ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2021, том 57, № 10, с. 1357-1370
УДК 547.316 + 547.326 + 547.382.3 + 547.385.3 + 547.451
СИНТЕЗ α,β-НЕНАСЫЩЕННЫХ АЛЬДЕГИДОВ
С Е-ТРИЗАМЕЩЁННОЙ КРАТНОЙ СВЯЗЬЮ
НА ОСНОВЕ РЕАКЦИЙ РАСКРЫТИЯ
ЦИКЛОПРОПАНОЛОВ
© 2021 г. В. С. Масюк, Ю. Ю. Козырьков, И. В. Минеева*
Белорусский государственный университет, Беларусь, 220047 Минск, просп. Независимости, 4
*e-mail: i.mineyeva@yandex.ru
Поступила в редакцию 20.05.2021 г.
После доработки 05.06.2021 г.
Принята к публикации 10.06.2021 г.
Впервые был разработан эффективный метод получения α,β-ненасыщенных альдегидов с (Е)-тризаме-
щенной кратной связью окислением смесей регио- и стереоизомерных аллилбромидов без их предва-
рительного разделения, доступных по реакции раскрытия сульфонатов 1,2-дизамещенных циклопропа-
нолов. Окисление с помощью N-метилморфолин-N-оксида аллилбромидов, имеющих в своем составе
дополнительные функциональные группы, протекало более эффективно. Реакции проводились в рас-
творителях, способствующих нуклеофильному замещению, повышение температуры и увеличение вре-
мени реакции приводило к увеличению доли (Е)-изомера конечного продукта и уменьшению количества
региоизомерного кетона.
Ключевые слова: α,β-ненасыщенные альдегиды, мезилаты циклопропанолов, аллилбромиды, (Е)-триза-
мещенная кратная связь, N-метилморфолин-N-оксид (NMMO), аллильное окисление, региоселективный
и стереоселективный синтез
DOI: 10.31857/S0514749221100013
ВВЕДЕНИЕ
Основные синтетические подходы к α,β-нена-
сыщенным альдегидам с тризамещенной кратной
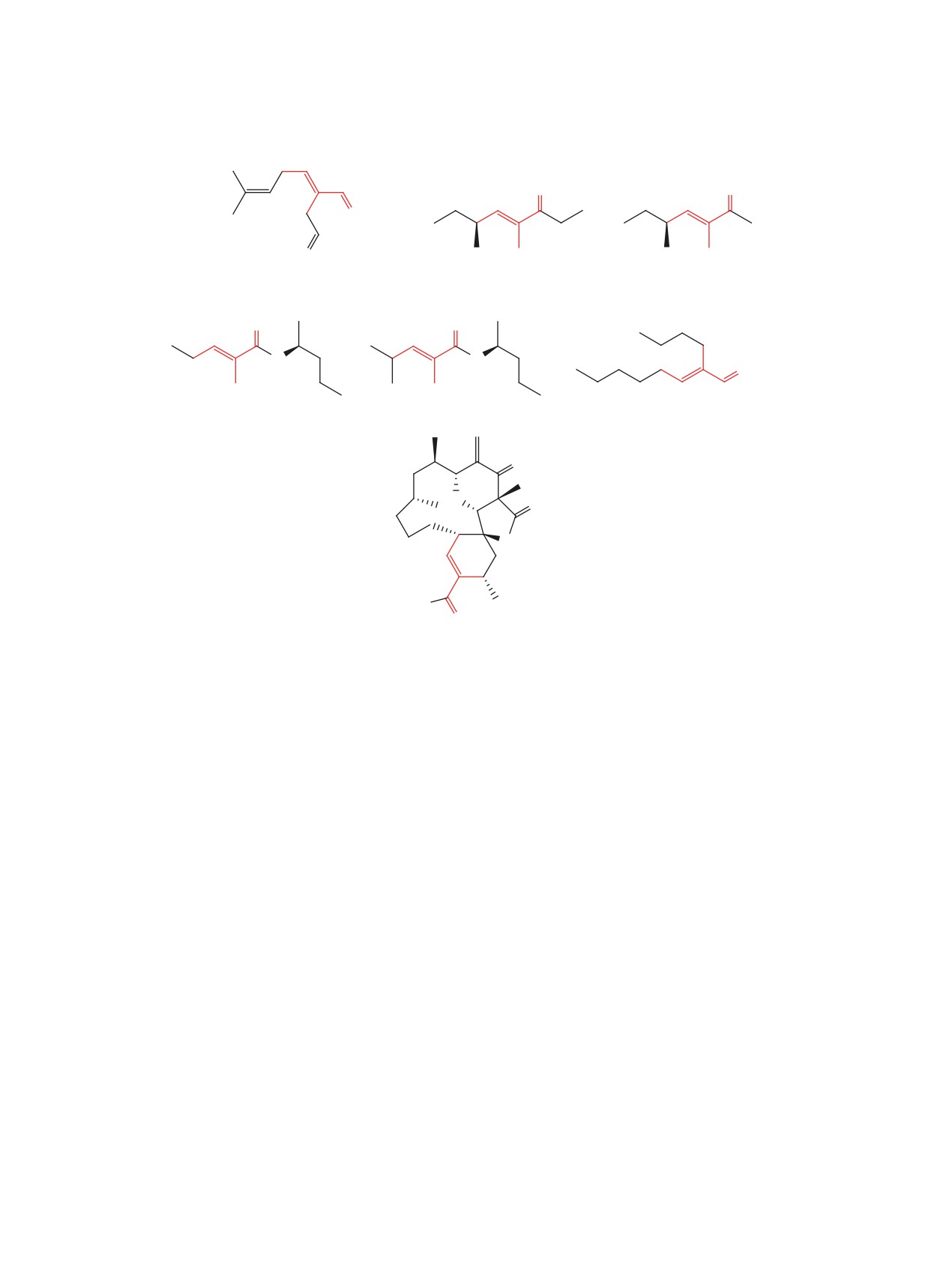
α,β-Ненасыщенные карбонильные соединения
связью включают в себя окисление аллиловых
являются важным классом природных соедине-
спиртов или восстановление карбоновых кислот
ний, среди которых биологически активные тер-
и их производных [6]; реакции алкенильных ме-
пеноиды, феромоны насекомых, сложные поли-
таллорганических реагентов с различными С1
кетиды, содержащие в своей структуре тризаме-
электрофилами, такими как ДМФА [7]; реакции
щенную кратную связь [1, 2] (схема 1). Подобные
кросс-метатезиса с акролеином и его гомолога-
вещества также являются ключевыми интермедиа-
ми [8, 9], перекрестные альдольные реакции с
тами в синтезе сложных природных соединений и
применением азаенолятов [10, 11], силиленоля-
лекарственных препаратов, а также ценными син-
тов [12], цис-2-этоксивиниллития [13], реакции
тетическими блоками в органическом синтезе [3,
Виттига [14, 15], гидроформилирование алкинов
4]. Соединения с тризамещенной кратной связью
по Бухвальду [16] и другие методы [17, 18].
участвуют во множестве биохимичеких процес-
сов, поэтому синтез таких соединений представля-
В общем случае α,β-ненасыщенные альдегиды
ет интерес с точки зрения изучения механизма их
могут быть также получены окислением по связи
действия и использования в косметической, пище-
углерод-галоген доступных галогенидов по мето-
вой промышленности и медицинской практике [5].
ду Корнблюма [19, 20], N-оксидами в различных
1357
1358
МАСЮК и др.
Схема 1
O
O
O
O
половой и агрегационный феромон
компоненты феромона тревоги Manica
Caloglyphus polyphyllae
O
O
O
O
O
агрегационные феромоны Rhyzopertha dominica
компонент феромона тревоги
Oecophylla longinoda
O
O
O
O
HO
O
(-)-Окилактомицин
противоопухолевый антибиотик
растворителях [21-27], соединениями хрома [28,
ным обменом этилацетата в присутствии алкенов
29], солями нитроалканов [30], по методу Крёнке
6а-d [39]. Проведение циклопропил-аллильной
[31], по реакции Соммле [32] и другими методами
изомеризации в мягких условиях позволило нам
[33-36] (схема 2).
добиться преимущественного образования пер-
вичных аллилбромидов 2а-d с (Е)-конфигурацией
В данной работе мы представляем наши ре-
кратной связи [39] (схема 3). Раскрытие мезилатов
зультаты исследований по разработке подхода к
циклопропанолов 4c, d проводилось в присут-
получению α,β-ненасыщенных альдегидов с (Е)-
ствии одного эквивалента основания Хюнига для
тризамещенной кратной связью (1) на основе ре-
предотвращения снятия лабильных в кислых сре-
акции окисления смесей изомерных аллилброми-
дах защитных групп. Состав образующихся сме-
дов 2-3 без их предварительного разделения, лег-
сей аллилбромидов, а также соотношение (E)- и
ко доступных по реакции раскрытия сульфонатов
(Z)-изомеров первичных аллилбромидов 2 опреде-
дизамещенных циклопропанолов (схема 2) [38, 39,
лялись из 1Н ЯМР-спектров реакционных смесей
40].
интегрированием характеристических сигналов
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
протонов при кратной связи. Конфигурация двой-
ной связи в образующихся аллилбромидах 2 была
Смеси регио- и стереоизомерных аллилброми-
также дополнительно [39] подтверждена спектро-
дов 2a-d, 3а-d были получены при взаимодей-
скопией ЯМР 1Н с применением ядерного эффекта
ствии сульфонатов 1,2-дизамещенных циклопро-
Оверхаузера (1D NOESY) (схема 3).
