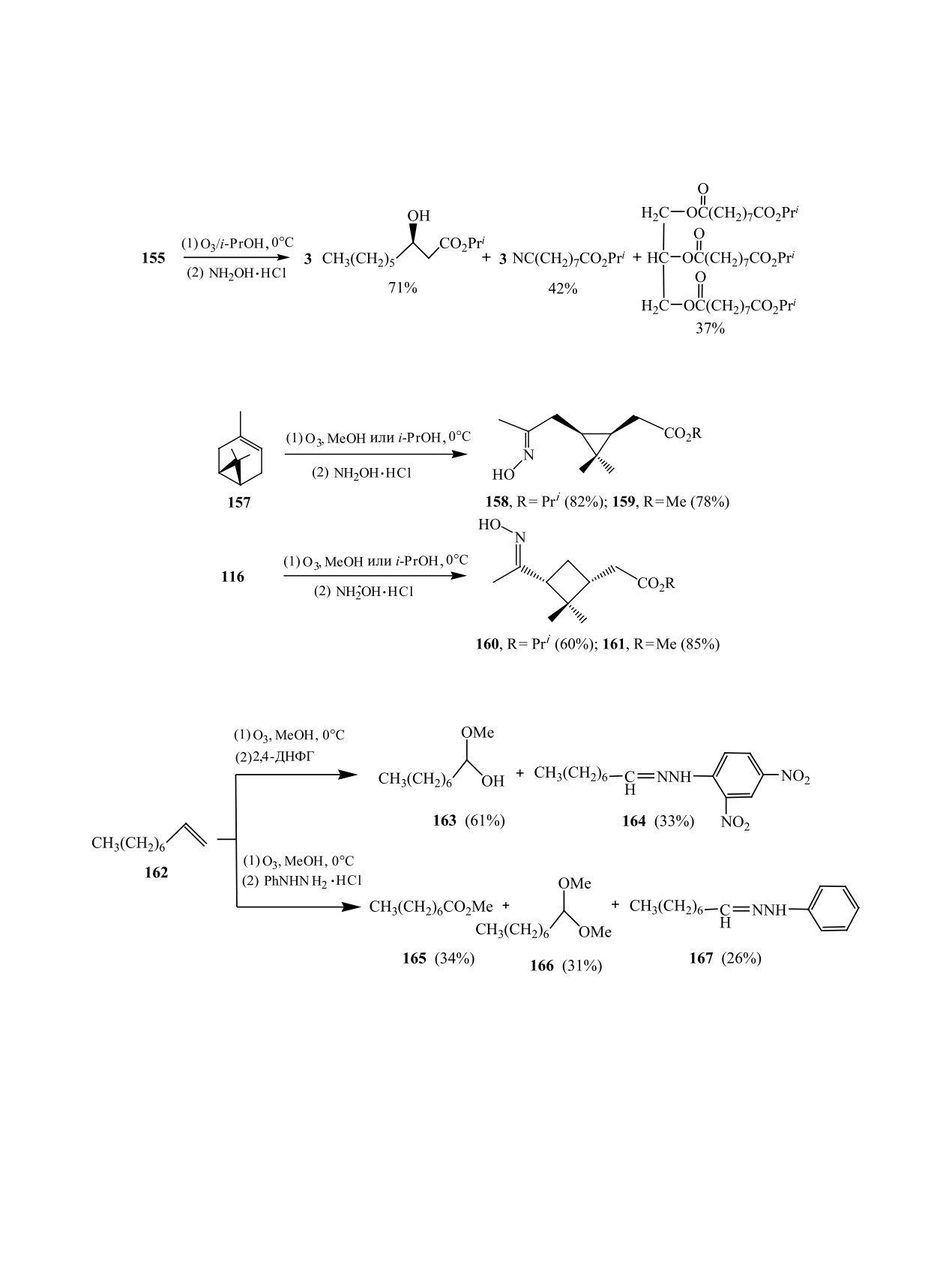
Использование солянокислого гидроксиламина
При исследовании гидрохлорида гидроксила-
для превращения перекисных продуктов озоно-
мина как восстановителя перекисных продуктов
лиза олефинов первоначально было ограничено
озонолиза олефинов в изопропаноле были отме-
Таблица 8
Алкен
Реагент
Метод (выход, %)
Продукт
O
BnO(H2C)3
морфолин
A (64)
H
N
BnO(H2C)3
H
O
AcO(H2C)8
морфолин
B (66)
H
N
AcO(H2C)8
H
Ph
A (72)
N
Ph(CH2)2NH2
B (62)
A (65)
Ph(CH2)2NH2
B (65)
Ph
N
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
90
МЯСОЕДОВА и др.
