Радиохимия, 2019, т. 61, N 1, c. 13-17
13
Растворимость нитрата радия в растворах азотной кислоты
© П. С. Буткалюк*а, И. Л. Буткалюка, Р. А. Кузнецов**б, А. С. Куприянова, Р. Г. Абдуллова
а Научно-исследовательский институт атомных реакторов, 433510, Димитровград Ульяновской обл.,
Западное шоссе, д. 9; * e-mail: orip@niiar.ru
б Димитровградский инженерно-технологический институт - филиал Национального исследовательского ядерного
университета «МИФИ», 433511, Димитровград Ульяновской обл., ул. Куйбышева, д. 294;
** e-mail: RAKuznetsov@mephi.ru
Получена 15.01.2018, после доработки 02.03.2018, принята к публикации 05.03.2018
УДК 621.039.3
Определена растворимость нитрата радия в растворах HNO3 высоких концентраций. Установлено,
что растворимость нитрата радия уменьшается от ~1·10-3 до ~2·10-5 моль/л при увеличении концентра-
ции HNO3 от 13.5 до 22 моль/л. Растворимость нитрата радия в исследованном диапазоне концентраций
HNO3 в 3-6 раз выше растворимости нитрата бария в аналогичных условиях.
Ключевые слова: радий-226, нитрат радия, азотная кислота.
DOI: 10.1134/S0033831119010039
Поведение нитрата радия в системе HNO3-
Корректность применяемой методики экспери-
Ra(NO3)2-H2O аналогично поведению нитратов
мента была подтверждена в серии предварительных
стронция и бария, хорошо растворимых в разбав-
экспериментов с использованием бария как имитато-
ленной и плохо растворимых в концентрированной
ра радия. Результаты этого эксперимента были ис-
HNO3 [1, 2]. Растворимость Ba(NO3)2 в HNO3 под-
пользованы и для выбора условий эксперимента с
робно изучена в работах [3-5]. По данным работы
радием.
[3], в водных растворах HNO3 она уменьшается от
Материалы и методы
9.2 мас% в дистиллированной воде до 1.9·10-5% в
92%-ной HNO3 при 25°С. Растворимость Ra(NO3)2 в
Для исследования использовали нитрат и карбо-
воде немного больше растворимости Ba(NO3)2 и
нат бария марки х.ч., дымящую HNO3 с массовым
составляет
12.2 мас%
(13.9 г/100 г воды, или
содержанием HNO3 99.65%. Массовую долю кисло-
0.388 моль/л) при 20°С [6]. Надежные количествен-
ты определяли потенциометрическим титрованием
ные данные о растворимости Ra(NO3)2 в концентри-
раствором NaOH с использованием программы НПП
рованных растворах HNO3 в доступной нам литера-
«СЕМИКО» [9], а также по известной зависимости
туре отсутствуют. Эти данные необходимы для оп-
плотности от температуры и концентрации раствора
ределения параметров разрабатываемого технологи-
[10].
ческого процесса производства 227Ac и 228Th, преду-
Аликвоты растворов отбирали весовым методом с
сматривающего облучение 226Ra в высокопоточном
использованием аналитических весов Sartorius
реакторе СМ и последующее отделение продуктов
ED224S (Sartorius AG, Германия) c ценой деления
активации от материала мишени методом кристал-
±0.0001 г. Значения pH растворов измеряли при по-
лизации малорастворимых нитратов [7, 8].
мощи рН-метра HANNA-HI 2210 со стеклянными
Целью данной работы является эксперименталь-
электродами Hi 1131B (Hanna Instruments). Калиб-
ное определение растворимости Ra(NO3)2 в системе
ровку pH-метра осуществляли при помощи стандарт-
HNO3-H2O при концентрациях HNO3 65-97 мас%.
ных буферных растворов c pH 9.18, 6.86 и 4.01 (ООО
Экспериментальная часть
«Химтитры»).
Экспериментальное определение растворимости
Содержание бария в растворе определяли c ис-
нитратов проводили с использованием известной
пользованием радиоактивного изотопа 133Ва (про-
методики определения растворимости (измерение
изводства НИИАР) c известной удельной активно-
концентрации элемента в насыщенном растворе),
стью. Объемную активность 133Ва определяли мето-
адаптированной к реализации в условиях радиацион-
дом γ-спектрометрии при помощи полупроводнико-
но-защитных боксов. Первая серия экспериментов
вого γ-спектрометра с коаксиальным Ge-Li-детек-
предусматривала достижение равновесия «сверху»,
тором типа ДГДК-В150 (энергетическое разрешени-
т.е. проводили кристаллизацию Ra(NO3)2 при увели-
ем 3.0 кэВ по линии 1.3 МэВ). Для градуировки спек-
чении концентрации HNO3 за счет добавления дымя-
трометра использовали образцовые радиоактивные
щей HNO3 к водному раствору Ra(NO3)2. Во второй
растворы (ОРР). Расшифровку γ-спектров проводили
серии экспериментов равновесие достигалось «сни-
в полуавтоматическом режиме при помощи стан-
зу», т.е. проводилось частичное растворение заранее
дартного программного обеспечения фирмы
сформированного осадка Ra(NO3)2.
