ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2019, том 55, № 12, с. 1942-1946
КРАТКИЕ СООБЩЕНИЯ
УДК 547.852.2
СИНТЕЗ НОВЫХ ПРОИЗВОДНЫХ ИМИНОСАХАРОВ
НА ОСНОВЕ (S)-(1,2,3,6-ТЕТРАГИДРОПИРИДАЗИН-
3-ИЛ)МЕТАНОЛА
© 2019 г. Ф. Н. Ахундоваa, *, М. М. Курбановаa, А. Э. Гусейнзадеa, М. Ж. Алвесb
a Бакинский Государственный Университет, 1148, Азербайджан, г. Баку, ул. З. Халилова 23
b Университет Миньо, 4710-057, Брага, Португалия
*e-mail: kurbanova1972@rambler.ru
Поступила в редакцию 21 мая 2019 г.
После доработки 21 октября 2019 г.
Принята к публикации 24 октября 2019 г.
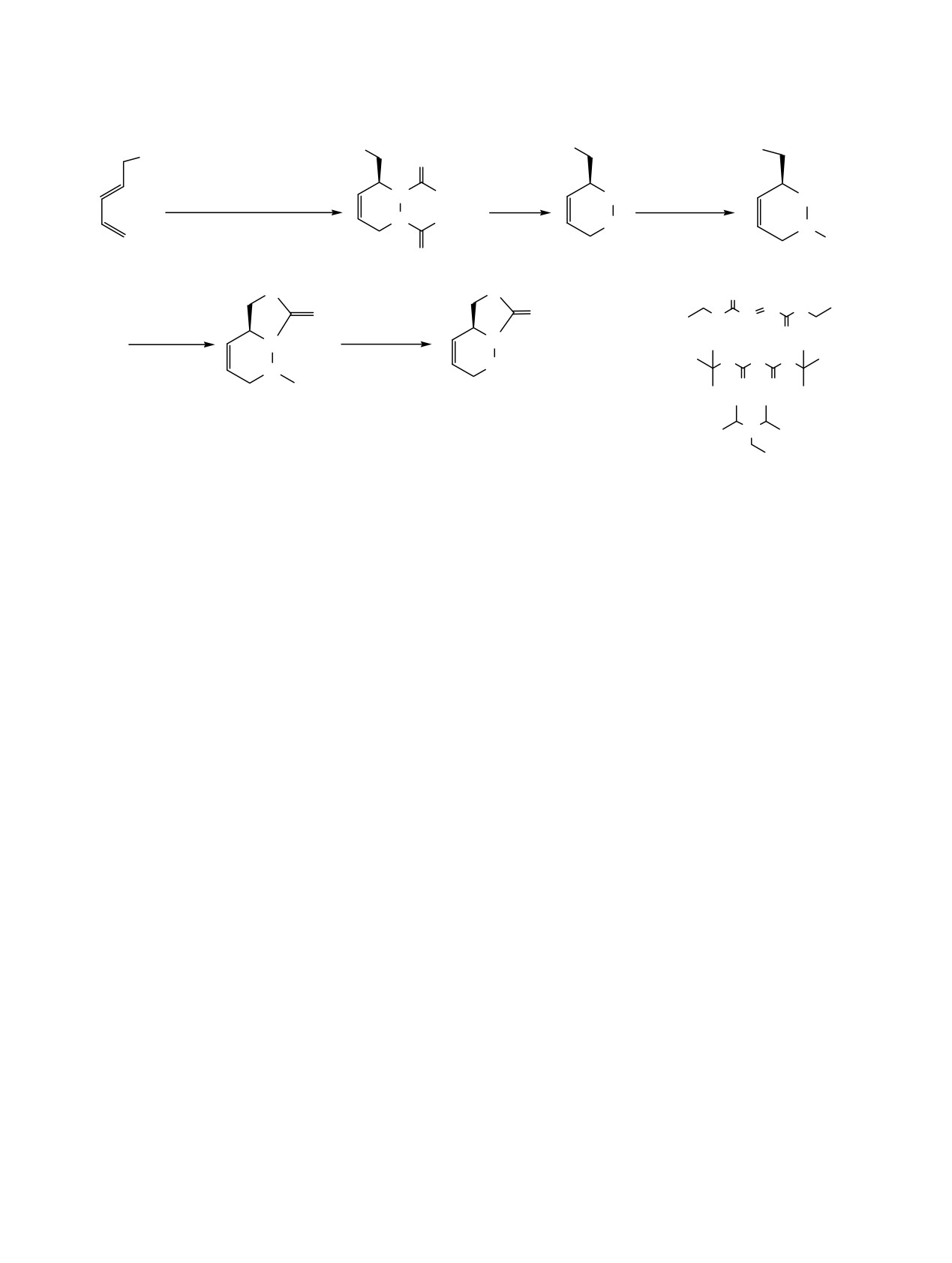
(S)-(1,2,3,6-Тетрагидропиридазин-3-ил)метанол синтезировали по реакции Дильса-Альдера в при-
сутствии хирального катализатора S-бинол в 2 стадии. Далее, блокировка N2 положения синтези-
рованного пиридазина Boc-группой с последующим карбонилированием гидроксильной группы и
удалением защитной Boc-группы приводит к образованию бициклических аналогов иминосахаров.
Структуры синтезированных продуктов были доказаны ЯМР, ИК, масс-спектроскопическими методами,
а также элементным анализом.
Ключевые слова: пиридазин, иминосахара, 1-азафагомин, реакция Дильса-Альдера, S-бинол, Boc-
блокировка.
DOI: 10.1134/S0514749219120206
Азасахара или иминосахара, как класс орга-
тонированной форме [8]. Этот факт способствовал
нических соединений с атомом азота в цикле,
синтезу новых различных аналогов иминосахаров,
являются предметом пристального интереса в
что привело к развитию методов синтеза ука-
настоящее время [1-3]. Причина этого заключается
занного класса соединений. Параллельно про-
