Журнал прикладной химии. 2020. Т. 93. Вып. 10
УДК 691.175.743:66.092-977
НАПРАВЛЕНИЯ ВТОРИЧНОЙ ХИМИЧЕСКОЙ ПЕРЕРАБОТКИ
ПОЛИВИНИЛХЛОРИДА (обзор). ЧАСТЬ 2
© Е. М. Захарян1, Н. Н. Петрухина1, Э. Г. Джабаров1, А. Л. Максимов1,2
1 Институт нефтехимического синтеза им. А. В. Топчиева РАН,
119991, г. Москва, Ленинский пр., д. 29
2 Химический факультет Московского государственного университета им. М. В. Ломоносова,
119991, г. Москва, Ленинский горы, д. 1
Поступила в Редакцию 3 июля 2020 г.
После доработки 21 июля 2020 г.
Принята к публикации 30 июля 2020 г.
В обзоре рассмотрены процессы гидролиза отходов поливинилхлорида в щелочных средах и органи-
ческих основаниях и процессы гидротермальной переработки в критических средах (вода, аммиак,
метанол, диоксид углерода) хлорсодержащих отходов. Описаны принципы и механизмы гидродехло-
рирования и гидроконверсии поливинилхлорида и смесей поливинилхлорид/вакуумный газойль, а также
газификации отходов поливинилхлорида и хлорсодержащих муниципальных отходов пластика. Анали-
зируются процессы совместных гидротермальной переработки и газификации отходов поливинилхло-
рида с биомассой и углем, а также различных специфических методов дехлорирования поливинилхло-
рида. Показано, что основным преимуществом гидротермальной переработки поливинилхлорида
в докритической воде является отсутствие в продуктах хлорированных органических соединений.
Основным достоинством гидродехлорирования поливинилхлорид-содержащих отходов является
высокая степень удаления органического хлора и высокое отношение Н/С в продуктах переработки.
Ключевые слова: поливинилхлорид; отходы поливинилхлорида; муниципальные отходы пластика;
дехлорирование; гидротермальная обработка; критические среды; биомасса; гидродехлорирование;
гидроконверсия; газификация; совместная переработка
DOI: 10.31857/S0044461820100011
Введение
рирование в присутствии оксидов металлов. Данные
методы имеют один общий недостаток: образование
Необходимость вторичной переработки хлорсо-
твердых трудноутилизируемых не находящих приме-
держащих полимеров не вызывает сомнения, однако
нения отходов — хлоридов металлов, хлорсодержа-
важное значение имеет выбор технологии перера-
щего кокса. Кроме того, зачастую жидкие продукты
ботки, на который влияет происхождение вторичного
такой переработки содержат хлорсодержащие соеди-
сырья (промышленные или бытовые отходы), его
нения и требуют облагораживания.
чистота, возможность реализации продуктов перера-
Цель работы — анализ данных литературы по
ботки. В работе [1] были рассмотрены возможности
проблемам в области переработки хлорсодержащих
механохимической переработки поливинилхлорида,
полимерных отходов для выявления основных тен-
пиролиз, низкотемпературное каталитическое дехло-
денций развития технологий дехлорирования, ана-
1370
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1371
лиза механизма дехлорирования в различных про-
зволило авторам [5] достичь 90% конверсии поливи-
цессах, оценки перспектив получения мономеров
нилхлорида уже при 70°С (6.25 М NaOH, 10 ч).
хлорсодержащих высокомолекулярных соединений.
Добавление катализатора Pd/Cакт и 1.76 М рас-
Во второй части обзора поставлена задача анализа
твора H2O2 в систему поливинилхлорид/1 М NaOH
экологически безопасных подходов к переработке
приводило к удалению 90% органического хлора
поливинилхлорида, позволяющих получать жидкие
из поливинилхлорида, тогда как в отсутствие пе-
углеводороды, не содержащие хлор: гидролиза, ги-
рекиси водорода это значение не превышало 17%
дротермальной переработки в до- и сверхкритической
(180°С) [6]. Варьирование концентрации NaOH в
воде, аммиаке, метаноле, совместной гидротермаль-
интервале 0.25-2.5 моль·л-1 при проведении процес-
ной переработки с биомассой и углем, гидродехлори-
са дехлорирования поливинилхлорида показало, что
рования, газификации отходов поливинилхлорида и
оптимальным является 1 М раствор, в среде которого
хлорсодержащих муниципальных отходов пластика,
наблюдалась наибольшая скорость дехлорирования
совместной газификации отходов поливинилхлорида
полимера. Дальнейшее повышение молярности рас-
с биомассой и углем.
твора щелочи способствовало росту вязкости жид-
кости, покрывающей активные участки полимерной
цепи и катализатора.
Использование оснований
Разложение поливинилхлорида в присутствии
в качестве катализаторов или сорбентов
H2O2 протекает двумя путями (схема 1): гидротер-
Как было отмечено ранее, основания NaOH и
мальное дегидрохлорирование и непосредственное
KОН являются эффективными сорбентами выде-
окисление частично дехлорированного поливи-
ляющегося хлороводорода в ходе разложения по-
нилхлорида до органической кислоты, CO2 и H2O2.
ливинилхлорида при его механохимической пере-
Степень полного окисления поливинилхлорида до-
работке [2]. Проведение процесса дехлорирования
стигала 50%, при этом было образовано до 37.5%
смеси поливинилхлорид/поливинилиденхлорид в
нетоксичных органических соединений, таких как
растворе приводило к удалению большего количе-
щавелевая кислота и ее производные.
ства органического хлора: конверсия составила 92%
Использование органического растворителя (эти-
по сравнению 22%-ной конверсией, достигаемой в
ленгликоль, полиэтиленгликоль, диэтиленгликоль ли-
твердой гомогенной среде [3]. Таким образом, сочетая
бо триэтиленгликоль) позволило проводить реакцию
два метода, механохимическую переработку и низ-
дегидрохлорирования поливинилхлорида при более
котемпературное дехлорирование, можно получать
низкой температуре [7, 10, 11], чем при дехлориро-
продукт с низким содержанием хлорированных сое-
вании полимера в водном растворе (T < 250°С) [4],
динений либо вовсе их исключая и без образования
а также значительно сократить длительность процес-
токсичных хлорсодержащих соединений (диоксины,
са. Константа дехлорирования при 200°С в 1 М рас-
фенолы и т. д.).
творе NaOH/этиленгликоль была примерно в 150 раз
Значительное удаление органического хлора из
больше, чем в 1 М растворе NaOH/Н2О [7].
поливинилхлорида в среде водного раствора NaOH
Процесс дехлорирования поливинилхлорида в
достигалось либо при повышенной температуре, ли-
присутствии щелочи NaOH, растворенной в этилен-
бо при большей концентрации щелочи [4]. Степень
гликоле [7], предполагал комбинацию механизмов
дехлорирования увеличивалась с повышением тем-
E2 (удаление HCl) и SN2 (замещение гидроксильной
пературы реакции (табл. 1): при обработке жесткого
группы) (схема 2). Элиминирование являлось доми-
поливинилхлорида раствором 5 М NaOH при тем-
нирующим, поскольку наличие ОН-групп в продукте,
пературе 200°С она составляла 68.9% (5 ч), а при
идентифицированное по ИК-спектрам, было незна-
обработке раствором 3 М NaOH при 250°С — около
чительно. Дехлорирование поливинилхлорида про-
100% (3 ч). Максимальная скорость дехлорирования
исходило на границе раздела фаз твердое вещество/
наблюдалась в 3 М растворе NaOH. При этом сна-
жидкость путем взаимодействия Cl- в структурном
чала происходило выщелачивание добавок поливи-
звене поливинилхлорида с гидроксильной группой
нилхлорида (до 4.3 мас%), таких как модификаторы,
растворителя. ОН-Группа реагирует с Cl- как на по-
стабилизаторы, пигменты и т. д., а затем — удаление
верхности полимерной частицы, так и внутри нее.
HCl. В результате реакции был получен пористый
Однако при высоких концентрациях NaOH происхо-
полукокс с размером пор до 2 мкм.
дит изменение поверхности частиц поливинилхлори-
Добавление раствора гидросульфата тетрабутил-
да, которое предотвращает дальнейшее проникнове-
аммония в качестве межфазового катализатора по-
ние ОН-групп внутрь частиц полимера.
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1373
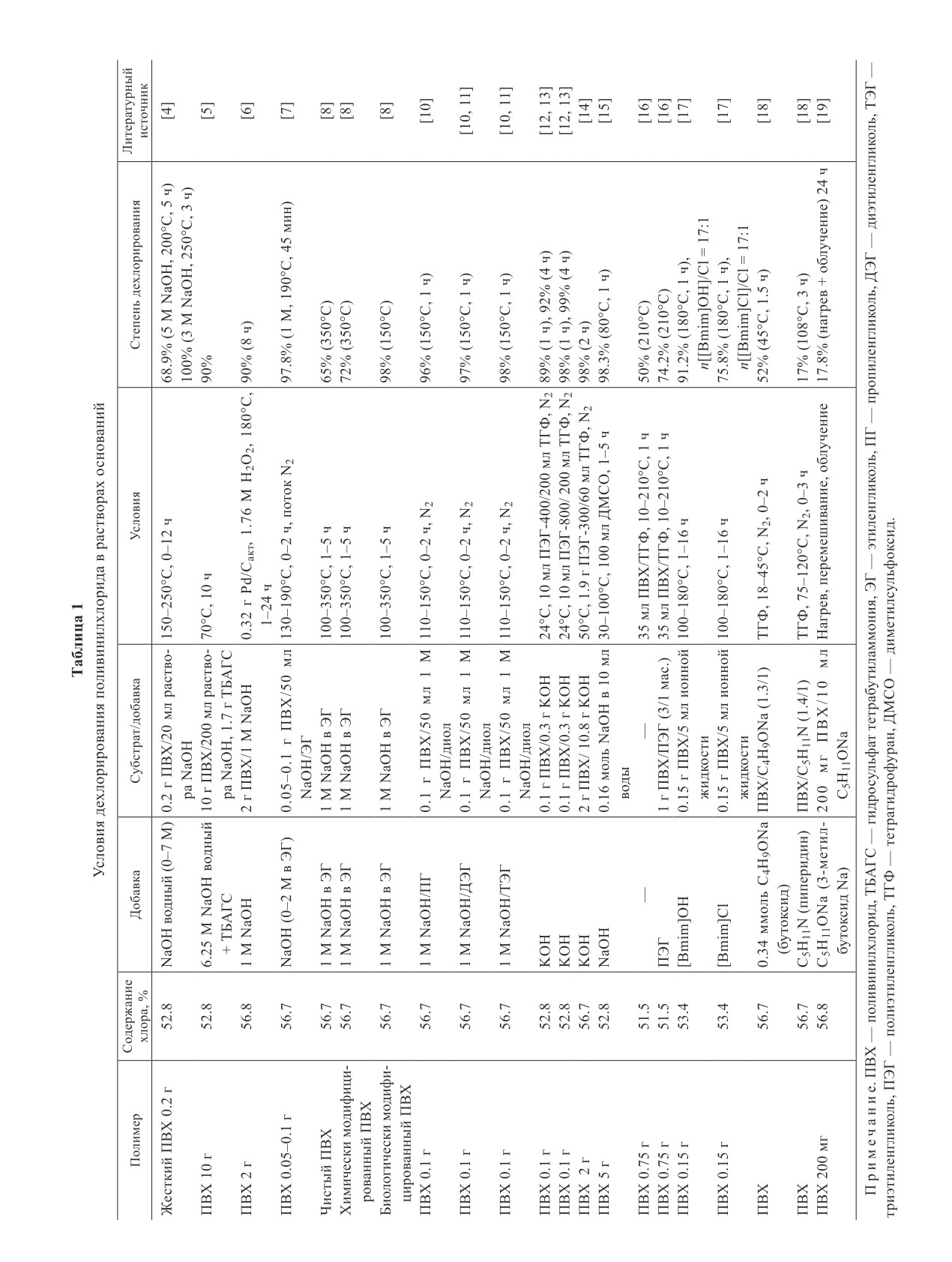
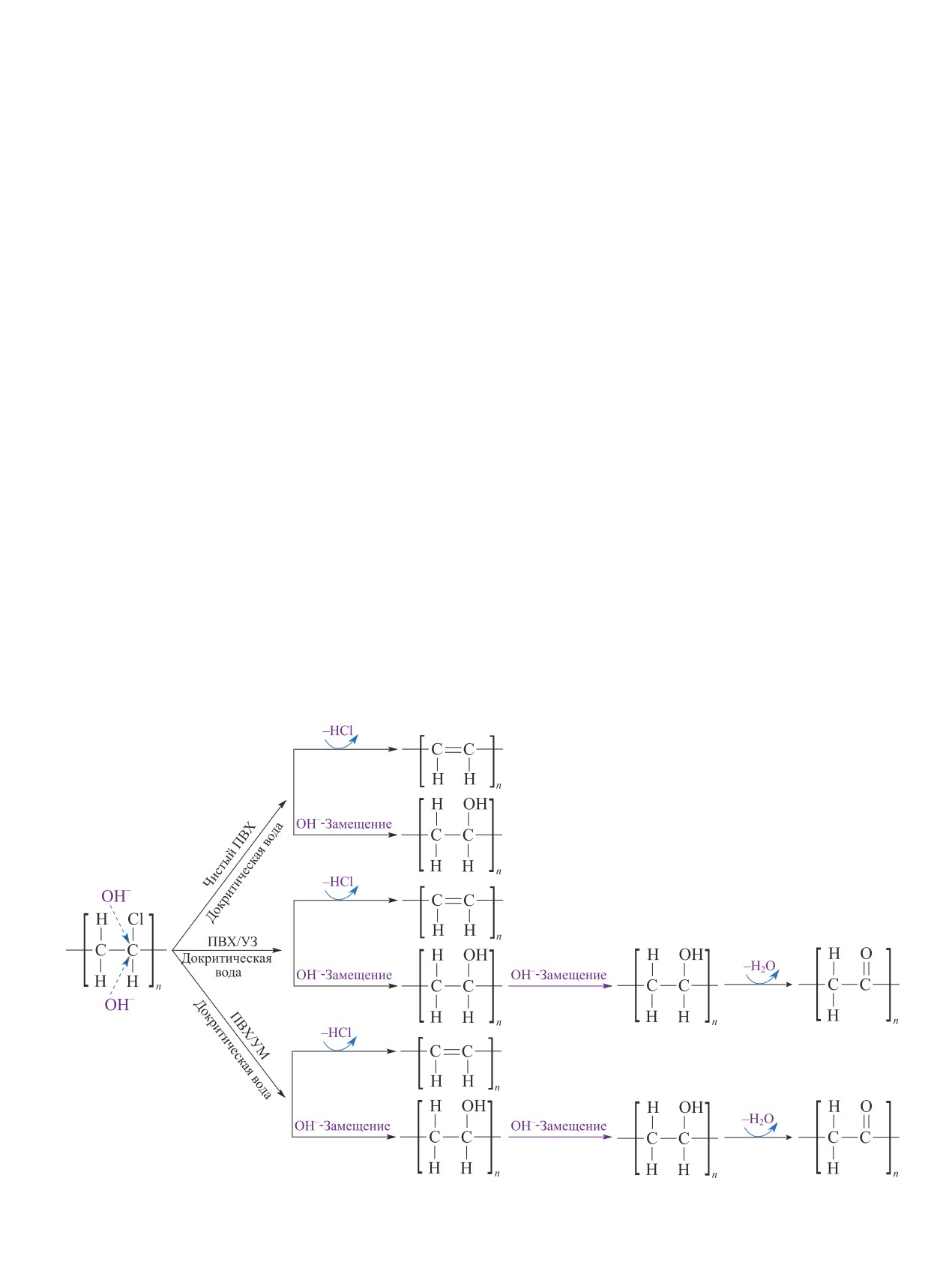
Схема 1
Предполагаемый механизм дехлорирования поливинилхлорида в отсутствие 1.76 М раствора H2O2 (а)
и с добавкой 1.76 М раствора H2O2 (б)
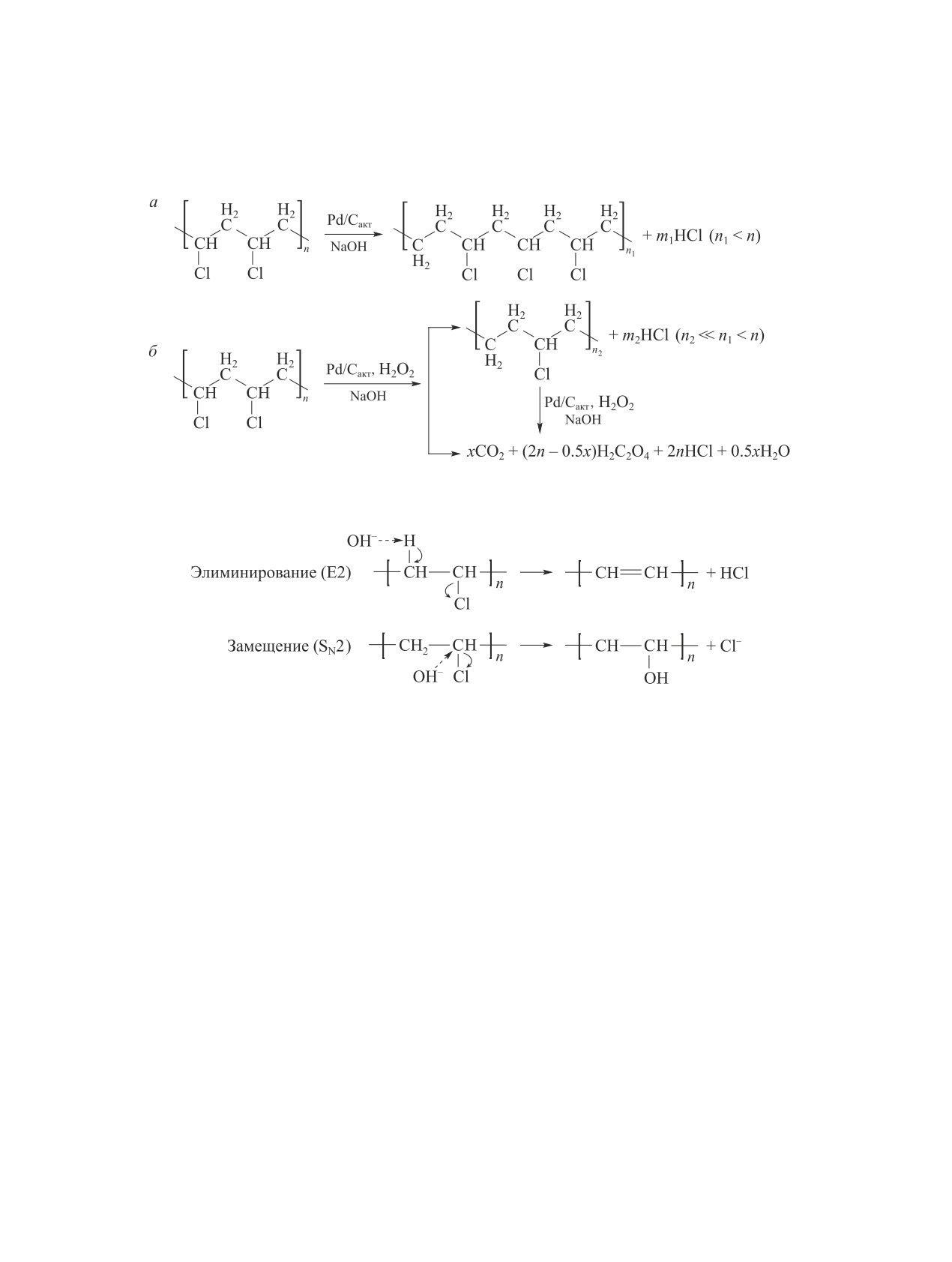
Схема 2
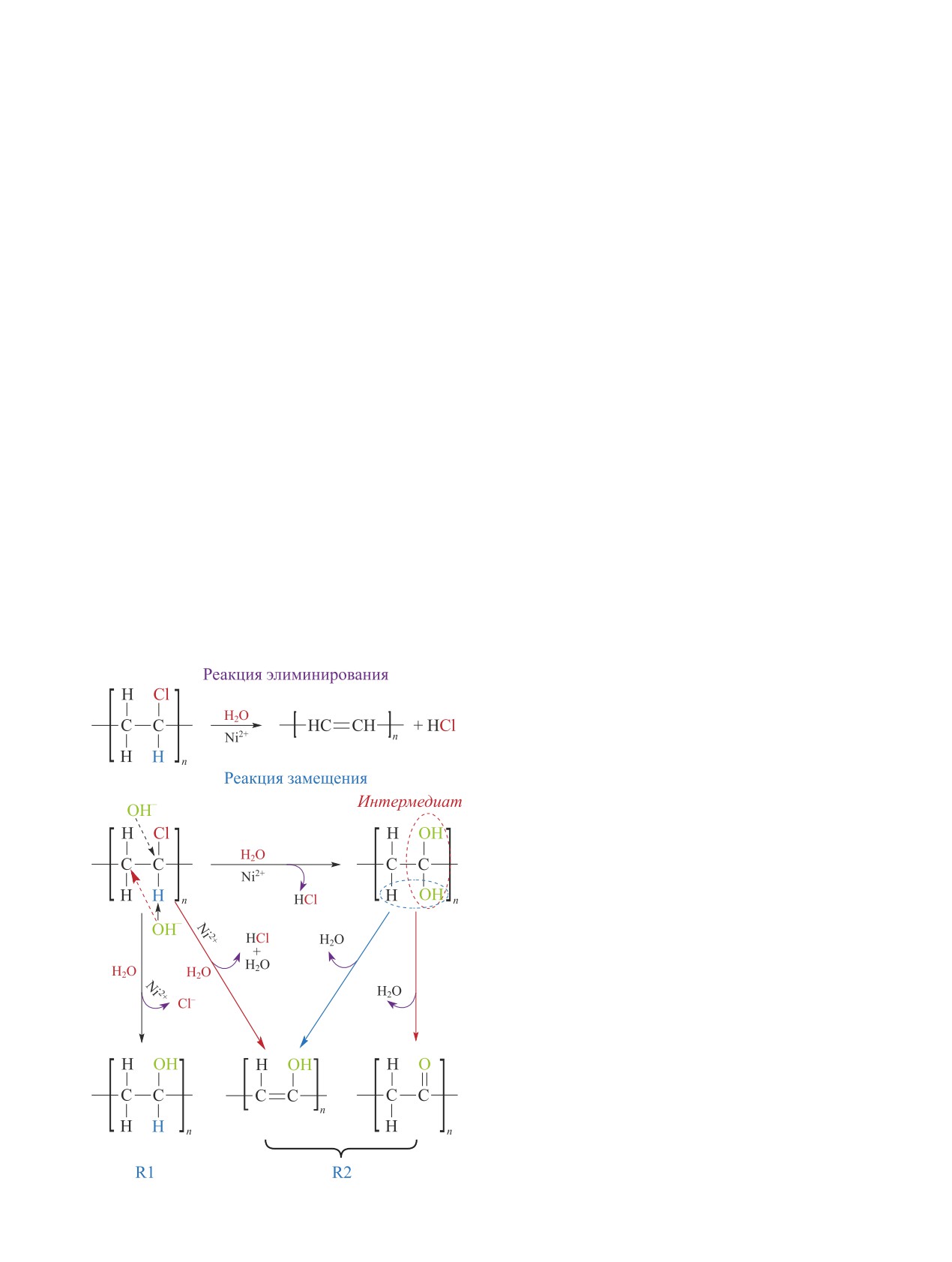
Механизм дехлорирования поливинилхлорида в NaOH/этиленгликоль
Степень дехлорирования росла с увеличением
химически модифицированного поливинилхлори-
длительности реакции в интервале концентраций
да. Средняя шероховатость росла в ряду поливи-
NaOH 0.2-2 моль·л-1, достигая максимума в 1 М
нилхлорид < химически модифицированный поли-
растворе щелочи (86%, 170°С, 80 мин). С повыше-
винилхлорид < биологически модифицированный
нием температуры до 190°С степень дехлорирования
поливинилхлорид с 9.67 до 23.36 нм, как и число
достигала максимального значения уже через 45 мин
зерен — с 17 в поливинилхлориде до 338 в биологи-
реакции — 97.8%, при 130°С она не превышала 5%.
чески модифицированном поливинилхлориде.
Влияние модификации поливинилхлорида на сте-
Максимальная степень дехлорирования биоло-
пень дехлорирования в среде смеси 1 М NaOH/эти-
гически модифицированного поливинилхлорида
ленгликоль исследовалось авторами [8]. Химическую
(98%) достигалась при 150°С, в то время как для
модификацию поливинилхлорида проводили в эти-
дехлорирования чистого и химически модифициро-
ленгликоле (180°С, 4 ч) путем его взаимодействия с
ванного поливинилхлорида требовалась более высо-
этиленкарбонатом и гидроксиэтилкарбаматом, обра-
кая температура. Столь высокое различие в степени
зованным в ходе реакции этиленгликоля с мочеви-
дехлорирования чистого и модифицированных по-
ной. Биологическую модификацию поливинилхло-
ливинилхлоридов, возможно, связано с различным
рида штаммами бактерий Pseudomonas fiuorescens и
влиянием атомов кислорода (химически модифици-
Аspergillus niger проводили согласно методике, при-
рованный) и азота (биологически модифицирован-
меняемой в [9].
ный) в структурах полимеров на скорость процесса
Биологически модифицированный поливинилхло-
дехлорирования.
рид имеет более неоднородную и шероховатую по-
Влияние природы органического растворителя
верхность в отличие от чистого и химически мо-
NaOH на дехлорирование поливинилхлорида иссле-
дифицированного поливинилхлорида благодаря
довали в [10, 11]. В качестве растворителя использо-
взаимодействию штаммов бактерий с поверхностью
вали различные диолы: диэтиленгликоль, триэтилен-
1374
Захарян Е. М. и др.