«ГринСтар».
14
П. С. Буткалюк и др.
Содержание 226Ra определяли с помощью α-спек-
Таблица 1. Результаты атомно-эмиссионного анализа
трометра на основе кремниевых PIPS-детекторов
раствора Ra(NO3)2
ПДПА-1К (ОАО «ИФТП», Дубна, Россия) и ампли-
Элемент
Содержание, % от массы 226Ra
тудного анализатора импульсов АЦП-8К-И2. Образ-
Al
0.08
цы (источники) для измерения α-излучения 226Ra го-
Ca
0.45
товили нанесением 10-25 мкл анализируемого рас-
Cr, Fe, Mg, Mn
<0.08
твора на подложку из нержавеющей стали с после-
Cu
<0.03
дующим упариванием и прокалкой на электроплитке
Ni, Ba, Pt
<0.25
с открытой спиралью для отгонки 222Rn. Перед изме-
Pb
0.50
Na
0.75
рением α-спектра подготовленный образец выдержи-
Si
0.25
вали в течение 1.5 ч, что необходимо для снижения
активности короткоживущих дочерних продуктов
133Ba ~4·106 Бк. Элюирование солей бария проводили
распада 222Rn. Для градуировки спектрометров ис-
раствором 0.02 моль/л H4ЭДТА, pH которого доводи-
пользовали образцовые спектрометрические α-ис-
ли раствором NH3 до значения 8.8 (условия разделе-
точники (ОСАИ).
ния взяты из работы [12]), затем элюировали радий
Содержание нерадиоактивных примесей в препа-
аналогичным раствором с pH 9.7. Аффинажную очи-
ратах 226Ra(NO3)2 определяли методом атомно-эмис-
стку 226Ra на второй колонке проводили путем сорб-
сионного анализа с использованием установки, пред-
ции из 0.02 моль/л (NH4)2ЭДТА в ацетатно-амми-
ставляющей собой искровой источник возбуждения
ачном буферном растворе с pH 4.5. Колонку промыва-
спектров ИВС-28, смонтированный внутри радиаци-
ли бидистиллированной водой, затем раствором
онно-защитного бокса, и спектрограф ИСП-1, соеди-
0.1 моль/л HNO3. Радий элюировали раствором
ненный с этим источником при помощи оптического
8 моль/л HNO3. Полученный раствор Ra(NO3)2 упари-
выхода.
вали досуха и нагревали в течение 30 мин при темпе-
ратуре 300°C для разложения следов солей аммония.
Методика очистки радия
Сухой остаток растворяли в 0.5 мл 2.5 моль/л HNO3.
В работе использовали препарат нитрата радия,
Содержание примесей в полученном препарате
выделенный из отработавших срок службы радиевых
определяли методом атомно-эмиссионного анализа,
источников γ-излучения. Вскрытие платиновых обо-
результаты которого приведены в табл. 1. Суммар-
лочек источников осуществляли путем растворения в
ное содержание примесей в полученном образце со-
царской водке. Полученный препарат радия содер-
ставило менее 3.1% относительно массы 226Ra. Со-
жал значительные количества солей платины и дру-
держание примесей Pb и Ba (~0.5 и <0.25%), нитраты
гих примесей. Очистку препарата от примесей про-
которых способны кристаллизоваться совместно с
водили в два этапа.
Ra(NO3)2 и, таким образом, влиять на результаты
На первом этапе проводили совместное осажде-
определения его растворимости, было незначитель-
ние Ra(NO3)2 с Pb(NO3)2 из раствора концентриро-
ным.
ванной HNO3 по методике, аналогичной описанной в
работе [11]. К 50 мл раствора Ra(NO3)2 (~10 мг) в
Определение растворимости Ba(NO3)2
в растворах HNO3
7 моль/л HNO3 добавляли в качестве носителя 500 мг
Pb(NO3)2, затем добавляли 50 мл 15.6 моль/л HNO3.
