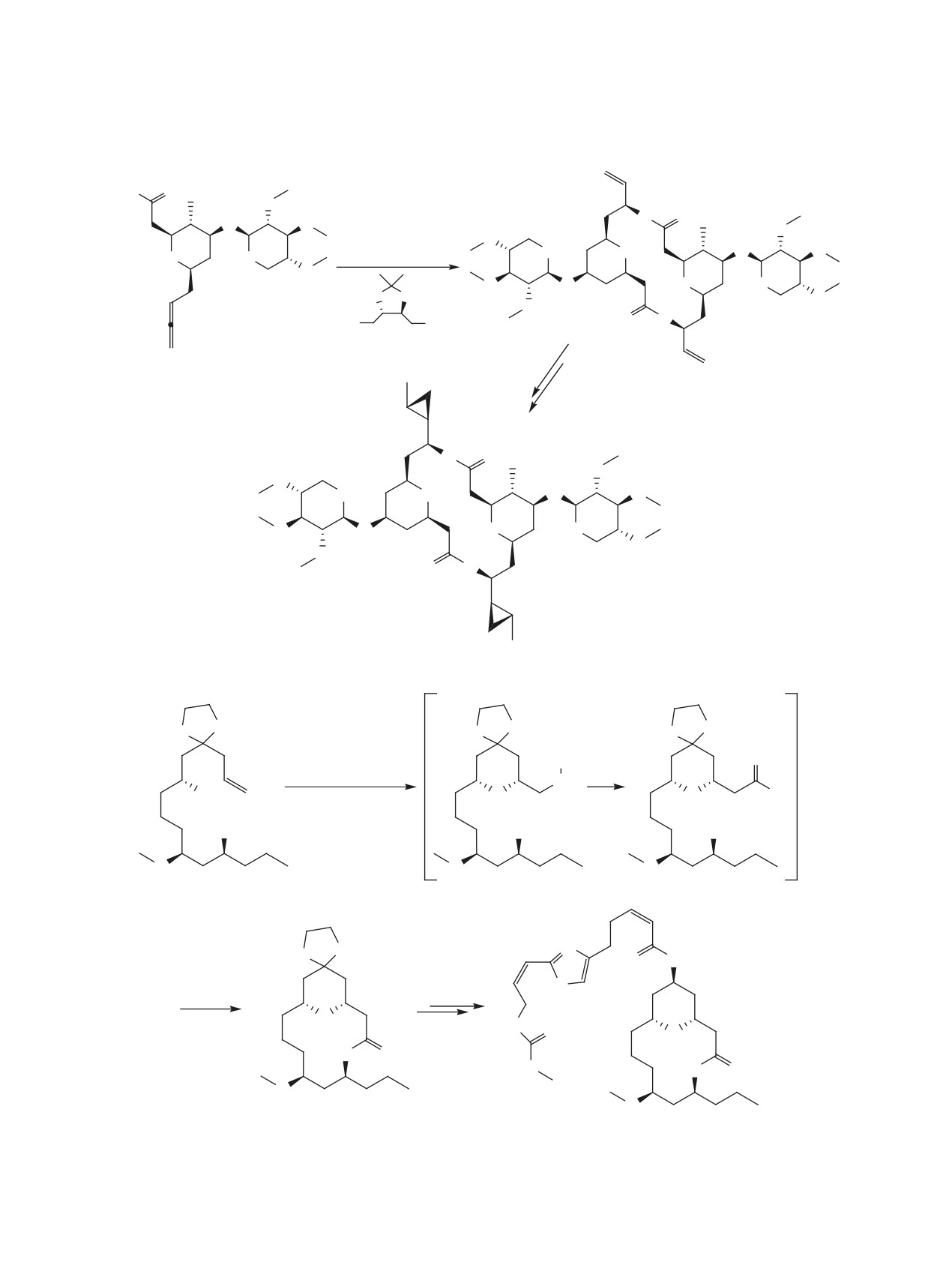
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2021, том 57, № 5, с. 607-660
ОБЗОРНАЯ СТАТЬЯ
УДК 547.47 + 547.89
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
В СИНТЕЗЕ ПРИРОДНЫХ И БИОЛОГИЧЕСКИ
АКТИВНЫХ СОЕДИНЕНИЙ
© 2021 г. М. П. Яковлева*, К. С. Денисова, В. А. Выдрина,
А. Г. Толстиков, Г. Ю. Ишмуратов
Уфимский Институт химии - обособленное структурное подразделение
ФГБНУ «Уфимского федерального исследовательского центра РАН»,
Россия, 450054 Уфа, просп. Октября, 71
*e-mail: insect@anrb.ru
Поступила в редакцию 26.01.2021 г.
После доработки 06.02.2021 г.
Принята к публикации 10.02.2021 г.
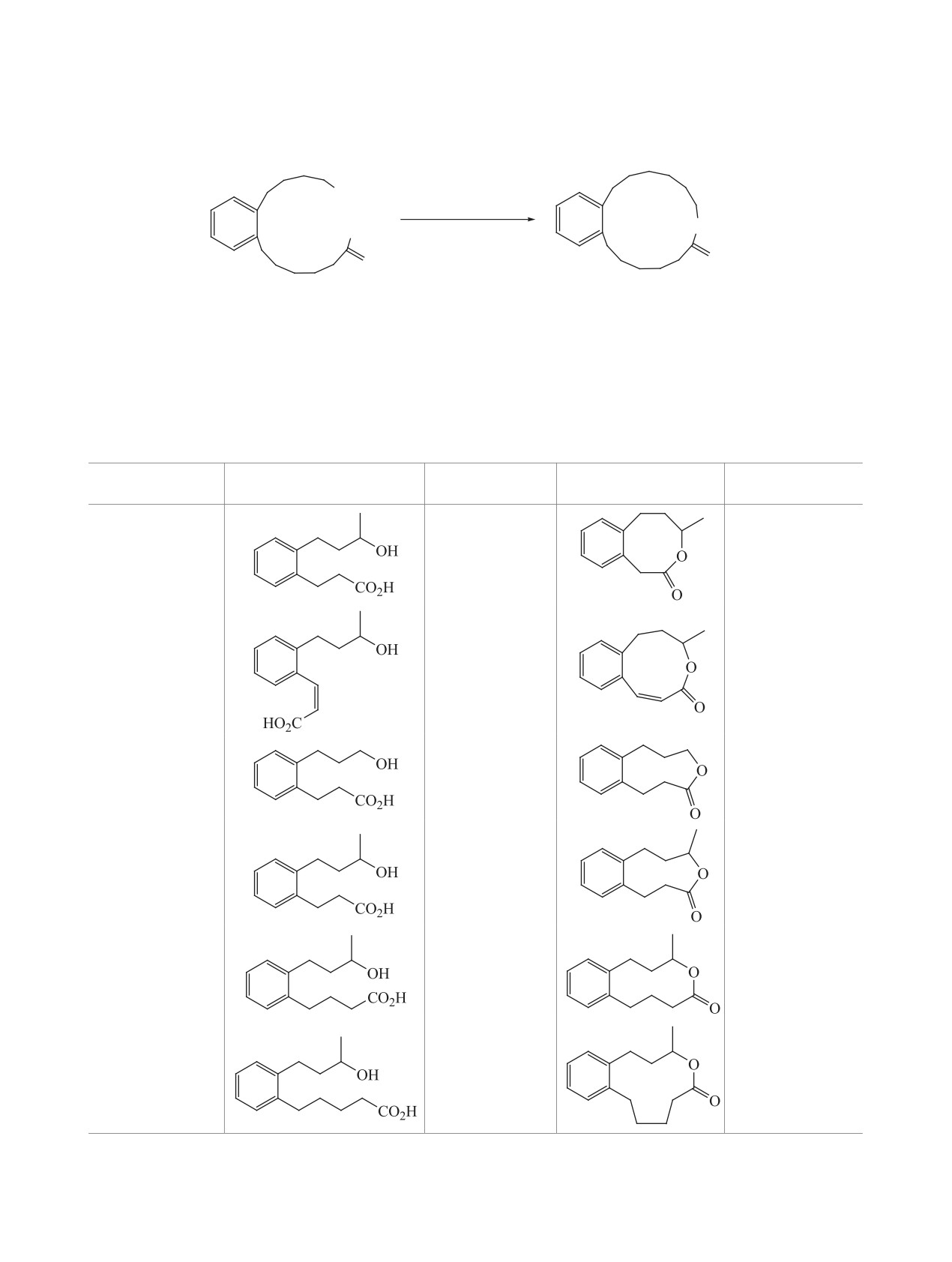
Описаны наиболее часто встречающиеся реакции макролактонизации, включающие активацию той
и(или) иной концевой функциональной групп секо-кислоты в синтезе природных соединений и веществ
с биологической активностью, а также проанализированы преимущества и недостатки вышеописанных
методов макролактонизации.
Ключевые слова: секо-кислота, макролактонизация, макролактоны, природные соединения, биологи-
ческая активность
DOI: 10.31857/S0514749221050013
ВВЕДЕНИЕ
1. МАКРОЛАКТОНИЗАЦИИ ЧЕРЕЗ АКТИВАЦИЮ КИСЛОТНОЙ ФУНКЦИИ
2. МАКРОЛАКТОНИЗАЦИЯ С АКТИВАЦИЕЙ ГИДРОКСИЛЬНОЙ ГРУППЫ
3. ОКИСЛИТЕЛЬНАЯ МАКРОЛАКТОНИЗАЦИЯ
4. ВЫВОДЫ
УСЛОВНЫЕ СОКРАЩЕНИЯ
Boc - трет-бутоксикарбонил; BOP-Cl - бис(2-оксо-3-оксазолидинил)фосфонийхлорид; BQ - бензо-
хинон; Cbz - карбобензилокси-; COD - циклооктадиен; CSA - камфорсульфоновая кислота; DEAD - ди-
этиловый эфир азодикарбоновой кислоты; DIAD - диизопропиловый эфир азодикарбоновой кислоты;
DMAP - 4-(диметиламино)пиридин; DMAPO - окись 4-(диметиламино)пиридина; DTBAD - ди-трет-бу-
тиловый эфир азодикарбоновой кислоты; EDC
-
1-этил-3-[3-(диметиламино)пропил]карбодиимид;
EOM - этоксиметил-; LA - кислота Льюиса; MEM - 2-метоксиэтоксиметил-; MOM - метоксиметил-;
MPM - 4-метоксибензил-; MP - п-метоксибензилиден-; PCC - хлорхромат пиридиния; PMB - 4-метокси-
бензил-; PMP - 4-метоксифенил-; PyBOP - (бензотриазол-1-илокси)трипирролидинфосфоний гексафтор-
фосфат; PyBroP - трис(пирродидино)бромфосфоний гексафторфосфонат; PPTS - пара-толуолсульфонат
пиридиния; TBS - трет-бутилдиметилсилил-; TFA - трифторуксусная кислота; THP - тетрагидропира-
нил-; TIPS - триизопропилсилил-; TMS - триметилсилил-; Δ - нагревание при температуре кипения.
607
608
ЯКОВЛЕВА и др.
ВВЕДЕНИЕ
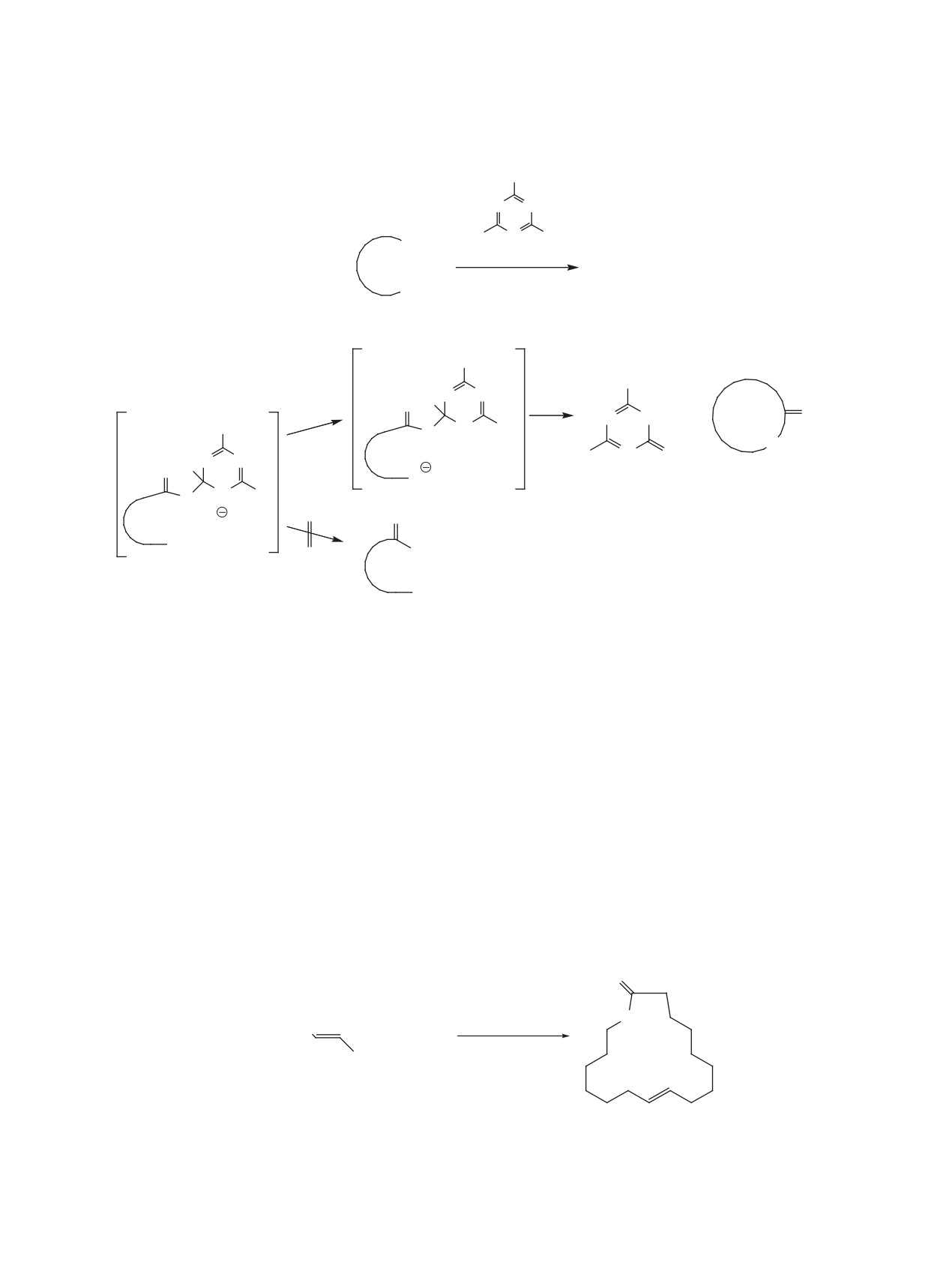
нечной функции секо-кислоты 2. Поскольку опи-
сать все известные схемы синтеза макроцикли-
В 1927 г. впервые из эфирного масла корней дя-
ческих лактонов с применением этого подхода в
гиля лекарственного Angelica archangelica был вы-
одном обзоре достаточно проблематично, приве-
делен 15-пентадеканолид (экзальтолид) (1) (рис. 1)
денные схемы синтезов лишь иллюстрируют пред-
[1], являющийся ценным душистым веществом и
лагаемые методы, на примерах получения природ-
широко используемый как компонент парфюмер-
ных и биологически активных соединений.
