ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2019, том 55, № 10, с. 1583-1591
УДК 547.551.42:543.544.5.068.7:615.22
СИНТЕЗ И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
НИТРАТА 2-МЕТИЛАНИЛИДА
N,N-ДИЭТИЛАМИНОУКСУСНОЙ КИСЛОТЫ
© 2019 г. Л. А. Чекрышкинаa, А. М. Дёминb, *, А. А. Тумашовb, c,
Е. А. Бабиковаd, Н. В. Слеповаa
a ФГБОУ ВО «Пермская государственная фармацевтическая академия Минздрава России»,
614990, Россия, г. Пермь, ул. Полевая 2
b ФГБУН «Институт органического синтеза им. И.Я. Постовского УрО РАН»,
620990, Россия, г. Екатеринбург, ул. С. Ковалевской 22/20
c ФГАОУ ВО «Уральский федеральный университет», Институт естественных наук и математики,
620002, Россия, г. Екатеринбург, ул. Мира 19
d ГБПОУ «Свердловский областной медицинский колледж (Фармацевтический филиал)»,
620014, Россия, г. Екатеринбург, ул. Бебеля 71
*e-mail: demin@ios.uran.ru
Поступила в редакцию 15 апреля 2019 г.
После доработки 14 августа 2019 г.
Принята к публикации 15 августа 2019 г.
Оптимизирован метод синтеза нитрата 2-метиланилида N,N-диэтиламиноэтановой кислоты (мономе-
каин), обладающего выраженными антиаритмическими свойствами. Разработана методика установления
подлинности мономекаина и контроля чистоты исследуемого соединения в субстанции с использованием
метода ВЭЖХ, установлен предел обнаружения примеси о-толуидина в образцах субстанции
мономекаина, который составил 0.02%. Для количественного определения содержания основного
вещества в субстанции мономекаина разработаны методики ВЭЖХ и экстракционного титрования и
проведена их валидация. Величина относительного стандартного отклонения (RSD) для мономекаина
свидетельствует о хорошей специфичности, линейности, прецизионности и правильности разработанных
методик.
Ключевые слова: мономекаин, синтез, ВЭЖХ, экстракционное титрование, валидация.
DOI: 10.1134/S0514749219100112
Заболевания сердечно-сосудистой системы
является актуальной задачей [1, 2]. С этой целью
занимают по летальности одно из первых мест в
исследуются соединения, относящиеся к различ-
мире. Согласно статистическим данным, каждый
ным классам. Наиболее распространённый класс
тринадцатый гражданин РФ страдает той или иной
анестезирующих и антиаритмических соединений
патологией сердца или сосудов, а смертность от
составляют производные N-фенилацетамида и
сердечно-сосудистых заболеваний в России
амидов ароматических карбоновых кислот [3-11],
составляет 53% от всех летальных исходов. Ука-
среди которых самым известным препаратом
занные заболевания, как правило, сопровождаются
является лидокаин [12]. Отдельный интерес пред-
аритмией
- нарушениями сердечного ритма,
ставляют соли 2-метиланилида N,N-диэтиламино-
которые могут усугублять течение основного забо-
уксусной кислоты с неорганическими кислотами.
левания. Хотя арсенал лекарственных антиарит-
Способ получения гидрохлорида 2-метиланилида
мических средств достаточно велик, многие из них
N,N-диэтиламиноуксусной кислоты описан в
имеют нежелательные побочные эффекты, поэтому
работах [9, 13]. Впоследствии были синтезированы
разработка и внедрение в медицинскую практику
различные соли 2-метиланилида N,N-диэтиламино-
новых антиаритмических лекарственных средств
уксусной кислоты с HBr, HI, H3PO4 [9] и салицилат
1583
1584
ЧЕКРЫШКИНА и др.
Таблица 1. Антиаритмическая активность солей 2-метиланилида N,N-диэтиламиноуксусной кислоты.
Антиаритмический индекс
Соединение
ЛД50, мг·кг-1
ЭД50, мг·кг-1
ЛД
50/ЭД50
Нитрат 2-метиланилида N,N-
65.0 (56-75)
1.4 (1.2-1.7)
46.4
диэтиламиноуксусной кислоты (мономекаин)
Гидрохлорид 2-метиланилида N,N-
46.0 (33-60)
1.4 (1.1-1.7)
32.8
диэтиламиноуксусной кислоты
Гидробромид 2-метиланилида N,N-
81.5 (71-84)
17.8 (15-21)
4.6
диэтиламиноуксусной кислоты
Гидроиодид 2-метиланилида N,N-
60.0 (48-74)
35.5 (29-42)
1.7
диэтиламиноуксусной кислоты
Дигидрофосфат 2-метиланилида N,N-
44.7 (37-59)
20.5 (18-24)
2.2
диэтиламиноуксусной кислоты
Лидокаина
39.5 (34-45)
7.7 (6-9)
5.1
а Структурный аналог мономекаина, применяющийся в качестве антиаритмического лекарственного средства.
2-метиланилида морфолинуксусной кислоты [10].
лекарственных формах, методик его анализа в
Была изучена антиаритмическая активность, пока-
биологических средах. Удобные методы контроля
зана перспективность дальнейшего исследования
его чистоты в субстанции, приемлемые для
данного класса соединений для внедрения в
разработки государственной фармакопейной
медицинскую практику. Как потенциальное
статьи на лекарственную форму, в литературе не
антиаритмическое средство представляет интерес
представлены. Поэтому целью данного исследо-
нитрат 2-метиланилида N,N-диэтиламиноуксусной
вания являлась оптимизация метода синтеза нит-
кислоты, получивший условное название моно-
рата
2-метиланилида N,N-диэтиламиноуксусной
мекаин. Установлено, что он проявил наиболее
кислоты, разработка методики установления его
выраженную активность среди выше указанных
подлинности и чистоты методом ВЭЖХ и коли-
солей 2-метиланилида N,N-диэтиламиноуксусной
чественное определение его содержания в лабора-
кислоты (табл.
