ПАРАЗИТОЛОГИЯ, 2020, том 54, № 6, с. 491-503.
УДК 595.122.2:575.174
GENETIC DIVERSITY IN MONOXENOUS
AND TRIXENOUS DIGENEANS SHARING
ONE MOLLUSCAN HOST SPECIES
© 2020 A. Gonchara, b, *
a Department of Invertebrate Zoology, St Petersburg University,
Universitetskaya emb., 7/9, Saint Petersburg, 199034 Russia
b Zoological Institute Russian Academy of Sciences,
Universitetskaya emb., 1, Saint Petersburg, 199034 Russia
* e-mail: anya.gonchar@gmail.com, a.gonchar@spbu.ru
Received 04.10.2020
Received in revised form 22.10.2020
Accepted 22.10.2020
Mudsnails Peringia ulvae serve as hosts for many digenean species; some of them differ a lot in
their life cycle pattern. In the north of Europe two contrasting examples are Cryptocotyle concava
(Heterophyidae) with a trixenous life cycle and Bunocotyle progenetica (Hemiuridae) with a mon-
oxenous life cycle. A ~ 870 base pairs long fragment of cox1 gene sequence was used to evaluate
and compare genetic structure within these two species. Our findings suggest that high dispersal of
C. concava keeps differentiation between different locations minimal and haplotype diversity quite
high. In B. progenetica two haplotypes dominate and have at least limited dispersal.
Keywords: Digenea, life cycle, intraspecific diversity, Bunocotyle progenetica, Cryptocotyle con-
cava
DOI: 10.31857/S1234567806060036
Monoxenous life cycles are not common in Digenea, but they occur and represent an
extreme limitation of transmission (Poulin, Cribb, 2002). Such a limitation may offer some
benefits, but also has significant drawbacks. One of the key concerns is the impact of
one-host life cycle on the genetic diversity in a parasite. This impact may seem straightfor-
ward, but it has never been experimentally tested.
When a single host remains in the digenean life cycle, it is always a first intermediate
host. Snails, the typical first intermediate hosts, usually harbour few clones of a parasite
491
following few infection events (Theron et al., 2004; Rauch et al., 2005; Keeney et al., 2007,
2008). Reproduction of the parthenitae - the sporocysts and rediae - does not produce ge-
netic exchange and is in effect asexual. When the life cycle runs within one snail up until
production of eggs or miracidia, these cannot be a result of mating between non-related
parasites. So, in species with a monoxenous life cycle we expect lack of advantage from
sexual reproduction and low genetic diversity.
Apart from sexual reproduction, intraspecific genetic diversity is also supported by spatial
distribution and associated mixing opportunities. The most vagile host in the life cycle -
for digeneans this generally means a vertebrate definitive host - largely determines genetic
structure of a parasite. Birds and mammals provide better dispersal and less structure than
fishes (Blasco-Costa, Poulin, 2013; Feis et al., 2015). Birds can even sustain connection
between parasite populations that are geographically isolated (Gonchar, Galaktionov, 2020).
In a monoxenous life cycle, the most vagile and the only host is a snail.
In the White Sea, the snails Peringia ulvae (along with their close relatives, Ecrobia
ventrosa) serve as hosts for Bunocotyle progenetica (Markowski, 1936) Chabaud & But-
tner, 1959 - the only monoxenous digenean in this region. These snails are also first in-
termediate hosts of other digeneans with more transmission events in their life cycles, for
example, several species from families Microphallidae and Notocotylidae, and a heterophyid
Cryptocotyle concava (Creplin, 1825) Lühe, 1899. The latter is an abundant parasite with
a trixenous life cycle that involves fish second intermediate host and bird definitive host
(a range of species with fish component in their diet, mainly gulls). It thus presents a con-
trasting example to B. progenetica regarding the expected genetic structure. Several studies
have compared these two parasites of P. ulvae (Levakin, 2004, 2005; Levakin et al., 2013),
but never in this particular aspect.
Here I for the first time assessed genetic structure of a monoxenous trematode
(B. progenetica) and that of a trixenous one (C. concava) found in the same region and in
the same molluscan host species.