ных композиций, отдушек для мыла и космети-
ческих изделий [2, 3]. С тех пор интерес к макро-
1. МАКРОЛАКТОНИЗАЦИИ ЧЕРЕЗ
циклическим лактонам только увеличился.
АКТИВАЦИЮ КИСЛОТНОЙ ФУНКЦИИ
Действительно, природные макролактоны
Циклизация через тиоэфиры является одним из
представлены широким спектром структур и боль-
самых популярных химических способов получе-
шим набором интересных свойств: от парфюмер-
ния макролактонов, а самая известная реакция с
ных, фитотоксичных, феромонных и инсектицид-
участием сложных тиоэфиров - метод «двойной
ных до лекарственных (антибиотических, цито-
активации» [5]. Механизм включает первоначаль-
токсических, противоопухолевых и др.). С момен-
ное образование 2-пиридинтиоэфира ω-гидрокси-
та синтеза первых макрогетероциклов в первой
кислоты 6 в результате окислительно-восстанови-
половине ХХ столетия [4] по настоящее время
тельной конденсации Мукаямы с помощью дипи-
известно более 5 тысяч сообщений по их синтезу.
ридилдисульфида (Py-S-S-Py) и PPh3, внутримо-
Значительная их часть посвящена получению ма-
лекулярный перенос протона в котором формирует
кролактонов, сложноэфирная функция в которые
промежуточное соединение 7, в котором активиро-
вводится либо до стадии циклизации, либо обра-
ваны как карбоксильная, так и гидрокси-функции,
зуется в процессе замыкания цикла.
что приводит к «электростатической» макролакто-
Несмотря на то, что на протяжении ряда лет
низации (схема 2).
были разработаны многие эффективные методы
«Классический» метод Кори-Николау был ис-
макроциклизации, такие как реакции метатези-
пользован, например, для циклизации секо-кисло-
са, Нозаки-Хияма-Киши (Nozaki-Hiyama-Kishi),
ты 10 в ендииновое производное 11 - полупродукт
Хорнера-Вудворда-Эммонса (Horner-Wadsworth-
в синтезе мощного противоопухолевого хромо-
Emmons) и др., лактонизация секо-кислот по-преж-
нему является одним из наиболее часто использу-
протеинового антибиотика С-1027 12 из культу-
рального бульона почвенной грамположительной
емых подходов для получения макроциклических
лактонов. Из-за энтропийных и энтальпийных
бактерии Streptomyces globisporus [6], а также для
факторов прямая циклизация α,ω-гидроксикисло-
циклизации гидроксикислоты 13 в синтезе макро-
ты 2 в лактон 3, как правило, невозможна без акти-
лида аплиолида A 14, выделенного из кожи мор-
вации либо концевой спиртовой группы через про-
ского моллюска Aplysia depilans и токсичного для
межуточное соединение 4, либо карбоксильной
обитающих в воде живых организмов [7], когда
группы через интермедиат 5 (схема 1).
другие методы были не эффективны. Следует от-
метить, что в синтезе аплиолида А 14 добавление
Цель данного обзора - описание наиболее ча-
каталитического количества триэтиламина для
сто встречающихся реакций макролактонизации,
уменьшения количества диолида и полимерных
включающих активацию одной и(или) другой ко-
побочных продуктов было признано необходимым
и привело к получению целевого соединения c вы-
O
ходом 78% (схема 3).Отмечаем, что для избежания
O
образования димеров, реакцию обычно проводят в
две стадии: сначала формируют тиоэфир при ком-
натной температуре, затем разбавляют и медленно
1
добавляют в большое количество кипящего толу-
Рис. 1. Структура 15-пентадеканолида (экзальтолида)
ола.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
609
Схема 1
O
Acid
activation
[Act]
OH
COOH
4
n > 7
O
OH
O
2
O
3
Carboxylate
attack
O
LG
5
Поскольку устранение побочных продуктов
ложили использовать диимидазоилсульфид 19, ко-
тиопиридона 9 и трифенилфосфинийоксида 8,
торый позволяет получать макролактоны в более
возникающих в результате активации системой
мягких условиях и с лучшими выходами: наличие
Py-S-S-Py/PPh3, в некоторых случаях может быть
трет-бутильной группы в 4-ом положении имеет
затруднено. Кори и Кларк разработали вариант
важное значение для предотвращения образования
этой реакции, в которой тиоэфир 15 сначала син-
нежелательного N-ацильного интермедиата.
тезируют взаимодействием кислоты 2 c 2-тиопи-
Реакция Кори в условиях Брунелле нашла мно-
ридилхлорформиатом 16 (схема 4) [8, 9].
гочисленное синтетическое приложение, напри-
Этот метод был использован в синтезе лактона
мер, при циклизации гидроксикислот 20 и 21 в
17 из арахидоновой кислоты 18, играющей уни-
синтезе макролидных антибиотиков - эритроми-
версальную роль в процессах жизнедеятельно-
цинов A 22 и B 23, продуцируемых бактериями
сти млекопитающих, в качестве составной части
Streptomyce serythreus (схема 6) [11].
фосфолипидов в мембранах и предшественника в
синтезе простаноидов, лейкотриенов и различных
Отмечено значительное увеличение скорости
гидроксикислот (схема 5) [9].
циклизации в присутствии ионов металлов (Ag и
После исследования в качестве реагента ряда
Cu) и предложено несколько объяснений для вы-
различных сульфидов Кори и Брунелле [10] пред-
яснения их роли в повышении электрофильности
Схема 2
H
PyS-SPy
O N
O
COOH
PPh3
Reflux
C
S
S
rt
N
OH
O
H
O
2
6
7
O
+ Ph3PO +
N
S
O
H
3
8
9
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
610
ЯКОВЛЕВА и др.
Схема 3
MPMO
MPMO
TBSO
OH
TBSO
O
O
O
O
1) PyS-SPy,
HO
HO
PPh3, ТГФ
O OMOM
O OMOM
2) Толуол, 120°C,
57%
Cl
O
Cl
O
O
OH
BocNH
BocNH
10
11
MeO
O
N
O
H
O
O O
Me2N
O
OH
O
OH
OH
O
Cl
O
NH2
12
O
O
PyS-SPy,
OH
PPh3,
NEt3 кат.
O
HO
Толуол, ∆,
78%
13
14
тиоэфира. Среди них комплексообразование ме-
позволяющая проводить некоторые реакции при
таллов с пиридином (структура 24), атомом серы
комнатной температуре. Так, в синтезе макрос-
фелида А 27 (ингибитора клеточной адгезии)
(структура 25) и, наконец, образование хелатных
[12] применение классических методов (реакции
комплексов, содержащих шестичленные кольца
Ямагучи, Кека и Мицунобу) потерпело неудачу из-
(структура 26) (рис. 2).
за β-элиминирования. При использовании метода
Наиболее распространенной является моди-
Мукаяма-Кори для циклизации гидроксикислоты
фикация Герлаха, использующая соли Ag (I) и
28 макролактон 29 был получен с низким выхо-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
611
Схема 4
RCOOH + Et3N +
SCOCl
SCOR + Et3NHCl
-CO2
N
N
2
16
15
Схема 5
O
COOH
16
Et3N
S
N
OH
OH
18
O
Толуол, 80°C
O
90%
17
дом. Его удалось увеличить до 40% добавлением
механизм этой реакции подобен макролактониза-
AgOTf при комнатной температуре (схема 7).
ции по Кори-Николау и включает двойную акти-
Цианурхлорид 30 впервые был введен в процесс
вацию. Альтернативный путь через ацилхлорид 31
макроциклизации Венкатараманом [13], причем
был исключен теми же авторами (схема 8) [14].
Схема 6
MP
i-Pr
N
O
S
PPh3
OH
R
N
t-Bu
OH
2
19
O
OH
толуол, ∆
R
OH O O
O O O
O
O
MP
O
O
20, 21
O
HO
NMe2
OH
OH
R
O
O
O
OMe
O
O
O
OH
22, 23
R = OH (20, 22), H (21, 23).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
612
ЯКОВЛЕВА и др.
O
O
R
R
S
S
N S
M
O N
M X
O
M
HO
H
24
25
26
M = Ag или Cu
Рис. 2. Структуры возможных продуктов взаимодействия ионов металлов с тиоэфиром
Хотя цианурхлорид 30 - дешевый коммерчески
Мукаяма 34 и триэтиламином давала макроцикли-
доступный реагент, эта методика не нашла широ-
ческие лактоны 41-45 с разумными, но неудовлет-
кого синтетического применения. Она использова-
ворительными выходами. В качестве побочных
на, например, при лактонизации транс-16-гидрок-
продуктов получались дилактоны 46-50, причем
си-9-гексадеценовой кислоты (32) в синтезе изо-
их доля уменьшалась с увеличением длины цепоч-
амбреттолида 33 [13], используемого в качестве
ки исходной кислоты (схема 11, табл. 1) [15].
пищевого ароматизатора (схема 9).
В тоже время лактонизация разветвленной би-
Применение 1-метил-2-хлорпиридиний йоди-
циклической кислоты 51 до трициклического про-
межуточного соединения 52 в синтезе природного
да (34) как эффективного агента для макролакто-
макролидного антибиотика (+)-тубелактомицина
низации ω-гидроксикислот 2 было предложено
53 протекает уже с выходом 96% (схема 12) [16].
Мукаяма [15]. Механизм реакции включает заме-
щение атома хлора на карбоксилат-ион с образова-
Принимая во внимание приведенные выше
нием сильно активированного ацилпиридиния 35,
факты, авторы [17] получили более стабильную
далее участвующего в заключительной циклиза-
пиридиниевую соль, 2-хлор-6-метил-1,3-дифенил-
ции (схема 10).
пиридинийтетрафторборат (54), и применили ее в
реакции лактонизации (рис. 3).
В этом способе синтеза обработка неразвет-
вленных гидроксикислот НО(СН2)nСООН [n =
В присутствии хлорида бензилтриэтиламмония
6 (36), 7 (37), 10 (38), 11 (39) и 14 (40)] солью
и стерически затрудненного основания, такого как
Схема 7
O
O
OMEM
1) PyS-SPy/PPh3,
O
толуол
OMEM
2) AgOTf
O
MEMO
MEMO
40%
O
OH
O O
O
O O
HO
O
28
29
O
OH
O
HO
O
O
O O
27
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
613
Схема 8
Cl
N
N
Cl
N
Cl
COOH
30
Et3N
OH
MeCN или ацетон
2
Cl
Cl
N
N
OCl
N NH
+
O
Cl
N
Cl
O
H
O
Cl
N
O
N
N
Cl
O
O
3
N Cl
O
O
OH
Cl
OH
31
2,4,6-трифенилпиридин, этот реагент дал лучшие
Например, взаимодействие дихлорангидрида
результаты в макролактонизации незамещенных-
малоновой кислоты с оптически активными ди-
секо-кислот до монолактонов при более низком
олами 55 и 56 в CH2Cl2 в присутствии пиридина
содержании соответствующих диолидов. Так,
преимущественно протекает как [1+1]-конденса-
12- 44 и 14- 45 -членные лактоны были получены
ция с образованием дилактонов 57 и 58 с выхода-
циклизацией гидроксикислот 39 и 40 с выходами
ми до 30%, выходы соответствующих тетралакто-
85 и 99% соответственно, в то время как реагент
нов 59 и 60 не превышали 6% (схема 14) [18].
Мукаяма 34 дал эти же макролактоны с выходами
При взаимодействии диолов 61 и 62 с дихлор-
69 и 84% соответственно (схема 13) [17].
ангидридом
2,6-пиридиндикарбоновой кисло-
Реакции α,ω-хлорангидридов дикислот с
ты добавка поташа несколько повышает выходы
α,ω-диолами для получения соответствующих ди-
краун-эфиров 63 и 64, очевидно, за счет действия
лактонов могут быть проведены несколькими спо-
иона калия как темплатного агента (схема 15)
собами.
[19].
Схема 9
O
HO(CH2)6
30
O
Et3N/ацетон
(CH2)7COOH
70%
32
33
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
614
ЯКОВЛЕВА и др.