1)
[9-11], поэтому его можно
торных сериях полученных субстанций методами
рассматривать как потенциальное лекарственное
ВЭЖХ и экстракционного титрования.
средство антиаритмического действия.
Ранее 2-метиланилид N,N-диэтиламиноуксусной
На доклиническом этапе исследования биоло-
кислоты (как ключевое соединение в синтезе моно-
гической активности соединения необходима его
мекаина) получали ацилированием о-толуидина
стандартизация, предполагающая разработку спо-
хлорацетилхлоридом в ледяной уксусной кислоте
соба получения, гарантирующего высокую чистоту
или в ацетоне (двухступенчатая методика) [13]. В
полученного соединения, эффективных способов
данной работе методика была упрощена: реакция
оценки его качества в субстанции и создаваемых
ацилирования проводилась в одну стадию в аце-
Схема 1.
O
H
H
H
Cl
N
NH2
Cl
N
N
2
Cl
4
N
C6H6 (сух.)
C6H6
(сух.),
O
O
1
3, 76.7%
5, 88.8%
H
HNO3, 67%
N
+
EtOH, 0oC
NH
_
NO3
O
6, 78.5%
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
СИНТЕЗ И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ НИТРАТА 2-МЕТИЛАНИЛИДА
1585
тоне или бензоле с добавлением к о-толуидину (1)
первого модельного раствора несущественно отли-
1-, 1.5- или 2-кратного мольного избытка хлор-
чалась от уровня шумов, тогда как в случае 0.02%
ацетилхлорида (2) (схема 1). Показано, что максима-
раствора о-толуидина соотношение сигнал/шум
льный выход достигается при проведении реакции
(S/N) составило 10.4 (рис. 1в). Таким образом, в
в бензоле (осушенном над Na) с использованием
соответствии с ОФС.1.1.0012.15 [14] предел коли-
1.5-кратного мольного избытка хлорацетилхлорида
чественного определения
примеси о-толуидина
2. Установлено, что реакция ацилирования проте-
кала практически в момент смешивания реагентов:
(а)
после добавления хлорацетилхлорида 2 к раствору
о-толуидина 1 сразу же выпадал белый осадок,
следов исходного амина при этом обнаружить не
удалось. На втором этапе проводили аминирование
2-метиланилида хлоруксусной кислоты 3 диэтил-
амином 4, а мономекаин 6 был получен после
добавления по каплям концентрированной HNO3 к
раствору 2-метиланилида N,N-диэтиламиноуксус-
ной кислоты 5 в EtOH при 0°С. Чистоту и строение
синтезированных соединений подтверждали дан-
ными спектров
1Н ЯМР, элементного анализа,
ВЭЖХ, ТСХ.
Согласно современным требованиям, при оценке
(б)
качества лекарственных средств актуальными
являются хроматографические методы, которые
могут использоваться на разных стадиях анализа
субстанций и лекарственных форм [14-16]. Для
контроля чистоты мономекаина в проект
фармакопейной статьи нами предложена методика
с использованием метода ВЭЖХ. Поскольку моно-
мекаин и основные возможные родственные при-
