Радиохимия, 2019, т. 61, N 1, c. 41-50
41
Изучение движения генетически связанных радионуклидов 221Fr
и 213Bi в хроматографической среде
© С. В. Ермолаев*, А. К. Скасырская
Институт ядерных исследований РАН, 117312, Москва, пр. 60-летия Октября, д.7а; * e-mail: ermolaev@inr.ru
Получена 22.12.2017, после доработки 24.01.2018, принята к публикации 31.01.2018
УДК 543.544.5+546.37
С целью получения хроматографических данных для разработки генератора 225Ac/213Bi, основанного
на отделении и распаде промежуточного 221Fr, рассмотрено движение 221Fr и 213Bi в сорбентах Actinide
Resin, Dowex 50×8 и AMP-PAN при их непрерывном отделении от материнского 225Ac, адсорбированно-
го на Actinide Resin. Предложена модель, описывающая концентрацию дочерних радионуклидов как
функцию времени элюирования и положения в объеме хроматографической системы. Эксперименталь-
но определены значения коэффициентов распределения k' Fr и Bi для указанных сорбентов.
Ключевые слова: франций-221, актиний-225, хроматография, непрерывное элюирование, разделе-
ние генетически связанных радионуклидов.
DOI: 10.1134/S0033831119010076
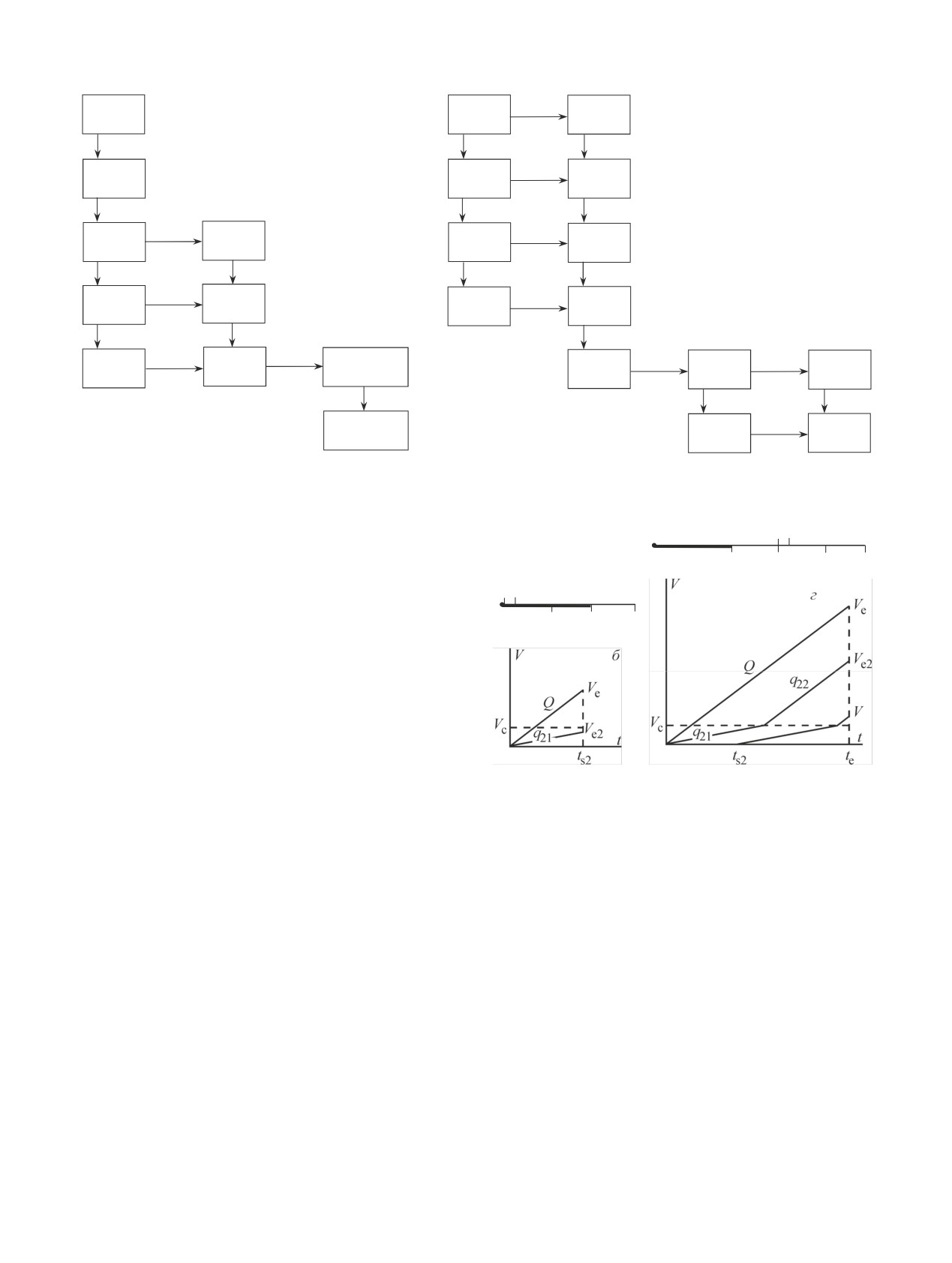
Хроматографическое поведение щелочных ме-
попадание которого в организм пациента недопусти-
таллов, за исключением Fr, хорошо изучено как для
мо. Схема распада 227Ac показана на рис. 1, б.
органических, так и для неорганических сорбентов.
Ограниченные хроматографические данные для
К органическим относятся главным образом сильно-
Fr объясняются отсутствием у этого элемента долго-
кислотные сульфосодержащие катионообменные
живущих изотопов. Наибольшим периодом полурас-
смолы Dowex 50, КУ-2, Duolite C-3 [1-5]. Неоргани-
пада обладает 223Fr (T1/2 = 22 мин). Этот изотоп обра-
ческие сорбенты, селективные к щелочным метал-
зуется из 227Ac с парциальной вероятностью канала
лам, включают нерастворимые гетерокислоты и их
распада 1.4% [17]. Для изучения 223Fr его периодиче-
соли, например фосфоромолибдат аммония [6, 7],
ски отделяют от 227Ac и затем приводят в контакт с
фосфаты циркония и некоторых четырехвалентных
исследуемым сорбентом [5, 18]. Пока 223Fr, используе-
элементов [8], смешанные ферроцианиды [9, 10] и
мый в опыте, распадается, из 227Ac накапливается но-
другие. Применяют также композитные сорбенты, в
вая порция 223Fr. В отличие от описанного метода нами
которых для улучшения характеристик сорбента се-
предложен метод, основанный не на периодическом, а
лективный компонент связывают с неорганическим
на постоянном отделении дочернего радионуклида от
[11, 12] или органическим носителем [13].
материнского, причем скорость образования дочер-
Франций интересен не только для фундаменталь-
него радионуклида соизмерима со скоростью его
ных исследований в хроматографии как самый тяже-
отделения и движения. Получаемое распределение
лый представитель щелочных металлов. В последнее
дочернего радионуклида в сорбенте и элюате позво-
время быстро развивается область ядерной медици-
ляет судить о его хроматографическом поведении.
ны, связанная с использованием α-излучающих
Радиоактивные превращения генетически связан-
радионуклидов для терапии различных онкологиче-
ных радионуклидов 1 λ1 2 λ2 ... n λn представляют
ских заболеваний. 225Ac (T1/2 = 9.9 сут) - один из наи-
собой пример последовательных реакций первого
более перспективных радионуклидов, может приме-
порядка, в которых константа скорости i-й стадии λi
няться и напрямую, и как материнский радионуклид
является постоянной распада i-го радионуклида. Ко-
в генераторе 213Bi (T1/2 = 46 мин) (рис. 1, а). Посколь-
личества дочерних радионуклидов Ni (i > 1), накоп-
ку 225Ac распадается во 221Fr (T1/2 = 4.9 мин), можно
ленных за время t, при условии, что в начальный
осуществить хроматографическую схему, в которой
момент их количества равны нулю, рассчитывают
213Bi извлекают посредством отделения и распада
по уравнению [19]
промежуточного короткоживущего 221Fr [14]. Полу-
чаемый элюат 213Bi содержит существенно меньше
i-1 i
i
225Ac по сравнению с непосредственным отделением
Ni = N10∏λj∑[exp(-λjt)/∏(λk - λj)],
(1)
j=1 j=1
k=1,
213Bi от 225Ac [15]. Это обстоятельство особенно важ-
k≠j
но в случае использования 225Ac, получаемого облу-
чением природного Th протонами средних энергий
где N0 - количество материнского радионуклида в
[16], поскольку одновременно с 225Ac образуется
начальный момент времени. Это уравнение описыва-
около 0.2% долгоживущего 227Ac (T1/2 = 21.8 года),
ет Ni только как функцию времени. Предложенный
42
С. В. Ермолаев, А. К. Скасырская
225Ac
227Ac
β-
227Th
9.9 сут
а
21.8 года
18.7 сут
б
98.6%
α
100%
α
1.4%
α
100%
221Fr
223Fr
β-
223Ra
4.9 мин
22 мин
99.994%
11.4 сут
α
100%
α
0.006%
α
100%
217At
β-
217Rn
219At
β-
219Rn
32.3 мс
0.007%
0.54 мс
56 с
6.4%
3.96 с
α
99.993%
α
100%
α
93.6%
α
100%
213Bi
β-
213Po
215Bi
β-
215Po
45.6 мин
97.8%
4.2 мкс
1.78 мс
7.6 мин
100%
α
2.2%
α
100%
α
100%
β-
209Pb
β-
209Bi
β-
β-
209Tl
211Pb
211Bi
211Po
2.2 мин
100%
3.3 ч
100%
1.9·1019 лет
36.1 мин
2.14 мин
0.516 с
100%
0.3%
α
100%
α
99.7%
α
100%
205Tl
207Tl
β-
207Pb
стабилен
4.77 мин
100%
стабилен
Рис. 1. Схемы распада 225Ac (а) и 227Ac (б) [17].