Для определения времени достижения равнове-
Полученный раствор упаривали до объема 75 мл,
сия между осадком Ba(NO3)2 и раствором была оп-
охлаждали в течение 4 ч. Затем добавляли 25 мл
ределена скорость изменения концентрации бария в
15.6 моль/л HNO3. Операции упаривания, охлажде-
растворе. В четыре пробирки вместимостью 10 мл
ния и добавления кислоты повторили еще 4 раза. По-
вносили по 0.50 мл раствора, содержащего 4.95 мг
лученный осадок, содержащий нитраты радия и
Ba(NO3)2, меченного радионуклидом 133Ba. Затем в
свинца, отделили от раствора декантацией и промы-
первые две пробирки добавляли 1.5749 и 1.5747 г
ли тремя порциями 15.6 моль/л HNO3.
деионизованной воды и 4.9440 и 4.9580 г дымящей
Второй этап очистки включал хроматографиче-
HNO3. В третью и четвертую пробирки добавляли
скую очистку радия от свинца и щелочноземельных
7.6089 и 7.6145 г 99.65%-ной дымящей HNO3. Во
элементов на двух колонках с катионообменной смо-
всех четырех пробирках выпал осадок. Растворы с
лой Dowex 50×8. Препарат растворяли в растворе
осадками в третьей и четвертой пробирках выдержи-
0.02 моль/л этилендиаминтетрауксусной кислоты
вали в течение 6 сут, от растворов отбирали аликво-
ты массой 0.9053 и 1.1487 г, затем в пробирки добав-
(H4ЭДТА) в ацетатно-аммиачном буферном растворе
ляли 2.4008 и 2.3205 г деионизованной воды соот-
с pH 4.5, из которого проводили сорбцию радия и
ветственно. Эксперименты проводили при темпера-
бария. Для удобства контроля процесса очистки ра-
дия к препарату перед подачей на колонку добавляли
туре 24.6 ± 0.9°С.
раствор, содержащий 133Ba(NO3)2, с активностью
От полученных растворов через заданные проме-
Растворимость нитрата радия в растворах азотной кислоты
15
жутки времени отбирали аликвоты для определения
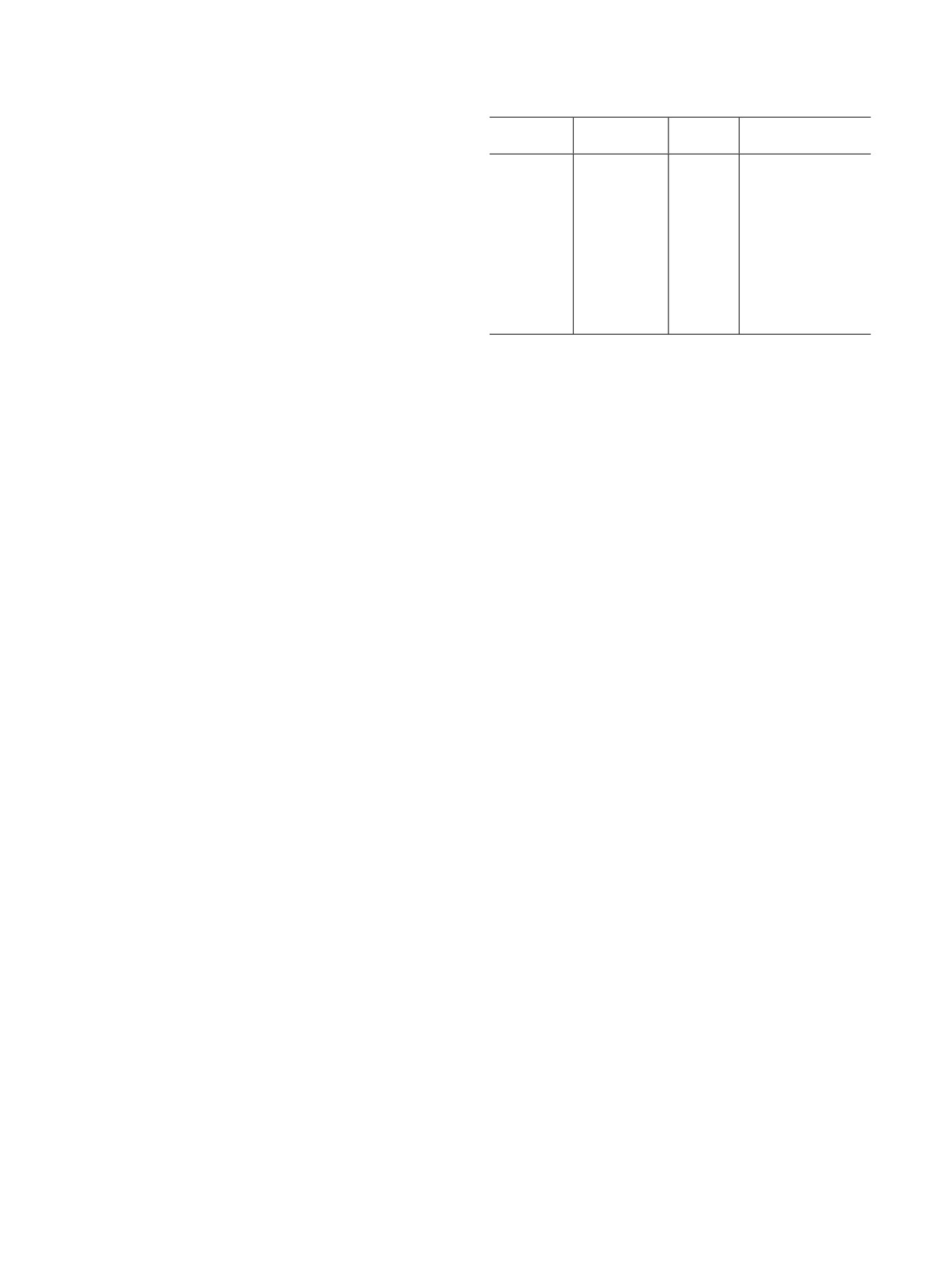
Таблица 2. Составы исходных растворов
содержания бария (по 133Ba). Перед отбором каждой
Номер
m(HNO3,
m(H2O), г
V[226Ra(NO3)2], мл
аликвоты содержимое пробирок центрифугировали
пробирки
99.65%), г
в течение 10 мин c ускорением ~1500g.
1
4.5620
0
0.100
Учитывая результаты, полученные для Ba(NO3)2,
2
4.1207
0.120
0.100
в последующих экспериментах исходили из того,
3
3.9404
0.250
0.100
4
3.6313
0.250
0.200
что равновесие при осаждении Ba(NO3)2 достигается
5
3.3204
0.350
0.200
за ~24 ч, а при разбавлении сформировавшегося
6
3.1817
0.450
0.200
осадка - менее чем за 2 ч.