меси [о-толуидин (1) и 2-метиланилид хлоруксус-
ной кислоты (3)] являются полярными соедине-
ниями, применение обращённо-фазовой ВЭЖХ
представлялось более предпочтительным. На
первом этапе исследования были найдены хрома-
(в)
тографические условия для анализа мономекаина и
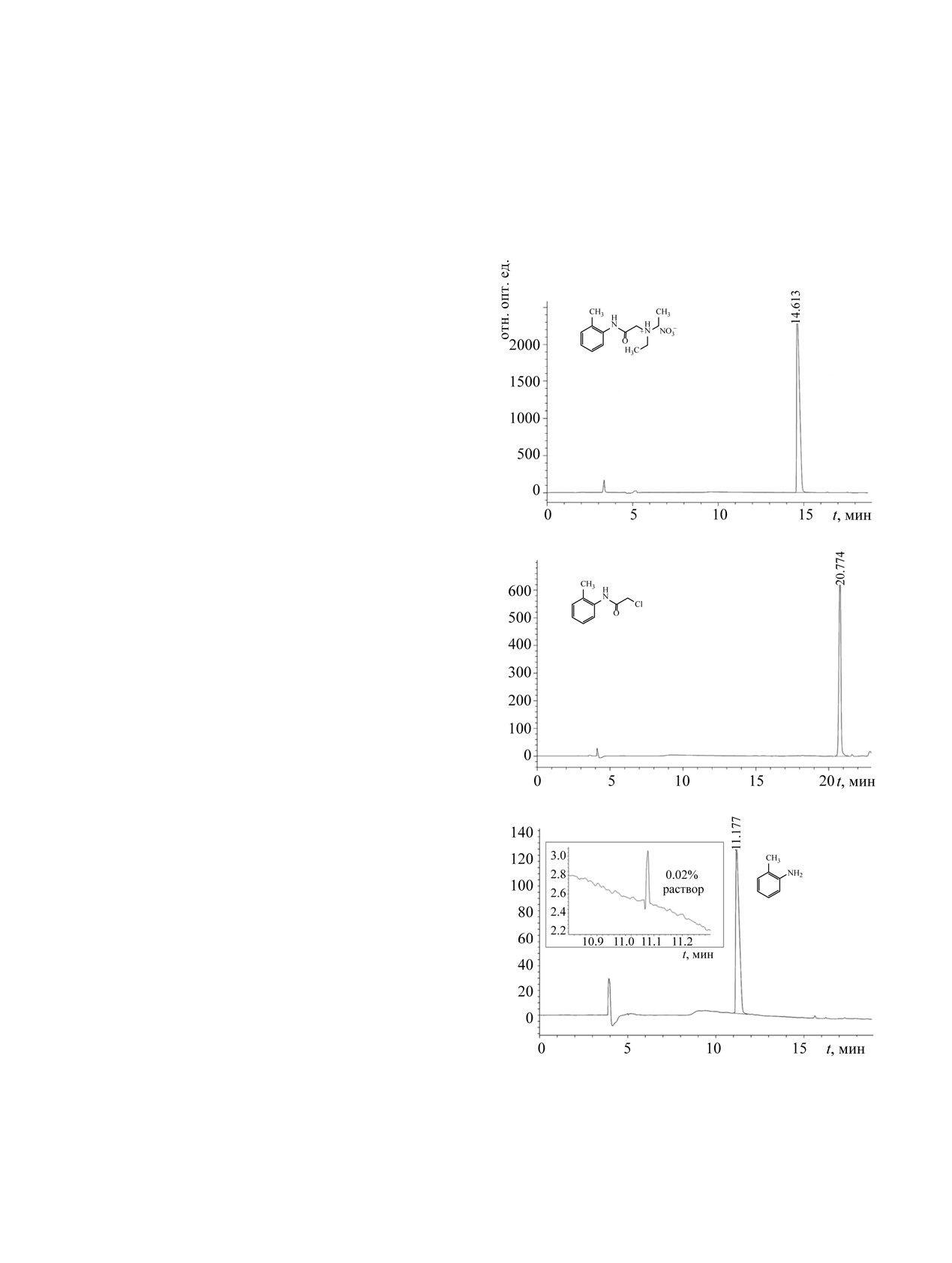
вышеуказанных примесей (рис. 1).
Показано, что полученные по разработанной
методике синтеза мономекаин и 2-метиланилид
хлоруксусной кислоты не содержали родственных
примесей. На следующем этапе с целью установ-
ления предела количественного определения
возможной примеси о-толуидина в образцах
субстанции мономекаина был проведен анализ
модельных растворов мономекаина, содержавших
0.01 и 0.02% о-толуидина.
Найденные хроматографические условия позво-
Рис. 1. Хроматограммы ВЭЖХ: (а) мономекаина, (б) 2-
лили успешно решить эту задачу ввиду значи-
метиланилида хлоруксусной кислоты, (в) 1 и 0.02%
тельного разрешения между пиками основного
раствора о-толуидина (во вставке). По оси абсцисс -
вещества и примеси (R = 8.8). Показано, что вели-
время, мин, по оси ординат - интенсивность сигнала,
чина пика 0.01% о-толуидина на хроматограмме
отн. опт. ед.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
1586
ЧЕКРЫШКИНА и др.
методом ВЭЖХ в образцах мономекаина был
ВЭЖХ также представлялось перспективным.
принят равным 0.02%. Анализ 4 лабораторных
Известно, что для использования методики ВЭЖХ
серий мономекаина, выполненный по разрабо-
в фармакокинетических исследованиях лекарст-
танной методике, выявил отсутствие примеси о-
венных форм мономекаина или для разработки
толуидина в исследуемых образцах. Данная
государственной фармакопейной статьи на его
методика также пригодна и для испытания
лекарственную форму необходима оценка её
мономекаина на подлинность из-за существенной
пригодности [15-17]. Поэтому в данной работе
разницы во времени удерживания мономекаина и
была выполнена валидация методики количест-
родственных соединений.
венного определения содержания основного
вещества в субстанции мономекаина. Для этого
Для количественного определения вышеуказан-
получали хроматограммы ВЭЖХ растворов
ного биологически активного соединения (БАС) в
мономекаина в различных концентрациях, а также
субстанции и потенциальных лекарственных
холостой пробы (без добавления мономекаина)
формах, разрабатываемых на стадии доклини-
(рис. 2). Валидацию аналитической методики коли-
ческих исследований, изучение метаболизма и фар-
чественного определения содержания мономекаина
макокинетики, возможности применения метода
в субстанции проводили по следующим харак-
теристикам: специфичность, линейность, анали-
тическая область, прецизионность и правильность.
