ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2019, том 55, № 10, с. 1510-1518
УДК 547.425.5
СИНТЕЗ И ОКИСЛЕНИЕ МИРТАНТИОЛА
И ЕГО ФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ
ДИОКСИДОМ ХЛОРА
© 2019 г. О. Н. Гребенкина, О. М. Лезина*, Е. С. Изместьев, Л. Л. Фролова,
С. А. Рубцова**, А. В. Кучин
ФГБУН «Институт химии Коми НЦ УрО РАН (Коми НЦ УрО РАН)»,
167000, Россия, Республика Коми, г. Сыктывкар, ул. Первомайская 48
e-mail: *lezina-om@yandex.ru, **rubtsova-sa@chemi.komisc.ru
Поступила в редакцию 28 марта 2019 г.
После доработки 10 августа 2019 г.
Принята к публикации 13 августа 2019 г.
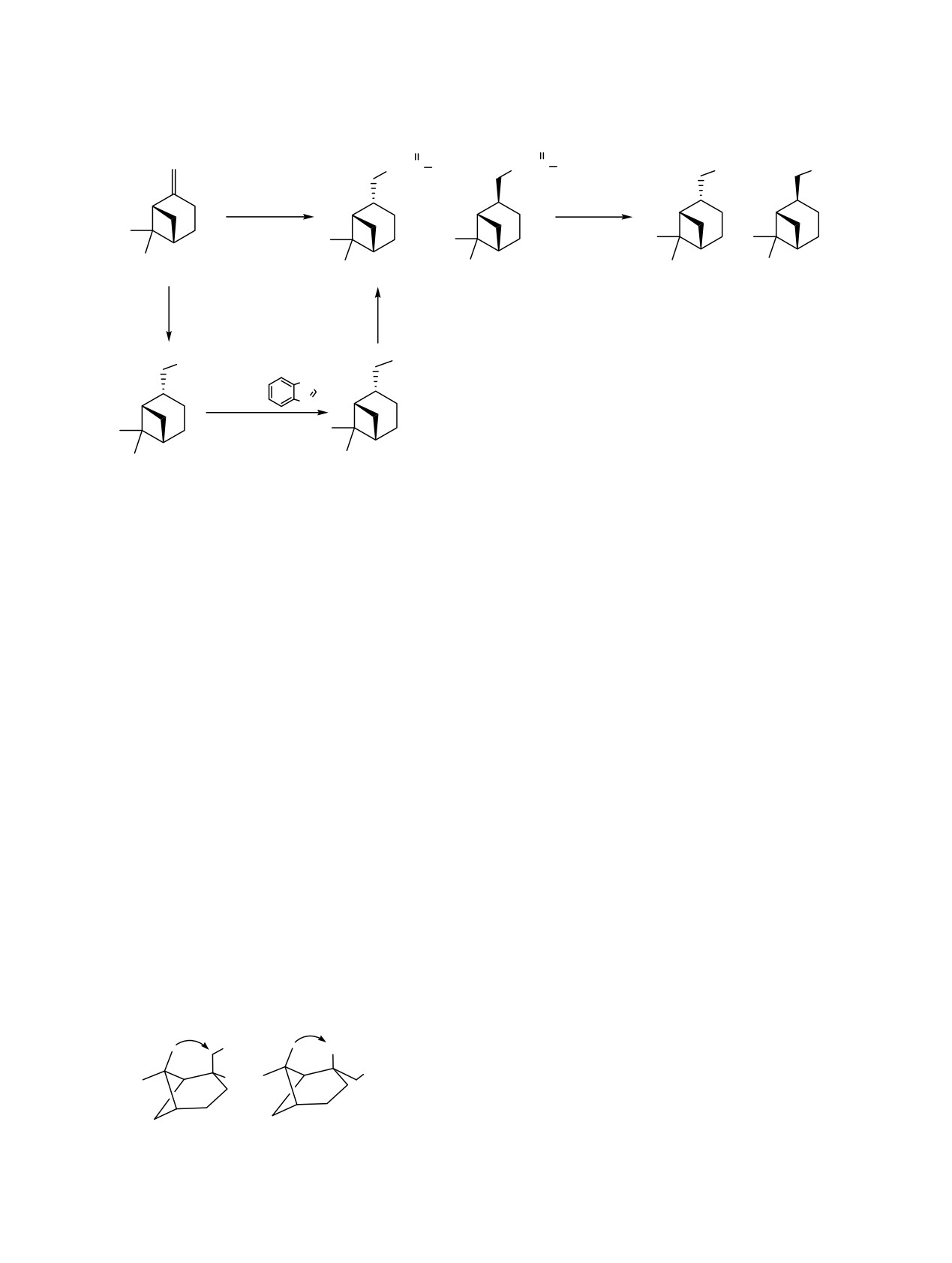
На основе
(-)-β-пинена синтезированы цис-миртантиол и смесь диастереомерных миртантиолов.
Окислением их диоксидом хлора получен ряд производных: дисульфиды, тиолсульфонаты,
сульфохлориды и сульфокислоты. Исследовано влияние условий реакции, таких как природа
растворителя, мольное соотношение реагирующих веществ, время реакции, наличие катализатора и
функциональных групп в молекуле субстрата, на реакционную способность цис-миртантиола.
Окислением тиолов в присутствии катализатора ацетилацетоната ванадила с количественным выходом
получен миртансульфохлорид. Подобраны оптимальные условия для количественного получения
миртансульфокислоты.
Ключевые слова: монотерпеноиды, селективное окисление, диоксид хлора, тиол, тиолсульфонат,
сульфохлорид, сульфокислота, катализ, ацетилацетонат ванадила.
DOI: 10.1134/S0514749219100045
Терпены являются природными биологически
реакции и функциональных групп в молекуле
активными соединениями, обладающими бакте-
субстрата на его реакционную способность.
рицидной, анальгетической и муколитической
Для синтеза тиола
1а первоначально был
активностью, используются в качестве фунгицидов
выбран метод, основанный на присоединении тио-
и противовирусных средств
[1]. Синтез новых
уксусной кислоты по двойной связи (-)-β-пинена
соединений, сочетающих терпеновый фрагмент с
(2) в присутствии кислоты Льюиса и последующем
различными серосодержащими фармакофорными
восстановлении полученного тиоацетата
3а до
группами, является перспективным направлением
тиола 1а (схема 1). Однако продуктом реакции
для получения биологически активных веществ [2].
