РАДИОХИМИЯ, 2022, том 64, № 4, с. 303-349
УДК 546.110.23:547.15/17
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА
В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
© 2022 г. В. П. Шевченко, И. Ю. Нагаев*, Н. Ф. Мясоедов
Институт молекулярной генетики НИЦ «Курчатовский институт»,
123182, Москва, пл. Курчатова, д. 2
*e-mail: nagaev@img.ras.ru
Поступила в редакцию 21.05.2021, после доработки 21.09.2021, принята к публикации 28.09.2021
Обобщены основные подходы к введению тритиевой и дейтериевой метки в разные классы биологически
активных соединений (липиды, нуклеотиды, аминокислоты, пептиды, низкомолекулярные
биорегуляторы и т.д.). Описано получение меченных изотопами водорода органических соединений
с использованием различных физико-химических методов. Рассмотрено влияние условий проведения
реакций на выход и содержание изотопов водорода в меченых препаратах. Большое внимание уделено
введению дейтерия и трития за счет изотопного обмена. Приведены возможные механизмы включения
изотопов водорода и связанное с этими процессами распределение метки в органических соединениях.
Показано, что при облучении препаратов, использовании посредников и других нововведений можно
значительно улучшить характеристики искомых меченых биологически активных соединений.
Ключевые слова: меченые соединения, тритий, дейтерий, синтез, изотопный обмен, гетерогенные и
гомогенные катализаторы, спилловер водорода.
DOI: 10.31857/S0033831122040013, EDN: FRGIYC
1. ВВЕДЕНИЕ
позволяют обнаруживать его на уровне ~10-14 моля.
Следует отметить также относительно невысокую
Изотопномеченные органические соединения
стоимость радиометрического оборудования.
нашли широкое применение в химии и биологии, в
В результате введения трития образуются мече-
медицине и сельском хозяйстве.
ные препараты с разным включением этого изото-
Для этих исследований можно использовать ра-
па. Если меченое соединение имеет молярную ра-
диоактивные изотопы углерода, иода, серы фтора,
диоактивность около 29.12 Ки/ммоль, это означает,
кислорода, фосфора и других элементов, но ис-
что в его молекулу произошло включение одного
пользование радиоактивного изотопа водорода -
атома трития. Получение препаратов с молярной
трития - для получения меченых органических сое-
радиоактивности 30 и более Ки/ммоль позволя-
динений имеет ряд преимуществ. Это связано с та-
ют проводить эксперименты, требующие высокой
кими параметрами этого изотопа, как мягкое излу-
чувствительности. В то же время для широкого
чение (максимальная энергия β-частиц 0.018 МэВ),
круга биологических исследований вполне доста-
удобный период полураспада (12.323 года) и высо-
точно использовать меченые соединения со значи-
кая молярная радиоактивность (29.12 кКи/г-атом).
тельно более низкой молярной радиоактивностью.
Введение этого изотопа возможно не только хими-
Поэтому синтез соединений с разным содержанием
ческими методами, но и изотопным обменом, с за-
трития имеет высокую востребованность. Выходы
меной атомов протия органического соединения на
при этом часто не являются определяющим пара-
тритий, поскольку водород присутствует практиче-
метром для использования той или иной методики
ски во всех органических соединениях. Предел об-
получения меченых соединений, так как их необхо-
наружения трития высок. При приемлемой степени
димые количества для проведения серии биологи-
включения трития в молекулы органического сое-
ческих экспериментов могут измеряться сотнями, а
динения современные радиометрические приборы
то и десятками микрокюри.
303
304
ШЕВЧЕНКО и др.
Кроме трития в природе существует еще ста-
Реакции с тритием или дейтерием проводят, как
бильный изотоп водорода - дейтерий. Для него
правило, при использовании катализаторов [2-10]
отсутствуют ограничения, которые необходимо со-
на основе палладия, платины, родия, рутения, ири-
блюдать при работе с радиоактивными материала-
дия, железа, кобальта, никеля и др. Есть новые све-
ми, что значительно упрощает проведение экспери-
дения о процессе введения изотопов водорода при
ментов. Подходы для отработки условий введения
использовании гомогенных катализаторов [6]. Ко
дейтерия и трития в органическое соединения, как
второй группе относятся реакции с изотопномечен-
правило, идентичны. Поэтому разработка техно-
ными водой, иодистым метилом, гидридами метал-
логий получения конкретного меченного тритием
лов и т.д., а также реакции, в которых донором изо-
препарата часто проводится с использованием га-
топа являются те или иные посредники.
зообразного дейтерия или дейтериевой воды. При
Очевидно, что роль катализаторов в различных
этом необходимо подчеркнуть, что соединения,
реакциях с участием изотопов водорода велика.
содержащие дейтерий, имеют самостоятельную
Поэтому в обзоре данные не только приведены по
ценность и широко используются, например, в ка-
их использованию, но и дана минимальная инфор-
честве реперов в масс-спектрометрии.
мация по их получению, если эти методики ориги-
В данном обзоре рассмотрены основные методы
нальны.
введения трития в различные органические соеди-
Реакции со 100%-ной тритиевой водой и реак-
нения. Естественно, учитывались публикации, при-
ции без использования растворителя (твердофаз-
веденные ранее в данном журнале [1]. Если методы
ные реакции) будут приведены в минимальном объ-
введения изотопов водорода каким-либо образом
еме, так как они довольно подробно рассмотрены в
дублировали разделы предыдущего обзора, то это
обзорах [1, 11]. Здесь необходимо подчеркнуть, что
связано с интересными новыми примерами их ис-
как следует из данных, приведенных в этих обзо-
пользования.
рах, часто успех при введении изотопов водорода в
органические соединения зависит от силы кислот-
Говоря о меченных тритием органических сое-
ных центров, которые образуются на поверхности
динениях, нужно отметить, что протий в различных
катализатора [6]. Также для понимания процессов,
группах органического соединения может иметь
которые приводят к тому или иному результату, не-
разную способность к обмену с протонами окру-
обходимо помнить о закономерностях, связанных
жающей среды. Водород, способный легко обме-
со спилловером активированных частиц изотопов
ниваться с протонными растворителями, например,
водорода по поверхности неорганических носите-
с водой, называют подвижным или лабильным.
лей и в пуле вещества, нанесенного на эту поверх-
Как правило, это водород полярных функциональ-
ность [3, 12, 13].
ных групп (гидроксильная, амино-, карбоксильная
и т.д.). В то же время водород, связанный с углеро-
Есть сведения, что на поверхности носителя
дом, с протонными растворителями не обменивает-
активированные частицы водорода существуют в
ся. Отрабатываются именно такие методики, кото-
виде пары электрон-протон [11]. При этом элек-
рые позволяют включать дейтерий или тритий при
трон связан с ионом металла, а протон - с ионом
углеродных атомах. И только такие соединения счи-
кислорода. Найдено несколько типов центров, где
таются мечеными препаратами. Препараты, мечен-
пары (Н+,ē) связаны с различными типами морфо-
ные тритием, как и другие радиоактивно меченные
логических дефектов поверхности носителя. Не
соединения, характеризуются молярной радиоак-
исключено, что межфазовые перемещения активи-
тивностью, радиохимической чистотой и распреде-
рованных на металле-катализаторе частиц изотопов
лением метки в их молекулах.
водорода связаны с возможностью туннелирования
электрона на поверхность носителя [14-16]. Такая
Все реакции, используемые для введения трития
интерпретация полученных данных подтверждена
(дейтерия), можно разделить на две группы: реак-
квантовомеханическими расчетами [17-20].
ции с использованием газообразного водорода (ре-
акции гидрогенолиза) [2-10] и реакции с использо-
По-видимому, молярная радиоактивность и вы-
ванием меченых соединений.
ход меченого соединения в значительной степени
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
305
зависит от взаимодействия вещества с активирован-
ся на самых современных достижениях синтетиче-
ными частицами изотопов водорода, т.е. от свойств
ской органической химии и радиохимии.
кластеров, которые образовались из этих частиц
К таким методам можно отнести восстановле-
[21, 22-25].
ние комплексными тритидами металлов и конден-
Данный обзор посвящен возможностям введе-
сацию соответствующих предшественников с ме-
ния изотопов водорода в разные по своей природе
чеными изотопами водорода реагентами [2, 31, 32].
соединения с использованием различных, уже отра-
Достоинством этого метода является то, что изотоп
ботанных и предложенных недавно методов [2, 4,
водорода присоединяется к определенному угле-
22]. Данные об использовании известных методов
родному атому в молекуле вещества, за участием
будут касаться только сведений, необходимых для
которого в химических реакциях или биологиче-
сравнения с обновленными методами. Механизмы
ских превращениях можно достоверно проследить.
включения трития и дейтерия будут приводиться
Недостатком метода, как уже отмечалось, можно
только для объяснения того или иного распределе-
считать необходимость получения часто трудно-
ния метки при получении меченых препаратов.
доступных и дорогостоящих предшественников, а
Так как тритийсодержащие соединения и побоч-
также сложность проведения реакции с количества-
ные продукты их получения являются радиоактив-
ми, измеряемыми микромолями, а часто и сотнями
ными веществами, после синтеза и использования
или десятками наномолей. Иногда получение ме-
их необходимо переводить в химически устойчи-
ченых препаратов этим методом можно значитель-
вую форму, которая сохраняет их стабильность в
но упростить, если предшественниками являются
течение времени хранения, минимизировать их
соединения, которые используются как реагенты в
объем, обеспечить надежность и безопасность тех-
других синтезах или являются природными веще-
нологий транспортировки, хранения, переработки и
ствами. Например, можно ввести изотопы водоро-
захоронения [26-30].
да в арахидоновую кислоту гидрированием дейте-
На схемах, рисунках и таблицах дейтерий обо-
рием или тритием ее ацетиленового производного,
используемого как ингибитор в биологических
значается символами 2H, тритий - 3H. На некото-
рых рисунках для большей наглядности дейтерий и
исследованиях, или в простагландин Е1 восстанов-
лением простагландина Е2, который получают в
тритий обозначены символами D и T соответствен-
но. Если положение изотопа водорода обозначается
препаративных количествах [2]. Повышение селек-
звездочкой *, это обозначает главное направление
тивности гидрирования в этом случае оказывается
включения дейтерия или трития. Если звездочка
решающим фактором при решении этой задачи.
приводится в скобках (*), это обозначает незначи-
Достижение этой цели возможно лишь при приме-
тельное включение метки в это положение.
нении самых современных разработок, в том числе
использования новых катализаторов.
Катализаторы, появившихся в последнее время,
2. ХИМИЧЕСКИЙ СИНТЕЗ
обладают уникальными свойствами. Их использо-
ИЗОТОПНОМОДИФИРОВАННЫХ
вание для получения меченых препаратов может
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
оказаться весьма перспективным. К сожалению,
они пока не нашли широкого применения для введе-
Известны различные методы введения метки в
ния изотопов водорода в органические соединения.
органические соединения. Те из них, которые про-
Обычно для селективного гидрирования алкинов
водятся с использованием химического синтеза,
используется катализатор Линдлара [33-37], хотя
требуют применения соответствующих предше-
есть примеры синтеза катализаторов с двухатомны-
ственников, из которых были получены меченые
ми активными центрами, которые после активации
соединения. Получение меченых реагентов и мече-
при нагревании в атмосфере водорода становятся
ных предшественников для синтеза биологически
способны к дейтерий-протиевому обмену [38].
активных соединений, по существу, в настоящий
момент превратилось в специфическую область
Для повышения селективности гидрирования
тонкого химического синтеза, который основывает-
используются также металлические катализаторы,
РАДИОХИМИЯ том 64 № 4 2022
306
ШЕВЧЕНКО и др.
которые модифицировали тиолами, серой, ами-
(Pd, Pt, Ru и т.д.) на поверхности неорганического
носодержащими компонентами
[34,
35,
39-52].
носителя [70].
Недостаток этих катализаторов заключается в том,
Способность гидрировать ненасыщенные двой-
что при решении проблемы селективности жертво-
ные связи в органических соединениях установле-
вали их стабильностью и каталитической активно-
на и для катализаторов на основе хрома и кобальта
стью.
(H2 или 2Н2) [71, 72]. Газообразный водород при
Поэтому параллельно с поверхностно-модифи-
взаимодействии с атомом металла диссоциирует.
цированными катализаторами для селективных ре-
Катион водорода связывается с атомом кислорода,
акций гидрирования проводили поиск альтернатив-
а гидрид-ион - с атомом металла. После адсорбции
ных методов их частичной дезактивации [53-65].
двойной связи на атоме кобальта сначала происхо-
Одним из таких подходов было использование
дит присоединение гидрид-иона, затем - катиона
носителей, где углеродная матрица была импрегни-
водорода.
рована атомами азота. В некотором смысле, как счи-
Продолжается поиск новых так называемых ге-
тают авторы, углерод, в структуру которого встрое-
терогенных металлоорганических катализаторов,
ны атомы азота, являлся для новых катализаторов
которые сочетают в себе свойства и гомогенных, и
тем же самым, что для катализатора Линдлара -
гетерогенных катализаторов [73].
ацетат свинца и хинолин [66].
Другим направлением модернизации катали-
Для изготовления таких катализаторов сначала
заторов является приготовление катализаторов, в
синтезировали палладий в виде нанокубиков (Pdнк)
которых частицы металла образуются внутри носи-
и равномерно наносили его на углеродные наново-
теля, и реакция при непосредственном контакте мо-
локнистые микросферы (NCM), в полупроводнико-
лекулы вещества с активным центром катализатора
вой структуре р-типа которого содержались атомы
становится невозможна [74]. В этих обстоятель-
азота [66].
ствах при проведении твердофазных реакций обра-
Pdнк(NCM) проявил высокую реакционную
зованию активированных частиц изотопов водоро-
способность и селективность при гидрировании
да не препятствуют молекулы вещества, в которые
алкинов до алкенов. Установлено, что электроны
вводится метка. Очевидно, что устойчивость таких
переносятся с поверхностного атома палладия на
катализаторов повышается, при этом в определен-
носитель и сольватируются на атомах азота [67].
ных условиях повышается эффективность и селек-
Следовательно, электронная плотность на кристал-
тивность включения дейтерия или трития [75].
ле палладия уменьшается, что приводит к более
слабому связыванию алкенов и предотвращает их
2.1. Синтез с использованием меченых
гидрирование [66].
реагентов
Никакого очевидного снижения селективно-
В первую очередь необходимо остановиться на
сти Pdнк(NCM) не наблюдалось даже через 9 ч,
что выгодно его отличает от поверхностно-моди-
использовании тритидов металлов, получившем
фицированных катализаторов типа катализатора
распространение в последние годы. В качестве
Линдлара [66].
исходного материала для получения комплексных
тритидов металлов обычно используют Li3H. Из
Продолжаются работы, связанные с нане-
него приготавливают LiAl3H4, LiEt3B3H, LiB3H4,
сением смеси металлов-катализаторов (Pd-Cu
LiB(OCH3)3H, Bu3Sn3H [31, 32, 76], которые затем
или Pd-Ag) на неорганический носитель [68, 69].
Лучшая активность и селективность при восста-
используют для получения меченого иодистого ме-
новлении акролеина до аллилового спирта наблю-
тила, а также для введения метки восстановлением
далась для 0.01% Pd с 8% Ag.
или заменой функциональных групп в молекулах
органических соединений на тритий (табл. 1) [77-
Значительно повысить селективность восстанов-
82].