MATERIALS AND METHODS
The mud snails Peringia ulvae were collected in 2018-2019 in two regions: the Chupa Inlet in
the Kandalaksha Bay of the White Sea (Russia) and the Varangerfjord in the south-western Barents
Sea (Norway). The distance between these regions is about 500 km directly and about 1000 km along
the shore line. In Varangerfjord, all the samples were taken from the same location, the large mudflat
in the head of the fjord. In the Chupa Inlet, sampling took place in four locations within 10 km from
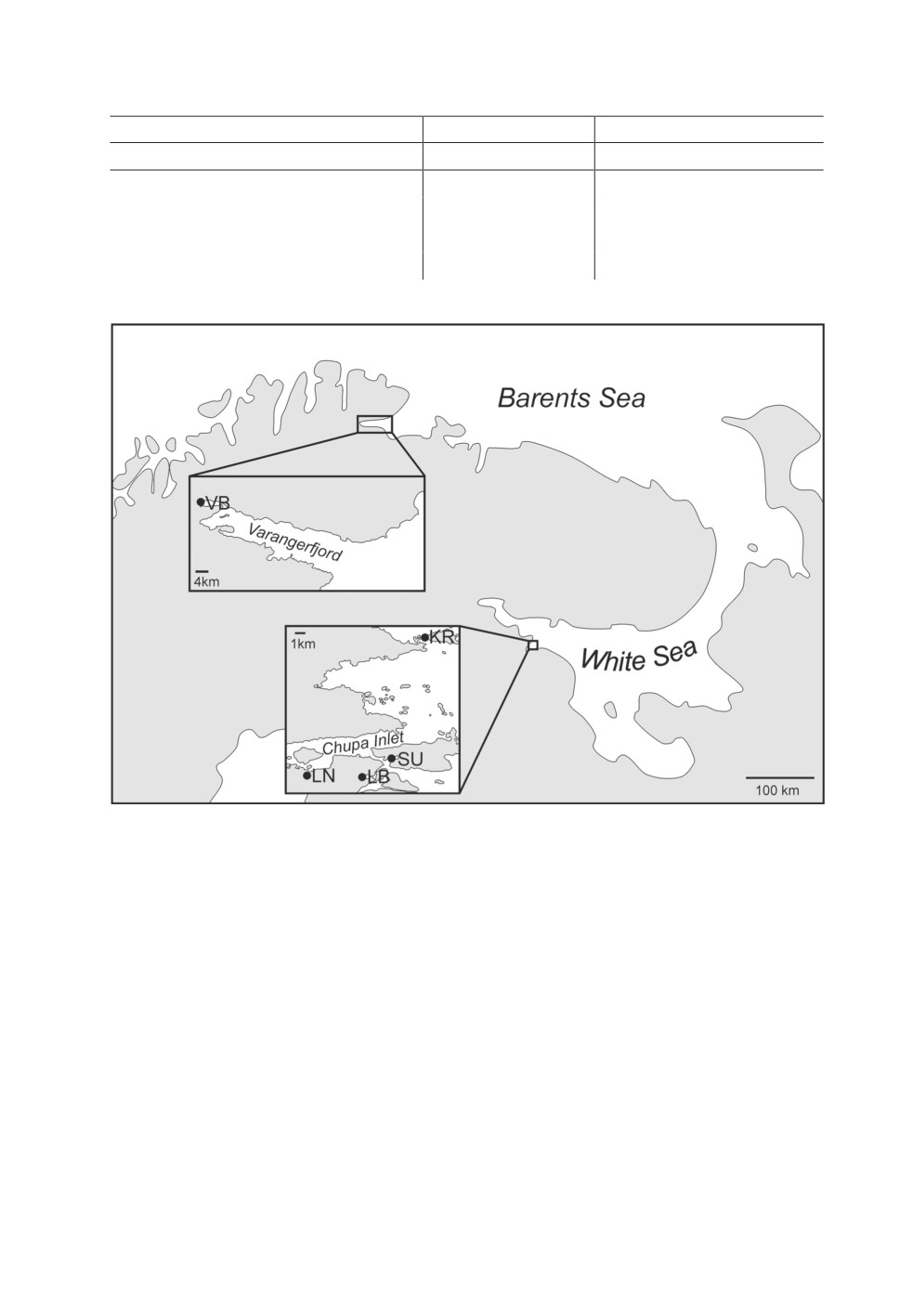
each other. Sampling sites are summarized in fig. 1 and table 1.
Mud snails were collected during low tide using a sieve with a 1 mm mesh size, taken to the
laboratory and dissected under a stereomicroscope to detect infection by digeneans. I selected rediae
of Cryptocotyle concava and Bunocotyle progenetica from an infected snail, rinsed them in sea water
and preserved in 96 % ethanol. Prevalence and intensity were not recorded.
492
Table 1. Collection sites
Region
Site
Coordinates
Varangerfjord, south-western Barents Sea
Varangerbotn
70°10′19.31″N, 28°33′52.42″E
Chupa Inlet, Kandalaksha Bay, White Sea
Sukhaya Salma Bay
66°18′42.12″N, 33°39′18.58″E
Lebyazhya Bay
66°17′39.95″N, 33°35′04.88″E
Levin Navolok Bay
66°17′50.57″N, 33°27′37.51″E
Krasnyi Island
66°25′06.74″N, 33°44′09.03″E
Figure 1. Schematic map showing collection sites in the Barents and White Seas.
Sampling locations are marked with circles: VB - Varangerbotn, LN - Levin Navolok Bay,
LB - Lebyazhya Bay, SU - Sukhaya Salma Bay, KR - Krasnyi Island.
To extract DNA, the ion exchange resin Chelex ® 100, 200-400 mesh, molecular biology grade (Bio-
Rad) was used. A single redia in a small drop of ethanol was transferred to a new 1.5 ml microtube;
any remaining ethanol was evaporated by incubating an opened tube at 35 °C for 1-3 min. Then 200 µl
of 5 % Chelex in Milli-Q water and 2 µl of proteinase K (20 mg/ml) were added to each tube and
samples were incubated at 56 °C for 16-18 hours while mixing at 750 rpm on thermomixer (Eppen-
dorf). Next, the samples were boiled at 90 °C for 8 min and centrifuged at 16,000 g for 10 min while
cooling to 4 °C. DNA in supernatant was carefully transferred to a new tube and stored at -20 °C.