7
3.1837
0.600
0.200
Эксперименты по определению растворимости
8
2.9976
0.750
0.200
Ba(NO3)2 при достижении равновесия «сверху» вы-
9
2.8646
0.700
0.400
полняли по следующей методике.
10
2.7256
0.800
0.400
В две пробирки вместимостью 10.0 мл вносили
но тринадцать последовательных измерений при
раствор, содержащий 19 мг Ba(NO3)2, меченный ра-
изменении концентрации HNO3 от 94 до 62%.
дионуклидом 133Ba, добавляли 6.2255 и 6.2247 г ды-
мящей HNO3 с массовой долей 99.65%. Растворы пе-
Определение растворимости Ra(NO3)2
ремешивали и оставляли для кристаллизации на 24 ч.
при достижении равновесия «сверху»
После центрифугирования отбирали от растворов
В 10 пробирок вместимостью 10 мл последова-
аликвоты объемом 0.100 мл и добавляли в каждую
тельно вносили навески 99.65%-ной HNO3, воды и
пробирку по 0.200 мл деионизованной воды. Содер-
раствора Ra(NO3)2 в 1 моль/л HNO3 с объемной ак-
жимое пробирок осторожно перемешивали и выдер-
тивностью 226Ra ~2.5 мКи/мл. Состав исходных рас-
живали в термостате при 20 ± 1°C в течение 4 ч. За-
творов приведен в табл. 2.
тем процедуру отбора пробы повторяли. Содержание
Ba в растворе определяли по активности 133Ba мето-
Пробирки выдерживали в течение 24 ч при тем-
дом γ-спектрометрического анализа, концентрацию
пературе 19.2 ± 0.5°С, поддерживаемой с помощью
HNO3 определяли методом потенциометрического
водяной бани. Затем от каждого раствора отбирали
титрования раствором NaOH. Всего было отобрано
аликвоту объемом 0.20 мл в центрифужную пробир-
10 проб, что соответствовало изменению концентра-
ку. Аликвоты центрифугировали в течение 10 мин c
ции HNO3 от 93 до 62%.
ускорением ~1500g для удаления следов твердой
фазы. От растворов в центрифужных пробирках от-
Определение растворимости Ra(NO3)2 при
бирали по 5 аликвот массой 40-60 мг каждая. Одну
достижении равновесия «снизу»
аликвоту из каждой центрифужной пробирки раз-
В две тарированные пробирки вместимостью
бавляли весовым методом и использовали для опре-
10 мл, содержащие 6.8062 и 6.8901 г 99.65%-ной
деления объемной активности 226Ra методом α-спек-
HNO3, вносили по 0.200 мл раствора, содержащего
трометрии, остальные использовали для определе-
2.4 мг 226Ra. Растворы перемешивали и оставляли
ния концентрации HNO3 методом потенциометриче-
для кристаллизации на 1 сут. Далее от растворов над
ского титрования раствором NaOH.