ления ненасыщенных углерод-углеродных связей
оказалось возможным при использовании катализа-
По схожим методикам готовили и реагенты, со-
торов с содержанием одиночных атомов металлов
держащие дейтерий [83]. Есть работы, в которых
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
307
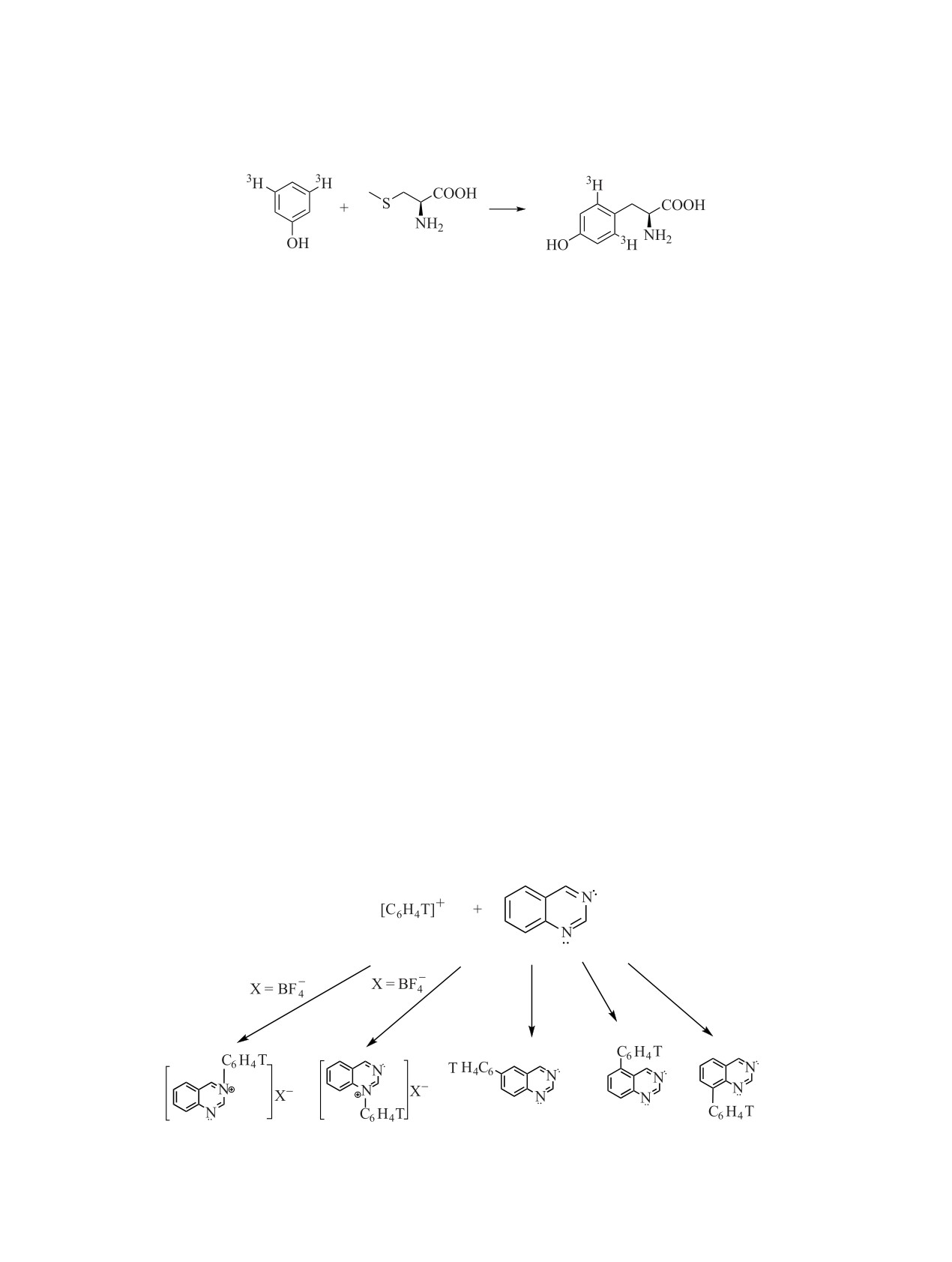
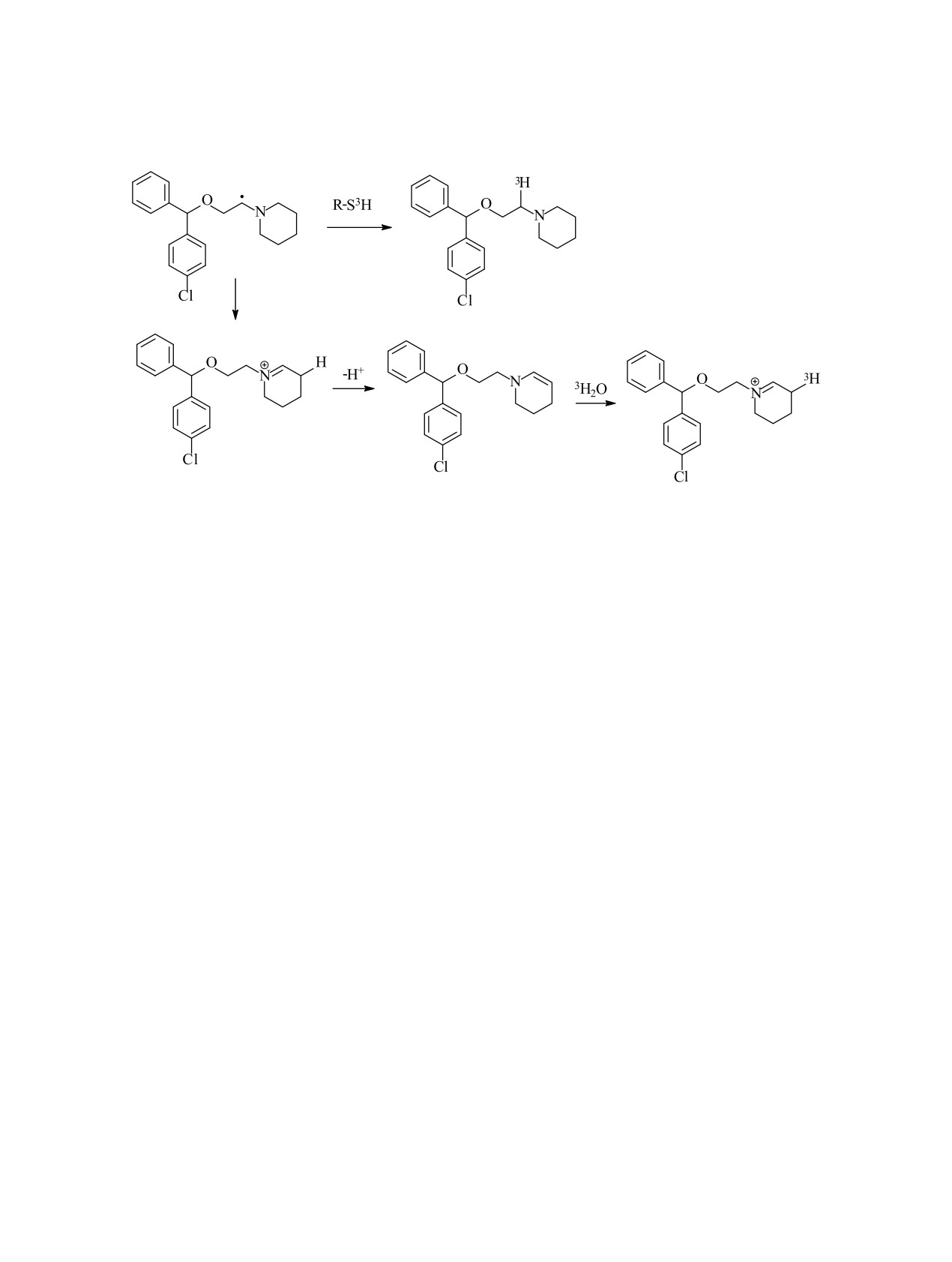
Схема 1.
проводят сравнение реагентов, наиболее подходя- Этот реагент восстанавливает альдегиды до спир-
щих для конкретного синтеза (табл. 2).
тов. Например, при использовании этого реагента
получен п-аминофенил-[3H]метанол с молярной ра-
При использовании дейтеридов получали за-
диоактивностью 24.3 Ки/ммоль (схема 2) [86].
мещенные амины восстановлением основания
Шиффа [84].
Синтез перечисленных выше меченых реаген-
тов представляет собой самостоятельную серьез-
В некоторых случаях к успеху приводило пред-
ную задачу. Ярким примером этого является синтез
варительное окисление препарата с последующим
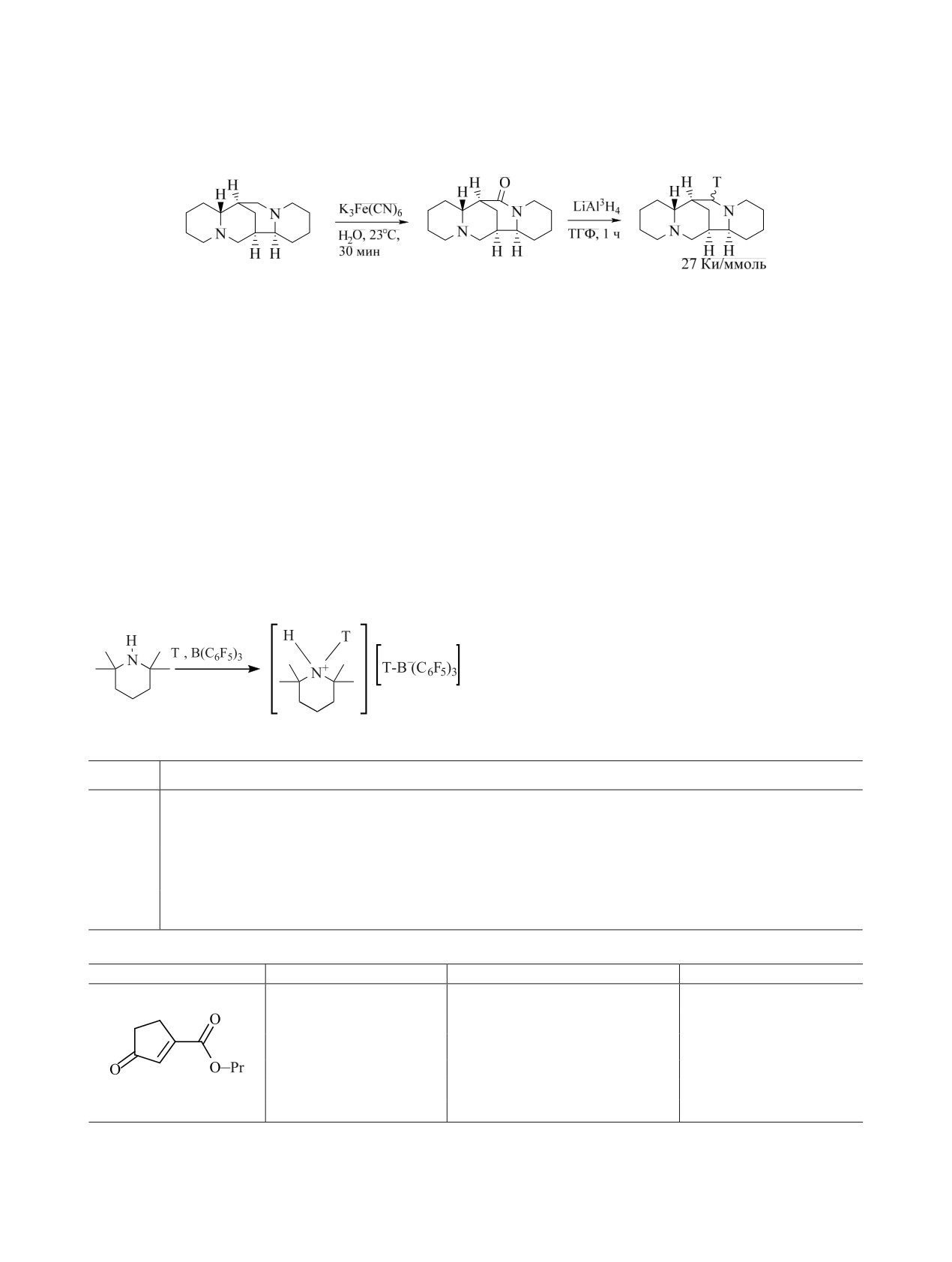
применением различных тритидов. Так, [3H]пахи- меченого иодистого метила, поиск новых методов
карпин получали окислением исходного соедине- получения которого продолжается и в настоящее
, а затем восстановлением литийа- время, хотя и сейчас их набор весьма разнообразен
ния K3Fe(CN)6
люмотритидом [85] (cхема 1).
(табл. 3) [4, 25, 87, 88].
Более экзотическим восстанавливающим реа-
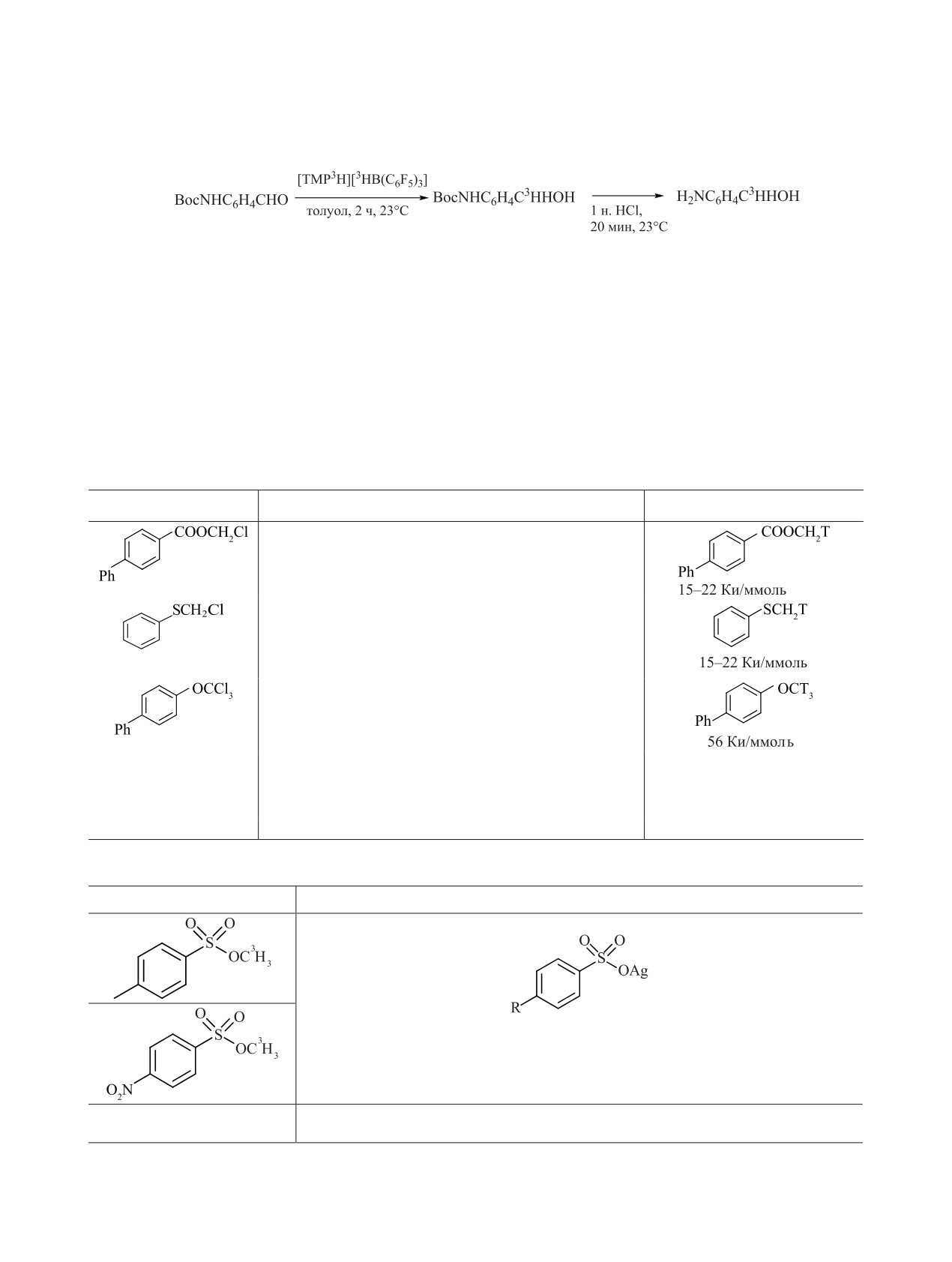
Из меченого иодистого метила получали другие,
гентом является [TMP3H][3НB(C6F5)3
]. Его полу- более удобные в использовании или хранении, ме-
чают с использованием газообразного трития и ченые реагенты (табл. 4) [89-91].
B(C6F5)3 (23°С, 1 ч) [86]:
TosОСН3Н может быть синтезирован и дебро-
мированием TosОСН2Br в атмосфере газообразно-
2
го трития (выход меченого препарата 50-60%, мо-
лярная радиоактивность [метил-3H]метилтозилата
15-17 Ки/ммоль) [97].
Таблица 1. Методики восстановления предшественников комплексными тритидами металлов.
Реагент
Условия реакции
NaB3H4
1.5 мл этанольного раствора 0.02 ммоля (60 Ки/ммоль) боротритида натрия, 0.1 ммоля альдегида в 2.5 мл
THF, 20 мин (или при 0°С, 30 мин), смесь разлагают кислотой, выход 75%, 12.6 Ки/ммоль.
Для восстановления эфиров карбоновой кислоты использовали кипячение в спирте 1 ч [77-79]
LiB3H4
Мольное соотношение LiB3H4 и амида 3 : 1 в CH2Cl2, 1 ч, 40°С, смесь разлагают кислотой, выход 43%,
54 Ки/ммоль [80]
LiAl3H4
Мольное соотношение LiAl3H4 и эфира карбоновой кислоты 1 : 2 в бензоле, 1 ч, затем с эфиром 15 мин,
смесь разлагают основанием и этанолом, выход 9% [81]
Таблица 2. Зависимость степени восстановления кетона до спирта от условий реакции (23С, 2 ч) [83]
Соединение
Реагент
Растворитель
Выход
Этанол
91
LiB(OMe)2Н
DMF
90
THF
88
Li2Н
THF
-
LiB2Н4
36
LiB(OMe)3На
95
а Молярная радиоактивность препарата 28.9 Ки/ммоль.
РАДИОХИМИЯ том 64 № 4 2022
308
ШЕВЧЕНКО и др.
Схема 2.
В биологически активное соединение, имею-
99-103]. В качестве производных меченого иоди-
щее в своей молекуле метильные группы, мож-
стого метила использовали [метил-3H]метилнози-
но ввести дейтерий или тритий с использованием
лат [101], [метил-3H]метилнозилат [89, 90, 102],
метилирующих агентов. Но необходимой стадией
C3H2=PPh3 [103]. Реакции обычно проводят при 23-
для этого является предварительное деметилиро-
60С в полярных растворителях (метанол, ацетон,
вание исходного соединения, например, синтез
DMF, DMSO и др.) в присутствии оснований (Et3N,
N-деметилированного антибиотика доксицикли-
i-Pr2EtN, Ag2O, K2CO3, NaОН, NaHCO3 и др.), в те-
на [98].
чение от 5 мин до нескольких часов.
Методики, связанные с использованием С3Н3I
Для синтеза (C2Н3)3CNH2 можно использовать
C2Н3MgCl, проводя реакцию с (C2Н3)2C=О с после-
и его производных, хорошо известны [4, 79, 87,
Таблица 3. Синтез реагентов в атмосфере трития для получения меченого иодистого метила*
Реагент
Условия реакции
Продукта
10% Pd/C, i-Pr2EtN, DMF, 23С, 1-12 ч
30% Pd/C, Et3N, EtOAc; 23С, 1 ч; иодистый бензил,
EtOAc, 140°C, 24 -48 ч
30% Pd/C, EtOAc; 23С, 2 ч
(ClCH2)2O
Раствор 0.3 ммоля вещества в 2.1 мл EtOAc, 0.6 ммоля
(TCH2)2O
Et3N, 7.7 мг 10% Pd/C, 1 ч
15 Ки/ммоль
CO2
Cu-Zn-Cr катализатор, 225-230С, 24 ч
CT3OH
CO2
LiAlТ4, 23С, 3 ч
CT3OH
а Меченый иодистый метил получали из предшественников реакцией с HI или иодистым бензилом.
Таблица 4. Метилирующие агенты - производные меченого иодистого метила.
Реагент
Условия реакции
R = CH3, NO2
0.4 ммоля С3Н3I, 0.5 ммоля тозилата серебра, 5 мл СН3CN, 80°C, 18 ч, выход 85%;
нозилат серебра, выход 51% [92-94]
0.8 ммоля Mg, 8 мкмоля I2, 5 мл Et2O, 0.6 ммоля (80 Ки/ммоль) С3Н3I, 10 мин, 23С,
C3H3MgI
45 мин, 30С [95, 96]
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
309
Рис. 1. Синтез меченых соединений с использованием (C2Н3)3CNH2.
дующей реакцией образовавшегося спирта с NaCN,
органических соединений имеет широкую область
2Н2SO4, С2Н3СОО2Н и KOH [104]. С использова-
применения. Основным недостатком этого направ-
нием этого меченого реагента были синтезированы
ления считается невысокая молярная радиоактив-
2Н9-мабутерол, 2Н9-бамбутерол и 2Н9-цимбутерол
ность многих реагентов, синтез которых к тому же
(рис. 1).
часто проводится в несколько стадий.
Из полностью меченного дейтерием ацетона
2.2. Предшественники для получения меченых
можно синтезировать не только (C2Н3)3CNH2, но и
соединений
(C2Н3)2CН-NH2. Для этого дейтерированный аце-
тон восстанавливают алюмогидридом лития, бро-
Для получения содержащих тритий или дей-
мируют трехбромистым фосфором, замещают бром
терий препаратов очень часто используют термин
на азот фталимидом калия с последующей обра-
1. (C2Н3)2CНNH2,
боткой образовавшегося продукта гидразингидра-
CHCl3 (этанол),
том [105].