оказалась смесь диастереомерных тиоацетатов по
Ранее нами было показано, что пинановые гид-
атому С2 (R)-3а и (S)-3b, которые не удалось раз-
рокситиолсульфонаты проявили антимикробную
делить методом колоночной хроматографии. Для
активность в отношении Candida albicans,
повышения стереоселективности образования тио-
Staphylococcus aureus и Cryptococcus neoformans
ацетата 3а варьировались такие условия реакции,
[3]. Сульфокислоты и сульфохлориды, как
как температура (-10÷20°С), растворитель (дихлор-
известно, активно используются в качестве
метан, пиридин) и кислоты Льюиса (ZnCl2, AlCl3,
полупродуктов в органическом синтезе лекарст-
LaCl3). Выявлено, что максимальное содержание
венных препаратов [4].
соединения 3а (de 75%) наблюдается при прове-
дении реакции при комнатной температуре в
В данной работе окислением миртантиола
дихлорметане в присутствии LaCl3 (схема 1).
диоксидом хлора (ClO2) синтезированы новые
хиральные S-, O-содержащие терпеноиды пина-
Согласно
[1],
поляризованный комплекс
новой структуры и исследовано влияние условий
«хлорид лантана - кислота» ориентируется около
1510
СИНТЕЗ И ОКИСЛЕНИЕ МИРТАНТИОЛА И ЕГО ФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ
1511
Схема 1.
O
O
S C
CH3
S C CH3
10
SH
SH
2
AcSH, LaCl3
LiAlH4
1
7
8
6
4
9
2
3a
3b
1а
1b
1) LiBH4, H2SO4
2) H2O2, NaOH
AcSK
OH
I
NH
Ph3P, I2,
N
4
5
двойной связи с пространственно доступной сто-
Пространственное взаимодействие протонов Н2
роны. Ввиду стерических затруднений, вызванных
и Н8 в молекуле тиола 1b приводит к значи-
метильными группами, подход объемного тиоаце-
тельному сдвигу сигналов протонов и атомов
татного фрагмента к четвертичному атому угле-
углерода в спектрах ЯМР в сильное поле относи-
рода С2 затруднен, поэтому на нем происходит коор-
тельно аналогичных сигналов соединения 1а. Так,
динация менее объемного атома водорода. При-
в спектре ЯМР 13С тиола 1b химические сдвиги
соединение комплекса происходит синхронно без
атомов С2 и С8 имеют значения 38.8 и 20.1 м.д., а у
образования карбкатионов, о чем свидетельствует
тиола 1а - 45.3 и 23.2 м.д. соответственно, в то
сохранение пинановой структуры, так как реакции
время как остальные сигналы аналогичных атомов
с участием частиц с локализованными зарядами
имеют разницу значений δ, не превышающую 1.0-
обычно приводят к продуктам изомеризации.
1.5 м.д.
При восстановлении смеси тиоацетатов 3a, b
После неудачных попыток получения диасте-
эквимолярным количеством LiAlH4
[5] также
реомерно чистого миртантиола
1а вышеопи-
образуется хроматографически неделимая смесь
санным способом нами был осуществлен встреч-
диастереомерных тиолов: (R)-1а и (S)-1b
(7:1
ный синтез. Из (-)-β-пинена 2 реакцией гидро-
соответственно, de 75%). Соотношение диастерео-
борирования-окисления был получен цис-миртанол
меров определяли методом ЯМР 1Н по интег-
4 [6], далее по модифицированной методике [7] -
ральным интенсивностям сигналов протонов Н10.
йодид 5, а затем - взаимодействием с AcSK [8] -
тиоацетат 3а, из которого по методике [5] - тиол 1а.
Конфигурация хиральных центров тиолов 1a и
1b доказана методом двумерной NOESY спект-
Окисление тиолов 1 диоксидом хлора прово-
роскопии по наличию в спектре кросс-пиков у
дили аналогично нашим работам [3, 9, 10], в
тиола 1a между протонами метиленовой Н10 и ме-
которых описаны реакции ClO2 с функциона-
тильной Н8 групп, а у тиола 1b - между протонами
льными производными миртантиола 1с, d. Диоксид
Н2 и Н8 (схема 2).
хлора использовали в виде промышленного
водного раствора или переводили в органические
Схема 2.
растворители.
NOE
NOE
SH
8
Окислению подвергались как индивидуальный
H
8
10
2
миртантиол 1а, так и смесь диастереомеров 1а, b с
SH
H
целью разделения их в виде производных методом
колоночной хроматографии. Основными продук-
тами окисления тиолов 1a, b в зависимости от
1а
1b
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
1512
ГРЕБЕНКИНА и др.
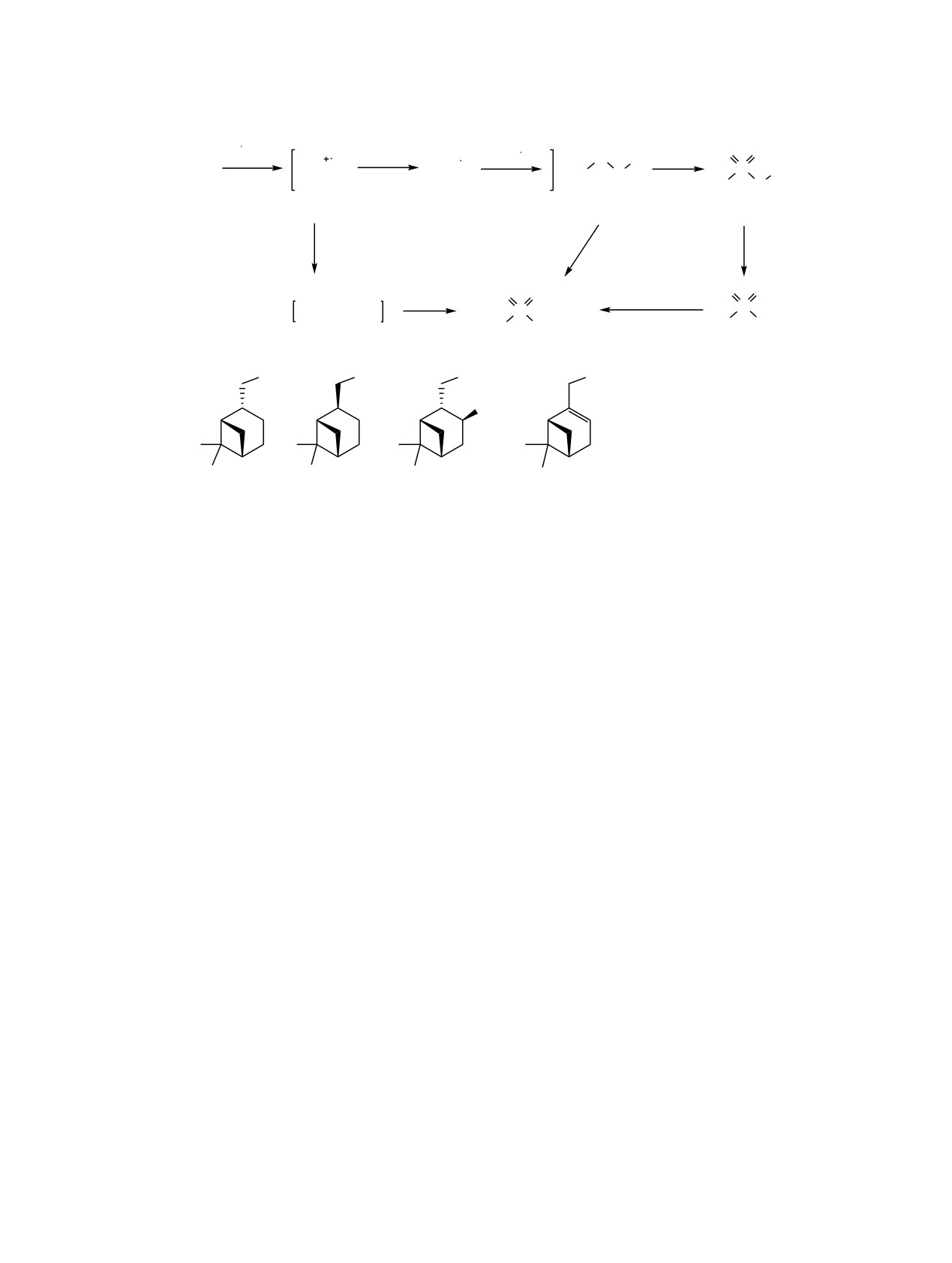
Схема 3.