Окончание элюирования
в
метод одновременно с изменением количества ра-
дионуклидов во времени рассматривает их распреде-
dV
Vc
ление в объеме. В данной работе рассмотрено разде-
Начало движения
ление генетически связанных 225Ac → 221Fr → 213Bi.
произвольного dV
N1
V
Ve2
Ve
Хроматографическое поведение 221Fr оценивали как
dV
а
непосредственно по его активности, так и из распре-
деления в сорбенте и элюате более долгоживущего
N1
Ve2
Vc
Ve
213Bi.
Описание движения генетически связанных
радионуклидов 1 → 2 → 3 → (одномерная модель)
Зафиксируем исходное вещество 1 в начальном
слое сорбента (среда 1) в точке V = 0. Объем сорбен-
та, а именно свободный объем, доступный для проте-
кания раствора, обозначим Vc. Количество вещества
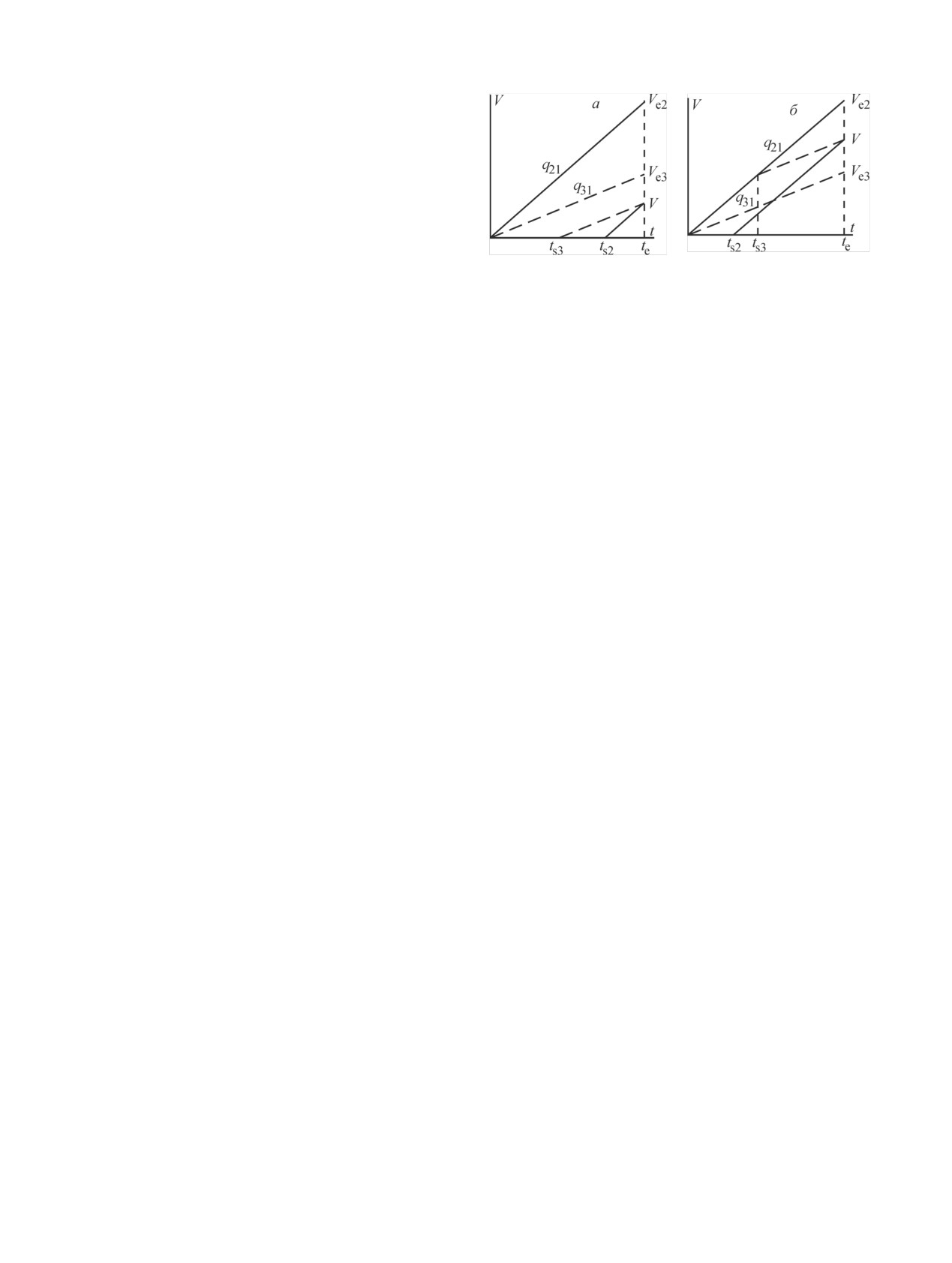
Рис. 2. Схема движения вещества 2: а, б - момент начала
1 в начальный момент времени равно N0, а количест-
движения наблюдаемого дифференциального элемента; в, г -
момент окончания элюирования.
ва дочерних продуктов N0 = 0 (i > 1). В этот момент
начинается движение раствора с объемной скоро-
составил Ve, а фронт дочернего вещества 2 достиг
стью Q. Положим, что скорости движения продуктов
Ve2. Скорость образования вещества 2 в точке V = 0
в сорбенте соотносятся как q31 ≤ q21 ≤ Q, где первый
в этот момент равна (dN2/dt)ts2
= λ1N1(ts2)
=
нижний индекс относится к продукту, а второй - к
λ1N0exp(-λ1ts2), а его концентрация в наблюдаемом
среде. Покинув сорбент, продукты движутся в сре-
dV равна (dN2/dV)ts2 = (λ1/q21)N0exp(-λ1ts2). За движе-
де 2 (элюат или другой сорбент) со скоростями q32 ≤
нием дифференциального элемента dV удобно сле-
q22 ≤ Q. При описании других соотношений скоро-
дить в координатах V-t (рис. 2, б), поскольку концен-
стей, а также большего количества продуктов или
трация движущегося вещества является функцией
хроматографических сред используются рассужде-
этих переменных.
ния, подобные приведенным ниже.
Потеряв связь с материнским веществом, движу-
1. Концентрация дочернего радионуклида 2
щееся вещество 2 убывает с постоянной распада λ2.
В момент te окончания элюирования дифференциаль-
Рассмотрим дифференциальный элемент dV
ный элемент окажется в произвольной точке V
(рис. 2, а), отделяющийся от вещества 1 спустя про-
(рис. 2, в, г), концентрация вещества 2 составит
извольное время ts2 после начала движения раствора
(начала элюирования) и двигающийся по сорбенту
dN2/dV = (λ1/q22)N0exp(-λ1ts2)exp[-λ2(te - ts2)].
(2)
(среда 1) со скоростью q21. К этому моменту объем
пропущенного через вещество 1 раствора (элюента)
Как видно из V-t-диаграммы (рис. 2, г), для диф-
Изучение движения генетически связанных радионуклидов 221Fr и 213Bi
43
ференциального элемента, покинувшего сорбент (V >
Vc), te - ts2 = Vc/q21 + (V - Vc)/q22. В случае V < Vc: te -
ts2
= V/q21. Преобразуя уравнение (2) и полагая, что
вещество 2, покинув сорбент, движется в среде 2 (в
элюате) со скоростью потока q22 = Q, приходим к
выражениям для концентрации вещества 2 в сорбен-
те (0-Vc)
dN
(3a)
2/dV = (λ1/q21)N0exp[-λ1te - (λ2 - λ1)V/q21]
и в элюате (Vc-Ve2)
Рис. 3. V-t-диаграммы движения веществ 2 и 3: а - в диапазо-
не 0-Ve3, б - в диапазоне Ve3-Ve2. Сплошные линии соответст-
вуют движению вещества 2, пунктирные - вещества 3; то же
dN2/dV = (λ1/Q)N0exp{-λ1te - (λ2 - λ1)[Vc/q21 + (V - Vc)/Q]}.
на рис. 4, 5.