63(23)°C, 5(2) ч
Полученный меченый реагент с успехом ис-
2. Борогидрид
пользовался для синтеза
2Н6-кленпроперола и
натрия, метанол
2Н6-циматерола с приемлемыми выходами и отлич-
(этанол), 23°C,
2(4) ч
ной химической чистотой (рис. 2) [105].
Для введения тритияиспользуютидругиемеченые
реагенты (C3HHI2, 3HCHO, 3HC3HO, 3HCO (СН3)2,
3HCOOCOСН3, C3HHN2, N-сукцинимидил-[2,3-3H]
пропионат, N-тритиоацетоксифталимид, 3HN=N3H,
C3H3COOН) [4].
Использование меченых реагентов для синте-
за дейтерированных или тритированных аналогов
Рис. 2. Получение 2Н6-кленпроперола и 2Н6-циматерола.
РАДИОХИМИЯ том 64 № 4 2022
310
ШЕВЧЕНКО и др.
Схема 3.
«предшественник». Под этим обычно понимают
и снятия защит получены меченые селанк и семакс
реагент, при обработке которого изотопом водорода
с выходами 15 и 35% соответственно [92, 93, 109].
получают меченый аналог биологически активного
Для получения производного уридина в качестве
соединения. В одних случаях предшественник со-
меченного тритием реагента может быть использо-
держит фрагменты, восстановлением которых мож-
вана меченая уксусная кислота (схема 3) [94].
но получить желаемый меченый препарат. В других
Такие же подходы использовали для синтеза ме-
случаях искомое меченое соединение получали
ченого антагониста тромбоксанового рецептора -
исходя из меченого предшественника. Синтезы с
(+)-S-145 [25]. Последний получали конденсацией
использованием меченых предшественников зна-
метилового эфира
(+)-{1S,[1,2(Z),3,4]}-7-(3-
чительно усложняют общую процедуру получения
амино-бицикло[2.2.1]гепт-2-ил)-5-гептената с [4-3H]
конечного соединения. Поэтому метку в этом слу-
бензол-сульфохлоридом (100 мКи, 23.8 Ки/ммоль)
чае предпочтительно вводить на последних стадиях
при молярном соотношении немеченого компо-
синтеза из соображений радиационной безопасно-
нента к меченому 43 : 1. Радиохимический выход
в этих условиях достигал 80%. После щелочного
сти и с целью уменьшения количеств используемо-
омыления был получен искомый продукт.
го, как правило, дорогостоящего меченого реагента.
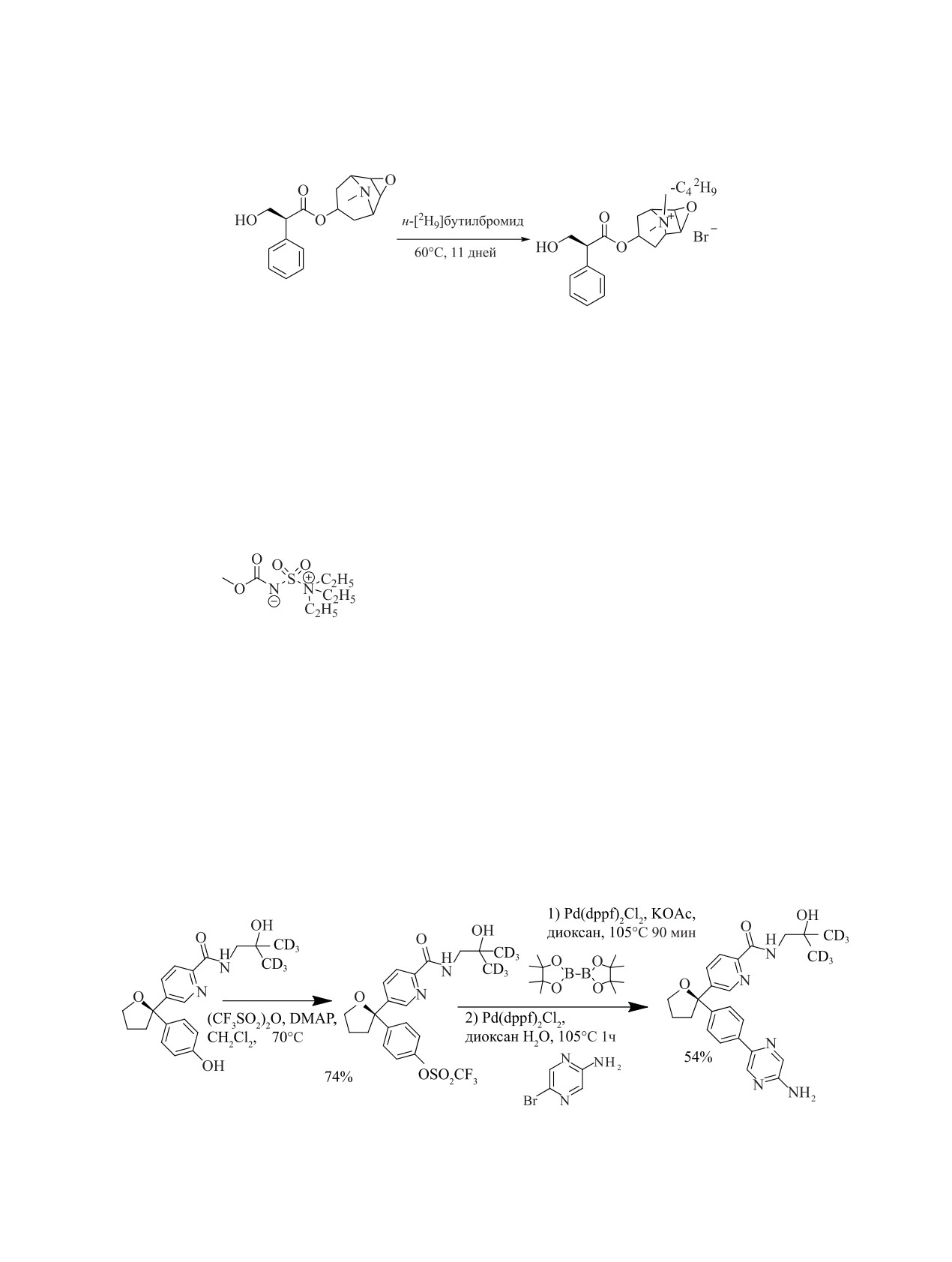
н-[2H9]Бутилбромид был исходным соединени-
Синтез меченного тритием эстронсульфата про-
ем при синтезе бутилового аналога скополамина
водили в два этапа: сначала твердофазным изотоп-
([2H9]бускопана), который является антихолинер-
ным обменом (5% Rh/Al2O3, 180C, 15 мин) полу-
гическим и антимускариновым лекарственным
чали [3H]эстрон (выход 32%, 158 Ки/ммоль), затем
средством, используемым для лечения боли и дис-
[3H]эстрон сульфатировали (пиридин, SO3-пири-
комфорта [95]. Конденсацию проводили, смешивая
дин, 12 ч, выход 65%, 154 Ки/ммоль) [106, 107].
раствор скополамина в безводном ацетонитриле с
Меченый эстрадиол также получали из [3H]эстро-
н-[2H9]бутилбромидом (схема 4).
на при восстановлении последнего борогидридом
Ацетонитрил заменяли безводным эфиром и
(этанол, 15 мин при 0C, 30 мин, 23C), выход 88%,
кипятили еще 30 мин. Выход искомого продук-
95 Ки/ммоль [106].
та 80%.
Меченую арахидоновую кислоту (210 Ки/ммоль)
Дейтерированные жирные кислоты послужи-
конденсировали с аминокислотами (силилирован-
ли источником дейтерия в синтезе более десятка
ный пролин, ацетонитрил, арахидоноил-изобутил-
дейтерированных церамидов [96]. Церамиды полу-
карбонат, 2 ч, 23C, 48 ч, 4C, выход 65%) и пепти-
чены с высокими выходами при использовании в
дами (семакс, DMF, Et3N, арахидоноил-изобутил-
качестве конденсирующего реагента PyBOP [(бен-
карбонат, 2 ч 23C, 48 ч, 4C, выход 65%) [108].
зотриазол-1-илокси)трипирролидинофосфонийгек-
После конденсации меченого пролина
сафторфосфата].
(60 Ки/ммоль) с Boc-Thr-Lys-Pro-Arg-Pro-Gly и
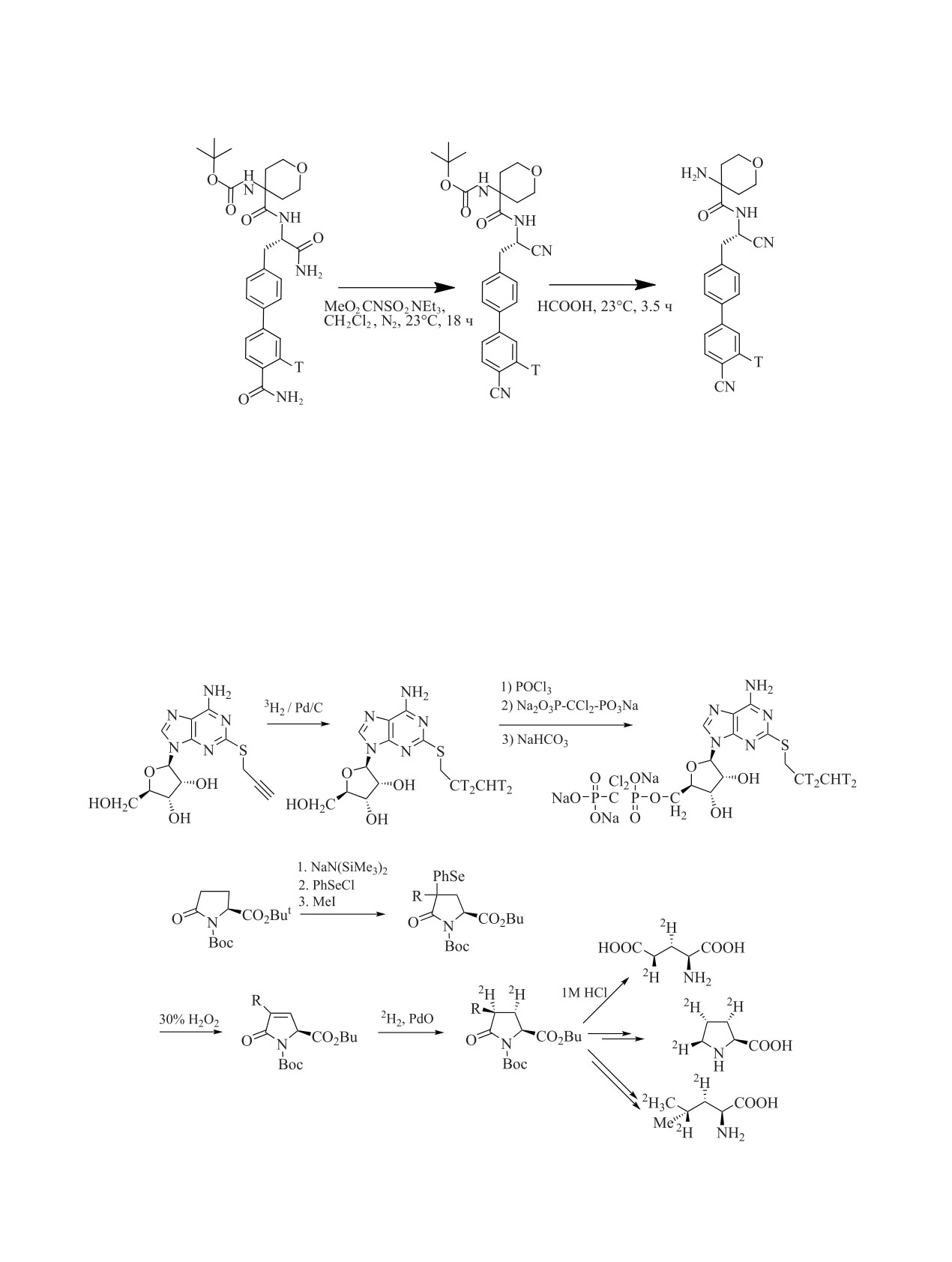
Для синтеза меченного дейтерием AZD6642
Boc-Met-Glu(O-tBu)-His-Phe-Pro-Gly (DMF, Et3N,
использовали [2H6]ацетон. Из [2H6]ацетона полу-
карбонилдитриазол, 40-80 мин, 23C, 30 мин, 50C)
чали (2Н3С)2С(ОН)СН2NН2, который конденсиро-
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
311
Схема 4.
n
вали с (R)5-(2-(4-(бензилокси)фенил)тетрагидрофу-
(12.3 Ки/ммоль). Необходимо отметить, что после
ран-2-ил)-2-бромопиридином и снимали бензиль-
двухстадийного синтеза происходило падение мо-
ную защиту. Дальнейшие превращения приведены
лярной радиоактивности препарата примерно на
на рис. 3 [110].
8%.
Еще одним примером использования меченых
Еще одним примером могут служить много-
предшественников для получения искомого препа-
стадийные синтезы аналогов нуклеотидов, име-
рата является синтез [3H]AZD5248 (рис. 4) [111].
ющих противотромбозную активность
([3H]AR-
Исходное меченое соединение обрабатывали
C67085MX) [82]. В этом случае лабильный фраг-
большим избытком метил-N-(триэтиламмоний-
мент этого соединения (полифосфатный остаток)
сульфонил)карбамата (реактив Бёрджесса):
конденсировали с меченым основанием (cхема 5).
Химическими методами получены и дру-
гие меченые соединения ([3H]BIBN 4096 [112],
[2,11,31-3H]ABT-578
[113],
[3H2]BIIL260
[114],
[3H4]BIIL260 [114]) не менее сложного строения ис-
ходя из меченых предшественников.
Этот реактив обычно используют для мягкой
дегидратации вторичных и третичных спиртов
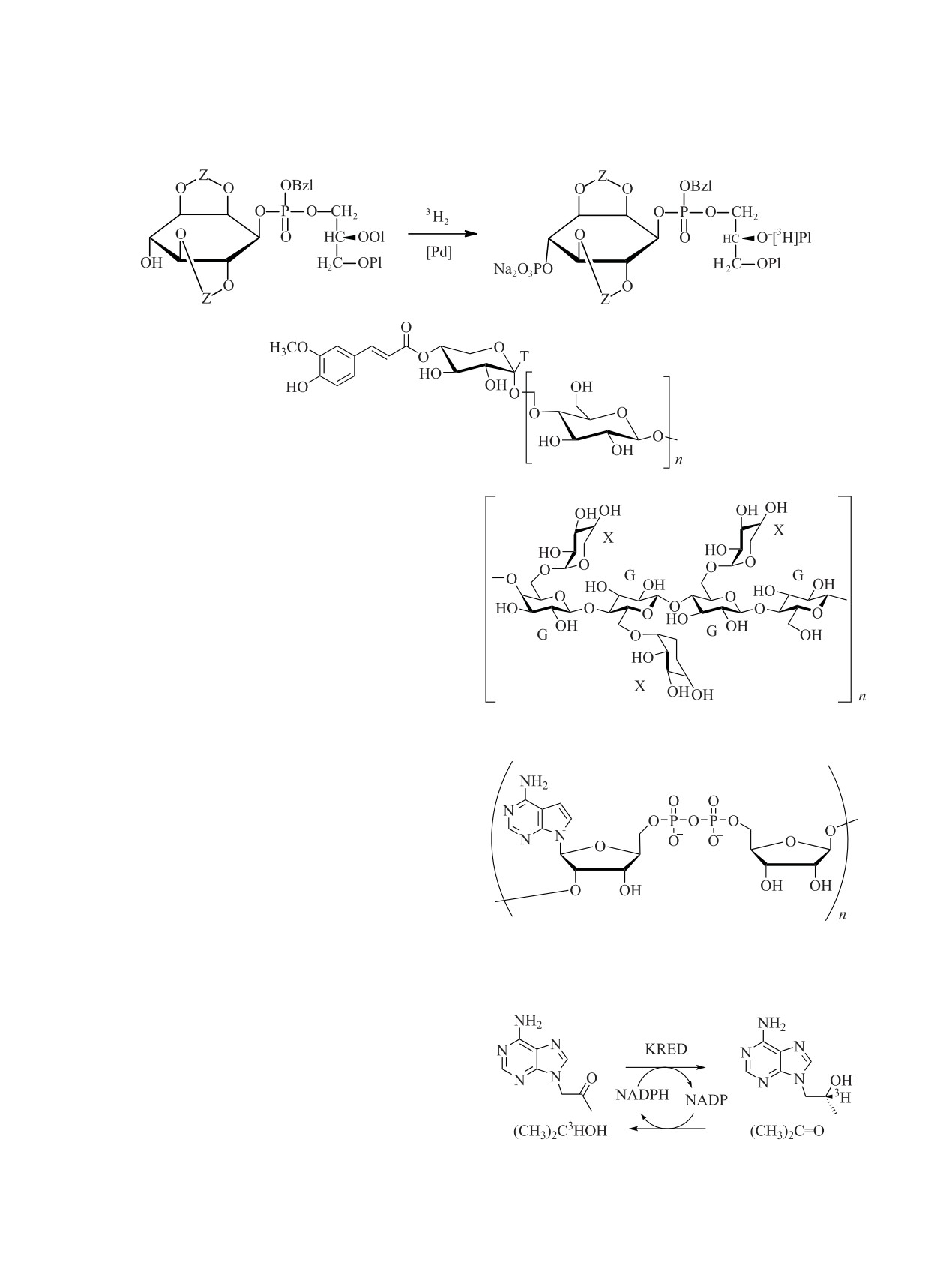
Получение [3,4,5-2Н]пролина, [3,4,5,5,5-2Н]лей-
до соответствующих алкенов, в этой же работе
цина, [3,4-2Н2]глутаминовой кислоты (R = H, Me,
его использовали для дегидратации -СОNН2 до
C2H3) [81] можно осуществить, исходя из меченого
-СN.
производного 5-oxo-Pro (схема 6).
Раствор трет-бутил-N-[4-[[(1S)-2-амино-1-[[4-
3,4-Дидегидропироглутамат получали обработ-
(4-карбамоил[3(5)-3H]фенил)фенил]метил]-2-ок-
кой 30%-ной перекисью водорода раствора в тетра-
со-этил]карбамоил]тетрагидропиран-4-ил]карба-
гидрофуране (23С, 60 мин) фенилселенилирован-
мата (13.3 Ки/ммоль, 2.5 мг, 4.9 мкмоль) и реакти-
ного производного трет-бутил-N-(трет-бутокси-
ва Бёрджесса (50 мг, 210 мкмоль) в CH2Cl2 (2 мл)
карбонил)пироглутамата. Дейтерированием с ок-
перемешивали в атмосфере азота 18 ч. После
сидом палладия в дейтерометаноле (23С, 30 мин)
снятия защитной группы получен [3H]AZD5248
получен
трет-бутил-N-(трет-бутоксикарбо-
–
Рис. 3. Синтез дейтерированного AZD6642. Pd(dppf)2Cl2 - [1,1ꞌ-бис(дифенилфосфино)ферроцен]дихлоропалладий(II).