в том, что исследования позволили обнаружить ин-
водились in vivo исследования, которые позволили
тересную структурную особенность данного класса
выявить наличие широкого спектра биологической
соединений. Было установлено, что незначите-
активности у данного класса соединений: проти-
льное перемещение азота в псевдоаномерное по-
вовирусной, антибактериальной, антипротозойной,
ложение (которое соответствует аномерному по-
противопаразитарной, антипсориатической, про-
ложению в моносахариде) в классическом имино-
тивогрибковой, нематоцидной, инсектицидной,
сахарингибиторе типа нойримицина способствует
противовоспалительной и противоопухолевой [10-
усилению ингибиторных свойств многократно, что
16]. Экспериментально было доказано, что инги-
в свою очередь способствовало выделению таких
биторы гликозидазы потенциально полезны при
типов ингибиторов глюкозидазы в отдельный класс,
лечении лизосомальных нарушений памяти
так называемые, 1-азасахара [4, 5]. Исследования
(болезни Гоше, Фабри, Сандхоффа или Тея-Сакса),
позволили выявить, что в ряду этого класса
а также могут использоваться в качестве анти-
соединений 1-азафагомин интенсивно ингибирует
диабетических, противомикробных, противо-
как α-, так и β-глюкозидазу [6, 7]. Причина био-
раковых и иммунодепрессивных препаратов [8, 9].
логической активности 1-азафагомина обусловлена
Очевидно, что область применения иминосахаров
тем, что он имитирует переходные состояния
стала очень интересной как для химических, так и
расщепления α-глюкозида и β-глюкозида в про-
для биологических исследований.
1942
СИНТЕЗ НОВЫХ ПРОИЗВОДНЫХ ИМИНОСАХАРОВ
1943
Схема 1.