осадками Ra(NO3)2 отбирали аликвоты объемом
Расчет растворимости
0.200 мл в пробирки, которые центрифугировали в
и оценка погрешности измерений
течение 10 мин c ускорением ~1500g для удаления
следов твердой фазы. От этих растворов отбирали по
Массовую долю Ra(NO3)2 в растворах рассчиты-
5 аликвот массой 40-60 мг каждая. Одну аликвоту
вали по формуле
использовали для определения объемной активности
ω[Ra(NO3)2] = 100%·M[Ra(NO3)2]A(226Ra)mразб/
226Ra методом α-спектрометрического анализа, ос-
/[M(226Ra)mαmаликв·3.7·1010],
тальные использовали для определения концентра-
где M[Ra(NO3)2]
- молярная масса Ra(NO3)2,
ции HNO3 методом потенциометрического титрова-
ния раствором Na2B4O7.
350.035 г/моль; M(226Ra)
- атомная масса 226Ra,
226.025 г/моль; A(226Ra) - активность 226Ra в источ-
В пробирки с Ra(NO3)2 вносили по 0.200 мл деио-
нике, Бк; mаликв - масса аликвоты до разбавления, г;
низованной воды и осторожно перемешивали содер-
mразб
- масса раствора после разбавления, г; mα -
жимое пробирок (покачиванием) в течение 1 мин.
масса препарата, нанесенного на источник для
Повторный отбор проб производили через 1-3 сут.
α-спектрометрии из разбавленного раствора, г.
Эксперименты проводили при температуре 25 ± 3°С.
Термостатирование в более узком диапазоне темпе-
Массовую долю HNO3 рассчитывали по формуле
ратур в условиях радиационно-защитного бокса в
ω(HNO3) = 100%·M(HNO3)VNaOHCNaOH/mT,
течение длительного (более двух недель) времени
было технически невозможно. Всего было проведе-
где M(HNO3) - молярная масса HNO3, 60.013 г/моль;
16
П. С. Буткалюк и др.
mT - масса аликвоты, использованной для титрова-
ния; CNaOH - концентрация раствора NaOH; VNaOH -
объем NaOH, использованного на титрование алик-
воты раствора.
Для вычисления молярных концентраций HNO3 и
Ra(NO3)2 использовали табличные данные зависимо-
сти плотности растворов HNO3 от массовой доли и
температуры ([13], табл. 2-66). Так как во всем ис-
следуемом диапазоне концентраций HNO3 раствори-
мость радия мала, отличие плотности насыщенного
раствора Ra(NO3)2 от плотности HNO3 считали пре-
небрежимо малым и при вычислении молярной кон-
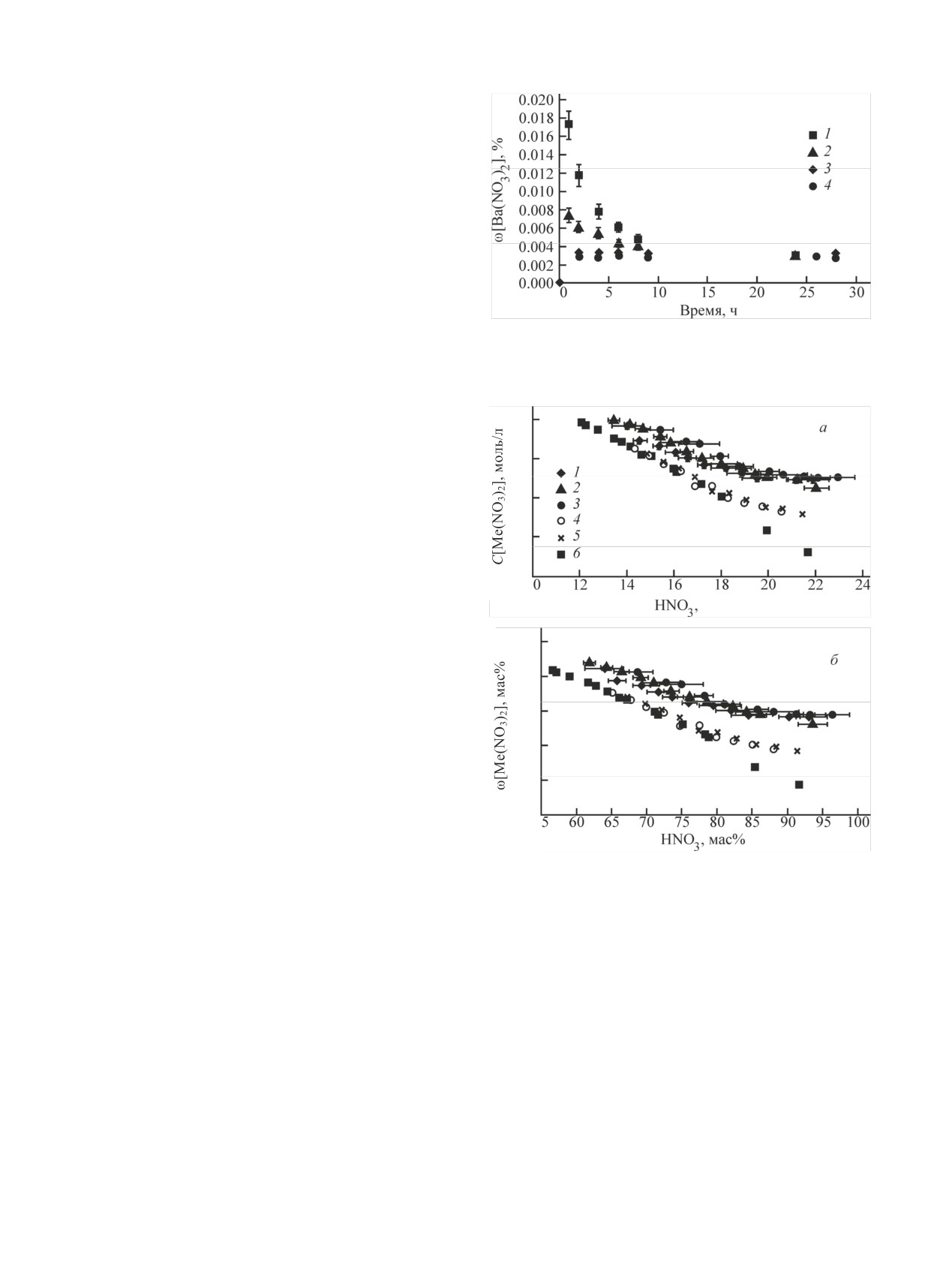