РАДИОХИМИЯ том 64 № 4 2022
312
ШЕВЧЕНКО и др.
Рис. 4. Синтез [3H]AZD5248.
нил)-[3,4-2Н2]пироглутамат. Кипячением с 1 М HCl
сыщенные предшественники (Z - циклогексили-
в течение 12-15 часов из трет-бутил-N-(трет-бу-
ден, Ol - олеоил, Pl - пальмитоил, Bzl - бензил)
токсикарбонил)-[3,4-2Н2]пироглутамата получали
(схема 7) [25].
[3,4-2Н2]глутаминовую кислоту. Дейтерированный
Фосфорилированием дикеталя мио-инозита
пролин получали из того же предшественника по
дифенилхлорфосфатом получали соединение,
методике [115]. Дейтерированный лейцин получа-
содержащее фосфатную группу в положении 1 мио-
ли из того же предшественника по методике [116].
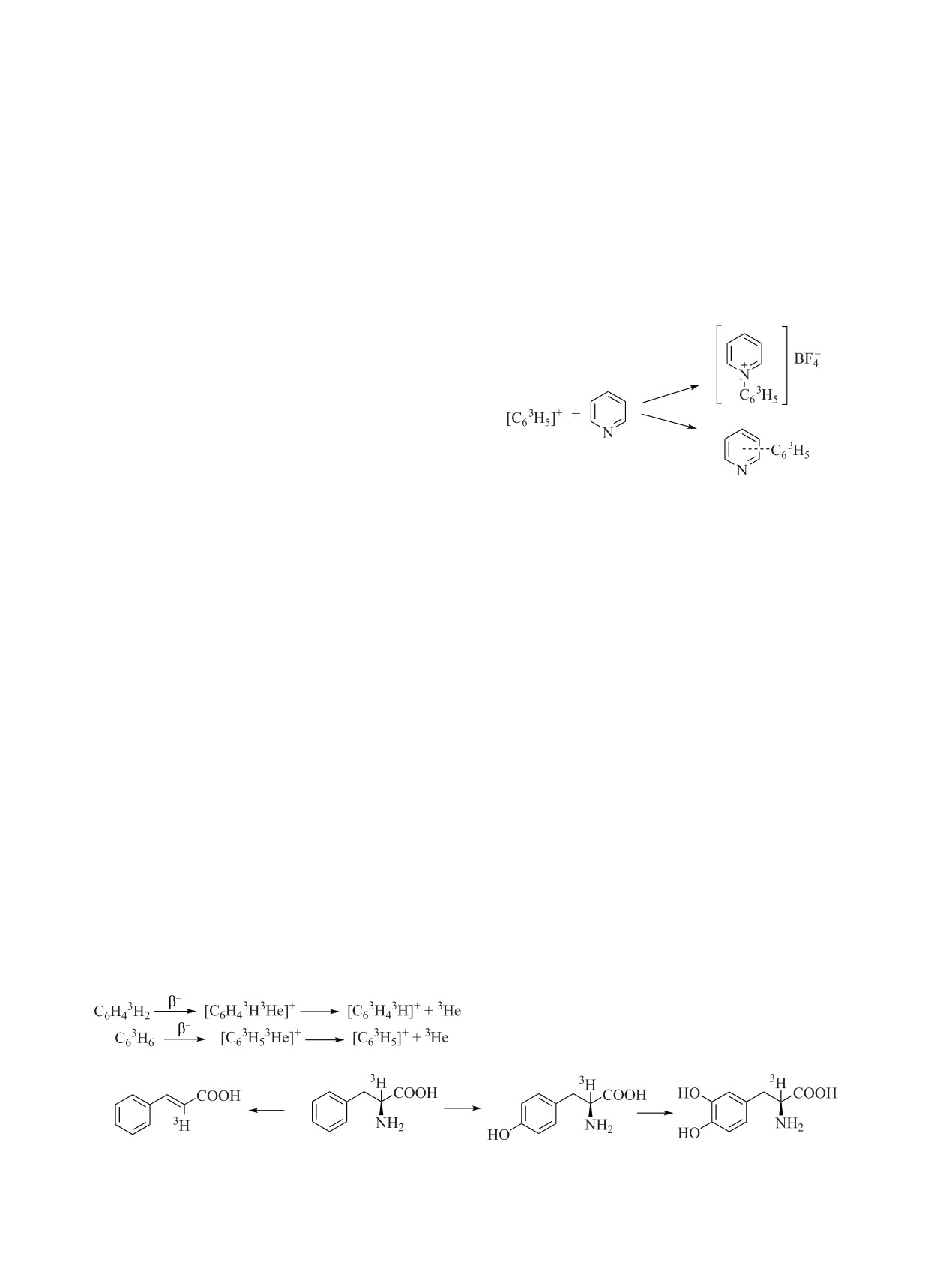
инозита. Положение 4 ацетилировали уксусным
Для получения меченых фосфатидилинозита
ангидридом, затем фенильные группы на фосфате
и дифосфоинозитида были синтезированы нена- снимали гидрогенолизом на платиновом катализаторе
Схема 5.
-
Схема 6.
t
t
t
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
313
Схема 7.
Схема 8.
с последующей обработкой фенилдиазометаном.
Дибензиловый эфир обрабатывали иодидом
натрия, затем нитратом серебра. Серебряную
соль конденсировали с
1-пальмитоил-2-олеоил-
sn-глицерин-3-иодгидрином, и образовавшееся
производное фосфатидилинозита деацетилировали
гидразингидратом.
Если ставилась задача получить фосфатидилино-
зит, защитные группы снимали анионным дебензи-
лированием и обработкой катионитом в H+-форме.
При использовании 3H-аденина получены по-
Для получения дифосфоинозитида сначала фос-
форилировали инозит по положению 4 избытком
ли-ADP-рибозы [117]:
хлороксида фосфора в пиридине, а затем снимали
защитные группы. На последней стадии синтеза
меченых препаратов проводили каталитическое
гидрирование ненасыщенного жирнокислотного
фрагмента в атмосфере газообразного трития [25].
Реже меченые предшественники используют-
ся для проведения ферментативных реакций, по-
зволяющих получать меченые соединения более
При использовании кеторедуктазы (KRED) и
сложного строения. Специфика этого направления
(СН3)2С3НОН удалось ввести тритий в (R)-9-(2-
существенно ограничивает возможности данно-
гидроксипропил)аденин [120]:
го метода. При использовании клеточных культур
оказалось возможным получение радиоактивно
меченых полимеров. При использовании Fesctuca
arundinacea и [3H]арабинозы получен арабинозосо-
держащий сахарид (схема 8) [117, 118].
Инкубацией с меченой глюкозой получены по-
лисахариды (Х - ксилоза, G - глюкоза) [119]:
РАДИОХИМИЯ том 64 № 4 2022
314
ШЕВЧЕНКО и др.
Используя меченый фенилаланин, можно полу-
В результате оказалось возможным получать ра-
чить коричную кислоту (при использовании фени-
нее неизвестные в классической химии органиче-
лаланин аммоний-лиазы) и тирозин (при использо-
ские соединения (рис. 5) [128].
вании фенилаланин 4ꞌ-монооксигеназы), а из тиро-
Таким образом, оказалось возможным разра-
зина - DOPA (тирозиназа в присутствии аскорбино-
ботать способы получения новых биологически
вой кислоты) (схема 9) [121].
активных соединений, меченных тритием, за счет
Используя тирозиндекарбоксилазу, можно из
прямого фенилирования атома азота в шестичлен-
DOPA синтезировать дофамин, а из него -норадре-
ных гетероциклических соединениях:
налин и адреналин, используя дофамин-β-гидрок-
силазу и фенилэтаноламин-N-метилтрансфера-
зу [121].
Меченый тирозин также можно синтезировать
с использованием β-тирозиназы из меченого фено-
ла (схема 10) [121].
Использование данного подхода позволило по-
лучать целые наборы оптически активных меченых
Этим ядерно-химическим методом в одну ста-
соединений с двойными связями, разными и по рас-
дию получены меченные тритием различные
положению, и по конформации. Получить такие со-
N-фенильные производные пиридина, хинолина,
единения другими способами крайне затруднитель-
акридина, фенантридина и бензохинолина, многие
но. Например, были получены простагландины,
из которых являются перспективными бактерицид-
тромбоксаны, лейкотриены, гидроксиэйкозатетра-
ными препаратами широкого спектра действия.
еновые кислоты, гидропероксиэйкозатетраеновые
Также имеются сведения об одностадийном полу-
кислоты, липоксины, гепоксилины, эпокситриено-
чении фенильных ониевых производных элементов
вые кислоты и др. В качестве исходного меченого
V-VII групп, меченных тритием. Ароматический
материала использовали как арахидоновую, так и
фрагмент мог включать метокси, этокси, метиль-
другие эйкозаполиеновые кислоты с молярными
ный, хлор, фтор и другие заместители (схема 11)
радиоактивностями выше 100 Ки/ммоль [25].
[122-127, 128-131].
Все ион-молекулярные реакции проводили в за-
2.3. Получение меченых соединений особым
паянных стеклянных ампулах, в которые были по-
способом инициирования реакции
мещены многократно меченные тритием аромати-
На применении дважды и более меченных три-
ческие соединения, немеченые нуклеофилы и кри-
тием предшественников основан новый нетрадици-
сталлы стабилизирующей соли (KBF4, KClO4, KI).
онный метод синтеза меченых фенилзамещенных
Ампулы с реакционной смесью выдерживали для
органических, элементоорганических и гетеро-
накопления продуктов реакции в количествах, до-
циклических соединений [122-127]. За счет превра-
статочных для их надeжного определения (не менее
щения одного из атомов трития в атом гелия образу-
30 сут), непрореагировавший меченый реагент от-
ется меченый реагент, который можно использовать
гоняли, а затем проводили выделение и идентифи-
для получения искомых меченых соединений:
кацию синтезированных меченых соединений. Для
изучения влияния условий проведения ион-молеку-
лярных реакций на выходы продуктов ядерно-хи-
мического синтеза их накопление осуществляли
Схема 9.
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
315
Схема 10.
как в твердой (-196°С; -5 и -10°С), так и в жидкой
При использовании метода гетерогенного изо-
фазе при температуре ~23°С [122-127].
топного обмена с газообразным тритием установ-
лено, что основная часть метки включается в пер-
Основные недостатки при использовании этого
вые часы реакции [25]. Поэтому необходимо под-
подхода однотипны недостаткам, которые присущи
бирать условия реакции, при которых насыщение
методу Вильцбаха (рис. 5). Например, при изуче-
активных центров металлов-катализаторов тритием
нии реакций с галогенпроизводными бензола ока-
происходит относительно медленно (использова-
залось, что атака фенил-катиона по неподеленной
ние частично дезактивированных катализаторов
паре электронов атома галогена приводит к образо-
и проведение реакций при пониженном давлении
ванию галогенониевого катиона. Стабилизация воз-
трития). Большое влияние на степень изотопного
никшего катиона возможна в двух направлениях:
обмена оказывают растворители. Молярные актив-
стабилизация комплексным анионом с образовани-
ности получаемых препаратов, как правило, низкие.
ем ониевого соединения и фрагментация катиона с
Однако в ряде случаев (например, введение трития
отщеплением одного из радикалов с образованием
в ароматические соединения или в соединения, где
меченого субстрата. Кроме того, атака фенил-катио-
возможна кето-енольная таутомерия), возникает
нов может происходить также по бензольному коль-
возможность получения высокомеченных соедине-
ний и этим методом.
цу в исходном соединении с образованием десятков
различных фенилзамещенных изомеров (о-, м-, п-)
При использовании гомогенных катализаторов
в более ранних работах в основном приводились
фенилгалогенидов. Поэтому этим методом пользу-
сведения, которые относились к иридиевым катали-
ются не часто.
заторам [2-5, 128-150]. В результате исследований
3. ВВЕДЕНИЕ МЕТКИ ИЗОТОПНЫМ
был выявлен целый ряд особенностей и закономер-
ОБМЕНОМ В РАСТВОРЕ С ИСПОЛЬЗОВАНИЕМ
ностей введения метки этим методом. Определены
ГАЗООБРАЗНОГО ДЕЙТЕРИЯ ИЛИ ТРИТИЯ
стабильность этих катализаторов, лучшие раство-
рители (хлористый метилен, ацетон и тетрагидро-
В предыдущем обзоре [1] рассматривалось вве-
фуран). Установлено, что метка в растворитель
дение метки в биологически активные соединения
практически не включается, некоторые заместите-
изотопным обменом при использовании как гетеро-
ли сильно ингибируют изотопный обмен, двойные
генных, так и гомогенных катализаторов.
связи, винильные двойные и сопряженные еноно-
Рис. 5. Электрофильные реакции гетероциклической системой хиназолина с меченым фенил-катионом.
РАДИОХИМИЯ том 64 № 4 2022
316
ШЕВЧЕНКО и др.
Схема 11.
вые связи гидрируются, а нитрогруппы, ароматиче-
активность даже близких по строению соединений
ские галоиды и насыщенные кетоны не восстанав-
может отличаться в десятки раз [4, 82, 135]. Она
ливаются.
могут быть меньше десяти и больше ста Ки/ммоль.
Естественно, те же закономерности наблюдаются и
Поэтому здесь будет приведен данные только по
при получении соединений, меченных дейтерием
гомогенному катализу на основе других металлов, а
[142]. Так как растворить некоторые препараты в
данные об иридиевых катализаторах будут приведе-
CH2Cl2 не удается, изучается возможность исполь-
ны в основном, чтобы показать отличие их от ката-
зования других растворителей [83, 157-161].
лизаторов на основе других металлов, например, в
плане распределения метки в молекулах органиче-
Влияние стерических факторов при использова-
ских соединений.
нии гомогенных катализаторов можно проследить
при введении трития в ароматические альдеги-
В последние годы особенно бурно развиваются
ды [162, 163]. Методика проведения этой реакции
работы, где в качестве гомогенных катализаторов
не отличалась от традиционной.
3,5-Ди-трет-
используются комплексы родия, рутения, иридия,
бутил-4-гидроксибензальдегид (10 мг) и (1,5-ци-
кобальта и железа [151-156]. Установлено, что при
клооктадиен)(пиридин)(трициклогексилфосфин)
использовании гомогенных катализаторов моляр-
иридий(I) гексафторфосфат (10 мг) в дихлорметане
ная активность препаратов колеблется в широких
(2 мл) перемешивали в атмосфере газообразного
пределах. Влияние оказывает природа как веще-
трития (10 Ки) при 23°C, 16 ч. Молярная радиоак-
ства, так и катализатора. Очевидно, от строения
тивность продукта 24 Ки/ммоль. Установлено, что
вещества и катализатора зависит вероятность об-
метильные группы уменьшали включение трития в
разования комплекса между ними, что необходимо
ароматическое кольцо на 19%, а трет-бутильные
для включения дейтерия или трития при изотопном
заместители в ароматическом кольце приводили к
обмене.
полному отсутствию метки в нем. Метка включа-
Номенклатура изотопномеченных соединений
лась только в формильную группу.
постоянно расширяется. Тритий и дейтерий вводят
в новые производные пиридина, стероиды, в произ-
3.1 Использование катализаторов на основе
водные анилина, нитрофенола, индолов, амидов и
железа
гетероциклических соединений [4, 135, 157-159].
Удалось синтезировать целый ряд катализаторов,
Поэтому развитие этого направления продолжает-
содержащих самые разные лиганды. В атмосфере
ся постоянно. В настоящее время используют как
водорода молекулы азота замещаются и возникают
фирменные катализаторы, так и катализаторы, ко-
связи атома железа с водородом (схема 12 )[152].
торые готовят непосредственно перед проведением
При использовании иридиевого катализатора
изотопного обмена [85].
дейтерий или тритий вводится в о-положения к
Полученные данные показывают, что молярная
заместителю в ароматическом кольце препарата, в
активность меченых препаратов сильно зависит от
то время как в присутствии катализатора на основе
строения соединений, их растворимости. Молярная
железа метка включается в м- и п-положения аро-
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
317
Схема 12.
i
i
i
i
i
i
(H4-iPrCNC)Fe(N2)2
(H4-iPrCNC)FeН2N2
(H4-iPrCNC)FeН4
-C6H3-4,5-H2-имидазол-2-илиден)2C5H3N)]Fe(N2)2
(H4-iPrCNC)Fe(N2)2 [2,6-(2,6-Pri
2
Схема 13.
t
t
i
i
i
i
t
t
[(NEt3)Ni(OPiv)2]2
[(iPrDI)Ni(μ2-H)]2
матического кольца. При этом включение дейтерия
же изменить распределение метки в молекулах этих
в отличие от иридиевых катализаторов мало зави-
соединений. При использовании атомов иридия и
сит от природы заместителя в ароматическом коль-
железа это продемонстрировано особенно нагляд-
це. Поэтому катализаторы на основе железа в ряде
но. Большое внимание при этом уделяется более
случаев оказались значительно эффективнее при
доступным, стабильным и устойчивым в условиях
введении дейтерия и трития изотопным обменом
реакции никелевым катализаторам (схема 13) [153].