_
ClO2
O O
ClO2
R S
S R
[O]
R SH
_
R SH
R S
S
R
_
R S
_ClO2
_HClO2
2HClO2
R S
1a_d
6a_d
7a_d
_
Py, H2O
Py, ClO2
_
[O]
2
ClO
O O
O O
H2O
[O]
R S(O)1,2H
S
S
R Cl
R OH
A
9a_d
8a_c
*
*
*
*
OH
R =
(a);
(b);
(c);
(d).
условий реакции являются соответствующие дису-
Реакции тиола
1а проводили в безводных
льфиды 6, тиолсульфонаты 7, сульфохлориды 8 и
гексане, дихлорметане, ацетонитриле и пиридине, а
сульфокислоты 9 (схема 3). Однако разделить (R)-
также с участием воды как сорастворителя при
и (S)-диастереомеры производных 6-9 колоночной
использовании водного раствора ClO2. Выявлено
хроматографией также не удалось ввиду их
влияние полярности и природы растворителя на
близкой хроматографической подвижности. Соот-
скорость реакции. При окислении тиола 1а двук-
ношение диастереомерных продуктов окисления
ратным избытком ClO2 в течение 0.5 ч в системах
6-9 такое же, как и в исходных тиолах, 7:1.
гексан-вода и дихлорметан-вода в реакционной
смеси остается
6-10% исходного тиола (см.
Строение и элементный состав соединений
таблицу, №№ 3, 10), при этом основным про-
подтверждены методами ЯМР и ИК спектрос-
дуктом является дисульфид 6а (57-59%), в то
копии, масс-спектрометрии и данными элемент-
время как в полярном водном ацетонитриле
ного анализа. В спектрах ЯМР 13С сигналы атомов
реакции тиола 1а с ClO2 приводят к образованию
С10 соединений 6-9 сдвигаются в слабое поле
продуктов глубокого окисления 8а, 9а (38 и 62%
относительно аналогичных сигналов атомов угле-
соответственно) (см. таблицу, « 6). Увеличение
рода тиолов 1. Так, значение химического сдвига
скорости реакции в зависимости от полярности
для атома С10 тиола 1a 31.4 м.д. увеличивается до
среды свидетельствует об образовании полярных
46.2, 70.4, 73.4, 59.1 м.д. для соединений 4а-9а
интермедиатов.
соответственно. В ИК спектрах соединений 7а-9а
наблюдаются характерные полосы поглощения
В водном пиридине в аналогичных условиях
группы SO2, соответствующие валентным симмет-
продуктами реакции являются дисульфид 6а и
ричным и асимметричным колебаниям (1128 и
кислота 9а с содержанием в смеси 18 и
82%
1321 см-1 для 7а; 1168 и 1377 см-1 для 8а; 1002,
соответственно, а в безводном - 38 и 62% соот-
1028, 1051 и 1167, 1215 см-1 для 9а).
ветственно (см. таблицу, №№ 16, 19). Увеличение
Исследовано влияние условий реакции на
времени синтеза до 2 ч приводит к количест-
реакционную способность миртантиола 1а с ClO2,
венному образованию сульфокислоты
9а (см.
таких как природа растворителя, мольное соотно-
таблицу, №№ 17, 20). Сульфохлорид 8а в пири-
шение реагирующих веществ, время реакции, нали-
дине не образуется из-за связывания хлорсо-
чие катализатора, а также влияние функциональ-
держащих ионов (продуктов восстановления ClO2)
ных групп в молекуле субстрата, с использованием
растворителем, и диоксид хлора, таким образом,
результатов, полученных ранее для тиолов 1с, d [3, 9].