Схема 10
I
N
Cl
Et3N
O
COOH
34
Et3N
O
N
O
+
MeCN, ∆
N O
OH
O
OH
I
2
35
3
Схема 11
O
34/Et3N
O
O (CH2)n
HO-(CH2)n-COOH
+
(CH2)
MeCN, ∆
n
(CH2)
O
O
n
O
36-40
41-45
46-50
Чуть выше (48 и 52%) выходы макроциклов
Этим методом успешно проведена макролакто-
66 и 67, используемых для комплексообразования
низация секо-кислоты 71 в синтезе выделенного
с ионами Cu (II), Co (II), Pb (II), Ag (I), Hg (II) и
из морских динофлагеллятов Amphidinium амфи-
Ni (II), наблюдаются при проведении реакции
динолида Х 72 с потенциальной цититоксической
[1+1]-конденсации диола 68 с дихлорангидрида-
активностью против раковых клеток различных
ми 2,6-пиридиндикарбоновой и 3-оксаглутаровой
линий (схема 18) [23].
кислот в ацетонитриле в присутствии триэтилами-
на с добавкой DMAP (схема 16) [20, 21].
Увеличение количества DMAP (до 30 экв) при-
водит к росту доли дилактона в смеси, что удачно
Макролактонизация с использованием реаген-
та Ямагучи - 2,4,6-трихлорбензоил хлорида (69) -
использовано при циклизации дигидроксикисло-
остается наиболее популярным методом [22]. В
ты 73 в синтезе вербалактона 74, выделенного из
классической процедуре смешанный ангидрид 70
корней коровяка волнистого Verbascum undulatum
предварительно формируют в ТГФ в присутствии
и проявляющего антибактериальную активность
триэтиламина. После фильтрования соли Et3N·HCl
(схема 19) [24].
и упаривания смешанный ангидрид 70 разбавляют
Существует много модификаций первоначаль-
толуолом и медленно добавляют в сильно разбав-
ленный раствор DMAP (2-5 экв) при высокой тем-
ной процедуры Ямагучи, две основные из которых
пературе (80°C или кипячении) (схема 17).
были разработаны Йонемицу [25].
Таблица 1. Зависимость выходов макроциклов от длины углеродной цепи исходных гидроксикислот
№ соединения
n
№ соединения
Размер цикла
Выход, %
№ соединения
Размер цикла
Выход, %
36
6
41
8
0
46
16
93
37
7
42
9
13
47
18
34
38
10
43
12
61
48
24
24
39
11
44
13
69
49
26
14
40
14
45
16
84
50
32
3
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
615
Схема 12
CO2Me
CO2Me
MOMO
MOMO
OH
34/Et3N
O
O
CO2H
MeCN, ∆
H
96%
H
H
H
OH
OH
51
52
CO2Me
HO
O
O
H
H
OH
53
В первой из этих двух модификаций, извест-
тон 79 в качестве основного продукта, а класси-
ной как «модифицированная Ямагучи», Йонемицу
ческая процедура Ямагучи - смесь 1:1 β/γ- 79 и
определил положительный эффект прямого добав-
β/γ- 80 лактонов (схема 21).
ления большого количества DMAP к предвари-
Несмотря на широкую распространенность
тельно приготовленному смешанному ангидриду
этих методов циклизации, до сих пор нет общего
при комнатной температуре без необходимости
правила о наилучших условиях для реализации
сильного разбавления. Это показано на примере
макролактонизации Ямагучи на конкретном суб-
синтеза стагонолида Е 75 - вторичного метабо-
лита фитопатогенного гриба Stagonospora cirsii,
страте. Однако общая тенденция, по-видимому,
используемого для биологической борьбы с мно-
заключается в использовании условий Йонемицу
голетним сорным растением бодяком полевым
на относительно крупных макроциклах и класси-
Cirsium arvense L. [26], когда мольное соотноше-
ческих условий на лактонах среднего кольца (для
ние секо-кислота 76-2,4,6-трихлорбензоилхлорид
предотвращения образования диолидов и олигоме-
(69)-DMAP составляло 1:1.5:10 (схема 20).
ров).
Во второй, известной как «условия Йонемицу»,
Ph
смешанный ангидрид предварительно не форми-
руется, и DMAP вводится в начале при комнатной
BF4
температуре. Эти менее основные условия оказа-
N Cl
лись весьма эффективными, например, в синте-
Ph
зе антибиотика рутамицина B 77 [27], когда при
54
циклизации гидроксикислоты 78 методики Кека,
Рис. 3. Структура 2-хлор-6-метил-1,3-дифенилпири-
Мукаяма и Кори давали несопряженный β/γ-лак-
динийтетрафторбората
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
616
ЯКОВЛЕВА и др.
Схема 13
54/PhCH2Et3N+Cl-
O
HO-(CH2)n-COOH
O
Ph
(
)
n
Ph
N
Ph
39, 40
44, 85%
ClCH2CH2Cl, ∆
45, 99%
Действительно, реагент Ямагучи также исполь-
двойных связей, а также чувствительных асим-
зовался при димеризации секокислот с образова-
метрических центров. Эти проблемы могут быть
нием природных диолидов. Это проиллюстриро-
решены макролактонизацией ацетиленовых ана-
ванно на примере синтеза антибиотика (+)-карпа-
логов секо-кислот с последующим гидрированием
ина 81 из гидроксикислоты 82 (схема 22) [28].
или использованием более мягких реагентов, ко-
торые обладают ненуклеофильными противоио-
Самым главным недостатком процедуры
нами, например, 2-бром-1-этилпиридиний тетраф-
Ямагучи является применение высокоосновного
торбората (рис. 4).
DMAP и высокой температуры. Эти факторы ино-
гда приводят к нежелательным побочным реак-
Использование 2-метил-6-нитробензойного ан-
циям, таким как Z/E-изомеризация сопряженных
гидрида (83) было впервые описано Шиина и
Схема 14
(
)n
O
O
O O
O
Cl
Cl
O
O
Py, CH2Cl2
O
O
O
(
)n
(
)n
(
)n
HO
HO
55, 56
57, 23%
58, 30%
O
O
O
O
(
)n
(
)n
O
O
+
O
O
(
)n
(
)n
O
O
O
O
59, 6%
60, 5%
n = 1 (55, 57, 59), 2 (56, 58, 60).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
617
Схема 15
O
O
Cl
Cl
N
O
O
O
O
O
O
K2CO3, CH2Cl2, ∆
O
O
44%
O
O
N
OH
HO
O
O
61
63
O
O
Cl
Cl
N
O
O
O
O
O
O
K2CO3, CH2Cl2,
∆
O
O
48%
O
O
N
O
O
OH OH
62
64
O
O
K +
O
O
O
HO
65
успешно применено к большому числу синтезов.
активностью против раковых клеток различных
Реакции макролактонизации секо-кислот проводят
линий (схема 23) [29].
в CH2Cl2 при комнатной температуре с примене-
Проведение лактонизации в присутствии оки-
нием избытка DMAP для ограничения образова-
си диметиламинопиридина (DMAPO) дает воз-
ния дилактонов.
можность повысить выход монолактона. Это ис-
Данный подход успешно применен при цикли-
пользовано при циклизации гидроксикислоты
зации секо-кислоты 84 в синтезе выделенного из
86 для эффективного построения ядра 29-член-
морских динофлагеллятов Amphidinium амфиди-
ного 2-гидрокси-24-оксооктакосанолида 87 - за-
нолида К 85 с потенциальной цититоксической
щитной слюнной секреции африканского тер-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
618
ЯКОВЛЕВА и др.
Схема 16
O
NH
HN
O
O
O
Cl
Cl
O
O
DMAP, Et3N, MeCN
O
O
O
52%
66
O
NH
HN
O
NH
HN
O
O
OH
HO
N
Cl
Cl
68
O
O
DMAP, Et3N, MeCN
N
48%
O
O
67
Схема 17
COCl
Cl
Cl
N
O
O
O
Cl
Cl
COOH
N
69
N
O
Et3N/ТГФ
толуол, 80°C или ∆
OH
N
Cl
Cl
O
Cl
O
OH
H
O
-
2
70
Cl
Cl
O
-H+
O
N
O
N
O
H
N
-
N
3
Схема 18
O
O
1. 69, Et3N/ТГФ
O O O
O O
HO
O
2. DMAP/толуол, ∆
O
COOH
62%
O
71
72
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
619
Схема 19
O
OH OH
1. 69, Et3N/ТГФ
H
HO
H
O
O
COOH
2. DMAP (30 экв)/толуол, ∆
H OH
H
60%
O
73
74
Схема 20
O
O
HOOC
OH
DMAP, 69
O
O
Et3N, толуол
65%
OMOM
OMOM
OH
76
75
Схема 21
COOH
α
O
OTBS
γ
OTBS
O
β
O
O
O
OTBS
O
O
OTBS
OH
69, DMAP, Et3N
OTBS
O
H
H
OTBS
бензол
86%
O
O
TBSO
TBSO
78
80
O
OH
O
OTBS
O
O
O
O
O
O
OH
OTBS
O
O
OH
OTBS
H
H
O
O
HO
TBSO
77
79
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
620
ЯКОВЛЕВА и др.
Схема 22
Cbz
N
O
HO
1. 69, Et3N/ТГФ
O
N
(CH
2. DMAP, толуол, ∆
2)7COOH
O
71%
Cbz
O
82
N
Cbz
H
N
O
O
O
O
N
H
81
Схема 23
NO2
O
O
NO2
O
O
O
83
OH
OH
HO
DMAP, CH2Cl2
O
71%
OH
O
O
84
85
мита Pseudacanthotermes spiniger (схема
24)
[30].
Описано также использование других смешан-
N
Br
ных ангидридов, но только с ограниченным синте-
Et
тическим применением.
BF4
Так, промежуточный ангидрид 88, полученный
Рис. 4. Структура
2-бром-1-этилпиридиний тетра-
фторбората
из пивалоилхлорида и секо-кислоты 89, использо-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
621
Схема 24
83/Et3N
DMAP или DMAPO
CH2Cl2/ТГФ, 50°C
OH
O
PMBO
PMBO
O OH
OH
O
OH
86
64% (с DMAP), 77% (с DMAPO)
O
HO
O
O
87
ван в синтезе 3-изоверрукарина А 90, как аналога
В работе [33] продемонстрировано, что для ги-
веррукарина 91 - сесквитерпеноидного вторич-
дроксикислот 98-103 с длиной углеродной цепи
ного метаболита плесневых грибков различных
7-9 основными продуктами являются диолиды
видов Fungi imperfecti, обладающего высокой ток-
110-112, с увеличением длины их цепочки доля
сичностью (схема 25) [31].
диолидов 113-115 уменьшается (схема 28, табл. 2).
Циклизация рицинолевой кислоты 92 проте-
Возможно также проведение прямой макро-
кает через образование смешанного ангидрида
лактонизации секо-кислот в присутствии TMSCl
93 с Boc2O [32], что приводит к смеси моно- 94 и
и TiCl2(OClO4)2 в дихлорметане при комнатной
ди- 95 -олидов. Замена триэтиламина и DMAP на
температуре или кипячении с хорошими выхода-
ми. Эта методика применена для синтеза лакто-
более основные диизопропилэтиламин и 4-пирол-
на 94 из (R)-рицинолевой кислоты 92 (схема 29)
лидинопиридин, соответственно, ведет к умень-
[34].
шению доли диолида 95 в смеси (схема 26).
Так как выходы среднецепочных макроциклов
В 1993 году Шиина и Мукаяма описали ма-
в реакциях лактонизации обычно невысоки, была
кролактонизацию силил ω-силилкарбоксилатов
применена новая стратегия [35], основанная на
96 при комнатной температуре в присутствии
Rh-катализированном образовании циклического
пара-трифторметилбензойного ангидрида (97) и
интермедиата 116 и дающая моно-лактоны 3 с вы-
Ti (IV) катализатора, полученного смешиванием
сокими выходами (схема 30).
TiCl4 и AgClO4 [33]. Применение силильных про-
изводных секо-кислоты 96 необходимо для пре-
Используя этот метод при циклизации секо-кис-
дотвращения дезактивации катализаторов титана
лот 117-121, удалось повысить выход моно-лакто-
(схема 27).
нов 104-108 до 90% (схема 31, табл. 3) [35].
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
622
ЯКОВЛЕВА и др.
Схема 25
O
O
H
H
H H
O
Cl
O
O
O
O
O
O
, Et3N, CH2Cl2
43%
O
O
O
O
O
O
OH
O
OH
O
THPO
THPO
OH
89
88
O
H H
H
O
H
H
O
O
O
O
1. DMAP, CH2Cl2
O
2. PPTS/EtOH
O
O
O
O
O
O
O
O
HO
O
HO
90
91
Схема 26
OH
OH
O O
Boc2O/Et3N
Me(CH2)5
(CH2)7COOH
Me(CH2)5
(CH2)7
O
O
92
93
Me(CH2)5
O
O
O
DMAP, толуол
O
+
90oC
O
Me(CH2)5
O (CH2)5Me
94, 44%
95, 24%
1. Boc2O/Et(i-Pr)2N
2. 4-пирролидинопиридин/толуол, 90°C
92
94 (56%) +
95 (15%)
Применение кислот Льюиса в качестве катали-
ω-гидроксикарбоновые кислоты 2 лактонизируют
заторов в макролактонизации смешанных анги-
в присутствии пара-нитробензойного ангидрида
дридов было исследовано Ямамото [36]. Используя
(122), получая соответствующие лактоны 3 с вы-
каталитические количества
(10-20%) Sc(OTf)3,
сокими выходами (схема 32).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
623
Схема 27
O O
O
O O
O
F3C
CF3
97
TMSO(CH2)n
O
(TiCl4 + 2AgClO4)
TMSO(CH2)n
OTMS
CF3
96
O
O
3
Схема 28
(CH2)n
O
O
97/TiCl4 + 2AgClO4
O
O
O
+
CH2Cl2
O
TMSO
(CH2)n
OTMS
(CH2)n
O
(CH
2)n
98-103
104-109
110-115
Схема 29
TMSCl/CH2Cl2
92
94
97/TiCl2(ClO4)2
83%
Результаты особенно впечатляющи для средне-
ции [37]. Высокие выходы реакций лактониза-
кольцевых лактонов 104-106, 124, при получении
ции наблюдаются как для неразветвленной 128,
которых основными продуктами обычно являются
так и α-метилзамещенных гидроксикислот 126,
диолиды 110-112, 125 (схема 33, табл. 4).