гликоль и пропиленгликоль. Степень дехлорирования
Дехлорирование поливинилхлорида в тех же усло-
поливинилхлорида в растворе 1 М NaOH (110°С,
виях, что и в работах [12, 13], с последующим окис-
2 ч) повышалась в ряду этиленгликоль < пропилен-
лением в среде кислорода (0.5-8 МПа) приводило к
гликоль < диэтиленгликоль < триэтиленгликоль с 8
образованию водорастворимых кислот (муравьиная,
до 50%, что объясняется различной растворимостью
фумаровая и малеиновая) при 100°С и к выделению
поливинилхлорида в растворителе. Чем лучше рас-
СО2 при Т > 150°С [14]. Связи С С в дехлорирован-
творимость поливинилхлорида в диоле, тем быстрее
ном поливинилхлориде легко окисляются на возду-
и лучше проникают молекулы растворителя в струк-
хе при высоких температурах — выше 80°С, а сам
туру поливинилхлорида и тем быстрее и эффективнее
дехлорированный поливинилхлорид разлагается при
протекает дегидрохлорирование полимера. С повы-
температуре выше 200°С до летучих соединений,
шением температуры и времени реакции значитель-
включая СО2 и воду. Выход водорастворимых сое-
но увеличивается степень дехлорирования поливи-
динений увеличивался с повышением давления O2
нилхлорида: уже спустя 1 ч она превышала 90%, а
до 8 МПа и достигал максимальных 82% (100°С,
спустя 2 ч реакции достигала 96-98%.
1 ч). С повышением температуры, а также времени
Авторы [12, 13] проводили дехлорирование поли-
реакции увеличивался выход уже СО2 за счет быст-
винилхлорида с добавлением KOH в смешанной го-
рого окисления двойных связей дехлорированного
могенной среде тетрагидрофуран/полиэтиленгликоль,
полиена.
(Мср = 200, 400, 800 г·моль-1) уже при комнатной
Мягкий щелочной гидролиз поливинилхлорида
температуре. Выбор данной системы растворите-
в диметилсульфоксиде приводил к образованию де-
лей обусловлен тем, что тетрагидрофуран является
гидрохлорированного полиена и поливинилового
хорошим растворителем поливинилхлорида, а поли-
спирта [15]. Поскольку поливинилхлорид гидроли-
этиленгликоль — неорганических оснований, таких
зуется в диметилсульфоксиде частично, добавление
как KOH. В данном процессе полиэтиленгликоль
небольшого количества воды (10 мл) способствовало
действует в качестве растворителя основания и ката-
большей эффективности процесса. Повышение тем-
лизатора реакции.
пературы и продолжительности процесса дехлориро-
На скорость дехлорирования значительно влияла
вания позволило достичь высоких результатов: так,
природа растворителя. Так, при использовании гли-
при 80°С степень дехлорирования составляла 98.3%
коля дехлорирование практически не протекало даже
(1 ч), тогда как при 60°C — 90.1 (5 ч). Исследование
после четырехчасовой реакции, поскольку гликоль
возможности рециркуляции реагентов показало, что
не проявлял каталитических свойств. Добавление
отфильтрованный и повторно использованный ди-
полиэтиленгликоля (800 г·моль-1) к гликолю либо
метилсульфоксид способствовал удалению до 74.6%
использование полиэтиленгликоля молярной массы
органического хлора из поливинилхлорида.
200, 400 или 800 г·моль-1 значительно ускоряло про-
Использование полиэтиленгликоля в качестве
цесс дехлорирования и приводило к конверсии 96,
катализатора дехлорирования поливинилхлорида и
49, 92 и 99% (4 ч) соответственно, т. е. длина цепи
реакционной среды в отсутствие неорганических ос-
полиэтиленгликоля определяла его каталитическую
нований (щелочей) было исследовано в [16], при этом
активность. При использовании полиэтиленгликоля
было исключено образование побочных продуктов,
молярной массы 400 и 800 г·моль-1 конверсия поли-
таких как хлориды натрия или калия.
винилхлорида составляла 89 и 98%, что объясняется
Высокая скорость дехлорирования в смеси поли-
возможностью полиэтиленгликоля не только рас-
винилхлорид/полиэтиленгликоль (400 г·моль-1) по
творять неорганическое основание, но и координи-
сравнению с чистым поливинилхлоридом обуслов-
роваться с катионами основания (в данном случае
лена взаимодействием полиэтиленгликоля и поли-
с K+) для образования короноподобных полимер-
винилхлорида. С добавлением полиэтиленгликоля
ных гидроксидов в качестве активного агента. При
начальная температура разложения поливинилхло-
использовании большего количества KОН возмож-
рида смещалась с 171.9 до 138.2°C, а пиковая темпе-
но протекание побочной реакции — замена Сl- на
ратура (Tmax) — с 289.1 до 250°C. Дехлорированный
гидроксильную группу. Чем больше молекулярная
поливинилхлорид, полученный при гидрообработке
масса полиэтиленгликоля, тем длиннее цепь сопря-
с добавлением полиэтиленгликоля (400 г·моль-1) в
женного полиена [n(C C) = 23-28], образованного
течение 1 ч, имел шероховатую поверхность с порис-
в результате дегидрохлорирования поливинилхлори-
тыми структурами, в то время как дехлорированный
да, и тем меньше вероятность протекания побочных
поливинилхлорид в отсутствие полиэтиленгликоля —
процессов.
гладкую поверхность, что соответствовало степени
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1375
их деградации. Стабильность полиэтиленгликоля
выведению HCl и замене атома хлора в структурном
доказывается 10-кратным повторным использованием
звене поливинилхлорида (схема 3).
в дехлорировании поливинилхлорида.
Cкорость дехлорирования повышалась при увели-
Использование ионных жидкостей, таких как
чении количества [Bmim]OH. При молярном соотно-
[Bmim]OH и [Bmim]Cl (гидроксид и хлорид 1-бу-
шении [Bmim]OH/Cl = 17:1 степень дехлорирования
тил-3-метилимидазолия), в качестве реакционной
достигала 21.1%, в то время как при молярном соот-
среды в [17] приводило к удалению до 91% органи-
ношении 67:1 — уже 43% (100°С, 1 ч), что было ана-
ческого хлора из поливинилхлорида, в то время как
логично результатам дехлорирования поливинилхло-
в отсутствие растворителя дехлорирование поливи-
рида в водном и спиртовом растворах NaOH [7].
нилхлорида не превышало 38% (180°С). Большая
Влияние органических оснований (н-бутоксида
основность гидроксида 1-бутил-3-метилимидазолия
натрия, 3-метилбутоксида натрия и пиперидина) и
по сравнению с [Bmim]Cl объясняет повышенную
температуры на дехлорирование поливинилхлорида
реакционную способность [Bmim]OH дехлориро-
исследовалось в [18, 19]. В ходе взаимодействия по-
вания поливинилхлорида. В [Bmim]OH вместе с ги-
ливинилхлорида с C4H9ONa и C5H11N в тетрагидро-
дроксид-ионом образуется карбен, способствующий
фуране осаждался дисперсный сопряженный полиен
[18]:
―CH2―CHCl―CH2―CHCl― + R2NH
―CH CH―CH2―CHCl― + R2NH2Cl,
―CH2―CHCl―CH2―CHCl― + RONa
―CH CH―CH2―CHCl― + ROH + NaCl.
Дегидрохлорирование поливинилхлорида проте-
Достоинством гидролиза поливинилхлорида в
кало по механизму бимолекулярного элиминирования
присутствии оснований является отсутствие в про-
(E2), в котором взаимодействие аллильного вторич-
дуктах токсичных хлорированных диоксинов, бен-
ного атома водорода и атома хлора с органическим
золов и фенолов. Значительную эффективность де-
основанием приводило к удлинению конъюгирован-
хлорирования поливинилхлорида в среде водного
ных полиеновых последовательностей в полимерной
щелочного раствора при пониженных температу-
цепи.
рах удалось достигнуть благодаря использованию
Процесс дехлорирования поливинилхлорида про-
межфазного агента [5] и катализатора Pd/Cакт с до-
текал в присутствии C4H9ONa быстрее по сравнению
бавлением раствора H2O2 [6]. Проведение процесса
с C5H11N, что обусловлено его основными свойства-
в диоловых растворах щелочи позволило снизить
ми (табл. 1). Дехлорирование поливинилхлорида в
температуру до 150°С и сократить время до 1 ч по
присутствии 3-метилбутоксида при нагревании и
сравнению с водным раствором [10, 11], а в смеси
облучении не превышало 17.8% [19].
тетрагидрофуран/полиэтиленгликоль — уже при ком-
натной температуре [12, 13].
Схема 3
Механизм дехлорирования поливинилхлорида в присутствии ионной жидкости
1376
Захарян Е. М. и др.
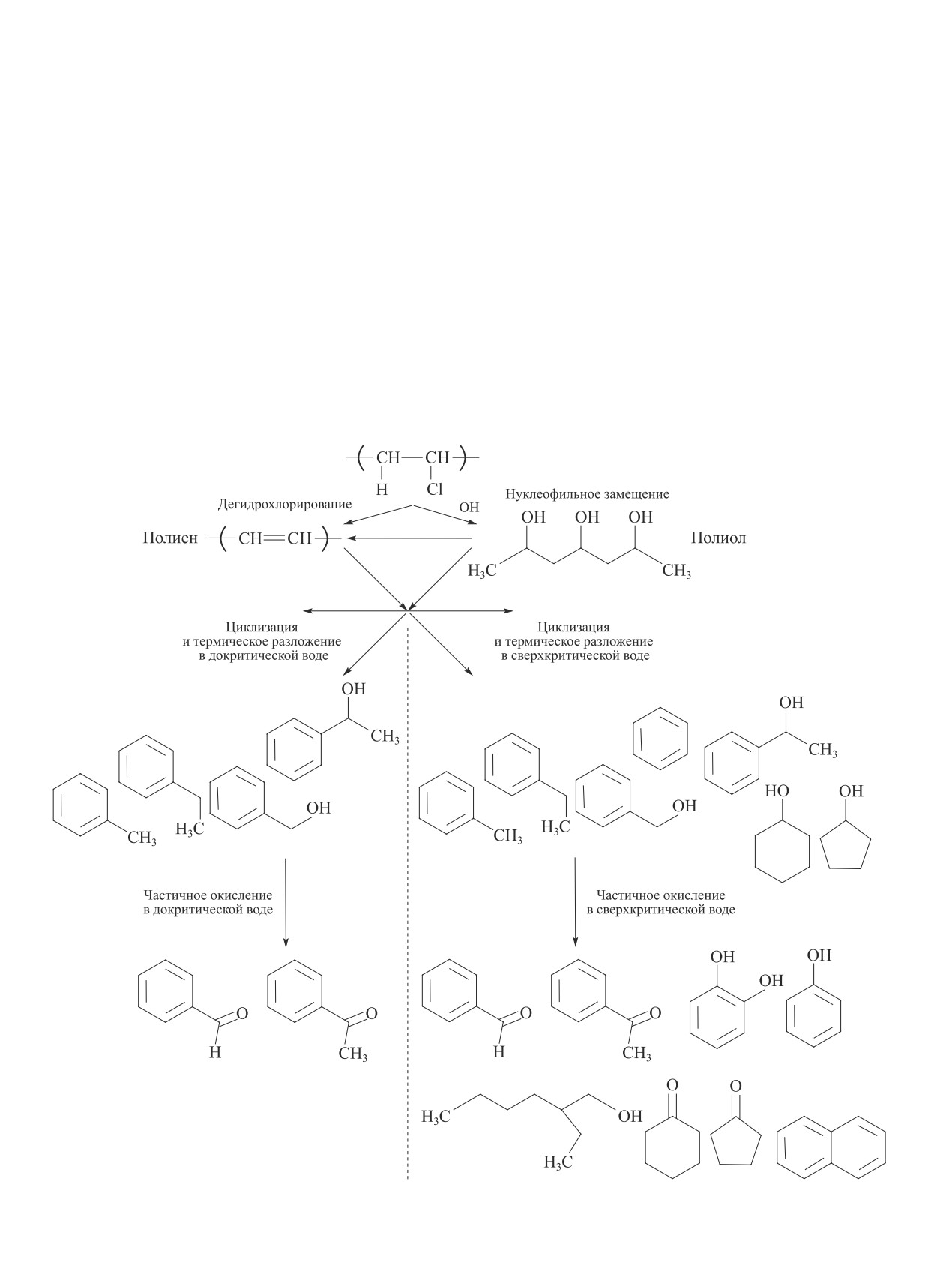
Гидротермальная обработка поливинилхлорида
в критических средах
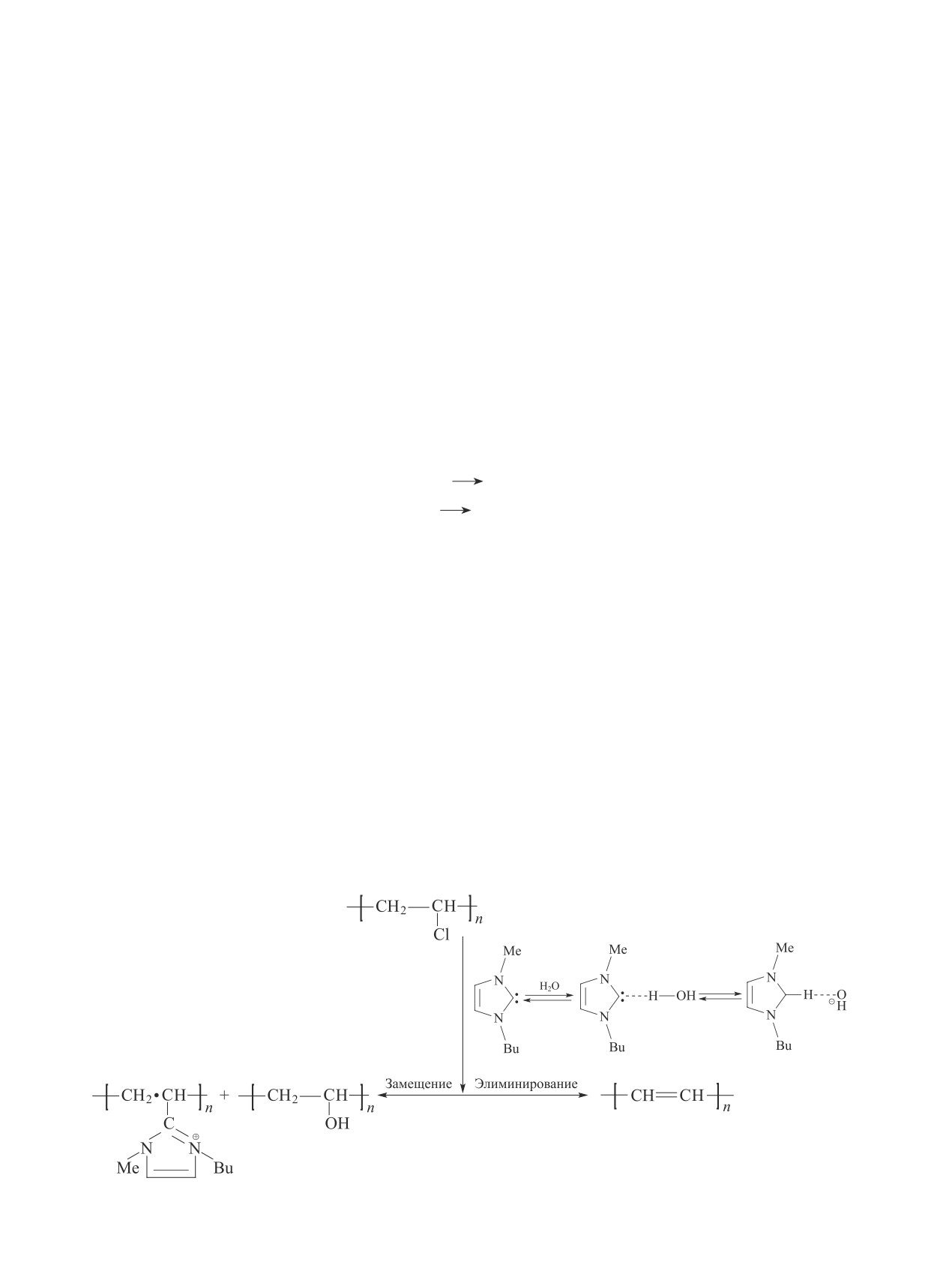
Разложение чистого поливинилхлорида и его от-
ходов в водных средах. Гидротермальную обработку
можно классифицировать на три типа в зависимости
от температуры процесса (T < 250°C, 250 < T < 350°С,
T > 350°C) и соответственно давления. Воду в указан-
ных температурных интервалах можно рассматривать
как пар, докритическую и сверхкритическую воду
(см. рисунок). Продуктами гидротермальной обра-
ботки сырья являются гидротермальные полукокс,
масло и газы [20, 21].
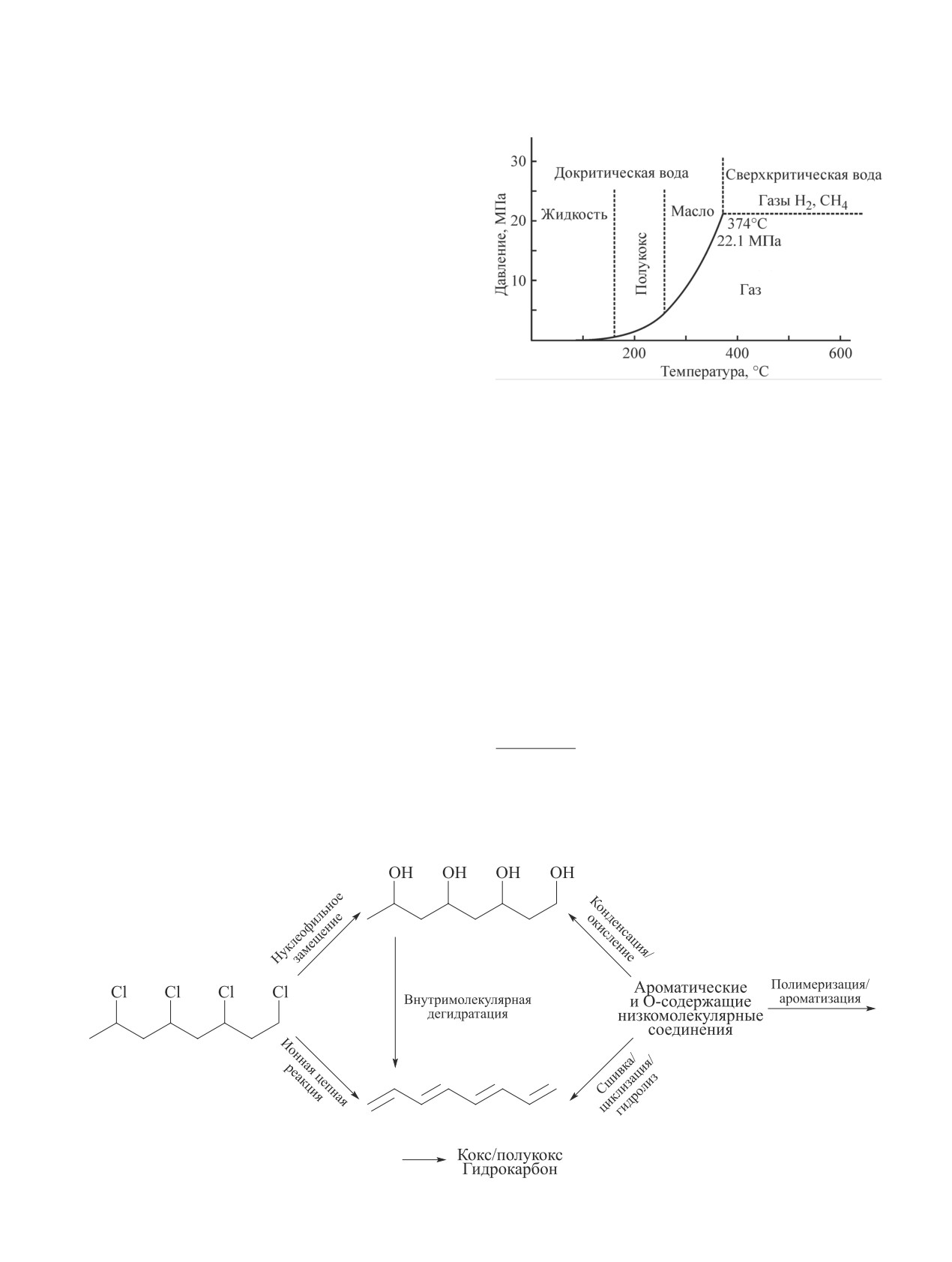
При высоких температурах (>400°С) разложение
Классификация гидротермальных процессов биомассы
поливинилхлорида протекает путем ионной цепной
в зависимости от давления и температуры на фазовой
реакции и крекинга, приводящего к образованию аро-
диаграмме воды [20, 21].*
матических и низкомолекулярных соединений. При
добавлении в систему воды нуклеофильное замеще-
ОН-нуклеофильному замещению в водной среде,
ние ОН-группы становится конкурентным процессом
действующей как нуклеофильный агент.
наряду с дехлорированием ионной пары с образова-
Как правило, при гидротермальной обработке по-
нием полиольных структур (схема 4) [22, 23].
ливинилхлорида полимер разлагается в два этапа:
При плотностях воды, превышающих критиче-
в первой области (T < 250°C) происходит дегидрох-
ское значение, повышение температуры приводит
лорирование поливинилхлорида с образованием по-
к более низкой скорости образования полиола, что
лиена, при более высокой температуре происходит
может быть связано с повышенной реакционной
разложение полиена на низкомолекулярные соеди-
способностью образованных полиеновых структур,
нения [23].
дегидратацией полиолов с образованием полиеновых
Обработка паром (до 75 об%) смолы и гибкого
структур и с разложением и гидролизом полиольных
поливинилхлорида проводилась в [24]. Основная
структур до соединений, имеющих более низкую
потеря массы смолы поливинилхлорида происходит
молекулярную массу, таких как три-, ди-, пропилен-
при Т > 210°С, гибкого поливинилхлорида — при
гликоли, которые в дальнейшем расщепляются до
гидроксиацетона. Таким образом, температура ни-
* Разрешение на публикацию получено 14.07.2020,
же 450°С и высокая плотность воды способствует
© 2015 Elsevier.
Схема 4
Предполагаемые пути разложения поливинилхлорида при гидротермальной обработке
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1377
170-180°С, что связано с потерей пластификатора
рования гибкого поливинилхлорида достигала 97%
диизононилфталата, составляющего до 50% массы
спустя 3 ч (230°С, пар 50 об%).
гибкого поливинилхлорида. В работе [25] при деги-
Разложение поливинилхлорида в докритической
дрохлорировании поливинилхлорида в среде смеси
воде [26] приводило к образованию полукокса, явля-
NaOH/этиленгликоль появление ОН-колебаний в ИК-
ющегося основным продуктом, в котором молярное
спектрах связывали с нуклеофильным замещением
соотношение С/Н составляло 1:1, что предполагало
Cl- гидроксигруппами (SN2). Но поскольку вода в
образование остатка из конъюгированных алкенов
газообразном состоянии [24] является более слабым
(—CH CH—CH CH—)n с высокой молекулярной
нуклеофилом, чем гидроксильная группа, авторы
массой. Содержание ароматических углеводородов
предположили, что замещение в данном случае про-
составляло ~1% от общей массы поливинилхлори-
текало по механизму SN1 (схема 5).
да. На термогравиметрических кривых при 200°С
С повышением температуры и давления пара
заметных потерь массы поливинилхлорида не на-
степень дехлорирования поливинилхлорида увели-
блюдалось. При повышении температуры реакции
чивалась в обоих случаях (смола и гибкий поливи-
до 250°С потеря массы составила 50%, при 300 и
нилхлорид), при этом она значительно превышала
350°С — 56%, что соответствовало удалению прак-
это значение при термическом разложении. Так, на-
тически всего количества органического хлора из
пример, при 250°С (50 об% пара, 1 ч) степень дехло-
полимерной структуры поливинилхлорида. До 80%
рирования смолы поливинилхлорида составляла 76%
всего хлора было обнаружено в виде хлорид-иона в
вместо 61%, достигнутой в атмосфере N2. При более
водной фазе.
высоких концентрациях воды в газообразном состо-
Дехлорирование поливинилхлорида большей
янии эффективное удаление органического хлора
массы в [27] в докритической воде по сравнению с
из смолы поливинилхлорида затруднительно ввиду
дехлорированием полимера в [26] позволило полу-
быстрого поверхностного образования полиена, ко-
чить полукокс с содержанием атомов хлора 3.03% при
торый в свою очередь вследствие экранирующего
260°С, при этом доля атомов углерода в нем состав-
эффекта препятствует дальнейшему дегидрохлориро-
ляла 82%, аналогично битуминозному углю. С повы-
ванию, что наблюдалось и при использовании различ-
шением температуры с 200 до 260°С увеличивалась
ных диолов в качестве растворителей и промоторов
степень дехлорирования, поскольку содержание ато-
[10, 11].
мов хлора падало с 42.7 до 3.03%, также снижался
В случае дехлорирования гибкого поливинилхло-
выход полукокса с 96.1 до 43.9%. С ростом темпе-
рида обработка паром также способствовала улуч-
ратуры обработки улучшались топливные качества
шению результатов: степень дехлорирования 69% по
полукокса, теплотворная способность повышалась
сравнению с 51% при термообработке в N2 (табл. 2).
до 36.87 МДж·кг-1, при этом высокая температура
Экранирующий эффект в данном случае не наблюдал-
приводила к интенсивной ароматизации и увеличе-
ся, что, возможно, связано с увеличением пористости
нию степени конденсации, а также снижению пло-
в результате улетучивания пластификатора, за счет
щади поверхности за счет разрушения полостей в
чего раскрывалась структура для проникновения па-
продукте.