панолов 4а-d с бромидом магния [38, 39] (схе-
ма 3). Мезилаты 4а-d в свою очередь легко до-
Аллилбромиды 2 и 3 обладают одинаковой хро-
ступны из циклопропанолов 5а-d, которые полу-
матографической подвижностью и полученные
чали по реакции циклопропанирования с лиганд-
смеси не могут быть разделены на отдельные ком-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
СИНТЕЗ α,β-НЕНАСЫЩЕННЫХ АЛЬДЕГИДОВ
1359
Схема 2
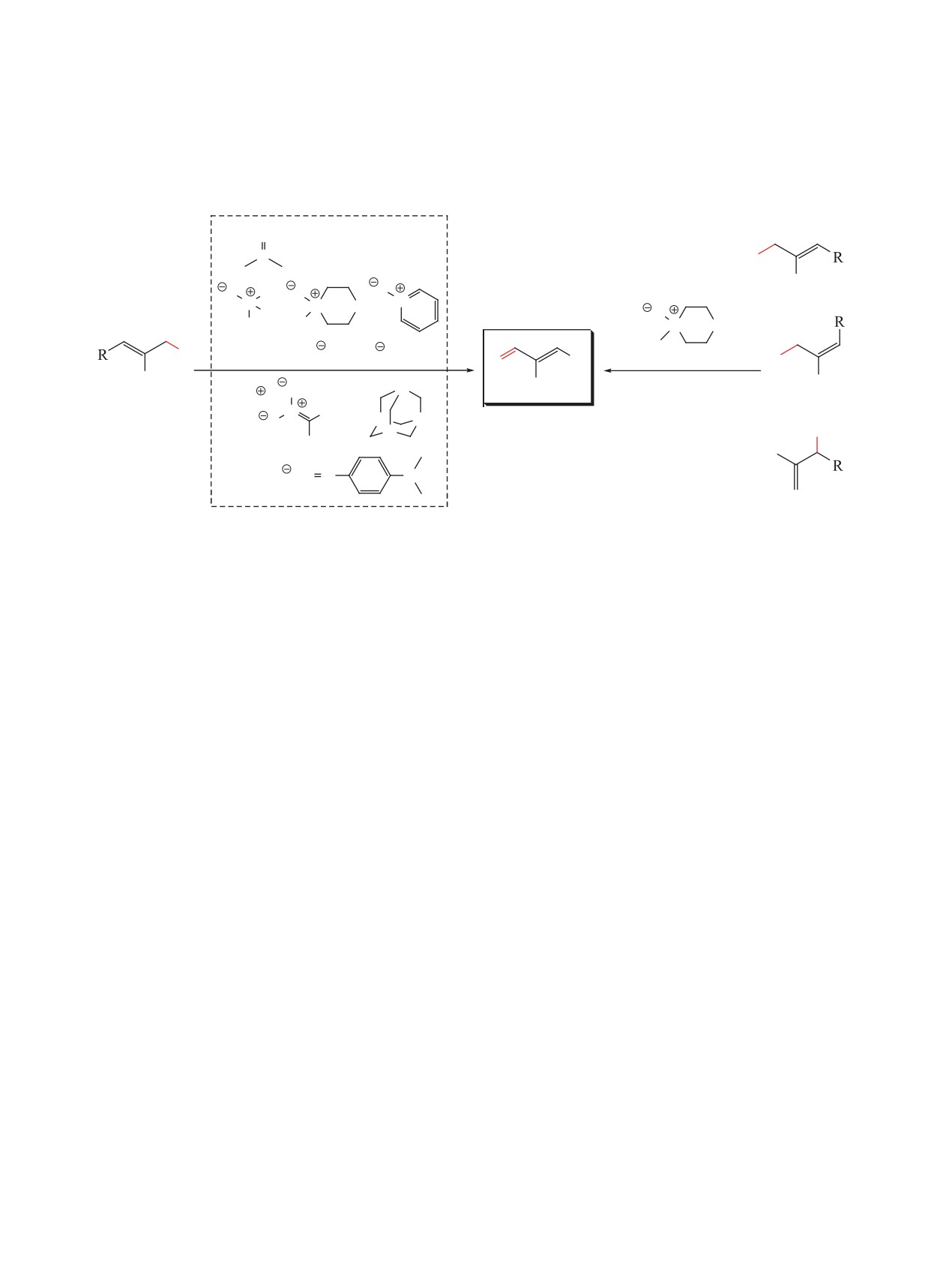
Общие методы окисления
аллилгалогенидов:
Данная работа:
O
, NaHCO3 (AgBF4, Et3N)
S
Br
R'
O
R
O
O
N
N
(E)-2
N
O
R
O
R
R
N
O
2
2
X
Cr2O7
, CrO4
ДМФА, 80°C
Br
O
R
R'
окисление неразделимой
R'
O
N
R'
Na
смеси аллилбромидов
(Z)-2
N R
(R' = CH3)
X= Hlg, OTs.
O
N
N
N
1
Br
R
R'
Py, OH , O
N
N
3
поненты. Однако мы предположили, что окисле-
ном аллилбромиде 3 с последующим окислением
ние региоизомерного бромида 3 может протекать с
образованного галогенида, что должно приводить
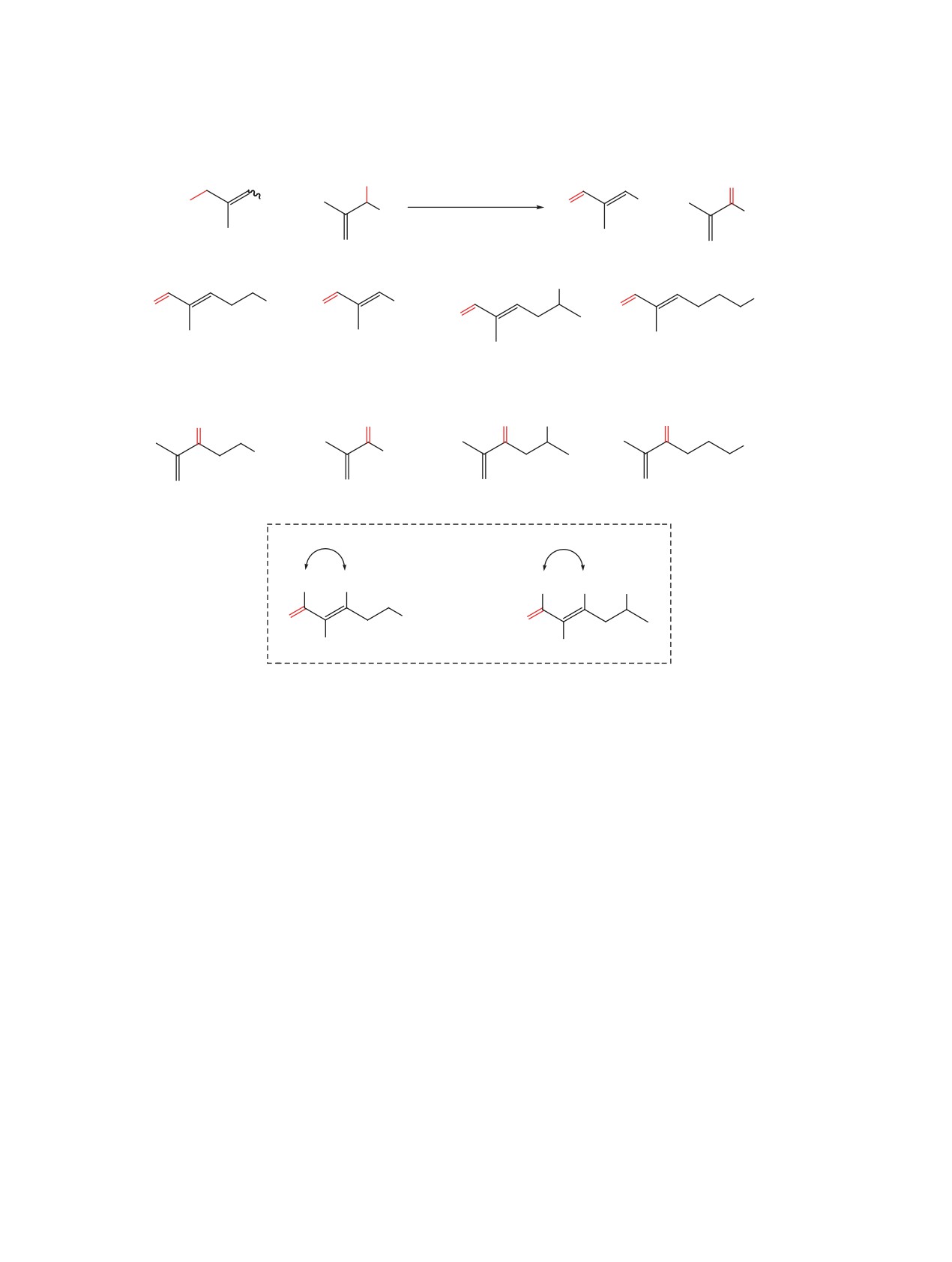
аллильной изомеризацией по механизму SN2'. При
к образованию целевого (Е)-изомера α,β-ненасы-
этом стереохимический результат окисления мо-
щенного альдегида 1 (схема 4).
жет зависеть от того, в какой конформации вторич-
В качестве модельного субстрата для поиска
ный аллилбромид вступит в реакцию (схема 4). С
подходящих условий окисления была выбрана
учетом этого мы начали проводить исследования
смесь аллилбромидов 2а и 3а. Результаты прове-
по поиску таких условий и реагентов, в которых
денной серии экспериментов приведены в табли-
окисление смесей регио- и стереоизомерных ал-
це. Варьируя температуру и время при проведении
лилбромидов 2 и 3 приводило бы исключительно к
реакции в диметилсульфоксиде, нам удалось до-
образованию α,β-ненасыщенных альдегидов с (Е)-
биться высокой стереоселективности получения
тризамещенной кратной связью 1 при отсутствии
ненасыщенного альдегида 1а (см. таблицу, экс-
(Z)-изомеров альдегида 1 и региоизомерных кето-
перименты 1-9). При использовании в качестве
нов 7, образование которых возможно при окисле-
растворителя диметилсульфоксида доля региои-
зомерного кетона 7а была достаточно высока. С
нии вторичных аллилбромидов 3 (схема 4).
целью решения этой проблемы было решено про-
В качестве окислителя нами был выбран ком-
водить реакцию в присутствии каталитических
мерчески доступный и удобный в использовании
количеств галогенидов металлов, таких как иодид
N-метилморфолин-N-оксид (NMMO), окисление
меди (I) и бромид лития [22] (см. таблицу, экспе-
под действием которого протекает по SN2 и SN2'
рименты 10-13). Однако, результаты эксперимен-
механизмам с последующим элиминированием
тов показали неэффективность данного подхода.