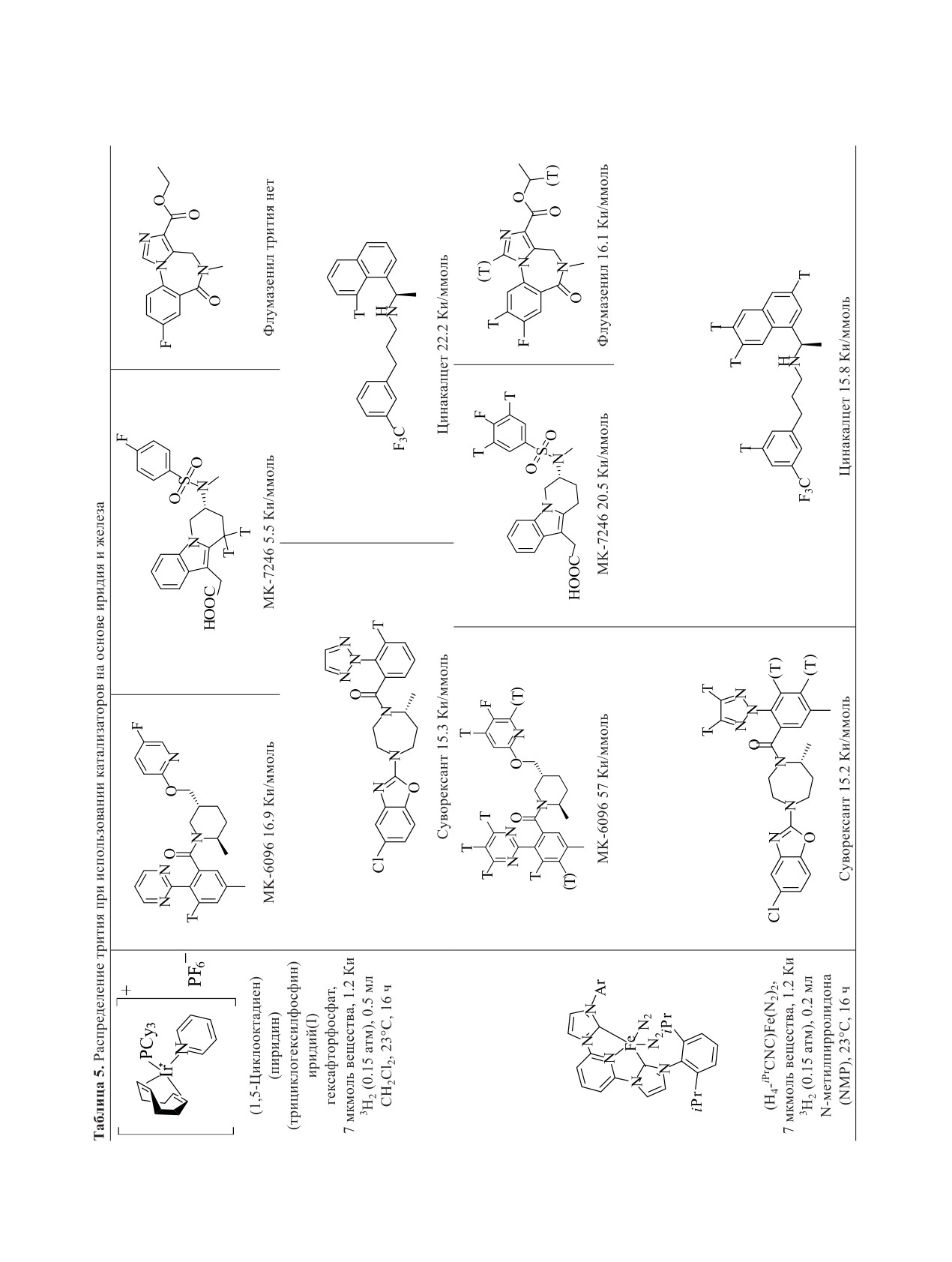
(табл. 5) [151, 152].
Стандартная процедура получения in situ гидри-
Как видно из приведенных данных (табл. 5),
да никеля [(iPrDI)Ni(μ2-H)]2 состояла в предваритель-
(H4-iPrCNC)Fe(N2)2 способствует включению три-
ном смешивании раствора 0.5 экв [(NEt3)Ni(OPiv)2]2,
тия, как правило, лучше, чем (1,5-циклооктадиен)
1.0 экв iPrDI
[N,Nꞌ-бис(2,6-диизопропилфенил)-2,3-
(пиридин)(трициклогексилфосфин)иридий(I)гек-
бутандиимина] и 6.0 экв HSi(EtO)3 в THF в течение
сафторфосфат. Достоинство (H4-iPrCNC)Fe(N2)2
3 ч. В результате образуется раствор никелевого ка-
особенно заметно, когда о-положения к направля-
тализатора, который можно сразу же использовать.
ющей группе не содержат атомов водорода. В этом
Активность катализатора сохраняется в течение не-
случае на иридиевом катализаторе включения изо-
скольких дней при хранении в морозильной камере.
топов водорода не происходит, в то время как ката-
Изотопный обмен проводили при использовании
лизатор на основе железа остается способен содей-
ствовать включению трития.
раствора [(iPrDI)Ni(μ2-H)]2 в 0.2 мл THF, содержа-
щего 25 мол% катализатора. В реакционную ампу-
3.2. Использование никелиевых катализаторов
лу (1 мл) помещали 2-3 мг препарата, 0.15 мл THF
Разработка гомогенных катализаторов на основе
и 50 мкл раствора никелевого катализатора. Ампулу
разных металлов позволяет увеличить количество
замораживали жидким азотом, вакуумировали и за-
препаратов, в которые можно ввести тритий, а так-
полняли газообразным тритием (1.0 Ки, 0.15 атм).
РАДИОХИМИЯ том 64 № 4 2022
318
ШЕВЧЕНКО и др.
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
319
Таблица 6. Эффективность введения трития в присутствии [(iPrDI)Ni(μ2-H)]2 (Ки/ммоль) [153, 164]
МК-6096 (23.3)
Варениклин (25.6)
MK-7622 (24.2)
MK-5395 (13.2)
Тропикамид (17.9)
Папаверин (20.9)
Эторикоксиб (18.0)
Фамцикловир (1.9)
Иматиниб (2.7)
Реакционную смесь перемешивали при 23°C в те-
Дейтерирование происходило в 1.6 раза быстрее
чение 20-24 ч (табл. 6).
с 4-CF3-замещенным пиридином по сравнению с
пиридином. Напротив, относительная константа
Молярная активность и распределение три-
скорости H/2Н-обмена в более богатом электрона-
тия в молекулах зависят от природы препаратов.
ми 4-пиколине оказалась в два раза меньше, чем в
Установлено, что эффективность включения изото- пиридине. Таким образом, в более электронодефи-
пов водорода зависит от того, донором или акцепто- цитных положениях изотопный обмен происходит с
ром электронов являются заместители в ароматиче- большей вероятностью.
ском кольце препарата (приведена степень изотоп-
3.3. Использование катализаторов на основе
ного обмена в минуту):
кобальта
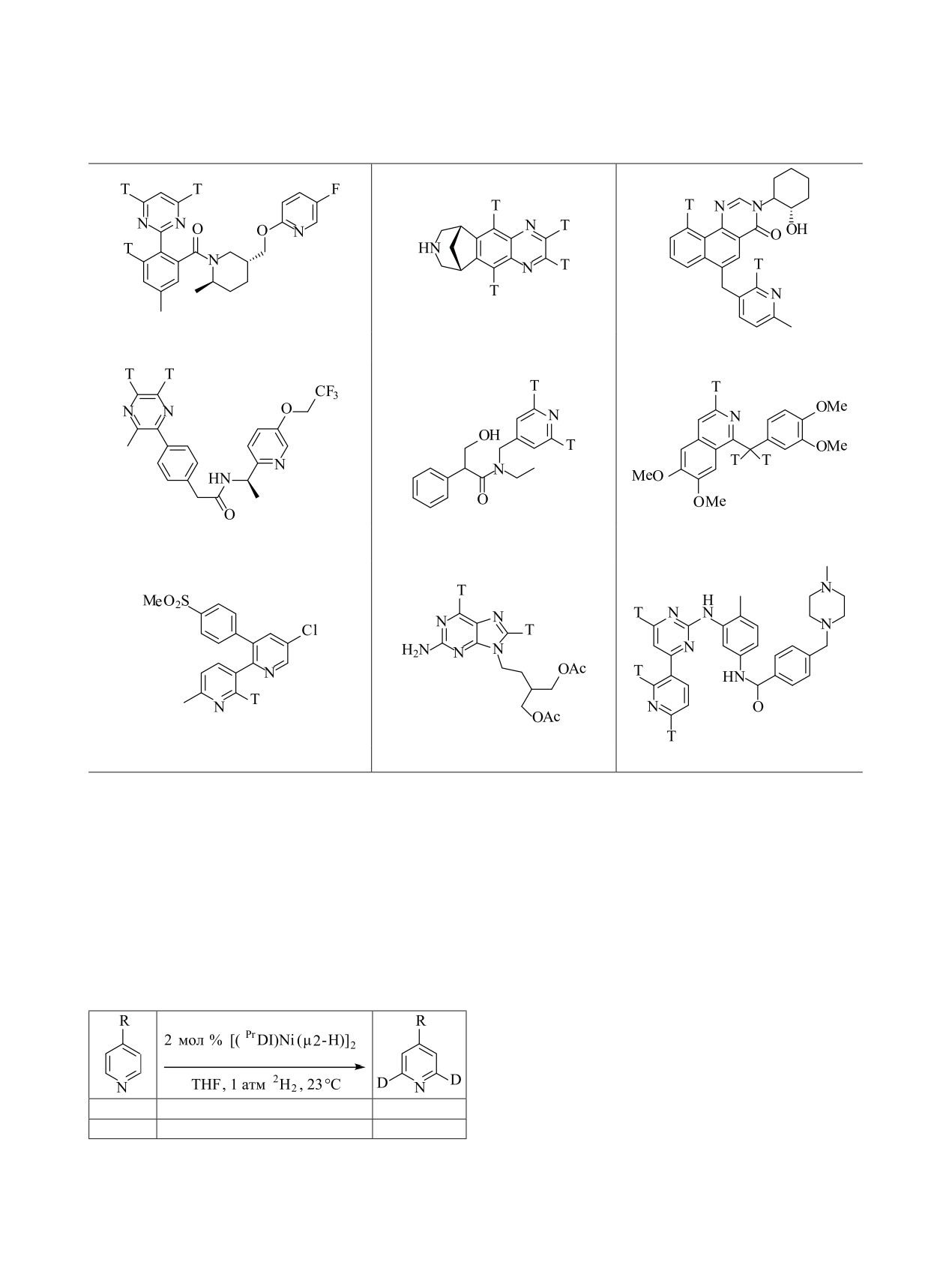
Для включения дейтерия не только в ароматиче-
i
ское кольцо, но и в алкильные заместители алкила-
ренов можно использовать различные кобальтовые
комплексы (схема 14) [154-156, 164-166].
R = CH3
R = H
R = CF3
Реакции с кобальтовыми катализаторами прово-
0.027
0.114
0.181
дили по следующей методике. Раствор алкиларена
РАДИОХИМИЯ том 64 № 4 2022
320
ШЕВЧЕНКО и др.
Схема 14.
i
i
i
i
i
i
i
i
(CyADI)Co(CH2SiMe3)2
(iPrDI)Co-(CH2SiMe3)2
(iPrDI)Co(η3-C3H5)
Схема 15.
i
в додекане перемешивали с 5 мол% катализатора
Электронодонорные заместители повышают сте-
при 4 атм 2Н2 в течение 24 ч при нагревании. При
пень обмена, а объемные группы (как заместители,
этом включение дейтерия происходило в несколь-
так и алкильные группы, в которые вводится дейте-
ко положений молекулы алкиларена. Наибольшая
рий) уменьшают степень обмена:
степень включения при использовании (CyADI)
i
Co(CH2SiMe3)2 наблюдалась у бензиловых свя-
зей С-Н (>95%), но м- и п-положения также были
дейтерированными
(11 и
14% соответственно).
Наблюдалось также восстановление арена (12%) до
При использовании кобальтовых катализаторов
метилциклогексана. Из этих данных очевидно, что
оказалось возможным вводить дейтерий изотопным
(CyADI)Co(CH2SiMe3)2 предпочтительно активиру-
обменом с протием, входящим в хиральный центр
ет бензильные C(sp3)-H положения (табл. 7).
молекулы алкиларена, при сохранении последнего
При использовании (iPrDI)Co(CH2SiMe3)2 также
(табл. 9).
наблюдались высокие уровни (>95%) включения
Реакции проводят с 0.55 ммоль алкиларена,
дейтерия в бензильные С-Н-связи с меньшим вос-
10 мол% (iPrDI)Co(CH2SiMe3)2 и 1 атм 2Н2 в 0.55 мл
становлением арена (8%) (схема 15).
гептана при 50°С в течение 24 ч. Процент включе-
При использовании (iPrDI)Co(η3-C3H5) изотоп-
ния дейтерия при разных атомах углерода указан в
ный обмен измерялся несколькими процентами, а
табл. 9. Влияние этой реакции на сохранение исход-
гидрирование вообще не происходило (табл. 7).
ного строения алкиларена проводили методом хи-
Установлено, что на изотопный обмен в при-
ральной газовой хроматографии.
сутствии (iPrDI)Co(CH2SiMe3)2 влияют как природа
Сохранение исходного строения субстрата при
заместителя, так и стерические факторы (табл. 8). введении дейтерия указывает на то, что изотопный
Таблица 7. Распределение дейтерия при использовании кобальтовых комплексов [164-166]
(CyADI)Co(CH2SiMe3)2
(iPrDI)Co(CH2SiMe3)2
(iPrDI)Co(η3-C3H5)
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
321
Таблица 8. Влияние стерических факторов и природы соединений на эффективность изотопного обмена и степень
замещения дейтерием в его ароматическом и алкильном фрагментах
Таблица 9. Введение дейтерия в оптически активные
4. ВВЕДЕНИЕ МЕТКИ ИЗОТОПНЫМ
соединения
ОБМЕНОМ С ИСПОЛЬЗОВАНИЕМ ТРИТИЕВОЙ
ИЛИ ДЕЙТЕРИЕВОЙ ВОДЫ
Многие из перечисленных в предыдущем разде-
ле катализаторов применимы и при использовании
в качестве источника изотопа водорода дейтерие-
вой или тритиевой воды. Это относится к гомоген-
ным и гетерогенным катализаторам. Но в отличие
от использования реакций, которые проходят в ат-
Оптическая чистота препарата до и после введения
мосфере газообразных дейтерия и трития, реакции
метки, %
с дейтериевой или тритиевой водой в ряде случа-
Немеченый
98
92
72
Меченый
98
92
72
ев могут происходить только при использовании
Отношение
98%
98%
98%
посредников [167-170]. Появление этих меченых
посредников повышает эффективность включения
дейтерия и трития в искомое соединение.
обмен осуществляется за счет внутримолекуляр-
4.1. Распределение дейтерия и трития
ного (SЕi) механизма электрофильного замещения,
в молекулах биологически активных соединений
(схема 16).
при изотопном обмене без использования
Важно отметить в связи с этим, что в данной ра-
катализаторов
боте при обработке бензилциклопропана (табл. 8)
Введение метки за счет изотопного обмена с
газообразным дейтерием не наблюдается раскры-
тритиевой водой, если вещество выдерживает на-
тия циклопропанового кольца. Это указывает на то,
гревание и среды с высокими значениями рН, мож-
что механизм активации связей C(sp3)-H не ради-
но эффективно проводить и в отсутствие катализа-
кальный.
торов [171-173].
Схема 16.
РАДИОХИМИЯ том 64 № 4 2022
322
ШЕВЧЕНКО и др.
Таблица 10. Эффективность изотопного обмена между
Таблица 11. Зависимость эффективности изотопного
дейтериевой водой и абсцизовой кислотой в зависимости
обмена между дейтериевой водой и абсцизовой кислотой
от температуры и времени реакции.а Используемое
от температуры и времени реакции. Используемое
основание - триэтиламин
основание - диизопропилэтиламин
Температура, С (время, мин)
Температура, С (время, мин)
2Н
2Н
110 (30)
160 (15)
200 (10)
200 (10)
200 (30)
220 (10)
220 (20) преп.а
0
91.06
69.12
45.42
0
39.73
8.32
2.51
5.85
1
6.27
15.97
25.29
1
28.79
29.42
13.06
18.64
2
2.98
8.85
9.72
2
9.11
30.87
24.74
29.79
3
0
4.11
6.64
3
6.51
16.95
19.59
25.29
4
0
1.87
6.14
4
6.37
9.18
16.62
13.93
5
0
0.12
2.73
5
5.22
3.42
12.27
4.34
6
0
0
2.33
6
3.17
1.04
6.62
1.57
7
0
0
1.60
7
0.99
0.75
2.65
0.50
8
0
0
0.19
8
0.16
0.08
1.42
0.14
Σ
0.11
0.54
1.30
Σ
1.45
2.08
3.25
2.45
а Приведено содержание изотопомера в дейтерированном пре-
а Препаративное получение дейтерированного препарата.
парате. Σ - среднее количество атомов дейтерия в молекуле ве-
щества. То же в табл. 11.