(3б)
Интегрируя эти уравнения в нужных пределах,
профиль концентраций вещества 3, формирующийся
получаем количество и активность A2 = λ2N2 веще-
в рамках этого временнóго интервала. Вещество 3,
ства 2 в разных частях хроматографической систе-
образующееся в момент начала элюирования, опре-
мы на момент окончания элюирования. Найдем,
деляет его фронт Ve3 (рис. 3), который разбивает
например, общую активность вещества 2, собирае-
весь объем существования вещества 3 на два диапа-
мую в элюате. На практике она складывается из
зона: 0-Ve3 и Ve3-Ve2.
активности, образующейся за время протекания
Диапазон 0-Ve3. Рассмотрим дифференциальный
элюента через вещество 1, и равновесной активно-
элемент dV, отделяющийся от начальной точки V = 0
сти, обычно вымываемой из сорбента в виде болю-
спустя произвольное время ts3 после начала элюиро-
са, т.е. в небольшом объеме элюента: A2 = A2 + A2,
вания и движущийся по сорбенту со скоростью q31
где верхние индексы f (flow) и b (bolus) означают
(рис. 3, а). В момент старта концентрация вещества
проточную и болюсную активности соответствен-
3 равна нулю. Спустя время t = te - ts3 накопления
но. Активность A2 определяем интегрированием
вещества 3 дифференциальный элемент достигает
уравнения (3б) в пределах Vc-Ve2. Равновесная ак-
точки V. В уравнении (4) материального баланса в
тивность, на начало элюирования равная A0 = A0×
этой точке скорость λ2dN2/dV образования вещества
λ2/(λ2 - λ1), к окончанию элюирования уменьшается
3 определяется уравнением
(2). Используя V-t-
до A2b= A10[λ2/(λ2 - λ1)]exp(-λ2te). В результате полу-
диаграмму на рис. 3, а: te = ts2 + V/q21 = ts3 + t и обо-
чаем
значая k1 = q31/q21, приводим уравнение (4) к виду
d2N3/dVdt + λ3dN3/dV = (λ2λ1/q21)N0exp(-λ1ts3)exp{t[-λ1 -
A2 = A10[λ2/(λ2 - λ1)]exp[-λ1te - (λ2 - λ1)Vc/q21].
(3в)
- k1(λ2 - λ1)]}.
(4a)
Таким образом, определяя общую собранную в
элюате активность вещества 2, из уравнения (3в)
Решая его относительно времени t накопления
находим скорость q21 его движения в сорбенте.
вещества 3, приходим к уравнению
Если проинтегрировать уравнение (3а) от 0 до Vc,
dN3/dV = [λ2λ1N0exp(-λ1ts3)]{q21[λ3 - λ1 - k1(λ2 - λ1)]}-1×
а (3б) - от Vc до Ve2 и затем сложить их, то придем к
×{exp[(-λ1 - k1(λ2 - λ1))t] - exp(-λ3t)}.
(5)
выражению N2
= N0[λ1/(λ2
- λ1)][exp(-λ1te)
-
exp(-λ2te)], являющемуся частным случаем уравне-
Обозначая Λ1 = λ3 - λ1 - k1(λ2 - λ1) и проводя под-
ния (1) для i = 2.
становку ts3 = te - t и t = V/q31, получаем концентра-
цию вещества 3 как функцию времени элюирования
2. Концентрация дочернего радионуклида 3
и объема
Изменение количества вещества 3 определяется
dN3/dV = [λ2λ1N0/(q21Λ1)]exp[-λ1te - (λ2 - λ1)V/q21][1 -
уравнением материального баланса в виде
- exp(-Λ1V/q31)].
(5a)
d2N3/dVdt = λ2dN2/dV - λ3dN3/dV.
(4)
Диапазон Ve3-Ve2. В точке Ve2 фронта вещества 2
непрерывно образуется вещество 3. Поскольку веще-
Поскольку скорость движения вещества 3 в сор-
ства 2 и 3 движутся с разными скоростями, концен-
бенте меньше скорости вещества 2: q31 < q21, то
трация вещества 3 в этой точке равна нулю. На
фронт вещества 2 первым достигнет границы сор-
рис. 3, б показано отделение дифференциального
бента объемом Vc спустя время Vc/q21 после начала
элемента dV от фронта вещества 2 спустя произволь-
элюирования.
ное время ts3 после начала элюирования и его движе-
2.1. Интервал времени te ≤ Vc/q21. Рассмотрим
ние со скоростью q31 в течение времени t = te - ts3 на-
44
С. В. Ермолаев, А. К. Скасырская
копления вещества 3. Рассуждая так же, как в случае
диапазона 0-Ve3, приходим к уравнению концентра-
ции вещества 3 в виде
dN3/dV = [λ2λ1N0/(q21Λ1)]exp(-λ2ts3){exp[(Λ1 - λ3)t] -
- exp(-λ3t)},
(6)
которое отличается от уравнения (5) только множи-
телем при ts3. Выражая с помощью V-t-диаграммы
(рис. 3, б) ts3 и t через te и V, находим профиль кон-
центрации вещества 3 в данном диапазоне на мо-
Рис. 4. V-t-диаграммы движения веществ 2 и 3: а - в ди-
мент окончания элюирования
апазоне Vc-Vs, б - в диапазоне Vs-Ve2.
dN3/dV = [λ2λ1N0/(q21Λ1)]exp[-λ1te - (λ2 - λ1)V/q21]{1 -
Диапазон Vs-Ve2. Дифференциальный элемент
exp[-Λ1(Ve2 - V)/((1 - k1)q21)]}.
(6a)
dV отделяется от фронта вещества 2, покинувшего
2.2. Интервал времени Vc/q21 ≤ te ≤ Vc/q31. Спустя
сорбент и движущегося во второй среде (рис. 4, б).
время Vc/q21 после начала элюирования фронт веще-
В остальном этот случай подобен рассмотренному
ства 2 выходит из сорбента и порождает вторичный
в разделе 2.1, диапазон Ve3-Ve2, и уравнение кон-
фронт Vs вещества 3 (рис. 4). В результате образуют-
центрации вещества 3 имеет вид, похожий на урав-
ся 4 диапазона существования вещества 3: 0-Ve3,
нение (6)
Ve3-Vc, Vc-Vs и Vs-Ve2.
dN3/dV = [λ2λ1N0/(q22Λ2)]exp(-λ2ts3){exp[(Λ2 - λ3)t] -
концентрация веще-
В диапазонах 0-Ve3 и Ve3-Vc
- exp(-λ3t)}.
(8)
ства 3 определяется уравнениями (5) и (6) соответст-
венно. Рассмотрим два оставшихся диапазона.
Выражая ts3 и t через te и V, получаем концен-
трацию вещества 3 как функцию времени элюиро-
Диапазон Vc-Vs. На рис. 4, а показано движение
вания и объема
дифференциального элемента dV, отделившегося от
фронта вещества 2 спустя произвольное время ts3
dN3/dV = [λ2λ1N0/(q22Λ2)]exp{-λ1te - (λ2 - λ1)[Vc/q21 + (V -
после начала элюирования. Сначала он движется в
- Vc)/q22]}{1 - exp[-Λ2(Ve2 - V)/((1 - k2)q22)]}.
(8а)
сорбенте со скоростью q31, и концентрация вещества
3 в нем описывается уравнением (6). Спустя время tc
2.3. Интервал времени te ≥ Vc/q31. Фронт Ve3 ве-
дифференциальный элемент достигает границы сор-
щества 3 достигает границы сорбента по прошест-
бента и приобретает скорость q32. В этот момент
вии времени, равного Vc/q31. В этот момент исчезает
концентрация вещества 3 равна
диапазон Ve3-Vc и вместо него появляется диапазон
(dN3/dV)tc = (q31/q32)[λ2λ1N0/(q21Λ1)]exp(-λ2ts3){exp[(Λ1 -
Vc-Ve3. В результате объем существования вещест-
ва 3 в данном интервале времени складывается из
- λ3)(tc - ts3)] - exp[-λ3(tc - ts3)]}.
диапазонов (рис. 5): 0-Vc [уравнение (5)], Vc-Ve3, Ve3-
Обозначая k2 = q32/q22, Λ2 = λ2 - λ1 - k2(λ2 - λ1) и
Vs [уравнение (7)] и Vs-Ve2 [уравнение (8)]. Получим
L = k1Λ2/(k2Λ1), приводим это уравнение к виду
выражение для профиля концентрации вещества 3 в
новом диапазоне.
(dN3/dV)tc = L[λ2λ1N0/(q22Λ2)]exp(-λ2ts3){exp[(Λ1 - λ3)(tc -
- ts3)] - exp[-λ3(tc - ts3)]}.
(6б)
Диапазон Vc-Ve3. Дифференциальный элемент dV
отделяется от начальной точки V = 0 спустя произ-
Затем дифференциальный элемент движется в
вольное время ts3 после начала элюирования (рис. 5).