[24]. Учитывая сказанное выше, было решено
При использовании в качестве добавки иодида
проводить окисление смесей аллилбромидов в
меди (I) уже при комнатной температуре реакция
растворителях, способствующих нуклеофиль-
протекала с образованием большого числа побоч-
ному замещению. Кроме того, мы планировали
ных продуктов (см. таблицу, эксперименты 10, 11).
проверить влияние добавления солей галогени-
При использовании бромида лития образования
дов на регио- и стереоселективность превраще-
побочных продуктов не наблюдалось, однако до-
ний. Использование неорганических галогенидов
биться значительного повышения региоселектив-
обосновывалось возможностью первоначального
ности реакции не удалось (см. таблицу, экспери-
протекания реакции SN2' замещения во вторич-
менты 12, 13).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
1360
МАСЮК и др.
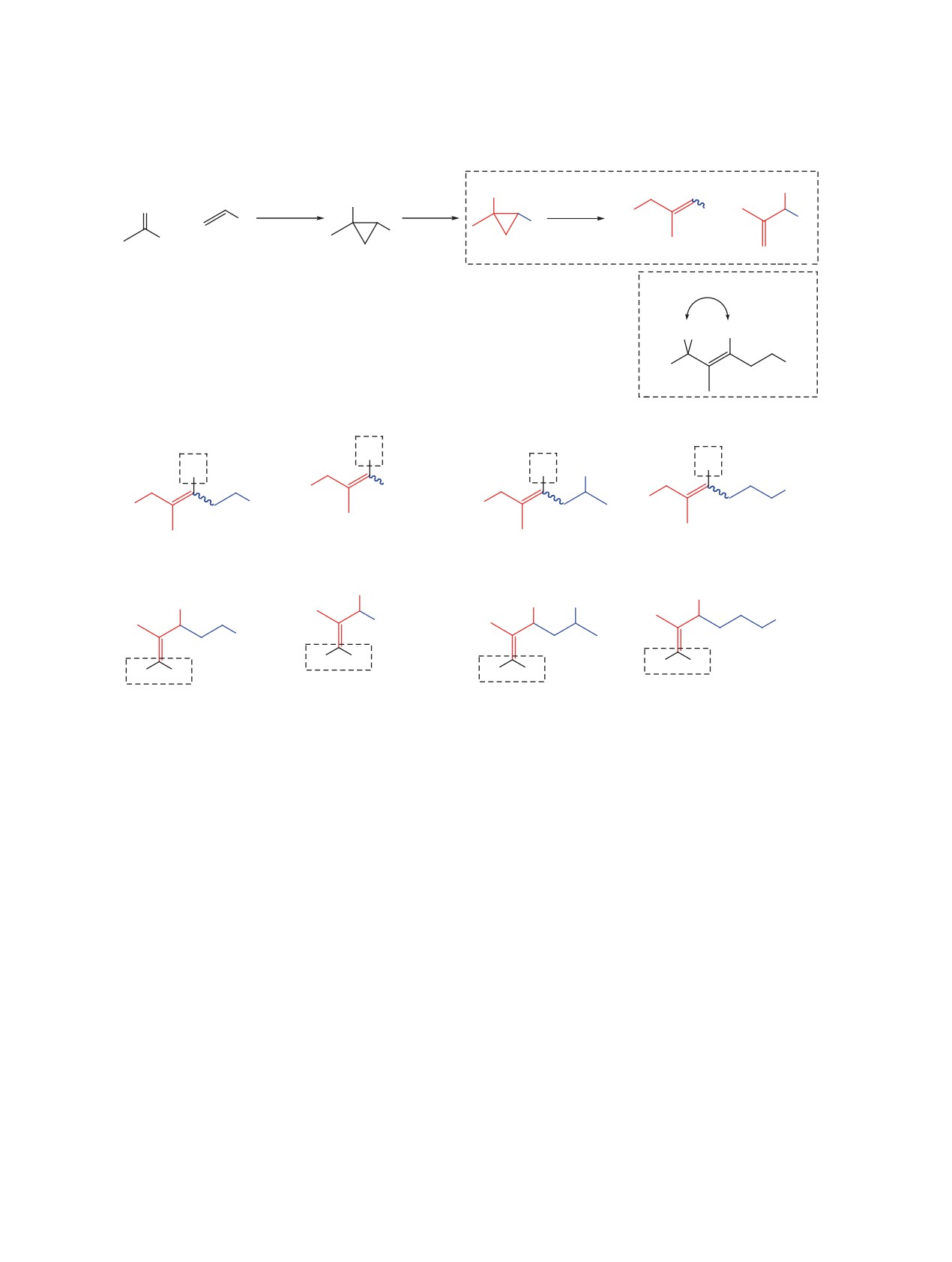
Схема 3
Br
MsCl
OMs
OH
O
CyMgCl
Et3N
MgBr2
Br
R
+
+
R
R
Ti(Oi-Pr)4
Et2O
R
Et2O
R
OEt
ТГФ-Et2O
4
2
3
6
5
R = CH2CH2Ph (6a)
5a (73%)
4a (96%)
nOe
н-C8H17 (6b)
5b (71%)
4b (96%)
(E)-2a
CH2CH(OТГП)CH3 (6c)
5c (62%)
4c (98%)
HH
H
CH2(CH2)2OTBS (6d)
5d (78%)
4d (99%)
Br
Ph
(E)-, δ = 5.63-5.67 м.д.
(E)-, δ = 5.58-5.63 м.д.
(E)-, δ = 5.58-5.66, 5.66-5.73 м.д. (E)-, δ = 5.54-5.59 м.д.
(Z)-, δ = 5.41-5.47 м.д.
(Z)-, δ = 5.35-5.43, 5.44-5.50 м.д.
H
H
H
H
OТГП
Br
C8H17
OTBS
Br
Br
Ph
Br
2b, E:Z = 99:1
2c, E:Z = 96:4
2d, E:Z = 99:1
2a, E:Z = 84:16
59%
77%
61%
44%
+
+
+
+
Br
Br
Br
Br OТГП
OTBS
C8H
17
Ph
H
H
H
H
H
H
H
H
3b, 19%
3d, 28%
3a, 16%
δ = 4.85-4.88 м.д.
3c, 13%
δ = 4.86-4.89 м.д.
δ = 4.91-4.93 м.д.
5.03-5.07 м.д.
δ = 4.84-4.93 м.д.
5.02-5.05 м.д.
5.04-5.08 м.д.
5.04-5.13 м.д.
При использовании в качестве растворителя ди-
При проведении реакции в присутствии СuBr
метилформамида уже при комнатной температуре
в ДМФА была велика доля кетона 7а и неудовлет-
реакция протекала с образованием незначитель-
ворительное соотношение Е:Z изомеров целевого
ных количеств кетона 7а (см. таблицу, эксперимент
альдегида 1 (см. таблицу, эксперимент 23). При
15). Этот факт может объясняться преимуществен-
добавлении LiI соотношение (E)-1а:(Z)-1а было
ным протеканием реакции в данном растворителе
не столь удачным, но в реакционной смеси теперь
как SN2' замещение атома галогена N-оксидом в
отсутствовал кетон (см. таблицу, эксперименты
молекуле вторичного аллилбромида 3. Увеличение
20-22). Окисление в диоксане также проходило
эффективно с минимальным содержанием кетона
времени реакции и повышение тепмературы до
только в случае присутствия бромида меди (см. та-
80°С позволило повысить стереоселективность
блицу, эксперименты 28-29), замена его на иодид
окисления (см. таблицу, эксперименты 16, 17). Мы
лития или полное удаление добавок солей ухуд-
предполагаем, что высокая стереоселективность
шало селективность и приводило к нарастанию
реакции в данных условиях является результатом
побочных продуктов (см. таблицу, эксперименты
процессов многократного присоединения-отще-
30-32).
пления по двойной углерод-углеродной связи мо-
лекулы альдегида 1а N-метилморфолина, являю-
При проведении экспериментов использовался
щегося сильным нуклеофилом и образующегося в
трехкратный избыток NMMO, как рекомендовано
ходе реакции окисления (схема 4).