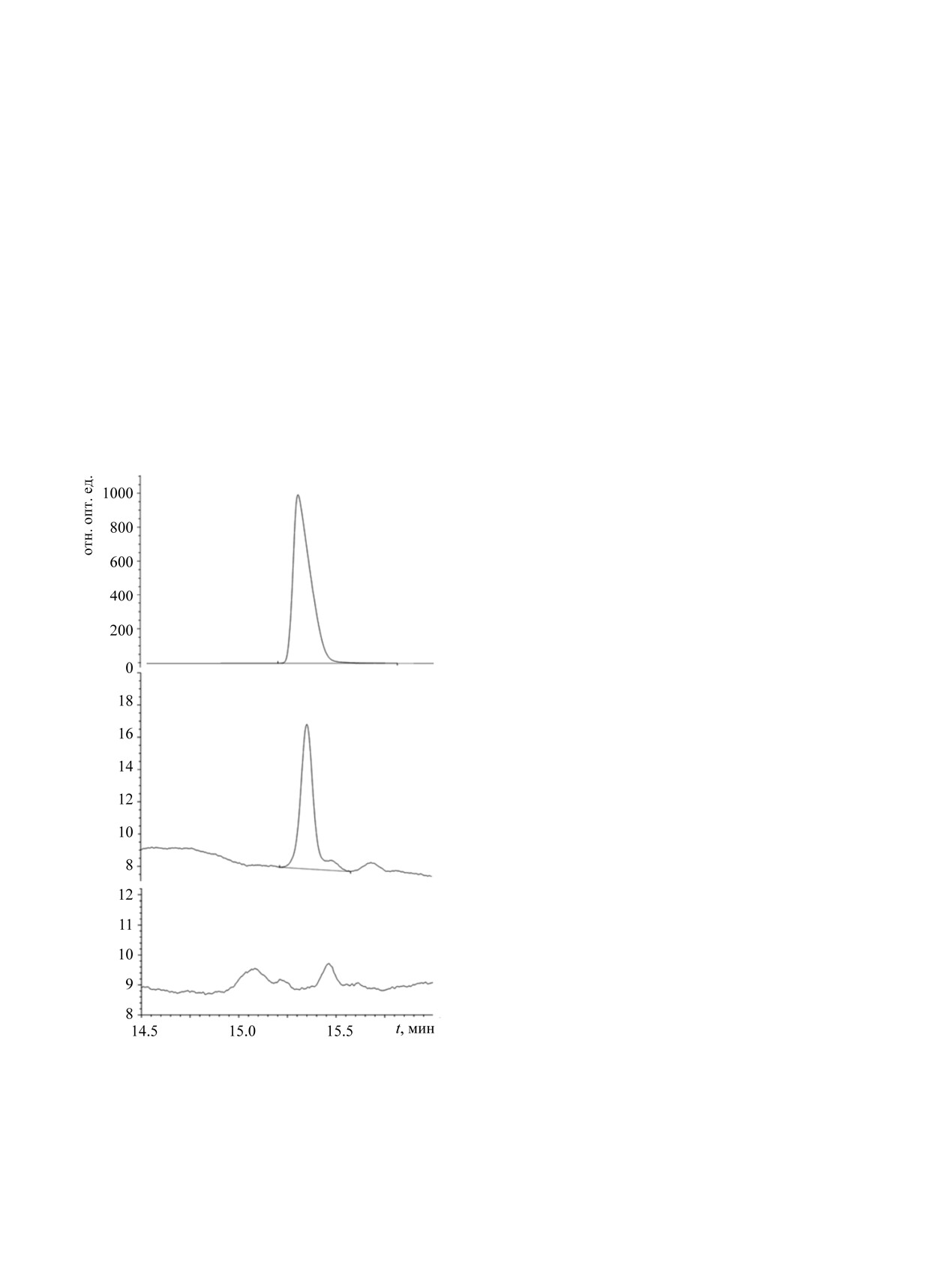
(а)
Для определения специфичности проводили
анализ подвижной фазы, используемой для приго-
товления испытуемых растворов субстанции моно-
мекаина. На хроматограмме образца подвижной
фазы не наблюдали пиков со временем удержи-
вания, соответствующим времени удерживания
анализируемого БАС (рис. 2в).
Для оценки линейности строили калибровочный
график (рис. 3) с использованием растворов суб-
(б)
станции мономекаина в
5 различных концент-
рациях: 0.001, 0.0107, 0.0535, 0.214 и 1.07 мг/мл.
Каждый раствор анализировали по описанной
выше методике в 3 параллелях. Установлено, что
зависимость площади хроматографического пика
от концентрации мономекаина в указанном диапа-
зоне концентраций линейна. Значение коэффи-
циента корреляции составляет 0.9997, а калибро-
вочный график описывается уравнением:
y = 26859.6 · x + 131
(1)
(в)
Аналитическую область методики определяли в
интервале от 80 до 120% от концентрации иссле-
дуемого вещества 1.00 мг/мл, принятой за 100%.
Для оценки прецизионности проводили статис-
тическую обработку результатов определения
концентраций мономекаина в разные рабочие дни.
Для этого выполняли анализ 3 модельных смесей,
Рис. 2. Хроматограммы ВЭЖХ растворов мономекаина
содержавших субстанцию мономекаина в различ-
(6) в концентрации: (а) 214 мкг/мл и (б) 2 мкг/мл, а
ных концентрациях - 80, 100, 120% от допустимого
также (в) холостой пробы, не содержащей мономе-
каина. По оси абсцисс - время, мин, по оси ординат -
содержания, что составляло 0.80, 1.00 и 1.20 мг/мл,
интенсивность сигнала, отн. опт. ед.
соответственно.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
СИНТЕЗ И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ НИТРАТА 2-МЕТИЛАНИЛИДА
1587
Для полученных значений концентраций были
рассчитаны величины стандартного отклонения
(Sδ) и относительного стандартного отклонения
(Sδ сред). Величина относительного стандартного
отклонения результатов количественного опреде-
ления содержания мономекаина в субстанции не
превышала 3% (табл. 2), что свидетельствует о
соответствии разработанной методики требуемым
критериям приемлемости.
Правильность методики оценивали на осно-
вании статистической обработки результатов ана-
лиза 3 лабораторных образцов субстанции моно-
мекаина. Рассчитывали и сравнивали с таблич-
ными значения критерия Стьюдента (t) при числе
Рис. 3. Калибровочный график зависимости площади
степеней свободы f = L - 1 = 4 и доверительной
пика, соответствующего мономекаину
(6), от его
концентрации в пробе. По оси абсцисс количество
вероятности Р = 0.95. Для всех образцов моно-
мономекаина, мг/мл, по оси ординат площадь
мекаина t < tтабл, следовательно, смещение незна-
хроматографического пика.