HO
HO
HO
OH
O
N OC2H5
NH
NH
DEAD
NaOH
NaOH, Boc2O
Me2Zn, (S)-BINOL, MeMgBr
N OC2H5
ТГФ
C2H5OH
NH
N
Boc
O
1
2
3
4
O
O
O
O
O
N O
DIPEA,
DEAD — O N
трифосген
CF3COOH
O
N
N
CH2Cl2
CH2Cl2
O O O
NH
N
Boc2О —
Boc
O O
5
6
DIPEA —
N
Учитывая важность иминосахаров, мы пос-
(Е)-2,4-Пентадиенол (1). Получен по известной
тавили цель синтезировать новые бициклические
методике [16]. Выход 5 г (31.4%). Спектр ЯМР 1H
производные 1-азафагомина на основе (S)-(1,2,3,6-
(CDCl3), δ, м.д.: 3.12 уш.с (1H, OH), 4.07 м (2H,
тетрагидропиридазин-3-ил)метанола (3).
CH2OH), 5.01 д (1H, CH, J 9.6 Гц), 5.13 д (1H, CH, J
OH, J 14.7, 5.7 Гц),
16.4 Гц), 5.74 д.т (1H, CH=CH2
Исходным продуктом синтеза соединения
3
6.25 м (2H, CH2=CH). Спектр ЯМР 13С (CDCl3), δ,
служит 2,4-пентадиенол, который получают из 2,4-
м.д.: 62.3 (CH2), 116.9 (CH2), 131.2 (CH),
132.5
пентадиеновой кислоты [17]. Дальнейшее цикло-
(CH), 136.2 (CH). Найдено, %: C 71.47; H 9.48.
присоединение диэтилазадикарбоксилата (DEAD)
C5H8O. Вычислено, %: C 71.42; H 9.52.
и
2,4-пентадиенола в присутствии хирального
катализатора S-BINOL приводит к оптически чис-
(S)-Диэтил-3-(гидроксиметил)пиридазин-1,2-
(3H,6H)-дикарбоксилат (2). Соединение 2 полу-
тому циклоаддукту 2 (конфигурация S) [18]. Уда-
чают из двух различных растворов (A и Б).
ление карбаматов в соединении 2 осуществляли
при кипячении в диоксане в присутствии 2 M
Раствор A. 1.2 M Раствор Me2Zn в толуоле
раствора гидроксида натрия в течение 3 ч с пос-
(991 мкл, 1.19 ммоль) добавляют к раствору 0.100 г
ледующей обработкой кислотной смолой (амбер-
(1.19 ммоль) пента-2,4-диен-1-ола
(1) в сухом
лит). Чистое соединение 3 было получено с выхо-
толуоле (6 мл) при 0°C и перемешивают в течение
дом 91% перекристаллизацией из ацетона (схема 1).
5 мин.
Чтобы сохранить положение N1 в качестве
Раствор Б. 1.4 M Раствор MeMgBr в толуоле/ТГФ
единственного нуклеофильного центра в молекуле
(849 мкл,
1.19 ммоль) добавляют к
0.340 г
для дальнейшей циклизации в реакции с три-
(1.19 ммоль) раствора (S)-бинол в сухом толуоле
фосгеном, положение N2 было блокировано Boc-
(6 мл) при 0°C и перемешивают в течение 5 мин.
группой, в результате чего получали новый про-
Затем раствор A разбавляют в сухом толуоле (10 мл),
дукт N-Boc-1-азафагомин 4 с выходом 93%. Обра-
добавляют к раствору Б, перемешивают в течение
ботка соединения 4 трифосгеном в присутствии
5 мин и затем охлаждают при -78°C. Далее к этой
DIPEA в инертной атмосфере дает новый целевой
смеси добавляют раствор диэтилазадикарбоксилата
продукт 5 всего за 5 мин с выходом 90% после
(543 мкл, 1.19 ммоль) в сухом толуоле (10 мл).