в большинстве методик. Далее была осуществлена
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
СИНТЕЗ α,β-НЕНАСЫЩЕННЫХ АЛЬДЕГИДОВ
1361
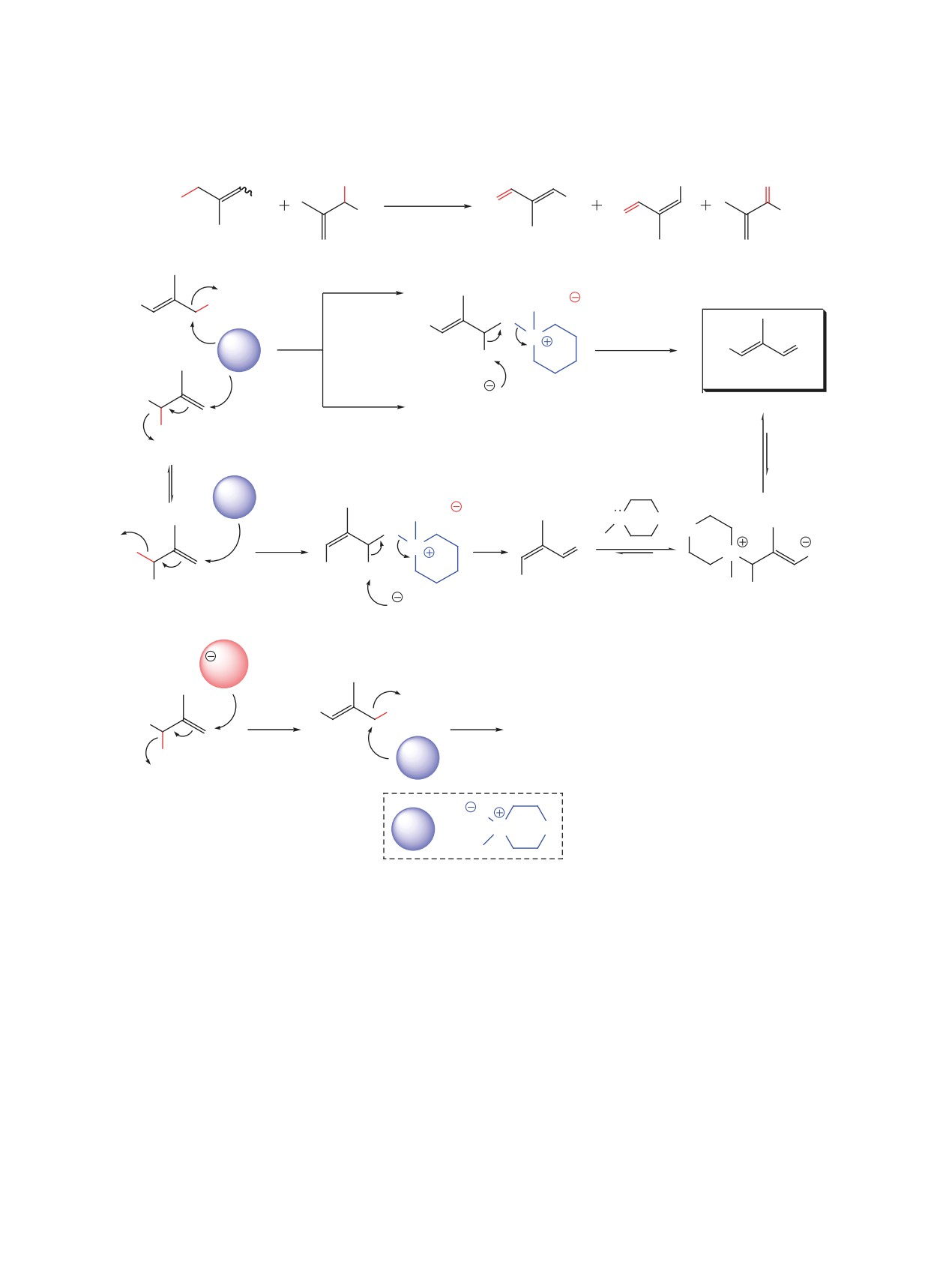
Схема 4
Br
R
O
окисление
Br
R
O
R
R
O
R
2
3
(E)-1
(Z)-1
7
SN2
R
Br
Br
R
O
2
N
R
O
Nu
H
(E)-1
SN2'
A
R
B
Br
3
Nu
Br
N
O
O
O
SN2'
N
O
Br
N
O
R H
R
R
R
Б
3
B
(Z)-1
Hlg
R
Hlg
SN2'
R
A
Br
Nu
3
O
Nu
=
N
O
оценка возможности понижения числа эквивален-
с использованием трех эквивалентов NMMO по-
тов окислителя. При использовании двух эквива-
зволило добиться протекания реакции с высокой
лентов окислителя реакция протекала с образова-
регио- и стереоселективностью. Так, соотношение
нием большого числа побочных продуктов неу-
E:Z изомеров продукта составляло 98:2, а доля
становленного состава (см. таблицу, эксперимент
кетона не превышала 2%. Целевой альдегид без
19). Было найдено, что при повышении количества
существенных осложнений был отделен от побоч-
NMMO до четырех эквивалентов не наблюдалось
ных продуктов реакции с помощью колоночной
ни улучшения конверсии исходных субстратов, ни
хроматографии с выходом 63% (см. таблицу, экс-
повышения регио- и стереоселективности реакции
перимент 17). В диоксане с добавлением 0.1 экв
(см. таблицу, эксперимент 18).
бромида меди был получен сходный результат, вы-
Таким образом, проведение реакции в диметил-
ход целевого альдегида составил 60% (см. табли-
формамиде в течение 24 ч при температуре 80°С и
цу, эксперимент 28).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
1362
МАСЮК и др.
Результаты экспериментов по окислению смеси аллилбромидов 2а и 3а
Br
O
Br
Ph
+
O
Ph
+
Ph
Ph
2a
3a
1a
7a
Соотношение
№
Реагент
Добавка
Растворитель
Т, °C
Время, ч
7а, мол %b,c
(E)-1а/(Z)-1аa
1
3 экв NMМO
-
ДМСО
20
24
91/9
8
2
3 экв NMМO
-
ДМСО-CH2Cl2
0
6
82/18d
0
3
3 экв NMМO
-
CH2Cl2
39
24
89/11
0 (5)
4
3 экв NMМO
-
ДМСО
50
3.5
85/15
13
50
6
5
3 экв NМMO
-
ДМСОe
91/9
14
20
48
6
3 экв NМMO
-
ДМСО
80
3.5
94/6
11
80
6
7
3 экв NМMO
-
ДМСОe
98/2
13
20
48
8
3 экв NМMO
-
ДМСО
80
8
97/3
11
9
3 экв NМMO
-
ДМСО
80
20
98/2
12
10
3 экв NМMO
CuI
ДМСО
20
4
86/14
7 (3)
11
3 экв NМMO
CuI
ДМСО
20
22
78/22
9 (25)
12
3 экв NМMO
LiBr
ДМСО
20
5
82/18
7
13
3 экв NМMO
LiBr
ДМСО
20
24
84/16
9
14
1.5 экв IBX
-
ДМСО
60-70
5
99/1
2 (12)
15
3 экв NMМO
-
ДМФА
20
20
86/14
3
16
3 экв NMМO
-
ДМФА
80
8
97/3
2
17
3 экв NMМO
-
ДМФА
80
24
98/2
2
18
4 экв NMМO
-
ДМФА
80
24
98/2
2
19
2 экв NMМO
-
ДМФА
80
24
97/3
2f
20
3 экв NMМO
2 экв LiI
ДМФА
50
6
80/20
0
21
3 экв NMМO
2 экв LiI
ДМФА
50
24
90/10
0 (6)
22
3 экв NMМO
2 экв LiI
ДМФА
80
24
90/10
0 (16)
23
3 экв NMМO
0.1 экв CuBr
ДМФА
50
6
89/11
7
24
3 экв NMМO
3 экв NaIg
ДМФА
80
24
50/50
0
25
1 экв NaIO4
-
ДМФА
150
2
-i
-
26
3 экв NMМO
-
СН3СN
80
2
91/9
2
27
3 экв NMМO
-
СН3СN
80
24
94/6
8 (27)
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
СИНТЕЗ α,β-НЕНАСЫЩЕННЫХ АЛЬДЕГИДОВ
1363
Таблица. (продолжение).
Br
O
Br
Ph
+
O
Ph
+
Ph
Ph
2a
3a
1a
7a
Соотношение
№
Реагент
Добавка
Растворитель
Т, °C
Время, ч
7а, мол %b,c
(E)-1а/(Z)-1аa
28
3 экв NMМO
0.1 экв CuBr
диоксан
50
6
97/3
2
29
3 экв NMМO
0.1 экв CuBr
диоксан
50
24
96/4
4
30
3 экв NMМO
2 экв LiI
диоксан
50
6
94/6
1.5 (10)
31
3 экв NMМO
-
диоксан
50
6
87/13
4 (5)
32
3 экв NMМO
-
диоксан
50
24
81/19
2 (7)
33
3 экв ТМАО
0.1 экв CuBr
диоксан
50
24
86/14
5
34
3 экв ТМАО
-
ДМФА
80
24
93/7
12
a определялось из анализа 1Н ЯМР-спектров реакционных смесей интегрированием сигналов протонов альдегидной группы с
δ 9.38 м.д. для E-изомера и δ 10.02 м.д. для Z-изомера
b определялось по 1Н ЯМР-спектрам реакционных смесей анализом интегральных интенсивностей сигналов в области с δ 5.76 и
5.95 м.д. соответствующих протонам терминальной кратной связи
c в скобках приведена доля альдегидов неустановленного строения в мол %
d вторичный аллилбромид не вступает в превращение
e реакция проводилась при нагревании с последующим выдерживанием реакционной смеси при комнатной температуре
f высокая доля побочных продуктов неустановленного состава
g первоначально длительное кипячение в ацетоне с иодидом натрия, а затем окисление
i продукты не были получены
Попытка замены NMMO на N-триметиламин-
ления смесей аллилбромидов к другим субстратам
N-оксид (ТMАO) в системах проявивших лучший
(схема 5). При окислении смеси аллилбромидов
результат оказалась неэффективной (см. таблицу,
2b и 3b, содержащих н-октильный заместитель,
эксперименты 33, 34), что возможно связано с
наблюдались определенные трудности. Как и в
малым размером амина и высокой скоростью его
случае субстратов 2а и 3а, реакция характеризова-
отщепления при изомеризации. Замена NMMO на
лась высокой регио- и стереоселективностью, од-
2-иодоксибензойную кислоту (IBX) [35] и окис-
нако ее протекание сопровождалось образованием
ление в условиях разработанных для бензильных
большого числа побочных альдегидов неустанов-
галогенидов привели к нарастанию побочных про-
ленного состава. Это обстоятельство значительно
дуктов неустановленного состава и низкому вы-
понижало выход целевого альдегида (схема 5).
ходу целевого соединения (30%), несмотря на вы-
Существенный интерес представляет собой
сокую стереоселективность реакции (см. таблицу,
вовлечение в описанные выше превращения смеси
эксперимент 14).
аллилбромидов, содержащих в себе дополнитель-
Окисление метапериодатом натрия в ДМФА,
ные функциональные группы. Окисление смеси
примененное для алифатических, бензильных и
аллилбромидов 2c и 3c, имеющих в своем составе
аллильных субстратов [33] привело к смеси неу-
ТГП-защищенную гидроксильную группу, про-
становленного состава, в которой целевой продукт
текало достаточно гладко, причем для окисления
не был обнаружен (см. таблицу, эксперимент 25).
данной смеси требовалось значительно меньше
Далее мы оценили применимость найденных
времени. Так, проведение реакции в диметилфор-
нами условий регио- и стереоселективного окис-
мамиде в течение 8 ч при температуре 80°С и с ис-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
1364
МАСЮК и др.