ра. Кроме того, стабилизатор ингибировал быстрое
С повышением температуры дехлорирования отхо-
дегидрохлорирование в начале реакции, позволяя
дов поливинилхлорида до 280°С (1 ч) [28] теплотвор-
пару проникать в поры полимера. Степень дехлори-
ная способность увеличивалась до 37.21 МДж·кг-1,
Схема 5
Механизм реакции SN1 и ионного элиминирования
1378
Захарян Е. М. и др.
а при большей длительности дехлорирования при
[26, 29]. Добавление лимонной кислоты не повлияло
250°С (90 мин) — до 38.54 МДж·кг-1. Авторы статьи
на дехлоририрование во всем диапазоне темпера-
также обнаружили, что увеличение длительности
тур обработки. pH раствора значительно снизился
процесса (более 60 мин) способствовало снижению
с нейтрального до рН 2.4 и рН 3.2 при 260 и 180°С
выхода золы при сжигании полученного в процессе
соответственно не только за счет выделения HCl и
гидротермальной обработки полукокса.
его растворения в воде, но и благодаря образова-
Обработка поливинилхлорида паром, как в [24], и
нию таких низкомолекулярных кислот, как уксусная,
в докритической, и сверхкритической воде, как в [26],
пропионовая, муравьиная и янтарная, полученных в
проводилась в [29]. В ходе дехлорирования полимера
результате окисления кратных связей. Доля атомов
давление в реакторе увеличивалось с повышением
хлора в полукоксе, полученном при 240°С, составила
температуры до 55.7 МПа при 600°С.
1.6 мас%, а в полукоксе, полученном при 260°С, —
Почти полное удаление органического хлора из
уже 0.24 мас% по отношению к доле органического
поливинилхлорида (степень дехлорирования 99%)
хлора в поливинилхлориде. Молярное отношение
достигалось уже при 280°С, что обусловлено полным
Н/С в полукоксе по сравнению с молярным отноше-
выделением HCl из поливинилхлорида при данной
нием Н/С в поливинилхлориде (1.46) снижалось от
температуре. Хлорсодержащих соединений в жидкой
1.3 при 180°С до 0.71 при 260°С.
и газовой фазах обнаружено не было; весь органиче-
Хлорированные углеводороды, в том числе хлор-
ский хлор, удаленный из поливинилхлорида, пред-
фенолы, могут быть определены только в следовых
ставлял собой растворенный в воде хлороводород.
количествах, поскольку некоторое количество не-
С ростом температуры и массы поливинилхлори-
удаленного органического хлора связано с sp2-ги-
да значительно повышался выход газа и снижалась
бридизованными атомами углерода в олефиновых
доля твердого остатка. При температуре от 250 до
и ароматических структурах, которые проявляют
350°С поливинилхлорид превращается в полиен в
химическую инертность по отношению к нукле-
твердом остатке и низкомолекулярные ароматиче-
офильному замещению гидроксильными ионами.
ские и алифатические соединения в жидкой и га-
Полихлорированные дибензодиоксины и дибензо-
зовой фракциях. При 300°С газовая фаза состоит
фураны обнаружены не были. В водной фазе был
только из водяного пара, СО2 и бензола, в то время
обнаружен широкий спектр полициклических аро-
как при 400°С в составе газа появляются предельные
матических углеводородов: от фенолов, диолов и
и непредельные углеводороды С1-С5. Дальнейшее
нафтолов до антрацена и фенантрена в суммарной
разложение полимера при температуре свыше 350°С
концентрации 140 мкг на 1 г поливинилхлорида при
в сверхкритической воде приводит к превращению
240°С, что согласовалось с данными, полученными
его в ацетон, фенол, бензол, производные бензола и
в [26, 29].
алифатические алканы и алкены благодаря процессам
В [30] был разработан процесс частичной окис-
циклизации, гидролиза и окисления кратных связей
лительной обработки гибкого поливинилхлорида
частично растворенным в воде кислородом.
в критической воде с целью его дехлорирования и
Доля атомов С и Н в твердом остатке повышалась
восстановления продуктов процесса в ценное хими-
до 88.8 и 7.5% при 300°С по сравнению с долей ато-
ческое сырье. С повышением температуры, как и в
мов С и Н в поливинилхлориде — 38.5 и 4.9% соот-
бескислородном процессе [27], значительно снижался
ветственно. Содержание атомов хлора в полукоксе
выход твердого остатка: с 80 при 200°С до 20 и 12%
составляло 1.1%, что является предельно допусти-
при 400°С. Разная доля твердого остатка при 400°С
мым в отходах после обработки поливинилхлори-
обусловлена различным соотношением твердого ве-
да. Энтальпия горения твердого остатка составила
щества и жидкости. Молекулы воды в критических
9270 ккал·кг-1, что превышает энтальпию горения
условиях могут диффундировать в поливинилхлориде
угля и кокса (4500-8000 и 6000-7500 ккал·кг-1 соот-
через пористую структуру, способствуя образованию
ветственно) и свидетельствует о том, что этот остаток
и удалению газовых продуктов из пор поливинилхло-
может быть использован в качестве топлива.
рида, тем самым ускоряя реакцию дегидрохлориро-
Исследование влияния лимонной кислоты на
вания и циклизации.
гидротермальную карбонизацию отходов поливи-
Доля хлорид-анионов в водной фазе, составляв-
нилхлорида в докритической воде проводили в [23].
шая при 200°С около 10%, при 250°С резко повы-
Эффективность дегидрохлорирования поливинилхло-
шалась до 85.5%, а при 300°С достигала 95%, что
рида увеличивалась с повышением температуры ре-
подтверждает тот факт, что практически весь орга-
акции и достигала почти 100% уже при 235°С, как и в
нический хлор из поливинилхлорида удаляется с во-
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1379
дой. Наличие добавок в гибком поливинилхлориде
продукты частичного окисления толуола, этилбензо-
несколько снижало выход хлороводорода при темпе-
ла, фенилкарбинола и 1-фенетилового спирта.
ратуре менее 300°С, что связано с ингибирующими
Процесс частичной окислительной обработки в
и стабилизирующими свойствами меркаптида олова,
критической воде разделяется на три этапа (схема 6).
входящего в состав гибкого поливинилхлорида, а
Первый этап — дегидрохлорирование поливинилхло-
при температурах свыше 300°С эти различия были
рида и образование полиена (200-300°С). Другой
незначительны.
возможный путь — преобразование поливинилхло-
Основным продуктом в бескислородном процессе
рида путем нуклеофильного замещения Cl- гидрокси-
при 300-400°С являлся ацетон [29], а за счет наличия
группой (ОН) в полиол с его последующей дегидра-
большего количества кислорода (массовое отношение
тацией до полиена. Второй этап — разложение и
поливинилхлорида к О2 было около 32) при этих же
циклизация полиена и полиола в критической воде.
температурах — бензальдегид и ацетофенон [30] —
Третья стадия — частичное окисление. Основными
Схема 6
Механизм частичного окисления в докритической и сверхкритической воде
1380
Захарян Е. М. и др.
реакциями процесса частичного окисления в докри-
пиренов практически не протекало при температурах
тической воде (ниже 374°C) являются разложение и
ниже 200°С и ускорялось с увеличением температуры
циклизация полиена на второй стадии с образовани-
до 275°С. Выход твердого остатка составлял 95.7,
ем толуола и этилбензола, которые были частично
85.3 и 83.9 мас% при 225, 250 и 275°С соответствен-
окислены до бензальдегида и ацетофенона на третьей
но, а содержание атомов брома в нем — 2.8, 2.3 и
стадии. В сверхкритической воде (выше 374°С) по-
1.4 мас%. Бром, выделявшийся в виде HBr, сразу же
мимо толуола и этилбензола были получены бензол
растворялся в воде. При 225°С было удалено 18.5%
и циклоалканы, которые в свою очередь превраща-
органического брома, присутствующего в антипире-
лись в бензальдегид, ацетофенон, фенол/фенольные
нах, при 250°С — 39.6%, а при 275°С — уже 63.6%.
производные, нафталин, алифатические алканолы и
Полученный полукокс с низким атомным содержани-
цикланоны на третьей стадии.
ем брома можно в дальнейшем подвергать пиролизу,
Увеличение температуры частичной окислитель-
минимизируя выбросы бромированных соединений,
ной обработки способствовало повышению реак-
поскольку при обычном пиролизе бромированных
ционной способности кислорода, участвующего в
антипиренов при 850°С в инертной атмосфере более
системе, и протеканию побочных реакций, приводя-
99% брома выделяется в форме HBr, Br2 и бромиро-
щих к образованию производных фенола, нафталина,
ванных соединений. Образование полициклических
алифатических алканолов и цикланонов.
ароматических углеводородов и бромированных фе-
Дебромирование бромсодержащих антипиренов,
нолов снижалось с увеличением температуры об-
входящих в состав печатных плат, было проведено в
работки (225, 250 и 275°С), при этом основными
докритической воде в [31]. Как и в случае дехлори-
соединениями являлись 2-, 3-, 4-Br-фенолы (12.8-
рования поливинилхлорида, дебромирование анти-
16.9 мг на 1 кг бромированных печатных плат).
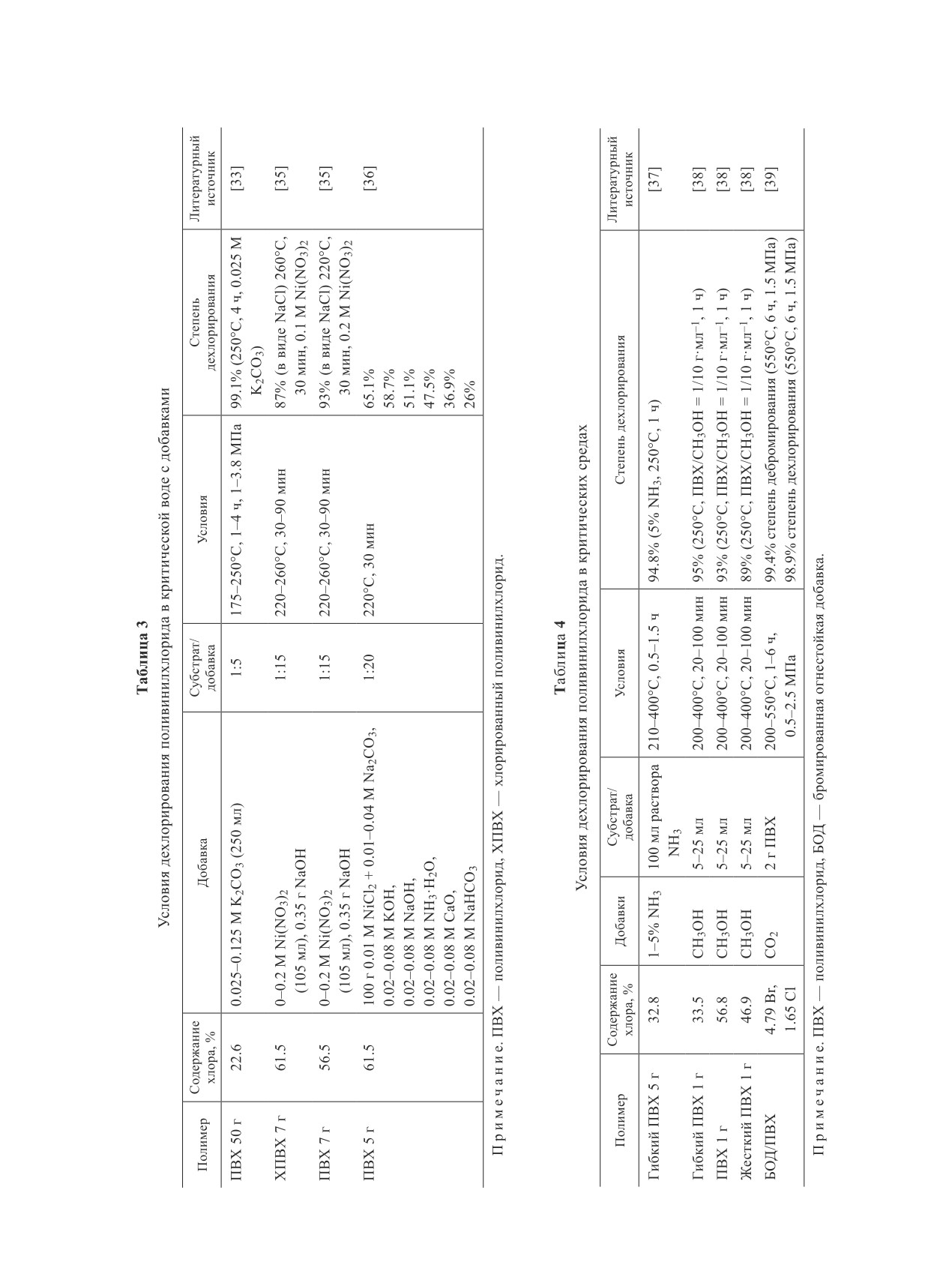
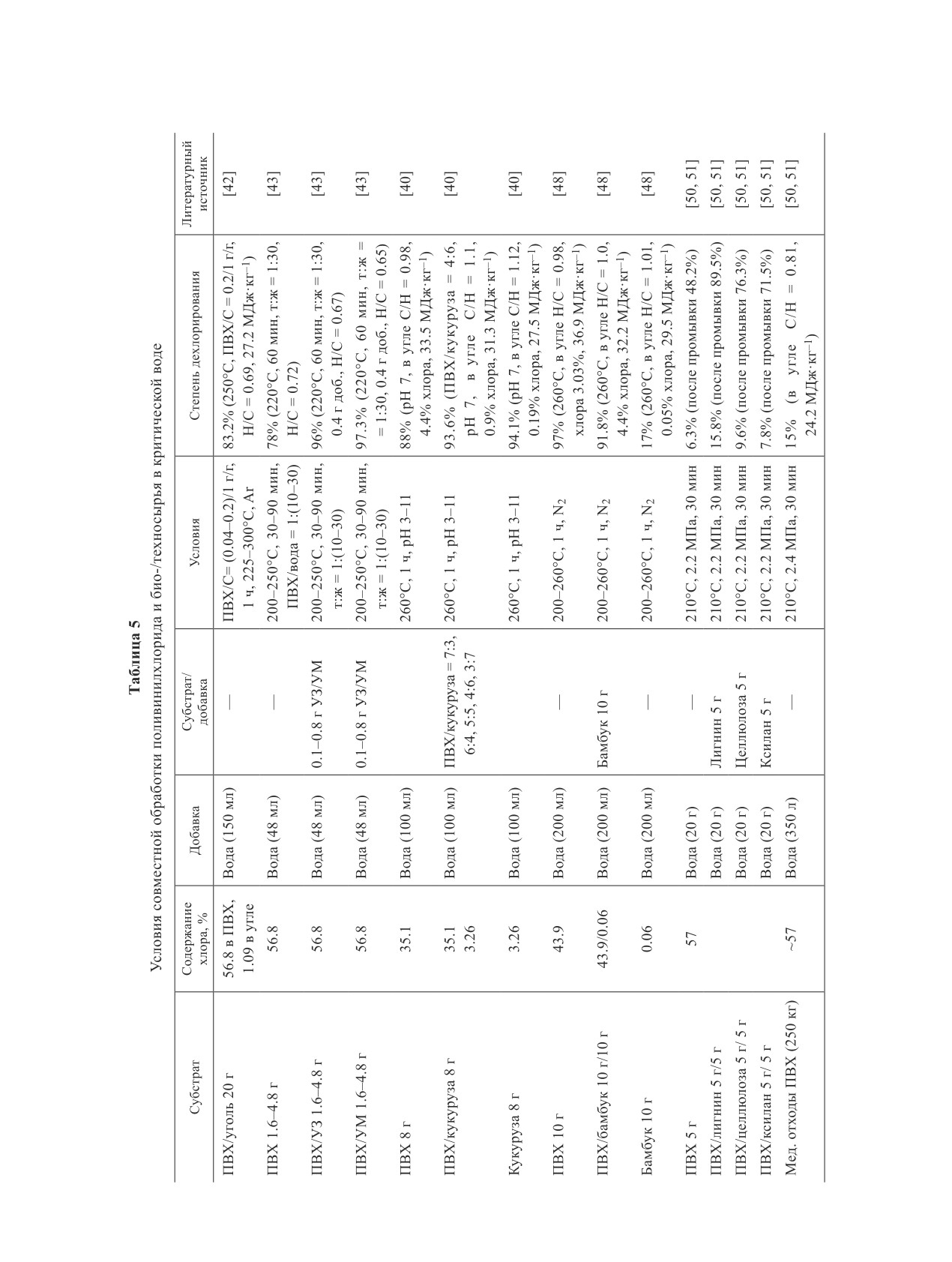
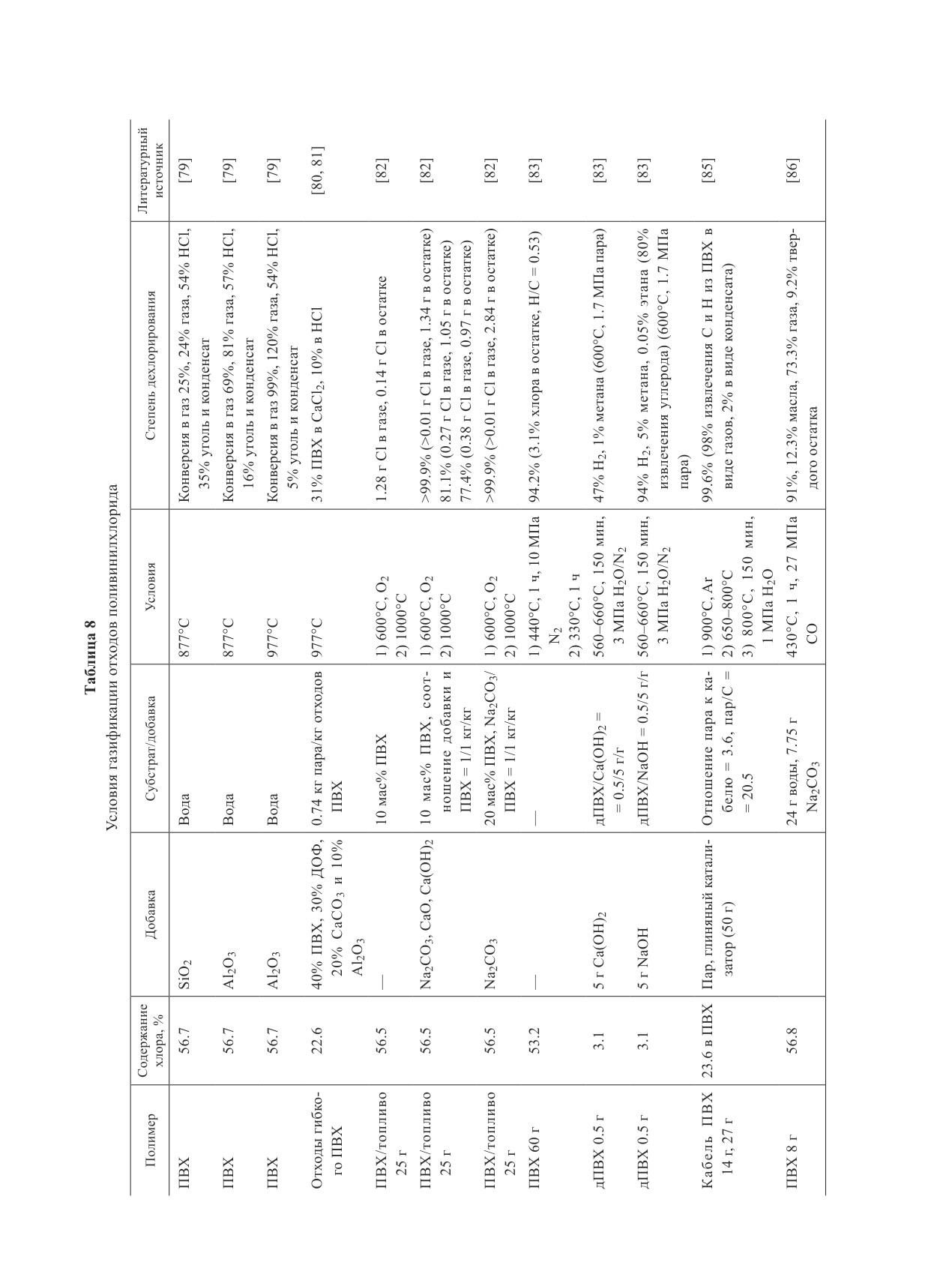
Таблица 2
Условия дехлорирования поливинилхлорида в критической воде
Содержание
Субстрат/
Литературный
Полимер
Добавка
Условия
Степень дехлорирования
хлора, %
добавка
источник
Смола ПВХ 0.5 г
56.7
Пар (0-75 об%)
170-250°С, 0-3 ч
82% (230°С, 50 об%, 3 ч)
[24]
76% (250°С, 50 об%, 1 ч)
Гибкий ПВХ 0.5 г
28.4
Пар (0-75 об%)
170-250°С, 0-3 ч
97% (230°С, 50 об%, 3 ч)
[24]
69% (250°С, 50 об%, 1 ч)
ПВХ 100 мг
56.6
Вода
3 мл воды
105-350°C, 1 ч
99% (350°C, 17 МПа,
[26]
0.13-17 МПа
1 ч)
ПВХ 10 г
56.9
Вода
90 мл воды
200-260°С, N2,
97% (260°C), в полукок-
[27]
1 ч
се 3.03% хлора
ПВХ 20 г
56.9
Вода
200 мл воды
220-280°С, N2,
99%
[28]
30-90 мин
ПВХ 10-200 мг
56.6
Вода
10 мл воды
200-600°С, 1.6-
99% (10-200 мг ПВХ,
[29]
55.7 МПа Н2О,
280°C, 1 ч, Ar)
Ar
ПВХ 25 мг
56.8
Вода
17 мл воды
180-260°С, 15 ч,
99% (235°C, 15 ч, Не)
[23]
Не
Гибкий ПВХ 3 г
32.8
Вода
200-300 мл
200-400°С, 1 ч,
95% (300°C, 1 ч, О2)
[30]
воды
ПВХ/О2 = 32/1
БПП 100 г
3.24 Br,
Вода
500 г воды
225-275°С, 2.5-
63.6% (275°С, 6 МПа)
[31]
0.067 Cl
6 МПа, 3 ч
Отходы ПП, ПЭ,
56.27 в ПВХ
Вода
100 мл воды
280°С, 1-2 ч, N2
—
[32]
ПВХ (20 г)
П р и мечан и е. ПВХ — поливинилхлорид, БПП — бромированные печатные платы, ПП — полипропилен, ПЭ —
полиэтилен.
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1381
Гидротермальная обработка отходов пластика,
изводные бензола и алифатические алканы и алкены
состоящего из полипропилена, полиэтилена и по-
за счет процессов циклизации, гидролиза и окисления
ливинилхлорида, при 280°С (2 ч) приводила лишь к
кратных связей частично растворенным в воде кис-
разложению и дехлорированию поливинилхлорида
лородом. При частичном окислении в докритической
[23]. Полипропилен и полиэтилен в результате дан-
воде основными продуктами являлись бензальдегид
ной обработки подвергались только изменению фи-
и ацетофенон, а в сверхкритической воде помимо них
зической формы: структура полимеров из гранулята
были образованы фенол и его производные, нафта-
переводилась в сшитую и блочную форму [32].
лин, алифатические алканолы и цикланоны [30].
Главным достоинством гидротермальной обра-
Дебромирование бромсодержащих антипиренов,
ботки чистого поливинилхлорида и его отходов в
входящих в состав технических отходов, в докритиче-
критической воде является отсутствие хлорирован-
ской воде позволило удалить до 63.6% органического
ных органических соединений в продуктах процес-
брома, получив полукокс с минимальным содержани-
са, поскольку практически весь органический хлор,
ем атомов Br 1.4 мас% [31].
удаляемый из поливинилхлорида, превращается в
Разложение чистого поливинилхлорида и его от-
растворимый в воде HCl. Докритическая вода дей-
ходов в водных средах с добавками. Добавление раз-
ствует в качестве реакционной среды и катализатора,
личных щелочных реагентов при дехлорировании по-
усиливающего растворение исходного сырья и его
ливинилхлорида в критических средах предполагало
реконденсацию в полукокс. В интервале температур
повышение эффективности удаления органического
180-280°C такая обработка применяется для произ-
хлора из полимера благодаря его большему связыва-
водства полукокса [26-29], который в дальнейшем
нию в щелочной среде.
можно использовать в качестве топлива. Так, напри-
Влияние K2CO3 в качестве щелочной добавки в
мер, в [27, 28] получали полукокс лучшего качества
гидротермальной обработке отходов поливинилхло-
по сравнению с битуминозным углем: при повы-
рида (полимерные покрытия кабеля, содержащие
шении температуры и длительности процесса был
82.28% поливинилхлорида и 17.2% полиэтилена)
образован продукт с большей теплотворной способ-
исследовалось в [33]. При изменении концентрации
ностью, до 37-38.5 МДж·кг-1.
от 0.025 до 0.125 моль·л-1 K2CO3 максимум дехло-
Обработка паром чистого поливинилхлорида и его
рирования отходов наблюдался в среде 0.05 моль·л-1
отходов [24] значительно улучшала качество полу-
раствора K2CO3 (200°С, 1-4 ч), что было впослед-
кокса по сравнению с обычным низкотемпературным
ствии выбрано в качестве оптимальной концентра-
дехлорированием.
ции. Повышение концентрации щелочной добавки
Разложение полимера при температуре выше
приводило, как и в [34], к блокированию процесса
350°С [29, 30] в сверхкритической воде приводило к
дехлорирования за счет дезактивации активных цен-
превращению полимера в ацетон, фенол, бензол, про-
тров на поверхности хлорид-ионами.
[―CH2―CHCl―] + K2CO3
[―CH CH―] + 2KCl + H2O + CO2.