очистки колоночной хроматографией. Удаление
Температуре позволяли постепенно повышаться до
Boc-группы от соединения 6 проводили в дихлор-
комнатной и реакционную смесь перемешивали в
метане добавлением ТГФ при комнатной темпе-
течение 18 ч. Реакцию гасили насыщенным раст-
ратуре, что, в конечном счете, приводило к целе-
вором NaHCO3 (1 мл), раствор фильтровали и про-
вому продукту.
мывали этилацетатом (3×20 мл). Далее фильтрат
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 12 2019
1944
АХУНДОВА и др.
объединяли и растворитель отгоняли под вакуу-
вали и тщательно промывали этанолом. Раствор кон-
мом, получая желтое масло. Очистку масла про-
центрировали, получая желтое твердое вещество.
водили колоночной хроматографией (петролейный
Впоследствии желтый осадок растворяли в ацетоне,
эфир-диэтиловый эфир, 1:1). (S)-бинол также реге-
фильтровали, раствор испаряли, получая желтое
нерировали в ходе очистки (0.200 г, 69%). Продукт
масло, выход 0.077 г (93%). [α]
20 +67° (конц. 0.78%
2 получают в виде желтого масла, выход
0.225 г
в дихлорметане). ИК спектр, ν, см-1: 3269 (OH), 1694
(73%). [α]
20 -23.4° (конц. 1.25% в CHCl3). ИК спектр,
(CO). Спектр ЯМР 1H (D2O), δ, м.д.: 1.50 с [9H,
ν, см-1 : 3483 (OH), 1707 (C=O). Спектр ЯМР 1H
C(CH3)3], 3.56-3.68 м (3H, H3, H3'), 3.96 уш.д (1H,
(CDCl3) δ, м.д.: 1.23-1.30 м (12H, 4CH3), 2.58 уш.с
H6, J 10.0 Гц), 4.09 уш.д (1H, H6, J 10.0 Гц), 5.90
(1H, OH), 3.35 д.д (1H, H3', A, J 12.3, 9.5 Гц), 3.45
уш.д (1H, H4, J 8.8 Гц), 5.96 уш.д (1H, H5, J 8.8 Гц).
д.д (1H, H3', B, J 12.0, 9.8 Гц), 3.56-3.69 м (2H, 2H3',
Спектр ЯМР 13С (D2O), δ, м.д.: 27.6 [C(CH3)3], 55.9
A + B), 3.77 д.д (1H, H6, A, J 13.5, 4.3 Гц), 3.91 уш.с
(C3), 61.8 (C3'), 82.7 (C=O), 125.0 (C5), 125.1 (С4),
(1H, H6, B), 4.11-4.26 м (8H, 4CH2, A + B), 4.30
156.8 (C=O). HRMS (ESI):
214.1317 (M
+ H).
т.д.д (1H, H6, B, J 6.0, 3.9, 2.2 Гц), 4.34-4.44 м (1H,
Найдено, %: C 56.11; H 8.37; N 13.05. C10H18N2O3.
H6, A), 4.72 уш.с (2H, H3, A + B), 5.66-5.88 м (4H,
Вычислено, %: C 56.07; H 8.41; N 13.08.
H4 + H5, A + B). Спектр ЯМР 13С (CDCl3), δ, м. д.:
Четвертичный бутил-(S)-7-оксо-4a,5-дигидро-
14.30 (CH3, A), 14.40 (CH3, B), 42.20 (C6, A), 43.60
7H-оксазоло[3,4-b]пиридазин-1(2H)-карбокси-
(C6, B), 55.90 (C3, A), 56.90 (C3, B), 61.90 (C3', A +
лата (5). К смеси 0.114 г (0.532 ммоль) соединения
B), 62.60, 62.70, 62.80, 62.90 (CH2, A + B), 123.40,
4 в сухом дихлорметане (10 мл) добавляли DIPEA
124.20, 124.60, 125.20 (C4 или C5, A + B), 154.90,
(10 экв., 0.93 мл, 0.532 ммоль) и трифосген (0.5 экв.,
155.70, 156.20, 156.30 (C=O, A + B). HRMS (ESI):
0.077 г, 0.532 ммоль). Смесь перемешивали при
281.1108 (M + Na). Найдено, %: C 51.21; H 6.91; N
комнатной температуре в течение 5 мин в среде
10.81. C11H18N2O5.Вычислено, %: C 51.16; H 6.97; N
азота. Далее растворитель отгоняли и остаток очи-
10.85.