Схема 5
Br
3 экв NMMO, ДМФА
O
80°C, 8-24 ч
Br
R
+
O
R
+
R
R
2
3
1
7
OТГП
OTBS
O
Ph O
C8H17
O
O
1a, E:Z = 98:2
1b, E:Z > 99:1
1c, E:Z = 99:1
1d, E:Z = 99:1
63%
39%
60%
65%
+
+
+
+
O
O
O OТГП
O
OTBS
Ph
C
8H17
7a, 2 мол %
7b, 4 мол %
7c, < 1 мол %
7d, < 1 мол %
nOe
nOe
H H
H H OТГП
O
Ph
O
CH3
CH3
пользованием трех эквивалентов окислителя по-
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
зволило добиться протекания реакции с высокой
Использованные в ходе работы реактивы
и
регио- и стереоселективностью: альдегид 1c был
растворители имели квалификацию «чистые»
и
получен в виде единственного стереоизомера при
«чистые для анализа». Оценку индивидуальности
отсутствии региоизомерного кетона 7c с выходом
синтезируемых веществ и наблюдение за ходом
60% (схема 5). Хорошие результаты были также
проводимых реакций осуществляли методом тон-
достигнуты при окислении силилсодержащих ал-
кослойной хроматографии (ТСХ) на пластинках
лилбромидов 2d и 3d, несмотря на высокую долю
«Sorbfil». В качестве элюента были использованы
вторичного аллилбромида 3d в смеси (схема 3).
смеси растворителей - петролейный эфир и этил-
В найденных условиях окисление данной смеси
ацетат в различных соотношениях. Выделение
также протекало с хорошим результатом: соот-
индивидуальных веществ осуществляли методом
ношение E:Z изомеров продукта превышало 99:1
колоночной хроматографии на силикагеле (70-
при отсутствии региоизомерного кетона 7d, а вы-
230 меш) производства фирмы Merck с исполь-
ход реакции составил 65% (схема 5). В описанных
зованием в качестве элюентов смесей тех же рас-
реакциях соотношение продуктов и стереоселек-
творителей. Спектры ЯМР 1H и 13С 5-10%-ных
тивность определялись из анализа спектров ЯМР
растворов синтезированных соединений в дейте-
1H реакционных смесей. Конфигурация кратной
рохлороформе (CDCl3) были получены на приборе
связи целевых альдегидов была дополнительно
Bruker Avanсe-500 (Германия) с рабочей частотой
подтверждена спектроскопией 1Н ЯМР с примене-
500 МГц и 125 МГц соответственно. Химические
нием ядерного эффекта Оверхаузера (1D NOESY)
сдвиги измерялись в шкале δ сигнала остаточ-
(схема 5).
ных протонов дейтерохлороформа (δ
7.26 и
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
СИНТЕЗ α,β-НЕНАСЫЩЕННЫХ АЛЬДЕГИДОВ
1365
77.0 м.д. для 1H и 13С, соответственно). ИК спек-
(С-О). Спектр ЯМР 1Н, δ, м.д.: 1.08 д (1.5Н, СН3, J
тры веществ записаны в пленке на спектрофотоме-
6.1 Гц), 1.19 д (1.5Н, СН3, J 6.1 Гц), 1.42-1.84 м (6Н,
тре Bruker FT - IR Alpha (Германия).
ОСНСН2СН2СН2), 2.11-2.37 м (2Н, СН2СН=СН2),
3.43-3.55 м (1Н, СН3СНОСН), 3.76-3.95 м (2Н,
4-Фенилбутен-1 (6а). К раствору бензилмаг-
CH2O), 4.54-4.78 м (1Н, ОСНO), 4.91-5.07 м (2Н,
нийхлорида (100 ммоль) в 80 мл абсолютного Et2O
CH2=), 5.71-5.86 м (1Н, CH=). Спектр ЯМР 13С,
добавили раствор 9.6 мл (110.0 ммоль) аллилбро-
δ, м.д.: 19.6, 19.9, 20.5, 21.1, 25.4, 25.5, 30.9, 31.1,
мида в 30 мл абсолютного Et2O и перемешивали до
40.7, 41.9, 62.3, 62.6, 70.8, 72.6, 97.9, 98.3, 116.6,
окончания реакции (контроль ТСХ). Реакционную
116.8, 134.8, 135.3.
смесь обработали
5%-ным водным раствором
H2SO4 до растворения осадка. Органический слой
трет-Бутил(диметил)(пент-4-ен-1-илокси)-
отделили, а продукт реакции из водного слоя про-
силан (6d). К раствору 0.52 г (6.0 ммоль) пент-
экстрагировали Et2O (3×20 мл). Объединенные
4-ен-1-ола в 4 мл CH2Cl2 при 0°С одной порцией
органические вытяжки промыли насыщенным
добавили последовательно 0.67 г (9.8 ммоль) ими-
водным раствором NaНСO3 и осушили Na2SO4.
дазола и 1.03 г (6.9 ммоль) t-BuMe2SiCl и выдер-
После удаления растворителя при понижен-
жали в холодильнике 12 ч. После обработки водой
ном давлении, продукт был выделен перегон-
(20 мл) органическую фазу отделили, продукт ре-
кой при пониженном давлении, т.кип 130-135°С
акции из водного слоя проэкстрагировали CH2Cl2
(80 мм рт.ст.). Выход 9.20 г (70%). ИК спектр, ν,
(3×10 мл), объединенные органические вытяжки
см-1: 1497 ср (С=С), 1454 ср (С=С). Спектр ЯМР
осушили MgSO4. После отгонки растворителя
1H, δ, м.д.: 2.48-2.54 м (2Н, CH2CH=CH2), 2.82-
при пониженном давлении продукт реакции вы-
2.86 м (2H, PhCH2), 5.11-5.21 м (2H, CH=CH2),
делили хроматографированием (элюент - смесь
5.95-6.05 м (1H, CH=CH2), 7.27-7.33 м (3H, Ph),
петролейного эфира и EtOAc, 200:1). Выход 1.12 г
7.37-7.48 м (2H, Ph). Спектр ЯМР 13С, δ, м.д.: 35.3,
(93%). ИК спектр, ν, см-1: 1098 с (С-О). Спектр
35.5, 114.8, 125.7, 128.2 (2C), 128.4 (2С), 137.9,
ЯМР 1H, δ, м.д.: 0.05 уш.с [6Н, (CH3)2SiC(CH3)3],
141.8.
0.90 уш.с [9Н, (CH3)2SiC(CH3)3], 1.59-1.64 м (2Н,
CH2CH2ОSi), 2.08-2.13 м (2H, CH2=CHCH2), 3.62
2-(1-Метил-3-бутенилокси)тетрагидро-2Н-
т (2Н, CH2CH2ОSi, J 6.4 Гц), 4.96-5.04 м (2H,
пиран
(6с). К раствору аллилмагнийбромида
CH2=CHCH2),
5.78-5.86 м
(1H, CH2=CHCH2).
(63.0 ммоль) в 50 мл абсолютного Et2O добавили
Спектр ЯМР 13С, δ, м.д.: -5.32 (2С), 18.3, 25.9,
раствор 2.20 г (50.0 ммоль) ацетальдегида в 15 мл
30.0, 32.0, 62.5, 114.5, 138.5.
абсолютного Et2O и перемешивали до окончания
реакции (контроль ТСХ). Реакционную смесь при
Общая методика получения циклопропано-
охлаждении обработали насыщенным водным
лов 5а-d. К раствору 4.40 г (50.0 ммоль) этилаце-
раствором NH4Cl. Органический слой отделили, а
тата, 7 мл (25.0 ммоль) Ti(Oi-Pr)4, 27 ммоль алкена
продукт из водного слоя проэкстрагировали Et2O
6а или 6b в 30 мл абсолютного Et2O (абсолютного
(3×15 мл). После удаления растворителя при ат-
ТГФ в случае алкенов 6c и 6d) при перемешива-
мосферном давлении получили 4.00 г (46.5 ммоль)
нии в течение 4.5 ч добавили раствор 125 ммоль
сырого продукта. Далее остаток растворили в
CyMgCl в 100 мл ТГФ. При комнатной темпера-
50 мл сухого CH2Cl2, добавили 4.68 г (55.7 ммоль)
туре, после чего реакционную массу оставили при
дигидропирана, каталитическое количество ППТС
перемешивании на 12 ч. Растворитель удалили при
и выдержали при комнатной температуре в тече-
пониженном давлении, к остатку при охлаждении
ние 12 ч. После обработки насыщенным раство-
добавили 120 мл Et2O и 5 мл насыщенного водного
ром NaHCO3 (25 мл), органический слой отделили
раствора NH4Cl. Полученную смесь декантирова-
и осушили безводным Na2SO4. После удаления
ли, осадок промыли Et2O (3×20 мл), объединенные
растворителя при пониженном давлении полу-
органические вытяжки промыли насыщенным во-
чили 7.80 г (88%) алкена 6с, который без допол-
дным раствором NaHCO3 (50 мл) и осушили без-
нительной очистки был введен в следующую
водным Na2SO4. После удаления растворителя при
стадию. ИК спектр, ν, см-1: 1261 с (С-О), 1021 с
пониженном давлении продукт выделили хрома-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
1366
МАСЮК и др.
тографированием (элюент - смесь петролейного
ср (С-О), 1096 с (С-О). Спектр ЯМР 1H, δ, м.д.:
эфира и EtOAc, 35:1).
0.02-0.07 м [7Н, (CH3)2SiC(CH3)3, CHциклопроп],
0.80-0.83 м (1H, CHциклопроп), 0.87 уш.с [9Н,
1-Метил-2-фенэтил-1-циклопропанол
(5а).
(CH3)2SiC(CH3)3], 0.93-0.99 м (1H, CHциклопроп),
Получен в виде смеси диастереомеров (≈ 1:1).