проявляет лишь окислительные свойства.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
СИНТЕЗ И ОКИСЛЕНИЕ МИРТАНТИОЛА И ЕГО ФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ
1513
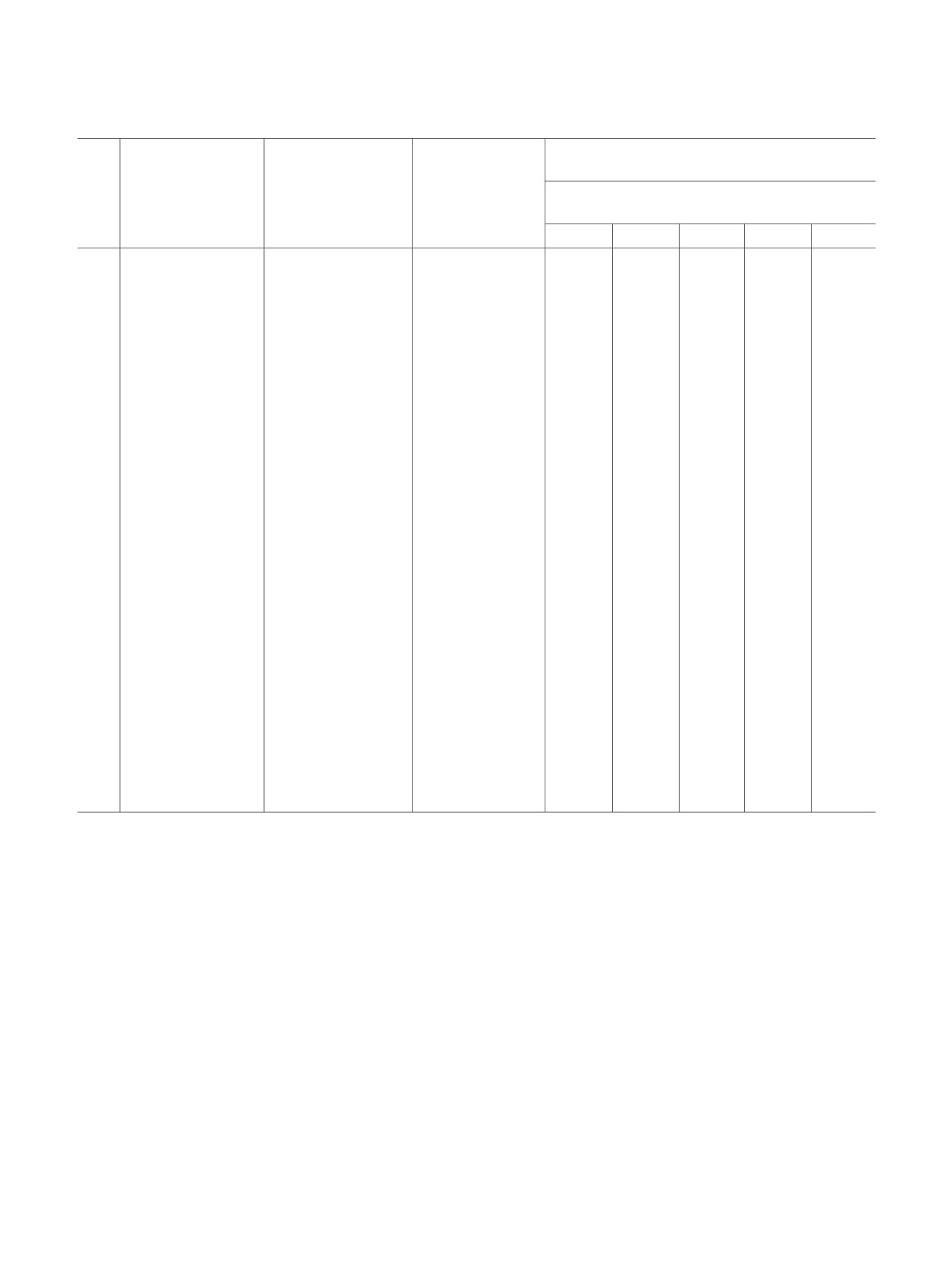
Результаты окисления тиола 1а диоксидом хлора в зависимости от условий реакции.
Соотношение продуктов реакции, %
(по спектрам ЯМР 1Н)
Мольное
№
Растворитель
соотношение
Время синтеза, ч
Cоединение
тиол:ClO2
1
6
7
8
9
1
C6H6
1:0.5
0.5
-
70
11
14
5
2
C6H6-H2O
1:0.5
0.5
29
48
10
4
9
3
C6H6-H2O
1:2
0.5
10
57
15
8
10
4
C6H6-H2O
1:2
1
-
53
21
4
22
5
CH3CN-H2O
1:1
0.5
-
-
21
54
25
6
CH3CN-H2O
1:2
0.5
-
-
-
38
62
7
CH3CN-H2O
1:1
1
-
-
14
48
38
8
CH3CN-H2O
1:2
1
-
-
-
33
67
9
CH2Cl2
1:2
0.5
-
-
26
33
41
10
CH2Cl2-H2O
1:2
0.5
6
59
16
10
9
11
CH2Cl2-H2O
1:2
2
-
-
49
29
22
12
CH2Cl2а
1:2
0.5
-
-
18
82
-
13
CH2Cl2а
1:2
2
-
-
-
98
2
14
CH2Cl2-H2Oа
1:2
2
-
-
41
45
14
15
C5H5N
1:0.5
0.5
-
47
6
-
47
16
C5H5N
1:2
0.5
-
38
-
-
62
17
C5H5N
1:2
2
-
-
-
-
98
18
C5H5N-H2O
1:0.5
0.5
-
33
-
-
67
19
C5H5N-H2O
1:2
0.5
-
18
-
-
82
20
C5H5N-H2O
1:2
2
-
-
-
-
98
а В присутствии катализатора VO(acac)2.
Вероятно, окисление в пиридине протекает, в
1:2 и времени реакции от 0.5 до 1 ч способствуют
основном, через образование сульфеновой и суль-
гидролизу сульфохлорида 8а до сульфокислоты 9а,
финовой кислот А, так как данные продукты
что подтверждается увеличением содержания кис-
устойчивы в основной среде пиридина. Нуклео-
лоты 9а с 25 до 67% и снижением содержания суль-
фильный характер растворителя также препятст-
фохлорида 8а с 54 до 33% (см. таблицу, №№ 5-8).
вует сольватации аниона ClO–, который образует-
При сравнении результатов окисления тиола 1а
ся на первой стадии окисления (схема 3), и делает
и его функциональных аналогов 1с [3], 1d [9] ClO2
его более активным, способствуя увеличению
установлено, что реакционная способность убы-
скорости реакции.
вает в ряду соединений 1с > 1а > 1d. При окис-
На состав продуктов оказывает также влияние
лении тиолов в среде гексан-вода в соотношении
мольное соотношение исходных веществ и время
тиол:ClO2, равном 1:0.5, в течение 0.5 ч конверсия
реакции. Так, в водном ацетонитриле увеличение
гидрокситиола 1с - полная, а тиолов 1а и 1d - 71 и
мольного соотношения тиол:окислитель от 1:1 до
39% соответственно (таблица, № 2).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
1514
ГРЕБЕНКИНА и др.
Наличие двойной связи у тиола 1d снижает
дисульфиды, тиолсульфонаты, сульфохлориды и
скорость окисления промежуточных соединений
сульфокислоты. Выявлено влияние среды на состав
6d, 7d, что способствует увеличению селектив-
и соотношение продуктов и подобраны опти-
ности их образования. При окислении тиола 1d
мальные условия для их получения. Дисульфид с
двукратным избытком ClO2 в течение 1 ч в водном
выходом 68% получен в малополярном гексане. В
ацетонитриле выход тиолсульфоната 7d достигает
среде дихлорметан-вода отмечается максимальное
85% [9], а при окислении тиола 1а основными
содержание тиолсульфоната
-
49%. Реакция в
продуктами являются соединения 8а, 9а, а тиол-
пиридине позволяет количественно получить суль-
сульфоната 7а не обнаружено (см. таблицу, строка 8).
фокислоту, а использование катализатора VO(acac)2
в дихлорметане - сульфохлорид. Показано, что нали-
В среде гексан-вода при окислении тиола 1d с
чие воды при окислении в присутствии VO(acac)2
количественным выходом образуется соответст-
препятствует каталитическому влиянию. В пири-
вующий дисульфид 6d, в то время как выходы
дине, напротив, вода ускоряет реакцию и участвует
соединения 6а (см. таблицу, № 4) и 6с [3] состав-
в образовании сульфокислоты.