Рис. 1. Скорость изменения концентрации Ba(NO3)2 в растворе
центрации не учитывали.
при достижении равновесия «сверху» (1, 2, увеличение кон-
учиты-
центрации HNO3 от 0 до 15.75 моль/л) и «снизу» (3, 4, разбав-
При вычислении массовой доли Ra(NO3)2
ление HNO3 от 22.09 до 16.25 моль/л).
вали следующие компоненты погрешности: погреш-
ность отбора аликвот из исходного и разбавленного
10-3
растворов
(~0.3-0.8%), погрешность разбавления
(0.08-0.2%), погрешность эталона 226Ra (~3.2%), по-
10-4
грешность обработки α-спектра (1-2%). Для сниже-
ния случайной погрешности, возникающей за счет
10-5
статистической природы α-распада, набор спектра
проводили до получения статистики в 105-106 им-
10-6
пульсов. Для снижения погрешности отбора аликвот
источники для α-спектрометрии готовили весовым
10-7
методом. При этом вводили поправку на испарение
1
раствора с поверхности источника за время взвеши-
моль/л
вания путем построения градуировочного графика
массы от времени и экстраполирования его на время
10-1
нанесения раствора на источник. Суммирование по-
грешностей отдельных величин проводили по зако-
10-2
ну сложения ошибок [14].
10-3
При вычислении массовой доли HNO3 учитывали
погрешности отбора аликвоты (0.2-0.7%), погреш-
10-4
ность определения концентрации (0.5%) и объема
титранта (0.9-1.8%). Случайную компоненту по-
10-5
грешности оценивали, проводя 3-4 параллельных
титрования для каждой точки. С учетом небольшого
10-6
5
числа параллельных титрований вклад случайной
компоненты в суммарную погрешность определения
массовой доли HNO3
был наибольший.
Рис. 2. Зависимость растворимости (а - моль/л, б - мас%)
Ra(NO3)2 (1-3) и Ba(NO3)2 (4, 5) от концентрации HNO3; 6 -
Иные потенциальные погрешности, связанные с
литературные данные [3] для Ва(NO3)2.
сорбцией радионуклида на стенках посуды, поверх-
во
ности наконечников пипеток, испарением HNO3
лученных данных видно, что при осаждении
время переноса раствора из пробирок в колбы, при-
Ba(NO3)2 дымящей HNO3 концентрация бария выхо-
нимали пренебрежимо малыми и в суммарную по-
дит на постоянный уровень через 24-30 ч. При раз-
грешность определения не включали.