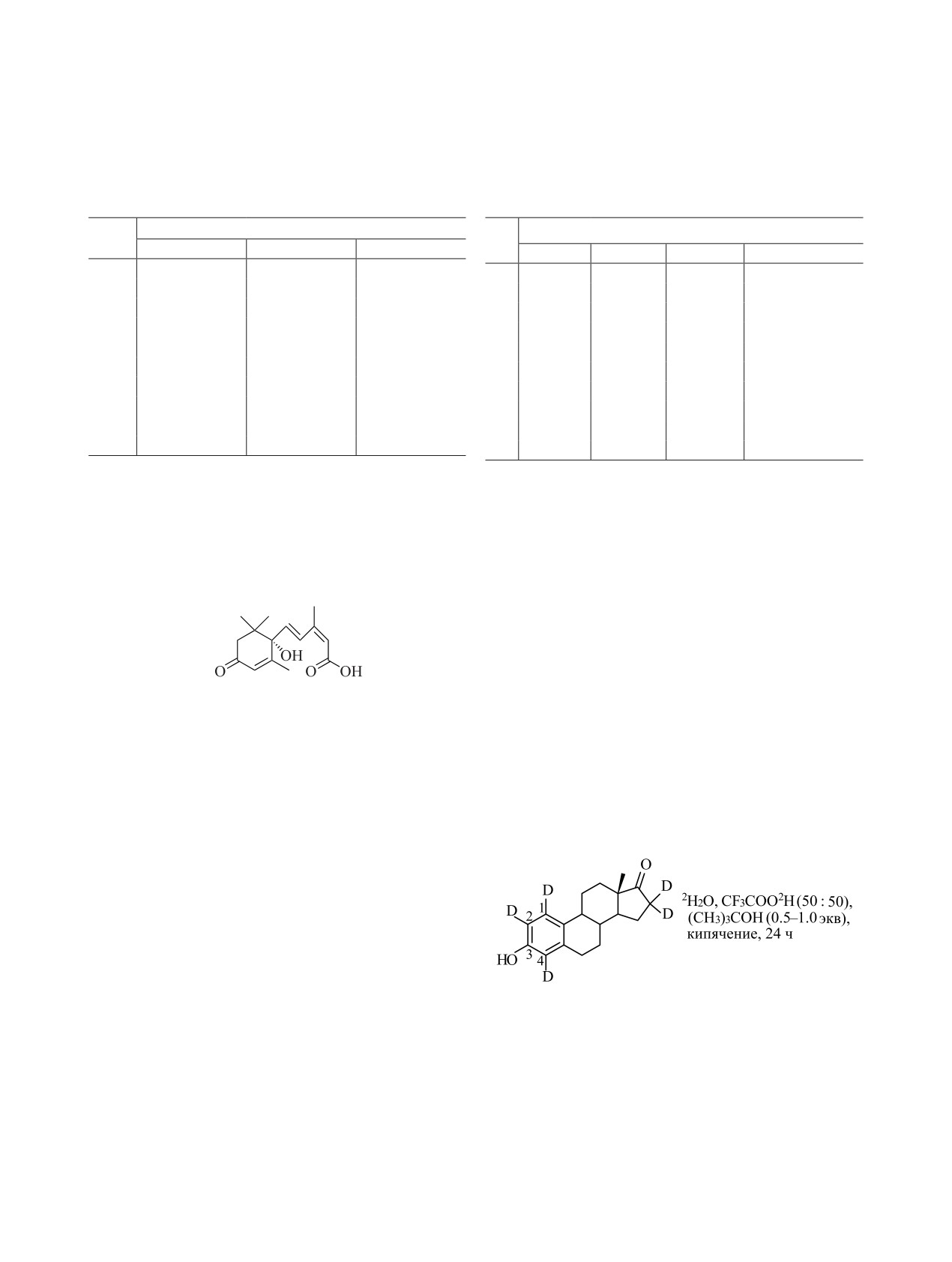
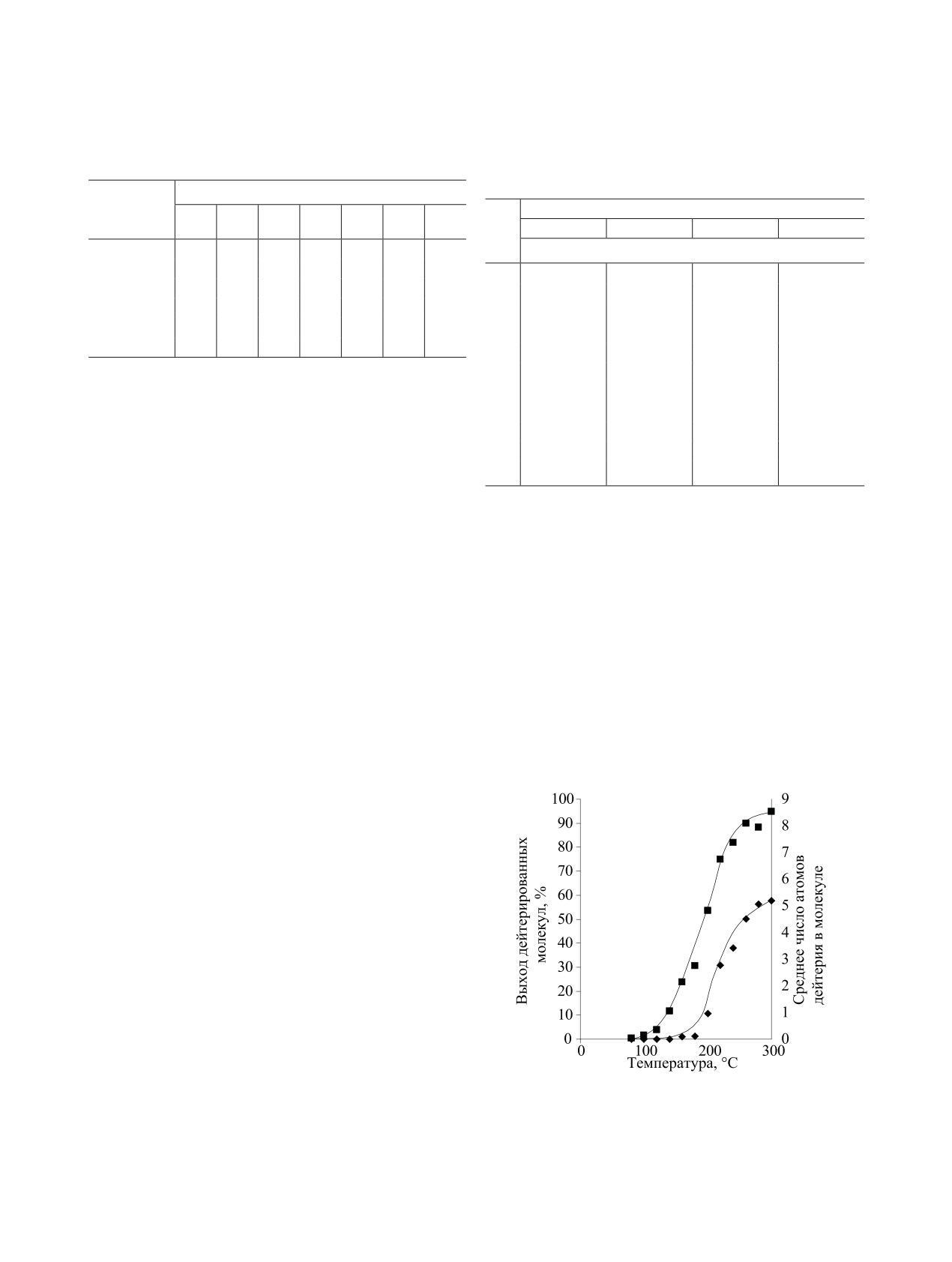
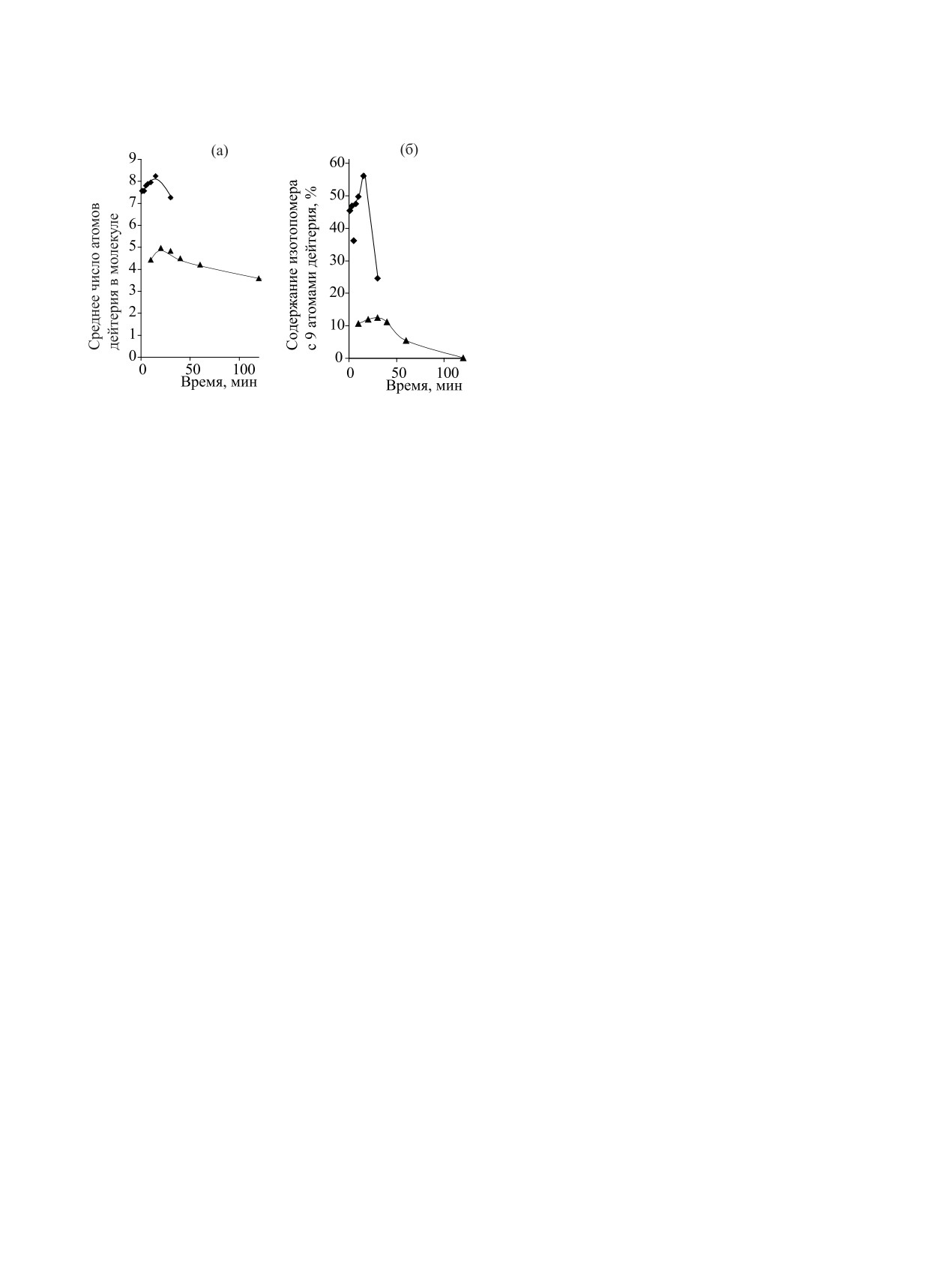
Например, при введении трития в абсцизовую
включать в ароматические соединения от 40-60%
кислоту оказалось, что это полиненасыщенное со-
метки в пересчете на молярную активность тритие-
единение может выдержать высокие температуры
вой воды [178, 179].
даже в щелочных условиях [174]:
Оказалось, что условия реакции могут влиять на
распределение изотопа водорода в молекуле биоло-
гически активного соединения. Например, при вве-
дении дейтерия в стероид при разном составе ре-
акционной смеси распределение изотопа водорода
В качестве основания использовали Et3N или ди-
было разным [180].
изопропилэтиламин (табл. 10, 11).
При введении дейтериевой метки в эстрон при
По данным масс-спектроскопии, основная часть
кислотном катализе в присутствии
0.5-1.0 экв
дейтерия включалась в кольцевую часть молеку-
трет-бутанола включение изотопной метки проис-
лы абсцизовой кислоты. Кроме -положений к ке-
ходило во все свободные положения ароматическо-
тогруппе часть дейтерия включается в метильные
го фрагмента эстрона и в -положения к кето-груп-
группы.
пе [180]:
При введении трития в абсцизовую кислоту 3Н2О
получали восстановлением PdO и 5% PdО/BaSO4
в диоксане газообразным тритием. Затем диоксан
и тритиевую воду перегоняли в ампулу, в которой
находилась абсцизовая кислота. Ампулу заполняли
аргоном, вносили диизопропилэтиламин и запаива-
ли. Реакцию вели 20 мин при 220C. После очистки
методом ВЭЖХ выход абсцизовой кислоты соста-
В отсутствие трет-бутанола включение дейте-
вил 50%, молярная активность - 30.5 Ки/ммоль.
рия в 1-положение не происходило. Причиной до-
полнительного включения дейтерия оказалось об-
Изотопный обмен с тритиевой водой можно так-
же проводить при кислотном катализе (3HHO/AlCl3,
разование неустойчивого трет-бутильного произ-
H3PO4/BF3, HCl, трифторуксусная кислота и др.) [4,
водного в 2-положении эстрона, с одновременным
175, 176] и при использовании кислот Льюиса [177-
включением дейтерия в 1- и 4-положения молекулы
179]. Смесь BF3/Et2O/3Н2О, BF3/3H3PO4 позволяет
эстрона (схема 17).
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
323
Схема 17.
На заключительной стадии реакции катион дей-
руживается в ароматических фрагментах органиче-
терия разрушает трет-бутильное производное
ского соединения [187]:
эстрона с отщеплением 2-метилпропена, в резуль-
тате происходит включение дейтерия во второе по-
ложение в ароматическом фрагменте эстрона.
4.2. Распределение изотопов водорода
в молекулах биологически активных соединений
при изотопном обмене в присутствии
гетерогенных катализаторов
Для работы со 100%-ной тритиевой водой обыч-
но применяют гетерогенные катализаторы, которые
При нагревании тритиевой воды и раствора ве-
устойчивы к радиолизу и тем самым не создают
щества в диглиме с платиновой чернью или 5% Pt/C
дополнительных трудностей при выделении мече-
молярная радиоактивность вещества составляла от
ных препаратов из реакционных смесей [181-184].
10 до 60% от молярной радиоактивности тритиевой
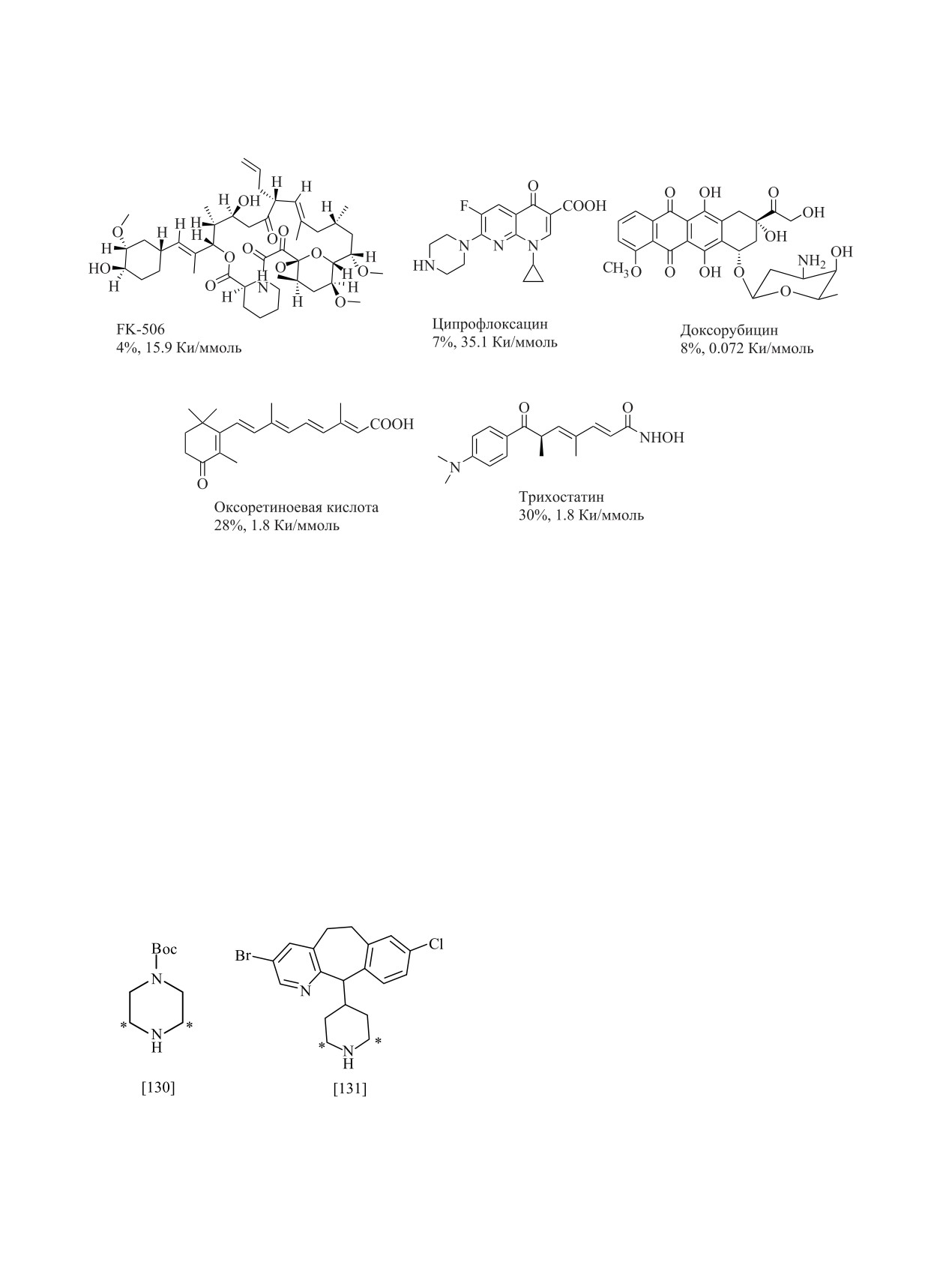
Есть данные о получении этим методом целого
воды [25, 190, 191]. И в этом случае вся метка вклю-
ряда стероидов (молярные радиоактивности от 5.4
чается в ароматический фрагмент органического
до
11.0 Ки/ммоль), сахаров
(6.5-7.0 Ки/ммоль),
соединения:
гиббереллинов (4.9-5.4 Ки/ммоль), фузикокцина Н
(10.8 Ки/ммоль) [25, 182, 185], т.е. данный метод
применим для введения метки практически в любое
соединение. Однако условия проведения реакции
необходимо корректировать с учетом устойчивости
препаратов. Ряд соединений, такие как доксору-
бицин, FK-506 и ципрофлоксацин, имеют выходы
ниже 10%, но, тем не менее, количества меченых
препаратов оказываются достаточными для прове-
Также установлено, что при использовании сме-
дения полного цикла биологических исследований
си Pt/C и Pd/C наблюдается синергетический эф-
(схема 18).
фект, когда включение изотопов водорода оказыва-
лось выше, чем включение изотопов водорода при
Изотопный обмен с тритиевой водой можно
раздельном использовании этих катализаторов.
проводить, используя катализаторы на основе пал-
ладия, платины, никеля, родия, рутения [186-190].
Кроме тритиевой воды, в качестве доноров три-
При проведении реакций изотопного обмена с три-
тия могут быть использованы: муравьиная кислота
тиевой водой при использовании этих катализато-
и ее соли, триэтилсилан, гидразин, циклогексен, те-
ров молярные активности препаратов составляли,
трагидрохинолин. Инициирование изотопного об-
как правило, 15-30% от молярной активности три-
мена этим методом можно осуществлять не только
тиевой воды.
нагреванием, но и другими способами, например,
При изотопном обмене с тритиевой водой в при-
микроволновым облучением реакционной смеси
сутствии никеля Ренея практически вся метка обна-
[192, 193].
РАДИОХИМИЯ том 64 № 4 2022
324
ШЕВЧЕНКО и др.
Схема 18.
4.3. Распределение дейтерия и трития
4.4. Использование рутениевых комплексов
в молекулах биологически активных соединений
для асимметрического восстановления
при изотопном обмене в присутствии гомогенных
кетонов и иминов
катализаторов
Получение оптически активных меченых соеди-
Для введения изотопов водорода изотопным об-
нений подразумевает как сохранение конформации
меном с тритиевой или дейтериевой водой можно
в результате введения трития или дейтерия [197], так
использовать относительно несложные гомогенные
и изменение конформации, например, когда ставит-
катализаторы, например, Ru(PPh3)3Cl2. При этом
ся задача введения метки с одновременным превра-
удавалось ввести метку в самые разнообразные со-
щением L-аминокислоты в D-аминокислоту [198].
единения (рис. 6) [142-144, 194-196].
Большой интерес также вызывает возможность об-
При использовании катализаторов более слож-
разования хиральных центров в результате восста-
новления двойных связей в присутствии асимме-
ного строения (раздел III.1.), применяя тот или иной
катализатор, можно получить препарат не только с
трических реагентов [199]. В качестве лигандов для
необходимым содержанием изотопа водорода, но
синтеза рутениевых катализаторов можно исполь-
и с необходимым для дальнейшего использования
зовать
(1R,2R)-(+)-1,2-дифенил-1,2-этандиамин и
распределением метки в молекулах этого соедине-
(1S,2S)-(-)-1,2-дифенил-1,2-этандиамин, в которых
ния (DG - направляющая группа) (схема 19) [165].