следующей среде в течение времени t = te - tc. Реша-
Подобно случаю, описанному в разделе 2.2, диапа-
ем уравнение (4) материального баланса относитель-
зон Vc-Vs, сначала он движется в сорбенте со скоро-
но t, используя уравнение (6б) в качестве граничного
стью q31, причем концентрация вещества 3 в нем
условия [t = 0; dN3/dV = (dN3/dV)tc]
описывается уравнением (5). Достигая границы сор-
dN3/dV = [λ2λ1N0/(q22Λ2)]exp[-λ2ts3 - λ3(te - ts3)]{exp[Λ1(tc -
бента спустя время tc, дифференциальный элемент
- ts3)][exp(Λ2(te - tc)) - (1 - L)] - L}.
(7)
приобретает скорость q32. В этот момент концентра-
ция вещества 3 равна
Выражая с помощью V-t-диаграммы (рис. 4, а)
ts3 и tc через te и V, находим профиль концентрации
(dN3/dV)tc = L[λ2λ1N0/(q22Λ2)]exp(-λ1ts3){exp[(Λ1 - λ3)(tc -
вещества 3 в диапазоне Vc-Vs на момент окончания
-ts3)] - exp[-λ3(tc - ts3)]}.
(5б)
элюирования
Затем дифференциальный элемент движется в
dN3/dV = [λ2λ1N0/(q22Λ2)]exp{-λ1te - (λ2 - λ1)[Vc/q21 + (V -
следующей среде в течение времени t = te - tc. Решая
Vc)/q22]}{1 - [1 - L(1 - exp[-Λ1(Vs - V)/((1 - k1)q32)])]×
уравнение (4) относительно t с использованием урав-
×exp[-Λ2(V - Vc)/q32]}.
(7а)
нения (5б) в качестве граничного условия, получаем
Изучение движения генетически связанных радионуклидов 221Fr и 213Bi
45
нов. Во втором интервале фронт вещества 2, поки-
нув сорбент, движется в среде 2 (другой сорбент или
элюат), в то время как фронт вещества 3 движется
еще в первой среде. В третьем интервале фронты
обоих веществ движутся во второй среде. Во втором
и третьем временны' х интервалах область существо-
вания вещества 3 состоит из четырех диапазонов.
Интегрирование уравнений концентрации вещества
3 во всех диапазонах любого интервала [например,
для интервала времени te ≥ Vc/q31 это уравнения (5а),
te
(9а), (7а) и (8а)] и сложение получаемых интегралов
Рис. 5. V-t-диаграммы движения веществ 2 и 3 в диапазоне
приводит к частному случаю уравнения (1) для i = 3.
Vc-Ve3.
Экспериментальная часть
Материалы. В экспериментах использовали сор-
бенты различных типов. Образцы экстракционно-
хроматографических смол, предоставленные фирмой
Triskem (Франция), включали DGA Resin (экстр-
агент - N,N,N',N'-тетра-н-октилдигликольамид), TRU
Resin (экстрагент - раствор октилфенил-N,N-диизо-
бутилкарбамоилметилфосфиноксида в трибутилфос-
фате) и Actinide Resin [экстрагент - ди-(2-этил-
гексил)метандифосфоновая кислота]. Размеры зерен
этих смол составляли 100-150 мкм. Образец компо-
зитного ионообменного сорбента AMP-PAN фирмы
Triskem представлял собой высокодисперсный фос-
форомолибдат аммония, внедренный в органиче-
скую матрицу на основе полиакрилонитрила для
улучшения механических и гидродинамических ха-
рактеристик. Размер гранул находился в пределах
100-600 мкм. Использовали также катионообмен-
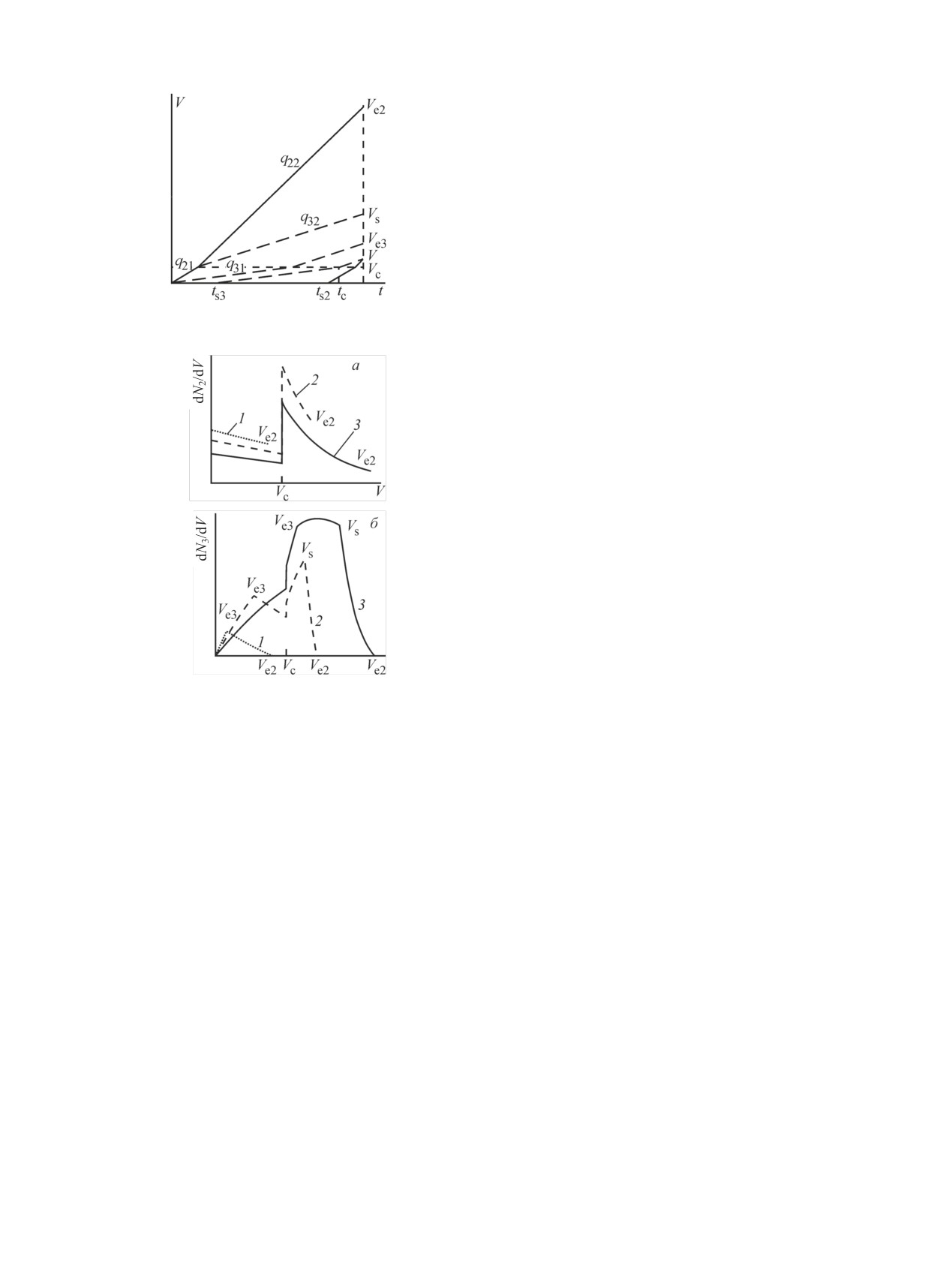
Рис. 6. Профили концентраций веществ 2 (а) и 3 (б) в интерва-
ную смолу Dowex 50×8, 100-200 меш (75-150 мкм)
лах времени: 1 - te ≤ Vc/q21, 2 - Vc/q21 ≤ te ≤ Vc/q31, 3 - te ≥ Vc/q31.
производства Dow Chemical (США).
Использованные в расчете значения: Vc = 10 мл, λ1 = 7.7·10-4,
Водные растворы готовили на основе деионизо-
λ2 = 3.3·10-3, λ3 = 9.9·10-4 c-1; q21 = 4, q31 = 0.8, q22 = 1, q32 =
0.6 мл/мин.
ванной воды. Чистота реактивов соответствовала
квалификации ос.ч. или х.ч.
dN3/dV = [λ2λ1N0/(q22Λ2)]exp[-λ1ts3 - λ3(te - ts3)]{exp[Λ1(tc -
Получение 225Ac. Пластины из металлического
- ts3)][exp(Λ2(te - tc)) - (1 - L)] - L}.
(9)
Th толщиной 0.05-1.1 мм облучали на линейном
ускорителе ИЯИ РАН [20] ускоренным пучком про-
Выражая ts3 и tc через te и V (рис. 5), находим
тонов с энергией 143 МэВ при токе 1-3 мкА. При
профиль концентрации вещества 3 в диапазоне Vc-
облучении ториевые пластины, завернутые в алюми-
Ve3 на момент окончания элюирования
ниевую фольгу, интенсивно охлаждали потоком во-
dN3/dV = [λ2λ1N0/(q22Λ2)]exp{-λ1te - (λ2 - λ1)[Vc/q21 + (V -
ды. Энергия протонов, приходящих на ториевую
- Vc)/q22]}{1 - [1 - L(1 - exp(-Λ1Vc/q31))]exp[-Λ2(V -
мишень, составляла около 115 МэВ. Поток протонов
– Vc)/q32]}.