1.14-1.21 м (1Н, CH2CH2CH2ОSi), 1.32-1.38 м (1Н,
Выход 3.2 г (73%). ИК спектр, ν, см-1: 3325 ш
CH2CH2CH2ОSi), 1.39 с (3H, CH3COH), 1.57-1.62
(ОН), 1074 ср (С-О). Спектр ЯМР 1H, δ, м.д.:
м (2Н, CH2CH2CH2ОSi), 2.21 уш.с (1H, OH), 3.63 т
0.04-0.11 м (1H, CHциклопроп), 0.85-0.90 м (1H,
(2Н, CH2CH2CH2ОSi, J 6.4 Гц). Спектр ЯМР 13С, δ,
CH2циклопроп), 0.97-1.08 м (1H, CH2циклопроп), 1.35 с
м.д.: -5.4 (2С), 18.2, 20.0, 20.3, 25.1, 25.8, 25.9 (3С),
(3H, CH3), 1.42-1.70 м (2H, CH2CH2Ph), 2.65-2.81
26.1, 55.4, 62.8.
м (2H, CH2Ph), 7.17-7.21 м (3H, Ph), 7.27-7.31 м
(2H, Ph). Спектр ЯМР 13С, δ, м.д.: 20.1, 20.4, 24.1
Общая методика получения мезилатов 4а-d.
(2С), 25.3, 25.4, 32.0, 35.5 (2С), 36.0, 55.7, 70.3,
К раствору 18.0 ммоль циклопропанолов 5а-d в
125.8 (2С), 128.3 (4С), 128.4 (4С), 142.2 (2С).
30 мл абсолютного Et2O, охлажденного до 0°С,
при перемешивании и охлаждении добавили 5 мл
1-Метил-2-октилциклопропанол-1 (5b). По-
(36.0 ммоль) Et3N и по каплям раствор 1.8 мл
лучен в виде смеси диастереомеров (≈ 1:1). Выход
(23 ммоль) MsCl в 10 мл абсолютного Et2O. После
2.6 г (71%). ИК спектр, ν, см-1: 3381 ш (ОН),
перемешивания в течение 1.5 ч реакционную мас-
1058 ср (С-О). Спектр ЯМР 1H, δ, м.д.: 0.03-0.06
су обработали насыщенным водным раствором
м (1H, CHциклопроп), 0.79-0.90 м [4H, CH3(CH2)7,
NaHCO3 (50.0 мл) и дополнительно перемешива-
CH2циклопроп], 0.93-1.00 м (1H, CH2циклопроп), 1.25-
ли в течение 40 мин. Органический слой отделили,
1.35 м [12H, CH3(CH2)6], 1.39 c (3H, CH3COH),
продукт реакции из водного слоя проэкстрагиро-
1.50-1.61 м [2H, CH3(CH2)6CH2], 2.01 уш.с (1H,
вали Et2O (3×15 мл), объединенные органические
OH). Спектр ЯМР 13С, δ, м.д.: 14.1 (2С), 20.2, 20.5,
вытяжки промыли насыщенным водным раство-
22.7 (2С), 25.6, 25.7, 29.3 (3С), 29.4, 29.5, 29.5, 29.6
ром NaHCO3 и осушили Na2SO4. Растворитель
(3С), 29.7, 29.9, 31.9 (2С), 32.8, 55.6, 63.0.
удалили при пониженном давлении, мезилаты без
1-Метил-2-(2-тетрагидро-2Н-2-пиранил-
дополнительной очистки ввели в следующую ста-
оксипропил)циклопропан-ол-1 (5c). Получен в
дию.
виде смеси диастереомеров (≈ 1:1:1:1). Выход 3.6 г
1-Метил-2-фенэтилциклопропил метансуль-
(62%). ИК спектр, ν, см-1: 3402 ш (ОН), 1134 с
фонат (4а). Получен в виде смеси диастереомеров
(С-О), 1076 с (С-О), 1022 с (С-О). Спектр ЯМР 1H,
(≈ 1:1). Выход 4.40 г (96%). ИК спектр, ν, см-1:
δ, м.д.: 0.02-0.09 м (1H, CHциклопроп), 0.78-0.87 м
1354 с (S=О), 1174 с (S=О), 1161 с (S=О). Спектр
(1H, CH2циклопроп), 0.92-1.02 м (1H, CH2циклопроп),
ЯМР 1H, δ, м.д.: 0.31-0.37 м (1H, CHциклопроп),
1.11 д (1.5 H, CH3CHOTГП, J 6.1 Гц), 1.21 д.д
1.31-1.44 м (1H, CH2циклопроп), 1.45-1.58 м (1H,
(1.5 H, CH3CHOTГП, J1 6.1, J2 1.3 Гц), 1.35 с
CH2циклопроп), 1.66 с (3H, CH3), 1.72-2.05 м (2H,
(3H, CH3COH), 1.03-1.88 м [8H, CH(CH2)3CH2O,
CH2CH2Ph), 2.71-2.81 м (2H, CH2Ph), 2.97 с (3Н,
CH2CHOTГП], 2.84 уш.с (1H, OH), 3.40-3.49 м
SO2CH3), 7.17-7.21 м (3H, Ph), 7.26-7.31 м (2H,
(1H, CH2OCHO), 3.72-3.84 м (1H, CHOTГП), 3.84-
Ph). Спектр ЯМР 13С, δ, м.д.: 18.3, 18.4, 23.4 (2C),
3.92 м (1H, CH2OCHO), 4.64-4.66 м (1H, OCHO).
24.8 (2С), 31.1, 32.7, 35.2, 38.7, 40.1 (2С), 57.3, 66.7,
Спектр ЯМР 13С, δ, м.д.: 19.0, 19.2, 19.8, 19.8 (2С),
125.9 (2С), 128.3 (4С), 128.4 (4С), 141.6 (2С).
19.9, 20.0, 20.1, 20.6 (2С), 20.6 (2С), 20.9, 21.4, 21.5,
1-Метил-2-октилциклопропил метансуль-
21.8, 22.0, 22.1, 22.8, 24.1, 25.4 (4С), 31.0 (2С), 31.2,
фонат (4b). Получен в виде смеси диастереомеров
35.4, 36.2, 36.5, 37.3, 37.5, 54.8 (2С), 55.0 (2С), 62.6,
(≈ 1:1). Выход 2.20 г (96%). ИК спектр, ν, см-1:
62.6 (2С), 62.8, 71.7, 71.8, 73.5, 73.6, 96.3, 96.4,
1355 с (S=О), 1177 с (S=О), 1162 с (S=О). Спектр
98.3, 98.4.
ЯМР 1H, δ, м.д.: 0.28-0.32 м (1H, CHциклопроп),
2-(3-{[трет-Бутил(диметил)силил]окси}-
0.87 т [3H, CH3(CH2)7, J 6.9 Гц], 1.13-1.46 м [14H,
пропил)-1-метилциклопропанол
(5d). Выход
CH2циклопроп, CH3(CH2)6], 1.65 c (3H, CH3COMs),
4.8 г (78%). ИК спектр, ν, см-1: 3422 ш (ОН), 1253
1.69-1.77 м [2H, CH3(CH2)6CH2], 2.97 c
(3H,
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
СИНТЕЗ α,β-НЕНАСЫЩЕННЫХ АЛЬДЕГИДОВ
1367
SO2CH3). Спектр ЯМР 13С, δ, м.д.: 14.0 (2C), 18.3,
NaHCO3 (50.0 мл) и осушили безводным Na2SO4.
18.4, 22.6 (2C), 23.7, 25.4, 29.0, 29.0, 29.1, 29.1, 29.2
После удаления растворителя при пониженном
(3С), 29.3 (2С), 29.4, 29.5, 31.8 (2С), 37.3, 40.1 (2С),
давлении продукты выделили хроматографирова-
66.9, 70.2.
нием (элюент - петролейный эфир).
1-Метил-2-(2-тетрагидро-2Н-2-пиранил-
(Е)-1-Бром-2-метил-5-фенилпентен-2
(2а).
оксипропил)циклопропил
метансульфонат
Получена смесь изомеров - 1-бром-2-метил-5-
(4c). Получен в виде смеси диастереомеров
фенилпентена-2 (2а) (E/Z = 84/16) (74%) и 3-бром-
(≈ 1:1:1:1). Выход 4.80 г (98%). ИК спектр, ν, см-1:
2-метил-5-фенилпентена-2 (3а) (26%) в количе-
1353 с (S=О), 1162 с (S=О), 1077 с (S=О), 1022 с
стве 2.6 г (60%). ИК спектр, ν, см-1: 1496 ср (С=С),
(S=О). Спектр ЯМР 1H, δ, м.д.: 0.32-0.45 м (1H,
1453 ср (С=С). Спектр ЯМР 1H, δ, м.д.: 1.68-1.72
CHциклопроп), 1.16 д.д (1.5H, CH3CHOTГП, J1 6.1,
м (3H, CH3), 2.35 д.т (2H, PhCH2CH2, J1 8.2, J2
J2 2.8 Гц), 1.26 д.д (1.5H, CH3CHOTГП, J1 6.1, J2
7.6 Гц), 2.68 т (2H, PhCH2, J 7.6 Гц), 3.96-3.97 м
4.6 Гц), 1.06-1.88 м [13H, CH2циклопроп, CH3COMs,
(2H, CH2Br), 5.63-5.67 м (1Н, CH=C), 7.18-7.22 м
CH(CH2)3CH2O, CH2CHOTГП],
2.97 c
(3H,
(3H, Ph), 7.28-7.32 м (2H, Ph).
SO2CH3), 3.43-3.51 м (1H, CH2OCHO), 3.79-3.95
(Е)-1-Бром-2-метилундецен-2 (2b). Получена
м (2H, CHOTГП, CH2OCHO), 4.64-4.73 м (1H,
смесь изомеров - (Е)-1-бром-2-метилундецена-2
OCHO). Спектр ЯМР 13С, δ, м.д.: 18.5 (4С), 18.6,
(2b)
(76%) и
3-бром-2-метилундецена-1
(3b)
18.7, 18.8, 18.9, 19.6, 19.7, 19.9, 20.0, 20.4, 20.4,
(24%) в количестве 1.6 г (78%). ИК спектр, ν,
20.5, 20.6, 21.5, 21.7, 23.4, 24.8, 25.4 (4С), 31.1.