129-131, а также для содержащей двойную связь
Z-конфигурации 127 (схема 34, табл. 5).
Хотя эта перспективная методология пока не
получила широкого применения в тотальном син-
В синтезе антибиотика цефалоспоролида D
тезе, неприродные ароматичные лактоны были
138 Шиина [38] описал использование Hf(OTf)4
синтезированы при использовании этой актива-
для циклизации гидроксикислоты 139 при получе-
Таблица 2. Зависимость выходов макроциклов от длины углеродной цепи исходных гидроксикислот
№
№
Число членов в
№
Число членов в
n
Выход, %
Выход, %
гидроксикислоты
монолактона
цикле
дилактона
цикле
98
5
104
8
0
110
16
50
99
6
105
9
0
111
18
40
100
7
106
10
33
112
20
47
101
8
107
11
70
113
22
23
102
10
108
13
75
114
26
7
103
13
109
16
89
115
32
4
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
624
ЯКОВЛЕВА и др.
Схема 30
O
O
Si
Si
H
RhCl(PPh3)3
OH
O
+
H
H
Si
Si
OH
OH
2
O
RhCl(PPh3)3
O Si
Me2Si(OTf)2
Si
n
O
+
O
80°C
n + 5
80°C
Si
O
O Si
116
3
Схема 31
1. RhCl(PPh3)3
O
2. 80°C
3. Me2Si(OTf)2, 80°C
104-108 + 110-114
HO
(CH2)n
OH
117-121
нии с выходом 67% восьмичленного лактона 140.
макроциклизациюсеко-кислоты 141 в 14-тичлен-
Выход в присутствии Sc(OTf)3 составил 44%, а
ный лактон 142 в полном синтезе антибиотика
процедура с катализаторами TMSCl и Ti (IV) ока-
нарбонолида 143 через образование смешанно-
залась неудачной (схема 35, табл. 6).
го ангидрида с использованием дифенилхлор-
фосфата 144 в качестве активирующего агента
Реагенты на основе фосфора, широко исполь-
(схема 36).
зуемые в синтезе пептидов, циклодепсипептидов
и пептидомиметиков, также нашли применение в
Так как смешанные углерод-фосфорные анги-
реакциях макролактонизации. Масамунэ [39] пер-
дриды 144, как правило, лабильны при нагревании
вым выявил потенциал углерод-фосфор-смешан-
и склонны к образованию симметричных анги-
ных ангидридов в синтезе макролактонов и описал
дридов 145 [39], Масамунэ впервые показал, что
Таблица 3. Зависимость выходов макроциклов от длины углеродной цепи исходных гидроксикислот
Число членов
Число членов
№ гидроксикислоты
n
№ монолактона
Выход, %
№ дилактона
Выход, %
в цикле
в цикле
117
5
104
8
85
110
16
4
118
6
105
9
87
111
18
8
119
7
106
10
88
112
20
8
120
8
107
11
83
113
22
10
121
10
108
13
87
114
26
10
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
625
Схема 32
O
O
OH
O
Sc
COOH
O2N
NO2
122
O
O
Sc(OTf)3/MeCN, ТГФ, ∆
OH
O
2
NO2
O
O
3
Схема 33
(CH2)n
O
O
O
122
O
O +
HO
(CH2)n
OH
Sc(OTf)3/MeCN, TГФ, ∆
O
(CH2)n
O
(CH2)n
117-119, 123
104-106, 124
110-112, 125
проводить макролактонизацию необходимо при
динемицина A 152 промежуточный ен-диино-
температурах ниже 80°С (схема 37).
вый трициклический макролактон 153 был полу-
чен из секо-кислоты 154 с выходом 51% с помо-
Реагент Паломо BOP-Cl 146 успешно использо-
щью PyBroP-опосредованной макролактонизации
вался при циклизации гидроксикислоты 147 в син-
с последующей внутримолекулярной реакци-
тезе хлоротриколида 148, являющегося агликоном
ей Дильса-Альдера при комнатной температуре
антибиотика хлоротрицина 149, когда методики
(схема 39) [41].
Ямагучи-Йонемицу и Боден-Кека давали очень
PyBOP 151 был использован при циклизации
низкие выходы (схема 38) [40].
гидроксикислоты 155 в синтезе (-)-спиносина A
Пептид-связывающие реагенты PyBroP 150 и
156, входящего в семейство инсектицидов с мощ-
PyBOP 151 также успешно применяются в син-
ной активностью на широком спектре насекомых
тезе макролактонов. Так, в синтезе антибиотика
(схема 40) [42].
Таблица 4. Зависимость выходов макроциклов от длины углеродной цепи исходных гидроксикислот
№
Число членов в
Число членов в
n
№ монолактона
Выход, %
№ дилактона
Выход, %
гидроксикислоты
цикле
цикле
117
5
104
8
71
110
16
<
118
6
105
9
52
111
18
3
119
7
106
10
87
112
20
1
123
13
124
16
99
125
32
1
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
626
ЯКОВЛЕВА и др.
Схема 34
OH
122
OH
Sc(OTf)3/MeCN, TГФ, ∆
O
O
O
126-131
132-137
Несмотря на то, что в этих исследованиях не
Дициклогексилкарбодиимид (DCC, 157) в при-
приведены детали механизма реакции макролак-
сутствии пиридина давно известный эффективный
тонизации, очевидно, что она протекает через пер-
этерифицирующий агент (рис. 5) [44]. В реакци-
воначальное образование ацил-оксифосфониевых
ях макроциклизации он используется обычно со-
интермедиатов [43].
вместно с DMAP. Данная методика применяется
Таблица 5. Зависимость выходов макроциклов от длины углеродной цепи исходных гидроксикислот
№
Гидроксикислота
№
Бензолактон
Выход, %
126
132
53
127
133
66
128
134
66
129
135
74
130
136
65
131
137
60
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
627
Схема 35
COOH
122/кат.
O
O
MeCN, ∆
BnO
HO
OH
OBn
O
O
139
140
138
Таблица 6. Зависимость выходов макроциклов от используемого катализатора
Катализатор
Выход 140, %
Hf(OTf)4
67
Sc(OTf)3
44
TMSCl и Ti (IV)
0
довольно редко, главным образом из-за образова-
160, а гексадеканолид 124 выделяется с выходом
ния нереакционноспособного побочного продукта
лишь 4% (схема 42) [44].
N-ацилмочевины 158 (схема 41).
В синтетических исследованиях по синтезу ан-
Действительно, основным продуктом макролак-
тибиотика кольлетодиола 161 Кек и Боден показа-
тонизации
15-гидроксипентадекановой кислоты
ли решающую роль стадии переноса протонов с
123 является побочный продукт N-ацилмочевина
использованием DMAP·HCl для предотвращения
Схема 36
O
O
O
O
1. (PhO)2 P
Cl
144
N, ТГФ, 0°С
Et3
OH
2. DMAP, бензол, 80°С
OH
O
O
O
O
O
O
O
O
O
O
141
142
143
Схема 37
O O
HO
OH
O
n
n
145
∆
O
O
O O
R
P
Cl
O
R
OH
R
O P
R
Et3N
O
OH
OH
2
144
3
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
628
ЯКОВЛЕВА и др.
Схема 38
TBDPSO
TBDPSO
COOH
O
O
O
O
O
P
N
N
O
O
O
Cl
O
O
146
O
HO
MOMO
MOMO
Et3N, толуол, 100°С
O
O
OH
O
O
CO
50%
2H
OH
SiMe3OMOM
SiMe3OMOM
147
148
CO2H
O
O
HO
O
O
MeO
Cl
H
O
O
O
H
HO
O
O
HO
O
149
образования нежелательного побочного продукта
заны мостиками по меньшей мере из 10 атомов
при циклизации секо-кислоты 162 и получения ди-
(схема 44) [47].
лактона 163 с выходом 82% [45] и гексадеканолида
Другая система DMAP-TsOH использовалась
124 - 95% (схема 43) [44].
для циклизации гидроксикислоты 166 при полу-
Потенциал этой так называемой методики
чении двух эпимерных депсипептидов турнагаи-
Боден-Кека был быстро выявлен и использовался
нолидов А 167 и В 168, выделенных из бактерий
в ряде синтезов биологически активных соедине-
Bacillus (схема 45) [48].
ний [46].
Основным недостатком, реагента DCC, кото-
Кроме того, несколько других источников про-
рый обычно используется в избытке и «гасится»
тонов были использованы в макролактонизации
метанолом в уксусной кислоте, является сложное
типа Боден-Кека.
удаление флеш-хроматографией побочного про-
Так, система DMAP-трифторуксусная кисло-
дукта - мочевины 159. Поэтому появилось не-
та была применена при циклизации секо-кислоты
сколько модификаций этерифицирующих реаген-
164 в синтезе анса-гликозида 165 - циклофана, в
тов, являющихся в основном водорастворимыми
котором пара-положения бензольного кольца свя-
мочевинами.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
629
Схема 39
O OMe
O Me
Br
P
N
H
H
N
N
PF6
3
H
150
H
HO
Et3N, CH2Cl2
O
HO2C
OMe
51%
H H
OMe
O
154
153
O Me
O HN
MeO2C
H
MeO
OMe
152
Схема 40
MeO
O P N
MeO
OMe
N
3
OMe
O
O
OMe
N
OMe
N PF6
151
O
O
O
CO2H
DMAP, CH2Cl2
OH
70%
O
TBSO
TBSO
Br
Br
155
MeO
OMe
O
O
NMe2
OMe
O
O
O
O
H
O
156
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
630
ЯКОВЛЕВА и др.
Высокоосновный DMAP иногда бывает вре-
ден для процесса макролактонизации. Например,
в синтезе антибиотика памамицина-607 175, вы-
N C N
деленного из бактерий Streptomyces alboniger и
S. aurantiacus, методики Кори-Николау-Герлаха,
Мукаяма и Ямагучи-Йонемицу при циклизации
секо-кислоты 176 не давали лактон 177, тогда как
157
«обычный» вариант Ямагучи приводил к полной
Рис. 5. Структура дициклогексилкарбодиимида
эпимеризации C2 центра. Система DCC-пиридин-
Так, Косиенски применял N-циклогексил-N'-(b-
PPTS была единственной, которая приводила к
[N-метилморфолино]-этил)-карбодиимид пара-то-
макродиолиду 177 с хорошим выходом (схема 48)
луолсульфонат (169) в присутствии DMAP-TFA
[51].
при циклизации секо-кислоты 170 в общем синте-
В макролактонизации также применялись де-
зе морского циклодепсипептида джаспамида 171
гидратирующие реагенты: N,N,N',N'-тетраметил-
(схема 46) [49].
хлорформомидиний хлорид
(178) и
1,3-диме-
1-(3-Диметиламинопропил)-3-этилкарбоди-
тил-2-хлоримидазолий хлорид (179) (рис. 6) [52].
имид (172), в виде его гидрохлорида используется
более широко, например, при циклизации гидрок-
Эти реагенты обычно синтезируют in situ на-
сикислоты 173 в синтезе антибиотика бафиломи-
греванием растворов соответствующих мочевин
цина 174 (схема 47) [50].
180 или 181 с оксалилхлоридом. После упарива-
Схема 41
H
O
N N
DCC
H
HO
OH
O
N N
OH
n-2
OH
n-2
n
O
O O
2
158
H
H
DCC-DMAP
N N
DMAP
O
O
O
159
HO
N
O
n
n-2
N
3
Схема 42
O
O
H C(CH2)14OH
157
O
HO(CH2)14COOH
+
N N
DMAP
O
123
160
124
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
631
Схема 43
O
O
O
O
OH
O
O
157, DMAP
O
O
CO
2H
DMAP-HCl
O
O
O
O
CHCl3
O
O
HO
82%
OH
162
163
161
Схема 44
O
O
O
O
157, DMAP
O
COOH
O
O
DMAP-TFA
O
O O
O
AcO
CHCl3
OH
O
AcO
AcO
54%
OAc
AcO
OAc
164
165
Схема 45
O
OH
O
N
COOH
DMAP-TsOH
O
H
CHCl3
N
HN
O O
H
O
HN
O O
O
O
NH
N
NH
H
N
H
166
167, 15%
O
O
N
+
H
HN
O O
O
O
NH
N
H
168, 33%
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
632
ЯКОВЛЕВА и др.
Схема 46
H
HO
H
HO
Br
N
N
N C N
Br
N
SO3
O
HO
NH
NH
169
N
N
O
DMAP, DMAP-TFA
O
O
O
CHCl3-ТГФ, ∆
OH
O
O
O
28%
O
NH
O NH
170
171
Схема 47
OMe
HO2C
OH
OH
OTBS OTBS S OHS
OMe
173
N
OMe
N C N
O
OH
172
O
DMAP, CH2Cl2 , ∆
OTBS OTBS S OHS
65%
OMe
OMe
O
OH
OH OH O
HO
O
OMe
174
ния растворителя и избытка оксалилхлорида оста-
получением соответствующих лактонов 3 с хоро-
ток растворяют в ацетонитриле и медленно добав-
шими выходами (схема 49).
ляют раствор коллидина и секо-кислот 2 в смеси
Так, при использовании N,N,N',N'-тетраметил-
эфир-ацетонитрил при комнатной температуре с
хлорформомидиний хлорида
(178) для макро-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
633
Схема 48
2
O
HO2C
HO
O
O O
NHBoc
O
176
O
DCC / Py / PPTS
O
Cl(CH2)2Cl, D
O
O O
NHBoc
56%
O
O
177
O
O
O
O
O O
N
O
175
циклизации 12-гидроксидодекановой (121) и 15-ги-
Старка, нашел применение в синтезе макролакто-
дроксипентадекановой (123) кислот выходы лак-
на 184 из 12-гидрокси-9Е-октадеценовой кислоты
тонов 108 и 124 составили 90 и 54% соответствен-
(185) (схема 52) [55].