одна из аминогрупп связана с SO2C6H4-p-CH3 или
SO2CH3. В рутениевый комплекс также входит ал-
килзамещенный бензол (схема 20).
При восстановлении кетонов оптическая чисто-
та достигала 92-99%, иминов - 77-96% [169, 170,
200-202].
4.5. Использование посредников для активации
изотопного обмена
Нередко включение метки происходит не непо-
Рис. 6. Распределение изотопа водорода в соединениях,
средственно при взаимодействии протонов молекул
меченных в присутствии Ru(PPh3)3Cl2.
органического соединения с активированными ча-
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
325
Схема 19.
Схема 20.
Схема 21.
стицами, генерируемыми катализатором при взаи-
терий реакцией со смесью меченных метанола и
модействии с газообразными изотопами водорода
воды. Обычно CН3O3Н получали при обработке
или дейтериевой и тритиевой водой, а при участии
раствора MeOH в ТHF 1 Ки 3Н2 (давление 0,13 атм.)
посредников. Этим методом пользуются, как пра-
в присутствии Pd/C. Раствор меченого метанола
вило, при необходимости дополнительного иници-
в ТHF добавляли к фосфониевой соли и карбона-
ирования процесса введения метки при изотопном
ту. Реакцию вели 3-15 ч. В результате образуется
обмене между источником дейтерия или трития и
Ph3PO [203-205], а изотопы водорода селективно
веществом.
включаются в 4-положение пиридинов и диазинов
(табл. 12).
4.5.1 Использование трифенилфосфина
4.5.2 Использование катализаторов
Изотопный обмен с производными пиридина и
на основе Pt(II)
диазина может быть инициирован трифенилфосфи-
ном (Х - атом азота или углерода) (схема 21) [193].
Изотопный обмен с тритиевой водой в присут-
ствии K2PtCl4 обычно проводят при нагревании в
Сначала получают трифенилфосфиновые про-
присутствии минеральных кислот [25, 206].
изводные пиридина и диазина, используя последо-
вательное добавление Tf2O, PPh3, NEt3 или DBU в
Механизм дейтерирования алканов в присут-
CH2Cl2 или EtOAc. Затем проводят обмен трифе-
ствии катализатора на основе Pt(II) можно предста-
нилфосфинового фрагмента на тритий или дей-
вить следующим образом:
РАДИОХИМИЯ том 64 № 4 2022
326
ШЕВЧЕНКО и др.
Таблица 12. Получение меченных тритием пиридинсодержащих лекарственных соединений с использованием
трифенилфосфина.
19.1 Ки/ммоль
18.4 Ки/ммоль
27.5 Ки/ммоль
21.1 Ки/ммоль
10.6 Ки/ммоль
В дальнейшем было разработано множество
других катализаторов с подобными структурами,
которые помогали избежать дезактивации катализа-
тора при проведении реакции (схема 22) [208].
4.5.3 Использование Et3B и DTBHN
При использовании K2PtCl4 изотопный обмен
Поскольку связь О-Н в молекуле воды инертна,
между группой С-Н вещества и дейтериевой во-
предпринимались попытки получить более реакци-
дой может происходить только в кислых условиях,
онноспособный посредник для включения дейте-
при которых сильно замедлен процесс диспропор-
рия или трития при использовании молекул воды.
ционирования двухвалентной платины и, следова- Одной из таких попыток можно считать активацию
тельно, дезактивации катализатора. Эта проблема реакции с дейтериевой или тритиевой водой при ис-
была минимизирована при использовании комплек- пользовании триэтилборана (Et3B) или ди-трет-бу-
са PtCl2 с бипиримидином и CF3
COО2Н в качестве тилгипонитрита (DTBHN) [209-213].
дейтерированной кислоты. Бипиримидиновый ли-
Для проведения этой реакции были предложены
ганд помогал избежать дезактивации катализатора две основные методики.
при проведении реакции. При низких значениях рН
Et3B (0.75 мл 2 M раствора в бензоле, 1.5 ммоль),
молекулы бипиримидина протонируются, что спо-
додекантиол (50 мкл 0.1 M раствора в бензоле,
собствует сдвигу электронной плотности от метал-
0.01 экв, 5 мол%) смешивали с раствором иодпро-
ла к лиганду [206, 207]:
изводного (0.5 ммоль) в 4 мл бензола и вносили
2Н2O (27 мкл, 1.5 ммоль). Сухой воздух (100 мл в
-
течение 2 ч, 50 мл/ч) пропускали под давлением.
Реакцию вели при перемешивании 1 ч.
-
К раствору Et3B (0.75 мл 2 M раствора в бензоле,
1.5 ммоль), додекантиола (50 мкл 0.1 M раствора в
В результате связь С-Н ослабевает, и обмен про- бензоле, 1 мол%), иодпроизводного (0.5 ммоль) и
тия на дейтерий становится более вероятным.
2Н2O (27 мкл, 1.5 ммоль) в 2.75 мл бензола в течение
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
327
Схема 22.
2 ч при 60С прибавляли DTBHN (87 мг, 0.5 ммоль)
образуется меченое соединение с более низким вы-
в 1 мл бензола. Реакцию вели при перемешивании
ходом (43%) [220].
еще 1 ч при 60С.
При исследовании реакционной активности
В результате рассмотрения нескольких альтер-
ряда вторичных алкилиодидов использовали моле-
натив был предложен радикальный механизм для
кулы этих соединений, в состав которых входили
этой реакции [214-216]. Оптимизацию процедуры
различные заместители (различные функциональ-
проводили с 4-иод-1-тозилпиперидином [217]. В ка-
ные группы) (табл. 14). Деиодированный продукт
честве посредника использовали додекантиол, ко-
образовывался с выходами 73-93% и с высокими
торый в результате ряда реакций взаимодействовал
уровнями включения дейтерия (80-96%). Таким
с дейтериевой водой с образованием n-C12H25S2H. В
образом, наличие функциональных групп (амиды,
качестве инициатора и реагента для переноса цепи
лактоны, силиловый эфир, бензильные и аллиль-
были выбраны Et3B и DTBHN.
ные системы), содержащих связи С-Н, которые, как
В отсутствие посредника включение дейтерия
считается, склонны к взаимодействию с тиольным
достигало лишь 32%. Использование 1 мол% доде-
радикалом, мало сказывается на выходе искомого
кантиола значительно увеличивало выход и вклю-
соединения. Аналогичные результаты были полу-
чение дейтерия (табл. 13) [217-219].
чены с третичными иодидами (табл. 14), но с более
При использовании в качестве инициатора су-
значительным колебанием уровня включения дей-
хого воздуха, а в качестве посредника - 1 мол%
терия (77-96%).
додекантиола получены следующие результаты
Механизм процесса деиодирования можно пред-
(табл. 14).
ставить следующим образом. Цепная реакция начи-
Как и следовало ожидать, при реакциях такого
нается с образования этильного радикала, генериру-
рода радикал из первичного иодистого алкила менее
емого реакцией Et3B с кислородом (стадия иниции-
устойчив, чем при деиодировании вторичных или
рования). Этот этильный радикал отщепляет атом
третичных иодистых алкилов (табл. 14). Поэтому
иода, образуя алкильный радикал. Взаимодействие
при дейтерировании первичных иодистых алкилов
алкильного радикала с додекантиолом-2Н дает дей-
Таблица 13. Оптимизация радикального деиодирования
Количество катализатора, мол%
Выход, %
Включение дейтерия, %
0
6
32
1
71
82
2
89
89
5
88
92
10
77
93
РАДИОХИМИЯ том 64 № 4 2022
328
ШЕВЧЕНКО и др.
Таблица 14. Влияние природы иодсодержащего соединения на радикальное замещение иода на атом дейтерия
(Et3B 3 экв, 2Н2O 3 экв, бензол, 23С, 3 ч)a
t
89 (92)
87 (95)
82 (91)
85 (85)
78 (96)
91 (94)
78 (83)
73 (93)
79 (77)
93 (96)
43 (91)
84 (96)
a Указан выход, % (в скобках - включение дейтерия, %).
терированный продукт и генерирует n-C12H25S•.
сится использование катализаторов, активируемых
Реакция последнего с Et3B поддерживает цепной
светом (схема 23) [167,168].
процесс образованием радикала Et•:
Эти катализаторы позволяют вводить изотопы
водорода даже в алифатические фрагменты органи-
1. n-C12H25SH + Et3B → n-C12H25SBEt2 + Et-Н
ческих соединений. Поэтому представляет интерес
2. n-C12H25SBEt2 + 2Н2О → n-C12H25S2H + Et2BО2Н
предложенный способ, позволяющий проводить
3. Et3B (воздух), или DTBHN → Et•
изотопный обмен между 2Н2O или 3Н2O и прото-
4. R-I + Et• → R• + Et-I
нами при алифатических углеродах, находящихся
5. R• + n-C12H25S2H → R-2H + n-C12H25S•
в α-положениях к аминогруппам (схема 24) [167,
6. n-C12H25S• + Et3B → n-C12H25SBEt2 + Et•
168].
Распределение метки при использовании этого
Таким образом, разработан метод получения
метода отличается от перечисленных ранее методов
в мягких условиях дейтерированных соединений
(● - положение метки при использовании катализа-
из иодистых алкилов по механизму радикально-
торов на основе иридия и железа, * - при использо-
го замещения с использованием 2Н2O в качестве
вании фотокатализаторов):
источника дейтерия. Метод оказался применим для
иодсодержащих соединений, имеющих фрагмен-
ты с широким спектром функциональных групп.
Использование этого метода позволило получить
монодейтерированные продукты с хорошими выхо-
дами и высоким уровнем включения дейтерия.
4.5.4 Использование катализаторов переноса
водорода
К специфическим методам введения метки с
участием дейтериевой или тритиевой воды отно-
Кломипрамин
Азитромицин
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
329
Схема 23.
Ir(ppy)2(dtbbpy)PF6
Ir[F-(CH
)ppy]2(dtbbpy)PF6 Ir[dF-(CF3)ppy]2(dtbbpy)PF6
4Cz-IPN
3
рpy - фенилпиридин
dtbbpy - 4,4ꞌ-ди-трет-бутил-2,2ꞌ-бипиридин
Схема 24.
Схема 25.
Согласно статистике, более 50% самых продава-
Два последних оказались наиболее перспек-
емых коммерческих субстанций содержат по край-
тивными, так как использование стерически за-
ней мере один алкиламиновый фрагмент. Поэтому
трудненных катализаторов переноса водорода
разработка методики изотопного обмена между
предотвращало связывание тиола с субстратом и,
тритием и алкиламинами может оказаться перспек-
следовательно, препятствовало образованию по-
тивной для введения метки в подобные препараты.
бочных продуктов. Использование 4Cz-IPN или
Последовательность событий при реализации дан-
Ir(F-(CH3)ppy)2(dtbbpy)PF6) и триизопропилси-
ной методики заключается в создании условий, при
лантиола в присутствии карбоната лития позволяет
которых образуется углеродсодержащий α-амино-
получать дейтерированные продукты с выходами
радикал. Последний взаимодействует с катализато-
70-80%, содержащие несколько атомов дейтерия
ром переноса водорода (HAT), в который включает-
(в зависимости от количества алкиламинов в моле-
ся дейтерий или тритий из 2Н2O или 3Н2O, в резуль-
куле соединения) (табл. 15). Напомним, что замена
тате чего образуется меченый продукт.
одного атома протия на тритий дает препарат с мо-
Для
активации
фотокатализатора
лярной радиоактивностью порядка 29 Ки/ммоль.
(Ir(ppy)2(dtbbpy)PF6,
Ir[F-(CH3)ppy]2(dtbbpy)PF6,
Как видно из табл. 15, дейтерий включается не
Ir[dF-(CF3)ppy]2(dtbbpy)PF6 и 4Cz-IPN использо-
только в α-, но и в другие положения молекул амино-
вали два источника света [синий светодиод (LED)
мощностью 34 Вт или встроенный фотореактор,
содержащих соединений. Поэтому были рассмотре-
разработанный в Merck]. В качестве HAT использо-
ны и другие возможные пути включения изотопов
вали серосодержащие соединения (схема 25).
водорода при использовании этого метода (схема 26).
РАДИОХИМИЯ том 64 № 4 2022
330
ШЕВЧЕНКО и др.
Схема 26.
В качестве растворителя при работе с тритием
комнатной температуре, образуется C2H6 в изме-
лучшим оказался NMP. Высокомеченную трити-
ряемых количествах [221]. Полученный результат
евую воду получали реакцией 1.0 Ки 3Н2 с PtO2.
позволил сделать вывод, что сила кислот Бренстеда
Выход тритиевой воды 61% (0.51 Ки, 8.8 ммоль).
на поверхности цеолита может быть решающим
Естественно, реакции с тритием проводили в мень-
фактором, определяющим эффективность восста-
ших масштабах (табл. 16).
новления C2H4. Степень восстановления C2H4 чет-
ко коррелировала со способностью цеолита образо-
5. ВВЕДЕНИЕ ТРИТИЕВОЙ МЕТКИ
вать кислотные центры Бренстеда. Реакция C2H4 с
БЕЗ ИСПОЛЬЗОВАНИЯ РАСТВОРИТЕЛЕЙ
активированным на кислотных центрах Бренстеда
Реакции без использования растворителя мож-
водородом имела самый низкий активационный ба-
но разделить на следующие группы: оба реагента,
рьер, и ее протекание наиболее вероятно при этих
нанесенные на носитель, являются нелетучими со-
условиях [222-224]. Следовательно, как активация
единениями; один из реагентов находится в газовой
C2H4 на кислотных центрах Бренстеда, так и гете-
фазе, а второй - нелетучее соединение; оба реагента
ролитическая диссоциация H2 на атомах алюминия
находятся в газовой фазе.
являются двумя критическими процессами, за счет
Классические топохимические реакции, когда
которых происходит гидрирование ненасыщенных
оба реагента, нанесенные на носитель, - нелетучие
соединений. В целом результаты, представлен-
соединения, для получения меченых соединений
ные в работе [221], имеют большое теоретическое
непригодны. Случай, когда оба реагента находятся
значение: они продемонстрировали важную роль
в газовой фазе, для введения дейтерия или трития
кислотных центров Бренстеда для реализации про-
можно использовать, но такие работы имеют пре-
цессов, связанных с взаимодействием молекуляр-
жде всего теоретический аспект. Они в основном
ного водорода с органическими соединениями.
используются для изучения механизма реакции.
Относительно низкая активность, проявляемая
Например, при исследовании реакций этилена и
при гидрировании C2H4 на цеолитах с различными
газообразного водорода на цеолитах рассматривали
соотношениями Si/Al, обусловлена небольшим ко-
условия гидрирования этилена без участия метал-
личеством и низкой реакционной способностью ак-
лов-катализаторов. При этом установлено, что, не-
тивных центров, на которых может осуществляться
смотря на то, что связи в молекулах как H2, так и
гетеролитическая диссоциация молекул водорода,
C2H4 при адсорбции на цеолите ослабевают, они не
адсорбированных на поверхности носителя. В тех
реакционноспособны [221]. В то же время при од-
же случаях, когда цеолит с нанесенным на него ве-
новременном присутствии H2 и этилена, даже при
ществом содержит переходные металлы, способ-
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
331
Таблица 15. Выход и содержание дейтерия в соединениях, содержащих алкиламины.
Кломипрамин
(±)-Верапамил
(+)-Эсциталопрам
4Cz-IPN, выход 76%,
4Cz-IPN, выход 84%,
4Cz-IPN, выход 71%,
7.22 2Н на молекулу
6.06 2Н на молекулу
7.76 2Н на молекулу
Кломипрамин гидрохлорид (1.41 г, 4.0 ммоль, 1.0 экв), 4Cz-IPN (63 мг, 0.08 ммоль, 0.02 экв), Li2CO3 (355 мг,
4.8 ммоль, 1.2 экв), NMP (16 мл, 0.25 M), триизопропилсилантиол (260 мкл, 1.2 ммоль, 0.3 экв) и 2Н2O (4 мл,
220 ммоль, 50 экв), реакционную смесь облучали синим светодиодом (LED) мощностью 34 Вт в течение 24 ч
(+)-цис-Дилтиазем
(±)-Клоперастин
Фенспирид
Ir[F-(CH3)ppy]2(dtbbpy)PF6,
Ir[F-(CH3)ppy]2(dtbbpy)PF6,
Ir[F-(CH3)ppy]2(dtbbpy)PF6,
выход 84%, 7.05 2Н на молекулу
выход 78%, 5.54 2Н на молекулу
выход 71%, 5.10 2Н на молекулу
Буспирон
(-)-Левофлоксацин
Арипипразол
4Cz-IPN,
Ir[F-(CH3)ppy]2(dtbbpy)PF6,
4Cz-IPN, выход 80%,
выход 88%, 5.52 2Н на молекулу
выход 75%, 6.53 2Н на молекулу
6.88 2Н на молекулу
ные активировать Н2, гидрирование протекает эф-
рбированным на поверхности носителя (например,
фективно [225, 226].
Al2О3), способствует более глубокой делокализации
связи Н-H, в результате чего становится возмож-
Таким образом, эта работа подтверждает данные
о том, что кластеры (Н+ē) на неорганическом носи-
ным образование кластера [H-Al3+O2- H+, который
взаимодействует с RСНН-С+НR1, что и приводит к
теле, образованные за счет спилловера водорода,
восстановлению двойной связи. Активированные
могут способствовать непосредственному участию
частицы водорода могут создавать условия для вза-
молекулярного водорода при восстановлении связей
С=С. Катион водорода протонирует двойные связи
имодействия молекул водорода с молекулами орга-
нического соединения.
ненасыщенного соединения, что приводит к появле-
нию карбокатиона (RСНН-С+НR1), а электрон при
Например, если Pt/Al2O3 или Ni/Al2O3 поме-
взаимодействии с молекулярным водородом, адсо- стить в ампулу, отделенную от носителя (SiO2 или
РАДИОХИМИЯ том 64 № 4 2022
332
ШЕВЧЕНКО и др.