The fragment of cytochrome c oxidase subunit I (cox1) gene sequence was amplified with PCR
in 20 µl reaction mixtures containing 13 µl Milli-Q water, 4 µl ScreenMix HS (Evrogen, Russia),
493
0.5 µl of each forward and reverse primer diluted to 10 pmol/µl and 2 µl of DNA template. I used
JB3 forward primer (TTTTTTGGGCATCCTGAGGTTTAT, Bowles et al., 1993) and trem.cox1.rrnl
(AATCATGATGCAAAAGGTA, Králová-Hromadová et al., 2008) or COIRtrema (CAACAAAT-
CATGATGCAAAAGG, Miura et al., 2005) reverse primer. PCRs were run on a Veriti thermal cycler
(Thermo Fisher Scientific) with the following thermal profile: initial denaturation at 94 °C for 3 min;
35 cycles with 40 s at 94 °C, 40 s at 48 °C (trem.cox1.rrnl) or 51 °C (COIRtrema) and 40 s at
72 °C; final elongation at 72 °C for 7 min; and cooling to 4 °C. PCR products were size-separated with
electrophoresis in a 1 % agarose gel (5 min at 60 V and 40 min at 80 V), stained with SybrGREEN
(Invitrogen); results were visualized and photographed using ChemiDoc MP (Bio-Rad). Sequencing
of fragments was performed directly from PCR mixture and with PCR primers in both directions on
the automated ABI 3500xl genetic analyzer (Applied Biosystems).
verse reads were assembled to verify resulting sequence, and the quality was checked by eye. Further
quality control involved ensuring no inappropriate stop codons in a translated sequence (translation
table 21), and testing for the functional effect of the amino acid substitutions using Provean with
a default threshold - 2.5 (Choi et al., 2012). I also used the translated sequence to search for similar
proteins using BLASTP 2.10.1 + (Altschul et al., 1997). The boundaries of cox1 gene were estimated
by aligning to the annotated mitochondrial genome of Metagonimus yokogawai (NC_023249) as
a reference. The alignment of all the obtained sequences was exported in *.nex format for further
analysis. To construct a haplotype network, I used Integer NJ Net method with reticulation tolerance
0.5 in PopART 1.7 (Leigh, Bryant, 2015). To estimate sequence divergence I used MEGA7 (Ku-
mar et al., 2016). Using DnaSP6 (Rozas et al., 2017) I calculated FST and Tajima’s D; built a mis-
match distribution graph using population growth-decline model and tested the fit to this model with
a Harpending’s raggedness index r. To test the significance of differences in haplotype diversity between
the Chupa Inlet and the Varangerfjord for C. concava I used the R script “genetic_diversity_diffs
v.1.0.6” (R Core Team, 2015; Alexander et al., 2016).
For comparison, I used partial cox1 sequences of European Cryptocotyle lingua isolates (Blake-
slee et al., 2008; EU876333-EU876430). These were aligned and analyzed in DnaSP6 and MEGA7,
as described above.
RESULTS
A total of 102 specimens were collected: 54 Cryptocotyle concava and 48 Bunocotyle
progenetica. In Varangerfjord only C. concava were found. In Chupa Inlet, Sukhaya Salma
and Lebyazhya Bays were major sampling sites, while from Levin Navolok Bay there were
only five isolates and from Krasnyi Island only one.
I obtained high quality sequences covering 3′-region of the cox1 gene and adjacent
tRNA-coding region for 40 C. concava and 32 B. progenetica isolates. High AT content was
observed in both species: 63.5 % in C. concava and 67.5 % in B. progenetica. Stop codon
was TAA in both species. All the sequences were submitted to GenBank under accession
numbers MT422274-MT422345.
494
Sequences of C. concava were 872-881 base pairs (b. p.) long after trimming, and the
resulting alignment contained 869 positions of which 27 were polymorphic (table 2). Average
genetic divergence within the species was 0.31 ± 0.09 %. In the translated sequence, the
majority of substitutions appeared synonymous, with two exceptions: Val Ile (position 56
of the amino acid alignment) and Ser Pro (236). None of these changes were predicted to
have functional effect. Position 264 was a stop codon at the end of the cox1 gene sequence.
The closest BLAST hit was Metagonimus suifunensis (QFS15968) with 87.6 % identity.