ляют около 50%.
Установлено влияние функциональных групп в
Установлено влияние растворителя и структуры
молекуле субстрата на реакционную способность,
тиола на устойчивость продуктов. Так, при окис-
которая уменьшается в ряду гидроксимиртантиол-
лении тиола
1d в дихлорметане двукратным
миртантиол-миртентиол. Снижение активности мир-
избытком ClO2 образуются неидентифици-
тентиола способствует селективному образованию
рованные продукты десульфуризации и раскрытия
промежуточных продуктов по сравнению с
цикла, в то время как окисление тиолов 1а, с в
миртан- и гидроксимиртантиолами: дисульфида -
дихлорметане протекает с образованием продуктов
98% в среде гексан-вода и тиолсульфоната - 85% в
7а, с-9а, с, а десульфуризации не наблюдается (см.
ацетонитриле. В дихлорметане реакции миртен-
таблицу, № 9).
тиола сопровождаются десульфуризацией и
Ранее было показано, что использование
раскрытием цикла, в то время как другие тиолы
VO(acac)2 в качестве катализатора при окислении
легко окисляются до сульфопроизводных с сохра-
гидроксикарановых тиолов
[3] и дифенилди-
нением пинановой структуры.
сульфида [11] увеличивает селективность образо-
вания соответствующих сульфохлоридов, поэтому
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
нами проведено каталитическое окисление тиолов
1а, d. При взаимодействии тиола 1а с двукратным
ИК спектры регистрировали на ИК-Фурье-
избытком ClO2 в дихлорметане в присутствии
спектрометре Shimadzu IR Prestige 21 (Япония) в
VO(acac)2 в течение 0.5 ч выход сульфохлорида 8а
тонком слое. Спектры ЯМР
1H и 13C регист-
увеличивается с 33 до 82%, в течение 2 ч - до 98%
рировали на спектрометре Bruker Avance-300
(см. таблицу, №№ 9, 12, 13). При этом выявлено,
(Германия) (300.17 МГц для 1Н и 75.48 МГц для
что в реакционной среде дихлорметан-вода
13С) в растворах CDCl3 и ДМСО-d6. Полное отне-
действие катализатора исчезает (выход 8а - 45%)
сение сигналов 1Н и 13С выполняли с помощью
(см. таблицу, № 14). В случае окисления тиола 1d в
двумерных гомо- (1H-1H COSY, 1H-1H NOESY) и
дихлорметане влияние катализатора отсутствует,
гетероядерных экспериментов
(1H-13C HSQC,
что связано, вероятно, с неустойчивостью интер-
HMBC). Масс-спектры регистрировали на высоко-
медиатов в среде растворителя.
эффективном жидкостном хроматографе с масс-
селективным детектором Thermo Finnigan LCQ
Реакции тиолов 1а-с в пиридине приводят к
Fleet (США) (растворители - H2O, CH3OH, CH3CN).
количественному образованию сульфоновых
Детектирование проводили по отрицательным
кислот 9а-с. Выход кислоты 9d снижается до 74%
ионам. Тонкослойную хроматографию выполняли
[9], что обусловлено образованием соответст-
на пластинах Sorbfil, используя системы раст-
вующей соли пиридиния.
ворителей: петролейный эфир-диэтиловый эфир,
Таким образом, синтезированы индивидуаль-
9:1 (для производных 1а, b, 6а, b-8а, b); в качестве
ный цис-миртантиол и смесь диастереомерных
проявителей
- раствор фосфорномолибденовой
миртантиолов, в результате окисления которых
кислоты в EtOH и водный раствор KMnO4, для
диоксидом хлора получены соответствующие
8а, b-9а, b - 0.2% раствор бромкрезолового зеле-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
СИНТЕЗ И ОКИСЛЕНИЕ МИРТАНТИОЛА И ЕГО ФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ
1515
ного в EtOH. Элементный анализ выполняли с
силикагеле, используя в качестве элюента
исполь-зованием автоматического анализатора
петролейный эфир. Целевой продукт содержит
марки ЕА 1110 CHNS-O. Все реакции проводили с
смесь диастереомеров по атому С2 (R)-3а и (S)-3b
использованием свежеперегнанных растворителей.
тиоацетатов в соотношении 7:1 соответственно.
Колоночную хроматографию выполняли на сили-
S-[(1S,2R,5S)-6,6-Диметилбицикло[3.1.1]геп-
кагеле Alfa Aesar (0.06-0.2 мм), используя те же
24
тан-2-ил]метилэтантиоат (3a). Выход 78%, [α]
системы растворителей, что и для ТСХ; для
–22.3° (с 0.4, CHCl3). Спектральные данные тио-
сульфокислот 9а, b - метанол. Соотношение про-
ацетата 3a идентичны данным, приведенным в
дуктов определяли методом спектроскопии ЯМР
литературе [5].
1Н по величинам интегральных интенсивностей
сигналов соответствующих протонов H10.
S-[(1S,2S,5S)-6,6-Диметилбицикло[3.1.1]геп-
тан-2-ил]метилэтантиоат (3b). Спектральные дан-
(-)-β-Пинен (2) - коммерческий продукт произ-
ные получены из спектра смеси с тиоацетатом 3а.
25
водства «Sigma Aldrich», чистота 99%, [α]
-22°.
Спектр ЯМР 1Н (CDCl3), δ, м.д.: 0.80 c (3H, H8),
Водный раствор ClO2 - продукт производства АО
0.84-0.93 м (1H, H7α), 1.20 c (3H, H9), 1.33-1.40 м
«Монди СЛПК» (Россия). Органический раствор
(1H, H3α), 1.74-1.79 м (2H, H4), 1.78-1.85 м (1H, H1),
ClO2 получали экстракцией водного раствора,
1.87-1.94 м (1H, H5), 2.05-2.16 м (2H, H2, H3β),
сушили над Na2SO4. Концентрацию раствора опре-
2.31-2.42 м (1H, H7β), 2.33 с (3H, COCH3), 2.80 д.д
деляли титрованием по методике [12].
(2Н, Н10, J 17.6, 7.4 Гц). Спектр ЯМР 13С (CDCl3), δ,
(1S,2R,5S)-2-(Иодометил)-6,6-диметилби-
м.д.: 20.04 (C8), 23.22 (C3), 24.19 (C4), 26.64 (C9),
30.61 (COCH3), 33.30 (C7), 34.95 (C10), 35.14 (C2),
цикло[3.1.1]гептан (5). В 7 мл толуола растворяли
0.154 г (1 ммоль) цис-миртанола 4, затем при пере-
38.65 (C6), 40.74 (C5), 45.00 (C1), 196.00 (COCH3).