чимо на фоне случайного разброса и принято
равным 0, относительная ошибка среднего резуль-
основания, мономекаин образует с титрантом
тата определения (εср) не превысила 3% (табл. 3).
ассоциат, который экстрагируется в органичес-
Полученные данные позволили сделать вывод о
кую фазу, в качестве которой используется
том, что методика дает правильные воспроизво-
хлороформ.
димые результаты.
Основными факторами, влияющими на экс-
В целом, по результатам валидации методики
тракцию вещества в процессе титрования лаурил-
количественного определения мономекаина на
сульфатом натрия, являются значение рН среды
основе обращённо-фазовой ВЭЖХ установлено,
водной фазы, соотношение водной и органической
что она характеризуется специфичностью, отвечает
фаз, выбранный индикатор. Оптимальные условия
требованиям линейности, прецизионности и
титрования, связанные с данными факторами, уста-
правильности и может быть использована для
навливали при изменении одного из параметров и
аналитических целей.
при постоянстве всех остальных. Найдено, что
Следует также отметить, что для количест-
оптимальными являются условия, при которых
венного определения мономекаина в субстанции
значение рН среды для титрования должно нахо-
ранее была разработана методика на основе метода
диться в интервале 2.18-2.60, что соответствует
неводной ацидиметрии
[18]. В данном иссле-
исходной концентрации хлористоводородной кис-
довании нами представлена ещё одна методика
лоты 0.02 М; соотношение водной и органической
количественного определения этого соединения
(CHCl3) фаз составляет 1:1 и 1:2 соответственно;
методом экстракционного двухфазного титрования
используют смешанный индикатор - смесь диме-
лаурилсульфатом натрия. Как соль органического
тилового желтого и метиленового синего (5:1), при
Таблица 2. Оценка прецизионности ВЭЖХ методики количественного определения основного вещества в субстанции
мономекаина (6) (серия 250115).
Метрологические характеристики (P = 95%, f = 4)
Модельный раствор субстанции, мг/мл
¯, мг/мл
Sδ, мг/мл
Sδ сред, %
Δ¯, мг/мл
0.80
0.79
0.016
2.00
0.044
1.00
1.01
0.021
2.10
0.059
1.20
1.20
0.023
1.91
0.064
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
1588
ЧЕКРЫШКИНА и др.
Таблица 3. Результаты статистической обработки данных анализа 2 серий субстанции мономекаина (6) методом
ВЭЖХ.
Метрологические характеристики (P = 95 %, f = 4)
Серия субстанции
¯, мг/мл
Sδ, мг/мл
Sδ сред, %
Δ¯, мг/мл
¯, %
250115
1.01
0.0095
0.94
0.026
2.61
170314
1.00
0.0071
0.71
0.020
1.97
050713
0.99
0.0095
0.96
0.026
2.66
Таблица 4. Оценка прецизионности результатов титрования мономекаина (6) (серия 050713).
Метрологические характеристики (P = 95 %, f = 6)
Уровень содержания, %
¯, мг/мл
Sδ, мг/мл
Sδ сред, %
Δ¯, мг/мл
80
99.46
0.091
0.091
0.084
100
99.28
0.088
0.089
0.082
120
99.36
0.089
0.090
0.082
этом наблюдается четкий переход окраски в точке
Доказано, что полученные результаты имеют
эквивалентности от светло-желтой до голубой.
приемлемый уровень правильности и прецизион-
ности.
Пригодность предлагаемой методики количе-
ственного определения мономекаина оценивали на
Оценку прецизионности результатов проводили
основании валидации по показателям: линейность
на основании статистической обработки выборок,
результатов, аналитическая область методики,
полученных в ходе количественного определения
прецизионность и правильность.
исследуемого БАС на 3 уровнях концентрации в
пределах рекомендуемой аналитической области
Для установления линейной зависимости
методики. Относительное стандартное отклонение
осуществляли статистическую обработку выборки,
(Sδ сред) результатов количественного определения
полученной в результате количественного опре-
мономекаина не превышало 0.09% (табл. 4), что
деления навесок исследуемого БАС на 7 уровнях
соответствует критериям приемлемости при содер-
концентрации. Показано, что регрессия эквива-
жании вещества в исследуемом объекте, близком к
лентного объема титранта от содержания вещества
100%.
строго линейна (коэффициент корреляции равен
Правильность методики оценивали на основа-
0.9999), а калибровочный график выражается урав-
нии статистической обработки результатов титро-
нением прямой:
вания 3-х лабораторных образцов мономекаина.
Полученные результаты (табл. 5) позволили сде-
y = 355.5 · x - 0.028
(2)
лать вывод о том, что методика дает правильные
Аналитическую область методики определяли в
воспроизводимые результаты, найденное содер-
интервале от 80 до 120% от количества иссле-
жание мономекаина близко к 100%, а величины
дуемого вещества 0.03 мг/мл, принятого за 100%.
относительных погрешностей невелики.
Таблица 5. Результаты титрования 3 серий мономекаина (6).
Метрологические характеристики (P = 95 %, f = 6)
Серия мономекаина (6)
¯, %
¯, мг/мл
Sδ, мг/мл
Sδ сред, %
Δ¯, мг/мл
0.17
250712
99.35
0.068
0.026
0.063
0.21
050713
99.28
0.086
0.033
0.080
080813
99.35
0.096
0.036
0.089
0.24
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
СИНТЕЗ И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ НИТРАТА 2-МЕТИЛАНИЛИДА
1589
Методика экстракционного титрования имеет
долях, константы спин-спинового взаимодействия
определенное преимущество перед ранее
(КССВ) J - в герцах. Элементный анализ образцов
предложенной методикой титрования в неводной
проводили на автоматическом CHN-анализаторе
среде. Она позволяет определять количественное
Perkin Elmer РЕ
2400, серия II (США). Для
содержание основного вещества в присутствии
обращённо-фазовой ВЭЖХ использовали хрома-
продуктов разложения, существенным фактором
тограф Agilent 1100 (США), колонку Kromasil 100-
при этом, кроме названных ранее, является
5C18 (Швеция), 250×4.6 мм, 5 мкм, подвижные
молярная масса определяемого вещества, которая
фазы (ПФ): А - 0.1% водный раствор CF3COOH и
должна составлять не менее
200 г·моль-1.