A total of 23 C. concava haplotypes were discovered; five of them were shared between
Varangerfjord and Chupa Inlet. The average number of nucleotide differences between these
populations was 2.885 and Fst was 0.12. The differences in haplotype diversity between the
populations is not significant (p = 0.195). The cox1-based haplotype network for C. concava
is in fig. 2a; it has reticulated structure and a repeated starburst pattern. Most haplotypes
differed by one or two substitutions; the maximum difference between the two neighbouring
haplotypes was four substitutions. Haplotype A was dominant in Varangerfjord; haplotype
B occurred in all five sampling sites; 17 haplotypes were unique. Mismatch distribution
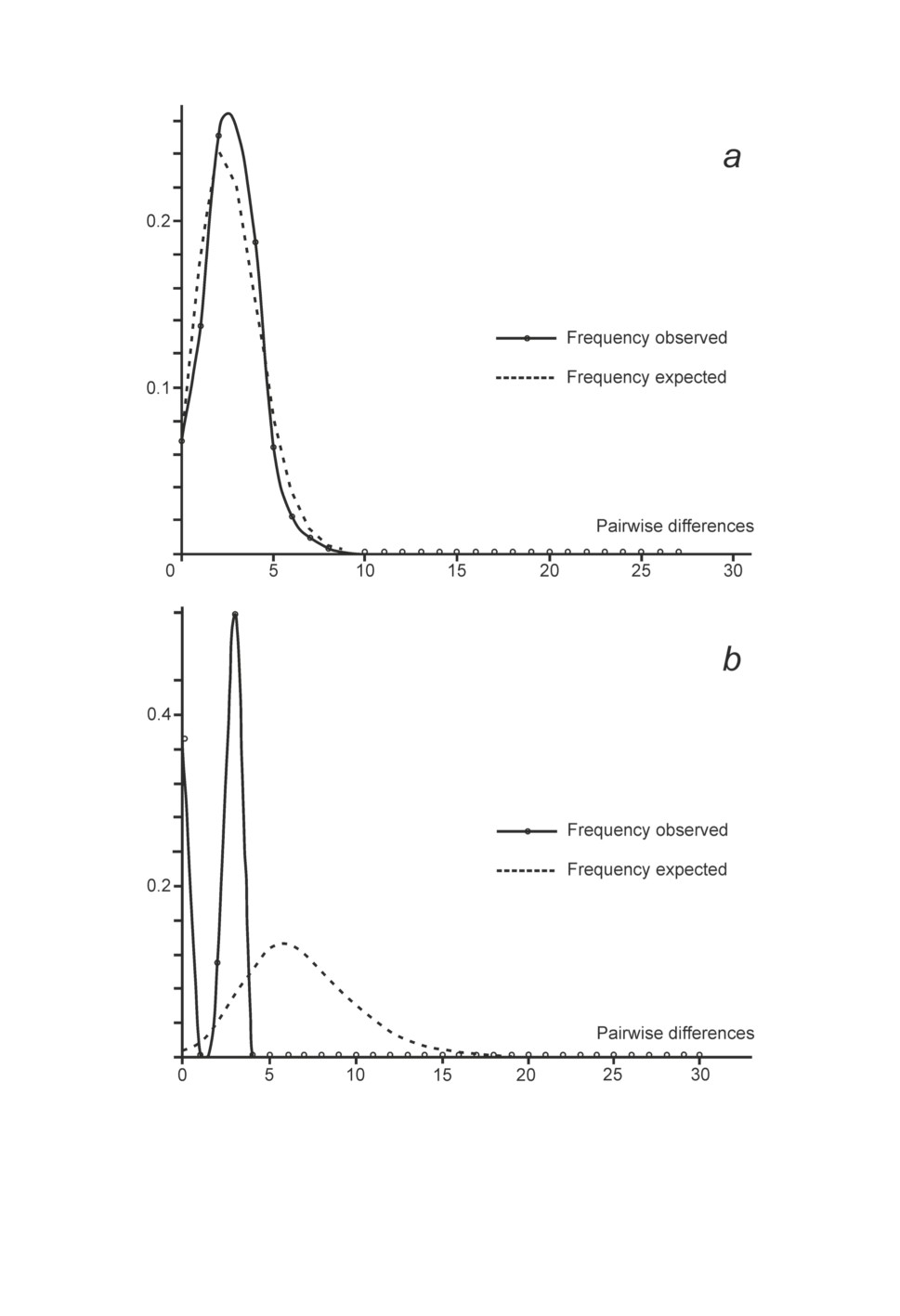
was unimodal (fig. 3a).
For B. progenetica sequences were 853-860 b.p., and the alignment was 853 b.p. Aver-
age pairwise distance per site between all sequences was 0.21 ± 0.1 %. Four positions were
polymorphic (table 2). In the translated sequence, three of them appeared non-synonymous:
Ser Asn (position three of the amino acid alignment), Ile Val (4), and Thr Met (239).
None of these changes were predicted to have functional effect. Position 260 was a stop
codon at the end of the cox1 gene sequence. The closest BLAST hit was Isoparorchis
eurytremum (BAO74170) with 74 % identity.
The cox1-based haplotype network for B. progenetica is in fig. 2b. A total of three hap-
lotypes were discovered, the neighbouring haplotypes differing by two or three substitutions.
Haplotypes C (frequency 16) and D (11) were dominant, and were found in both major
sampling sites (Sukhaya Salma and Lebyazhya Bays). Haplotype E (frequency 5) occurred
in all the three sampling sites. Mismatch distribution was bimodal (fig. 3b).