щали колоночной хроматографией (EtOAc-петро-
(S)-(1,2,3,6-Тетрагидропиридазин-3-ил)мета-
лейный эфир, 1:1), получая желтое масло, выход
нол (3). К раствору 0.119 г (0.461 ммоль) цикло-
0.077 г (90%). [α]
20 +30° (конц. 0.4% дихлорметан).
аддукта 2 в ТГФ (2 мл) добавляли 2 мл 2 M
ИК спектр, ν, см-1: 1717 (C=O). Спектр ЯМР 1H
раствора NaOH. Смесь кипятили в течение 3 ч. По
(ДМСО-d6), δ, м.д.: 3.46 д (1H, J 11.2, 7.2 Гц), 3.55
истечении времени смесь охлаждали и добавляли
д.д (1H, J 11.2, 4.8 Гц), 3.71 уш.с (1H), 4.02 д.д (2H,
ТГФ (2 мл) и суспензию Амберлита (H+) в воде.
J 4.8, 2.4 Гц), 5.86 д.д.д (1H, J 8.4, 4.8, 2.0 Гц), 5.89
Смесь перемешивали, а затем сразу же фильтро-
д.м (1H, J 8.8 Гц). Спектр ЯМР 13С (ДМСО-d6), δ,
вали в вакууме. Далее растворитель отгоняли, в
м.д.: 27.4, 27.7, 27.8, 27.9 (CH3), 43.3, 43.8 (CH2N),
результате получали желтое масло, выход 0.048 г
53.0, 55.6, 56.2 (CH), 59.9, 60.4 (CH2O), 81.6, 83.0
(91.0%). [α]
20 -20° (конц. 0.3% в EtOH). ИК спектр,
(C=O), 123.8-124.5 (CHаром), 152.6 (CO), 153.6 (CO).
ν, см-1: 3422 (N-H), 1643 (C=C). Спектр ЯМР 1H
HRMS (ESI): 240.0793 (M + H). Найдено, %: С
(D2O), δ, м.д.: 3.24 д.д.д (1H, H6, J 2.6, 3.2, 17.2 Гц),
55.05; H 6.63; N 11.69. C11H16N2O4. Вычислено, %:
3.35 д.д.д (1H, H6, J 2.8, 5.2, 17.6 Гц), 3.48-3.54 м
C 55.00; H 6.66; N 11.66.
(1H, H3), 3.57-3.65 м (2H, H3'), 5.79 д.д.д (1H, H4, J
(S)-1,2,4a,5-Тетрагидро-7H-оксазоло[3,4-b]пи-
2.0, 4.4, 10.4 Гц), 6.01 д.д.д (1H, H5, J 2.4, 5.6, 10.4 Гц).
ридазин-7-он (6). К раствору 0.138 г (0.574 ммоль)
Спектр ЯМР 13С (D2O) δ, м.д.: 44.2 (C6), 54.8 (C3),
соединения 5 в дихлорметане (11 мл) добавляли
62.5 (C3'), 125.0 (C4), 127.4 (C5). HRMS (ESI):
2.76 мл (0.574 ммоль) ТФА и далее перемешивали
114.0793 (M + H). Найдено, %: C 52.67; H 8.74; N
в течение 2 ч при комнатной температуре. По окон-
24.51. C5H10N2O. Вычислено, %: C 52.63; H 8.77; N
чании времени реакции добавляли насыщенный
24.56.