но [53].
Применение дистанноксанов 186 (X = Cl) и 187
1,3-Диметил-2-хлоримидазолий хлорид
(179)
(X = NCS) (20%) описано в работе [56]. Интересно,
нашел применение в селективной димеризации се-
что в отличие от реакций, катализируемых Bu2SnO,
ко-кислоты 182 в синтезе гликолипида цикловира-
процесс практически необратим, так что аппарат
цина В1 183, обладающего высокой антивирусной
Дина-Старка не требуется, и реакция циклизации
активностью. В работе [54] показано, что добавле-
секо-кислот 123 и 188 в макролактоны 124 и 189
ние катиона калия имеет решающее значение для
может быть проведена в условиях умеренного раз-
селективности: в этом случае макродилактониза-
бавления (схема 53).
ция, как полагают, протекает за счет формирова-
ния вокруг него полости определенного размера
Cl
Cl
(схема 50).
N
Cl
В 1980-х гг. был разработан протекающий с
N
N N
двойной активацией метод макролактонизации (и
Cl
этерификации) с применением для дегидратации
178
179
Bu2SnO (схема 51) [55].
Рис. 6. Структуры N,N,N',N'-тетраметилхлорформо-
Этот метод, проводимый в кипящем мезити-
мидиний хлорида (178) и 1,3-диметил-2-хлоримидазо-
лене (165°С) с использованием аппарата Дина-
лий хлорида (179)
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
634
ЯКОВЛЕВА и др.
Схема 49
O
Cl
O
(COCl)2
Коллидин, MeCN, Et2O
N
Cl
N N
N
O
Cl(CH2)2Cl
R'
n
O
R R'
65°C
R
HO
OH
n
2
180, 181
178, 179
3
R, R' = H (180, 178); R, R' = CH2CH2 (181, 179).
Термолиз диоксоленона 190 является извест-
Эта методология нашла синтетическое приме-
ным процессом получения производных β-ацетил-
нение в демонстрирующей впечатляющую регио-
кетена 191 в относительно мягких условиях (при
селективность циклизации секо-кислоты 193, со-
кипячении в толуоле). Промежуточный кетено-
держащей несколько гидроксильных групп, в вось-
вый продукт 191 может быть внутримолекулярно
мичленный лактон 194 - полупродукт в синтезе
схвачен кислородным нуклеофилом с получением
пиран-полуацеталь содержащего цитотоксическо-
соответствующего макролактона 192 (схема 54)
го макролида каллипелтозида А 195, выделенного
[57].
из морской губки Callipelta sp. (схема 55) [58].
Схема 50
OBn
OBn
(H2C)11
BnO
OBn
(CH2)11
BnO
OBn
O
O
O
O
CO2H
OH
179
O O
K
DMAP, KH
O
O
OH
CO2H
CH2Cl2
O
O
O O
54%
BnO
OBn (CH2)11
BnO
OBn
(CH2)11
OBn
OBn
182
OH
(CH2)11
HO
OH
O
O
O O
O
O
O O
HO
OH (CH2)11
OH
183
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
635
Схема 51
O
O
O
SnBu2
O
O
3
H2O
O H
O
Bu2SnO
COOH
O
O Sn
OH
Bu2
2
Схема 52
H
H
(CH2)5Me
(CH2)5Me
Ph2S2 , hν
OH
Bu2SnO 10%
O O
92
mesitylene, ∆
O
44%
OH
185
184
Схема 53
186
HO(CH2)14CO2H
O
декан, ∆
81%
O
123
124
(CH2)3Me
(CH2)3Me
187
O O
декан, ∆
HO (CH2)10CO2H
63%
188
189
BuX
Bu Bu
Sn O
Sn
O
Bu
Bu
O Sn
O Sn
Bu BuX Bu
X = Cl (186), NCS (187).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
636
ЯКОВЛЕВА и др.
Схема 54
O
O
O O
O O
Толуол, ∆
O
O
OH
OH
190
191
192
Циклизацию ациклического предшественника
нов, что продемонстрировано циклизацией его в
196 возможно провести в присутствии гидрида
макролактон 200 в синтезе агликона каллипелто-
натрия в ТГФ даже при комнатной температуре,
зида 201 (схема 57) [60].
получая лактон 197 - полупродукт в синтезе са-
Удивительная региоселективность образования
лицилигаламида A 198 - цитотоксического макро-
восьми- 202, а не 15-членного лактона наблюдает-
лида, выделенного из морской губки Haliclona sp.
ся при макролактонизации по Бекману соединения
(схема 56) [59].
203 (схема 58) [61].
Интермедиат алкилкетена также может быть
Кислотно-катализируемая термодинамически
получен из β-кетоэфира 199 вместо диоксолено-
управляемая транс-лактонизация гидроксилакто-
Схема 55
OH
OH O O
O
Толуол, ∆
O
53%
OH
OH
MeO
O
O
MeO
OH
OH
OH
193
194
O
O
NH
MeO
O
O
O
H OH
MeO
O
O
Cl
195
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
637
Схема 56
OH
HO
H
O
O
O
NaH, ТГФ
OPMB
O
64%
O
OPMB
196
197
O
HN
OH
H
O
O
OH
198
Схема 57
OTBS O O
OTBS
OEt
OPMB
Толуол, ∆
O
OPMB
73%
MeO
OH
MeO
O O
TIPS
TIPS
199
200
OH
O
H OH
MeO
O O
Cl
201
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
638
ЯКОВЛЕВА и др.
Схема 58
H
MeO
N
H
N
MeO
N
N
O
O
O
O
OH
NaH, ТГФ
O HO
O
O
30%
OH
O
O
O
O
203
202
Схема 59
O
O
TsOH, CH2Cl2
O
O
n = 3, 97%
OH
OH
n
204
205
нов 204 была изучена Кори и Николау [62]. Она
Описаны также основная и кислотная транс-
проходила с высокими выходами для n = 1-3, с уве-
лактонизации с сокращением цикла природных
личением n скорость взаимопревращений умень-
соединений. Эти подходы нашли применение для
получения 12-тичленного макролида - антибио-
шалась, так как все бóльшие размеры кольца фор-
тика эритромицина А 209 [64] - из 14-тичленного
мируются в переходном состоянии. Так, 9-тичлен-
предшественника 210 при дополнительном воз-
ный лактон 204 превращается в 12-тичленный с
действии микроволнового облучения (схема 61,
выходом 97% (схема 59).
табл. 7).
Аналогичный процесс при катализе камфор-
Методология двухэтапного процесса Гауса и
сульфокислотой, протекающий с 11-тичленнымти-
Кита-Троста основана на синтезе винилового эфи-
олактоном 206, дает 12-тичленный 207 с хорошим
ра с последующей макролактонизацией, катализи-
выходом, что использовано в синтезе метинолида
руемый кислотой.
208 - агликона антибиотика метимицина (схема 60)
В методике Гауса виниловый эфир 211, полу-
[63].
ченный взаимодействием карбоновой кислоты 2 с
Схема 60
O
O
O
OBn
CSA
OBn
OH
бензол, 70°С
S
HO
63%
O
O
HS
O
O
HO
O
206
207
208
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
639
Схема 61
OH
N
N
O
HO
HO
HO
O
O
O
O
O
метод А или В
HO
OH O
O
O
OMe
O
OMe
O
O
OH
OH
O
O
210
209
Таблица 7. Зависимость выхода макроцикла (209) от условий проведения реакции
Метод
Реагенты
Мощность микроволнового излучения, В
Температура, °С
Выход, %
K2CO3, Bu4NBr, ДМФА
200
130
58
А
Bu4NBr, ДМФА
200
130
72
Al2O3 (pH 6.5-7.5)
400
180
63
B
SiO2
400
180
76
4-(диметиламино)бут-3-ин-2-оном 212, обрабаты-
В макролактонизации Кита-Троста винило-
вают камфорсульфокислотой (1-5%), получая со-
вые эфиры 216 образуются через рутений-ката-
лизируемые реакции [66] карбоновых кислот 2 с
ответствующий лактон 3 (схема 62) [65].
коммерчески доступным этоксиацетиленом 217.
Эта методика использовалась для циклизации
Выделенный хроматографически виниловый эфир
гидроксикислоты 213 до макролактона 214 в син-
216 далее лактонизуется в кислой среде (CSA или
тезе брефельдина А 215 - антибиотика, произ-
TsOH, 10 мол %) (схема 64) [67].
водимого грибками Eupenicillium brefeldianum
Эта методика применялась в макролактониза-
(схема 63) [65].
ции секо-кислоты 218 до макролактона 219 в раз-
Схема 62
O
O
N
O
HO
212
O
O
HO n CO2H
n
HO
n
O N
O N
2
211
O
O
H+ или LA
N
O
O
n
3
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
640
ЯКОВЛЕВА и др.
Схема 63
O
O
COOH
1. 212, ТГФ, 0°C (96%)
O
MEMO
2. CSA (1%),
MEMO
O
OH
толуол, 80°C (74%)
213
214
HO
H
O
HO
O
215
Схема 64
OEt
O
217
O
HO n
HO
COOH
n
Trost’s protocol:
Trost’s protocol:
O OEt
O
[RuCl2(p-cymene)]2 (2 мол %), толуол
CSA, толуол
n
Kita’s protocol:
Kita’s protocol:
[RuCl2(p-cymene)]2 (0.5 мол %), ацетон
TsOH, ClCH2CH2Cl
3
2
216
Схема 65
OBn
OMe
(2%)
1. 217, [RuCl2(p-cymene)]2
COOH
толуол, 0°C
OMe O
OBn OH OH
2. CSA, толуол, 80°C
OH
O
63%
BnO
OBn
218
219
OH
O
O
NH
O
O
OH
220
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
641
Схема 66
OH
Me(CH2)5
N
Ts
92
CH2Cl2
O
N
(CH
2)6
Ts
O
H H
221
H
Me(CH2)5
O:
H+
–H+
95 + 95
CH2Cl
2
O
N
(CH2)6
Ts
-
N
Ac
Ts
O
H H
H+
мер-селективном синтезе природного соединения
Нарасака описал использование (метилтио)ме-
апикуларена A 220 [68] с цитотоксическими свой-
тиловых эфиров в макролактонизации. В синтезе
ствами (схема 65).
пирролизидинового алкалоида интегеррмина 222,
выделенного из Crotalaria incana, исходный эфир
Недавно сообщалось [69] о еще одном синте-
223 «активируется» окислением, а затем спирто-
зе макролактонов через промежуточные вини-
вая функция депротонируется с получением соот-
ловые эфиры. На примере рицинолевой кислоты
ветствующего макролактона 223 (схема 67) [70].
92 показано, что ее трансформация в эфир 221 с
последующей кислотной обработкой, приводит
Берк разработал методику использования три-
преимущественно к образованию моно-лактона
хлорэтиловых эфиров в присутствии основания
94. Показано, что в данной реакции эффективны
в синтезе дигидропиранильных макролактонов.
кислоты Льюиса и Бренстеда, но наилучшие ре-
Применение солей калия, в частности К2СО3, не-
зультаты достигнуты при использовании TsOH
обходимо для активации процесса макролактони-
(схема 66, табл. 8).
зации секо-предшественника 225 до макролактона
Таблица 8. Зависимость выходов лактонов от используемого катализатора
Катализатор Н+
Выход моно-лактона 94, %
Выход ди-лактона 95, %
Sc(OTf)3
23
5
Yb(OTf)3
19
7
Cu(OTf)2
35
6
CuOTf
32
7
Zn(OTf)2
43
10
Bi(OTf)3
42
8
CSA
63
13
Fe(OTf)3
34
18
PTSA·H2O
91
0
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
642
ЯКОВЛЕВА и др.
Схема 67
OMOM
MOMO O
OH
O
S
O
O
O
1. H2O2, Mo(VI)
кат.
O
H
2. BuLi
O
O
O
OH
H
H
40%
O
O
N
N
N
O
223
224
222
Схема 68
H
H
H
H
O
BocHN
O
CO2CH2CCl3
BocHN
O
O
O
HO
O
O
O
NHBoc
K2CO3, ТГФ
NHBoc
H
H
H
H
88%
O
O
O
O
O
O
H
H
H
H
O
O
BocHN
BocHN
225
226
226 как представителя класса ионофоров. В при-
бисдипиридилзамещенного кетона 229. Несмотря
сутствии карбонатов натрия или лития циклизация
на то, что полученный из него и гидроксикислоты
не происходит (схема 68) [71].
2 оксимоэфир 230 пространственно сближает ги-
Цианометиловый эфир 227 был зациклизован
дрокси- и карбокси-функции, он является стабиль-
Панеком в промежуточный лактон 228 в синтезе
ным и без дополнительной активации не лактони-
апикуларена А 220 (схема 69) [72].
зуется. Лишь обработка промежуточного медьсо-
В макроциклизации также широко использует-
держащего комплекса 231 фторидом пиридиния
ся реагент Паломо, представляющий собой оксим
приводит к макроциклу 3 (схема 70).
Схема 69
OBn
OBn
NaH, ТГФ, ∆
O
O
O
63%
O
NC
O
HO
O
MOMO
OMOM
227
228
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
643
Схема 70
N
N
HO
N
N
O
O
N
OH
229
O
EDC-DMAP
O
N
OH
H
2
230
TfO
OTf
Cu
N
O
O
Cu(OTf)2, MeCN, ∆
N
HF-Py, ТГФ
O
O
H
O
N
231
3
Этот подход был применен при циклизации
эфиров, была использована внутримолекулярно
секо-кислоты 232 в синтезе депсипептида гапола-
в общем синтезе 25-членного мощного иммуно-
зина 233, обладающего противоопухолевым дей-
супрессивного (-)-патеамина А из морских губок
ствием (схема 71) [73].