см-1: 1466 ср (С=С). Спектр ЯМР 1H, δ, м.д.: 0.88
(3С), 32.6, 35.7 (2С), 36.4, 36.9, 38.7, 39.9, 40.0
т [3H, CH3(CH2)7, J 6.9 Гц], 1.19-1.43 м [12H,
(2С), 62.5, 62.7, 62.8, 66.2, 66.4, 66.6, 66.7, 70.6,
CH3(CH2)6], 1.73-1.76 м (3H, CH3C=), 1.96-2.05 м
70.7, 73.2, 73.6, 81.3, 95.8, 96.0, 98.5, 99.0.
Br), 5.58-5.63
[2H, CH3(CH2)6CH2], 3.97 c (2H, CH2
2-(3-{[трет-Бутил(диметил)силил]окси}-
м (1H, C=CH).
пропил)-1-метилциклопропил метансульфонат
Общая методика раскрытия лабильных
(4d). Выход 5.74 г (99%). ИК спектр, ν, см-1: 1355
мезилатов 4c, d при действии бромида маг-
с (S=О), 1154с (S=О). Спектр ЯМР 1H, δ, м.д.:
ния. К свежеприготовленному раствору MgBr2
0.03 уш.с
[6Н, (CH3)2SiC(CH3)3],
0.30-0.33 м
(25.0 ммоль) в 30 мл абсолютного Et2O при 0°С
(1H, CHциклопроп), 0.86-0.90 м (1H, CH2циклопроп),
и интенсивном перемешивании внесли рас-
0.85-0.88 м [10Н, (CH3)2SiC(CH3)3, CH2циклопроп],
твор мезилата 4c или 4d (12.0 ммоль) и 1.55 г
1.25-1.35 м (1Н, CH2CH2CH2ОSi), 1.38-1.47 м (1Н,
(12.0 ммоль) i-Pr2NEt в 25 мл абсолютного Et2O.
CH2CH2CH2ОSi), 1.60-1.69 (2Н, CH2CH2CH2ОSi),
1.65 c (3H, CH3COMs), 2.96 c (3H, SO2CH3), 3.63 т
По окончании реакции (контроль ТСХ), реакци-
онную смесь обработали водой до растворения
(2Н, CH2CH2CH2ОSi, J 6.1 Гц). Спектр ЯМР 13С, δ,
м.д.: -5.4 (2С), 18.2, 18.4, 23.4, 23.5, 25.5, 25.9 (3С),
осадка. Органический слой отделили, а продукт
32.1, 40.1, 62.4, 66.8.
реакции из водного слоя проэкстрагировали Et2O
(3×15 мл). Объединенные органические вытяжки
Общая методика раскрытия мезилатов 4а,
промыли насыщенным раствором NaHCO3 и осу-
b при действии бромида магния. К свежеприго-
шили безводным Na2SO4. После удаления раство-
товленному раствору MgBr2 (45.0 ммоль) в 50 мл
рителя при пониженном давлении продукты вы-
абсолютного Et2O при 0°С и интенсивном переме-
делили хроматографированием (элюент - смесь
шивании внесли раствор 18 ммоль метансульфона-
петролейного эфира и EtOAc, 200:1).
тов 4а или 4b в 25 мл абсолютного Et2O. По окон-
чании реакции (контроль ТСХ), в реакционную
2-[(Е)-5-Бром-1,4-диметил-3-пентенилокси]-
смесь добавили 5%-ный водный раствор H2SO4
тетрагидро-2Н-пиран (2c). Получена смесь изо-
до растворения осадка. Органический слой отде-
меров - 2-[5-бром-1,4-диметил-3-пентенилокси]-
лили, продукт реакции из водного слоя проэкстра-
тетрагидро-2Н-пирана (2c) (E/Z = 96:4) (83%) и
гировали Et2O (3×15мл). Объединенные органиче-
2-(3-бром-1,4-диметил-4-пентенилокси)тетра-
ские вытяжки промыли насыщенным раствором
гидро-2Н-пирана (3c) (17%) в количестве 3.0 г
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
1368
МАСЮК и др.
(90%) в виде диастереомерной смеси (≈1:1). ИК
(Е)-2-Метилундецен-2-аль (1b). Выход 28 мг
спектр, ν, см-1: 1133 с (С-О), 1076 с (С-О), 1022
(39%). Спектральные характеристики совпали с
с (С-О). Спектр ЯМР 1H, δ, м.д.: 1.09 д (1.5H,
приведенными в литературе [42].
CH3CHOTГП, J 6.1 Гц), 1.20 д (1.5H, CH3CHOTГП,
(Е)-2-Метил-5-тетрагидро-2Н-2-пиранил-
J 6.1 Гц), 1.43-1.88 м [6H, CH(CH2)3CH2O], 1.76 c
окси-2-гексаналь (1c). Получен в виде смеси
(1.5H, CH3C=), 1.77 c (1.5H, CH3C=), 2.09-2.40 м
диастереомеров (≈ 1:1). Выход 55 мг (60%). ИК
(2H, CH2CHOTГП), 3.43-3.54 м (1H, CH2OCHO),
спектр, ν, см-1: 1689 с (С=О), 1646 с (С=О), 1133 с
3.76-3.94 м (2H, CH2OCHO, CHOTГП), 3.97 c (1H,
(С-О), 1076 с (С-О), 1022 с (С-О). Спектр ЯМР 1H,
CH2Br), 3.98 c (1H, CH2Br), 4.61-4.67 м (0.5H,
δ, м.д.: 1.15 д (1.5H, CH3CHOTHP, J 6.1 Гц), 1.25
OCHO), 4.68-4.73 м (0.5H, OCHO), 5.58-5.65 м
д (1.5H, CH3CHOTHP, J 6.1 Гц), 1.51-1.87 м [6H,
(0.5H, C=CH), 5.66-5.73 м (0.5H, C=CH).
CH(CH2)3CH2O], 1.74 c (3H, CH3C=), 2.46-2.64 м
{[(4E)-6-Бром-5-метилгекс-4-ен-1-ил]окси}-
(2H, CH2CHOTHP), 3.44-3.50 м (1H, CH2OCHO),
(трет-бутил)диметилсилан
(2d).
Получена
3.78-3.92 м (1H, CH2OCHO), 3.92-4.02 м (1H,
смесь изомеров - {[(4E)-6-бром-5-метилгекс-4-ен-
CHOTHP), 4.63-4.67 м (0.5H, OCHO), 4.67-4.71
1-ил]окси}(трет-бутил)диметилсилана (2d) (69%)
м (0.5H, OCHO), 6.50-6.56 м (0.5H, C=CH), 6.60-
и [(4-бром-5-метилгекс-5-ен-1-ил)окси](трет-бу-
6.66 м (0.5H, C=CH), 9.40 c (0.5H, CHO), 9.41 c
тил)диметилсилана (3d) (31%) в количестве 3.28 г
(0.5H, CHO). Спектр ЯМР 13С, δ, м.д.: 9.4 (2С),
(89%). ИК спектр, ν, см-1: 1466 ср (С=С). Спектр
19.3, 19.6, 19.7, 21.5, 25.3, 25.4, 30.9, 31.0, 35.6,
ЯМР 1H, δ, м.д.: 0.07 уш.с [6Н, (CH3)2SiC(CH3)3],
36.8, 62.7 (2С), 70.4, 71.9, 96.4, 98.1, 140.5, 140.8,
1.05 уш.с
[9Н, (CH3)2SiC(CH3)3],
1.58-1.56 м
151.1, 151.7, 188.7, 189.4.
(2Н, CH2CH2CH2ОSi), 1.75 c (3H, CH3C=), 2.11-
(2E)-6-{[трет-Бутил(диметил)силил]окси}-
2.17 м (2Н, =CCH2CH2CH2О), 3.63-3.71 м (2Н,
2-метилгекс-2-еналь (1d). Выход 72 мг (65%).
CH2CH2CH2ОSi), 3.93-3.97 м (2H, CH2Br), 5.54-
Спектральные характеристики совпали с приве-
5.59 м (1H, C=CH).
денными в литературе [9].
Общая методика окисления изомерных сме-
ВЫВОДЫ
сей аллилбромидов 2a-d, 3а-d в ненасыщен-
ные альдегиды 1а-d. К раствору 0.46 ммоль
Целевые α,β-ненасыщенные альдегиды с Е-три-
смеси изомерных аллилбромидов 2a-d, 3а-d в
замещенной кратной связью были синтезированы
2 мл ДМФА внесли 80 мг (0.69 ммоль) NMMO.
окислением смесей регио- и стереоизомерных ал-
Реакционную смесь перемешивали при 80°С в ат-
лилбромидов, доступных по реакции раскрытия
мосфере аргона в течение 1 ч. По истечении этого
сульфонатов
1,2-дизамещенных циклопропано-
времени добавили ещё 80 мг (0.69 ммоль) NMMO
лов без их предварительного разделения, с по-
и перемешивали в тех же условиях 23 ч. По окон-
мощью NММО. В ходе исследования нами были
чании реакции реакционную смесь разбавили 3 мл
найдены подходящие условия, в которых реакции
CH2Cl2 и обработали охлажденным до 0°С полу-
окисления протекали с высокой регио- и стерео-
насыщенным раствором NaCl. Органический слой
селективностью. Реакции проводились в раство-
отделили, а продукт реакции из водного слоя про-
рителях способствующих нуклеофильному заме-
экстрагировали CH2Cl2 (3×5мл). Объединенные
щению (ДМФА, ДМСО), повышение температуры
органические вытяжки промыли насыщенным
и увеличение времени реакции способствовало
раствором хлорида натрия и сушили безводным
увеличению доли Е-изомера конечного продук-
Na2SO4. После удаления растворителя при пони-
та и уменьшению количества региоизомерного
женном давлении продукт выделили хроматогра-
кетона. Добавление каталитических количеств
фированием (элюент - смесь петролейного эфира
галогенидов металлов не повышало регио- и сте-
и EtOAc, 200:1).