мешивании добавляли 0.314 г (1.2 ммоль) трифе-
Найдено, %: С 67.91; Н 9.41; S 15.16. C12H20OS.
Вычислено, %: С 67.87; Н 9.49; S 15.10.
нилфосфина, 0.236 г (2 ммоль) бензимидазола и
0.305 г (1.2 ммоль) йода. Полученную смесь кипя-
Миртантиолы 1a, b получены по методике [5].
тили с обратным холодильником в течение 1 ч,
Спектральные характеристики тиола 1а идентичны
после чего добавляли порциями насыщенный
приведенным в литературе [5], [α]
22 -38.5° (с 0.2,
раствор Na2S2O3 до исчезновения окраски йода.
CHCl3). Выход 72%.
Экстрагировали полученную смесь хлороформом,
[(1S,2S,5S)-6,6-Диметилбицикло[3.1.1]гептан-
органическую фазу сушили над Na2SO4. Далее
2-ил]метантиол (1b). Спектральные данные полу-
отфильтровывали раствор и удаляли растворитель.
чены из спектра смеси с тиолом 1а. Спектр ЯМР 1Н
Остаток хроматографировали на силикагеле,
(CDCl3), δ, м.д.: 0.83 c (3H, H8), 0.87-0.95 м (1H,
используя в качестве элюента петролейный эфир.
22
H7α), 1.22 c (3H, H9), 1.26-1.36 м (2H, SH, H3α),
Выход 90%, [α]
-42.8° (с 0.2, CHCl3). Спектр
1.72-1.84 м (2H, H4), 1.86-1.99 м (2H, H1, H5), 2.01-
ЯМР 1Н (CDCl3), δ, м.д.: 0.92 д (1H, H7α, J 9.3 Гц),
2.14 м (2H, H2, H3β), 2.33-2.43 м (1H, H7β), 2.39-2.46
1.01 c (3H, H8), 1.24 c (3H, H9), 1.43-1.60 м (1H,
м (2Н, Н10). Спектр ЯМР 13С (CDCl3), δ, м.д.: 20.09
H3α), 1.85-2.00 м (3H, H4, H5), 2.01-2.17 м (2H, H1,
(C8), 23.27 (C3), 24.20 (C4), 26.65 (C9), 30.56 (C10),
H3β), 2.33-2.54 м (2H, H2, H7β), 2.29 д.д (2Н, Н10, J
33.27 (C7), 38.63 (C6), 38.82 (C2), 40.89 (C5), 44.65
7.9, 4.0 Гц). Спектр ЯМР 13С (CDCl3), δ, м.д.: 15.33
(C1).
(C10), 23.09 (C8), 23.38 (C3), 25.87 (C4), 27.84 (C9),
33.17 (C7), 38.59 (C6), 41.20 (C5), 44.30 (C2), 46.81
Методика окисления тиолов ClO2. Общий
(C1).
объем реакционной среды рассчитывали для моль-
ной концентрации тиола 0.02 моль/л. Контроль за
Методика синтеза миртантиоацетатов из β-
ходом реакции проводили методом ТСХ.
пинена. К 0.136 г (1 ммоль) β-пинена в 5 мл ди-
хлорметана при перемешивании добавляли 0.076 г
а. К водному/органическому раствору 0.135 г
(1 ммоль) тиоуксусной кислоты, затем прибавляли
(2 ммоль) ClO2 добавляли при перемешивании
0.021 г (0.1 ммоль) LaCl3. Смесь перемешивали 2 ч,
органический раствор 0.17 г (1 ммоль) тиола 1а
добавляли
20 мл воды, экстрагировали хлоро-
или смеси тиолов 1а, b. Время синтеза 0.5-2 ч.
формом (3×15). Объединенные органические слои
Водную и органическую фазы разделяли. Раство-
сушили над Na2SO4, удаляли растворитель при
рители отгоняли при пониженном давлении.
пониженном давлении. Остаток делили на
Остаток органической фазы, содержащий соеди-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
1516
ГРЕБЕНКИНА и др.
нения
6-8, хроматографировали на силикагеле.
Найдено, %: С 64.91; Н 9.19; S 17.34. С20H34О2S2.
Сульфокислоты
9 извлекали из водной фазы
Вычислено, %: С 64.82; Н 9.25; S 17.30.
выпариванием воды.
[(1S,2R,5S)-6,6-Диметилбицикло[3.1.1]гептан-
б. К раствору 0.17 г (1 ммоль) тиола 1а или
2-ил]метансульфохлорид
(8а). б. Соотношение
смеси тиолов 1а, b в дихлорметане при переме-
1а:ClO2 = 1:2. Растворитель дихлорметан. Время
шивании добавляли 0.027 г (0.1 ммоль) VO(acac)2,
синтеза 2 ч. Выход 96%, прозрачная вязкая жид-
затем раствор 0.135 г (2 ммоль) ClO2 в дихлор-
кость, [α]
23 -28.3° (с 0.5, CHCl3). ИК спектр (KBr),
метане. Время синтеза 2 ч. Растворитель отгоняли.
ν, см-1: 1377 (SO2)a, 1168 (SO2)s. Спектр ЯМР 1Н
Сухой остаток органической фазы хроматогра-
(СDCl3), δ, м.д.: 1.01-1.12 м (1H, H7α), 1.05 c (3H,
фировали на силикагеле.
H8), 1.26 c (3H, H9), 1.64-1.81 м (1H, H3α), 1.90-2.08
м (3H, H4, H5), 2.06-2.17 м (1H, H1), 2.18-2.35 м
1,2-бис(1S,2R,5S)-(6,6-Диметилбицикло[3.1.1]-
(1H, H3β), 2.39-2.51 м (1H, H7β), 2.89-3.03 м (1H,
гептан-2-ил)метилдисульфид (6а). а. Соотноше-
H2), 3.85 д (2Н, Н10, J 6.5 Гц). Спектр ЯМР 13С
ние 1а:ClO2 = 1:0.5. Растворитель гексан. Время
(CDCl3), δ, м.д.: 21.37 (C3), 23.09 (C8), 25.61 (C4),
синтеза 0.5 ч. Выход 68%. Спектральные харак-
27.42 (C9), 32.31 (C7), 36.60 (C2), 38.36 (C6), 40.52
теристики дисульфида 6а аналогичны приведен-
(C5), 46.09 (C1), 73.38 (C10). Найдено, %: С 50.79; Н
ным в литературе [13].
S. Вычислено, %: С 50.73;
7.19; S 13.61. С10H17СlО2
1,2-бис(1S,2S,5S)-(6,6-Диметилбицикло[3.1.1]-
Н 7.24; S 13.54.