I analyzed 98 cox1 sequences of C. lingua from GenBank. The samples originated from
16 sites in Europe: seven in southern Scandinavia, six in the UK and three along the conti-
nental coast of central Europe. The alignment was 1043 b.p. long, overlapping by 372 b. p.
with the 5′-region of the cox1 alignment for C. concava. There were 19 non-synonymous
substitutions across the whole alignment. The fragment overlapping for C. lingua and
C. concava included 123 amino acids and had 10 fixed differences between the species.
The total number of cox1 haplotypes for C. lingua was 72, with haplotype and nucleotide
diversity values Hd = 0.984 and π = 0.00379.
495
Table 2. Genetic diversity indices based on cox1 sequences
Dataset
b. p.
N
S
H
Hd
π
K
Tajima’s D
B. progenetica
all
853
32
4
3
0.627
0.00208
1.97
n.s.
C. concava
all
869
40
27
23
0.932
0.00312
2.715
-1.95**
VB
869
20
14
12
0.879
0.00249
2.163
-1.65*
CH
869
20
18
15
0.942
0.00335
2.911
-1.6*
VB - Varangerfjord, CH - Chupa Inlet, b.p. - length of alignment, N - number of samples,
S - Number of polymorphic sites, H - number of haplotypes, Hd - haplotype diversity, π- nucleotide diversity,
K - average number of nucleotide differences, * p < 0.05, ** p < 0.005, n.s. not significant.
Figure 2. Haplotype networks based on cox1 sequence data for (a) Cryptocotyle concava, n=40;
and (b) Bunocotyle progenetica, n=32. Circles indicate haplotypes, their frequencies correspond
to circle sizes. Haplotypes connected by line differ by one (no hatch marks) or more (number
of hatch marks) substitutions. Black dot illustrates a missing haplotype. Capital letters A-E mark
haplotypes mentioned in the text. Sampling locations are represented by different fill patterns.
Levin - Levin Navolok Bay, Krasnyi - Krasnyi Island, Lebyazhya - Lebyazhya Bay,
Sukhaya - Sukhaya Salma Bay.
496
Figure 3. Mismatch distributions based on cox1 haplotypes for (a) Cryptocotyle concava
and (b) Bunocotyle progenetica. Solid lines show observed frequencies,
dashed lines show expected frequencies.
497
DISCUSSION
This study dealt with two species of trematodes that share the same first intermediate
host, Peringia ulvae, but have a contrasting life cycle strategy. I obtained 72 sequences of
a ~ 870 b.p. long fragment that included a 3′-region of cox1 gene. For Cryptocotyle con-
cava this was the first molecular genetic evidence. Data on Bunocotyle progenetica were,
to the best of our knowledge, the first intraspecific genetic diversity data for a monoxenous
digenean. The study had relatively small sample sizes, relied on one DNA marker and pro-
vided non-exhaustive geographic range of sampling. The latter was especially critical for
B. progenetica which I did not find in northern Norway. Despite these limitations, the results
contribute to building a global picture of genetic diversity in digeneans.
For C. concava, the samples from two distant locations (Varangerfjord and Chupa Inlet)
have shown lack of genetic differentiation. The haplotype network illustrates this well: five
haplotypes are shared between the two locations, and haplotype pattern does not match
a location pattern (fig. 2a). To test these observations I calculated Fst measure. It infers ge-
netic differentiation from comparing genetic diversity within and among (sub)populations
(Holsinger, Weir, 2009). In C. concava, Fst = 0.12 indicates gene flow between Varanger-
fjord and Chupa. This gene flow is consistent with the vagility of definitive hosts: birds can
maintain long-distance dispersal of the parasite. This type of life cycle is called allogenic;
the opposite is autogenic when the definitive hosts are fish, and colonization potential is
smaller (Esch et al., 1988). Meta-analysis has also found that genetic structuring is lower
in allogenic trematodes (Blasco-Costa, Poulin, 2013). The study of the two trixenous trema-
tode species - autogenic and allogenic - in a marine environment gave the same result
(Feis et al., 2015). Our results are thus consistent with the established ideas.
The results of this study were compared with those on three other representatives of
Heterophyidae. All of them have a three-host life cycle where a definitive host is a fish-
eating bird or mammal (including humans). First, I re-analyzed the published dataset for
a closely related marine species C. lingua in Europe (Blakeslee et al., 2008), and proved no
differentiation across sampling locations (Fst = 0). Then, the freshwater species Metagonimus
suifunensis in the Russian Far East also follows this trend (Fst = 0.05, excluding the only
sample from the northernmost location which had a highly diverged haplotype) (Tatonova
et al., 2019). However, Haplorchis taichui forms several isolated populations in Vietnam
(Dung et al., 2013) and, at the broader scale, in the mainland Southeast Asia (Thaenkham
et al., 2017). These examples highlight that, although some trends in population genetics
of trematodes have been identified, the case of every species adds new information.
Demographic history of C. concava inferred from sequence data suggests recent popu-
lation expansion. The first evidence is the haplotype network that has star-like features,
showing many unique haplotypes closely related to few central ones (structure similar to
498
type ii “complex star” in Jenkins et al., 2018). Population expansion can also be detected
by neutrality tests, for example Tajima’s D that compares average pairwise differences with
the number of segregating sites. For populations that underwent bottleneck recently it takes
negative value, which is the case for C. concava. Further evidence is the unimodal mismatch
distribution (fig. 3a). Probably the traces of bottleneck event that are observed in C. concava
and C. lingua (Tajima’s D = -2.59) date from the last glacial maximum.