С повышением температуры от 175 до 250°C сте-
С повышением температуры и времени реакции
пень дехлорирования поливинилхлорида возрастала
увеличивалась доля твердого остатка и соотношения
с 4.7 до 92.6% при соотношении K2CO3/Cl = 1:25
в нем С/Н. Так, при 250°С (4 ч) потеря массы соста-
(0.05 М K2CO3), что связано с полным удалением
вила 48.8%, половина от которой связана с удалением
органического хлора при данной температуре. С уве-
органического хлора из поливинилхлорида (0.05 М
личением времени реакции в 0.05 М растворе K2CO3
K2CO3). Теплотворная способность полученного по-
от 1 до 4 ч степень дехлорирования поливинилхло-
лукокса составила 34.51 МДж·кг-1, что предполагает
рида повышалась только при 250°С с 8 до 92.6%,
использование его в виде высококачественного то-
что связано с высоким давлением пара, приводяще-
плива для доменной печи или сырья для производства
го к увеличению пористости полимера, способст-
активированного угля [27].
вующей проникновению ОН-нуклеофилов и проте-
Добавление Ni2+-содержащей воды также спо-
канию быстрой карбонизации полимера [24]. При
собствовало значительному ускорению процесса
остальных температурах она не превышала 20-45%.
дехлорирования поливинилхлорида [35]. При 220°С
Уменьшив соотношение K2CO3/Cl до 1:50 (0.025 М
степень дехлорирования резко возрастала с 10.5 до
K2CO3), авторы смогли достичь 99.1% извлечения
59% после добавления 0.01 М Ni(NO3)2. С увели-
атомов хлора.
чением концентрации раствора Ni(NO3)2 эффектив-
1382
Захарян Е. М. и др.
ность дехлорирования росла: степень дехлорирова-
CaO и NaHCO3 проводили в [36]. Высокая степень
ния хлорированного и чистого поливинилхлорида
дехлорирования, составляющая 65.1%, наблюдалась
при добавлении 0.2 М раствора Ni2+ достигала 78
при использовании Na2CO3, самая низкая — 26% —
и 93% (220°С) соответственно. Добавление 0.1 М
при NaHCO3. Продукт, полученный при добавлении
Ni2+-содержащей воды при 220°С приводило к уда-
Na2CO3, имел структуру более пористую и рыхлую,
лению 76% органического хлора, а при 260°С — 87%
с большей площадью поверхности, что способство-
(табл. 3). Дальнейшее повышение температуры спо-
вало ускорению процесса дехлорирования поливи-
собствовало усилению процесса коррозии.
нилхлорида. Степень дехлорирования полимера па-
Ni2+ способствовал расширению пор в поливи-
дала в ряду Na2CO3 > KOH > NaOH > NH3·H2O >
нилхлориде и замене групп Cl- на OH- (схема 7),
> CaO > NaHCO3. При добавлении NaOH и KOH в
при этом он использовался в качестве катали-
Ni2+-содержащую воду степень дехлорирования сни-
затора, не расходуясь в данном процессе, что
жалась с повышением концентрации щелочи с 0.02
доказывало возможность повторного использования
до 0.08 моль·л-1: с 62 до 40.3% для NaOH и с 62.9
Ni2+-содержащей воды. Увеличение концентрации
до 40.7% для KOH, что связано с поверхностным за-
Ni2+ до 0.2 моль·л-1 не способствовало повышению
грязнением активной фазы в результате образования
эффективности дехлорирования поливинилхлорида,
частиц хлоридов металлов.
что обусловлено реакцией терминации, включающей
Выход твердого продукта составлял 63.4-79.6%
циклизацию и сшивание с ростом полиена, что пре-
в зависимости от природы добавки. Органический
дотвращало тем самым полное дехлорирование.
хлор, удаляемый из поливинилхлорида при его дехло-
Гидротермальное дехлорирование поливинилхло-
рировании в виде хлорид-анионов, способствовал
рида в докритической 0.01 М Ni2+-содержащей во-
снижению скорости извлечения твердого вещества.
де с добавлением Na2CO3, KOH, NaOH, NH3·H2O,
Большая часть органического хлора из поливи-
нилхлорида была превращена в водорастворимый
хлор в жидкой фазе, т. е. HCl, до 65% при добавлении
Схема 7
Na2CO3 и до 34% при добавлении NaHCO3. Доля
Механизм дехлорирования ПВХ в Ni2+-содержащей
воде
атомов хлора в твердых остатках составляла от 34
(Na2CO3) до 54% (NaHCO3). В ИК-спектрах твердого
остатка фиксировались пики, относящиеся не только
к сопряженным С С-связям, но и к ароматической
структуре, что свидетельствует о протекании реакции
циклизации/ароматизации.
Добавление щелочных реагентов, дополнительно
связывающих выделяющийся из поливинилхлорида
хлороводород, приводило к повышению эффектив-
ности процесса не только за счет более полного уда-
ления органического хлора, но и за счет снижения
температуры и сокращения длительности процесса.
При 220°С степень дехлорирования поливинилхлори-
да резко возрастала с 10.5 до 59% после добавления
0.01 М Ni(NO3)2 [35]. А добавление 0.2 М раствора
Ni(NO3)2 приводило к удалению 93% органического
хлора из поливинилхлорида (220°С, 30 мин), тогда
как в работе [27] сопоставимая степень дехлори-
рования (97%) без добавок достигалась при 260°С
(1 ч). В полупромышленном подходе (50 г отхо-
дов поливинилхлорида) авторам удалось получить
полукокс с высокой теплотворной способностью
(34.51 МДж·кг-1) и высокой степенью извлечения
хлора (99.1%) путем добавления карбоната калия
[33].
Разложение поливинилхлорида в других крити-
ческих средах. Гидротермальная обработка гибкого
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1383
поливинилхлорида в критическом 1-5%-ном водном
разницей, что при 400°C доля атомов хлора в полу-
растворе NH3 исследовалась в [37]. Эффективность
коксе составила 1.9%, а в воде — 98.1% (отходы 1).
дехлорирования возрастала с увеличением концен-
Благодаря существенной разнице температур ки-
трации аммиака и при низких температурах. Так,
пения представляется возможным отделение плас-
при 210°C степень дехлорирования увеличивалась с
тификаторов (дибутилфталата, диоктилфталата и
30.8 до 65.2% с ростом концентрации NH3 от 1 до 5%
диметилфталата) и стабилизаторов (гексадекановой
(табл. 4). При дальнейшем повышении температуры
и октадекановой кислот) от метанола с помощью
до 250°C степень дехлорирования достигала 94.8%
процесса дистилляции. Примечательным является тот
(5% NH3), а уже при 300°C наблюдалась 100%-ная
факт, что метанол можно повторно использовать в дан-
эффективность дехлорирования.
ном процессе, поскольку чистота регенеративного
При низкой температуре (210°С) содержание Cl
метанола достигает 99.5% после пятикратного цикла.
в твердом продукте и водной фазе составляло 34.8 и
Отходы пластиковых корпусов и электронного
65.2% соответственно (5% NH3, 1 ч). С повышением
дисплея, имеющие в своем составе поливинилхлорид
температуры реакции до 400°С доля атомов хлора в
и антипирен тетрабромбисфенола в качестве огне-
твердой фазе снизилась до 0.2%, а в водной, наобо-
стойкой добавки, обрабатывали сверхкритическим
рот, возросла до 99.8%.
CO2 [39]. Эффективность дебромирования варьиро-
Авторами [38] был разработан процесс дехлори-
валась от 1.1 (200°С, 3.5 ч, 0.5 МПа) до 99.4% (550°С,
рования медицинских отходов поливинилхлорида
6 ч, 1.5 МПа), эффективность дехлорирования — от 3
[трубка для переливания (отходы 1), коллектор для
до 98.9% в аналогичных условиях. Степени деброми-
мочи (отходы 2)] в околокритическом метаноле, дей-
рования и дехлорирования значительно повышались
ствующем в качестве реакционной среды и реагента.
с ростом температуры. Первоначальное давление
Как и в [26], резкое увеличение степени дехлориро-
2.5 МПа повышалось до 9.8 МПа при 550°С, что
вания (до 90%) наблюдалось при повышении тем-
означало достижение сверхкритического состояния
пературы до 250°С, а уже при 300°С она составила
системы, в которой достигается низкая вязкость, вы-
95%. Извлечение атомов хлора из образцов падало
сокий коэффициент переноса, высокая диффузионная
в ряду отходы 1 > чистый поливинилхлорид > от-
способность и высокая растворимость органических
ходы 2. Наличие пластификаторов, содержащихся
веществ. Кроме того, коэффициент набухания, пла-
в составе отходов 1 (гибкого поливинилхлорида),
стичность, реакционная способность сверхкрити-
приводило к снижению температуры дехлорирования
ческого CO2 повышались при высоких давлениях и
полимера, что способствовало практически полному
температурах, что приводило к значительному по-
удалению органического хлора уже при 250°С, в то
вышению эффективности дебромирования и дехло-
время как термостабилизаторы в отходах 2 (жест-
рирования.
ком поливинилхлориде) вели себя в качестве инги-
Выход твердого продукта увеличивался с темпе-
биторов, сдерживая удаление HCl из поливинилхло-
ратурой. Содержание атомов C в твердых продуктах
рида.
повысилось с 31.3 до 81.3% в интервале температур
Эффективность удаления органического хлора
375-550°С. Это подтверждает тот факт, что более
значительно увеличивалась с повышением соотноше-
высокие температуры способствовали образованию
ния твердого вещества и жидкости, что обусловлено
твердого углерода. Масло, полученное обработкой
влиянием давления внутри реактора. Молекулы кри-
сверхкритического CO2 при 375°С, в основном со-
тической жидкости с более высоким давлением могут
стояло из соединений, полученных путем разложения
легче диффундировать в частицы твердых отходов.
антипирена тетрабромбисфенола. С повышением
Кроме того, повышение давления внутри реактора
температуры до 550°С доля этих компонентов масла
также может способствовать диффузии газа HCl, т. е.
снижалась за счет преобразования ароматических
более высокое давление в реакционной системе мо-
углеводородов в углеродные материалы. В газовой
жет ускорить удаление HCl из поливинилхлорида и
фазе основными составляющими были диоксид угле-
способствовать процессу дегидрохлорирования. При
рода, водород, монооксид углерода, метан и этан, ко-
этом оптимальным соотношением твердого вещества
торые могут быть использованы в качестве топлива.
и жидкости с учетом расхода растворителя и эффек-
По сравнению с водной средой критические амми-
тивности оказалось 1:10 г:мл (степень дехлориро-
ак и метанол оказались более эффективными в дехло-
вания >90%). Зависимость атомного распределения
рировании отходов поливинилхлорида [37, 38]. Почти
хлора в твердой и жидкой фазах была аналогична
полное удаление органического хлора достигалось
зависимости, полученной в работе [37], с той лишь
уже при 250°С, тогда как в докритической воде это
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1385
происходило при 300-350°С. При этом доля атомов
уголь = 0.04/1 г/г и до 83.2% при соотношении поли-
хлора в полукоксе, полученном при обработке гиб-
винилхлорид/уголь = 0.2/1 г/г.
кого поливинилхлорида при 400°С, составляла 0.2%
Аналогичная зависимость степени выщелачивания
в случае аммиачной среды [37] и 1.9% — в метаноле
от соотношения субстрат/добавка наблюдалась и при
[38]. Дополнительным преимуществом обработки
удалении натрия. Наименьшая степень выщелачива-
отходов поливинилхлорида в метаноле является воз-
ния наблюдалась при 225°C (44.5-46.4%) и наимень-
можность его повторного использования, поскольку
шем массовом соотношении. Степень выщелачивания
чистота метанола даже после пятикратного цикла
повышалась с ростом температуры и массы поливи-
достигала 99.5%, а также возможность отделения
нилхлорида в смеси, до 83.4% при 300°С и соотно-
полученных пластификаторов и стабилизаторов в
шении поливинилхлорид/уголь = 0.2/1 г/г.
процессе разложения отходов поливинилхлорида
Максимальный массовый выход полукокса состав-
от метанола посредством дистилляции. Высокую
лял 91% при 225°С и с повышением температуры
эффективность дегалогенирования полимерных от-
снижался до 84% (300°С). При добавлении поли-
ходов показал и сверхкритический диоксид углерода
винилхлорида к высокощелочному углю и с увели-
благодаря высокой диффузионной способности и
чением его доли выход твердого остатка падал до
высокой растворимости в нем органических соеди-
75.4% (300°С), что обусловлено разложением по-
нений [39].
ливинилхлорида и взаимодействием со щелочным
углем. Содержание воды в полукоксе резко сокра-
щалось по сравнению с содержанием воды в необра-
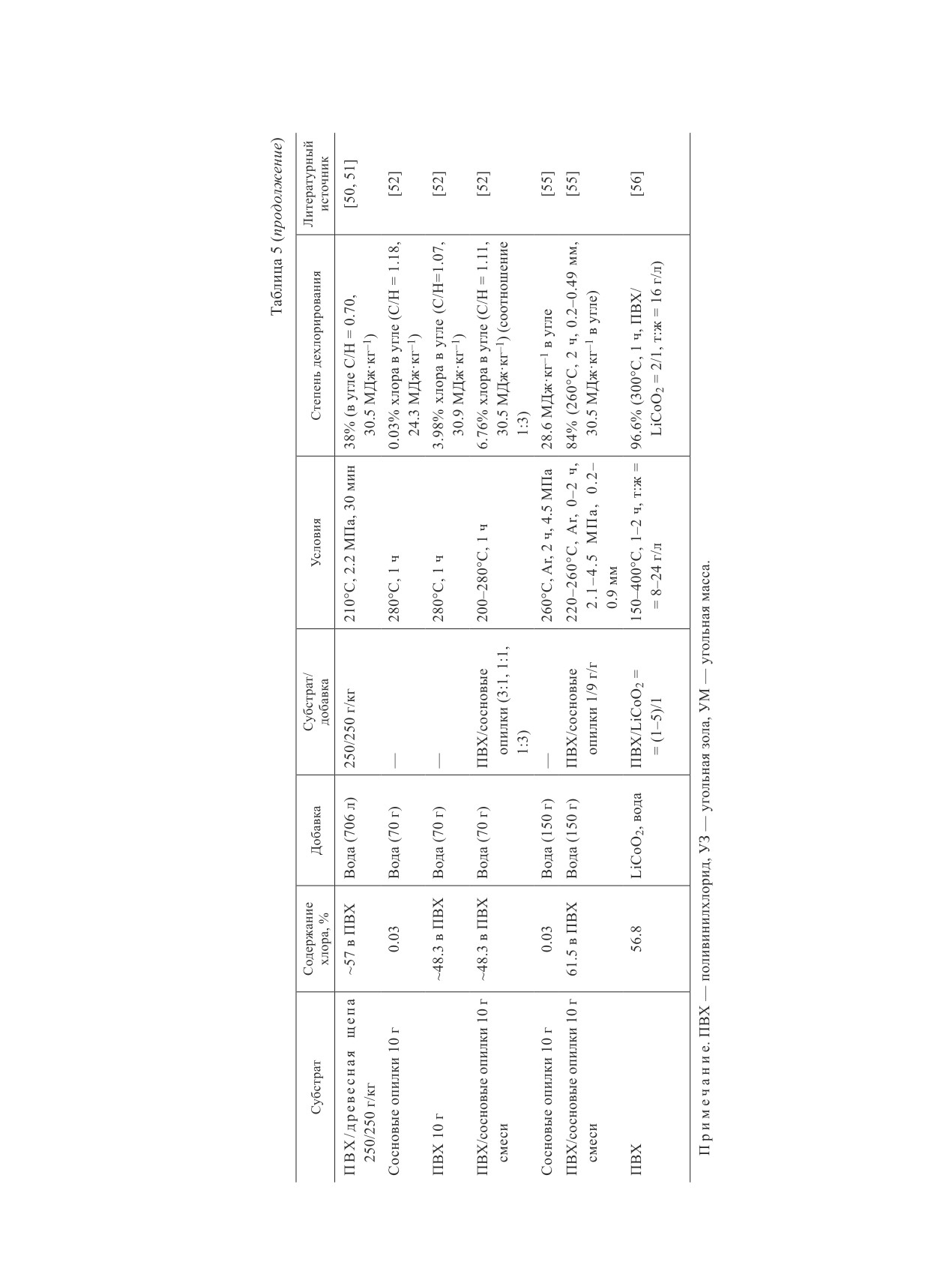
Совместная гидротермальная карбонизация
ботанном угле как следствие действия температуры.
поливинилхлорида в критических средах
Теплотворная способность угольного гидрокарбона
Совместная гидротермальная обработка, как и
увеличивалась с повышением температуры и соотно-
совместный пиролиз, позволяет в одном процессе
шения поливинилхлорид/уголь до 27-29 МДж·кг-1 по
переработать два различных вида отходов, поливи-
сравнению с теплотворной способностью необрабо-
нилхлорид и биосырье [40, 41], поливинилхлорид и
танного угля (24.3 МДж·кг-1), что позволяет исполь-
угольные компоненты [42, 43] или поливинилхлорид
зовать его в качестве синтетического топлива.
и технические отходы, тем самым снижая нагрузку
Совместная переработка поливинилхлорида с
на экологию.
угольной золой и угольной массой, используемыми
Авторами [42] был предложен процесс совмест-
в качестве усилителей процесса, в докритической
ной гидротермальной карбонизации высокощелоч-
воде исследовалась в [43]. Сравнение цвета твердого
ного угля и поливинилхлорида с целью удаления
остатка при обработке чистого поливинилхлорида
хлорида натрия. Доля атомов хлора в необработан-
и его смесей с угольными компонентами при 200°С
ном угле снижалась с 32.4 до 8.8% при увеличении
показало, что присутствие золы и угля способство-
отношения поливинилхлорид/уголь с 0.04 до 0.2 г/г.
вало снижению температуры дехлорирования по-
Степень дехлорирования чистого поливинилхлорида
ливинилхлорида и ускорению процесса. Данный
при 225°С составляла 78.8%, что было значительно
синергический эффект угольных добавок обуслов-
ниже, чем в работе [23], где практически 100%-ное
лен влиянием каталитических свойств оксидов SiO2,
удаление органического хлора достигалось при
Al2O3, Fe2O3, TiO2 и сорбирующих свойств щелоч-
235°С. Данный факт можно объяснить лишь дли-
ных CaO, K2O, входящих в состав угольных ком-
тельностью процесса: в работе [23] — 15 ч, в данном
понент. Большему удалению органического хлора
исследовании — 1 ч. С повышением температуры
из поливинилхлорида способствовали повышение
эффективность дехлорирования увеличивалась до
температуры и увеличение доли усилителя в смеси.
95.5 и 96% при 250 и 300°C. Небольшое различие в
Степень дехлорирования при добавлении угольной
степенях дехлорирования обусловлено тем, что боль-
золы/угольной массы достигала 96 и 97.3% (220°С)
шая часть хлороводорода выделялась уже при 250°C.
соответственно, что оказалось эффективнее крити-
Добавление высокощелочного угля к поливинилхло-
ческого аммиака [37] и Ni2+-содержащей воды [35].
риду приводило к значительному снижению степе-
С увеличением длительности процесса до 90 мин
ни дехлорирования с 78.8 до 20.8-22.4% при 225°С
извлечение атомов хлора снижается за счет усиления
в зависимости от массового соотношения поливи-
карбонизации полиена и образования пористой струк-
нилхлорид/уголь. Повышение температуры до 250°C
туры, облегчающей повторную фиксацию удаленного
способствовало резкому скачку дехлорирования до
хлора в остатке поливинилхлорида, т. е. атомы хлора
63% при массовом соотношении поливинилхлорид/
«закрываются» внутри.
1386
Захарян Е. М. и др.
Доля атомов О в остатках чистого поливинилхло-
люлозы до ксилозы с последующим образованием
рида, смеси поливинилхлорид/угольная зола и по-
фурфурола [45]. Длинноцепочечная целлюлоза при
ливинилхлорид/угольная масса составляла 20, 20.8
220°С расщепляется до глюкозы и затем до 5-гек-
и 21.8% соответственно, что доказывает протека-
саметилфурфурола или фурфурола [46]. В процессе
ние нуклеофильного замещения хлорид-группы на
гидролиза лигнина при 250°С образуются фенольные
гидроксильную из воды (схема 8) во время гидро-
соединения [47]. Таким образом, гидроксильная груп-
термальной обработки. Наличие угольных добавок
па в образованных фурфуролах и фенолах замещается
способствовало внутримолекулярной дегидратации
на Cl- в поливинилхлориде, тем самым способствуя
и окислению продуктов гидроксильного замещения
процессу дехлорирования полимера.
поливинилхлорида, в результате чего были образова-
Исследование влияния среды водного раствора по-
ны более стабильные углерод-кислородные двойные
казало, что среди всех добавок наиболее эффективной
связи в структуре. В ходе процесса при всех темпе-
оказалась лимонная кислота с максимальной эффек-
ратурах жидких органических продуктов образовано
тивностью дехлорирования, равной 95.2% при рН 5,
не было, а основная часть хлора была переведена в
самая низкая степень дехлорирования — 87.1% при
водную фазу в виде хлорид-анионов.
рН 11 при добавлении Na2CO3 (табл. 5). Аналогичная
Положительный синергический эффект на дехло-
зависимость наблюдалась в работе [33], в которой эф-
рирование поливинилхлорида в докритической
фективность дехлорирования сначала увеличивалась,
воде оказала и добавка биосырья (кукуруза) [40].
а затем уменьшалась с повышением концентрации
Эффективность дехлорирования смеси кукурузного
Na2CO3 от 0.01 до 0.04 моль·л-1.
биосырья и поливинилхлорида была выше (88-94%),
Доля атомов углерода в полученном полукоксе
чем при разложении поливинилхлорида (87.4%).
варьировалась от 68.8 до 75.9%. Уменьшение со-
Кукурузное сырье в основном состоит из целлюло-
держания биосырья в смеси приводило к резкому
зы (C6H10O5)n, гемицеллюлозы (C5H8O4)n и лигнина
снижению доли атомов углерода, а увеличение доли
[44]. Во время процесса гидротермальной карбони-
биосырья способствовало снижению выхода полу-
зации происходит растворение и гидролиз гемицел-
кокса, поскольку биосырье легче разлагается на газы
Схема 8
Предполагаемые пути дехлорирования поливинилхлорида в докритической воде
ПВХ — поливинилхлорид, УЗ — угольная зола, УМ — угольная масса
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1387
и гидролизат, чем поливинилхлорид. Теплотворная
туры процесса при соотношении поливинилхлорид/
способность полученного полукокса достигала 29.2-
сосновые опилки, равном 1:1, росла эффективность
32.8 МДж·кг-1.
дехлорирования и достигала 93% при 280°С, при этом
В отличие от добавки кукурузы [40] добавка бам-
выход гидрокарбона снижался с 72.6 (200°С) до 42.6%
бука способствовала ускорению процесса дехлориро-
(280°С). С повышением доли поливинилхлорида в
вания поливинилхлорида только при 200°С, а не во
смеси с опилками выход полукокса увеличивался с 44.4
всем температурном диапазоне [48]. При 230 и 260°C
до 51%, при этом повышение доли соляной кислоты в
степени дехлорирования смеси поливинилхлорид/
жидкой фазе приводило к снижению выхода полукокса.
бамбук были ниже, чем при гидротермальной об-
Структура частиц полукокса, полученного из сос-
работке чистого поливинилхлорида, что свидетель-
новых опилок, была однородная с гладкой поверх-
ствует об отрицательном синергическом эффекте
ностью, в то время как у дехлорированного поливи-
данной биомассы. Это связано с тем, что получен-
нилхлорида, наоборот, неоднородна и многослойна.
ные посредством реконденсации и полимеризации
При добавлении поливинилхлорида к опилкам на-
нерастворимого лигнина при высокой температуре
блюдалась агрегация наночастиц, размер которых
фенольные гидрокарбоны [49] покрывают поверх-
повышался с увеличением доли поливинилхлорида
ность образующегося полукокса поливинилхлорида,
в смеси. Размер пор/пористости в полукоксе связан
забивая его поры, тем самым затрудняя удаление
с гидролизом гемицеллюлозы и лигнина [41, 53], в
«внутреннего» органического хлора жидкой фазой.
ходе процесса воздуху легче получить доступ к вну-
С повышением температуры процесса, как и ожида-
тренней структуре полукокса. Исследование влияния
лось, снижался выход полукокса до 38-42% и улуч-
температуры на дехлорирование поливинилхлорида в
шались его топливные свойства. По теплотворной
смеси показало, что наименьшее содержание органи-
способности гидрокарбон, полученный из бамбука,
ческого хлора в гидрокарбоне было достигнуто при
был аналогичен бурому углю, тогда как гидрокарбон
280°С и составляло 3.98%.
из поливинилхлорида и смеси поливинилхлорид/
Основными компонентами жидких продуктов при
бамбук — полибитуминозному.
гидротермальной обработке опилок являлись фур-
Исследование влияния модельных компонентов
фурол (32.5%) и 5-гидроксиметилфурфурол (15.3%),
биомассы на дехлорирование поливинилхлорида по-
полученные посредством гидролиза лингноцеллюлоз-
казало, что большую эффективность проявлял лигнин
ной биомассы [46] с дальнейшей дегидратацией и
[50, 51]. Степень дехлорирования падала в ряду лиг-
изомеризацией. Поскольку жесткий поливинилхло-
нин/поливинилхлорид > целлюлоза/поливинилхло-
рид содержит в своем составе большое количество
рид > гемицеллюлоза/поливинилхлорид > поливи-
пластификатора, основными компонентами жидких
нилхлорид с 15.8 до 6.3% при 210°C. Дальнейшее
продуктов оказались продукты гидролиза и пере-
промывание водой полученных гидрокарбонов спо-
этерификации диоктилфталата — 2-этил-1-гексанол
собствовало повышению эффективности от 48.2
(26%) и фенол (9.6%). При совместной обработке
(поливинилхлорид) до 89.5% (поливинилхлорид/
поливинилхлорида и биосырья 5-гидроксиметил-
лигнин).