реоселективность реакции. Окисление аллилбро-
(Е)-2-Метил-5-фенилпентен-2-аль (1a). Вы-
мидов, имеющих в своем составе дополнительные
ход 50 мг (63%). Спектральные характеристики
функциональные группы, протекало более гладко.
совпали с приведенными в литературе [41].
Полученные результаты показывают работоспо-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
СИНТЕЗ α,β-НЕНАСЫЩЕННЫХ АЛЬДЕГИДОВ
1369
собность предложенного нами метода и возмож-
11.
Corey E.J., Enders D., Bock M.G. Tetrahedron Lett.
ность его дальнейшего применения в синтезе бо-
1976, 17, 7-10. doi 10.1016/S0040-4039(00)71308-6
лее сложных молекул.
12.
Duhamel L., Gralak J., Bouyanzer A. Tetrahedron
Lett.
1993,
34,
7745-7748. doi
10.1016/S0040-
ИНФОРМАЦИЯ О ВКЛАДЕ АВТОРОВ
4039(00)61554-X
В.С. Масюк - выполнение эксперимента, напи-
13.
Wollenberg R.H., Albizati K.F., Peries R. J. Am. Chem.
сание рукописи статьи.
Soc. 1977, 99, 7365-7367. doi 10.1021/ja00464a051
14.
Zhou G., Hu Q.-Y., Corey E.J. Org. Lett. 2003, 5,
Ю.Ю. Козырьков - постановка задачи.
3979-3982. doi 10.1021/ol035542a
И.В. Минеева - выполнение эксперимента, на-
15.
Nagamitsu T., Takano D., Fukuda T., Otoguro K.,
писание рукописи статьи, общее руководство.
Kuwajima I., Harigaya Y., Omura S. Org. Lett. 2004, 6,
1865-1867. doi 10.1021/ol049356w
ИНФОРМАЦИЯ ОБ АВТОРАХ
16.
Johnson J.R., Cuny G.D., Buchwald S.L. Angew.
Масюк Владимир Сергеевич, ORCID: http://
Chem., Int. Ed. 1995, 34, 1760-1761. doi 10.1002/
orcid.org/0000-0002-5175-2419
anie.199517601
Козырьков Юрий Юрьевич, ORCID: http://
17.
Bruch A., Gebert A., Breit B. Synthesis. 2008, 2169-
orcid.org/0000-0001-6952-6420
2176. doi 10.1055/s-2008-1067140
18.
Sato T., Okazaki H., Otera J., Nozaki H. Tetrahedron
Минеева Ирина Владимировна, ORCID: http://
Lett.
1988,
29,
2979-2982. doi
10.1016/0040-
orcid.org/0000-0002-6422-1967
4039(88)85063-9
КОНФЛИКТ ИНТЕРЕСОВ
19.
Babler J.H., Coghlan M.J., Feng M., Fries P. J. Org.
Chem. 1979, 44, 1716-1717. doi 10.1021/jo01324a030
Авторы заявляют об отсутствии конфликта ин-
20.
D’Aniello F., Mattii D., Taddei M. Synlett. 1993, 1993,
тересов.
119-121. doi 10.1055/s-1993-22369
СПИСОК ЛИТЕРАТУРЫ
21.
Mukaiyma S., Inanaga J., Yamaguchi M. Bull.
1. Wichard T., Göbel C., Feussner I., Pohnert G. Angew.
Chem. Soc. Jpn. 1981, 54, 2221-2222. doi 10.1246/
Chem. Int. Ed. 2005, 44, 158-161. doi 10.1002/
bcsj.54.2221
anie.200460686
22.
Suzuki S., Onishi T., Fujita Y., Misawa H., Otera J. Bull.
2. Das B., Banerjee J., Chowdhury N., Majhi A., Mahen-
Chem. Soc. Jpn. 1986, 59, 3287-3288. doi 10.1246/
der G. Helv. Chim. Acta. 2006, 89, 876-883. doi
bcsj.59.3287
10.1002/hlca.200690090
23.
Hayashi T. Tetrahedron Lett. 1990, 31, 4155-4158. doi
3. Taylor R.E., Paquette W.D. Org. Lett. 2004, 6, 103-
10.1016/S0040-4039(00)97568-3
106. doi 10.1021/ol0361397
24.
Ganem B., Godfrey A. Tetrahedron Lett. 1990, 31,
4. Smith A.B., Basu K., Bosanac T. J. Am. Chem. Soc.
4825-4826. doi 10.1016/S0040-4039(00)97742-6
2007, 129, 14872-14874. doi 10.1021/ja077569l
25.
Griffith W.P., Jolliffe J.M., Ley S.V., Springhorn K.F.,
5. Dawson G.W., Pickett J.A., Smiley D.W.M. Bioorg.
Tiffin P.D. Synth. Commun. 1992, 22, 1967-1971. doi
Med. Chem. 1996, 4, 351-361. doi 10.1016/0968-
10.1080/00397919208021328
0896(96)00012-0
26.
Chandrasekhar S., Sridhar M. Tetrahedron Lett. 2000,
6. Escher I., Glorius F. Science of Synthesis. Ed.
41, 5423-5425. doi 10.1016/S0040-4039(00)00874-1
R. Brückner. Thieme: Stuttgart, 2007, 25, 733-777.
27.
Chen D.X, Ho C.M., Rudy Wu Q.Y., Wu P.R.,
7. Olah G.A., Arvanaghi M. Angew. Chem., Int. Ed. 1981,
Wong F.M., Wu W. Tetrahedron Lett. 2008, 49, 4147-
20, 878-879. doi 10.1002/anie.198108781
4148. doi 10.1016/j.tetlet.2008.04.124
8. Crimmins M.T., King B.W. J. Am. Chem. Soc. 1998,
28.
Cardillo G., Orena M., Sandri S. Chem. Commun.
120, 9084-9085. doi 10.1021/ja9817500
1976, 6, 190. doi 10.1039/C39760000190
9. Sirasani G., Paul T., Andrade R.B. Tetrahedron. 2011,
29.
Cardillo G., Orena M., Sandri S. Tetrahedron Lett. 1976,
67, 2197-2205. doi 10.1016/j.tet.2011.01.080
17, 3985-3986. doi 10.1016/S0040-4039(00)92554-1
10. Wittig G., Reiff H. Angew. Chem., Int. Ed. 1968, 7,
30.
Suzuki S., Onishi T., Fujita Y., Otera J. Synth. Commun.
7-14. doi 10.1002/anie.196800071
1985, 15, 1123-1129. doi 10.1080/00397918508077254
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021
1370
МАСЮК и др.
31. Krohnke F. Angew. Chem. Int. Ed. Engl. 1963, 2, 380-
38. Kozyrkov Yu.Yu., Kulinkovich O.G. Synlett. 2002,
393. doi 10.1002/anie.196303801
2002, 443-446. doi 10.1055/s-2002-20461
32. Karamé I., Jahjah M., Messaoudi A., Tommasino M.L.,
39. Kananovich D.G., Hurski A.L., Kulinkovich O.G.
Lemaire M. Tetrahedron Asymmetry. 2004, 15, 1569-
Tetrahеdron Lett. 2007, 48, 8424-8429. doi 10.1016/
1581. doi 10.1016/j.tetasy.2004.03.023
j.tetlet.2007.09.172
33. Das S., Panigrahia A.K., Maikap G.C. Tetrahedron
40. Гацак Е.Л., Козырьков Ю.Ю. Сборник работ 69-й
Lett.
2003,
44,
1375-1377. doi
10.1016/S0040-
научной конференции студентов и аспирантов
4039(02)02885-X
Белорусского государственного университета, 14-
34. Li C. Angew. Chem. Int. Ed. 2003, 42, 5063-5066. doi
17 мая 2012 г., Минск, 307-310.
10.1002/anie.200351902
41. Podunavac M., Lacharity J., Jones K.E., Zakarian A.
35. Moorthy J.N., Singhal N., Senapati K. Tetrahedron Lett.
2006, 47, 1757-1761. doi 10.1016/j.tetlet.2006.01.039
Org. Lett.
2018,
20,
4867-4870. doi
10.1021/
acs.orglett.8b02011
36. Tang J., Zhu J., Shen Z., Zhang Y. Tetrahedron Lett.
2007, 48, 1919-1921. doi 10.1016/j.tetlet.2007.01.084
42. Rountree S.M., Taylor S.F.R., Hardacre C., Lagu-
37. Sofiyev V., Navarro G., Trauner D. Org. Lett. 2008, 10,
nas M.C., Davey P.N. Appl. Catal. A Gen. 2014, 486,
149-152. doi 10.1021/ol702806v
94-104. doi 10.1016/j.apcata.2014.08.032
Synthesis of α,β-Unsaturated Aldehydes with E-Trisubstituted
Double Bond via Cyclopropanol Ring Cleavage Reactions
U. S. Masiuk, Yu. Yu. Kozyrkov, and I. V. Mineyeva*
Belarusian State University, prosp. Nezavisimosti, 4, Minsk, 220030 Belarus
*e-mail: i.mineyeva@yandex.ru
Received May 20, 2021; revised June 5, 2021; accepted June 10, 2021
For the first time, an efficient procedure for the synthesis of α,β-unsaturated aldehydes with (E)-trisubstituted
double bond via oxidation of inseparable mixtures of regio- and stereoisomeric allyl bromides, readily avail-
able by the ring-opening reaction of 1,2-disubstituted cyclopropanols sulfonates, was developed. Oxidation of
allyl bromides, bearing additional functional groups, under the action N-methylmorpholine-N-oxide was more
effective. The reactions were carried out in solvents promoting nucleophilic substitution. Rising temperature
and increased reaction time contributed to an increase in the share (E)-isomer of the final product and a decrease
in the amount of regioisomer ketone.
Keywords: α,β-unsaturated aldehydes, cyclopropanol mesylates, allylbromides, (E)-trisubstituted double bond,
N-methylmorpholine-N-oxide (NMMO), allylic oxidation, regioselective and stereoselective synthesis
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 10 2021