раствор NaHCO3 и полученный раствор экстра-
Четвертичный бутил-(S)-3-(гидроксиметил)-
гировали EtOAC (3×30 мл). Впоследствии орга-
3,6-дигидропиридазин-1(2H)-карбоксилат (N-Boc-
нический слой сушили MgSO4 и отгоняли раство-
1-азафагомина) (4). К смеси 0.044 г (0.385 ммоль)
ритель, получали желтое масло. Применение коло-
соединения 3 в этаноле (10 мл) добавляли 0.015 г
ночной хроматографии позволяет более глубоко
(0.385 ммоль) NaOH и 0.015 г (0.385 ммоль) Boc2O.
очистить полученный продукт (EtОAc-петролей-
Реакционную смесь перемешивали при комнатной
ный эфир, 2:1), выход 0.061 г (76%). ИК спектр, ν,
температуре в течение 1 ч. Далее смесь фильтро-
см-1: 2926 (N-H), 1788 (C=C), 1729 (C=O). Спектр
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 12 2019
СИНТЕЗ НОВЫХ ПРОИЗВОДНЫХ ИМИНОСАХАРОВ
1945
ЯМР 1H (CDCl3), δ, м.д.: 3.90-4.03 д.д.д (1H, J 18.4,
5. Ichikawa Y., Igarashi Y., Ichikawa M., Suhura Y.
3.6, 2.0 Гц), 4.20-4.26 м (2H), 4.45-4.50 м (1H),
J. Am. Chem. Soc. 1998, 120, 3007. doi 10.1021/ja985509x
4.52-4.58 м (1H), 4.62 д.д (1H, J 8, 1.2 Гц), 5.87
6. Bols M., Hazell R., Thomsen I. Chem. Eur. J. 1997, 3,
уш.д (1H, J 8.4 Гц, HC), 6.00-6.10 м (1H, HD).
940. doi 10.1002/chem.19970030616
Спектр ЯМР 13С (CDCl3), δ, м.д.: 39.7 (CH2N), 47.6
7. Ernholt B.V., Thomsen I.B., Lohse A., Jensen K.B.,
(CH2N), 52.9, 53.7 (CH), 67.10, 67.12 (CH2O), 124.3,
Hazell R.G., Plesner I., Liang X., Jacobsen A., Bols M.
Chem. Eur. J. 2000, 6, 278. doi 10.1002/(SICI)1521-
124.7, 125.8, 125.9 (CH), 156.4-156.6 (CO). HRMS
3765(20000117)6:2<278::AID-CHEM278>3.0.CO;2-6
(ESI): 140.0586 (M + H). Найдено, %: С 51.42; H
8. Compain P., Martin O.R. Iminosugars: From Synthesis
5.75; N 19.99. C6H8N2O2. Вычислено, %: C 51.48; H
to Therapeutic Applications. Chichester: Wiley,
5.81; N 19.91.
2007.
Все реактивы (Merck), используемые в данной
9. Karpas A., Fleet G.W.J., Dwek R.A., Fellows L.E.,
работе, использовались без дополнительной очист-
Tyins A.S., Petursson S., Namgoong S.K., Ramsden N.G.,
ки. Ход реакций контролировали методом ТСХ на
Jacob G.S., Radenincher T.W. PNAS. 1988, 85, 9229.
doi 10.1073/pnas.85.23.9229
пластинах Silufol UV-254. Удельное вращение
определяли поляриметром AUTOPOL III. Элемент-
10. Lee R.E., Smith M.D., Nash R.J., Griffiths R.C.,
McNeil M., Grewal R.K., Yan W., Besra G.S.,
ный анализ проводили на анализаторе Carlo Erba
Brennan P.J., Fleet G.W.J. Tetrahedron Lett. 1997, 38,
1108. ЯМР-эксперименты проводили на ЯМР-
6733. doi 10.1016/S0040-4039(97)01539-6
спектрометре BRUKER FT AVANCE 400 (Bruker,
11. Veerapen N., Yuan Y., Sanders D.A.R., Pinto B.M.
Карлсруэ, Германия) [400 (1H) и 100.6 (13C) МГц].