гептан-2-ил)метилдисульфид (6b). а. Соотноше-
ние смесь диастереомеров 1а, b:ClO2 = 1:0.5. Раст-
[(1S,2S,5S)-6,6-Диметилбицикло[3.1.1]гептан-
воритель гексан. Время синтеза 0.5 ч. Выход смеси
2-ил]метансульфохлорид
(8b). б. Соотношение
диастереомеров 6а, b (7:1) 68%. Спектральные
смесь диастереомеров 1а, b:ClO2 = 1:2. Раство-
данные получены из спектров смеси с диастерео-
ритель дихлорметан. Выход смеси диастереомеров
мером 6а. Спектр ЯМР 1Н (CDCl3), δ, м.д.: 0.87 c
8а, b (7:1) 96%. Время синтеза 2 ч. ИК спектр
(6H, H8), 1.02-1.09 м (2H, H7α), 1.24 c (6H, H9),
(KBr), ν, см-1: 1377 (SO2)a, 1168 (SO2)s. Спектр ЯМР
1.28-1.36 м (2H, H3α), 1.76-1.87 м (4H, H4), 1.87-
1Н (СDCl3), δ, м.д.: 0.92 c (3H, H8), 0.98-1.10 м (1H,
2.06 м (6H, H1, H2, H5), 2.04-2.13 м (2H, H3β), 2.37-
H7α), 1.11 c (3H, H9), 1.60-1.78 м (1H, H3α), 1.80-
2.45 м (2H, H7β), 2.63 д.д (4Н, Н10, J 6.9, 3.0 Гц).
1.91 м (2H, H4), 1.94-2.03 м (3H, H1, H3β, H5), 2.36-
Спектр ЯМР 13С (CDCl3), δ, м.д.: 20.12 (C8), 23.41
2.48 м (1H, H7β), 2.83-2.91 м (1H, H2), 3.70 д (2Н,
(C3), 24.26 (C4), 26.66 (C9), 32.58 (C7), 38.66 (C6),
Н10, J 6.6 Гц). Спектр ЯМР 13С (CDCl3), δ, м.д.:
40.29 (C2), 40.86 (C5), 44.61 (C1), 45.83 (C10).
20.01 (C8), 23.20 (C3), 25.61 (C4), 26.50 (C9), 32.30
Найдено, %: С 71.03; Н 10.04; S 18.90. С20H34S2.
(C7), 32.62 (C2), 39.74 (C6), 40.20 (C5), 45.08 (C1),
Вычислено, %: С 70.94; Н 10.12; S 18.94.
72.53 (C10). Найдено, %: С 50.79; Н 7.19; S 13.61.
С10H17СlО2S. Вычислено, %: С 50.73; Н 7.24; S
S-{[(1S,2R,5S)-6,6-Диметилбицикло[3.1.1]-
13.54.
гептан-2-ил]метил}-((1S,2R,5S)-6,6-диметилби-
цикло[3.1.1]гептан-2-ил)метансульфонотиоат
[(1S,2R,5S)-6,6-Диметилбицикло[3.1.1]гептан-
(7а). а. Соотношение 1а:ClO2 = 1:2. Растворитель
2-ил]метансульфоновая кислота (9а). а. Соотно-
дихлорметан-вода. Время синтеза 2 ч. Выход 45%.
шение 1а:ClO2 = 1:2. Растворитель пиридин-вода.
ИК спектр (KBr), ν, см-1: 1321 (SO2)a, 1128 (SO2)s.
Время синтеза 2 ч. Выход 98%, прозрачная вязкая
23
Спектр ЯМР 1Н (CDCl3), δ, м.д.: 0.87-0.96 м (1H,
жидкость, [α]
-9.6° (с 0.3, CHCl3). ИК спектр
H7'α), 1.00-1.09 м (1H, H7α), 1.03 c (3H, H8), 1.06 c
(KBr), ν, см-1: 1215, 1167 (SO2)a, 1051,1028,1002
(3H, H8'), 1.23 c (3H, H9), 1.25 c (3H, H9'), 1.47-1.59
(SO2)s. Спектр ЯМР 1Н (ДМСО-d6), δ, м.д.: 0.85-
м (1H, H3α), 1.62-1.74 м (1H, H3α'), 1.81-2.14 м (9H,
0.94 м (1H, H7α), 0.94 c (3H, H8), 1.14 c (3H, H9),
H1, H1', H3β, H4, H4', H5, H5'), 2.15-2.30 м (1H, H3β'),
1.48-1.67 м (1H, H3α), 1.75-1.95 м (3H, H4, H5),
2.33-2.50 м (2H, H7β, H7β'), 2.75-2.98 м (2H, H2, H2'),
1.91-2.01 м (2H, H3β, H1), 2.20-2.38 м (1H, H7β),
3.22 д (2Н, Н10', J 7.9 Гц), 3.40-3.51 м (2Н, Н10).
2.41-2.53 м (1H, H2), 2.75 д (2Н, Н10, J 6.6 Гц), 7.18
Спектр ЯМР 13С (CDCl3), δ, м.д.: 21.70 (C3'), 21.87
уш.с (1H, OH). Спектр ЯМР 13С (ДМСО-d6), δ, м.д.:
(C3), 23.13 (C8, C8'), 25.80 (C4'), 25.89 (C4), 27.53
22.12 (C3), 23.34 (C8), 26.22 (C4), 28.13 (C9), 32.66
(C9'), 27.79 (C9), 32.47 (C7'), 33.18 (C7), 35.29 (C2'),
(C7), 36.83 (C2), 38.46 (C6), 40.88 (C5), 46.27 (C1),
36.00 (C2), 37.07 (C6'), 37.36 (C6), 40.64 (C5'), 41.05
59.51 (C10). Масс-спектр (ESI, 5 кВ), m/z (Iотн, %):
(C5), 42.68 (C10'), 45.14 (C1'), 46.50 (C1), 70.28 (C10).
217.29 (100) [M - H]-, 97.00 (48) [C7H13]. Найдено,
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
СИНТЕЗ И ОКИСЛЕНИЕ МИРТАНТИОЛА И ЕГО ФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ
1517
%: С 55.47; Н 8.36; S 14.55. С10H18О3S. Вычислено,
3. Гребенкина О.Н., Лезина О.М., Изместьев Е.С.,
%: С 55.05; Н 8.26; S 14.68.
Судариков Д.В., Пестова С.В., Рубцова С.А.,
Кучин А.В. ЖОрХ. 2017, 53, 844. [Grebyonkina O.N.,
[(1S,2S,5S)-6,6-Диметилбицикло[3.1.1]гептан-
Lezina O.M., Izmest’ev E.S., Sudarikov D.V., Pestova S.V.,
2-ил]метансульфоновая кислота (9b). а. Соот-
Rubtsova S.A., Kutchin A.V. Russ. J. Org. Chem. 2017,
ношение смесь диастереомеров 1а, b:ClO2 = 1:2.