фурфурол, полученный из лигнина, дополнительно
Совместная гидротермальная обработка меди-
дегидратируется с образованием левулиновой и му-
цинских отходов поливинилхлорида с древесной
равьиной кислот, которые в свою очередь вступают в
щепой в пилотной установке промышленного масш-
реакцию этерификации со спиртами до превращения
таба (масса смеси отходов составила 500 кг) приве-
в метиллевулинат (49.9%) [54].
ла к удалению 38% органического хлора из поли-
Влияние температуры, времени процесса и раз-
винилхлорида, тогда как степень дехлорирования
мера частиц сосновых опилок на гидротермальное
чистого поливинилхлорида составила всего 15% в
дехлорирование поливинилхлорида при соотношении
тех же условиях. Дальнейшее промывание дистил-
поливинилхлорид/опилки, равном 1:9, исследовали
лированной водой повышало эффективность до 80%,
в [55]. Степень дехлорирования, как и в работах [40,
а конденсированной — до 83%. При добавлении дре-
43, 52], увеличивалась с повышением температуры
весной щепы к поливинилхлориду теплотворная спо-
и длительности процесса, а в данном исследовании
собность полученного полукокса повышалась с 24.2
еще и с уменьшением размера частиц биосырья за
до 30.5 МДж·кг-1.
счет тепло- и массопереноса: степень дехлорирова-
Влияние добавки сосновых опилок при дехлориро-
ния составляла 79.1% при размере частиц опилок,
вании жесткого поливинилхлорида в докритической
равном 0.22-0.49 мм, и 71.1 — при размере, равном
воде исследовалось в [52]. С повышением темпера-
0.6-0.9 мм (260°С, 30 мин).
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1389
1390
Захарян Е. М. и др.
Увеличение температуры, времени реакции и
низкомолекулярных соединений, полученных путем
размера частиц способствовало не только сниже-
разрыва получающихся полиенов.
нию числа кислородсодержащих функциональных
Основным преимуществом совместной гидротер-
групп, приводящему к ослаблению абсорбционной
мальной обработки, как и совместного пиролиза, яв-
способности гидрокарбона по отношению к неорга-
ляется переработка/утилизация двух различных видов
ническим соединениям металлов, но и повышению
отходов, поливинилхлорида и биосырья [40-55] либо
теплотворной способности (до 29.4-30.2 МДж·кг-1),
технических отходов [56]. Добавление угольных ком-
аналогичной битуминозному углю. Добавление по-
понент (высокощелочной уголь [42], угольные зола и
ливинилхлорида в опилки повысило кислотность
масса [43]) и биомассы (кукуруза [40], бамбук [48],
массы, что способствовало эффективности удаления
древесная щепа [50, 51] и сосновые опилки [52, 55])
металлов K, Na, Al, Ca и Mg.
способствовало снижению температуры разложения
Совместную гидротермальную обработку отра-
поливинилхлорида и значительному ускорению про-
ботанных литий-ионных батареек (LiB) и поливи-
цесса его дехлорирования. Совместная переработка
нилхлорида в докритической и сверхкритической
поливинилхлорида и высокощелочного угля [42] и
воде проводили в [56]. Уникальность данного метода
литий-ионных батареек [56] приводила к одновремен-
заключается в том, что выделяющийся хлороводород
ному дехлорированию поливинилхлорида и выщела-
выщелачивает металл в критической воде, не образуя
чиванию металлов из данных отходов с образованием
при этом хлорорганических соединений.
соответствующих хлоридов. Во всех исследованиях
Эффективность выщелачивания Co и Li (до 10%)
увеличению эффективности удаления органического
была незначительна при температурах совместной
хлора из поливинилхлорида способствовало повы-
обработки ниже 200°C за счет неэффективного дехло-
шение температуры, длительности процесса, а также
рирования поливинилхлорида при этих температурах.
снижение размера частиц биосырья [55] и подкис-
Удаление Co и Li резко возрастало до 61.4 и 93.9%
ление реакционной среды [40]. Полученные таким
уже при 250°C благодаря их взаимодействию с со-
образом гидрокарбоны по теплотворной способности
ляной кислотой, полученной при растворении в
аналогичны битуминозному углю, что предполагает
воде выделяющегося HCl из поливинилхлорида.
их использование в качестве твердого топлива.
Поскольку полное дехлорирование поливинилхлори-
да происходит, как правило, при 350°C [57], при этой
Специфические способы дегидрохлорирования
температуре удалялось максимальное количество Co
поливинилхлорида
(86.9%) и Li (98.6%). Дальнейшее повышение темпе-
ратуры ускоряло гидролиз LiCl и CoCl2, тем самым
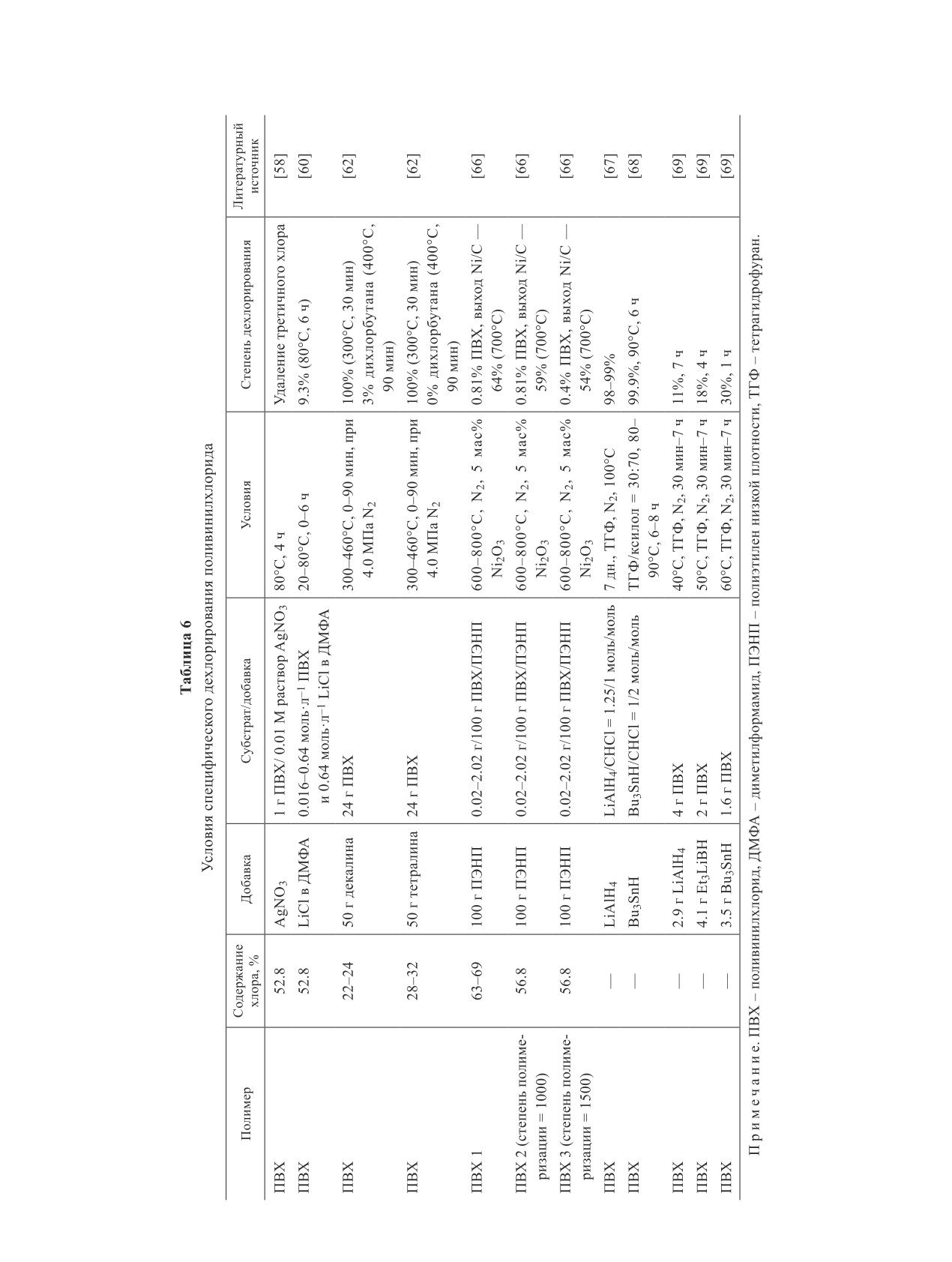
Авторы [58] в своем исследовании показали,
ухудшая выщелачивание металлов.
что AgNO3 в водном растворе взаимодействует ис-
Небольшой атомный радиус, высокие поляризуе-
ключительно с хлораллильными группами в поли-
мость и активность Li в кристаллах LiCoO2 способ-
винилхлориде и его низкомолекулярных фракциях,
ствовали его взаимодействию с Cl- с образованием
растворенных в хлороформе и ацетоне, т. е. AgNO3
LiCl, при этом его выщелачивание превышало 90%
не реагирует с хлором, расположенным в концевых
при всех соотношениях поливинилхлорид/LiCoO2, в
группах типа —СН2—СН СН—СН2С1 (схема 9).
то время как до 90% Co удалялось при соотношении
Термическое разложение поливинилхлорида в
поливинилхлорид/LiCoO2 = (3-5)/1 (350°C, 1 ч), т. е.
диметилформамиде проводили в [59]. Во время раз-
увеличение массового соотношения поливинилхлори-
ложения газообразный HCl не выделялся: он рас-
да в смеси способствовало образованию избыточного
творялся в диметилформамиде, превращаясь в ком-
количества HCl и, как следствие, выщелачиванию
плекс, разлагающийся с образованием гидрохлорида
обоих металлов в докритическую воду.
диметиламина (схема 10), который в свою очередь
Рентгенограммы твердых продуктов после сов-
оказался катализатором дехлорирования поливи-
местной обработки при 350°С показали появление
нилхлорида.
аморфного дифракционного пика при 10-30°С —
При использовании диметилформамида в каче-
остатка графита из катодного материала. Основными
стве растворителя появляются полиеновые структу-
органическими продуктами являлись производные
ры, состоящие из приблизительно 13 сопряженных
бензола, образованные согласно механизму [27] по
двойных связей (максимум поглощения при 478 нм)
межмолекулярной и внутримолекулярной циклизации
и 4-8 двойных связей (максимум поглощения при
Дильса-Альдера, гидролиза, пиролиза и окисления
320-340 нм).
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1391
Схема 9
Дехлорирование поливинилхлорида
Схема 10
Образование гидрохлорида диметиламина
Добавление LiC1 к поливинилхлориду в диметил-
в качестве катализаторов, значительно повышая ско-
формамиде [60] позволило снизить температуру про-
рость дехлорирования, в 20 и 30 раз соответственно.
цесса до 20-80°С (схема 11). Оптическое поглощение
Термическое разложение поливинилхлорида в тет-
характеризовало распределение длины полиеновой
ралине приводило к незначительному образованию
последовательности; можно предположить, что об-
хлорорганических соединений, что свидетельствует
разуется большее число полиенов и сопряженных
о предотвращении хлорирования вакуумных остатков
двойных связей.
и жидких продуктов благодаря лабильности водорода
С использованием LiCl в чистом диметилформа-
в структуре тетралина, при этом наблюдался высокий
миде были получены продукты фиолетового цвета с
выход жидкого продукта и высокое отношение Н/С в
пиком поглощения при 540 нм, а с LiBr — пурпурно-
вакуумном остатке [62]. В качестве основного хлор-
го с пиком при 450 нм.
органического соединения был получен 1,4-дихлор-
Исследование влияния различных добавок на
бутан. Полное дехлорирование поливинилхлорида
скорость дехлорирования поливинилхлорида в ди-
в тетралине и декалине завершалось уже при 300°С
метилформамиде показало [61], что при увеличе-
(30 мин), а максимальный выход HCl достигался при
нии концентрации диметилформамида в 10 раз до
400°С.
1.6 моль·л-1 скорость дехлорирования поливинилхло-
Доля вакуумного остатка в декалине составляла
рида увеличилась в 3 раза (120°С, 2 ч). Диметиламин,
40-50, газообразных продуктов — 10-15%, среди
серная кислота, хлориды цинка и кадмия действовали
которых доля HCl — 35-50% (300°С), которая повы-
в качестве ингибиторов, в то время как HCl и NaCl —
шалась с увеличением времени реакции. Выход ор-
ганического хлора из вакуумного остатка был более
Схема 11
8% при 300°С, быстро снижающийся с увеличением
длительности процесса. Основным органическим
Механизм взаимодействия поливинилхлорида
с LiCl в диметилформамиде
компонентом жидкого продукта являлся бензол, до
2.4 мас% за 30 мин. Доля вакуумного остатка в тетра-
лине составляла 70%, газообразные продукты, как и
в декалине, — 10-15%, доля HCl — 10% при 300°С
(15 мин), повышающаяся с увеличением времени
реакции до 50% (90 мин).
Влияние О3 на кинетику и процесс деградации
поливинилхлорида было изучено в [63]. Молекулы
озона, полученного посредством воздействия элект-
рического разряда на кислород, взаимодействовали
1392
Захарян Е. М. и др.
с насыщенными и ненасыщенными звеньями поли-
новительной электрохимической деградации при
винилхлорида, причем продукты в первом случае
сравнительно низких отрицательных потенциалах,
усиливали процесс дехлорирования, а во втором —
что приводило к дегалогенированию макромолекул,
никак не влияли. Предварительная обработка поливи-
распаду радикала и образованию полимера с трехмер-
нилхлорида озоном при 80°С способствовала значи-
ной структурой на поверхности катодного материала
тельному повышению скорости выделения HCl при
(схема 12). Электролиз чистого поливинилхлорида и
разложении полимера в атмосфере N2 (Т = 175°C).
поливинилхлорида, пластифицированного диоктил-
Авторы [64] проводили дехлорирование твердого
фталатом, проводили при катодных потенциалах, кон-
поливинилхлорида и поливинилхлорида, растворен-
тролируемых в диапазоне E = -(1.6-2.5) В, в растворе
ного в диметилформамиде, с помощью электролиза
диметилформамида с добавлением 0.1 М раствора
(10-12 ч). Поливинилхлорид подвергался восста-
иодида тетрабутиламмония.
Схема 12
Механизм электролиза поливинилхлорида
Растворенный и мелкодисперсный непластифи-
разложить пленки по истечении всего 30 дней (потеря
цированный поливинилхлорид восстанавливали на
массы до 19%).
платиновом и стеклоуглеродном катодном материа-
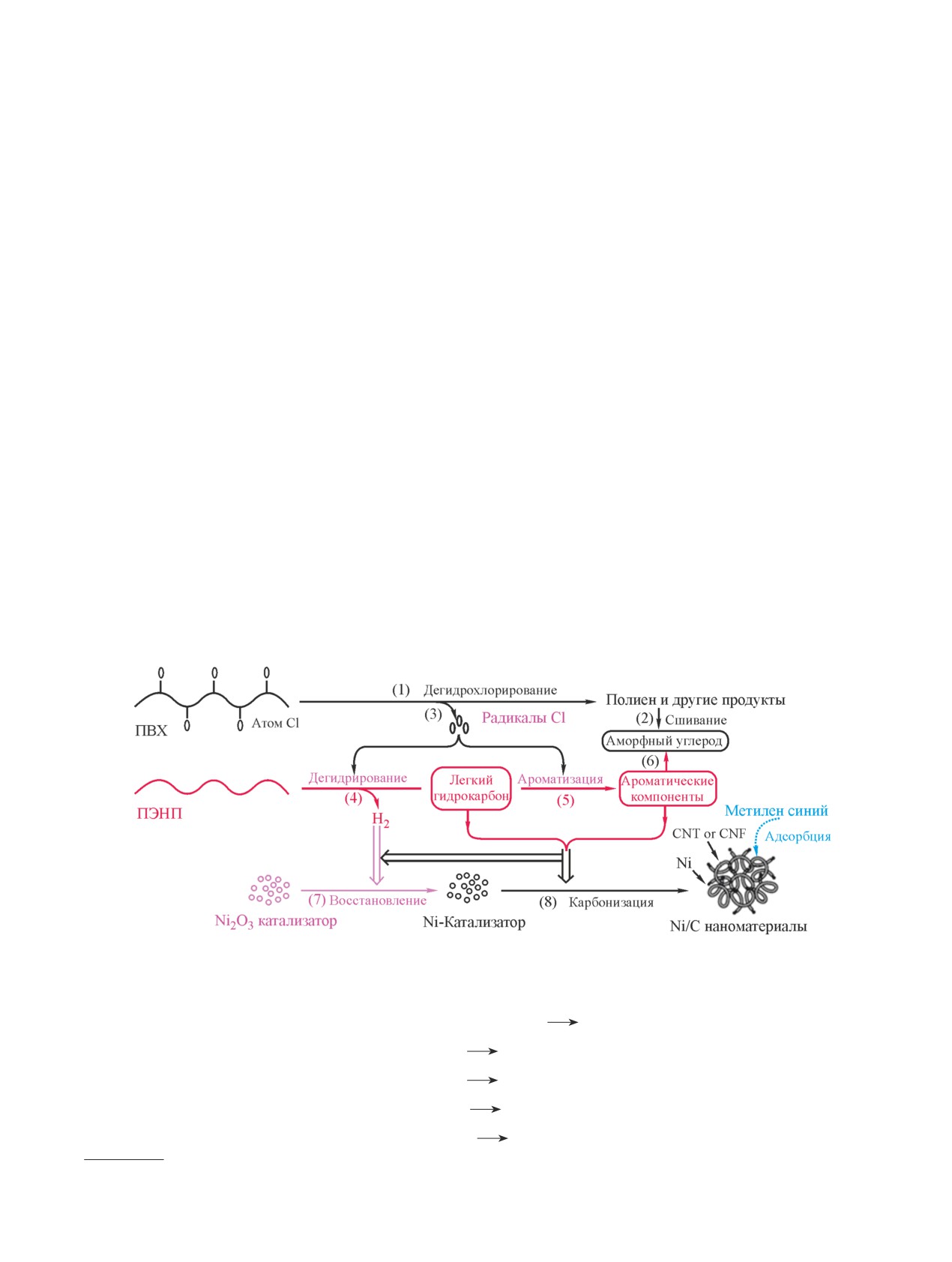
Путем каталитической карбонизации полиэтилена
лах с Е = -(2.0-2.2) В. Последовательно протекают
низкой плотности над Ni2O3 в присутствии различ-
два процесса: деградация (область кристаллического
ных видов смолы поливинилхлорида получали маг-
полимера) и вторичная полимеризация — сшивание
нитные наноматериалы Ni/C [66]. Независимо от типа
макроцепей (аморфная область).
смолы поливинилхлорида (табл. 6) выход углеродно-
При электролизе поливинилхлорида, пластифи-
го наноматериала сначала резко увеличивался, а затем
цированного диоктилфталатом, наблюдаются изме-
снижался в зависимости от количества добавленного
нения, характерные и для непластифицированного
поливинилхлорида (700°С). Выход магнитного ма-
полимера, но потенциал электролиза уменьшали до
териала без добавления поливинилхлорида в смесь
Е = -(1.6-1.8) В, что связано с электролизом диок-
полиэтилена и оксида никеля не превышал и 5%, что
тилфталата.
доказывает положительное влияние добавок поли-
Целью работы [65] было изучение биоразлагаю-
винилхлорида на выход данного продукта, которое,
щих cвойств пяти штаммов бактерий [Pseudomonas
возможно, обусловлено катализирующим действием
chlororaphis (DSM 50083), Pseudomonas citronellolis
выделяющегося HCl при разложении смолы поливи-
(DSM 50332), Bacillus subtilis (DSM 15029), Bacillus
нилхлорида на карбонизацию полиэтилена.
flexus (DSM 1320) и Chelatococcus daeguensis (DSM
Продукт карбонизации (700°С) представлял со-
22069)] в отношении полимерных пленок (полиэти-
бой длинноцепочечный углерод трубчатой формы
лен, полипропилен, полистирол, поливинилхлорид)
с небольшим количеством аморфного углерода при
в аэробных условиях.
небольших добавках поливинилхлорида. Наружный
Отбор проб проводился после 45- и 90-дневной
диаметр полученных углеродных наноматериалов
инкубации для оценки роста бактерий, формирования
варьировался в диапазоне 40-60 нм. При увеличении
биопленки и биодеградации пленки. Только штаммы
содержания поливинилхлорида до 1.62 мас% углерод-
P. citronellolis и B. Flexus проявили биоразлагающую
ные наноматериалы состояли в основном из коротких
способность в отношении пленок и порошка поливи-
углеродистых цепей и аморфного углерода, тогда как
нилхлорида. Уменьшение средней молекулярной мас-
полученный продукт в отсутствие поливинилхлорида
сы (Mn) от 100 до 90.87 ± 4.54 и 93.48 ± 4.67% спустя
являлся аморфным углеродом.
90 дней с P. citronellolis и B. Flexus соответственно
Аналогично результатам, полученным при исполь-
говорит о способности штаммов атаковать поливи-
зовании пиролизованных материалов при 700°C, не-
нилхлорид и расщеплять некоторые цепи, ведущей
зависимо от вида смолы поливинилхлорида выходы
к образованию более мелких фрагментов. Штамм
углеродного наноматериала посредством пиролиза
P. citronellolis с добавлением глюкозы смог частично
при 600 и 800°С сначала увеличивались, достигая
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1393
1394
Захарян Е. М. и др.
максимального значения, а затем уменьшались при
материал проявил высокую адсорбционную способ-
дальнейшем добавлении смолы поливинилхлорида.
ность в удалении красителя метиленового синего из
Высокий выход углеродных материалов при 600°С
воды (до 165.5~175.2 мг·г-1), что предполагает его
при более низком содержании поливинилхлорида
применение в качестве хорошего адсорбента в очист-
в смеси по сравнению с выходом продуктов, полу-
ке сточных вод.
ченных при пиролизе при 700°С, связан с тем, что
Восстановительное дехлорирование поливи-
снижение температуры пиролиза замедлило скорость
нилхлорида авторы [67-69] проводили в присутствии
разложения поливинилхлорида смолы на HCl и поли-
гидридов LiAlH4, Et3LiBH и Bu3SnH. Используя
ен и одновременно скорость диффузии выделяемого
LiAlH4 в качестве восстановителя поливинилхло-
хлороводорода, большее количество которого способ-
рида, авторы [67] добились 98-99% восстановления
ствует дегидрированию и ароматизации продуктов
полимера.
разложенного полиэтилена низкой плотности. При
При замене восстановителя на Bu3SnH и добав-
низком содержании поливинилхлорида дегидриро-
лении к тетрагидрофурану растворителя ксилола ав-
вание преобладает над ароматизацией, что повы-
торам [68] удалось дехлорировать поливинилхлорид
шает долю газообразных продуктов. При высоком
на 99.9% за 8 ч (80°С). Поскольку восстановительное
содержании поливинилхлорида, наоборот, основным
дехлорирование поливинилхлорида протекало по
процессом является ароматизация с преобладающими
радикальному пути (схема 14), авторы использовали
ароматическими соединениями в составе продуктов
азобисизобутиронитрил в качестве инициатора про-
процесса (схема 13).
цесса.
Повышение температуры реакции способствовало
Содержание органического хлора в начале процес-
повышению каталитической активности никелевых
са стремительно падало, затем уменьшалась скорость
катализаторов. Полученный магнитный Ni/C нано-
реакции, что обусловлено быстрым распадом инициа-
Схема 13
Механизм карбонизации полиэтилена низкой плотности в присутствии поливинилхлорида
и Ni2O3 (600-800°С, N2)*
Схема 14
Радикальный механизм восстановления поливинилхлорида
Азобисизобутиронитрил
R*
R* + Bu3SnH
RН + Bu3Sn*
Bu3Sn* + [—CH2—CHCl—]
Bu3SnCl + [—CH2—C*H—]
[—CH2—C*H—] + Bu3SnH
[—CH2—CH2—] + Bu3Sn*
Bu3Sn* + [—CH2—CHCl—]
* Разрешение на публикацию получено 17.07.2020. © 2015 Elsevier.
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1395
тора, скорость полураспада которого при 80°C состав-
в 3 раза при повышении температуры до 440°С в
ляет 1 ч. Добавление второй порции инициатора спу-
некаталитическом процессе и в 560 раз под действием
стя 4 ч реакции приводило к удалению оставшегося
Pd/Al2O3 (350°С), снижающееся до 182 ppm при
органического хлора в поливинилхлориде. При повы-
440°С. Наличие катализатора способствовало росту
шении температуры реакции до 90°С восстановление
доли углеводородов С1-С4 в газовой фазе до 3.9-
завершалось спустя 6 ч, содержание атомов хлора в
7.9 мол%, тогда как повышение температуры бла-
извлеченном продукте составляло менее 0.1%.
гоприятствовало образованию метана и в меньшей
Сравнение восстановительных свойств гидри-
степени этана.
дов LiAlH4, Et3LiBH и Bu3SnH проводилось в [69].
Авторы [71] проводили гидродехлорирование
Степень дехлорирования поливинилхлорида в при-
предварительно дехлорированного поливинилхло-
сутствии Bu3SnH достигала 30% (1 ч), тогда как при
рида, в котором благодаря термическому разложению
Et3LiBH — 18% (4 ч) и при LiAlH4 — 11% (7 ч).
удалялось до 40% HCl, в тетралине (300°С, 24 ч [72]).
Наибольшую активность проявил Bu3SnH, что обу-
Водород, переносимый из растворителя и газообраз-
словлено не только природой гидрида, но и темпера-
ного Н2, способствует большему разложению дегид-
турой реакции.
рохлорированного поливинихлорида и снижению ко-
личества остаточного хлора в продуктах разложения.