Carbohydr. Res.
2004,
339,
2205. doi
10.1016/
ИК спектры регистрировали на Bomem MB 104.
j.carres.2004.07.012
Спектр снимали в диапазоне 4000-400 см-1 при
12. Maddry J.A., Bansal N., Bermudez L.E., Comber R.N.,
комнатной температуре. Спектры МС записывали
Orme I.M., Suling W.J., Wilson L.N., Reynolds R.C.
на спектрометре Vary 500-MS LC Ion Trap Mass и
Bioorg. Med. Chem. Lett. 1998, 8, 237. doi 10.1016/
VG Autopsic M.
S0960-894X(98)00017-1
13. Marques E.T.A., Ichikawa Jr.Y., Strand M., August J.T.,
КОНФЛИКТ ИНТЕРЕСОВ
Hart G.W., Schnaar R.L. Glycobiology. 2001, 11, 249.
doi 10.1093/glycob/11.3.249
Авторы заявляют об отсутствии конфликта
14. Evans S.V., Gate house A.M.R., Fellows L.E. Entomol.
интересов.
Exp. Appl.
1985,
37,
257. doi
10.1111/j.1570-
7458.1985.tb03483.x
СПИСОК ЛИТЕРАТУРЫ
15. Bols M., Hazell R.G., Thomsen I.B. Chem. Eur. J.
1. Heightman T.D., Vasella A.T. Angew. Chem. Int. Ed.
1997, 3, 940. doi 10.1002/chem.19970030616
1999, 38, 750. doi 10.1002/(SICI)1521-3773(19990315)
16. Shen C., Bullens D., Kasran A., Maerten P., Boon L.,
38:6<750::AID-ANIE750>3.0.CO;2-6
Aerts J.M.F.G., Assche G. van, Geboes K., Rutgeerts P.,
2. Zechel D.L., Withers S.G. Acc. Chem. Res. 2000, 33,
Ceuppens J.L. Int. Immunopharmacol. 2004, 4, 939.
11. doi 10.1021/ar970172+
doi 10.1016/j.intimp.2004.04.008
3. Stütz A.E. Iminosugars as Glycosidase Inhibitors:
17. Tang C.C., Wilmington D. US Патент
4526993,
Nojirimycin and Beyond. Weinheim: Wiley-VCH, 1999.
1982.
4. Bols M. Acc. Chem. Res. 1998, 31, 1. doi 10.1021/
18. Duarte Vera C.M., Alves Maria J., Fortes António Gil.
ar970058r
Synlett. 2014, 25, 1751. doi 10.1055/s-0034-1378227
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 12 2019
1946
АХУНДОВА и др.
Synthesis of New Derivatives of Imino Sugars Based
on (S)-(1,2,3,6-Tetrahydropyridazin-3-yl)methanol
F. N. Axundovaa, M. M. Kurbanovaa, *, A. E. Huseynzadaa, and M. J. Alvesb
a Baku State University, AZ 1148, Azerbaijan, Baku, ul. Z. Khalilova 23
b Universidade do Minho de Gualtar, 4710-057 Braga, Portugal
*e-mail: kurbanova1972@rambler.ru
Received May 21, 2019; revised October 21, 2019; accepted October 24, 2019
(S)-(1,2,3,6-tetrahydropyridazin-3-yl) methanol was synthesized by the Diels-Alder reaction in the presence of
the chiral catalyst S-BINOL in two stages. Further, the blocking of the N2 position of the synthesized pyridazine
by the Boc group, followed by carbonylation of the hydroxyl group and the removal of the protective Boc group,
leads to the formation of bicyclic analogues of imino sugars. The structures of the synthesized products were
proved by NMR, IR, mass spectroscopic methods, as well as elemental analysis.
Keywords: pyridazine, imino sugars, 1-azafagomine, Diels-Alder reaction, S-BINOL, Boc- protection
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 55 № 12 2019