(9а)
определяли с помощью фольг-мониторов из Al и Cu
по образованию соответственно 22Na и 62Zn, сечения
Таким образом, эволюция распределения вещест-
которых достаточно точно известны [21].
ва 3 проходит в трех сменяющих друг друга времен-
ны' х интервалах. Профили концентраций веществ 2 и
Процедура извлечения 225Ac из облученной ми-
3, возникающие в каждом временнóм интервале, по-
шени, подробно описанная в работе [22], заключа-
казаны на рис. 6. Значения постоянных распада и ско-
лась в растворении Th в растворе 6 моль/л HNO3 с
ростей движения веществ в разных средах подобраны
добавлением каталитических количеств HF и после-
так, чтобы продемонстрировать своеобразие профи-
дующей экстракции раствором ди-2-этилгексил-
лей концентраций. В первом интервале область суще-
фосфорной кислоты (Д2ЭГФК) в толуоле, при этом
ствования вещества 3 находится внутри сорбента
225Ac оставался в водной фазе. На следующей стадии
(среда 1), в начальном слое которого зафиксировано
225Ac вместе с фракцией РЗЭ извлекали из водной
материнское вещество 1, и состоит из двух диапазо-
фазы, адсорбируя на смоле DGA Resin и отделяя от
46
С. В. Ермолаев, А. К. Скасырская
основной массы радионуклидов. После десорбции
вило, она находилась в пределах 10-15%). По хроно-
раствором 0.01 моль/л HNO3 проводили окончатель-
метру отмечали начало и окончание элюирования и
ную очистку 225Ac с помощью смолы TRU Resin в
начало измерения каждого образца. γ-Спектро-
среде 3 моль/л HNO3. Активности образцов 225Ac
метрические измерения проводили с использовани-
составляли около 1 МБк в пересчете на окончание
ем спектрометра с детектором из сверхчистого Ge
облучения.
(ORTEC GEM15P4-70). Дочерние радионуклиды
идентифицировали по наиболее интенсивным
Методика эксперимента. Раствор с 225Ac упари-
γ-квантам
[17]: 221Fr - 218.1 кэВ (интенсивность
вали, остаток растворяли в 0.2-0.3 моль/л HNO3. За-
11.4%), 213Bi - 440.4 кэВ (25.9%). Активность гене-
тем 225Ac адсорбировали на хроматографической
тически связанных радионуклидов в образцах рас-
колонке, заполненной смолой Actinide Resin объе-
считывали с учетом их радиоактивных превращений
мом 0.5 мл (здесь и далее использовали колонки диа-
как в течение измерения, так и за время, прошедшее
метром 3.5 мм). Скорость движения 221Fr в смоле
с окончания элюирования до начала измерения. На
определяли, пропуская через колонку растворы HCl,
следующий день после каждого элюирования по-
HNO3 и HClO4 различной концентрации со скоро-
вторно измеряли все образцы для определения в них
стью 0.7-1.2 мл/мин и собирая элюат порциями по
225Ac по активностям пришедших в равновесие 221Fr
1-1.5 мл в течение 20-25 мин. В каждой порции сра-
зу после отбора измеряли активность 221Fr. Подачу
и 213Bi.
растворов и контроль скорости их пропускания осу-
Результаты и обсуждение
ществляли с помощью перистальтического насоса,
герметично соединенного с колонкой. Для оценки
При непрерывном отделении дочерних коротко-
скорости движения 213Bi соединяли колонку, содер-
живущих радионуклидов от материнского, зафикси-
жащую материнский 225Ac, с несколькими последо-
рованного на сорбенте, в течение времени, сопоста-
вательно соединенными колонками, заполненными
вимого со временем их накопления, дочерние радио-
Actinide Resin, либо напрямую, либо посредством
нуклиды распределяются между сорбентом (сор-
стеклянной трубки диаметром 4 мм и объемом
бентами) и элюатом в зависимости от скоростей их
44 мл. В первом случае объем смолы в каждой из
движения в среде. Возникающие профили концен-
последовательно соединенных колонок составлял
траций описываются моделью движения генетически
0.25 мл, во втором - 0.15 мл. Через сборку колонок
связанных радионуклидов как функции времени
пропускали раствор 0.25 моль/л HNO3 в течение не
элюирования и положения в объеме хроматографи-
менее 3 ч со скоростью 1.2 мл/мин.
ческой системы. Применение модели рассмотрено на
Для изучения скорости движения 221Fr и 213Bi в
примере цепочки 225Ac → 221Fr → 213Bi. Интегрирова-
среде Dowex 50×8 действовали сходным образом:
ние функций по объему в выбираемых пределах свя-
одну либо несколько последовательно соединенных
зывает скорости движения дочерних продуктов с
колонок, заполненных этой смолой, присоединяли к
величинами их активностей, определяемыми экспе-
колонке с материнским 225Ac и в течение около 4 ч
риментально.
через сборку колонок пропускали раствор 0.25 моль/л
Полученные данные использованы для оценки
HNO3. В опыте с одной колонкой Dowex 50×8 объем
коэффициента распределения k', представляющего
смолы составлял 0.5 мл, скорость пропускания -
собой по определению отношение времен пребыва-
0.6 мл/мин. В опыте с несколькими колонками объ-
ния частиц отделяемого вещества в адсорбирован-
ем смолы в каждой из них и скорость пропускания
ном (tads) и свободном (tsol) состоянии ([23], с. 32).
составляли 0.12 мл и 1.1 мл/мин соответственно.
Поскольку эти времена связаны со скоростями дви-
В опытах с AMP-PAN использовали три колонки
жения отделяемого вещества q и элюента Q соотно-
с разным количеством смолы: 0.12, 0.27 и 0.40 мл.
шением q/Q = tsol/(tads + tsol), выражение, используе-
Каждую колонку по очереди присоединяли к колон-
мое в расчетах, принимает вид k' = (1 - q/Q)/(q/Q).
ке с материнским 225Ac и в течение 3.5 ч пропускали
Определение k' Fr(I) и Bi(III) на смоле Actinide
раствор 0.25 моль/л HNO3 со скоростью 1.15 мл/мин.
Resin. Для проведения длительных элюирований
По окончании элюирований, длившихся несколь-
требуется сорбент, хорошо удерживающий 225Ac,
ко часов, измеряли активности 221Fr и 213Bi во всех
поэтому мы остановились на Actinide Resin. Значе-
частях хроматографической системы: в элюате, в
ния k' Ac(III) для растворов минеральных кислот с
материнской и исследуемых колонках, а также в рас-
концентрацией около 1 моль/л достигают 104, а в
творе из соединительной стеклянной трубки, когда
диапазоне концентраций 0.1-0.3 моль/л превышают
она была использована. Дополнительно трубку про-
105 [24, 25]. Скорость движения 221Fr определяли из
мывали 2-3 порциями по 2 мл раствора 1 моль/л
уравнения (3в), в котором величина A2 является сум-
HCl, по которым оценивали долю 213Bi, адсорбиро-
мой активностей 221Fr в пробах элюата, приведенных
ванного на внутренней поверхности трубки (как пра-
на окончание элюирования, а Vc - это объем в сор-
Изучение движения генетически связанных радионуклидов 221Fr и 213Bi
47
1
3
2
4
3
4
Рис. 9. Расчет распределения 213Bi между частями хроматогра-
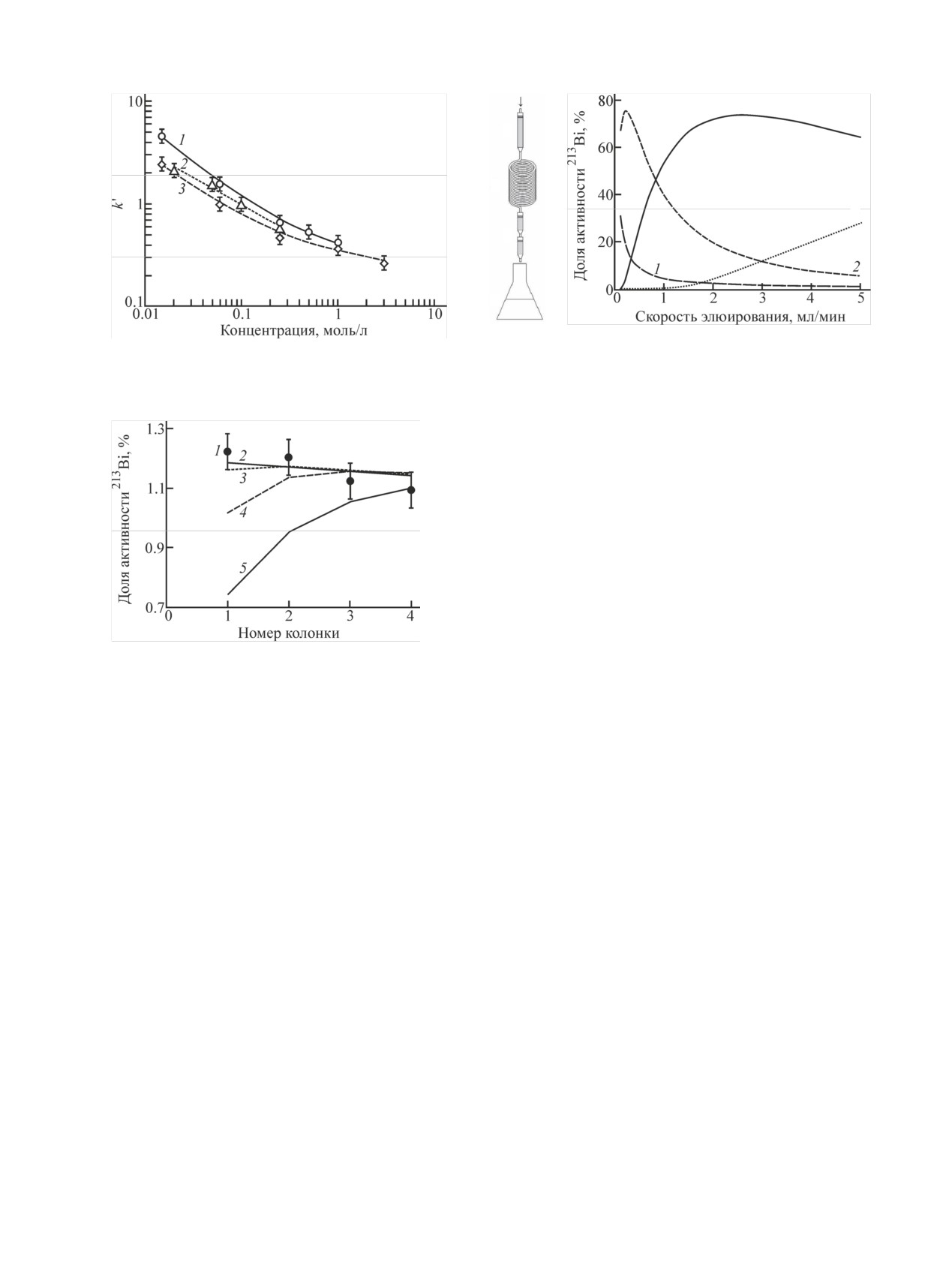
Рис. 7. Экспериментальная зависимость коэффициентов рас-
фической системы в зависимости от скорости элюирования
пределения k' Fr(I) при сорбции на смоле Actinide Resin от
(скорость движения 213Bi в среде Actinide Resin равна 0, время
концентрации кислот. 1 - HCl, 2 - HClO4, 3 - HNO3.