Схема 46.
чены пониженные скорости превращений альде-
силамина в метаноле [83] или изопропаноле [77],
гид → альдоксим → нитрил → сложный эфир и
соответственно (схема 50) .
переэтерификации триглицеридной группы касто-
Авторами [84] представлено применение арил-
рового масла 155 в сравнении с таковыми в мета-
производных гидразина в качестве восстановите-
ноле (схема 49) [77].
лей пероксидных продуктов озонолиза алкенов.
Предложен препаративный однореакторный
Так, 2,4-динитрофенилгидразин, а также соляно-
метод получения хиральных циклопропан-(ци-
кислый фенилгидразин оказались эффективными,
клобутан)-содержащих строительных блоков для
но неселективными реагентами. Озонолиз 1-ноне-
природных биологически активных веществ: ме-
на 162 и последующая обработка 2,4-динитрофе-
тиловых 159, 161 и изопропиловых 158, 160 ке-
нилгидразином привели к смеси полуацеталя 163
токсимоэфиров с транс-конфигурацией двойных
и 2,4-динитрофенилгидразона 164. При использо-
связей оксимных групп обработкой пероксидов из
вании солянокислого фенилгидразина для превра-
монотерпенов 116 и 157 гидрохлоридом гидрок-
щения перекисных продуктов озонолиза алкена
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
91
Схема 47.
Схема 48.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
92
МЯСОЕДОВА и др.
Схема 49.
Схема 50.
Схема 51.
162 получена смесь эфира 165, диметилацеталя
последующую обработку in situ образующихся пе-
166 и фенилгидразона 167 (схема 51).
роксидных продуктов смесью (1 : 2) солянокислого
В работе [85] предложен однореакторный озоно-
фенилгидразина с ацетатом натрия (схема 52).
литический метод получения моно- и дифенилги-
Предложенная методика была успешно при-
дразонов общей формулы 168 из алкенов, предпо-
менена в синтезе фенилгидразонов из линейных,
лагающий озонолиз олефина в метаноле при 0°С и
моно- и бициклических алкенов (табл. 9).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
93
Схема 52.
Схема 53.
Схема 54.
Показано, что тозилгидразид также являет-
ходами от 40 до 86%. Следует отметить, что при
ся эффективным восстановителем пероксидных
проведении реакции в смеси AcOH-CH2Cl2 наря-
продуктов озонолиза олефинов в протонных рас-
ду с тозилгидразонами образуются, в зависимо-
творителях, что легло в основу однореакторного
сти от субстрата, соответствующие карбоновые
озонолитического метода синтеза моно- [86] и
кислоты.
дитозилгидразонов [87, 88]. Общая схема синте-
На примере монотерпеновых субстратов 116 и
за включает в себя озонолиз алкенов при 0°С в
157 показана возможность получения циклопро-
протонодонорных растворителях (MeOH, i-PrOH,
пан- и циклобутан-содержащих кетокарбоксиль-
AcOH-CH2Cl2) и восстановление образующихся
ных производных: метиловых 118, 171 и изопро-
пероксидов тозилгидразидом (схема 53). В зави-
пиловых эфиров 169, 172, карбоновых кислот 170,
симости от исходного алкена и используемого
173 при проведении озонолиза и последующей об-
растворителя тозилгидразоны получаются с вы-
работке солянокислым семикарбазидом в MeOH
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
94
МЯСОЕДОВА и др.
Таблица 9
№
Исходный алкен
Продукт
Выход, %
1a
80
2a
76
3a
74
4a
73
5b
72
6b
70
7b
51
Условия: 1. O3, MeOH, 0ºС; 2. NH2NHPh-HCl/AcONa (1 : 2), 20ºC;
a 2 экв. cмеси (1 : 2) NH2NHPh-HСl/AcONa по отношению к исходному алкену;
b 3.5 экв. cмеси (1 : 2) NH2NHPh-HСl/AcONa по отношению к исходному алкену.
[83], PriOH [77] и смеси AcOH-CH2Cl2 [78], соот-
ных хрущаков, красной калифорнийской щитовки
ветственно (схема 54).
и т.д.).