бавлении раствора (частичное растворение осадка)
равновесие устанавливается менее чем через 2 ч.
Результаты и обсуждение
На рис. 2 представлена зависимость растворимо-
Определение растворимости Ba(NO3)2
сти Ba(NO3)2 от концентрации HNO3 в системе
в растворах HNO3
Ba(NO3)2-H2O-HNO3. Удовлетворительное соответ-
На рис. 1 представлен график изменения концен-
ствие полученных результатов с данными работы [3]
трации Ba(NO3)2 в растворе после добавления дымя-
свидетельствует о корректности используемой нами
щей HNO3 и после разбавления полученного насы-
методики определения растворимости нитратов ба-
щенного раствора дистиллированной водой. Из по-
рия и радия.
Растворимость нитрата радия в растворах азотной кислоты
17
Таблица 3. Растворимость Ra(NO3)2 в системе Ra(NO3)2-H2O-HNO3
Номер
Условия
Концентрация HNO3,
Растворимость Ra(NO3)2
T, °С
серии
равновесия
мас%
моль/л
мас%
моль/л
24.0
93.1 ± 2.5
21.9 ± 0.7
(6.8 ± 0.4)·10-4
(2.89 ± 0.17)·10-5
24.0
90.3 ± 1.5
21.2 ± 0.4
(7.0 ± 0.4)·10-4
(2.94 ± 0.17)·10-5
26.5
84.5 ± 2.5
19.5 ± 0.7
(7.6 ± 0.4)·10-4
(3.17 ± 0.17)·10-5
26.5
82.0 ± 2.3
18.8 ± 0.6
(1.02 ± 0.06)·10-3
(4.22 ± 0.25)·10-5
25.8
79.6 ± 2.2
18.2 ± 0.6
(1.45 ± 0.08)·10-3
(6.0 ± 0.3)·10-5
1
«Снизу»
24.0
76.1 ± 1.1
17.3 ± 0.3
(1.71 ± 0.10)·10-3
(7.0 ± 0.4)·10-5
28.0
73.7 ± 1.5
16.6 ± 0.4
(2.55 ± 0.15)·10-3
(10.3 ± 0.6)·10-5
26.0
71.8 ± 1.8
16.1 ± 0.5
(3.49 ± 0.20)·10-3
(14.1 ± 0.8)·10-5
27.0
69.3 ± 1.2
15.4 ± 0.3
(5.3 ± 0.3)·10-3
(2.11 ± 0.12)·10-4
23.0
65.9 ± 1.3
14.6 ± 0.4
(7.3 ± 0.4)·10-3
(2.89 ± 0.16)·10-4
23.0
64 ± 3
14.0 ± 0.7
(1.72 ± 0.10)·10-2
(6.8 ± 0.4)·10-4
24.0
93.6 ± 2.1
22.0 ± 0.6
(4.29 ± 0.25)·10-4
(1.82 ± 0.11)·10-5
24.5
90.8 ± 1.7
21.3 ± 0.5
(6.6 ± 0.4)·10-4
(2.78 ± 0.16)·10-5
26.5
86.1 ± 1.6
19.9 ± 0.4
(8.1 ± 0.5)·10-4
(3.36 ± 0.19)·10-5
26.5
84.2 ± 1.9
19.4 ± 0.5
(9.6 ± 0.5)·10-4
(3.99 ± 0.21)·10-5
25.8
82.3 ± 1.2
18.9 ± 0.3
(1.38 ± 0.08)·10-3
(5.7 ± 0.3)·10-5
24.0
78.8 ± 2.4
18.0 ± 0.7
(1.82 ± 0.11)·10-3
(7.5 ± 0.5)·10-5
2
«Снизу»
28.0
76.1 ± 1.8
17.2 ± 0.5
(2.57 ± 0.15)·10-3
(10.5 ± 0.6)·10-5
26.0
73.5 ± 1.1
16.6 ± 0.3
(3.71 ± 0.22)·10-3
(15.0 ± 0.9)·10-5
27.0
71.1 ± 1.7
15.9 ± 0.5
(6.4 ± 0.4)·10-3
(2.59 ± 0.14)·10-4
23.0
69.1 ± 1.1
15.4 ± 0.3
(9.2 ± 0.5)·10-3
(3.69 ± 0.20)·10-4
23.0
66.4 ± 1.2
14.7 ± 0.3
(1.42 ± 0.08)·10-2
(5.7 ± 0.3)·10-4
23.0
64.3 ± 0.8
14.1 ± 0.2
(1.88 ± 0.11)·10-2
(7.4 ± 0.4)·10-4
23.0
61.8 ± 0.9
13.5 ± 0.2
(2.45 ± 0.14)·10-2
(9.6 ± 0.6)·10-4
19.2
96.4 ± 2.5
22.9 ± 0.8
(7.7 ± 0.5)·10-4
(3.29 ± 0.19)·10-5
19.2
93.3 ± 2.1
22.1 ± 0.6
(7.6 ± 0.4)·10-4
(3.26 ± 0.19)·10-5
19.2
91.2 ± 1.1
21.5 ± 0.3
(8.2 ± 0.5)·10-4
(3.47 ± 0.20)·10-5