In the Chupa Inlet, C. concava and Bunocotyle progenetica were collected in the same
sites, and even though sample sizes were not equal, the dataset is suitable for comparison.
The number of detected haplotypes (H), haplotype diversity (Hd) and nucleotide diversity
(π) were higher in C. concava (table 2). The difference is also evident from the haplotype
network (fig. 2). Low intraspecific variability in B. progenetica is likely due to its life cycle
that runs within one mollusc individual. This favours local expansion of single haplotypes
but limits dispersal and genetic exchange.
Dispersal of B. progenetica relies on dispersal of its only host, the mollusc. P. ulvae
may spread at the larval stage, but veligers cannot carry a parasite. Juveniles and adults
of mud snails can float at the water surface and thus move much faster than by crawling
(reviewed in Anderson, 1971; Armonies, Hartke, 1995). They may also be able to disperse
once ingested by a bird, passing through the gut still alive (Haase et al., 2010; Cadée, 2011;
van Leeuwen et al., 2012). At the local scale, either of these processes must be happen-
ing in the Chupa Inlet, as we find the same haplotypes of B. progenetica in different spots
throughout the inlet (fig. 2). At the larger scale, biogeographic data on P. ulvae suggest
that gene flow is quite high between the Baltic and the White Sea, probably due to disper-
sal by birds (Wilke, Davis, 2000). It is thus likely that B. progenetica was transferred to
White Sea also from the Baltic, which is consistent with the records of this parasite there
(Markowski, 1936; Reimer, 1961).
Since three B. progenetica haplotypes were detected at the White Sea, at least three
P. ulvae infected with genetically distinct B. progenetica had arrived here at some point. This
could have happened on a single occasion (for example, if snails were carried by the same
bird) or as several independent events. The number of such events may be underestimated
judging from present data: some parasite haplotypes may be unsampled; some could have
become extinct at the White Sea; and some arriving snails could carry genetically identical
parasites.
In any case, we assume that divergence of B. progenetica into the three haplotypes
differing by 2-3 substitutions could not happen within the White Sea. It is a geologically
young water body which formed and was colonized after the end of the last glacial maximum
(LGM), not before 20 kya (Svendsen et al., 2004; Hughes et al., 2016). Considering the
estimate of mutation rate for cox1 gene in digeneans as 2.5 % per Ma (Attwood et al., 2008),
499
the three haplotypes could not have formed at the White Sea. The Baltic Sea was also under
ice during the LGM; the mud snails and parasites could spread here from the refugia at the
Atlantic coast of Europe where B. progenetica is also documented now (Deblock, 1978).
Summing up, our findings on the genetic diversity in C. concava and B. progenetica
illustrate some differences that would be expected between a tri- and monoxenous digenean
species. Data on C. concava expand understanding of dispersal in species with an avian
definitive host. As for B. progenetica, further study of intraspecific variation patterns could
clarify more on its dispersal and persistence abilities. This will require sampling at a larger
geographic scale, and sampling across several years to test whether the same haplotypes
continue to dominate.
ACKNOWLEDGEMENTS
Funding for this study was provided by the Russian Foundation for Basic Research,
grant number 18-34-01001. The study was carried out using the resources of the Educational
and Research Station “Belomorskaia” of St Petersburg University (SPbU) and the White
Sea Biological Station “Kartesh” of the Zoological Institute of Russian Academy of Sci-
ences (ZIN RAS). Sampling was in part supported by ZIN RAS according to the research
programme number AAAA-A19-119020690109-2. The study relied on equipment of the
research resource centre “Molecular and Cell Technologies” SPbU. The author is grateful
to Peter Smirnov for his great assistance with material collection, Prof. Andrei Granovitch
for making sampling in Varangerfjord possible, and to Drs. Anna Romanovich and Aleksey
Masharsky for excellent sequencing support.
REFERENCES
Alexander A., Steel D., Hoekzema K., Mesnick S.L., Engelhaupt D., Kerr I., Payne R., Baker C.S. 2016. What
influences the worldwide genetic structure of sperm whales (Physeter macrocephalus)? Molecular Ecology
25: 2754-2772.
Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. 1997. Gapped BLAST and
PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 25: 3389-3402.
Anderson A. 1971. Intertidal activity, breeding and the floating habit of Hydrobia ulvae. Journal of Marine Bio-
logical Association UK 51: 423-437.
Armonies W., Hartke D. 1995. Floating of mud snails Hydrobia ulvae in tidal waters of the Wadden Sea, and its
implications in distribution patterns. Helgoländer Meeresuntersuchungen 49: 529-538.
Attwood S.W., Fatih F.A., Upatham E.S. 2008. DNA-sequence variation among Schistosoma mekongi populations
and related taxa; phylogeography and the current distribution of Asian schistosomiasis. PLoS Neglected
Tropical Diseases 2 (3): e200.