Б - CH3CN, скорость элюирования 0.8 мл/мин.
Применение данной методики перспективно на
Подвижные фазы и времена удерживания соеди-
стадии разработки лекарственной формы моно-
нений (τ, мин) указаны в конкретных методиках.
мекаина, в частности, раствора для инъекций.
Для ТСХ использовали пластинки Sorbfil (OOO
«Имид», Россия), которые проявляли в УФ-свете и
Таким образом, оптимизирован метод синтеза
парами йода. Коммерчески доступные о-толуидин,
нитрата
2-метиланилида N,N-диэтиламиноуксус-
хлорацетилхлорид и диэтиламин с чистотой ч.д.а
ной кислоты, в частности, упрощена методика
использовали без дополнительной очистки.
синтеза 2-метиланилида хлоруксусной кислоты -
исходного соединения для получения солей
2-
2-Метиланилид хлоруксусной кислоты
(3).
метиланилида N,N-диэтиламиноуксусной кислоты.
Растворяли 1 мл (9.32 ммоль) о-толуидина в 10 мл
бензола (сух.), охлаждали до 0°С и по каплям
Показано, что максимальный выход
2-метил-
анилида хлоруксусной кислоты достигается при
добавляли 1.11 мл (14.0 ммоль) хлорацетилхло-
проведении реакции в 1 стадию в бензоле (осушен-
рида, при этом сразу выпадал белый осадок. Реак-
ционную массу упаривали, осадок промывали
ном над Na) при использовании
1.5-кратного
мольного избытка хлорацетилхлорида. Чистота и
водой до нейтрального значения pH и сушили.
строение синтезированных соединений подтвер-
Выход 1.31 г (76.7%), белые кристаллы, т.пл. 111-
113°С (102-104°С [13]). Спектр 1Н ЯМР, δ, м.д.:
ждены данными 1Н ЯМР спектров, элементного
анализа, ВЭЖХ и ТСХ. Разработана методика
2.20 с (3Н, СН3), 4.30 с (1Н, СН2), 7.10 д (1Н, Ar, J
установления подлинности мономекаина и конт-
7.3 Гц), 7.15 д (1Н, Ar, J 7.5 Гц), 7.20 д (1Н, Ar, J
7.4 Гц), 7.40 д (1Н, Ar, J 7.8 Гц), 9.65 с (1Н, NH). Най-
роля его чистоты в субстанции методом ВЭЖХ,
установлен предел обнаружения примеси о-
дено,
%: С
58.86; H
5.48; N
7.65; Cl
19.22.
C9H10ClNO. Вычислено, %: С 58.86; H 5.49; N 7.63;
толуидина в лабораторных образцах исследуемого
Cl 19.31. ВЭЖХ, режим элюирования: 0-3 мин 100%
соединения, который составил 0.02%. Для коли-
чественного определения содержания основного
ПФ А; 3-20 мин от 0 до 100 % ПФ Б; 20-22 мин 60%
ПФ Б, τ 20.77. ТСХ: бензол-EtOAc, 9:1, Rf 0.57.
вещества в субстанции мономекаина предложены
методики ВЭЖХ и экстракционного титрования.
2-Метиланилид N,N-диэтиламиноуксусной
Проведена валидация разработанных методик.
кислоты (5). Растворяли 9.10 г (49.56 ммоль) 2-
Показано, что величина относительного стандарт-
метиланилида хлоруксусной кислоты (3) при на-
ного отклонения (RSD) для мономекаина свиде-
гревании в 110 мл сухого бензола и добавляли
тельствует о хорошей специфичности, линейности,
12.64 мл (123.9 ммоль) диэтиламина. Реакционную
прецизионности и правильности предлагаемых
массу кипятили 5 ч. Выпавший осадок гидро-
аналитических методик. Это позволяет исполь-
хлорида диэтиламина отфильтровывали, раствор
зовать их для разработки государственной фарма-
упаривали. Полученный продукт использовали в
копейной статьи на лекарственную форму моно-
дальнейшем синтезе без дополнительной очистки.
мекаина.
Выход
9.69 г
(88.8%), коричневое подвижное
масло. Спектр
1Н ЯМР, δ, м.д.: 1.05 д
(6Н,
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2СН2CH3, J 7.13 Гц), 2.23 с (3Н, СН3), 2.61 д (4Н,
2СН2CH3, J 7.1 Гц), 3.14 с (2Н, СН2), 7.03 д.д (1Н,
Спектры ЯМР (δ, м.д., J, Гц) регистрировали на
Ar, J 7.5, 1.1 Гц), 7.18 д.д (1Н, Ar, J 7.6, 1.1 Гц),
приборе Bruker Avance
500
(500 МГц для 1Н,
7.22 д (1Н, Ar, J 7.4 Гц), 7.90 д (1Н, Ar, J 7.9 Гц),
Германия) ДМСО-d6 с SiMe4 в качестве внутрен-
9.47 с (Н, NH). Найдено, %: С 70.46; H 9.06; N
него стандарта при комнатной температуре. Хи-
13.53. C13H20N2O. Вычислено, %: C 70.87; H 9.15; N
мические сдвиги (δ) приведены в миллионных
12.72. ТСХ: бензол-EtOAc, 9:1, Rf 0.40.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
1590
ЧЕКРЫШКИНА и др.