Продуктами процесса являлись газы, жидкие
Гидродехлорирование поливинилхлорида
масла, гексан-растворимые и гексан-нерастворимые
и хлорсодержащих отходов
соединения и остаток. При 440°С доли гексан-раст-
Каталитическое гидродегалогенирование — это
воримых соединений (20.7 и 20.8%) и гексан-нераст-
«зеленый» подход к удалению органического хлора
воримых соединений (48.8 и 46.4%) в присутствии
из хлорсодержащих соединений и полимеров, вклю-
NiMo/Al2O3 и в его отсутствие оказались практиче-
чающий водородное расщепление, т. е. гидрогенолиз,
ски равными, существенная разница заключалась
одной или нескольких связей C—X, где Х — галоген,
в выходе других жидких продуктов и остатка, доля
благодаря чему снижается токсичность образуемо-
которого при добавлении NiMo/Al2O3 не превышала
го продукта и становится возможным производство
3.4%. Это говорит о том, что катализатор способству-
повторно используемого сырья. В качестве донора
ет производству жидкого продукта. Соотношение Н/С
водорода может использоваться газообразный Н2,
фракций гексан-растворимых и гексан-нераствори-
растворитель, например тетралин, смесь СО/Н2О и
мых соединений в присутствии катализатора были
низкокипящая фракция (вакуумный газойль).
немногим выше, чем в его отсутствие.
Повышение температуры процесса (470°С) спо-
RX + Н2
R—H + HX.
собствовало снижению доли гексан-нерастворимых
соединений за счет большего образования жидких
Авторы [70] проводили гидродехлорирование чис-
продуктов и гексан-растворимых продуктов, причем
того поливинилхлорида над Pd/Al2O3, присутствие
при использовании катализатора это снижение оказа-
которого приводило к заметному усилению скорости
лось значительным — до 9.4% по сравнению с 34.9%
разложения с большей потерей массы и значитель-
(без катализатора). С использованием NiMo/Al2O3
ному удалению органического хлора из полимера
доля гексан-растворимых компонентов составила
за меньшее время, что говорит о дехлорирующем
42%, а жидких продуктов — 43.2%.
действии катализатора.
Основными компонентами жидких продуктов
В процессах дехлорирования и гидрокрекинга
являлись нафталин, 1-метилиндан, бутилбензол и
образовывались жидкость (масла, С5-С30), твердый
декалины, образованные путем дегидрирования тет-
остаток и газы (Н2, HCl, С1-С4). В обоих случаях
ралина, изомеризации, гидрокрекинга и гидриро-
(каталитический и некаталитический гидрокрекинг)
вания, а бензол и толуол — из дехлорированного
повышение температуры процесса с 350 до 440°С
поливинилхлорида.
смещало распределение продукта в пользу больше-
Исследование разложения чистого поливинил-
го образования жидкости и газа за счет разложения
хлорида и поливинилхлорид-содержащих отходов
твердого остатка. Проведение реакции при 440°С
в атмосфере водорода проводилось в [73]. Гидро-
приводило к снижению содержания хлорированных
дехлорирование чистого поливинилхлорида привело
соединений в жидкостях и газах путем их концен-
к образованию 2% масла, 38% газообразных продук-
трации в остатке. Так, количество хлорсодержа-
тов и 52% твердого остатка (табл. 7). Основными
щих соединений в жидком продукте уменьшалось
газообразными соединениями помимо HCl являлись
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1397
1398
Захарян Е. М. и др.
насыщенные алканы — 52% метана, 27% этана,
пиролиза (350°С) происходит выделение хлороводо-
18% пропана и оставшаяся часть — бутан/бутен.
рода из поливинилхлорида, а на второй (430°С) —
Масла содержали до 52.5% ароматических соеди-
гидрокрекинг дехлорированного поливинилхлорида.
нений, таких как бензол, толуол и т. д., полученных
Гидропиролиз чистого поливинилхлорида в присут-
в результате циклизации и сшивания по механизму
ствии Fe2O3/C приводил к выделению 50.6% HCl,
Дильса-Альдера образованного в процессе разложе-
что составляло 92.3% по весовому соотношению из
ния поливинилхлорида полиена, среди которых доля
поливинилхлорида, 36.6% нерастворимых в тетра-
ароматических соединений, имеющих в своей струк-
гидрофуране органических соединений, 6.8% рас-
туре одно кольцо, составляла 18.1%, два — 25.9% и
творимых в гексане и 4.3% — в толуоле. Доля газо-
три — 8.7%.
образных продуктов помимо HCl не превышала 1.6%.
Авторы также исследовали гидродехлорирование
Переработка чистого вакуумного газойля (т. кип.
различных отходов смешанных пластиков: модель-
325-500°С) над ZSM-5 приводила к образованию
ную смесь (12.2% поливинилхлорида), муниципаль-
жидкого масла, доля гексан-растворимых соединений
ные отходы пластика из Германии (Duales System
в котором составляла 70.5%, толуол-растворимых —
Deutschland, DSD), муниципальные отходы пластика
4.2% и нерастворимых в тетрагидрофуране — 9.5%.
из Бельгии (Fost Plus). Благодаря наличию в пласти-
Совместный гидропиролиз вакуумного газойля и
ковых отходах различных видов полимеров и доба-
поливинилхлорида предполагал значительное обра-
вок и соответственно взаимодействию продуктов их
зование жидких продуктов, растворимых в гексане,
разложения между собой при сжижении в Н2 была
которые можно было бы использовать в качестве
получена значительная доля жидких продуктов и
транспортного топлива. Пиролизные масла, получен-
твердого остатка, тогда как доля газов не превышала
ные из смеси, состояли из 55-65.9% гексан-раство-
2.2-4.9% в зависимости от типа отходов пластика.
римых, 6.2-8.5% толуол-растворимых, 1.3-2.6% те-
Более низкий выход масел и высокий выход остатка
трагидрофуран-растворимых соединений и 9.5-16%
при гидродехлорировании смеси DSD был обуслов-
нерастворимых в тетрагидрофуране соединений в за-
лен наличием значительного количества бумаги и
висимости от используемого катализатора. Удаление
грязи по сравнению с выходом масел и полукокса,
органического хлора из поливинилхлорида по массо-
полученного из смеси Fost Plus, состоящей из твер-
вому соотношению составляло 49.4-54%. Наиболее
дых бытовых отходов с низкой плотностью.
эффективным катализатором дехлорирования по-
Помимо хлороводорода, полученного при разло-
ливинилхлорида оказался ZSM-5, степень дехло-
жении из поливинилхлорида, и оксида и диоксида
рирования смеси при его использовании достигала
углерода, образованных из полиэтилентерефталата,
98.5%. Добавка вакуумного газойля в каталитическом
газообразные продукты содержали низшие алканы
гидропиролизе приводила к значительному образова-
и алкены С1-С4 — 20-38% метана, 10-38% этана,
нию газов по сравнению с продуктами, полученными
25-30% пропана в зависимости от вида отходов пла-
из чистого поливинилхлорида. Доля газообразных
стика. При гидродехлорировании модельной смеси
продуктов каталитического гидропиролиза смеси по-
отходов большую часть газообразных продуктов со-
ливинилхлорид/вакуумный газойль достигала 12.8%,
ставлял этан (38%), при гидродехлорировании смеси
помимо хлороводорода, благодаря крекингу газойля.
DSD — метан (38%) и при гидродехлорировании
Среди используемых катализаторов катализато-
смеси Fost Plus — пропан (30%).
ры каталитического крекинга (FCC) и гидрокрекин-
Основными компонентами жидких масел муни-
га (HC-1) проявили наибольшую эффективность в
ципальных отходов пластика являлись насыщенные
производстве жидких топливных (растворимых в
С6-С15 и ненасыщенные С7-С13, доля алканов была
гексане) фракций, к тому же они способствовали
в разы больше, чем соединений с кратными связями.
образованию меньшей доли газов и нерастворимых
Лишь 24.1-24.9% содержалось в жидких продуктах
в тетрагидрофуране соединений.
ароматических соединений: 20% с одним кольцом,
В работе [75] гидродехлорирование смеси по-
2.7-4.8% с двумя и 0.6-1.8% с тремя. Конверсия одно-
лиэтилен (20 мас%)/ жесткий поливинилхлорид
атомных ароматических соединений составляла 6.5%
(5 мас%)/вакуумный газойль (т. кип. 242-578°С,
от общей массы всего пластика DSD и 14% от общей
2.5 мас% S) проводили в два раздельных этапа.
массы смеси Fost Plus.
Двустадийное дегидрохлорирование смеси приводи-
Двустадийный каталитический пиролиз чистого
ло к образованию 90.6% остатка, содержание хлори-
поливинилхлорида и смеси поливинилхлорид/ва-
рованных соединений в котором составляло 700 ppm,
куумный газойль проводили в [74]. На первой стадии
и 0.5% жидкости. Таким образом удавалось удалить
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1399
92.1% органического хлора от общего количества
Для обеих смесей, как и в [75], выход жидких
исходного хлора в поливинилхлориде. Последующий
продуктов гидрокрекинга увеличивался с повыше-
гидрокрекинг полученной смеси полностью исклю-
нием температуры до 425°С, которая является мак-
чал образование хлорорганических соединений в
симальной температурой разложения полиэтилена,
жидких продуктах, но при этом были получены серо-
и уменьшался при 450°С с использованием катали-
содержащие соединения, 41-117 ppm в зависимости
затора DHC-8 и в некаталитическом гидрокрекинге
от условий процесса. Увеличение времени процесса
смеси.
с 1 до 2 ч на всем температурном диапазоне в при-
Использование катализатора привело к неболь-
сутствии DHC-8 (коммерческий кремнезем-глинозем)
шому снижению выхода газа для ПВХ/ПЭ/ВГО при
и в его отсутствие способствовало незначительному
всех температурах, тогда как для ПВХ/ПП/ВГО при
повышению доли газообразных продуктов и сниже-
425 и 450°С — к повышению этой доли. Аналогичная
нию доли асфальтенов, образующихся из вакуумного
зависимость наблюдалась и в случае образования
газойля. Добавка дехлорированного поливинилхло-
кокса. В случае гидрокрекинга дехлорированного
рида в смесь полиэтилен/вакуумный газойль спо-
ПВХ/ПП/ВГО при 450°С каталитический процесс
собствовала значительному снижению содержания
производил больше газовых продуктов (58.8%), чем
асфальтенов по сравнению с их долей, образованных
тепловой (51.4%), тогда как при гидрокрекинге смеси
в ходе гидропиролиза смеси полиэтилен/вакуумный
полипропилен/вакуумный газойль, наоборот, тепло-
газойль. Использование катализатора при 400 и 450°С
вой прогон приводил к образованию 34.8% газов,
приводило к образованию 3-6% кокса, тогда как в
каталитический — 19.2%.
некаталитическом гидропиролизе при этих темпера-
При гидрокрекинге дехлорированной смеси
турах кокс был обнаружен только в следовых коли-
ПВХ/ПЭ/ВГО при 425°С жидкость, полученная в
чествах.
результате каталитического процесса, содержала 32%
Повышение температуры до 425°С способство-
нафты и 10% среднего дистиллята, тогда как в тер-
вало увеличению образования жидких продуктов на
мическом гидрокрекинге 5% образованной жидко-
20-30%, до 72-77% масел получено в некаталитиче-
сти составляла нафта. В процессе, проведенном при
ском гидропиролизе (1, 2 ч). Дальнейшее повышение
450°С, наоборот, больше количество углеводородов
(до 450°С) приводило к незначительному снижению
с низкими температурами кипения в термическом
доли жидких продуктов, при этом не было образовано
процессе (75%). В результате каталитического и тер-
асфальтенов, только 1.3% в присутствии катализатора
мического гидрокрекинга дехлорированной смеси
(2 ч). При гидрокрекинге (425°С) около 37% жидко-
ПВХ/ПП/ВГО было получено около 60% нафты и
сти, получаемой при каталитическом процессе, имело
15% среднего дистиллята.
температуру кипения ниже 200°С, тогда как в случае
В [77] проводили совместную переработку муни-
термического процесса — только 7%. Тепловые про-
ципальных отходов пластмасс, содержащих 2 мас%
гоны при 450°С приводили к образованию большего
поливинилхлорида, и вакуумного газойля (т. кип.
количества низкокипящих углеводородов, чем катали-
242-578°С, 2.5 мас% S), исследуя каталитическую
тический гидрокрекинг, — 75 и 55% соответственно.
активность катализаторов HZSM-5, DHC-8 и Со/Сакт.
Основными газообразными продуктами (450°С, 1 ч,
Авторы провели два типа экспериментов: непосред-
DHC-8) являлись С1 (59%) и С2 (23.6%).
ственный гидрокрекинг смеси муниципальные от-
Влияние полимерных составляющих на продукты
ходы пластика/вакуумный газойль и двухступенча-
гидрокрекинга исследовалось в [76] на примере сме-
тый процесс переработки с дехлорированием смеси
сей ПВХ/ПЭ/ВГО и ПВХ/ПП/ВГО, где ПВХ — по-
согласно указанной в [75] методике, приводящий к
ливинилхлорид, ПЭ — полиэтилен, ПП — полипро-
образованию 91.2% (120 ppm хлорированных соеди-
пилен, ВГО — вакуумный газойль. Дехлорирование
нений) и 2.1% жидкости, с последующим гидрокре-
смесей ПВХ/ПЭ/ВГО и ПВХ/ПП/ВГО, проводимое
кингом. Гидрокрекинг недехлорированной смеси над
по методике, предложенной в [75], приводило к обра-
HZSM-5 в отличие от гидрокрекинга дехлорирован-
зованию 90.6% остатка (700 ppm хлорированных со-
ной приводил к большему образованию газов, выход
единений) и 0.5% жидкого масла из смеси ПВХ/ПЭ/
которых увеличивался с повышением температуры
ВГО и 88.5 (600 ppm) и 1.5 из смеси ПВХ/ПП/ВГО.
до 75.7% (450°С). Среди всех катализаторов именно
Выделяющийся при разложении поливинилхлорида
в его присутствии достигался наибольший выход
HCl катализировал частичное разложение полиэти-
жидкости при низких температурах — 38.3% (425°С),
лена и полипропилена, способствуя снижению их
снижающийся до 10.8% при 450°С. Такое поведение
температуры разложения.
катализатора определяется его кислотными свойства-
1400
Захарян Е. М. и др.
ми, поскольку наличие большего количества более
позволило большую часть органического хлора пе-
сильных кислотных центров приводит к большей эф-
ревести в твердый остаток [70]. Гидроконверсия раз-
фективности при деградации полимеров, способствуя
личных муниципальных отходов пластика, содержа-
разрыву углерод-углеродных связей с образованием
щих различные виды отходов и разное количество
газообразных продуктов.
органического хлора, приводила соответственно к
DHC-8, являющийся аморфным кремнезем-глино-
различному соотношению выходов масла, газов и
земом (ASA), имеющим кислотные сайты Льюиса и
твердого остатка [73]. Предварительное дехлориро-
Бренстеда, в отличие от HZSM-5 с повышением тем-
вание отходов поливинилхлорида и последующий
пературы приводил к росту доли газов и жидкостей за
гидрокрекинг полученного сырья с вакуумным га-
счет крекинга парафиновых углеводородов. Большая
зойлем значительно улучшал качество получаемых
разница в выходах этих фракций между дехлориро-
продуктов [75-77]. Коммерческие катализаторы, ис-
ванной и недехлорированной смесями наблюдалась
пользуемые в гидрокрекинге смеси муниципальные
при 435°С, тогда как при 450°С она исчезала.
отходы пластика/вакуумный газойль [77], каждый в
Деградация смеси муниципальные отходы пла-
своем температурном режиме способствовали обра-
стика/вакуумный газойль над нейтральным Со/Сакт,
зованию достаточного выхода жидкого продукта с
протекающая по радикальному механизму в отличие
высоким содержанием нафты (более 50%) и среднего
от ионного в случае кислотного катализа (HZSM-5
дистиллята (15-18%).
и DHC-8), была схожа с термическим некаталити-
ческим крекингом. В обоих случаях термический
Газификация поливинилхлорида
процесс и крекинг недехлорированной смеси над
и хлорсодержащих отходов
Со/Сакт приводил к образованию большего количе-
ства жидких продуктов и меньшему количеству газов,
Газификация — процесс преобразования органи-
чем при крекинге дехлорированной смеси.
Поскольку вакуумный газойль содержит до 2.5%
веществ в горючие газы при высокотемпературном
серы, процесс гидрокрекинга не исключает образо-
вание серосодержащих соединений: их доля росла в
ряду DHC-8 > Со/Сакт/некаталитический процесс >
мости от применяемого сырья получают различные
> HZSM-5 с 1.01 до 2.1%.
виды газообразного топлива: синтез-газ, водяной,
При самой низкой температуре (425°С) наиболь-
воздушный или смешанный газы.
шее количество жидкого топлива, содержащего 66%
Газификация отходов поливинилхлорида. Превра-
нафты и 17% фракции среднего дистиллята, получали
щение поливинилхлорида при высоких температурах
с использованием HZSM-5. Использование DHC-8
в атмосфере пара в хлористый водород, смесь угле-
приводило к образованию большого количества жид-
водородов и синтез-газа называется паровой газифи-
кости (53% нафты и 18% среднего дистиллята) при
кацией. Разложение поливинилхлорида в инертной
450°С. Максимальный выход жидкости (47.8%) был
атмосфере начинается при 200°C с процесса деги-
получен в присутствии Co/Сакт (435°C) — 55% нафты
дрохлорирования с образованием промежуточного
и 18% среднего дистиллята. При некаталитическом
полиена, который продолжает разрушаться, с одной
гидрокрекинге смеси больший выход жидких про-
стороны, в результате выделения ароматических со-
дуктов достигался при 435°С (50.2%).
единений, с другой — в результате сшивания. Таким
Основными достоинствами гидродехлорирования
образом, продуктами разложения будут газообраз-
поливинилхлорид-содержащих отходов являются вы-
ный HCl, газообразные и жидкие углеводороды и
сокая эффективность удаления органического хлора
уголь. В атмосфере пара при высоких температурах
посредством его связывания подаваемым водородом
углеводородная фракция будет превращаться в оксид
и облагораживание полученных продуктов, поскольку
углерода(II) и водород (схема 15) [79].
наличие водорода значительно улучшает качество
Оксид углерода(II) далее реагирует с паром:
продукта (более высокое отношение H/C и более
CO + H2O
CO2 + H2.
низкое содержание ароматики). Источником водорода
в такого типа процессах может быть сам атомарный
Поскольку при высоких температурах дегидро-
водород [70], растворитель [71] либо низкокипящая
хлорирование (1) протекает быстро, а образование
фракция (ВГО) [74]. Каталитическое гидродехлори-
ароматических соединений (3) и реакция газифика-
рование поливинилхлорида в полупромышленном
ции (4 и 5) — значительно медленнее, продуктом про-
масштабе (до 100 г) в присутствии 1.2% Pd/Al2O3
цесса будет смесь HCl, оксида и диоксида углерода,
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1401
Схема 15
Схема разложения поливинилхлорида
водорода, газообразные и жидкие углеводороды, а
(температура и время пребывания). В данной работе
также уголь, при этом точный состав продукта бу-
процесс газификации поливинилхлорида представлен
дет сильно зависеть от технологических параметров
следующей реакцией:
(C2H3Cl)n + 2.74nH2O
nHCl + 3.44nH2 + 1.08nCO + 0.83nCO2 + 0.15nCH4.
Образующийся хлороводород можно использовать
впоследствии отделялись от газового потока с по-
в производстве соляной кислоты или поливинилхло-
мощью циклона и рукавного фильтра. Полученный
рида на установке оксихлорирования, а синтез-газ —
таким образом синтез-газ, содержащий (в пересче-
в регенерации энергии, в синтезе метанола, синтезе
те на сухое вещество и без HCl) 63% Н2, 20% СО,
Фишера-Тропша и т. д.
15% СО2 и 3% СН4, имел относительно низкую те-
Использование каталитического неактивного твер-
плотворную способность — 8.6 МДж·Нм-3 (табл. 8).
дого кварца в качестве материала слоя приводило
Образующийся CaCl2 добавляли и рециркулировали
к образованию большого количества полукокса и
в растворе HCl до 40 мас% CaCl2 с последующей экс-
смолы уже в начале эксперимента. Конверсия угле-
трактивной дистилляцией. 100 мас% HCl извлекали,
рода в газ над SiO2 не превышала 25%, тогда как
сушили и сжимали до 7 бар, подходящих для процес-
конверсия над активным Al2O3 достигала уже 69%
са оксихлорирования поливинилхлорида.
(877°C). Основными газообразными компонентами в
Авторы [82] представили возможность регене-
обоих случаях являлись H2, CO, CO2 и CH4 в равном
рации энергии из отработанного поливинилхлори-
процентном соотношении, лишь над SiO2 было обра-
да в смеси с топливом, полученным из различных
зовано больше СО (25%), а над Al2O3 — больше CO2
отходов (Cl < 0.1%, теплотворная способность =
(19%). Газообразные углеводороды с большей моле-
= 17.5 МДж·кг-1), путем газификации в двухступен-
кулярной массой (С2-С5) обнаружены не были, фрак-
чатом реакторе. Без использования сорбентов ор-
ция смолы и конденсирующихся веществ состояла
ганического хлора из поливинилхлорида его доля
примерно из 20 ароматических соединений, при этом
в синтез-газе составляла 1.28, а в остатке — 0.14 г.
хлорорганические соединения ни в газовой фракции,
Удаление органического хлора из образующейся
ни в смолах и конденсате обнаружены не были. Столь
смеси газов путем сгорания в кислороде топлива и
большая разница в конверсии между двумя катализа-
отходов поливинилхлорида проводилось путем про-
торами была обусловлена более высокой каталитиче-
хождения смеси газа через двойной слой щелочных
ской активностью и большей площадью поверхности
наполнителей. Сравнение щелочных наполнителей
оксида алюминия. Увеличение времени контакта не-
показало, что наиболее эффективным оказался кар-
значительно влияло на конверсию углерода в газ и со-
бонат натрия (доля хлора в синтез-газе — менее 0.01,
став продуктов, в том числе газообразных. Лишь по-
в остатке — 1.34 г), удаление органического хлора
вышение температуры до 977°C позволило увеличить
в ряду Na2CO3 > CaO > Ca(OH)2 падало до 0.38 г
конверсию до 99% за счет глубокого крекинга смолы
в синтез-газе и 1.03 г в остатке. Даже двукратное
и конденсатов, отношение СО2/СО падало до 0.8.
увеличение содержания поливинилхлорида в смеси
Проведение газификации отходов поливинилхло-
(до 20 мас%) при прохождении через слои Na2CO3
рида по двухреакторной концепции Hitachi [80, 81]
не привело к повышению доли хлорированных сое-
позволило избежать образования CaCl2 на поверх-
динений в синтез-газе, также менее 0.01 г, тогда как
ности катализатора, тем самым не дезактивируя
в остатке количество хлорсодержащих соединений
его. Поток газа, выходящий из реактора, содержал
повысилось до 2.84 г. Такой газ, согласно итальян-
захваченный материал из слоя оксида алюминия,
ским нормативам, можно было использовать непо-
полукокса и твердых и жидких неорганических сое-
средственно для получения электроэнергии и (или)
динений, таких как CaO и CaCl2. Твердые вещества
локального отопления. Состав образующегося син-
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1403
1404
Захарян Е. М. и др.
тез-газа был практически одинаков с использованием
ностью данной системы является то, что сверхкри-
наполнителей и в их отсутствие: до 72% водорода,
тическая вода в условиях процесса представляет
10% СО, 12% СО2 и 6% метана.
собой отличный растворитель, имеющий высокую
В [83] переработку поливинилхлорида проводи-
диффузионную способность и низкую поверхность
ли в два этапа: предварительное термическое деги-
напряжения, благодаря образованию водорода в ре-
дрохлорирование поливинилхлорида с последующей
зультате взаимодействия СО и воды улучшается ка-
газификацией над щелочами в потоке пара и азо-
чество продуктов.
та. Преобладающим продуктом в процессе газифи-
CО + H2O
CO2 + H2.
кации являлся водород —до 47% при добавлении
Сa(OН)2 и до 94% при добавлении NaOН (600°С), в
Поскольку такой метод включает использование
меньших количествах были получены метан, этан и
щелочных катализаторов, большая часть органиче-
углекислый газ. Столь большое различие в выходах
ского хлора удаляется в виде соли NaCl, а хлорорга-
водорода обусловлено тем, что для полной газифи-
нические соединения не образуются.
кации в присутствии Сa(OН)2 были недостаточны
Влияние карбонатов лития, натрия и калия на па-
температура и парциальное давление пара, уже при
ровую газификацию отходов гибкого поливинилхло-
700°С происходил резкий скачок в выходе Н2 [84],
рида (кабель) было исследовано в [87]. При паровой
тогда газификация дегидрохлорированного поливи-
газификации коммерческого поливинилхлоридного
нилхлорида ускорялась посредством добавления ги-
кабеля в присутствии расплавленных карбонатов 96%
дроксида натрия. Выходы Na2CO3, твердого остатка
хлороводорода было превращено в неорганическую
и CH4 в присутствии NaOН составляли 64.1, 8.8 и
соль (хлориды) в карбонатном остатке, около 3%
5.3% соответственно, т. е. полное извлечение угле-
хлороводорода было удалено при помощи восста-
рода достигло 80%. Процесс образования водорода
новленного пара. В отсутствие пара и в присутствии
непосредственно взаимодействием щелочи с паром
расплавленных карбонатов доля хлороводорода, улав-
и углеродом обусловливает ускорение щелочью про-
ливаемого расплавленными карбонатами, все еще
цесса газификации:
была высокая — до 87%, 2% хлороводорода раство-
рялось в воде, оставшаяся доля образованного хлора
C + 2NaOH + H2O
Na2CO3 + 2H2.
была представлена газообразными хлорсодержащими
соединениями. В отсутствие карбоната, но в присут-
Конверсия и выход газообразного водорода уве-
ствии пара при температуре 650°С 35% органическо-
личивались с ростом парциального давления пара,
го хлора удалялось с паром и восстановленной водой,
а также молярного отношения NaOH/C (до 3-4).