элюирования 6 ч). 1 - колонка с 225Ac (объем смолы 0.5 мл),
2 - трубка для распада 221Fr (44 мл), 3 - колонки для концен-
трирования 213Bi (объем смолы в одной колонке 0.15 мл), 4 -
элюат.
четырьмя дополнительными последовательно соеди-
ненными колонками, заполненными Actinide Resin, и
затем пропускали раствор в течение 370 мин. Резуль-
таты эксперимента в сравнении с расчетными данны-
ми представлены на рис. 8. Вследствие высокой ско-
рости движения Fr(I) в среде Actinide Resin доли ак-
тивности 213Bi, накопленной на четырех дополни-
тельных колонках, оказались чуть больше 1%. Экс-
периментальные данные хорошо согласуются с рас-
четными для значений k' Bi(III) выше 104, что соот-
Рис. 8. Доля активности 213Bi, накопленной на последовательно
ветствует случаю, описанному в разделе 2.2, когда
соединенных колонках, заполненных Actinide Resin. 1 - экспе-
риментальные значения; 2-5 - расчетные данные для различ-
фронт 221Fr движется вместе с элюатом, а фронт 213Bi
ных значений k' Bi(III): 2 - 1.5·104, 3 - 3·103, 4 - 1.5·103, 5 -
еще находится в сорбенте. На основании этих дан-
7.5·102.
ных можно заключить, что в пределах ошибки экспе-
римента k' Bi(III) не ниже, чем 3·103.
бенте, доступный для протекания элюата, равный
объему сорбента в колонке, умноженному на долю
Чтобы повысить точность определения, между
свободного объема в нем. Для Actinide Resin и ряда
материнской и дополнительными колонками добави-
других экстракционно-хроматографических смол
ли стеклянную трубку. Ее объем выбирали таким,
доля ε свободного объема находится в пределах
чтобы 221Fr успевал распасться за время движения.
0.65-0.69 [26-28]. Полученные коэффициенты рас-
213Bi, накапливающийся в трубке и движущийся с
пределения k' Fr(I) при сорбции на смоле Actinide
потоком элюата, концентрировался в первой допол-
Resin из растворов HCl, HClO4 и HNO3 различной
нительной колонке. Схема эксперимента и расчет
концентрации (рис. 7) оказались близкими. Они сви-
распределения 213Bi между частями хроматографиче-
детельствуют о слабой сорбции Fr(I): при концентра-
ской системы показаны на рис. 9. В расчете приняли
ции кислоты выше 0.1 моль/л их значения не превы-
в качестве первого приближения, что скорости дви-
шали единицу.
жения 221Fr и 213Bi в среде Actinide Resin q21 = q23 =
0.7Q и q31 = q33 = 0; время элюирования 6 ч.
Распределение Bi(III) изучали в среде 0.25 моль/л
HNO3. В случаях, когда хроматографическая система
Видно, что зависимость доли 213Bi, накопленного
включала только колонку, содержащую материнский
на дополнительной колонке, от скорости пропуска-
225Ac, и элюат, не удалось оценить скорость движе-
ния раствора проходит через максимум. Он объясня-
ния 213Bi по сорбенту, можно было только утвер-
ется тем, что при низких скоростях значительная
ждать, что 213Bi адсорбирован и неподвижен. При
часть 213Bi остается в растворе в трубке между ко-
измерениях колонки большую погрешность вносил
лонками, а при высоких - объем трубки недостато-
213Bi, успевший накопиться из 225Ac, а в пробах элюа-
чен для распада 221Fr, заметная часть которого распа-
та обнаруживали только 213Bi от попавшего туда
дается уже в элюате. При скорости пропускания вы-
221Fr. Поэтому материнскую колонку соединяли с
ше 1 мл/мин более 50% 213Bi скапливается на первой
48
С. В. Ермолаев, А. К. Скасырская
видно из рис. 10, в эксперименте наблюдали плавное
нарастание активности 213Bi в отличие от резкого
подъема, демонстрируемого данными расчета. Веро-
ятные причины заключаются в диффузионном раз-
мытии профиля концентраций 213Bi в первой допол-
нительной колонке, а также в том, что при данной
концентрации HNO3 в растворе могут присутство-
вать различные ионные формы Bi(III), такие как
BiNO32+ и Bi(NO3)+.
Таким образом, рассмотрены возможности изуче-
ния хроматографического поведения генетически
Рис. 10. Зависимость доли активности 213Bi, переместившейся
связанных 221Fr и 213Bi в среде Actinide Resin при их
на вторую дополнительную колонку с Actinide Resin, от дли-
непрерывном длительном отделении от материнско-
тельности элюирования. 1 - экспериментальные значения; 2-
4 - расчетные данные для различных значений k' Bi(III): 2 -
го 225Ac. Схема накопления 213Bi на дополнительной
3.7·103, 3 - 3.8·103, 4 - 3.9·103.
колонке в результате распада 221Fr в соединительной
трубке (рис. 9) может быть полезной для разработки
а
генератора 213Bi. Кроме того, благодаря слабому удер-
живанию Fr(I) колонка с Actinide Resin, содержащая
225Ac, может служить источником 221Fr для оценки его
хроматографического поведения на других смолах. О
1-2
2-3
3-4
4-5
поведении 221Fr также можно судить по распределе-
нию его дочернего 213Bi, что бывает удобнее, так как
период полураспада 213Bi почти на порядок больше.
б
Как показано в разделе 2, в уравнениях, описываю-
щих концентрацию 213Bi, содержатся скорости и 213Bi,
и 221Fr, определяя которые можно получить коэффи-
циенты распределения этих элементов.