Mycale sp. [76]. Исходные условия для алкоголи-
Ранее описанный олово-содержащий реагент
за (NaHMDS в ТГФ) лактама 238 были слишком
187 являлся эффективным для переэтерификации
основными и поэтому заменялись на более мягкие
метилового эфира гидроксикислоты 234 в её се-
условия Паломо (CH2Cl2 с Et4NCN) в качестве
лективной циклодимеризации в соответствующий
растворимого источника цианида, с получением
дилактон 235 (схема 72) [74].
соответствующего макролактона 239 с выхода-
ми 59-68% через промежуточный ацилцианид
Пептид-связывающий реагент BID-Npy
236
(схема 74).
также использовался в макролактонизации раз-
личных ω-гидроксикислот 2 с хорошими выхода-
Используя липазу Pseudomonas при 40°С в не-
ми через образование активированного эфира 237
полярных растворителях, была выполнена пере-
(схема 73) [75].
этерификация HO(CH2)nCO2Me для n = 12, 13, 14
Cтратегия, основанная на расщеплении лак-
и 15 с выходами 38, 64, 78 и 80% соответственно
тамов спиртами до соответствующих β-амино-
[77].
Схема 71
H
CO2
O O
OH
1. 228, EDC/DMAP, CH2Cl2
2. Cu(OTf)2, MeCN, ∆
O
3. HF-Py
Me(CH2)6
Me(CH2)6
OTBSO
O N
N
54%
O
O
O OH Ph
Ph
232
233
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
644
ЯКОВЛЕВА и др.
Схема 72
OMe
O
OMe
O
187
HO
CO2Me
Cl, ∆
C6H5
O
75%
O
OMe
234
235
Схема 73
O2N
N
O
N
SO2
NO2
O
O
O
236
HO
OH
HO
n = 10-15
n
Et3N
O
O
N
n
68-88%
n
2
237
3
В статье [78] описан оригинальный способ по-
которые могут реагировать с электронодефицит-
лучения макролактона 240 с хорошим выходом
ными алкенами [79]. Внутримолекулярный вари-
с помощью фотолактонизации ацетата о-хинона
ант этой реакции позволил расширить макроцикл
на два углеродных атома. Для этого исходный лак-
241 через промежуточный диен-кетен 242 в син-
тон 244 сначала преобразуется в моноакриловый
тезе метаболита лишайника (+)-аспицилина 243
эфир дикарбоновой кислоты 245. Присутствие
(схема 75).
этих двух функций необходимо для заключитель-
Позднее было установлено, что при декарбок-
ной макролактонизации в лактон 246, который, в
силировании карбоновых кислот в фотолитиче-
свою очередь, способен к тем же превращениям
ских условиях образуются концевые радикалы,
(схема 76).
Схема 74
S
S
Br
Br
N
N
O
Et4NCN
O
O
CH2Cl2
O
BocHN
BocN
O
59-68%
O
OH
O
238
239
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
645
Схема 75
O
SO2Ph
O
OAc
hν 340 нм
PhO2S
(CH2)9
OH
N-Me-имидазол
CCl4
69%
(CH2)9
OH
241
242
OH
O
SO2Ph
OAc
OH
O
OH
O
O
243
240
2. МАКРОЛАКТОНИЗАЦИЯ С АКТИВАЦИЕЙ
и трифенилфосфина [80]. В механизме реакции
ГИДРОКСИЛЬНОЙ ГРУППЫ
ключевым промежуточным звеном является полу-
чаемая in situ алкоксифосфониевая соль 249, при
В 1976 году Мицунобу описал методику по-
лучения макролактонов 247, основанную на ак-
этом макролактонизация протекает через внутри-
тивации спиртовой части секо-кислот 248 с ис-
молекулярную SN2 реакцию с инверсией конфигу-
пользованием диэтилазодикарбоксилата (DEAD)
рации спирта (схема 77).
Схема 76
O
OMOM
1. KOH, EtOH, ∆
COOH
O
2. MOMCl, Py, DMF
84%
244
O
O
1. Акрилоил хлорид
NEt3, CH2Cl2
O
hν 300 нм, фенантрен
O
2. MgBr2(OEt2), Et2O
1,4-дицианобензол, NaOH
COOH
CH3CN, H2O
84%
245
246
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
646
ЯКОВЛЕВА и др.
Схема 77
H
RO2C N
N CO2R
O
OH
PPh3
PPh3
PPh3
O
O
COOH
+
RO2C-N=N-CO2R
OH
R1
CHOOH
n
n
1
R1
DEAD, R = Et
R
COO
n
DIAD, R = i-Pr
1
R
248
n
249
O=PPh3
247
H
H
RO2C
N
N CO
2R
DEAD-H2 или DIAD-H2
Изначально, как правило, диолиды были ос-
зовании классической методики Мицунобу для
новными продуктами для среднецепочечных лак-
циклизации секо-кислоты 251 в качестве главного
тонов [81], и реакция Мицунобу издавна рассма-
продукта был получен диолид 252 (40%), а жела-
тривалась как селективный метод их получения.
емый макролактон 253 с выходом лишь 2%. В то
Модификация была введена Стегличем в 1991 году
же время при медленном добавлении секо-кисло-
при синтезе аналогов комбрестатина D-2 (250),
ты 251 к смеси DEAD-PPh3 монолактон 253 стал
относящихся к классу природных фенолов и при-
сутствующих в коре южноафриканской кустарни-
основным (59%), а диолид 252 образовался в сле-
ковой ивы Combretum caffrum [82]. При исполь-
довых количествах (< 1%) (схема 78).
Схема 78
OMe
O
OMe
O
O
O
DEAD, PPh3
253
+
толуол
OH
OH
OMe
O
O
O
O
251
O
O
O
MeO
252
OH
O
O
O
250
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
647
Схема 79
CO2Et
EtO2C
NH
N
PPh3, DEAD
OBn
ТГФ
85%
CO2H
254
OBn
PPh3, DIAD
OBn
OH
толуол, -10°С
O O
95%
CO2H
255
256
Реакция Мицунобу в классических условиях
флэш-хроматографии. В синтезе девятичленного
(PPh3-DEAD в бензоле, толуоле, или THF при ком-
тиолактонового ядра соединения 257 - полупро-
натной температуре) имеет некоторые недостатки,
дукта в синтезе антибиотика гризевиридина -
например, такой как образование дигидразида
при циклизации секо-кислоты 258 использова-
254. Эванс столкнулся с этой проблемой в общем
лись PPh2Py и ди-трет-бутилдиазодикарбоксилат
синтезе антибиотика лономицина А и решил ее,
(DTBAD), так как побочно образующийся Ph2(Py)
применяя для циклизации гидроксикислоты 255 в
PO водорастворим и может быть удален кислотной
макролактон 256 более затрудненный диизопропи-
обработкой, а DTBAD-H2 спонтанно разлагается
лазодикарбоксилат (DIAD) в неполярном раство-
на изопрен и СО2 (схема 80) [84].
рителе (толуоле) (схема 79) [83].
Было также описано применение полимерных
Еще одним недостатком реакции Мицунобу
реагентов, которые обеспечивают псевдовысокое
является трудоемкое удаление DEAD-H2 или
растворение и легкое удаление побочных продук-
DIAD-H2 и побочного продукта Ph3PO при
тов при фильтрации. Сообщалось, что нанесенный
Схема 80
Me
O
O
O
Ph
N
H
S
PPh2Py, DTBAD
CO2Et
O CO
2H
OH
бензол
257
+
S
68%
Ph
N
N O
H
CO2Et
P
258
+
DTBAD-H2
+ CO2 + N2H4
DTABD = t-BuO2C-N=N-CO2t-Bu
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
648
ЯКОВЛЕВА и др.
Схема 81
OMe
OPMB
DEAD, resin-PPh3 43%
CO2H
DIAD, PPh3
25%
MOMO OH
resin-DEAD, PPh3 0%
OMe O
OMOM
O
OPMB
260
259
на полимер трифенилфосфин давал наибольший
циклодепсипептида R-901375 264 [88], решающее
выход макролактона 259 при циклизации гидрок-
значение сыграло добавление TsOH, поднявшее
сикислоты 260 в формальном синтезе цитоток-
выход целевого макролида 265 из гидроксикисло-
сических макролидов салицилигаламидов A и В
ты 266 до 62%, в то время как методология Боден-
198, выделенных из морской губки Haliclona sp.
Кека давала лишь 5%-ный выход (схема 83).
(схема 81) [85].
Использование реагента этерификации Эшен-
Аналогичная циклизация секо-кислоты
261,
моузера 267 для макроциклизации ω-гидрокси-
нанесенной на полимер, в макролид 262 была ис-
кислот позволяет получать лактоны с умеренным
пользована в синтезе лактона резорциловой кисло-
выходами (40-50%). Только этот синтетический
ты 263 (схема 82) [86].
подход к полному синтезу алкалоида декалина 268
из секо-кислоты 269 приводил к циклизации с низ-
Применение аллиловых эфиров в макролак-
ким (10%) выходом, тогда как реакции Келлога и
тонизации Мицунобу является затрудненным,
Мицунобу не давали результата (схема 84) [89].
поскольку преимущественно протекает элими-
нирование активированного алилльного спирта.
Макролактонизация
ω-бромкарбоксильных
Первоначально предполагалось, что это связано с
кислот Br-(СН2)n-CO2H в присутствии карбоната
сильно напряженной структурой секо-кислот, но
калия была впервые описана в 1947 году [90] и да-
позднее [87] была предложена в качестве альтер-
вала с хорошими выходами (56-96%) 9-17-член-
нативного объяснения возможность побочных SN1
ные лактоны. Эта методология затем была подроб-
реакций. Для устранения этих проблем в синтезе
но изучена [91] и установлено, что наибольшие
Схема 82
OEOMO
OEOMO
HO
O
OH
DIAD, PPh
3
EOMO
OBz
EOMO
OBz
O
O
S
O
S
O
261
262
OH
O
O
MeO
OBz
OH
OH
263
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
649
Схема 83
O
O
O
O
O
O
N
N
N
H
H
H
O NH
OH
O
NH
O
NH
PPh3, DIAD
O
O
TsOH·H2O
OH
O
O
O
NH
ТГФ
NH
NH
62%
TrtS
NH
TrtS
NH
S
NH
O
O
O
TrtS
TrtS
S
266
265
264
выходы достигаются при использовании карбона-
Активация спирта переводом в мезилат 272 и
та цезия в ДМФА или карбоната калия в ДМСО.
последующая макролактонизация в присутствии
карбоната цезия была продемонстрирована в
Четвертичные аммониевые соли
270a-с из
синтезе зеараленона 273, являющегося мощным
2-пирролидона также эффективны в получении
эстрогенным метаболитом, вырабатываемым не-
макролактонов, причем направление циклизации
бромкислоты 271 (до моно- 43 или ди- 48 лактона)
которыми видами анаморфных аскомицетовых
напрямую зависит от заместителя в аммонийной
грибов Fusarium, с полной инверсией ассиметри-
соли (схема 85, табл. 9) [92].
ческого центра по SN2-механизму (схема 86) [93].
Схема 84
OMe
MeO
O
N
O
OMe
Me
O
MeO
N
O
H
O
Me
268, 10%
266
O
+
толуол, 110°С
OMe
N
MeO
OH
H
CO2H
O
269
N
OCHO
NMe2
H
H
O
28%
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
650
ЯКОВЛЕВА и др.
Схема 85
O
N
O
NR4
O
270a-c
O
(CH2)10
Br-(CH2)10-COOH
(CH2)10
+
DMF
(CH2)10
O
O
O
271
43
48
Таблица 9. Зависимость выходов лактонов от заместителя в аммонийной соли 270
R
Выход 43, %
Выход 48, %
Et
25
64
Me(CH2)3
93
7
Me(CH2)7
0
66
Яркий пример макролактонизации с раскры-
циклизация ацетиленового спирта 277 давала же-
тием эпоксидного кольца было описан в полном
лаемый лактон 278 с выходом лишь 39%. Реакция
синтезе природного макролида с противоопу-
протекала через образование промежуточного ви-
холевой активностью (-)-дактилолида 274 [94].
нилиденового комплекса 279 и оксигенирование
Межмолекулярная методика Шарплесса, заключа-
его в кетен 280. Добавка каталитического коли-
ющаяся в Ti(Oi-Pr)4-опосредованном региоселек-
чества трифлата иттербия для усиления электро-
тивном открытии кольца эпоксида 275, давала со-
фильности кетена позволяет повысить выход до
ответствующий макролактон 276 с выходом 40%
70%. В дополнение к эксплуатационной простоте,
(схема 87).
эта макролактонизация протекает при относитель-
3. ОКИСЛИТЕЛЬНАЯ
но высокой концентрации, исключая необходи-
МАКРОЛАКТОНИЗАЦИЯ
мость в высоком разбавлении или медленном при-
бавлении (схема 88).
Окислительная лактонизация α,ω-диолов пред-
ставляется привлекательной и прямой стратегией
В последнее время основное внимание уделя-
получения макролактонов, однако объем ограни-
ется последним достижениям в разработке нетра-
чен небольшими (< 8) лактонами или нефункцио-
диционных методов каталитической макролакто-
нализованными крупными лактонами.
низации (без предварительной активации карбок-
сильной или спиртовой функций). Особое внима-
Недавно [95] была разработана каталитическая
макролактонизация путем окислительной цикли-
ние уделяется каталитической C-H макролакто-
зации терминальных ω-ацетиленовых спиртов при
низации, энантиоселективной Rh-катализируемой
синергетическом катализе переходными металла-
окислительно-восстановительной макроциклиза-
ми и кислотами Льюиса. В присутствии родиевого
ции алленовых кислот и каталитической карбони-
катализатора и N-оксида пиридина окислительная
лированной макролактонизации.