59% вымывалось водой.
Побочный метан был образован двумя путями: пря-
Повышение температуры с 625 до 675°С способ-
мое тепловое разложение дегидрохлорированного
ствовало большему удалению органического хлора,
поливинилхлорида при нагревании и вторичная ре-
используемого в паровой газификации, доля которого
акция полученных в процессе углерода и водорода.
соответственно увеличивалась — до 5% в восстанов-
В [85] авторы путем паровой газификации при
ленной и до 0.5% в промывочной воде.
850°С полностью переработали многопроволочный
Сравнение материала слоя в паровой газифика-
медный луженый кабель с поливинилхлоридной
ции поливинилхлорида показало, что наличие SiO2
изоляцией, состоящей из пластифицированного по-
в псевдоожиженном слое реактора приводило к зна-
ливинилхлорида, полиэтилентерефталата и хлопка.
чительному выходу смолы и полукокса уже в начале
Извлечение углерода и водорода из поливинилхло-
эксперимента, тогда как каталитически активный
рида составило более 98% посредством пропуска-
оксид алюминия способствовал превращению до
ния образованных при газификации газов над слоем
69% углерода в синтез-газ [79]. Повышение темпе-
обожженной глины, используемой в качестве катали-
ратуры процесса на 100°С приводило практически к
затора и сорбента. Было обнаружено, что 28% хлоро-
100% конверсии сырья. Газификация отходов поли-
водорода прореагировало с образованием CaCl2, 71%
винилхлорида по концепции Hitachi [80, 81] позво-
осталось в водном конденсате и только 0.6% было
ляла получать обогащенный водородом синтез-газ
поглощено щелочным скруббером. Поливинилхлорид
(63%), при этом полностью удаляя органический хлор
и другие углеводородные составляющие были полно-
из полимера с последующим использованием его в
стью удалены из образца кабеля.
производстве соляной кислоты и HCl для оксихлори-
Сжижение поливинилхлорида в системе СО/H2O
рования поливинилхлорида. Исследование влияния
[86] позволило достичь конверсии 91%. Особен-
шелочных сорбентов в двустадийной газификации
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1405
смеси поливинилхлорид/топливо показало, что наи-
из реактора с псевдоожиженным слоем, содержащим
большую эффективность в удалении органического
SiO2, Fe2O3, MgO, Al2O3 и CaO, и зоны крекинга смо-
хлора проявил Na2CO3 [82]. Предварительное дехло-
лы, в которую для полного удаления органического
рирование поливинилхлорида с последующей гази-
хлора из смеси добавляли кальцинированные напол-
фикацией полученного сырья в присутствии NaOН
нители и активированный уголь в качестве смоляной
в потоке пара и азота позволило получить до 94%
присадки.
водорода уже при 600°С [83]. Для полного удаления
Добавки на основе Са способствовали не только
органического хлора из отходов поливинилхлорида
улавливанию выделяемого из поливинилхлорида хло-
и получения высокого выхода водорода авторы [86]
роводорода, но и каталитическому крекингу образу-
использовали атмосферу СО/H2O. Аналогично [82],
ющихся в процессе газификации смол. Добавление
хорошие результаты в процессе газификации отхо-
негашеной извести, раковин моллюсков и доломита в
дов гибкого поливинилхлорида получили благодаря
полимерную смесь приводило к увеличению выхода
использованию в качестве сорбентов хлороводорода
Н2 с 11 до 12-14%, СО — с 3 до 9-12% по сравнению
смеси карбонатов щелочных металлов [87].
с выходами продуктов газификации, проведенной в
Газификация муниципальных отходов пласти-
отсутствие каких-либо добавок, и снижению выхода
ка, содержащих поливинилхлорид. Для проведения
CO2 благодаря его связыванию с кальцинированными
газификации муниципальных отходов пластика и
добавками с 9 до 6-7% (табл. 9). Содержание HCl
отходов, содержащих поливинилхлорид, необходи-
в генераторном газе значительно уменьшилось, до
мо изначально исследовать поведение основных со-
<1 ppm по сравнению с содержанием хлороводорода,
ставляющих муниципальных отходов пластика при
образованного в процессе газификации в отсутствие
термическом разложении в инертной атмосфере (пи-
добавок (7.5 ppm). Доля хлорированных соединений
ролиз) и СО2 (газификация). Авторы [88] проводили
в маслах резко падала с использованием добавок на
разложение полиэтилена, полипропилена и поли-
основе кальция с 32 789 (без) до 1234 ppm (раковины
винилхлорида с помощью термогравиметрического
моллюсков). При добавлении раковин моллюсков в
анализатора в среде N2 и CO2.
качестве сорбента хлороводорода большее количе-
В атмосфере N2 температурные пики кривых дери-
ство хлора в полукоксе было образовано за счет об-
вативной термогравиметрии полиэтилена и полисти-
разования твердого хлорида кальция. По сравнению
рола наблюдались при 477 и 417°C соответственно,
с другими сорбентами HCl применение раковин мол-
в то время как в атмосфере CO2 — 473 и 413°C, что
люсков имело дополнительное преимущество — пе-
было немного ниже, чем в N2. Процессы пиролиза и
реработка таким образом их отходов, поскольку боль-
газификации поливинилхлорида разделены на две
шая часть (70%) раковин моллюсков утилизируется
стадии — 250-380 и 400-550°С. В атмосфере N2
путем сброса их на побережье, что вызывает загряз-
температурные пики кривой деривативной термо-
нение окружающей среды и подземных вод.
гравиметрии для поливинилхлорида наблюдались
Добавление активированного угля в зоне крекинга
при 287 и 467°С, тогда как в атмосфере CO2 — 299
значительно повысило выход водорода в генератор-
и 469°C соответственно. Сравнивая кривые термо-
ном газе до 30% за счет снижения доли газов, выход
гравиметрии для поливинилхлорида в двух средах,
полукокса составил 6.3% в отличие от 0.1-0.5% в
можно обнаружить, что в интервале 250-380°C CO2
процессе с добавлением Са-наполнителей, при этом
способствовал процессу термического разложения
содержание HCl в нем не превышало <1 ppm, как и в
первой стадии, что связано с выделением большого
случае кальцинированных добавок. Эффективность
количества хлороводорода.
удаления смолы была увеличена до 84%. Процессы,
Переработку смеси пластиковых отходов поли-
протекающие над активированным углем, представ-
этилен/полипропилен/поливинилхлорид в [89] про-
лены ниже.
водили в двухступенчатом газификаторе, состоящем
Паровой риформинг: CnHm + nH2O
nCO + (n + m/2)H2,
(1)
Сухой риформинг: CnHm + nCO2
2nCO + ½mH2,
(2)
Термический или каталитический крекинг: CnHm
C + CxHy + H2,
(3)
где CnHm углеводороды относятся к смолам (соеди- ла); CxHy — углеводород с меньшим числом атомов
нения с большей молекулярной массой, чем у бензо- углерода, чем CnHm.
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1407
1408
Захарян Е. М. и др.
В [90] авторы проводили воздушную газификацию
повышению концентрации Н2 и СО в генераторном
фракции смешанных пластиковых отходов, состоя-
газе — с 14.9 до 26.1 и с 9.4 до 16.8% соответственно
щей из полиэтилена, полипропилена, полистирола,
и, как следствие, снижению содержания смолы на
поливинилхлорида, полиметилметакрилата и поли-
96% по сравнению с ее выходом при добавлении
этилентерефталата, в двухступенчатом газификаторе,
кварцевого песка. Повышение температуры, согласно
исследуя влияние активированного угля и доломита
принципу Ле Шателье, способствовало еще больше-
в переработке смолы и полукокса. В зависимости от
му повышению доли водорода и монооксида углеро-
температурного режима, добавления наполнителя
да — 28 и 18.4%. Как и в исследовании [90], большая
(его количества), скорости подачи сырья выходы ге-
часть органического хлора оседала в полукоксе —
нераторного газа, полукокса, конденсатной жидкости
26 700-31 000 ppm, тогда как в генераторном газе его
и смолы, образуемой на оборудовании, достигали
количество не превышало 17.9 ppm.
87-94%, 3-8%, 2-4% и 1-3% соответственно.
В [92] в качестве материала псевдоожиженно-
Снижение скорости подачи сырья приводило к
го слоя использовали оливин [смесь оксидов крем-
повышению отношения его к кислороду воздуха, что
ния, железа(III), магния, кальция и алюминия].
способствовало протеканию реакций окисления, т. е.
Увеличение добавленного количества активирован-
увеличивались концентрации в генераторном газе
ного угля приводило к значительному повышению
CO2 и H2O за счет окисления СО и Н2, концентрации
концентрации H2 и CO в генераторном газе и количе-
которых снижались до 5 и 12.6%, выход CH4 резко
ства органического хлора в нем (табл. 9). Благодаря
падал с 15.7 до 3.6%. С повышением температуры
крекирующим свойствам угля достигалась 98%-ная
верхнего реактора концентрации H2 и CO увеличива-
эффективность удаления смолы: с 2170 до 5 мг·Нм-3.
лись с 13.5 до 18.2% и с 5.5 до 7.2% соответственно.
Проведение газификации без электростатического
Добавление активированного угля и увеличение
осадителя незначительно повысило концентрацию
массы добавки способствовало значительному росту
водорода в генераторном газе, при этом содержание
выхода водорода с 11 (в отсутствие активированного
хлорсодержащих соединений в нем резко возросло
угля) до 21.3% (630 г), что связано с каталитическими
до 25.4 ppm, а в конденсатной жидкости снизилось
свойствами угля по отношению к реакциям крекин-
до 13 123 ppm. Использование фильтра с активиро-
га смолы. Использование доломита по сравнению с
ванным углем вместо электростатического осадителя
активированным углем повышало концентрацию H2
также позволило достичь высокой концентрации H2
незначительно, до 13.2%. Преимуществом активи-
(30.6%), но, как и при использовании электроста-
рованного угля являлось и значительное снижение
тического осадителя, ухудшив качество генератор-
массы образующейся в процессе смолы, до 20 г, тог-
ного газа (11.3 ppm хлорированных соединений).
да как в «чистом» прогоне она составляла 50 г, а в
Увеличение длительности процесса до 3 ч приводило
присутствии доломита — 32-35% в зависимости от
к сильной дезактивации активированного угля и спо-
количества наполнителя.
собствовало снижению содержания хлорорганиче-
Удаление хлороводорода путем его сорбции на-
ских соединений в генераторном газе и конденсатной
полнителями приводило к образованию хлоридов,
жидкости — 2.4 и 9187 ppm соответственно.
входящих в состав полукокса (28 932 ppm при ис-
Использование кальцинированного оливина либо
пользовании активированного угля и 33 310 ppm —
натурального оливина с никелированным распреде-
доломита). Хлороводород частично сорбировали са-
лителем в смеси с активированным углем (900 г) в
ми наполнители (397 ppm в активированном угле и
качестве добавки для крекинга смолы позволило до-
583 ppm в доломите).
стичь максимальной концентрации водорода — 26.7
В [91] воздушную газификацию фракции сме-
и 27.3% [93] соответственно. При этом наблюдалась
шанных пластиковых отходов проводили при одно-
значительная доля хлорированных соединений в ге-
временном использовании активированного угля в
нераторном газе (19-44.7 ppm) и в конденсатной жид-
качестве добавки для крекинга смолы и прокален-
кости (13 000-23 400 ppm), тогда как при использова-
ного доломита в качестве материала слоя в реакторе.
нии в качестве добавки кальцинированного доломита
Добавка прокаленного доломита приводила к повы-
(1500 г) доля органического хлора в генераторном
шению содержания H2 на 5% и уменьшению коли-
газе и конденсатной жидкости была минимальной
чества смолы на 73% по сравнению с ее выходом
(менее 1 ppm и 149 ppm), и водорода было получе-
при использовании кварцевого песка: 42 и 170 г·кг-1
но значительно меньше (17.3%). Не ухудшая состав
сырья. Добавление активированного угля и увеличе-
генераторного газа, т. е. не снижая выход водорода,
ние его количества способствовали значительному
снизить долю органического хлора в генераторном
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1409
газе и конденсатной жидкости удалось посредством
Газификацию смесей хлорсодержащих отходов
совместного использования кальцинированного оли-
и муниципальных отходов пластика, как правило,
вина и никелированного распределителя.
проводят в двухступенчатом реакторе, где первой
Паровая газификация твердых бытовых отходов с
стадией является пиролиз полимеров, второй — зона
добавлением CaO и в его отсутствие была исследо-
крекинга смолы и полукокса в воздушной [89-94]
вана в периодическом реакторе [94]. Процесс пере-
либо паровой атмосфере [95]. В качестве каталити-
работки муниципальных отходов пластика состоял
ческого слоя в реакторе крекинга смолы использо-
из стадии пиролиза и последующей газификации.
вали обычный кварцевый песок [89], смесь песка
Повышение температуры на стадии газификации
и доломита [90], доломита и активированного угля
полукокса с 700 до 800°С приводило к увеличе-
[91], дробленые раковины моллюсков [89], содержа-
нию молярной концентрации H2 на 30%, а с 800 до
щие до 93% CaCO3, натуральный и кальцинирован-
900°С — к снижению на 10%. Аналогичная зависи-
ный оливин [93]. Исследование влияния различных
мость наблюдалась и в присутствии сорбента СаО,
кальцинированных добавок и активированного угля
также способствующего образованию большей кон-
показало [89], что наибольшую эффективность в уда-
центрации H2 (70%, 800°С). Выход Н2 также значи-
лении органического хлора проявили измельченные
тельно увеличивался с повышением соотношения
раковины моллюсков по сравнению со слоем песка,
CaO/муниципальные отходы пластика.
при этом значительная часть органического хлора
Добавка CaO, имеющего хорошую адсорбционную
благодаря сорбции наполнителями была обнаружена
способность в отношении CO2, приводила к образо-
в полукоксе в виде хлоридов кальция. Использование
ванию обогащенного водородом синтез-газа. Доля
активированного угля в зоне крекинга смолы позво-
СО и СО2 снижалась при 700 и 800°С, дальнейшее
ляло значительно снизить выход смолы и увеличить
повышение температуры до 900°С ухудшало погло-
концентрацию водорода в генераторном газе [90].
тительные свойства оксида за счет разложения обра-
Большая по весу добавка активированного угля и
зовавшегося ранее СаСО3. Кальциевые катализаторы
доломита способствовала повышению выхода Н2
благоприятствовали обширному разрыву длинных
до 26.1% [91], а замена кварцевого песка в псевдо-
цепей, производящих компоненты с цепями средней
ожиженном слое на оливин с добавкой Сакт — до
длины на стадии инициации радикала, действующие
30.2% [92]. Дополнительным преимуществом нату-
с целью уменьшения вероятности разветвления цепи.
рального и кальцинированного оливина в [92, 93] по
Авторы [95] предложили систему непрерывного
сравнению с кварцевым песком в [90, 91] являлось
производства водорода из муниципальных отходов
минимальное количество органического хлора в гене-
пластика, включающую процесс газификации му-
раторном газе (<1 ppm). Добавка СаО в газификации
ниципальных отходов пластика в псевдоожижен-
муниципальных отходов пластика [94] позволила по-
ном слое в сырой газ, содержащий CO, H2, CH4, его
высить выход Н2 с 58 до 70% уже за 10 мин процесса.
очистку паром и над катализатором на основе Ni и
Совместная газификация отходов, содержащих
сорбентом углекислого газа и хлороводорода CaO,
поливинилхлорид, и муниципальных отходов пластика
паровой риформинг. На этапе газификации выход
и биомассы, угля. Как и в случае совместных пиро-
СО, углеводородов и HCl в неочищенном газе уве-
лиза и гидротермальной обработки отходов пластика
личивался с повышением температуры. Отношение
и биомассы/угля, в совместной газификации также
CO/H2 в диапазоне 1.15-1.77 увеличивалось с повы-
наблюдался положительный синергический эффект
шением температуры, как и CO/CH4 (3.71-4.27). CO2
благодаря каталитическому взаимодействию образу-
в неочищенном генераторном газе обнаружен не был.
емых в процессе соединений.
При снижении температуры в реакторе риформинга
Исследование совместной паровой газификации
значительно увеличивалось удаление CO2 путем его
рисовой соломы, полиэтилена и поливинилхлорида
сорбции CaO, тем самым обеспечивая получение
проводилось в [96]. Добавление поливинилхлорида к
высокочистого H2. Также CaO способен восстанав-
рисовой соломе и повышение его доли (до 75%) спо-
ливать HCl из неочищенного генераторного газа при
собствовало увеличению выхода водорода с 42.2 до
538-788°С до 93%. Одновременное использование
54.2% (900°С) и снижению доли CO и CO2 с 32.6 до
Ni-катализатора и пара приводило к риформингу
22.6% и с 15.9 до 11% соответственно. Аналогичным
углеводородов до H2. Таким образом, сочетание обо-
образом повышалась теплотворная способность газа
их реакторов позволило получить около 88.4% водо-
с 11.9 до 13.9 МДж·кг-1. Варьирование соотношения
рода (без H2O и N2) при температуре газификации
рисовая солома/полиэтилен/поливинилхлорид в га-
818 и риформинга 583-706°С.
зификации тройной смеси показало, что наибольший
1410
Захарян Е. М. и др.
выход водорода и, как следствие, большая теплотвор-
мальным являлся 0.3, достигались максимальный
ная способность были получены, когда смесь состо-
выход водорода и СО и наибольшая теплотворная
яла из 40% полиэтилена, 40% поливинилхлорида и
способность генераторного газа, конверсия углерода
20% рисовой соломы — 49.1% и 15.3 МДж·кг-1.
в газ составила 70%.
Увеличение выхода газа происходило в резуль-
Авторы [99] разработали трехступенчатую си-
тате более высокой скорости конверсии во время
стему совместной газификации смешанных отходов
совместной газификации, в процессе которой благо-
с высокощелочным углем в качестве катализатора
даря взаимодействию продуктов разложения пласт-
крекинга образующейся в процессе смолы и сор-
масс биомасса начинает разлагаться позже, чем при
бента выделяющегося HCl. Модельную смесь под-
газификации чистой рисовой соломы, высвобождая
вергали пиролизу на первой стадии (450-650°С),
большее количество летучих соединений посред-
образующийся сырой синтез-газ частично окисляли
ством разрыва ковалентных связей при термическом
на второй стадии (800°С) и затем полученную в про-
разложении компонентов смеси. Образующиеся та-
цессе газификации смолу восстанавливали высоко-
ким образом свободные радикалы реагировали не
щелочным углем на последнем этапе (800-1000°С).
только с органическим веществом биомассы, но и
Исследование влияния температурных режимов гази-
с пластиком, способствуя процессам разложения и
фикации показало, что наибольший выход синтез-газа
окисления/газификации. Наличие щелочных метал-
(1.57 Нм3·кг-1) с максимальной концентрацией во-
лов, присутствующих в биомассе, таких как Na, K и
дорода (41.9%) достигался при 550°С в пиролизе и
Ca, также способствовало гетерогенной газификации.
800°С при восстановлении с углем. Данные условия
Добавление древесных пеллетов к хлорсодержа-
также способствовали высокой конверсии углеро-
щим отходам легкой фракции пластиков и муници-
да — 90.4%. Дополнительное количество H2 и CO бы-
пальным отходам пластика приводило к снижению
ло образовано в результате каталитического крекинга
выхода водорода в процессе газификации сырья по
смолы, осуществляемого путем взаимодействия со
сравнению с добавлением чистых пеллетов и отходов
щелочами и щелочноземельными металлами в угле
легкой фракции [97]. Доля СО и СО2 также снижа-
на третьей стадии восстановления.
лась с повышением доли легкой фракции пластиков
Повышение температуры на стадии пиролиза
с 20%, образованных при переработке пеллетов, до
приводило к большему разложению образующихся
17%, полученных при переработке легкой фракции
углеводородов и, как следствие, образованию боль-
пластиков, а доля метана, как и теплотворная спо-
шего количества смолы (до 215 мг·кг-1) и полукокса
собность образующегося генераторного газа, наобо-
(0.215 г·кг-1 сырья), а повышение на стадии восста-
рот, повышались с 10 до 23% и с 13 до 18 МДж·кг-1
новления — к снижению концентрации H2 и СО и
соответственно (табл. 10). Посредством пара и воз-
эффективности удаления смолы (до 86.3%).
душного потока генераторный газ очищали от обра-
Основная часть органического хлора выделялась
зующегося в процессе газификации хлороводорода.
в виде газообразного хлороводорода в процессе пи-
Производство генераторного газа из смеси пласти-
ролиза поливинилхлорида в смеси отходов, содер-
ковых отходов и древесной щепы и его регенерация
жание которого после второй стадии варьировалось
в электроэнергию были исследованы в газифика-
в интервале 255-260 мг·кг-1. Благодаря сорбции ор-
торе с подвижной решеткой мощностью 500 кВт,
ганического хлора CaO и MgO, содержащихся в угле,
в который входила установка риформинга смолы с
удалось добиться снижения содержания HCl до 93%,
последовательными процессами очистки газа и си-
т. е. до 17.6 мг·кг-1 в очищенном синтез-газе. С рос-
стема выработки электроэнергии газовым двигателем
том температуры восстановления за счет быстрой
[98]. Мощность обработки отходов в газификато-
зашлакованности угля при высоких температурах
ре составляла более 80 кг·ч-1, система очистки —
снижалась эффективность удаления органического
~300 Нм3·ч-1, двигатель синтез-газа — 30 кВт·ч-1.
хлора до 72.9%.
Процесс газификации был разделен на три этапа: ста-
Добавление к хлорсодержащим отходам и му-
дия сушки, пиролиза и газификации угля. В качестве
ниципальным отходам пластика биосырья (рисовой
газифицирующего агента был использован чистый
соломы [96], древесных пеллетов [97] и щепы [98]),
кислород, позволяющий практически полностью
а также высокощелочного угля [99] благодаря поло-
избавиться от образующихся в процессе пиролиза
жительному синергическому эффекту приводило к
отходов смолы.
выработке более обогащенного водородом генера-
Исследование влияния коэффициента избытка
торного газа с большей теплотворной способностью
кислорода (0.15-0.65) показало, что наиболее опти-
и меньшим выходом смолы, наличие которой спо-
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1411
1412
Захарян Е. М. и др.
собствовало образованию кокса, засорению фильтров
поскольку наличие водорода значительно улучшает
и конденсации в холодных местах установки, что
качество продукта (более высокое отношение H/C и
может привести к серьезным сбоям в работе газифи-
более низкое содержание ароматических соединений).
кационных реакторов.
Летучие компоненты, высвобождаемые из сырья в
процессе гидрокрекинга, взаимодействовали с водоро-
дом с образованием значительной доли таких аромати-
Заключение
ческих соединений, как бензол, толуол, этилбензол и
В данном обзоре обобщены результаты работ по
ксилолы, используемые в качестве химического сырья
вторичной химической переработке чистого поливи-
для производства различных ценных производных.
нилхлорида, его отходов и хлорсодержащих муни-
— Посредством газификации отходов поливи-
ципальных отходов пластика, такие как шелочной/
нилхлорида и муниципальных отходов пластика в
основный гидролиз, гидротермальная переработка в
атмосфере воздуха, пара, смеси СО/Н2О, СО2 и кис-
до- и сверхкритической воде, аммиаке, метаноле, со-
лорода можно получать обогащенный водородом ге-
вместная гидротермальная переработка с биомассой
нераторный газ с низким содержанием органического
и углем, гидродехлорирование, включающее гидро-
хлора, позволяющий использовать его в регенерации
очистку продуктов пиролиза поливинилхлорида и
энергии, а выделяемый HCl — в процессе хлорсодер-
одностадийный процесс гидроконверсии, газифика-
жащих полимеров — в производстве соляной кислоты
ция отходов поливинилхлорида и хлорсодержащих
либо в процессе оксихлорирования поливинилхлори-
муниципальных отходов пластика, совместная гази-
да. Использование кальцинированных добавок и акти-
фикация отходов поливинилхлорида с биомассой и
вированного угля, а также замена кварцевого песка в
углем. Анализ данных литературы позволяет сделать
псевдоожиженном слое на натуральный либо кальци-
следующие выводы.
нированный оливин в газификации хлорсодержащих
— Совмещая преимущества двух методов, меха-
отходов и муниципальных отходов пластика приводи-
нохимической переработки и низкотемпературного
ли к образованию большего количества водорода в ге-
дехлорирования, в щелочном/основном гидролизе
нераторном газе благодаря процессу каталитического
отходов поливинилхлорида можно получать продукт
крекинга образуемой в процессе смолы и эффективно-
с низким содержанием хлорированных соединений
му удалению органического хлора путем его сорбции.
без образования токсичных хлорсодержащих орга-
— Основным преимуществом совместной гидро-
нических соединений (диоксины, фенолы и т. д.).
термальной обработки, как и совместного пиролиза,
Замена водной среды на органические растворители
как и совместной газификации, является переработка/
и основания позволят значительно снижать темпера-
утилизация двух различных видов отхода, полимер-
туру и длительность процесса.
ного поливинилхлорида и биосырья, либо техни-
— Главным достоинством гидротермальной об-
ческих отходов, либо высокощелочного угля, что
работки чистого поливинилхлорида и его отходов
значительно улучшает экологическую обстановку.
в докритической воде является отсутствие хлори-
рованных органических соединений, поскольку
Финансирование работы
практически весь хлор, выделяемый из поливи-
Работа выполнена в рамках государственного за-
нилхлорида, превращается в растворимый в воде
дания Института нефтехимического синтеза РАН.
HCl. Докритическая вода действует в качестве ре-
акционной среды и катализатора, усиливающего
растворение исходного сырья и реконденсацию в
Конфликт интересов
полукокс. Разложение полимера в сверхкритической
А. Л. Максимов является главным редактором
воде приводило к превращению его в ацетон, фе-
Журнала прикладной химии, у остальных соавторов
нол, бензол, производные бензола и алифатические
конфликт интересов, требующий раскрытия в данной
алканы и алкены за счет циклизации, гидролиза и
статье, отсутствует.