Нитрат 2-метиланилида N,N-диэтиламиноук-
образца мономекаина, %; Р - содержание основ-
сусной кислоты (6). Растворяли 9.69 г (43.98 ммоль)
ного вещества в субстанции стандартного образца
2-метиланилида N,N-диэтиламиноуксусной кис-
мономекаина, %.
лоты (5) в 9 мл EtOH, охлаждали до 0°С и добав-
Методика количественного определения нит-
ляли по каплям 2.96 мл (43.98 ммоль) конц. HNO3
рата 2-метиланилида N,N-диэтиламиноуксусной
(67%), в течение 5 мин выпадал белый кристал-
кислоты
(6) методом экстракционного двух-
лический осадок. Реакционную массу упаривали,
фазного титрования лаурилсульфатом натрия.
кристаллический продукт перекристаллизовывали
Для проведения анализа 0.03 г образца субстанции
из ацетона, сушили. Выход 9.03 г (78.5%), белый
мономекаина (6), предварительно высушенного до
кристаллический продукт, т.пл.
138.5-139.5°С.
постоянной массы при температуре 100°С, поме-
Спектр 1Н ЯМР, δ, м.д.: 1.24 д (6Н, 2СН2CH3, J
щали в коническую колбу вместимостью 100 мл,
7.2 Гц), 2.23 с (3Н, СН3), 3.23 д (4Н, 2СН2CH3, J
растворяли в 10 мл 0.02 М HCl, добавляли 10 мл
6.9 Гц), 4.15 с (2Н, СН2), 7.16 д.д (1Н, Ar, J 7.4,
CHCl3, 2 капли раствора смешанного индикатора
1.2 Гц), 7.22 д.д (1Н, Ar, J 7.5, 1.2 Гц), 7.27 д (1Н,
(диметиловый желтый - метиленовый синий, 5:1) и
Ar, J 7.5 Гц), 7.42 д (1Н, Ar, J 7.7 Гц), 9.45 уш.с
титровали при энергичном встряхивании 0.01 М
(1Н, HNО3), 9.96 с (1Н, NH). Найдено, %: С 55.23;
раствором лаурилсульфата натрия до изменения
H 7.73; N 14.70. C13H21N3O4. Вычислено, %: C
окраски органического слоя от светло-желтой до
55.11; H 7.47; N 14.83. ВЭЖХ, режим элюирова-
голубой. Анализ проводили в 2 параллелях.
ния: 0-3 мин. 5% ПФ Б; 3-20 мин от 5 до 60%
ПФ Б; 20-22 мин 60% ПФ Б, τ 14.61. ТСХ: бензол-
БЛАГОДАРНОСТИ
диизопропилэтиламин, 3:0.05, Rf 0.30.
Методика количественного определения нит-
Авторы выражают благодарность к.х.н. М.И.
рата 2-метиланилида N,N-диэтиламиноуксусной
Кодессу за регистрацию спектров ЯМР.
(6) кислоты методом ВЭЖХ. В мерную колбу
вместимостью
25 мл переносили
0.025 г суб-
ФОНДОВАЯ ПОДДЕРЖКА
станции мономекаина (6), колбу заполняли смесью
95% ПФ А и 5% ПФ Б на 2/3 объема и переме-
Работа выполнена в рамках темы государствен-
шивали до полного растворения субстанции. Объем
ного задания (АААА-А19-119011790130-3) с исполь-
раствора доводили до метки смесью 95% ПФ А и
зованием оборудования Центра коллективного
5% ПФ Б, перемешивали и фильтровали через
пользования «Спектроскопия и анализ органичес-
мембранный фильтр с размером пор 0.45 мкм, от-
ких соединений» (ЦКП «САОС», ИОС УрО РАН).
брасывая первые порции фильтрата. Вводили 20
мкл полученного раствора в колонку
КОНФЛИКТ ИНТЕРЕСОВ
аналитического жидкостного хроматографа. Режим
элюирования: 0-3 мин 95% ПФ А - 5% ПФ Б; от 3
Авторы заявляют об отсутствии конфликта
до 20 мин изменение состава ПФ от 5 до 60% Б;
интересов.
20-23 мин 40% ПФ А - 60% ПФ Б, τ мономекаина
составляет 14.9-15.4 мин (рис. 2).
СПИСОК ЛИТЕРАТУРЫ
Содержание мономекаина рассчитывали по
1. Camm A.J. Int. J. Cardiol. 2017, 237, 71. doi 10.1016/
формуле:
j.ijcard.2017.03.056
S1 · a0 · 25 · P
S1 · a0 · P
,
(3)
2. Nadimi A.E., Ebrahimipour S.Y., Afshar E.G.,
X =
=
S0 · a1 · 25
S0 · a1
Falahati-Pour S.K., Ahmadi Z., Mohammadinejad R.,
Mohamadi M. Eur. J. Med. Chem. 2018, 157, 1153. doi
- среднее значение площади пика моно-
где S1
10.1016/j.ejmech.2018.08.080
мекаина на хроматограмме испытуемого раствора;
3. Kalinin D.V., Pantsurkin V.I., Syropyatov B.Ya.,
- среднее значение площади пика мономекаина
S0
Kalinina S.A., Rudakova I.P., Vakhrin M.I.,
на хроматограмме раствора стандартного образца
Dolzhenko A.V. Eur. J. Med. Chem. 2013, 63, 144. doi
субстанции мономекаина; a0
- навеска стандартного
10.1016/j.ejmech.2013.02.003
образца субстанции мономекаина; a1
- навеска
4. Neumeyer J.L., Perianayagam C., Ruchirawat S.,
субстанции испытуемого образца; Х - содержание
Feldman H.S., Takman B.H., Tenthorey P.A. J. Med.