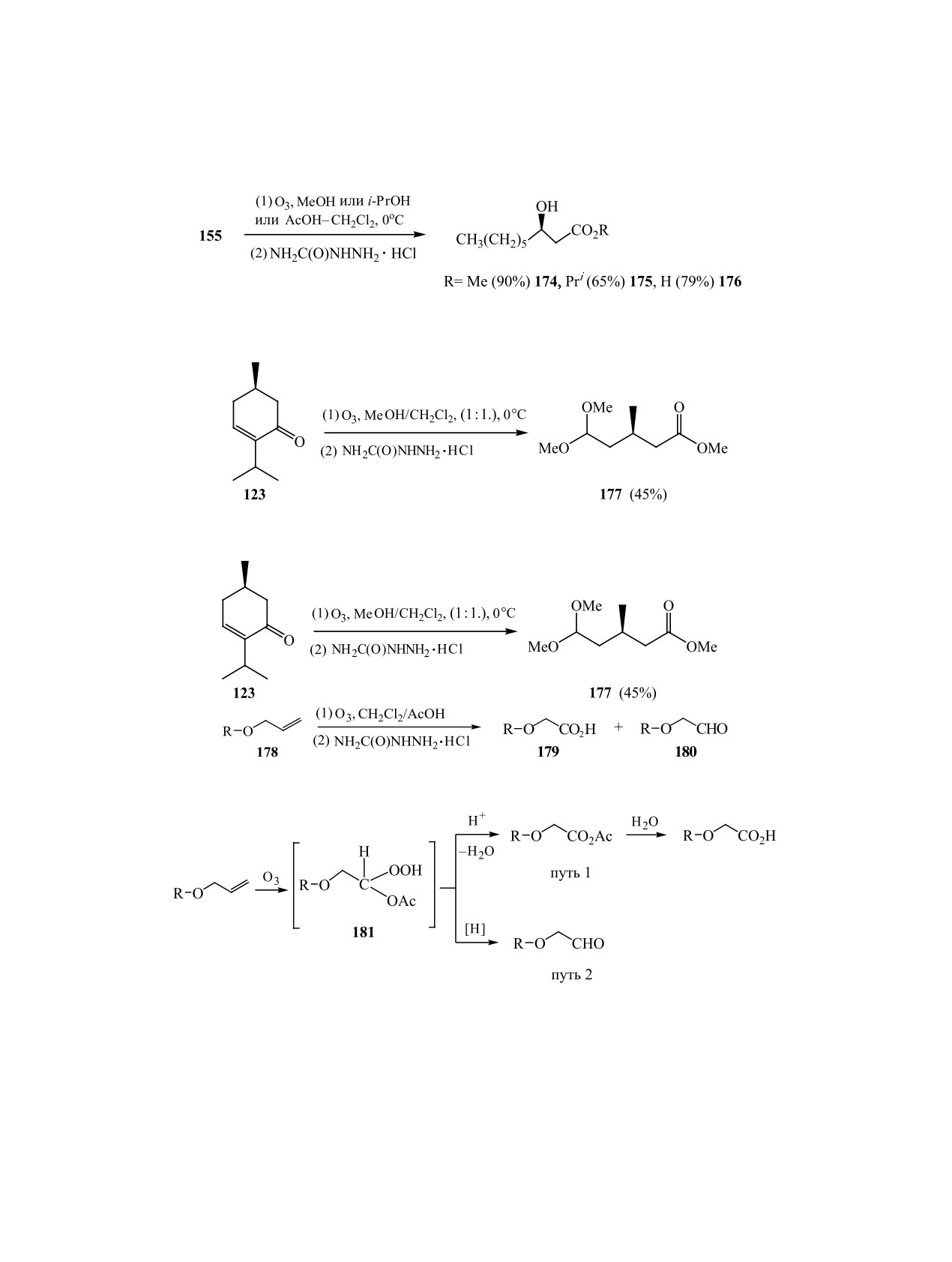
Разработаны эффективные синтезы биологи-
Низкотемпературный озонолиз простых или
чески активной (3R)-гидроксинонановой кисло-
сложных аллиловых эфиров 178 в среде АсОН/
ты 176 и ее сложноэфирных производных 174 и
CH2Cl2 с последующей обработкой гидрохлори-
175 на основе хемоселективных озонолитических
дом семикарбазида позволяет получать алкоксиук-
превращений касторового масла 155 в спиртовых
сусные кислоты с хорошими выходами без выде-
ления промежуточных пероксидов (схема 57) [90].
растворителях и уксусной кислоте с применением
Отмечается, что селективность восстановления
на стадии восстановления пероксидных продуктов
образующегося на первой стадии ацетоксигидро-
солянокислого семикарбазида (схема 55) [89].