1-2
2-3
3-4
4-5
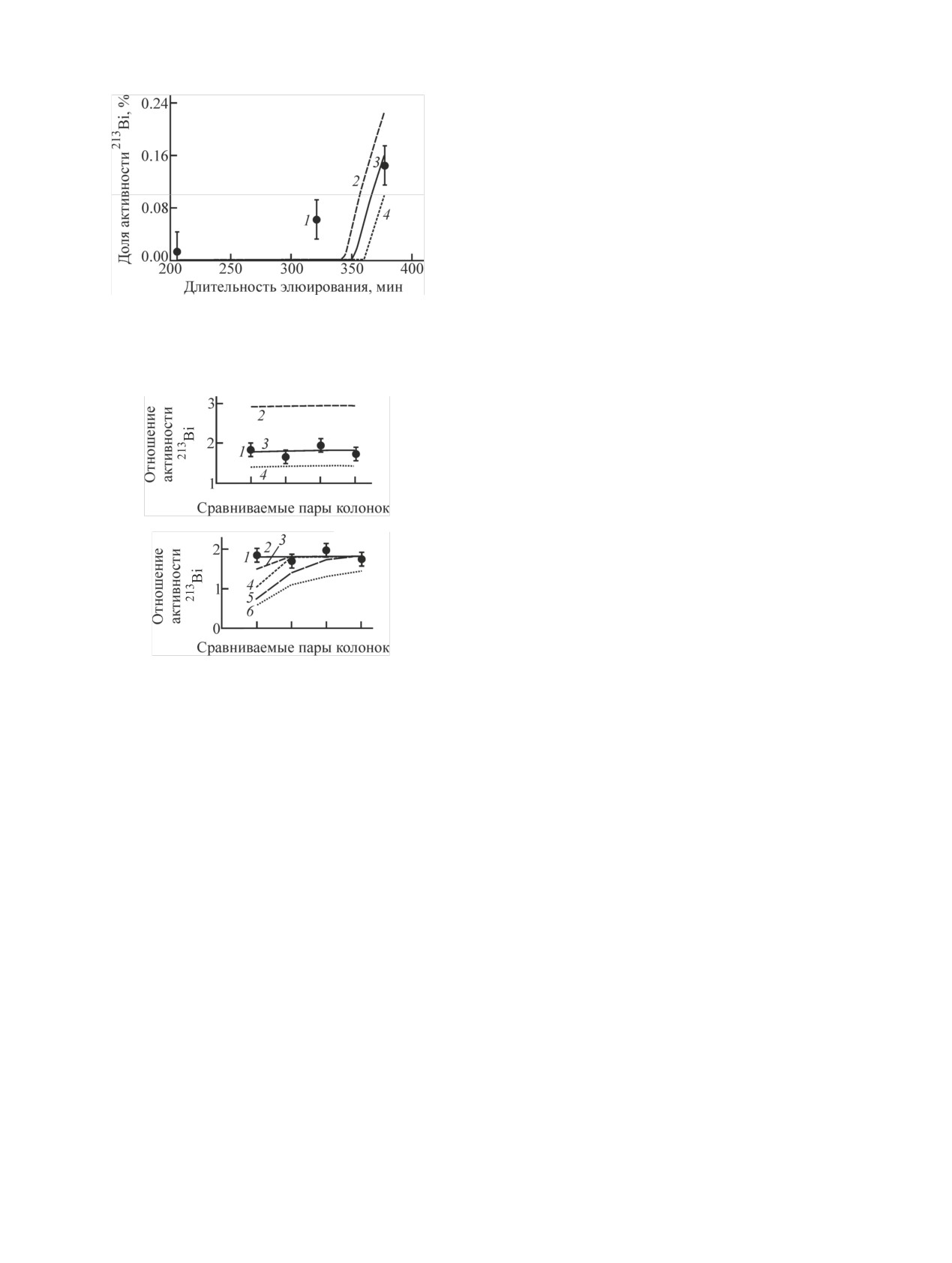
Определение k' Fr(I) и Bi(III) на смоле Dowex
50×8 в среде 0.25 моль/л HNO3. Колонку с Actinide
Рис. 11. Отношение значений активности 213Bi в сравниваемых
Resin, содержащую 225Ac, присоединяли к колонке,
попарно колонках, заполненных Dowex 50×8. 1 - эксперимен-
заполненной Dowex 50×8 (0.5 мл), и пропускали рас-
тальные значения. а - расчетные данные, полученные для фик-
твор в течение времени, достаточного для установле-
сированного отношения скоростей движения 213Bi и 221Fr в
ния радиоактивного равновесия (около 4 ч). Распре-
среде Dowex 50×8 q32/q22 = 0.001 и переменного отношения
q22/Q: 2 - 0.006, 3 - 0.01, 4 - 0.017. б - расчетные данные, полу-
деление активности 221Fr и 213Bi на момент оконча-
ченные для фиксированного отношения скорости движения
ния элюирования составило (%):
221Fr в среде Dowex 50×8 и скорости потока q22/Q = 0.01 и пе-
Нук-
ременного отношения q32/q22: 2 - 0.001, 3 - 0.01, 4 - 0.025, 5 -
Колонка с 225Ac Колонка с Dowex 50×8 Элюат
0.05, 6 - 0.1.
лид
221Fr
12.4 ± 1.4
86 ± 9
1.2 ± 0.3
дополнительной колонке. Поскольку в действитель-
213Bi
9.8 ± 1.0
89 ± 9
1.2 ± 0.3
ности скорость движения 213Bi в смоле больше нуля,
На основании этих данных можно оценить значе-
при достаточной длительности элюирования часть
ния коэффициентов распределения k' Fr(I) (~80-110)
213Bi переместится, «проскочит» на вторую дополни-
и Bi(III) (>6·104). При расчете использовали величи-
тельную колонку. Провели серию экспериментов,
ну доли свободного объема ε = 0.38 ([29], с. 129).
увеличивая время опыта при постоянной скорости
Основную неточность в оценку k' вносит погреш-
пропускания. Пока время элюирования не превыша-
ность активности в элюате, связанная со сложной
ло 4 ч, на второй дополнительной колонке наблюда-
геометрией измерения. Поэтому, аналогично экспе-
ли фоновую (несколько сотых процента) активность
рименту с Actinide Resin, к материнской колонке
213Bi, образующуюся при протекании оставшегося
присоединяли пять колонок с Dowex 50×8 неболь-
221Fr. После ~5 ч элюирования заметили рост актив-
шого (0.12 мл) объема. Исходя из сделанной оценки
ности
213Bi на второй дополнительной колонке
k' Fr(I) размер колонок и скорость пропускания рас-
(рис. 10). Сравнение результатов эксперимента с рас-
твора выбирали такими, чтобы добиться уменьше-
четными данными, полученными для разных скоро-
ния активности 221Fr и 213Bi примерно в два раза от
стей движения 213Bi, позволяет оценить коэффициент
колонки к колонке. Результаты эксперимента и со-
распределения k' Bi(III) на уровне ~4·103, что согла-
провождающего его расчета представлены на рис. 11
суется с предыдущим экспериментом (рис. 8). Как
в виде отношения значений активности 213Bi в срав-
Изучение движения генетически связанных радионуклидов 221Fr и 213Bi
49
ниваемых попарно колонках с Dowex 50×8: первой и
второй; второй и третьей и т.д.
Отношения экспериментально полученных значе-
ний активности 213Bi в каждой паре колонок оказа-
лись одинаковыми и равными в среднем 1.8. Расчет-
ные данные на рис. 11, а показывают, что при отно-
шении скоростей движения 213Bi и 221Fr в среде
Dowex 50×8 q32/q22 ≤ 0.001 наилучшая сходимость с
экспериментом достигается при q22/Q = 0.01. При
более низкой скорости 221Fr (q22/Q = 0.006) актив-
Рис.
12. Доля активности
221Fr и 213Bi в элюатах после
ность 213Bi в каждой предыдущей колонке больше,
длительных (3.5 ч) элюирований сборок Actinide Resin (225Ac)-
AMP-PAN в зависимости от объема смолы AMP-PAN. 1, 3 -
чем в последующей, в ~3 раза, а при более высокой
экспериментальные значения активностей; 2, 4 - расчетные
скорости отношение активностей приближается к
значения активностей 221Fr и 213Bi соответственно.
единице, распределение активности по колонкам
выравнивается. Рис. 11, б демонстрирует, как влияет
элюент через сборку из материнской и исследуемой
скорость движения 213Bi в среде Dowex 50×8 на от-
колонок, обнаружили (рис. 12) рост активности 221Fr
ношение его активностей: при увеличении скорости
и 213Bi в элюате с уменьшением объема смолы в ис-
213Bi успевает переместиться из колонки в колонку,
следуемой колонке. На основании этих данных оце-
и его количество в первой, а затем и последующих
нили коэффициенты распределения k' Fr(I) (250) и
колонках уменьшается. Исходя из полученной таким
Bi(III) (>3·104). Необходимые для расчета параметры
образом скорости движения 221Fr можно уточнить
AMP-PAN взяты из работы [13].
значение коэффициента распределения k' Fr(I) (97 ±
10), при этом оценка k' Bi(III) (>6·104) осталась преж-
Таким образом, на примере цепочки 225Ac →
ней.