Схема 86
OMe
OMe
O
CO2H
Cs2CO3, DMF
OMs
O
O O
92%
MeO
MeO
O
272
273
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
651
Схема 87
O
HO
O
OH
OTBS
O
OTBS
HO
Ti(Oi-Pr)4
O
CH2Cl2
HO
40%
O
O
275
276
O
O
O
O
Me
O
274
Значительный прогресс был достигнут в обла-
действием комплекса Pd(OAc)2/PhS(O)-S(O)Ph
сти прямой функционализации аллильных C-H
проходила активация аллильного C-H фрагмента с
связей прекурсоров без спиртовой части. Вместо
образованием карбоксилата π-аллилпалладия 283,
использования секо-кислот в этой стратегии в ка-
последующая обработка которого бензохиноном
честве предшественников макролактона 281 вы-
(BQ) позволяла региоселективно формировать ал-
ступали линейные ω-алкеновые кислоты 282. Под
лильные макроэфиры 281. Эта методика позволя-
Схема 88
Rh(COD)2BF4
(4-F-C6H4)3P
O
O
N O
O
O
O
O
OH MeCN, 80oC
O
O
O
O
277
278
[M]
O
279
280
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
652
ЯКОВЛЕВА и др.
Схема 89
O
O
O
Pd(II)/сульфоксид
OH
кат.
O
O
BQ
[Pd]
H
282
283
281
ет получить 14-19-членные лактоны с хорошими
зованием линейных ω-алкиновых кислот 287 в
(52-60%) выходами без необходимости высокого
качестве предшественников. В реакции было ис-
разбавления (схема 89) [96, 97].
пользовано Rh-катализируемое окислительно-вос-
становительное окисление пропаргильной C-H
Данная стратегия была использована для син-
связи с целью изомеризации концевой алкиновой
теза 6-дезоксиэритронолида B 284 - агликона эри-
части субстрата 287 в аллен 288, гидрометалли-
тромициновых антибиотиков - на стадии внутри-
зация которого давала карбоксилат π-аллилродия
молекулярного окисления непредельной кислоты
289, который превращается в желаемый макро-
285, приводящей к макролактону 286 с выходом
лактон 281 через образование связи C-O внутри
56% с высокой регио- и стереоселективностью
сферы. В целом терминальный алкин действует
(схема 90) [98, 99].
как внутренний окислитель и восстанавливается в
В работах [98, 99] сообщалось о еще одной
терминальный олефин, а для протекания реакции
окислительной C-H макролактонизации с исполь-
не требуется внешний окислитель (схема 91).
Схема 90
PMP
H
O
Ph S
S Ph
PMP
O O
Pd(OAc)2
O
O
O
O O H
O
бензохинон, CH2Cl2, 45°C
HO
56%
O
O
O
O
285
286
O
OH
O
OH
O
OH
284
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
653
Схема 91
[Rh(COD)Cl]2
H H
ClCH2CH2Cl, 70oC
281
Rh H
Rh
PPh2
PPh2
OH
O
O
O
O
O
O
287
288
289
Применение этого метода может быть проил-
что показано при циклизации кислоты 293 в лак-
люстрировано [98, 99] циклизацией ацетиленовой
тон 295 с хорошими выходами и высокой энантио-
кислоты 290 в макролактон 291 в синтезе эпотило-
селективностью (схема 93, табл. 10).
на D 292 - соединения с мощной противораковой
Основным побочным продуктом реакции ока-
активностью. Диастереоселективность процес-
зался димерный макролидный продукт, что ис-
са образования нового асимметрического центра
пользовано в элегантном полном синтезе [98, 99]
контролировалась субстратом и составила
4:1
морского природного продукта клавозолида A 296.
(схема 92).
В этом случае ω-алленил-замещенная карбоновая
Для повышения диостереоселективности реак-
кислота 297, несущая желаемый углеводный фраг-
мент, подверглась димеризации в соединение 298
ции макролактонизации в качестве субстрата была
с выходом 72% и диастереоселективностью 92:8,
использована ω-алленовая кислота 293 с добавле-
демонстрируя эффективность хиральной добавки
нием в реакционную смесь хирального лиганда
294. (R)- или (S)-Конфигурацию вновь образую-
(схема 94).
щегося центра можно регулировать, используя
Еще одна привлекательная стратегия для фор-
различные диастереомеры хирального лиганда,
мирования макролидов предполагает использова-
Схема 92
[Rh(COD)Cl]2
бензойная кислота
ClCH2CH2Cl, 70°C
H
OTBS
OTBS
PPh2
PPh2
HO
O
O
O OTBS O
O OTBS O
43% (dr 4:1)
290
291
S
H
OH
N
O
O OH O
292
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
654
ЯКОВЛЕВА и др.
Схема 93
O
O
O
O
N
[Rh(COD)acac]
N
H
H
OH
ClCH2CH2Cl
O
294
*
293
295
Таблица 10. Зависимость выходов макролида 295 и диастереоселективности реакции от конфигурации хирального
лиганда 294
Конфигурация хирального лиганда 294
Выход макролида 295, %
Диастереоселективность реакции
99
92:8
56
0:100
ние прекурсоров, не содержащих карбоксилатные
макроцикла 303 в виде единственного цис-изомера
группы. В синтезе макролида 9-деметилнеопель-
с выходом 58% (схема 95).
толида 299 с мощным противоопухолевым дей-
Окислительное расщепление цис-конденсиро-
ствием [98, 99] окисление непредельного диола
ванных бициклических β-гидрокситетрагидрофу-
300, активированного Pd (II), в присутствии окиси
ранов 304a-d каталитическим количеством RuO4
углерода протекало с образованием алкилпалла-
с использованием RuCl3 в качестве его источника
дия 301, перегруппировывающегося в его аналог
и NaIO4 как стехиометрического соокислителя,
302. Образующаяся реакционноспособная частица
является мягким и селективным методом полу-
связывалась со спиртовой группой с образованием
чения 9-членных лактонов 305a-d. Селективное
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
655
Схема 94
HO
O
O
O
O
O
O
[Rh(COD)Cl]2
O
Cs2CO3
O
O
O
O
O
O
O
ClCH2CH2Cl, 0oC
O
O
O
O
O
O
O
O
O
O O
Ph2P
PPh
2
297
298
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O O
296
Схема 95
O
O
O
O
O
O
Pd(OAc)2
CO, CuCl2
CO
O
4A MS, ClCH2CH2Cl
Pd
OH
O
O
Pd
58%
OH
OH
OH
O
O
O
300
301
302
O
N
O
O
O
O
O
HN
O
O
O O
O
O O
O
O
303
299
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
656
ЯКОВЛЕВА и др.
Схема 96
O
R1
R1
OH
OH
R2
RuCl3, NaIO4
R1
H2O, CCl4, MeCN
O
O
O
R2
R2
OH
O
304a-d
306a-d
305a-d
Таблица 11. Зависимость выходов макролактонов от строения исходных молекул
Опыт
R1
R2
Выход макролидов 305, %
a
Н
Me
55
b
Me
Me
81
c
H
t-Bu
69
d
Me
t-Bu
82
Схема 97
MeO
O
H
O
O
H
H
O
H
O
PCC, CH2Cl2
OTBS
OTBS
H
O
65%
O
O
O
O
307
308
окисление соединений 304a-d дает возможность
ФОНДОВАЯ ПОДДЕРЖКА
получить лактолы 306a-d, которые далее без вы-
Работа выполнена при финансовой поддерж-
деления подвергаются расщеплению по третичной
ке программы РАН «Фундаментальные основы
C-C связи, ведущему к расширению кольца и по-
химии», тема № 8 «Хемо-, регио- и стереоселек-
лучению макролактонов 305a-d с хорошими выхо-
тивные превращения терпеноидов, стероидов и
дами (схема 96, табл. 11) [100].
липидов в направленном синтезе низкомолекуляр-
Проведено окислительное расщепление под
ных биорегуляторов» (№ госрегистрации ААА-
действием реагента Кори тетрациклического про-
А-А17-117011910023-2, 2017 г.)
изводного 307, в котором конденсированы цикло-
КОНФЛИКТ ИНТЕРЕСОВ
гексановое, тетрагидропирановое и лактольное
Авторы заявляют об отсутствии конфликта ин-
кольца, с получением 10-тичленного лактона 308
тересов.
с хорошим выходом (схема 97) [101].
СПИСОК ЛИТЕРАТУРЫ
4. ВЫВОДЫ
1. Kerschbaum M. Ber. Dtsch. Chem. Ges. (A and B Series).
Представлены данные по методам макролакто-
1927, 60, 902-909. doi 10.1002/cber.19270600411
низации секо-кислот в синтезе природных и био-
2. Хейфиц Л.А., Дашунин В.М. Душистые вещества и
логически активных соединений. Рассмотрены
другие продукты для парфюмерии. М.: Химия, 1994.
стратегии, основанные на активации карбоксиль-
[Xeifits L.A., Dashunin V.M. Dushistye veshhestva
ной или/и гидроксильной групп, а также на окис-
i drugie produkty dlya parfyumerii (Fragrances and
лительной лактонизации, проанализированы пре-
Other Products for Perfumery). M.: Khimiya, 1994.]
имущества и недостатки вышеописанных методов
3. Войткевич С.А. 865 душистых веществ для пар-
макролактонизации.
фюмерии и бытовой химии. М.: Пищевая промыш-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
657
ленность,
1994.
[Vojtkevich S.A. 865 dushistykh
22.
Inanaga J., Hirata K., Saeki H., Katsuki T., Yamagu-
veshchestv dlya parfyumerii i bytovoi khimii
(865
chi M. Bull. Chem. Soc. Jpn. 1979, 52, 1989-1993. doi
Fragrances for Perfumery and Household Chemicals).
10.1246/bcsj.52.1989
M.: Pishchevaya promyshlennost’, 1994.]
23.
Fürstner A., Kattnig E, Lepage O. J. Am. Chem. Soc.
4.
Шилл Г. Катенаны, ротаксаны и узлы. Ред. Р.Г. Ко-
2006, 128, 9194-9204. doi 10.1021/ja061918e
стяновского. М.: Мир, 1973. [Schill G. Catenanes,
24.
Salunke G.B., Shivakumar I., Gurjar M.K.
Rotaxanes and Knots. New York and London:
Tetrahedron Lett. 2009, 50, 2048-2049. doi 10.1016/
Academic Press, 1971.]
j.tetlet.2009.02.062
5.
Corey E.J., Nicolaou K.C. J. Am. Chem. Soc. 1974, 96,
25.
Hikota M., Tone H., Horita K., Yonemitsu O. J. Org.
5614-5616. doi 10.1021/ja00824a073
Chem. 1990, 55, 7-9. doi 10.1021/jo00288a004
6.
Sasaki T., Inoue M., Hirama M. Tetrahedron Lett. 2001,
26.
Dey S., Sudalai A. Tetrahedron Asymmetry. 2015, 26,
42, 5299-5303. doi 10.1016/S0040-4039(01)00959-5
344-349. doi 10.1016/j.tetasy.2015.03.001
7.
Hansen T.V., Stenstrom Y. Tetrahedron Asymmetry.
27.
Evans D.A., Ng H.P., Rieger D.L. J. Am. Chem. Soc.
2001,
12,
1407-1409. doi
10.1016/S0957-
1993, 115, 11446-11459. doi 10.1021/ja00077a049
4166(01)00250-6
28.
Sato T., Aoyagi S., Kibayashi C. Org. Lett. 2003, 5,
8.
Corey E.J., Clark D.A. Tetrahedron Lett. 1979, 20,
3839-3842. doi 10.1021/ol030088w
2875-2878. doi 10.1016/S0040-4039(01)86439-X
29.
Wehlan H., Dauber M., Mujica F.M.T., Schuppan J.,
9.
Corey E.J., Iguchi S., Albright J.O., De B. Tetrahedron
Mahrwald R., Ziemer B., Juarez G.M.E., Koert U.
Lett.
1983,
24,
37-40. doi
10.1016/S0040-
Angew. Chem. Int. Ed. 2004, 43, 4597-4601. doi
4039(00)81320-9
10.1021/ol802829n
10.
Corey E.J., Brunelle D.J. Tetrahedron Lett. 1976, 17,
30.
Shiina I., Kikuchi T., Sasaki A. Org. Lett. 2006, 8,
3409-3412. doi 10.1016/S0040-4039(00)93057-0
4955-4968. doi 10.1021/ol062011o
11.
Kochetkov N.K., Sviridov A.F., Ermolenko M.S.,
31.
Jeker N., and Tamm C. Helv. Chim. Acta. 1988, 71,
Yashunsky D.V., Borodkin V.S. Tetrahedron. 1989, 45,
1895-1903. doi 10.1002/hlca.19880710808
5109-5136. doi 10.1016/S0040-4020(01)81090-5
32.
Nagarajan M., Kumar V.S., Rao B.V. Tetrahedron. 1999,
12.
Kusaka S.-I., Dohi S., Doi T., Takahashi, T.
55, 12349-12360. doi 10.1016/S0040-4020(99)00718-
Tetrahedron Lett. 2003, 44, 8857-8859. doi 10.1016/
8
j.tetlet.2003.09.186
33.
Mukaiyama T., Izumi J., Miyashita M., Shiina I. Chem.
13.
Venkataraman K., Wagle D.R. Tetrahedron Lett. 1980,
Lett. 1993, 22, 907-910. doi 10.1246/cl.1993.907
21, 1893-1896. doi 10.1016/S0040-4039(00)92809-0
34.
Shiina I. Tetrahedron. 2004, 60, 1587-1599. doi
14.
Venkataraman K., Wagle D.R. Tetrahedron Lett. 1979,
10.1016/j.tet.2003.12.013
20, 3037-3040. doi 10.1016/S0040-4039(00)71006-9
35.
Mukaiyama T., Izumi J., Shiina I. Chem. Lett. 1997, 26,
15.
Mukaiyama T., Usui M., Saigom K. Chem. Lett. 1976,
187-188. doi 10.1246/cl.1997.187
5, 49-50. doi 10.1246/cl.1976.49
36.
Ishihara K., Kubota M., Kurihara H., Yamamoto H.
16.
Motozaki T., Sawamura K., Suzuki A., Yoshida K.,
J. Org. Chem. 1996, 61, 4560-4567. doi 10.1021/
Ueki T., Ohara A., Munakata R., Takao K., Tadano K.
jo952237x
Org. Lett. 2005, 7, 2265-2267. doi 10.1021/ol050763x
37.