пероксида зависит как от температуры осущест-
Использование солянокислого семикарбазида
вления процесса, так и природы субстрата. Пони-
для превращения перекисного продукта озоноли-
жение температуры благоприятствует протеканию
за ментенона 123 в смеси (1 : 1) CH2Cl2-MeOH
кислотного гидролиза и образованию карбоновой
дало с высоким выходом ацеталеэфир 177 (схема
кислоты. При более высоких температурах (-45°С
56) [77] - ключевой синтон в синтезе целого ряда
или -20°С) озонолиз эфиров 178 протекает менее
низкомолекулярных биорегуляторов насекомых
селективно: помимо кислот 179 были выделены
(половых феромонов малого и большого муч-
соответствующие альдегиды 180.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
95
Схема 55.
Схема 56.
Схема 57.
Схема 58.
Наблюдаемое снижение селективности авторы
вой кислоты в результате кислотного гидролиза
связывают с механизмом восстановления перокси-
(путь 1), либо восстановление при действии соля-
дов. При озонировании олефинов в системе АсОН/
нокислого семикарбазида до соответствующего
CH2Cl2 промежуточным соединением, чаще всего,
альдегида (путь 2) (схема 58). При -70°С, вероятно,
является ацетоксигидропероксид 181 [91], для ко-
преимущественно протекает кислотный гидролиз, а
торого возможны либо расщепление до карбоно-
при более высокой температуре (-45°С или -20°С)
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
96
МЯСОЕДОВА и др.
процессы расщепления и восстановления проходят
7.
Ишмуратов Г.Ю., Легостаева Ю.В., Боцман Л.П.,
одновременно, что приводит к смеси продуктов.
Насибуллина Г.В., Муслухов Р.Р., Казаков Д.В.,
Толстиков Г.А. ЖОрХ. 2012, 48 (1), 26. [Ishmuratov G.
Преобладание альдегида либо карбоновой кислоты
Yu., Legostaeva Yu.V., Botsman L.P., Nasibullina G.V.,
связано, видимо, со строением субстратов.
Muslukhov R.R., Kazakov D.V., Tolstikov G.A. Russ.
Таким образом, озонолиз является современ-
J. Org. Chem. 2012. 48 (1), 18.]
ным и перспективным методом функционализа-
8.
Ишмуратов Г.Ю., Легостаева Ю.В., Гарифул-
ции олефинов различной природы, причем его
лина Л.Р., Боцман Л.П., Муслухов Р.Р., Толсти-
синтетический потенциал неисчерпаем, особенно
ков Г.А. ЖОрХ. 2014, 50 (12), 1765. [Ishmuratov
на стадии превращения промежуточных перокси-
G.Y., Legostaeva Y.V., Garifullina L.R., Botsman L.P.,
Muslukhov R.R., Tolstikov G.A. Russ. J. Org. Chem.
дов.
2014. 50 (12), 1746.]
9.
Ишмуратов Г.Ю., Легостаева Ю.В., Гарифулли-
ФОНДОВАЯ ПОДДЕРЖКА
на Л.Р., Муслухов Р.Р., Боцман Л.П., Козлова Г.Г.
ХПС. 2015, 1, 71. [Ishmuratov G.Y., Legostaeva Y.V.,
Работа выполнена при финансовой поддерж-
Garifullina L.R., Muslukhov R.R., Botsman L.P.,
ке программы РАН “Фундаментальные основы
Kozlova G.G. Chem. Nat. Compd. 2015, 51 (1), 71.]
химии”, тема №8 “Хемо-, регио- и стереоселек-
10.
Раскильдина Г.З., Легостаева Ю.В., Гарифулли-
тивные превращения терпеноидов, стероидов и
на Л.Р., Султанова Р.М., Ишмуратов Г.Ю., Злот-
липидов в направленном синтезе низкомолекуляр-
ский С.С. Докл. АН. 2015, 462 (3), 307. [Raskil’di-
ных биорегуляторов” (№ госрегистрации ААА-
na G.Z., Legostaeva Yu.V., Garifullina L.R.,
А-А17-117011910023-2, 2017 г.).
Sultanova R.M., Ishmuratov G.Yu., Zlotskii S.S.
Doklady Chemistry. 2015. 462 (1), 127.]
КОНФЛИКТ ИНТЕРЕСОВ
11.
Yadav J.S., Krishna V.H., Srilatha A., Somaiah R.,
Subba Reddy B.V. Synthesis. 2010, 17, 3004.
Авторы заявляют об отсутствии конфликта ин-
12.
Fisher T.J., Dussault P.H. Tetrahedron Lett. 2010, 51,
тересов.