Blakeslee A.M., Byers J.E., Lesser M.P. 2008. Solving cryptogenic histories using host and parasite molecular
genetics: the resolution of Littorina littorea’s North American origin. Molecular Ecology 17 (16): 3684-3696.
500
Blasco-Costa I., Poulin R. 2013. Host traits explain the genetic structure of parasites: a meta-analysis. Parasitol-
ogy 140 (10): 1316-1322.
Bowles J., Hope M., Tiu W.U., Liu X., McManus D.P. 1993. Nuclear and mitochondrial genetic markers highly
conserved between Chinese and Philippine Schistosoma japonicum. Acta Tropica 55 (4): 217-229.
Cadée G.C. 2011. Hydrobia as “Jonah in the Whale”: shell repair after passing through the digestive tract of
shelducks alive. Palaios 26 (4): 245-249.
Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P. 2012. Predicting the functional effect of amino acid sub-
stitutions and indels. PLoS ONE 7 (10): e46688.
Deblock S. 1978. Distribution géographique des cercaires parasites des Mollusques du genre Hydrobia Hartman
des côtes de France. Annales de Parasitologie Humaine et Comparée 53 (6): 577-593.
Dung D.T., Hop N.T., Thaenkham U., Waikagul J. 2013. Genetic differences among Vietnamese Haplorchis taichui
populations using the COI genetic marker. Journal of Helminthology 87 (1): 66-70.
Esch G.W., Kennedy C.R., Bush A.O., Aho J.M. 1988. Patterns in helminth communities in freshwater fish in
Great Britain: alternative strategies for colonization. Parasitology 96: 519-532.
Feis M.E., Thieltges D.W., Olsen J.L., de Montaudouin X., Jensen K.T., Bazaïri H., Culloty S.C., Luttikhuizen
P.C. 2015. The most vagile host as the main determinant of population connectivity in marine macropara-
sites. Marine Ecology Progress Series 520: 85-99.
Gonchar A., Galaktionov K.V. 2020. New data support phylogeographic patterns in a marine parasite Tristriata
anatis (Digenea: Notocotylidae). Journal of Helminthology 94: e79, 1-5.
Haase M., Naser M.D., Wilke, T. 2010. Ecrobia grimmi in brackish Lake Sawa, Iraq: indirect evidence for long-
distance dispersal of hydrobiid gastropods (Caenogastropoda: Rissooidea) by birds. Journal of Molluscan
Studies 76 (1): 101-105.
Holsinger K.E., Weir, B.S. 2009. Genetics in geographically structured populations: defining, estimating and
interpreting FST. Nature Reviews Genetics 10 (9): 639-650.
Hughes A.L., Gyllencreutz R., Lohne Ø.S., Mangerud J., Svendsen J.I. 2016. The last Eurasian ice sheets-a
chronological database and time-slice reconstruction, DATED-1. Boreas 45 (1): 1-45.
Jenkins T.L., Castilho R., Stevens, J.R. 2018. Meta-analysis of northeast Atlantic marine taxa shows contrasting
phylogeographic patterns following post-LGM expansions. PeerJ 6: e5684.
Keeney D.B., Waters J.M., Poulin, R. 2007. Clonal diversity of the marine trematode Maritrema novaezealandensis
within intermediate hosts: the molecular ecology of parasite life cycles. Molecular Ecology 16 (2): 431-439.
Keeney D.B., Bryan-Walker K., King T.M., Poulin R. 2008. Local variation of within-host clonal diversity coupled
with genetic homogeneity in a marine trematode. Marine Biology 154 (1): 183-190.
Králová-Hromadová I., Špakulová M., Horáčková E., Turčeková L., Novobilský A., Beck R., Koudela B., Marinculić
A., Rajský D., Pybus M. 2008. Sequence analysis of ribosomal and mitochondrial genes of the giant liver
fluke Fascioloides magna (Trematoda: Fasciolidae): intraspecific variation and differentiation from Fasciola
hepatica. Journal of Parasitology 94 (1): 58-67.
Kumar S., Stecher G., Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger
datasets. Molecular Biology and Evolution 33 (7): 1870-1874.
Leigh J.W., Bryant D. 2015. PopART: Full-feature software for haplotype network construction. Methods in Ecol-
ogy and Evolution 6 (9): 1110-1116.
Levakin I.A. 2004. The influence of infections with trematodes Bunocotyle progenetica (Hemiuridae) and Cryp-
tocotyle cancavum (Heterophiidae) onto mortality of littoral molluscs Hydrobia ulvae (Gastropoda: Proso-
branchia) in condition of extremely high temperature. Parazitologiia 38 (4): 352-358.
501
Levakin I.A. 2005. The influence of infections with trematodes Bunocotyle progenetica (Hemiuridae) and Cryp-
tocotyle cancavum (Heterophyidae) onto mortality of littoral mollusks Hydrobia ulvae (Gastropoda: Proso-
branchia) after freezing. Parazitologiia 39 (5): 407-413.
Levakin I.A., Nikolaev K.E., Galaktionov K.V. 2013. A case study of singular spectrum analysis application in
parasitology: dynamics of prevalence of Cryptocotyle concavum and Bunocotyle progenetica trematode
parthenitae in Hydrobia ventrosa snails at the White Sea. Parazitologiia 47 (1): 23-37.