Métay E., Léonel E., Condon S., Nédélec J.-Y.
17.
Narasaka K., Maruyama K., Mukaiyama T. Chem. Lett.
Tetrahedron.
2006,
62,
8515-8524. doi
10.1016/
1978, 7, 885-888. doi 10.1246/cl.1978.885
j.tet.2006.06.071
18.
Riala M., Chronakis N. J. Org. Chem. 2013, 78, 7701-
38.
Shiina I., Fukuda Y., Ishii T., Fujisawa H., Mukaiya-
7713. doi 10.1021/jo4013173
ma T. Chem. Lett. 1998, 27, 831-832. doi 10.1246/
19.
Kowalska E., Phopase J., Gathergood N. Aust. J. Chem.
cl.1998.831
2010, 63, 1348-1357. Doi 10.1071/CH10112
39.
Kaiho T., Masamune S., Toyoda T. J. Org. Chem. 1982,
20.
Gao M.Z., Reibenspies J.H., Wang B., Xu Z.L.,
47, 1612-1614. Doi 10.1021/jo00347a061
Zingaro R.A. J. Heterocycl. Chem. 2004, 41, 899-908.
40.
Roush W.R., Sciotti R.J. J.Am. Chem. Soc. 1998, 120,
doi 10.1002/jhet.5570410609
7411-7419. doi 10.1021/ja980611f
21.
Chen Y., Gao M., Tan S., Reibenspies J.H., Zinga-
41.
Wood J.L., Porco J.A., Taunton J., Lee A.Y., Clar-
ro R.A. Heterocycles. 2009, 78, 891-897. doi 10.3987/
dy J., Schreiber S.L. J. Am. Chem. Soc. 1992, 114,
COM-08-11583
5898-5900. doi 10.1021/ja00040a085
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
658
ЯКОВЛЕВА и др.
42.
Frank S.A., Roush W.R. J. Org. Chem. 2002, 67, 4316-
61.
Shen R., Lin C.T., Bowman E.J., Bowman B.J., Por-
4324. doi 10.1021/jo025580s
co J.A. Jr. J. Am. Chem. Soc. 2003, 125, 7889-7901.
doi 10.1021/ja0352350
43.
Coste J., Frerot E., Jouin P. J. Org. Chem. 1994, 59,
2437-2446. doi 10.1021/jo00088a027
62.
Corey E.J., Brunelle D.J., Nicolaou K.C. J. Am. Chem.
Soc. 1977, 99, 7359-7360. doi 10.1021/ja00464a047
44.
Boden E.P., Keck G.E. J. Org. Chem. 1985, 50, 2394-
2395. doi 10.1021/jo00213a044
63.
Vedejs E., Buchanan R.A., Watanabe Y.J. J. Am. Chem.
Soc. 1989, 111, 8430-8438. doi 10.1021/ja00204a016
45.
Keck G.E., Boden E.P., Wiley M.R. J. Org. Chem.
1989, 54, 896-906. doi 10.1021/jo00265a033
64.
Bao K., Zhang W., Zhang C., Qu Y., Tian L., Wu L.,
Zhao X., Cheng M. Molecules. 2007, 12, 2123-2129.
46.
Ишмуратов Г.Ю., Яковлева М.П., Мингалеева Г.Р.,
doi 10.3390/12092123
Толстиков А.Г. Макрогетероциклы. 2011, 4, 270-
65.
Gais H.J. Tetrahedron Lett. 1984, 25, 273-276. doi
310. [Ishmuratov G.Yu., Yakovleva M.P., Mingalee-
va G.R., Tolstikov A.G. Macroheterocycles. 2011, 4,
10.1016/S0040-4039(00)99860-5
270-310.] doi 10.6060/mhc2011.4.06
66.
Kita Y., Maeda H., Omori K., Okuno T., Tamura Y.
Synlett. 1993, 1, 273-274. doi 10.1055/s-1993-22428
47.
Schulz T., Eicher T. Synthesis. 2003, 8, 1253-1261. doi
10.1055/s-2003-39404
67.
Ohba Y., Takatsuji M., Nakahara K., Fujioka H.,
Kita Y. Chem. Eur. J. 2009, 15, 3526-3537. doi
48.
Li D., Carr G., Zhang Y., Williams D.E., Amlani A.,
10.1002/chem.200801548
Bottriell H., Mui A.L.-F., Andersen R.J. J. Nat. Prod.
2011, 74, 1093-1099. doi 10.1021/np200033y
68.
Petri A.F., Bayer A., Maier M.E. Angew. Chem. Int. Ed.
2004, 43, 5821-5823. doi 10.1002/anie.200460760
49.
Ashworth P., Broadbelt B., Jankowski P., Kociens-
ki P., Pimm A., Bell R. Synthesis. 1995, 199-206. doi
69.
Yang M., Wang X., Zhao J. ACS Catal. 2020, 10, 5230-
5235. doi 10.1021/acscatal.0c00523
10.1055/s-1995-3870
70.
Narasaka K., Sakakura T., Uchimaru T., Guědin-
50.
Hanessian S., Ma J., Wang W. J. Am. Chem. Soc. 2001,
Vuong D. J. Am. Chem. Soc. 1984, 106, 2954-2961.
123, 10200-10206. doi 10.1021/ja025105b
doi 10.1021/ja00322a036
51.
Lee E., Jeong E.J., Kang E.J., Sung L.T., Hong S.K.
71.
Burke S.D., McDermott T.S., O’Donnell C.J. J. Org.
J. Am. Chem. Soc. 2001, 123, 10131-10132. doi
Chem. 1998, 63, 2715-2718. doi 10.1021/jo970942v
10.1021/ja016272z
72.
Su Q., Panek J.S. J. Am. Chem. Soc. 2004, 126, 2425-
52.
Fujisawa T., Mori T., Fukumoto K., Sato T. Chem. Lett.
2430. doi 10.1021/ja037957x
1982, 11, 1891-1894. doi 10.1246/cl.1982.1891
73.
Palomo C., Oiarbide M., Garcia J.M., González A.,
53.
Ishmuratov G.Yu., Yakovleva M.P., Vydrina V.A.,
Pazos R., Odriozola J.M., Baňuelos P., Tello M., Lin-
Shutova М.А., Ishmuratova N.M., Tolstikov A.G.
den A. J. Org. Chem. 2004, 69, 4126-4134. doi
Macroheterocycles. 2017, 10, 345-379. doi 10.6060/
10.1021/jo0497499
mhc161066i
74.
Su Q., Beeler A.B., Lobkovsky E., Porco J.A.,
54.
Fürstner A., Ruiz-Caro J., Prinz H., Waldmann H.
Panek J.S. Org. Lett. 2003, 5, 2149-2152. doi 10.1021/
J. Org. Chem. 2004, 69, 459-467. doi 10.1021/
ol034608z
jo035079f
75.
Ahmed A., Taniguchi N., Fukuda H., Kinoshita H.,
55.
Steliou K., Poupart M.-A. J. Am. Chem. Soc. 1983, 105,
Inomata K., Kotake H. Bull. Chem. Soc. Jpn. 1984, 57,
7130-7138. doi 10.1021/ja00362a018
781-786. doi 10.1246/bcsj.57.781
56.
Otera J., Yano T., Himeno Y., Nozaki H. Tetrahedron
76.
Rzasa R.M., Shea H.A., Romo D. J. Am. Chem. Soc.
Lett.
1986,
27,
4501-4504. doi
10.1016/S0040-
1998, 120, 591-592. doi 10.1021/ja973549f
4039(00)84989-8
77.
Yamada H., Ohsawa S., Sugai T., Ohta H., Yoshika-
57.
Reber K.P., Tilley S.D., Sorensen E. Chem. Soc. Rev.
wa S. Chem. Lett. 1989, 18, 1775-1776. doi 10.1246/
2009, 38, 3022-3034. doi 10.1039/B912599J
cl.1989.1775
58.
Hoye T.R., Danielson M.E., May A.E., Zhao H.J. J. Org.
78.
Quinkert G., Küber F., Knauf W., Wacker M., Koch U.,
Chem. 2010, 75, 7052-7060. doi 10.1021/jo101598y
Becker H., Nestler H.P., Dürner G., Zimmermann G.,
59.
Holloway G.A., Hьgel H.M., Rizzacasa M.A. J. Org.
Bats J.W., Egert E. Helv. Chim. Acta. 1991, 74, 1853-
Chem. 2003, 68, 2200-2204. doi 10.1021/jo026798h
1923. doi 10.1002/hlca.19910740828
60.
Marshall J.A., Eidam P.M. Org. Lett. 2007, 10, 93-96.
79.
Yoshimi Y., Masuda M., Mizunashi T., Nishikawa K.,
doi 10.1021/ol702024b
Maeda K., Koshida N., Itou T., Morita T., Hatana-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
МЕТОДЫ МАКРОЛАКТОНИЗАЦИИ СЕКО-КИСЛОТ
659
ka M. Org. Lett. 2009, 11, 4652-4655. doi 10.1021/
91.
Talma A.G., Jouin P., De Vries J.G., Troostwijk C.B.,
ol9019277
Buning G.H.W., Waninge J.K., Visscher J., Kel-
80.
Swamy K.C.K., Kumar N.N.B., Balaraman E.,
logg R.M. J. Am. Chem. Soc. 1985, 107, 3981-3997.
Kumar K.V.P.P. Chem. Rev. 2009, 109, 2551-2651.
doi 10.1021/ja00299a038
doi 10.1021/cr800278z
92.
Shono T., Ishige O., Uyama H., Kashimura S. J. Org.
81.
Ohta K., Mitsunobu O. Tetrahedron Lett. 1991, 32,
Chem. 1986, 51, 546-549. doi 10.1021/jo00354a030
517-520. doi 10.1016/S0040-4039(00)79484-6
93.
Kruizinga W.H., Kellogg R.M. J. Am. Chem. Soc.
82.
Justus K., Steglich W. Tetrahedron Lett. 1991, 32,
1981, 103, 5183-5189. doi 10.1021/ja00407a039
5781-5784. doi 10.1016/S0040-4039(00)93554-8
94.
Hoye T.R., Hu M.J. J. Am. Chem. Soc. 2003, 125,
83.
Evans D.A., Ratz A.M., Huff B.E., Sheppard G.S.
9576-9577. doi 10.1021/ja035579q
J. Am. Chem. Soc. 1995, 117, 3448-3467. doi 10.1021/
95.
Zhang W.-W., Gao T.-T., Xu L.-J., Li B.-J.
ja00117a014
Org. Lett.
2018,
20,
6534-6538. doi
10.1021/
84.
Marcantoni E., Massaccesi M., Petrini M., Bartoli G.,
acs.orglett.8b02858
Bellucci M.C., Bosco M., Sambri L. J. Org. Chem.
96.
Fraunhoffer K.J., Prabagaran N., Sirois L.E.,
2000, 65, 4553-4559. doi 10.1021/jo000116d
White M.C. J. Am. Chem. Soc. 2006, 128, 9032-
85.
Herb C., Bayer A., Maier M.E. Chem. Eur. J. 2004, 10,
9033. doi 10.1021/ja063096r
5649-5660. doi 10.1002/chem.200400617
97.
Li Y., Yin X., Dai M. Nat. Prod. Rep. 2017, 34, 1175-
86.
Dakas P.-Y., Jogireddy R., Valot G., Barluenga S.,
1246. doi 10.1039/c7np00038c
Winssinger N. Chem. Eur. J. 2009, 15, 11490-11497.
doi 10.1002/chem.200901373
98.
Stang E.M., White M.C. Nat. Chem. 2009, 1, 547-
551. doi 10.1038/nchem.351
87.
Rychnovsky S.D., Hwang K. J. Org. Chem. 1994, 59,
5414-5418. doi 10.1021/jo00097a052
99.
Sengupta S., Mehta G. Org. Biomol. Chem. 2020,
1-42. doi 10.1039/C9OB02765C
88.
Chen Y., Gambs C., Abe Y., Wentworth P.Jr., Jan-
da K.D. J. Org. Chem. 2003, 68, 8902-8905. doi
100.
Ferraz H.M.C., Longo L.S. Jr. J. Org. Chem. 2007,
10.1021/jo034765b
72, 2945-2950. doi 10.1021/jo0626109
89.
Shishido K., Tanaka K., Fukumoto K., Kametani T.
101.
Файзуллина Л.Х., Тагиров А.Р., Салихов Ш.М.,
Chem. Pharm. Bull. 1985, 33, 532-539. doi 10.1248/
Валеев Ф.А. ЖОрХ. 2019, 55, 1834-1842. [Faizulli-
cpb.33.532
na L.K., Tagirov A.R., Salikhov S.M., Valeev F.A.
90.
Hunsdiecker H., Erlbach H. Chem. Ber. 1947, 80, 129-
Russ. J. Org. Chem. 2019, 55, 1832-1839.] doi
137. doi 10.1002/cber.19470800207
10.1134/S1070428019120042
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021
660
ЯКОВЛЕВА и др.
Methods for Macrolactonization of Seco Acids in the Synthesis
of Natural and Biologically Active Compounds
M. P. Yakovleva*, K. S. Denisova, V. A. Vydrina, A. G. Tolstikov, and G. Yu. Ishmuratov
Ufa Institute of Chemistry, Ufa Federal Research Center of the Russian Academy of Sciences,
prosp. Oktyabrya, 71, Ufa, 450054 Russia
*e-mail: insect@anrb.ru
Received January 26, 2021; revised February 6, 2021; accepted February 10, 2021
The review describes the most common macrolactonization reactions, including the activation of one and (or)
another terminal functional group of seco-acid in the synthesis of natural compounds and substances with bio-
logical activity, and also analyzes the advantages and disadvantages of the above-described macrolactonization
methods.
Keywords: seco-acid, macrolactonization, macrolactones, natural compounds, biological activity
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 57 № 5 2021