5615.
13.
Amarante G.W. Synlett. 2009, 1, 155.
СПИСОК ЛИТЕРАТУРЫ
14.
Cheng P., Shao W., Clive D.L.J. J. Org. Chem. 2013, 78
(23), 11860.
1. Зверева Т.И., Куковинец О.С., Касрадзе В.Г.,
15.
Schneider M.-A., Dötterl S., Siefert K. Chemistry and
Казакова О.Б.
ЖОрХ.
2010,
46
(10),
1431.
Biodiversity. 2013, 10, 1252.
[Zvereva T.I., Kukovinets O.S., Kasradze V.G.,
16.
Hoegenauer E.K., Thomas E.J. Org. Biomol. Chem.
Kazakova O.B. Russ. J. Org. Chem. 2010, 46 (10),
2012, 10, 6995.
1431.]
17.
Pearson A.J., Kim E.H., Sun H. Tetrahedron. 2010, 66,
2. Ишмуратов Г.Ю., Легостаева Ю.В., Боцман Л.П.,
4943.
Толстиков Г.А. ЖОрХ.
2010,
46
(11),
1591.
18.
Kulkarni M.G., Dhondge A.P., Borhade A.S.,
[Ishmuratov G.Yu., Legostaeva Yu.V., Botsman L.P.,
Gaikwad D.D., Chavhan S.W., Shaikh Y.B.,
Tolstikov G.A. Russ. J. Org. Chem. 2010. 46 (11),
Nigdale V.B., Desai M.P., Birhade D.R., Shinde M.P.
1593.]
Eur. J. Org. Chem. 2009, 3875.
3. Van Ornum S.G., Champeau R.M., Pariza R. Chem.
19.
Tian W.-S., Dung K., Sun Y.-S., Chen L., Lei Z.
Rev. 2006, 106, 2990.
Synthesis. 2013, 45, 438.
4. Kukovinets O.S., Zvereva T.I., Kabalnova N.N.,
20.
Yadav J.S., Krishana G.C., Kumar S.N. Tetrahedron.
Kasradze V.G., Salimova E.V., Khalitova L.R.,
2010, 66, 480.
Abdullin M.I., Spirikhin L.V. Mendeleev Commun.
2009, 19, 106.
21.
Perlikowska W., Mikołajczyk M. Tetrahedron: Asym.
5. Schiaffo C.E., Dussault P.H. J. Org. Chem. 2008, 73,
2011, 22, 1767.
4688.
22.
Strikrishna A., Gowri V. Synlett. 2011, 18, 2652.
6. Suwancharoen S., Pornpakakul S., Muangsin N.
23.
Chavan S.P., Lasonkar P.B., Chavan P.N. Tetrahedron:
Tetrahedron Lett. 2012, 53, 5418.
Asym. 2013, 24, 1473.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
97
24.
Ren L., Piers E. Tetrahedron Lett. 2012, 53, 3329.
Chattopadhayay S. J. Org. Chem. 2013, 78 (15), 7406.
25.
Giesbrecht H.E., Knight B.J., Tanguileg N.R., Emerson
49.
Laventin D.M., Davies M., Evinson E.L., Jenkins P.R.,
C.R., Blakemore P.R. Synlett. 2010, 3, 0374.
Cullins P.M., García M.D. Tetrahedron. 2009, 65, 4766.
26.
Ishmuratov G.Yu., Yakovleva M.P., Shutova M.A.,
50.
Lewis K., Vivier L., Clacens J.-M., Brandhorst M.,.
Muslukhov R.R., Ishmuratova N.M., Tolstikov G.A.
Dubois J.-L, De Oliveira Vigier K., Pouilloux Y. Green
Chem. Nat. Compd. 2014, 50 (4), 658.
Chem. 2014, 16, 96.
27.
Paioti P.H.S., Rezende P., Coelho F. J. Braz. Chem.
51.
Kersten L., Hilt G. Adv. Sinth. Catal. 2012, 354, 863.
Soc. 2012, 23 (2), 285.
52.
Edvinsson S., Johansson S., Larsson A. Tetrahedron
28.
Hilt G., Arndt M., Weske D.F. Synthesis. 2010, 8, 1321.
Lett. 2012, 53, 6819.
29.
Hilt G. Synlett. 2011, 12, 1654.
53.
Smits G., Zemribo R. Org. Lett. 2013, 15 (17), 4406.
54.
Debnar T., Wang T., Menche D. Org. Lett. 2013, 15
30.