221Fr → 213Bi рассмотрено движение 221Fr и 213Bi в
хроматографической среде при непрерывном отде-
В литературе для ионообменных смол часто ис-
лении от материнского радионуклида, зафиксиро-
пользуют массовый коэффициент распределения kd,
ванного на сорбенте. Предложена модель, описы-
связанный в первом приближении с k' соотношением
вающая возникающие профили концентраций до-
kd = k'*ε/ρapp, где ρapp - насыпная (кажущаяся) плот-
черних радионуклидов как функции времени элюи-
ность сорбента. Для Dowex 50×8 ρapp = 0.8 г/мл ([29],
рования и положения в объеме. С использованием
с. 376), тогда kd Fr(I) равен 46 ± 5 мл/г. Сравнение
смолы Actinide Resin, хорошо удерживающей 225Ac,
полученного результата со значениями kd (мл/г,
испытаны варианты формирования различных рас-
0.25 моль/л HNO3) ионов остальных щелочных ме-
пределений 221Fr и 213Bi в хроматографической сис-
таллов, определенными в работе [2] для смолы AG
теме. Вариант накопления 213Bi на колонке, соеди-
50W×8, являющейся аналогом Dowex 50×8, приведе-
ненной с материнской колонкой промежуточным
но ниже:
Fr, может служить
объемом раствора для распада 221
Li(I)
Na(I)
K(I)
Rb(I)
Cs(I)
Fr(I)
прототипом генератора 213Bi. Колонку с Actinide
15
24
45
52
64
46 ± 5
Resin, содержащую 225Ac, использовали как источ-
ник 221Fr для оценки его хроматографического пове-
Учитывая, что данные в работе [2] получены в
дения на смолах Dowex 50×8 и AMP-PAN. В резуль-
статических условиях (время контакта раствора и
тате были получены значения коэффициентов рас-
сорбента 24 ч), а значение kd Fr(I) определено в ди-
пределения для трех различных по отношению к Fr
намических условиях, сходимость результатов мож-
сорбентов.
но считать удовлетворительной.
Авторы выражают благодарность Ш. Хаппелю
Определение k' Fr(I) и Bi(III) на смоле AMP-
(S. Happel) (Triskem, Франция) за предоставленные
PAN в среде 0.25 моль/л HNO3. Смола AMP-PAN
образцы экстракционно-хроматографических смол,
содержит в качестве активного компонента фосфо-
а также научному сотруднику ИЯИ РАН А. Н. Ва-
ромолибдат аммония, высокая селективность кото-
сильеву за помощь при подготовке статьи.
рого к ионам тяжелых щелочных металлов, в част-
ности цезия, известна [6]. Введение фосфоромолиб-
Работа выполнена с использованием оборудова-
дата аммония в органическую PAN-матрицу улуч-
ния ЦКП «Ускорительный центр нейтронных иссле-
шило размеры, пористость и структуру зерен смолы
дований структуры вещества и ядерной медицины
AMP-PAN [13]. Для оценки коэффициентов распре-
ИЯИ РАН» при поддержке Министерства образова-
деления Fr(I) и Bi(III) к колонке с материнским 225Ac
ния и науки РФ в рамках Соглашения о предоставле-
по очереди присоединяли колонки, заполненные
нии субсидии (N 14.621.21.0014 от 28.08.2017), уни-
исследуемой смолой разного объема. Пропуская
кальный идентификатор RFMEFI62117X0014.
50
С. В. Ермолаев, А. К. Скасырская
[6] Nonaka N., Sato K., Higuchi H., Hamaguchi H. // Radioiso-
Приложение: список обозначений
topes. 1976. Vol. 25, N 10. P. 599-602.
- постоянная распада i-го (i = 1-n) радионук-
[7] Van Smith J. R. // Nature. 1958. Vol. 181. P. 1530-1531.
λi
[8] Inoue Y. // J. Inorg. Nucl. Chem. 1964. Vol. 26, N 12.
лида, с-1; Ni, Ni0 - количество i-го радионуклида в
P. 2241-2253.
произвольный и начальный моменты времени; Ai,
[9] Huys D., Baetslé L. H. // J. Inorg. Nucl. Chem. 1964. Vol. 26,
Ai0 - активность i-го радионуклида в произвольный
N 7. P. 1329-1331.
и начальный моменты времени, Бк; Af, Aib - проточ-
[10] Krtil J. // J. Inorg. Nucl. Chem. 1965. Vol. 27, N 1. P. 233-
ная и болюсная активность i-го радионуклида, Бк;
236.
[11] Воронина А. В., Семенищев В. С., Ноговицына Е. В., Бете-
Q - скорость движения раствора (элюента), мл/мин
неков Н. Д. // Радиохимия. 2012. Т. 54, N 1. С. 66-70.
(м3/с); qij
- скорость движения i-го вещества
[12] Voronina A. V., Semenishchev V. S., Nogovitsyna E. V., Be-
(радионуклида) в j-й среде, мл/мин (м3/с); t - время
tenekov N. D. // J. Radioanal. Nucl. Chem. 2013. Vol. 298.
(переменная), с; te
- время окончания элюирования,
P. 67-75.
[13] Sebesta F., Stefula V. // J. Radioanal. Nucl. Chem. 1990.
с; ts2, ts3 - время начала движения дифференциаль-
Vol. 140, N 1. P. 15-21.
ного элемента dV со скоростью q21 или q31 соответ-
[14] Ермолаев С. В., Скасырская А. К. // Тез. докл. VIII Всерос.
ственно, с; tс - время достижения дифференциаль-
конф. по радиохимии «Радиохимия-2015». Железногорск
ным элементом dV границы среды, с; tads, tsol
- вре-
Красноярского края, 2015. С. 442.
мя пребывания частиц вещества в адсорбирован-
[15] Morgenstern A., Bruchertseifer F., Apostolidis C. // Curr.
Radiopharm. 2011. Vol. 4. P. 295-305.
ном и свободном состоянии, с; V - объем (перемен-
[16] Ermolaev S. V., Zhuikov B. L., Kokhanyuk V. M. et al. // Ra-
ная), мл (м3); Ve - объем элюата, мл (м3); Vс - сво-
diochim. Acta. 2012. Vol. 100. P. 223-229.
бодный объем сорбента (хроматографической сре-
[17] National Nuclear Data Center, Brookhaven National Labora-
ды 1), мл (м3); ε - доля свободного объема сорбен-
[18] Nelson F. // J. Chromatogr. 1964. Vol. 16. P. 538-540.
та; Ve2, Ve3 - фронты движения веществ 2 и 3, мл
[19] Bateman H. // Proc. Cambridge Phil. Soc. 1910. Vol. 15.
(м3); Vs - вторичный фронт вещества 3 в хромато-
P. 423-427.
графической среде 2, мл (м3); kj = q3j/q2j
- отноше-
[20] Zhuikov B. L., Kokhanyuk V. M., Konyakhin N. A., Vin-
ние скоростей движения веществ 3 и 2 в j-й среде;
cent J. // Nucl. Instr. Meth. Phys. Res. Sect. A.
1999.
Vol. 438. P. 173-179.
k' - коэффициент распределения; kd - массовый ко-
[21] Charged particle cross-section database for medical radioiso-
эффициент распределения, мл/г (м3/кг); ρapp - на-
tope production; diagnostic radioisotopes and monitor reac-
сыпная (кажущаяся) плотность сорбента, г/мл
tions: coordinated research project: IAEA-TECDOC-1211.
(кг/м3); Λj = λ3 - λ1 - kj(λ2 - λ1) - вспомогательная
[22] Aliev R. A., Ermolaev S. V., Vasiliev A. N. et al. // Solvent
константа, с-1; L = (k1Λ2)/(k2Λ1) - вспомогательная
Extr. Ion Exch. 2014. Vol. 32, N 5. P. 468-477.
константа.
[23] Сакодынский К. И., Бражников В. В., Волков С. А. и др.
Аналитическая хроматография. М.: Химия, 1993. 464 с.
Список литературы
[24] Horwitz E. P., Chiarizia R., Dietz M. L. // React. Funct.
Polym. 1997. Vol. 33. P. 25-36.
[1] Strelow F. W. E. // Anal. Chem. 1960. Vol. 32, N 9. P. 1185-
[25] Wu C., Brechbiel M. W., Gansow O. A. // Radiochim. Acta.
1188.
1997. Vol. 79. P. 141-144.
[2] Strelow F. W. E., Rethemeyer R., Bothma C. J. C. // Anal.
[26] Horwitz E. P., Chiarizia R., Dietz M. L. // Solvent Extr. Ion
Chem. 1965. Vol. 37, N 1. P. 106-111.
Exch. 1992. Vol. 10. P. 313-336.
[3] Tsubota H. // Bull. Chem. Soc. Jpn. 1963. Vol. 36, N 8.
[27] Horwitz E. P., Dietz M. L., Chiarizia R., Diamond H. // Anal.
P. 1038-1043.
Chim. Acta. 1992. Vol. 266. P. 25-37.
[4] Лилова O. M., Преображенский Б. К. // Радиохимия. 1960.
[28] Horwitz E. P., Dietz M. L., Chiarizia R. et al. // Anal. Chim.
Т. 2. С. 728-730.
Acta. 1995. Vol. 310. P. 63-78.
[5] Nelson F., Michelson D. C., Phillips H. O., Kraus K. A. //
[29] Мархол М. Ионообменники в аналитической химии. М.:
J. Chromatogr. 1965. Vol. 20. P. 107-121.
Мир, 1985. 548 с.