53, 860.] doi 10.1134/S1070428017060082
Растворитель пиридин-вода. Выход смеси диасте-
4. Котегов Н.А., Белогурова Н.В., Зырянов В.А.,
реомеров 9а, b (7:1) 98%. ИК спектр (KBr), ν, см-1:
Тупикина В.Г., Петров А.Ю. Пат. 2119332 (1995).
1215, 1167 (SO2)a, 1051,1028,1002 (SO2)s. Спектр
РФ. Б.И. 1998, № 27.
ЯМР 1Н (ДМСО-d6,), δ, м.д.: 0.81 c (3H, H8), 0.93-
5. Banach A., Ścianowski Ja., Ozimek P. Phosphorus,
1.02 м (1H, H7α), 1.16 c (3H, H9), 1.55-1.65 м (1H,
Sulfur, Silicon Rel. Elements. 2014, 189, 274. doi
10.1080/10426507.2013.819867
H3α), 1.65-1.73 м (2H, H4), 1.76-1.85 м (1H, H3β),
1.92-1.99 м (1H, H1), 2.20-2.38 м (2H, H5, H7β),
6. Кучин А.В, Фролова Л.Л. Изв. АН. Сер хим. 2000, 9,
1658. [Kutchin A.V., Frolova L.L. Russ. Chem. Bull.,
2.35-2.41 м (1H, H2), 2.59 д (2Н, Н10, J 6.6 Гц), 7.18
Int. Ed. 2000, 49, 1647.] doi 10.1007/BF02495177
уш.с (1H, OH). Спектр ЯМР 13С (ДМСО-d6), δ, м.д.:
7. Garegg P.J., Samuelsson B. J. Chem. Soc. Perkin Trans. 1.
20.45 (C8), 23.30 (C3), 24.49 (C4), 27.03 (C9), 31.94
1980, 2866. doi 10.1039/P19800002866
(C2), 32.66 (C7), 38.69 (C6), 40.66 (C5), 45.15 (C1),
8. Zheng T.-C., Burkart M., Richardson D.E. Tetrahedron
58.00 (C10). Масс-спектр (ESI, 5 кВ), m/z (Iотн, %):
Lett. 1999, 40, 603. doi 10.1016/s0040-4039(98)02545-3
217.29 (100) [M - H]-, 97.00 (48) [C7H13]. Найдено,
9. Лезина О.М., Гребенкина О.Н., Судариков Д.В.,
%: С 55.55; Н 8.41; S 14.61. С10H18О3S. Вычислено,
Крымская Ю.В., Рубцова С.А., Кучин А.В. ЖОрХ.
%: С 55.05; Н 8.26; S 14.68.
2015,
51,
1391.
[Lezina O.M., Grebenkina O.N.,
Sudarikov D.V., Krymskaya Yu.V., Rubtsova S. A.,
ФОНДОВАЯ ПОДДЕРЖКА
Kutchin A.V. Russ. J. Org. Chem. 2015, 51, 1359.] doi
10.1134/S1070428015100012
Работа выполнена при финансовой поддержке
10. Изместьев Е.С., Лезина О.М., Гребенкина О.Н.,
проекта УрО РАН № 18-3-3-17 с использованием
Патов С.А., Рубцова С.А., Кучин А.В. Изв. АН. Сер.
оборудования Центра коллективного пользования
Хим. 2014, 9,
2067.
[Izmest’ev E.S., Lezina O.M.,
(ЦКП) «Химия» Института химии Коми НЦ УрО
Grebyonkina O.N.,
Patov S.A., Rubtsova S.A.,
РАН.
Kutchin A.V. Russ. Chem. Bull., Int. Ed. 2014, 63,
2067.] doi 10.1007/s11172-014-0702-8
КОНФЛИКТ ИНТЕРЕСОВ
11. Лезина О.М., Рубцова С.А., Кучин А.В. ЖОрХ. 2011,
47, 1230. [Lezina O.M., Rubtsova S. A., Kutchin A.V.
Авторы заявляют об отсутствии конфликта
Russ. J. Org. Chem. 2011, 47, 1249.] doi 10.1134/
интересов.
S1070428011080239
СПИСОК ЛИТЕРАТУРЫ
12. Петренко Н.Ф., Мокиенко А.В. Диоксид хлора:
применение в технологиях водоподготовки. Одесса:
1. Paduch R., Kandefer-Szerszen M., Trytek M., Fiedurek J.
Оптимум, 2005, 371.
Arch. Immun. Ther. Exp. 2007, 55, 315. doi 10.1007/
13. Пестова C.В., Изместьев Е.С., Шевченко О.Г.,
s00005-007-0039-1
Рубцова С.А., Кучин А.В. Изв. АН. Сер. Хим. 2015,
2. Nikitina L.E., Artemova N.P., Startseva V.A.,
64, 723. [Pestova S.V., Izmest’ev E.S., Shevchenko O.G.,
Fedyunina I.V., Klochkov V.V. Chem. Nat. Comp.
Rubtsova S.A., Kuchin A.V. Russ. Chem. Bull., Int. Ed.
2017, 53, 811. doi 10.1007/s10600-017-2131-z
2015, 64, 723.] doi 10.1007/s11172-015-0926-2
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
1518
ГРЕБЕНКИНА и др.
Synthesis and Oxidation of Myrthanyl Thiol
and Its Functional Derivatives by Chlorine Dioxide
O. N. Grebyonkina, O. M. Lezina*, E. S. Izmest’ev, L. L. Frolova,
S. A. Rubtova**, and A. V. Kutchin
Institute of Chemistry of Komi SC UB of the RAS of FSBIS FRC “Komi SC UB of the RAS”,
167000, Russia, Republic of Komi, Syktyvkar, Pervomaiskaya 48
e-mail: *lezina-om@yandex.ru, **rubtsova-sa@chemi.komisc.ru
Received March 28, 2019; revised August 10, 2019; accepted August 13, 2019
Proceeding from (-)-β-pinene, we synthesized cis-myrthanethiol as well as a mixture of diastereomeric
myrthanethiols. A number of their derivatives: disulfides, thiosulfinates, sulfochlorides, and sulfonic acids were
obtained by oxidation with chlorine dioxide. The effect of reaction conditions, such as the nature of a solvent, a
molar ratio of reactants, the reaction time, the presence of a catalyst and functional groups in the substrate
molecule, on the reactivity of cis-myrtanethiol was investigated. Myrthane sulfochloride was produced in quan-
titative yield by the oxidation of thiols with ClO2 in the presence of a vanadyl acetylacetonate catalyst. Optimal
conditions for quantitative transformation of thiols into myrthane sulfonic acid were matched.
Keywords: monoterpenoids, selective oxidation, chlorine dioxide, thiol, thiosulfonate, sulfochloride, sulfonic
acid, catalyze, vanadyl acetylacetonate
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019