Heo G.S., Cho S., Wooley K.L. Polym. Chem. 2014, 5,
(11), 2774.
3555.
55.
Luna-Freire K.R., Scaramal J.P.S., Resende J.A.L.C.,
31.
Reynaud S., Giorgi M., Doucet H., Santelli M.
Tormena C.F., Oliveira F.L., Aparicio R., Coelho F.
Tetrahedron. 2010, 66, 4101.
Tetrahedron. 2014, 70, 3319.
32.
Reynaud S., Giorgi M., Doucet H., Santelli M.
56.
Guduguntla S., Faňanás-Mastal M., Feringa B.L.
Tetrahedron Lett. 2009, 50, 3385.
J. Org. Chem. 2013, 78 (17), 8274.
33.
Zhylitskaya H., Litvinovskaya R., Drach S., Khripach V.
57.
Iskakova M.M., Biktagirov I.M., Faizullina L.Kh.,
Tetrahedron Lett. 2011, 52, 5267.
Salikhov Sh.M., Safarov M.G., Valeev F.A. Russ.
34.
Budaev A.S., Mikhailova R.L., Spirikhin L.V.,
J. Org. Chem. 2014, 50 (1), 105.
Baltina L.A. Chem. Nat. Compd. 2016, 52 (3), 448.
58.
Lentsch C., Fürst R., Muzler J., Rinner U. Eur. J. Org.
35.
Budaev A.S., Michailova L.R., Spirikhin L.V.,
Chem. 2014, 919.
Baltina L.A. Chem. Nat. Compd. 2014, 50 (2), 302.
59.
Reynaud C., Doucet H., Santelli M. Synthesis. 2010,
36.
Laventin D.M., Davies M., Evinson E.L., Jenkins P.R.,
11, 1787.
Cullins P.M., García M.D. Tetrahedron. 2009, 65, 4766.
60.
Pujol A.R., Ratel-Ramond N., Gourdon A. Tetrahedron.
37.
Dalby S.M., Goodwin-Tindall J., Paterson I. Angew.
2013, 69, 9139.
Chem. Int. Ed. 2013, 52, 6517.
61.
Reunaud C., Fall Y., Feuersten M., Doucet H., Santelli
38.
Ho S., Bucher C., Leighton J.L. Angew. Chem. Int. Ed.
M. Tetrahedron. 2009, 65, 7440.
2013, 52, 6757.
62.
Kulciţki V., Bourdelais A., Schuster T., Baden D.
39.
Florence G.J., Morris J.C., Murrey R.G., Vanga R.R.,
Tetrahedron Lett. 2010, 51, 4079.
Osler J.D., Smith T.K. Chem. Eur. J. 2013, 19, 8309.
63.
Халилова Ю.А., Спирихин Л.В., Салихов Ш.М., Ва-
40.
Aldrich L.N., Berry C.B., Bates B.S., Koncol L.C., So
леев Ф.А. ЖОрХ. 2014, 50 (1), 125. [Khalilova Yu.A.,
M., Lindsley C.W. Eur. J. Org. Chem. 2013, 4215.
Spirikhin L.V., Salikhov Sh.M., Valeev F.A. Russ. J.
41.
Zhan W., Jiang Y., Sharma S., Brodie P.J., Bane S.,
Org. Chem. 2014. 50 (1), 117.]
Kingston D.G.I., Liotta D.C., Snyder J.P. Chem. Eur. J.
2011, 17, 14792.
64.
Molander G.A., Cooper D.J. J. Org. Chem. 2007, 72,
3558.
42.
Holmes M.T., Britton R. Chem. Eur. J. 2013, 19, 12649.
65.
Stallforth P., Matthies S., Adibekain A., Gilligham D.G.,
43.
Miege F., Meyer C., Cossy J. Chem. Eur. J. 2012, 18,
Hilvert D., Seeberger P.H. Chem. Commun. 2012, 48,
7810.
11987.
44.
Tietze L.F., Biller S., Wolfram T. Synlett. 2010, 14,
2130.
66.
Xu S., Toyama T., Nakamura J., Arimito H. Tetrahedron
Lett. 2010, 51, 4534.
45.
Bielitza M., Pietruszka J. Chem. Eur. J. 2013, 19, 8300.
46.
Kosaki Y., Ogawa N., Kobayashi Y. Tetrahedron Lett.
67.
Xu S., Arimoto H., Uemura D. Angew. Chem. 2007,
2010, 51, 1856.
119, 5848.
47.
LaPorte M.G., Tsegay S., Hong K. B., Lu C., Fang C.,
68.