Markowski S. 1936. Über die Trematodenfauna der Baltischen Mollusken aus der Umgebung der Halbinsel Hel.
Bull. Acad. Polon. Sci. Lett. B 2: 311.
Miura O., Kuris A.M., Torchin M.E., Hechinger R.F., Dunham E.J., Chiba S. 2005. Molecular-genetic analyses
reveal cryptic species of trematodes in the intertidal gastropod, Batillaria cumingi (Crosse). International
Journal for Parasitology 35 (7): 793-801.
Poulin R., Cribb T.H. 2002. Trematode life cycles: short is sweet? Trends in Parasitology 18 (4): 176-183.
R Core Team. 2015. R: a language and environment for statistical computing. Vienna, Austria, R Foundation for
Rauch G., Kalbe M., Reusch T.B.H. 2005. How a complex life cycle can improve a parasite‘s sex life. Journal of
Evolutionary Biology 18 (4): 1069-1075.
Reimer L. 1961. Die Stufen der Progenesis bei dem Fischtrematoden Bunocotyle cingulata Odhner, 1928. Wiadomości
Parazytologiczne 7 (4-6): 843-849.
Rozas J., Ferrer-Mata A., Sánchez-DelBarrio J.C., Guirao-Rico S., Librado P., Ramos-Onsins S.E., Sánchez-Gracia
A. 2017. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Molecular Biology and Evolu-
tion 34: 3299-3302.
Svendsen J.I., Alexanderson H., Astakhov V.I., Demidov I., Dowdeswell J.A., Funder S., Gataullin V., Henriksen
M., Hjort C., Houmark-Nielsen M., Hubberten, H.W., Ingólfsson O., Jakobsson M., Kjær K.H., Larsen
E., Lokrantz H., Lunkka J.P., Lyså A., Mangerud J., Matiouchkov A., Murray A., Möller P., Niessen F.,
Nikolskaya O., Polyak L., Saarnisto M., Siegert C., Siegert M.J., Spielhagen R.F., Stein R. 2004. Late
Quaternary ice sheet history of northern Eurasia. Quaternary Science Reviews 23: 1229-1271.
Tatonova Y.V., Besprozvannykh V.V., Shumenko P.G., Nguyen H.M., Solodovnik D.A. 2019. First description of
genetic diversity for the genus Metagonimus using the complete cox1 gene sequence. International Journal
for Parasitology 49 (12): 985-992.
Thaenkham U., Phuphisut O., Nuamtanong S., Yoonuan T., Sa-Nguankiat S., Vonghachack Y., Belizario V.Y.,
Dung D.T., Dekumyoy P., Waikagul J. 2017. Genetic differences among Haplorchis taichui populations in
Indochina revealed by mitochondrial COX1sequences. Journal of Helminthology 91 (5): 597-604.
Theron A., Sire C., Rognon A., Prugnolle F., Durand P. 2004. Molecular ecology of Schistosoma mansoni trans-
mission inferred from the genetic composition of larval and adult infrapopulations within intermediate and
definitive hosts. Parasitology 129 (5): 571-580.
van Leeuwen C.H.A., van der Velde G., van Lith B., Klaassen M. 2012. Experimental quantification of long distance
dispersal potential of aquatic snails in the gut of migratory birds. PLoS ONE 7 (3): e32292.
Wilke T., Davis G.M. 2000. Infraspecific mitochondrial sequence diversity in Hydrobia ulvae and Hydrobia ven-
trosa (Hydrobiidae: Rissooidea: Gastropoda): do their different life histories affect biogeographic patterns
and gene flow? Biological Journal of the Linnean Society 70 (1): 89-105.
502
ВНУТРИВИДОВАЯ ГЕНЕТИЧЕСКАЯ ИЗМЕНЧИВОСТЬ
У ТРЕМАТОД С МОНО- И ТРИКСЕННЫМ ЦИКЛОМ
ИЗ ОДНОГО ВИДА МОЛЛЮСКОВ-ХОЗЯЕВ
А. Г. Гончар
Ключевые слова: Digenea, трематоды, жизненный цикл, внутривидовая генетиче-
ская изменчивость, Bunocotyle progenetica, Cryptocotyle concava
РЕЗЮМЕ
Моллюски Peringia ulvae служат хозяевами для многих видов трематод, которые при этом
могут сильно различаться по структуре своих жизненных циклов. На севере Европы два кон-
трастных примера - это виды Cryptocotyle concava (Heterophyidae) с триксенным жизненным
циклом и Bunocotyle progenetica (Hemiuridae) с моноксенным. Мы секвенировали фрагмент гена
сох1 длиной ~ 870 пар нуклеотидов для оценки и сравнения генетической структуры внутри
этих видов. Наши результаты свидетельствуют о том, что активное распространение C. concava
ограничивает дифференциацию между различными географическими регионами и поддержи-
вает довольно высокое разнообразие гаплотипов. У B. progenetica два гаплотипа доминируют
и имеют по крайней мере некоторый потенциал для распространения.
503