основного вещества в субстанции испытуемого
Chem. 1977, 20, 894. doi 10.1021/jm00217a005
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019
СИНТЕЗ И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ НИТРАТА 2-МЕТИЛАНИЛИДА
1591
5. Corbo F., Franchini C., Lentini G., Muraglia M.,
kova I.P., Syropyatov B.J. Pharm. Chem. J. 2017, 51,
Ghelardini C., Matucci R., Galeotti N., Vivoli E.,
641.] doi 10.1007/s11094-017-1667-3
Tortorella V. J. Med. Chem. 2007, 50, 1907. doi
11. Гашкова О.В. Дис. … канд. фарм. наук. Пермь.
10.1021/jm061078e
2009.
6. Franchini C., Corbo F., Lentini G., Bruno G.,
12. Mao J., Chen L.L. Pain. 2000, 87, 7. doi 10.1016/
Scilimati A., Tortorella V., Camerino D.C., De Luca A.
S0304-3959(00)00229-3
J. Med. Chem. 2000, 43, 3792. doi 10.1021/jm000931l
13. Кудряшова Н.И., Ремизов А.Л., Хромов-Борисов Н.В.
7. Хайруллина В.Р., Тарасов Г.П., Герчиков А.Я.,
ЖОХ. 1959, 29, 1240.
Зарудий Ф.С., Тюрина Л.А. Биомед. хим. 2010, 56,
14. ОФС.1.1.0012.15. Валидация аналитических мето-
471. [Khairullina V.R., Tarasov G.P., Gerchikov A.Ya.,
дик. Государственная фармакопея Российской
Zarydiy F.S., Tyurina L.A. Biochem. Moscow Suppl.
Федерации, ХIV изд. Т.I. М.: НЦЭСМП. 2018.
Ser. B. 2010, 4, 130.] doi 10.1134/S1990750810020034
15. Эпштейн Н.А. Хим.-фарм. ж.
2004,
38,
40.
8. Шепель Ф.Г., Зозуля Р.Н., Шепель Д.Ф., Макаев Ф.З.
[Épshtein N.A. Pharm. Chem. J. 2004, 38, 212.] doi
Хим.-фарм. ж. 2010, 44, 3. [Shepel’ F.G., Zozulya R.N.,
10.1023/B:PHAC.0000038422.27193.6c
Shepel’ D.F., Makaev F.Z. Pharm. Chem. J. 2010, 44,
469.] doi 10.1007/s11094-010-0493-7
16. Pendela M., Kahsay G., Baekelandt I., Van Schepdael A.,
Adams E. J. Pharm. Biomed. Anal. 2011, 56, 641. doi
9. Гашкова О.В., Панцуркин В.И., Рудакова И.П.,
10.1016/j.jpba.2011.06.028
Сыропятов Б.Я., Вахрин М.И. Хим.-фарм. ж. 2008,
42, 8. [Gashkova O.V., Pantsurkin V.I., Rudakova, I.P.,
17. Zivanovic Lj., Zecevic M., Markovic S., Petrovic S.,
Syropyatov B.J., Vahrin M.I. Pharm. Chem. J. 2008, 42,
Ivanovic I. J. Chromatogr. A. 2005, 1088, 182. doi
665.] doi 10.1007/s11094-009-0217-z
10.1016/j.chroma.2005.04.049
10. Гашкова О.В., Рудакова И.П., Сыропятов Б.Я.
18. Чекрышкина Л.А., Бабикова Е.А., Слепова Н.В.
Хим.-фарм. ж. 2017, 51, 9. [Gashkova O.V., Ruda-
Фундам. исслед. 2015, 2, 3333.
Synthesis and Quantification
of N,N-Diethylaminoacetic Acid 2-Methylanilide Nitrate
L. A. Chekryshkinaa, A. M. Deminb, *, A. A. Tumashovb, c,
Е. А. Babikovad, and N. V. Slepovaa
a Perm state pharmaceutical Academy Ministry of health of Russia, 614990, Russia, Perm, ul. Polevaya 2
b Postovsky Institute of Organic Synthesis of RAS (Ural Branch), 620990, Russia, Yekaterinburg, ul. S. Kovalevskoy 22/20
c Institute of Natural Sciences and Mathematics, Ural Federal University, 620002, Russia, Yekaterinburg, ul. Mira 19
d Sverdlovsk Regional Medical College (Pharmaceutical branch), 620014, Russia, Yekaterinburg, ul. Bebelya 71
*e-mail: demin@ios.uran.ru
Received April 15, 2019; revised August 14, 2019; accepted August 15, 2019
The synthetic method of N,N-diethylaminoacetic acid 2-methylanilide nitrate (named as “monomekaine”), a
promising antiarrhythmic drug, has been optimized. We developed the method for purity determination of this
compound using a high performance liquid chromatography (HPLC). A detection limit of o-toluidine as impurity
in monomekaine samples was 0.02%. For monomekaine quantification in substance we suggested technics based
on HPLC and on extraction titration methods, and validated them. The relative standard deviation (RSD) values
for monomekaine indicated a good specifity, linearity, precision and accuracy of the developed technics.
Keywords: N-diethylaminoacetic acid 2-methylanilide, monomekaine, synthesis, HPLC, extraction titration,
validation
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 10 2019