Xu S., Unabara D., Uemura D., Arimoto H. Chem.
Wang L., Xie X.-Q., Floreancig P.E. ACS Comb. Sci.
Asian. J. 2014, 9, 367.
2013, 15, 344.
69.
White J.D., Grether U.M., Lee C.-S. Org. Synthesis.
48.
Kumar K.S.A., Rathee J.S., Subramanian M.,
2005, 82, 108.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
98
МЯСОЕДОВА и др.
70.
Garifullina L.R., Salimova E.V., Kasradze V.G.,
Muslukhov R.R., Tolstikov G.A. Russ. J. Org. Chem.
Spirikhin L.V., Kukovinets O.S. Chem. Nat. Compd.
2013. 49 (10), 1415.]
2012, 48 (5), 791.
81.
Ишмуратов Г.Ю., Легостаева Ю.В., Гарифулли-
71.
Tyagi V., Gupta A.K. Synth. Commun. 2012, 42, 843.
на Л.Р., Боцман Л.П., Муслухов Р.Р., Толстиков Г.А.
Бутлеров. сооб. 2014, 38 (6), 129.
72.
Савченко Р.Г., Уразаева Я.Р., Костылева С.А., Одино-
82.
Ишмуратов Г.Ю., Шаяхметова А.Х., Яковлева М.П.,
ков В.Н. ЖОрХ. 2013, 49 (4), 626. [Savchenko R.G.,
Легостаева Ю.В., Шитикова О.В., Галкин Е.Г., Тол-
Urazaeva Ya.R., Kostyleva S.A., Odinokov V.N. Russ.
стиков Г.А. ЖОрХ. 2007, 43 (8), 1125. [Ishmuratov
J. Org. Chem. 2013, 49 (4), 610.]
G.Yu., Shayakhmetova A.Kh., Yakovleva M.P.,
73.
Ишмуратов Г.Ю., Баннова А.В., Латыпова Э.Р., Тух-
Legostaeva Yu.V., Shitikova O.V., Galkin E.G.,
ватшин В.С., Куковинец О.С., Муслухов Р.Р., Тол-
Tolstikov G.A. Russ. J. Org. Chem. 2007. 43 (8), 1114.]
стиков Г.А. ЖОрХ. 2013, 49 (1), 52. [Ishmuratov G.
83.
Ишмуратов Г.Ю., Легостаева Ю.В., Боцман Л.П.,
Yu., Bannova A.V., Latypova E.R., Tukhvatshin V.S.,
Яковлева М.П., Шаханова О.О., Муслухов Р.Р., Тол-
Kukovinets O.S., Muslukhov R.R., Tolstikov G.A.,
стиков Г.А. ХПС. 2009, 3, 272. [Ishmuratov G.Yu.,
Russ. J. Org. Chem. 2013. 49 (1), 42.]
Legostaeva Yu.V., Botsman L.P., Yakovleva M.P.,
74.
Савченко Р.Г., Уразаева Я.Р., Шафиков Р.В., Одино-
Shakhanova O.O., Muslukhov R.R., Tolstikov G.A.
ков В.Н. ЖОрХ. 2009, 45 (8), 1163. [Savchenko R.G.,
Chem. Nat. Compd. 2009, 45 (3), 318.]
Urazaeva Ya.R., Shafikov R.V., Odinokov V.N. Russ. J.
84.
Ишмуратов Г.Ю., Легостаева Ю.В., Боцман Л.П.,
Org. Chem. 2009. 45 (8), 1149.]
Муслухов Р.Р., Яковлева М.П., Талипов Р.Ф. Вестн.
75.
Kyasa S.K., Fisher T.J., Dussault P.H. Synthesis. 2011,
Баш. ун-та. 2009, 1, 27.
21, 3475.
85.
Легостаева Ю.В., Гарифуллина Л.Р., Наза-
76.
Wu C.-Y., Lin H.-C., Wu H.-J. Tetrahedron. 2012, 68,
ров И.С., Ишмуратов Г.Ю. ЖОрХ. 2018, 54 (1), 56.
2100.
[Legostaeva Yu.V., Garifullina L.R., Nazarov I.S.,
Ishmuratov G.Yu. Russ. J. Org. Chem. 2018. 54 (1),
77.
Ишмуратов Г.Ю., Легостаева Ю.В., Гарифулли-
51.]
на Л.Р., Боцман Л.П., Идрисова З.И., Муслухов Р.Р.,
Ишмуратова Н.М., Толстиков Г.А. ЖОрХ. 2013,
86.