окисления кратных связей частично растворенным в
воде кислородом.
Информация об авторах
— Основными достоинствами гидродехлорирова-
ния поливинилхлорид-содержащих отходов являют-
Захарян Елена Михайловна, к.х.н., м.н.с. ИНХС
ся высокая эффективность удаления органического
хлора посредством его связывания подаваемым во-
Петрухина Наталья Николаевна, к.т.н., с.н.с. ИНХС
дородом и облагораживание полученных продуктов,
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1413
Джабаров Эдуард Геннадьевич, м.н.с. ИНХС РАН,
Polym. Degrad. Stab. 2016. V. 123. P. 80-87. https://
Максимов Антон Львович, д.х.н., чл.-корр. РАН,
[10]
Kameda T., Imai K., Grause G., Mizoguchi T.,
Yoshioka T. Kinetics of the dehydrochlorination of
директор ИНХС РАН, профессор химического фа-
poly(vinyl chloride) in the presence of NaOH and
культета МГУ им. М. В. Ломоносова, ORCID: http://
various diols as solvents // Polym. Degrad. Stab. 2009.
orcid.org/0000-0001-9297-4950
Список литературы
[11]
Yoshioka T., Kameda T., Grause G., Imai K. Effect of
[1] Захарян Е. М., Петрухина Н. Н., Максимов А. Л.
compatibility between solvent and poly(vinyl chloride)
Направления вторичной химической переработки
on dechlorination of poly(vinyl chloride) // J. Polym.
поливинилхлорида (обзор). Часть 1 // ЖПХ. 2020.
Res. 2010. V. 17. P. 489-493.
Т. 93. № 9. С. 1219-1263.
[12]
Guo L., Shi G., Liang Y. Poly(ethylene glycol)s
[Zakharyan E. M., Petrukhina N. N., Maksimov A. L.
catalyzed homogeneous dehydrochlorination of
Pathways of chemical recycling of polyvinyl chloride:
poly(vinyl chloride) with potassium hydroxide //
Part 1 // Russ. J. Appl. Chem. 2020. V. 93. N 9. P. 1271-
Polym. J. 2001. V. 42. N 13. P. 5581-5587.
[2] Miyazaki M., Kamitani M., Kano J., Saito F., Inoue T.
[13]
Guo L., Shi G., Liang Y. High-quality polyene
Dechlorination of polyvinyl chloride by its grinding
films prepared by poly(ethylene glycol)s catalyzed
with KOH and NaOH // Adv. Powder Technol. 2005.
dehydrochlorination of poly(vinyl chloride) with
V. 16. N 1. P. 27-34.
potassium hydroxide // Eur. Polym. J. 1999. V. 35.
N 2. P. 215-220.
[3] Okada T., Sutoh S., Sejima K., Tomohara H.,
Mishima Sh. A useful method for thorough dehydro-
[14]
Okuda K., Yanagisawa K., Moritaka Sh.-I., Onda A.,
chlorimation of poly(vinylidene chloride-co-vinyl
Kajiyishi K. Solubilization of dechlorinated poly(vinyl
chloride) using zinc(II) oxide // Polym. Degrad. Stab.
chloride) in water by wet oxidation // Polym. Degrad.
[4] Shin Sh.-M., Yoshioka T., Okuwaki A. Dehydro-
[15]
Yoshinaga T., Yamaye M., Kito T., Ichiki T., Ogata M.,
chlorination behavior of rigid PVC pellet in NaOH
Chen J., Fujino H., Taminamura T., Yamanobe T.
solutions at elevated temperature // Polym. Degrad.
Alkaline dechlorination of poly(vinyl chloride) in
Stab. 1998. V. 61. N 2. P. 349-353.
organic solvents under mild conditions // Polym.
Degrad. Stab. 2004. V. 86. N 3. P. 541-547. https://
[5] Owen E. D., Shan M., Twigg M. V. Phase transfer
catalyzed degradation of poly(vinyl chloride). I: Product
[16]
Wu Y.-H., Zhou Q., Zhao T., Deng M.-L., Zhang J.,
characterization and handling // Polym. Degrad. Stab.
Wang Yu-Zh. Poly(ethylene glycol) enhanced
1996. V. 51. N 2. P. 151-158.
dehydrochlorination of poly(vinylchloride) // J.
Hazard. Mater. 2009. V. 163. N 2-3. P. 1408-1411.
[6] Lv B., Zhao G., Li D., Liang Ch. Dechlorination and
oxidation for waste poly(vinylidene chloride) by
[17]
He X.-L., Zhou Q., Li X.-Y., Yang P., van Kasteren J. M. N.,
hydrothermal catalytic oxidation on Pd/Ac catalyst
Wang Y.-Zh. Dechlorination of poly(vinyl chloride)
// Polym. Degrad. Stab. 2009. V. 94. N 7. P. 1047-
by 1-butyl-3-methylimidazoliumhydroxide //
1052.
Polym. Degrad. Stab. 2012. V. 97. N 2. P. 145-148.
[7] Yoshioka T., Kameda T., Imai Sh., Okuwaki A.
[18]
Ghaemy M., Gharaebi I. Study of dehydrochlorination
Dechlorination of poly(vinyl chloride) using NaOH in
of poly(vinyl chloride) in solution and the effect of
ethylene glycol under atmospheric pressure // Polym.
synthesis conditions on graft copolymerization
Degrad. Stab. 2008. V. 93. N 6. P. 1138-1141. https://
with styrene // Eur. Polym. J. 2000. V. 36. N 9.
P. 1967-1979.
[8] Singh R., Pant D. Bio-inspired dechlorination of poly
vinyl chloride // Chem. Eng. Res. Des. 2018. V. 132.
[19]
Avila A., Sanchez E. I., Gutierrez M. I. Optimal
P. 505-517.
experimental design applied to the dehydrochlorination
of poly(vinyl chloride) //Chemom. Intell. Lab. Syst.
[9] Singh R., Pant D. Polyvinyl chloride degradation
2005. V. 77. N 1-2. P. 247-250.
by hybrid (chemical and biological) modification //
1414
Захарян Е. М. и др.
[20]
Kambo H. S., Dutta A. A comparative review of
water: Preparation of benzaldehyde/acetophenone and
biochar and hydrochar in terms of production,
dechlorination // J. Clean. Prod. 2018. V. 196. P. 331-
physico-chemical properties and applications //
Renew. Sustain. Energy Rev. 2015. V. 45. P. 359-378.
[31]
Soler A., Conesa J. A., Ortuno N. Emissions of
brominated compounds and polycyclic aromatic
[21]
Shen Y. A review o hydrothermal carbonization of
hydrocarbons during pyrolysis of E-waste
biomass and plastic wastes to energy products //
debrominated in subcritical water // Chemosphere.
Biomass Bioenergy. 2020. V. 134. ID 105479. https://
2017. V. 186. P. 167-176.
[22]
Nagai Y., Smith Jr.R.L., Inomata H., Arai K. Direct
[32]
Lin Y., Ma X., Peng X., Yu Zh. Hydrothermal
obserbvation of polyvinylchloride degradation in
carbonization of typical components of municipal solid
water at temperature up to 500C and at pressures
waste for deriving hydrochars and theirs combustion
up to 700 MPa // J. Appl. Polym. Sci. 2007. V. 106.
behavior // Bioresource Technol. 2017. V. 243. P. 539-
[23]
Poerschmann J., Weiner B., Woszidlo S., Koehler R.,
[33]
Gandon-Rose G., Soler A., Aracil I., Gomez-Rico M. F.
Kopinke F.-D. Hydrothermal carbonization of
Dechlorination of polyvinyl chloride electric wires by
poly(vinyl chloride) // Chemosphere. 2015. V. 119.
hydrothermal treatment using K2CO3 in subcritical
P. 682-689.
water // Waste Manage. 2020. V. 102. P. 204-211.
[24]
Fonseca J. D., Grause G., Kameda T., Yoshioka T.
[34]
Lu J., Borrjigin S., Kumagai Sh., Kameda T., Saito Yu.,
Effects of stream on the thermal dehydrochlorination
Yoshioka T. Practical dechlorination of polyvinyl
of poly(vinyl chloride) resin and flexible poly(vinyl
chloride wastes in NaOH/ethylene glycol using an
chloride) under atmospheric // Polym. Degrad. Stab.
up-scale ball mill reactor and validation by discrete
element method simulations // Waste Manage. 2019.
V. 99. P. 31-41.
[25]
Lu J., Borrjigin S., Kumagai Sh., Kameda T., Saito Yu.,
Yoshioka T. Practical dechlorination of polyvinyl
[35]
Zhao P., Li Zh., Li T., Yan W., Ge Sh. The study of
chloride wastes in NaOH/ethylene glycol using an
nickel effect on the hydrothermal dechlorination of
up-scale ball mill reactor and validation by discrete
PVC // J. Clean. Prod. 2017. V. 152. P. 38-46.
element method simulations // Waste Manage. 2019.
V. 99. P. 31-41.
[36]
Zhao P., Li T., Yan W., Yuan L. Dechlorination of PVC
wastes by hydrothermal treatment using alkaline
[26]
Kubanova A., Lagadec A. J. M., Hawthorne S. B.
additives // Environ. Technol. 2017. V. 39. N 8. P. 977-
Dechlorination of lindaine, dieldrin, tetrachloroethane,
985.
trichloroethene and PVC in subcritical water //
Environ. Sci. Technol. 2002. V. 36. N 6. P. 1337-1343.
[37]
Xiu F.-R., Wang Y., Yu X., Li Y., Lu Y., Zhou K., He J.,
Song Zh., Gao X. A novel safety treatment strategy of
[27]
Yao Zh., Ma X. A new approach to transforming
DEHP-rich flexible polyvinyl chloride waste through
PVC waste into energy via combined hydrothermal
low-temperature critical aqueous ammonia // Sci. Total
carbonization and fast pyrolysis // Energy. 2017.
Environ. 2020. V. 708. ID 134532.
V. 141. P. 1156-1165.
[38]
Qi Y., He J., Li Y., Yu X., Xiu F.-R., Deng Y., Gao X.
[28]
Ning X., Teng H., Wang G., Zhang J., Zhang N.,
A novel treatment method of PVC-medical waste by
Huang Ch., Wang Ch. Physiochemical, structural
near-critical methanol: Dechlorination and additives
and combustion properties of hydrochar obtained
recovery // Waste Manage. 2018. V. 80. P. 1-9.
by hydrothermal carbonization of waste polyvinyl
chloride // Fuel. 2020. V. 270. ID 117526.
[39]
Zhang C.-C., Zhang F.-Sh. Enhanced dehalogenation
and coupled recovery of complex electronic display
[29]
Takeshita Y., Kato K., Takahashi K., Sato Y., Nishi S.
housing plastics by sub/supercritical CO2 // J. Hazard.
Basic study on treatment of waste polyvinyl chloride
Mater. 2020. V. 382. ID 121140.
plastics by hydrothermal decomposition in subcritical
and supercritical regions // J. Supercrit. Fluids. 2004.
[40]
Lu X., Ma X., Chen X., Yao Zh., Zhang Ch. Co-
V. 3. N 2. P. 185-193.
hydrothermal carbonization of polyvinyl chloride and
corncob for clean solid fuel production // Bioresource
[30]
Qi Y., He J., Nie W., Chen M. Partial oxidation
Technol. 2020. V. 301. ID 1222763.
treatment of waste polyvinyl chloride in critical
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1415
[41]
Shen Y. A review on hydrothermal carbonization
[52]
Zhang X., Zhang L., Li A. Co-hydrothermal
of biomass and plastic wastes to energy products //
carbonization of lignocellulosic biomass and waste
Biomass Bioenergy. 2020. V. 134. ID 105479.
polyvinyl chloride for high-quality solid fuel
production: Hydrochar properties and its combustion
[42]
Zhao P., Huang N., Li J., Cui X. Fate of sodium and
and pyrolysis behaviors // Bioresource Technol. 2019.
chlorine during the co-hydrothermal carbonization
V. 294. ID 1222113.
of high-alkali coal and polyvinyl chloride // Fuel
Process. Technol. 2020. V. 199. ID 106277. https://
[53]
Qian Sh., Wang H., Zarei E., Sheng K. Effect of
hydrothermal pretreatment on the properties of moso
[43]
Xiu F.-R., Yu X., Qi Y. A high-efficiency and low-
bamboo particles reinforced polyvinyl chloride
temperature subcritical water dechlorination strategy
composites // Composites. Part B. 2015. V. 82. N 1.
of polyvinyl chloride using coal fly ash (CFA) and
P. 23-29.
coal gangue (CG) as enhancers //J. Clean. Prod. 2020.
V. 260. ID 121085.
[54]
Li H., Fang Zh., Luo J., Yang S. Direct conversion of
biomass components to the biofuel methyl levulinate
[44]
Ahmad F., Silva E. L., Varesche M. B. A. Hydrothermal
catalyxed by acd-base bifunctional zirconia-zeolites //
processing of biomass for anaerobic digestion —
Appl. Catal. B: Environmental. 2017. V. 200. P. 182-
A review // Renew. Sustain. Energy Rev. 2018.
V. 98. P. 108-124.
[55]
Huang N., Zhao P., Ghosh S., Fedyukhin A. Co-
hydrothermal carbonization of polyvinyl chloride and
[45]
Tekin K., Karagoz S., Bektas S. A review of
moist biomass to remove chlorine and inorganic for
hydrothermal biomass processing // Renew. Sustain.
clear fuel production // Appl. Energy. 2019. V. 240.
Energy Rev. 2014. V. 40. P. 637-687.
P. 882-892.
[46]
Wang T., Zhai Y., Zhu Y., Li C., Zeng G. A review
[56]
Liu K., Zhang F.-Sh. Innovative leaching of cobalt
of the hydrothermal carbonization of biomass
and lithium from spent lithium-ion batteries and
waste for hydrochar formation: Process conditions,
simultaneous dechlorination of polyvinyl chloride
fundamentals, and physicochemical properties //
in subcritical water // J. Hazard. Mater. 2016. V. 316.
Renew. Sustain. 2018. V. 90. P. 223-247.
P. 19-25.
[47]
Kang S., Li X., Fan J., Chang J. Characterization of
[57]
Yu J., Sun L., Ma Ch., Qiao Y., Yao H. Thermal
hydrochars produced by hydrothermal carbonization
degradation pf PVC: A review // Waste Manage. 2016.
of lignin, cellulose, d-xylose, and wood meal // Ind.
V. 48. P. 300-314.
Eng. Chem. Res. 2012. V. 51. N 26. P. 9023-9031.
[58]
Vymazal Z., Mastny L., Vymazalova Z. Study of the
[48]
Yao Zh., Ma X. Characteristics of co-hydrothermal
effect of the chloroallyl group content on the thermal
carbonization on polyvinyl chloride wastes with
dehydrochlorination of PVC // Eur. Polym. J. 1985.
bamboo // Bioresource Technol. 2018. V. 247. P. 302-
V. 21. N 8. P. 747-755.
[49]
Barbier J., Charon N., Loppinet-Serani A.,
[59]
Bengough W. I., Grant G. F. The thermal degradation
Mane L., Ponthus J., Courtiade M., Ducrozet A.,
of polyvinylchloride in solution-V. Effect of
Quoinead A.-A., Cansell F. Hydrothermal
dimethylformamide as solvent // Eur. Polym. J. 1968.
conversion of lignin compounds. A detailed study of
V. 4. N 4. P. 521-535.
fragmentation and condensation reaction pathways //
Biomass Bioenergy. 2012. V. 46. P. 479-491. https://
[60]
Hollander A., Zimmermann H., Behnisch J. Chemical
dehydrochlorination of poly(vinyl chloride) by LiCl
[50]
Shen Y. Dechlorination of poly(vinyl chloride) wastes
in dimethylformamide // Polym J. 1992. V. 33. N 3.
via hydrothermal carbonization with for clean solid
P. 637-642.
fuel production // Ind. Eng. Chem. Res. 2016. V. 55.
N 44. P. 11638-11644.
[61]
Фихман В. Д., Вайман Э. Я., Пакшвер А. Б.,
Минскер К. С. Кинетика дегидродехлорирования
[51]
Shen Y., Yu Sh., Chen X., Ge X., Chen M. Hydrothermal
поливинилхлорида в растворах в диметилформами-
carbonization of medical wastes and lignocellulosic
де // Высокомолекуляр. соединения. 1972. Т. 14A.
biomass for solid fuel production from lab-scale and
№ 11. С. 2376-2384 [Fikhman V. D., Vaiman E. Ya.,
pilot-scale // Energy. 2017. V. 118. P. 312-323.
Pakshver A. B., Minsker K. S. Dehydrochlorination
kinetics of polyvinylchloride (PVC) solutions in
1416
Захарян Е. М. и др.
dimethylformamide // Polym. Sci. Ser. A. 1972. V. 14.
N 11. P. 2376-2384.
[72]
Kamo T., Kondo Y., Kodera Y., Sato Y., Kushiyama S.
[62]
Kamo T. Thermal decomposition of poly(vinyl
Effect of solvent on degradation of poly(vinyl chloride)
chloride) in organic solvents under high pressure
// Polym. Degrad. Stab. 2003. V. 81. N 2. P. 187-196.
// Polym. Degrad. Stab. 2013. V. 98. N 2. P. 502-
[73]
Williams P. T., Slaney E. Analysis of products from the
pyrolysis and liquefaction of single plastics and waste
[63]
Abdullin M. I., Gatallin R. F., Kefeli K. S.,
plastic mixtures // Resour. Conver. Recy. 2007. V. 51.
Rasumovskii S. D. Effect of ozone on poly(vinyl
N 4. P. 754-769.
chloride) degradation // Eur. Polym. J. 1978. V. 14.
N 10. P. 811-816.
[74]
Ali M. F., Siddiqui M. N. Thermal and catalytic
decomposition behavior of PVC mixed plastic waste
[64]
Шаповал Г. С., Томилов А. П., Пуд А. А., Бацало-
with petroleum residue // J. Anal. Appl. Pyrolysis.
ва К. В. Электрохимическая восстановительная де-
2005. V. 74. P. 282-289.
струкция поливинилхлорида // Высокомолекуляр.
соединения. 1987. Т. 29A. № 7. С. 1424-1430
[75]
Karayildirim T., Yanik J., Ucar S., Saglam M.,
[Shapoval G. S., Tomilov A. P., Pud A. A.,
Yuksel M. Conversion of plastics/HVGO mixtures to
Batsalova K. V. The electrochemical reductive
fuels by two-step processing // Fuel Process. Technol.
degradation of polyvinyl chloride // Polym. Sci. 1987.
2001. V. 73. N 1. P. 23-35.
Ser. A. V. 29. N 7. P. 1564-1572.
[76]
[65]
Giacomucci L., Raddadi N., Soccio M., Lotti N.,
Conversion of polymer to fuels in a refinery stream
Fava F. Polyvinyl chloride biodegradation by
// Polym. Degrad. Stab. 2002. V. 75. N 1. P. 161-171.
Pseudomonas citronellolis and Bacillus flexus // New
biotechnol. 2019. V. 52. P. 35-41.
[77]
Karagöz S., Karayildirim T., Uçar S., Yuksel M.
Liquefaction of municipal waste plastics in VGO over
[66]
Gong J, Yao K., Liu J., Wen X., Chen X., Jiang Zh.,
acidic and non-acidic catalysts // Fuel. 2003. V. 82.
Mijowska E., Tang T. Catalytic conversion of linear
N 4. P. 415-423.
low density polyethylene into carbon nanomaterials
under the combined catalysis of Ni2O3 and poly(vinyl
[78]
Lopez G., Artetxe M., Amutio M., Alvarez J., Bilbao J.,
chloride) // Chem. Eng. J. 2013. V. 215-216. P. 339-
Olazar M. Recent advances in the gasification of waste
plastics. A critical overview // Renew. Sustain. Energy
[67]
Starnes W. H., Schilling F. C., Plitz K. B.,
Rev. 2018. V. 82. N 1. P. 576-596.
Hartless R. L., Bovey F. A. Structural selectivities in
the reduction of poly(vinyl chloride) with lithium
[79]
Slapak M. J., van Kasteren J. M. N., Drinken-
aluminium hydride and tri-n-butylin hydride //
burg B. A. A. H. Hydrothermal recycling of PVC
Macromolecules. 1979. V. 12. P. 13-19.
in a bubbling fluidized bed reactor: The influence
[68]
Hjertberg Th., Wendel A. Reduction of poly(vinyl
of bed material and temperature // Polym. Adv.
chloride) with tri-n-butilin hydride // Polym. J. 1982.
V. 23. P. 1640-1645.
[69]
Contreras J. M., Martinez G., Millan J. Local chain
configuration dependence of the mechanisms of
[80]
Slapak M. J., van Kasteren J. M. N., Drinken-
chemical reactions of PVC.8. New results from the
burg B. A. A. H. Design of a process for steam
reductive dechlorination reaction // Polym. J. 2001.
gasification of PVC waste // Resour. Conver. Recy.
V. 42. N 25. P. 9867-9876.
2000. V. 30. N 2. P. 81-93.
[70]
Keane M. A. Review. Catalytic conversion of waste
[81]
Pat. Japan 7244546 (publ. 1972). Gasification of waste
plastics: Focus on waste PVC // J. Chem. Technol.
plastics.
Biotechnol. 2007. V. 82. P. 787-795.
[82]
Borgianni C., De Filippis P., Pochetti F., Paolucci M.
Gasification process of waste containing PVC // Fuel.
[71]
Kamo T., Kodera Y. Effect of hydrogen transferred
2002. V. 81. N 14. P. 1827-1833.
from solvent and gaseous hydrogen on thermal
decomposition of dehydrochlorinated poly(vinyl
[83]
chloride) // Polym. Degrad. Stab. 2005. V. 87. N 1.
of steam and sodium hydroxide for the production of
Направления вторичной химической переработки поливинилхлорида (обзор). Часть 2
1417
hydrogen on gasification of dehydrochlorinated poly(vinyl
[92]
Cho M.-H., Mun T.-Y., Kim J.-S. Production of low-
tar producer gas from air gasification of mixed plastic
waste in a two-stage gasifier using olivine combined
[84]
Lin S.-Y., Suzuki Y., Hatano H. Hydrogen production
with activated carbon // Energy. 2013. V. 58. P. 688-
from hydrocarbon by integration of water-carbon
reaction and carbon dioxide removal (HyPr-RING
[93]
Cho M.-H., Mun T.-Y., Choi Y.-K., Kim J.-S. Two-stage
method) // Energy and Fuels. 2001. V. 15. N 2. P. 339-
air gasification of mixed plastic waste: Olivine as the
bed material and effects of various additives and a
[85]
nickel-plated distributor on the tar removal // Energy.
Recovery of copper from PVC multiwire cable waste
2014. V. 70. P. 128-134.
P. 488-496.
[94]
Zhou Ch., Stuermer Th., Gunarathne R., Yang W.,
Blasiak W. Effect of calcium oxide on high-
[86]
Sivakumar P., Jung H., Tierney J.W., Wender I.
temperature steam gasification if municipal solid
Liquefaction of lignocellulosic and plastic wastes with
waste // Fuel. 2014. V. 122. P. 36-46.
coal using carbon monoxide and aqueous alkali // Fuel
Process. Technol. 1996. V. 49. N 1-3. P. 219-232.
[95]
Dou B., Wang K., Jiang B., Song Y., Zhang Ch.,
Chen H., Xu Y. Fluidized-bed gasification combined
[87]
Zhang Sh., Yu Y. Dechlorination behavior on the
continuous sorption-enhanced steam reforming
recovery of useful resources from WEEE by the
system to continuous hydrogen production from waste
steam gasification in the molten carbonates // Procedia
plastic // Int. J. Hydrogen Energy. 2016. V. 41. N 6.
Environ. Sci. 2016. V. 31. P. 903-910.
P. 3803-3810.
[88]
Chen Sh., Meng A., Long Y., Zhou H., Li Q., Zhang Y.
[96]
Baloch H. A., Yang T., Li R., Nizamuddin S., Kai X.,
TGA pyrolysis and gasification of combustible
Bhutto A. W. Parametric study of co-gasification
municipal solid waste // J. Energy Institute. 2015.
of ternary blends of rice straw, polyethylene and
V. 88. N 3. P. 332-343.
polyvinylchloride // Clean Technol. Environ. Policy.
2016. V. 18. P. 1031-1042.
[89]
Cho M.-H., Choi Y.-K., Kim J.-S
. Air gasification of
PVC (polyvinyl chloride)-containing plastic waste
[97]
in a two-stage gasifier using Ca-based additives and
and biomass in a dual fluidized-bed steam gasifier:
Ni-loaded activated carbon for the production of clean
Possible interactions of fuels // Energy Fuels. 2013.
V. 27. N 6. P. 3261-3273.
P. 586-593.
[98]
Lee J. W., Yu T. U., Lee J. W., Moon J. H., Jeong H. J.,
[90]
Kim J.-W., Mun T.-Y., Kim J.-O., Kim J.-S. Air
Park S. Sh., Yang W., Lee U. D.
Gasification of
gasification of mixed plastic for the production of
mixed plastic wastes in a moving-grate gasifier and
producer gas with low tar and a high caloric value //
application of the producer gas to a power generation
Fuel. 2011. V. 90. N 6. P. 2266-2272.
engine // Energy Fuels. 2013. V. 27. N 4. P. 2092-
[91]
Cho M.-H., Mun T.-Y., Kim J.-S. Air gasification of
[99]
Hu B., Huang Q., Buekens A., Y., Yan J.
Co-gasification
mixed plastic wastes using calcined dolomites and
of municipal solid waste with high alkali coal char in a
activated carbon in a two-stage gasifier to reduce tar //
Energy. 2013. V. 53. 299-305.