19.2
88.0 ± 3.7
20.7 ± 1.0
(9.3 ± 0.5)·10-4
(3.92 ± 0.23)·10-5
19.2
85.7 ± 1.6
20.0 ± 0.4
(1.12 ± 0.07)·10-3
(4.7 ± 0.3)·10-5
3
«Сверху»
19.2
81.1 ± 2.3
18.8 ± 0.6
(1.50 ± 0.09)·10-3
(6.3 ± 0.4)·10-5
19.2
78.1 ± 1.4
17.9 ± 0.4
(2.75 ± 0.16)·10-3
(11.4 ± 0.7)·10-5
19.2
75 ± 3
17.1 ± 0.9
(5.8 ± 0.3)·10-3
(2.37 ± 0.14)·10-4
19.2
72.8 ± 2.1
16.5 ± 0.6
(6.5 ± 0.4)·10-3
(2.65 ± 0.16)·10-4
19.2
68.7 ± 2.3
15.4 ± 0.6
(1.32 ±·0.08)·10-2
(5.3 ± 0.3)·10-4
Определение растворимости Ra(NO3)2
[3] Greene C. H. // J. Am. Chem.
Soc. 1937. Vol. 59, N 7.
в растворах HNO3
P. 1186-1188.
[4] Мишина Н. Е., Зильберман Б. Я., Кольцова Т. И. и др. //
Экспериментальные данные по растворимости
Радиохимия. 2014. Т. 56, N 3. С. 214-222.
Ra(NO3)2 в HNO3 представлены на рис. 2 и в табл. 3.
[5] Пузиков Е. А., Мишина Н. Е., Зильберман Б. Я. // Радиохи-
мия. 2016. Т. 58, N 5. С. 409-414.
Результаты выполненных экспериментов показали,
[6] Erbacher O. // Ber. Deutsch. Chem. Ges. (Ser. A, B). 1930.
что растворимость Ra(NO3)2 уменьшается от ~1·10-3
Bd 63, Hf. 1. S. 141-156.
до ~2·10-5 моль/л при увеличении концентраций
[7] Кузнецов Р. А., Буткалюк П. С., Тарасов В. А. и др. // Ра-
HNO3 от 13.5 до 22 моль/л. Данные, полученные при
диохимия. 2012. Т. 54, N 4. С. 352-356.
достижении равновесия «сверху» и «снизу», удовле-
[8] Кузнецов Р. А., Буткалюк П. С., Буткалюк И. Л. и др. // Изв.
творительно (в пределах погрешности определения)
Самар. науч. центра РАН. 2014. Т. 16, N 6. С. 129-135.
[9] Программа для проведения ионометрического и потен-
согласуются между собой. Растворимость Ra(NO3)2
циометрического титрования [электронный ресурс]. URL:
(моль/л) в исследованном диапазоне концентраций
HNO3 в 3-6 раз больше растворимости Ba(NO3)2 в
[10] Зинченко А. В., Изотова С. Г., Румянцев А. В. и др. Новый
аналогичных условиях.
справочник химика и технолога. Химическое равновесие.
Свойства растворов. СПб.: Профессионал, 2004. 998 с.
Список литературы
[11] Кузнецов Р. А., Тарасов В. А., Романов Е. Г. и др. // Изв.
[1] Вдовенко В. М., Дубасов Ю. В. Аналитическая химия ра-
Самар. науч. центра РАН. 2014. Т. 16, N 6. С. 136-141.
дия. Сер.: Аналитическая химия элементов. Л.: Наука,
[12] Nelson F. // J. Chromatogr. A. 1964. Vol. 16. P. 403-406.
1973. 191 с.
[13] Perry’s Chemical Engineers’ Handbook / Eds R. H. Perry,
[2] Проценко П. И., Разумовская О. Н., Брыкова Н. А. Спра-
D. W. Green, J. O. Maloney. McGraw-Hill, 1997. 7th ed.
вочник по растворимости нитритных и нитратных соле-
[14] Дёрффель К. Статистика в аналитической химии / Пер. с
вых систем. Л.: Химия, 1971. 272 с.
нем. Л. Н. Петрова. М.: Мир, 1994. 268 с.