Легостаева Ю.В., Гарифуллина Л.Р., Назаров И.С.,
Кравченко А.А., Кравченко Л.В., Ишмуратов Г.Ю.
49 (10),
1433.
[Ishmuratov G.Y., Legostaeva Y.V.,
ЖОрХ. 2016,
52
(11),
1712.
[Legostaeva Yu.V.,
Garifullina L.R., Botsman L.P., Idrisova Z.I.,
Garifullina L.R., Nazarov I.S., Kravchenko A.A.,
Muslukhov R.R., Ishmuratova N. M., Tolstikov G.A.
Ishmuratov G.Yu. Russ. J. Org. Chem. 2016. 52 (11),
Russ. J. Org. Chem. 2013. 49 (10), 1409.]
1708.]
78.
Ишмуратов Г.Ю., Легостаева Ю.В., Гарифул-
87.
Легостаева Ю.В., Гарифуллина Л.Р., Назаров И.С.,
лина Л.Р., Боцман Л.П., Муслухов Р.Р., Толсти-
Кравченко А.А., Ишмуратова Н.М., Ишмура-
ков Г.А. ЖОрХ. 2014, 50 (8), 1095. [Ishmuratov G.Y.,
тов Г.Ю. ХПС. 2017, 5, 758. [Legostaeva Yu.V.,
Legostaeva Y.V., Garifullina L.R., Botsman L.P.,
Garifullina L.R., Nazarov I.S., Kravchenko A.A.,
Muslukhov R.R., Tolstikov G.A. Russ. J. Org. Chem.
Ishmuratova N.M., Ishmuratov G.Yu. Chem. Nat.
2014. 50 (8), 1075.]
Compd. 2017, 53 (5), 891.]
79.
Ишмуратов Г.Ю., Легостаева Ю.В., Гарифулли-
88.
Легостаева Ю.В., Гарифуллина Л.Р., Назаров И.С.,
на Л.Р., Боцман Л.П., Муслухов Р.Р., Ишмурато-
Нуриева Э.Р., Колотова М.А., Ишмуратов Г.Ю.
ва Н.М., Толстиков Г.А. ЖОрХ. 2014, 50 (7), 948.
Бутлеров. сооб. 2017, 52 (11), 18.
[Ishmuratov G.Y., Legostaeva Y.V., Garifullina L.R.,
89.
Ишмуратов
Г.Ю.,
Легостаева
Ю.В.,
Botsman L.P., Muslukhov R.R., Ishmuratova
Гарифуллина Л.Р., Боцман Л.П., Насибуллина Г.В.,
N.M.,Tolstikov G.A. Russ. J. Org. Chem. 2014. 50 (7),
Газетдинов Р.Р. Вестн. Баш. ун-та. 2014, 19 (1), 29.
928.]
90.
Raskil’dina G.Z., Legostaeva Yu.V., Garifullina L.R.,
80.
Ишмуратов Г.Ю., Легостаева Ю.В., Гарифуллина
Sultanova R.M., Ishmuratov G.Yu., Zlotskii S.S. Lett.
Org. Chem. 2016, 13 (9), 652.
Л.Р., Боцман Л.П., Муслухов Р.Р., Толстиков Г.А.
ЖОрХ.
2013,
49
(10),
1439.
[Ishmuratov G.Y.,
91.
Zabicky J., Rappoport Z. (Ed). Chichester: Wiley &
Legostaeva Yu.V., Garifullina L.R., Botsman L.P.,
Sons, 2006, 597-773. Sons, 2006, 597-773.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019
ПРЕВРАЩЕНИЯ ПЕРОКСИДНЫХ ПРОДУКТОВ ОЗОНОЛИЗА АЛКЕНОВ
99
Transformations of Peroxide Products of Alkenes Ozonolysis
Yu. V. Myasoedova*, I. S. Nazarov, and G. Yu. Ishmuratov
Ufa Institute of Chemistry, Ufa Researcher Centre, RAS,
450054, Russia, Republic of Bashkortostan, Ufa, pr. Oktyabrya 71
*е-mail: legostaevayuv@yandex.ru
Received June 13, 2018
Revised June 26, 2018
Accepted July 2, 018
The review article systematizes and describes the data over the past 10 years on the transformations of the peroxide
products of the ozonolysis of alkenes in various variants (“cleavage” reactions, under the action of reducing agents and
N-containing compounds), as well as the use of these reactions in targeted syntheses.
Keywords: ozonolysis, peroxide products, N-containing compounds, reducing agents
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 1 2019