Таблица 16. Молярная радиоактивность соединений, содержащих алкиламины
Кломипрамин 4Cz-IPN,
(±)-Верапамил 4Cz-IPN,
(+)-Эсциталопрам 4Cz-IPN,
40.2 Ки/ммоль
43.5 Ки/ммоль
35.1 Ки/ммоль
В реакции использовали 4Cz-IPN и триизопропилсилантиол. Облучение проводили в фотореакторе 4 ч. Реакционная
смесь имела активность 257.6 мКи. После удаления лабильного трития проводили очистку ВЭЖХ на колонке
Gemini NX C18 10 × 250 мм
(±)-цис-Дилтиазем, 34.5 Ки/ммоль,
(±)-Клоперастин, 40.2 Ки/ммоль,
Фенспирид, 25.2 Ки/ммоль,
Ir[F-(CH3)ppy]2(dtbbpy)PF6
Ir[F-(CH3)ppy]2(dtbbpy)PF6
Ir[F-(CH3)ppy]2(dtbbpy)PF6
Буспирон, 21.5 Ки/ммоль, 4Cz-IPN
(-)-Левофлоксацин, 22.5 Ки/ммоль,
Арипипразол 4-CzIPN,
Ir[F-(CH3)ppy]2(dtbbpy)PF6
11.5 Ки/ммоль
Al2O3) пористой перегородкой, и нагреванием в ат-
Явление спилловера водорода активно иссле-
мосфере водорода создать на носителе центры типа
дуется и в настоящее время [24, 75, 227-238].
[H-Al3+O2-]H+, то даже после удаления катализато-
Получаемые данные могут открыть новые перспек-
ра такой носитель, насыщенный активированными
тивы в использовании твердофазного метода для
частицами водорода, достаточно долго сохранял
синтеза меченных тритием соединений.
способность к реакциям гидрогенизации и гидроге-
5.1. Твердофазный изотопный обмен
нолиза в атмосфере водорода, т.е. активированные
Основные закономерности, влияющие на эф-
частицы водорода на носителе делают возможным
фективность введения трития в молекулы орга-
непосредственное взаимодействие молекул водо-
нических соединений твердофазным методом,
рода с органическим соединением. В таких усло-
можно проиллюстрировать на примере введения
виях проводили гидрирование этилена, ацетилена,
трития в D-рибозу, аденин, 5-oxo-Pro-His-Pro-NH2,
бензола, а также крекинг кумола при температуре
ACTH(6-9)PGP (His-Phe-Arg-Trp-Pro-Gly-Pro), 4-фе-
200-270С. В то же время на данном носителе без
нил-бензойную кислоту и N-метил-N-(2-метил-4-
предварительной активации молекулярный водород
метокси-5-трифторметилфенил)-N΄-метилмочевину
с этими соединениями не реагировал.
[21, 239-243].
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
333
Таблица 17. Зависимость распределения дейтерия во фрагментах 5-oxo-Pro-His-Pro-NH2 от температуры при
изотопном обмене (время реакции 5 мин)
5% Rh/Al2O3
5% Pd/Al2O3
Т, C
а
5-oxo-Proа
Hisа
Pro-NH
2
[2Н]His, % б
5-oxo-Proа
Hisа
Pro-NH2а
[2Н]His,б
160
0.16
0.75
0.15
74
0.14
0.82
0.12
76
180
0.26
1.43
0.25
70
0.29
1.74
0.27
76
200
0.43
1.74
0.37
70
0.44
1.90
0.37
70
220
0.56
2.26
0.44
69
0.59
2.41
0.49
70
240
0.68
2.60
0.57
68
0.59
2.60
0.55
69
260
0.70
2.73
0.67
67
0.69
2.77
0.63
68
а Число атомов дейтерия в соответствующем фрагменте.
б [2Н]His - доля дейтерия, приходящаяся на фрагмент гистидина.
Таблица 18. Зависимость выхода 5-oxo-Pro-His-Pro-NH2 и содержания в нем и в его фрагментах дейтерия от времени
проведения реакции (5% Rh/Al2O3)
Содержание дейтерия в пептиде и его фрагментах
Т, C
Время, мин
Выход, %
[2Н]His, % а
5-oxo-Pro-His-Pro-NH
2
5-oxo-Pro
His
Pro-NH2
180
5
1.94
0.26
1.43
0.25
65
73
10
2.22
0.31
1.64
0.27
58
74
5
2.54
0.43
1.74
0.37
62
68
10
3.13
0.50
2.18
0.45
52
70
200
15
3.36
0.54
2.34
0.48
48
70
20
3.44
0.54
2.40
0.50
46
70
30
3.84
0.66
2.58
0.60
38
67
аДоля дейтерия в %, приходящегося на данный фрагмент
Зависимость включения дейтерия или трития от
тет, а ее распределение между аминокислотами из-
температуры, времени проведения реакции и при-
меняется незначительно. Так, при проведении реак-
роды катализатора исследовали, используя 5-oxo-
ции при 180°C в течение 5-30 мин в His включа-
Pro-His-Pro-NH2.
лось 74% метки, а при 200°C в течение 5-30 мин - в
Зависимость содержания дейтерия в
5-oxo-
среднем 69% метки (табл. 18) [16]. Таким образом,
Pro-His-Pro-NH2 от температуры приведена в
оптимальным условием введения метки в 5-oxo-
табл. 17 [16, 240]. Из этих данных следует, что
Pro-His-Pro-NH2 является проведение реакции при
включение метки в основном происходит в His, при
200°C в течение 15 мин при использовании смеси,
этом с ростом температуры вклад гистидина в сум-
в которой соотношение катализатор : вещество :
марное количество метки в пептиде падает. Можно
Al2O3 равно 2 : 1 : 30.
отметить, что хотя включение метки во фрагменты
Для оценки изотопных эффектов препаративный
5-oxo-Pro и Pro-NH2 примерно одинаково, в первый
синтез с дейтерием и тритием проводили в анало-
фрагмент метки включается больше, несмотря на то,
гичных условиях, использовалась ампула одного
что в его молекуле на два атома водорода меньше.
объема, с одним и тем же количеством смеси. При
Возможно, это связано с тем, что в процессе вклю-
использовании дейтерия был получен меченый
чения дейтерия в Pro-NH2 могут принимать участие
пептид, содержащий 2.93 атома дейтерия. При про-
протоны амидной группы. В результате включение
ведении реакции с газообразным тритием получен
дейтерия в этот пролиновый остаток падает.
меченый пептид с молярной радиоактивностью бо-
При исследовании зависимости выхода 5-oxo-
лее 70 Ки/ммоль, т.е. он содержал примерно 2.5 ато-
Pro-His-Pro-NH2 и содержания в нем и в его фраг-
ма трития. Следовательно, эффективность изотоп-
ментах дейтерия от времени проведения реакции
ного обмена при переходе от дейтерия к тритию
оказалось, что со временем включение метки рас-
падает примерно на 15%.
РАДИОХИМИЯ том 64 № 4 2022
334
ШЕВЧЕНКО и др.
Таблица 19. Зависимость выхода 5-oxo-Pro-His-Pro-NH2 и содержания в нем дейтерия от температуры и природы
инициатора спилловера водорода (5% Rh/Al2O3 и 5% Pd/Al2O3, время реакции 5 мин) [16, 240]
5% Rh/Al2O3
5% Pd/Al2O3
Т, C
Включение 2Hа
Выход, %
Включение 2H
Выход, %
160
1.06
76
1.08
82
180
1.94
65
2.30
70
200
2.54
62
2.71
62
220
3.26
43
3.49
43
240
3.85
22
3.74
22
260
4.10
12
4.09
13
а Среднее количество атомов дейтерия в молекулах 5-oxo-Pro-His-Pro-NH2.
Влияние
природы металла-катализатора
10 мин. Выход ПAM-43 23 %, а молярная радиоак-
на эффективность изотопного обмена между
тивность 132 Ки/ммоль [244].
5-oxo-Pro-His-Pro-NH2 и дейтерием показана в
Степень влияния природы носителя была уста-
таблице 19 [16, 240].
новлена при использовании в качестве носителя
Из приведенных данных видно, что при работе
Al2O3, цеолита, СаСО3, наноалмазов (НА), SiO2 и
с механической смесью катализатора с пептидом,
графена [240].
нанесенным на носитель, влияние разных металлов
Содержание дейтерия отличалось между Al2O3 и
платиновой группы на выходы и среднее количе-
графеном почти в 5 раз, выход пептида - примерно
ство атомов дейтерия в молекулах 5-oxo-Pro-His-
в 2.5 раза. Масс-спектрометрический анализ пока-
Pro-NH2 незначительно.
зал, что хотя во всех случаях основная часть метки
Сравнение катализаторов 5% Rh/Al2O3 и 5%
включалась в гистидин, ее количество в гистидине
Pd/Al2O3 проводилось и при введении дейтерия
существенно менялось на разных носителях. При
в ПAM-43 (препарат, облегчающий обучаемость,
использовании графена в этой аминокислоте со-
улучшающий память, обладающий нейропротек-
держалось 89% метки, цеолита - 81%, Al2O3 - 71%,
торным действием, который может быть использо-
СаСО3 - 62%.
ван для лечения болезни Альцгеймера):
Большое значение имеет не только природа но-
сителя, но и его дисперсность. Это показано при
проведении изотопного обмена дейтерия с пепти-
дом His-Phe-Arg-Trp-Pro-Gly-Pro [ACTH(6-9)PGP]
на 5% Rh/Al2O3 или 5% Pd/Al2O3, нанесенным
на Al2O3 трех размеров: 50-100, 10 и <0.05 мкм
(Sigma-Aldrich) [239].
При температуре 180С наибольшее включение
дейтерия оказалось при использовании в качестве
При этом оказалось, что при использовании
носителя Al2O3 50-100 мкм (1.42 атома дейтерия),
этих катализаторов характеристики ПAM-43 от-
на втором месте Al2O3 <0.05 мкм (1.23 атома дейте-
личались мало. В случае 5% Rh/Al2O3 выход при
рия), затем идет Al2O3 10 мкм (0.95 атома дейтерия).
возрастании температуры падал от 80 до 10%, в
Выход меченого ACTH(6-9)PGP составил 7 (Al2O3
случае 5% Pd/Al2O3 - от 90 до 20%. Если принять
50-100 мкм), 8% (10 мкм) и 9% (<05 мкм). При
среднее количество дейтерия, находящегося в мо-
более высоких температурах на Al2O3 10 мкм вы-
лекуле ПAM-43, за 100%, то в 2,3-дигидробензофу-
ход меченого ACTH(6-9)PGP составил 70 (190С),
рановых фрагментах содержалось 80% дейтерия, в
46 (200С) и 4% (210С), а ACTH(6-9)PGP в этих
центральном фрагменте - 20% дейтерия.
условиях при использовании в качестве носителя
Введение трития проводили при соотношении
Al2O3 50-100 мкм и Al2O3 <0.05 мкм деградировал.
5% Pd/Al2O3 : ПAM-43 = 20 : 1 при 150С в течение
Таким образом, использование в качестве носителя
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
335
Al2O3 10 мкм и проведение реакции при температу-
Изотопным обменом без использования раство-
ре 200С оказалось наиболее целесообразным для
рителей удавалось введение трития в самые разноо-
введения дейтерия в ACTH(6-9)PGP.
бразные соединения [2, 11, 101, 239-243, 245-250].
Как правило, при увеличении соотношения ка-
Выход при этом был в пределах 10-60% с моляр-
тализатор-вещество выход меченого препарата
ной радиоактивностью, достигающей 100 и более
уменьшается, а его молярная радиоактивность воз-
Ки/ммоль.
растает. В качестве иллюстрации этой закономер-
Интересные результаты были получены при
ности выбраны рибоза и аденин, нанесенные на
введении метки в даларгин (Tyr-D-Ala-Gly-Phe-
носитель разной степени дисперсности [241, 245].
Leu-Arg) - синтетический аналог эндогенного опи-
Известно, что сольватация активированных частиц
оидного пептида [Leu5]энкефалина (Tyr-Gly-Gly-
трития может происходить не только на носителе,
Phe-Leu), используемый в гастроэнтерологии [251,
но и в пуле вещества [11]. Если вероятность сольва-
252]. В работе показано влияние носителя с разной
тации активированных частиц трития на носителе
структурой углеродного наноматериала (оксид гра-
выше, чем в пуле вещества, то чем больше будет
фена, восстановленный оксид графена, однослой-
поверхность носителя, тем более вероятен обмен
ные углеродные нанотрубки) на распределение изо-
между протонами вещества и тритием. Если обра-
топов водорода в даларгине.
зование сольватированных активированных частиц
Оказалось, что строение углеродного носителя
трития в пуле вещества происходит эффективно,
заметно влияет на включение трития в тот или иной
то изотопный обмен будет идти во всем объеме
аминокислотный остаток (рис. 7) [251].
вещества и влияние дисперсности носителя бу-
Как видно из рис. 7, наибольшее влияние струк-
дет не столь очевидно. В случае D-рибозы и аде-
тура углеродного наноматериала оказывала на
нина обнаружено влияние дисперсности носителя.
включение трития в тирозин и лейцин. Наибольшая
Следовательно, для этих соединений сольватация
подвижность протия и, следовательно, наиболь-
активированных частиц трития на носителе выше,
шая вероятность изотопного обмена наблюдается
чем в пуле вещества.
у связей С-Н глицина, адсорбированного на вос-
Логично предположить, что увеличить зоны, в
становленном оксидграфене (рис. 7, б). Авторы
которых создаются благоприятные условия для по-
попытались связать полученные закономерности с
лучения меченых препаратов исходного строения (с
разными структурами молекул даларгина на разных
низким содержанием активированных частиц три-
носителях [247, 248].
тия или дейтерия), можно за счет не только исполь-
Структуры молекулы даларгина построены при
зования дополнительного носителя, но и уменьше-
использовании метода молекулярной механики в
ния содержания металла-катализатора в используе-
силовом поле AMBER (рис. 8).
мом катализаторе. При введении трития в N-метил-
Моделирование в рамках этого метода показало,
N-(2-метил-4-метокси-5-трифторметилфенил)-
что структура молекулы даларгина без участия под-
N΄-метилмочевину использовали и один, и другой
ложки формируется за счет образования внутримо-
подход [242]:
лекулярных водородных связей между остатками
аргинина и глицина, а также остатками аргинина и
фенилаланина (рис. 8, а) [252]. На поверхности гра-
фенового носителя внутримолекулярные водород-
ные связи заменяются водородными связями между
При этом оказалось, что и тот, и другой под-
атомами кислорода поверхности и NH-группами ар-
ходы благоприятствовали получению мечено-
гинина, лейцина и тирозина. За счет этого молекула
го препарата исходного строения. Но в данном
даларгина принимает более плоскую форму. При
случае более хорошие результаты получены при
этом молекула даларгина деформируется (рис. 8, б).
использовании низкопроцентного катализатора
Так, при адсорбции на поверхности оксида графена
(0.2% Pd/SiO2). Молярная радиоактивность пре-
даларгина в молекуле глицина расстояние во фраг-
парата 28-30 Ки/ммоль.
менте -NH-СН2С(О)- между атомами азота и угле-
РАДИОХИМИЯ том 64 № 4 2022
336
ШЕВЧЕНКО и др.
Рис. 7. Распределение трития в аминокислотных остатках даларгина при использовании разных углеродных носителей:
С - активированный уголь, ОГ - оксид графена, вОГ - восстановленный оксид графена, УНТ - углеродные нанотрубки.
(а) молярная активность аминокислотных остатков относительно молярной активности пептида в процентах; (б) молярная
активность аминокислотных остатков относительно молярной радиоактивности пептида в процентах при пересчете на чис-
ло атомов протия при углеродных атомах (С-Н) в аминокислотном остатке.
(а)
(б)
(в)
Рис. 8. Структура свободной молекулы даларгина (а), даларгина на поверхности оксида графена (б) и на поверхности
углеродных нанотрубок (в).
рода карбонильной группы увеличивается с 2.49 Å
При контакте даларгина с углеродной нанотруб-
до 2.68 Å. В результате протий в остатке глицина
кой на поверхности нанотрубок, как и на оксиде гра-
становится более доступен для взаимодействия с
фена, образуются внутримолекулярные водородные
тритием и эффективность изотопного обмена воз-
связи между остатками аргинина и фенилаланина,
растает (рис. 7). Естественно, при адсорбции да-
аргинина и глицина, а также тирозина и аланина.
ларгина на поверхности оксида графена и другие
В результате молекула даларгина «обворачивается»
остатки аминокислот в даларгине подвергаются до-
вокруг нанотрубки (рис. 8, в). При этом все амино-
статочно сильной деформации при взаимодействии
кислотные остатки испытывают одинаковые стери-
с атомами подложки. Так, из-за уменьшения угла
ческие и другие затруднения для реакции с тритием.
между ароматическим кольцом фенилаланина и
По-видимому, с этим связано то, что распределение
алкильным радикалом лейцина со 107° (свободная
трития по остаткам аминокислот в даларгине в этом
случае становится более равномерным (рис. 8, б).
молекула) до 93° (на оксиде графена) возможно не-
которое экранирование атомов водорода в молекуле
Комплексное исследование зависимости содер-
лейцина, что заметно снижало включение трития в
жания дейтерия от температуры, давления и време-
эту аминокислоту (рис. 7).
ни проведения твердофазных реакций проводили,
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
337
Таблица 20. Изменение изотопного состава в 4-фенил-
Таблица 21. Изменение изотопного состава в 4-фенил-
бензоате натрия в зависимости от температурыа
бензоате натрия в зависимости от давления дейтерия
Количество
Температура, °С
(5% Pd/C, 10 мин, 280°С)а
атомов
Давление, мм рт. ст.
80
100
120
140
160
180
200
дейтерия
N
300
150
75
37
2Н0
98.1
90.4
78.8
53.6
40.2
36.2
20.5
Распределение дейтерия по изотопомерам, %
2Н1
1.2
6.1
10.7
16.4
8.9
7.1
4.6
0
4.9
5.5
3.7
4.4
2Н2
0.6
3.0
7.4
13.3
12.8
7.7
4.2
1
0
0
0
0.1
2Н3
0.04
0.3
2.1
9.2
10.9
9.4
4.8
2
0.1
0.2
0.2
0.5
2Н4-2Н9
0.06
0.2
1.0
7.5
27.2
39.6
65.9
3
0.3
0.2
0.2
2.6
Σ
96.8
97.9
95.3
83.8
54.5
37.9
17.1
4
0.4
0.3
0.8
8.3
а Содержание атомов дейтерия в %; Σ - отношение суммарного
5
0.7
0.9
3.7
18.7
содержания изотопомеров с 1-3 атомами дейтерия к суммар-
6
1.4
4.2
12.3
24.4
ному содержанию всех изотопомеров, содержащих дейтерий,
7
10.3
16.7
24.8
22.9
в %.
8
32.0
36.5
34.0
14.2
используя 4-фенилбензоат натрия. Из данных, по-
9
50.2
35.8
20.4
4.0
лученных ранее, включение основного количества
∑
7.95
7.63
7.26
5.92
дейтерия можно ожидать в о- и п-положениях фе-
а N - число атомов дейтерия в молекуле изотопомера, ∑ - сред-
нильного фрагмента 4-фенилбензоата натрия (элек-
нее число атомов дейтерия на молекулу.
трофильное замещение). Из этого следует, что при
Из полученных данных видно, что образова-
более низкой температуре содержание дейтерия в
ние изотопомера с девятью атомами дейтерия при
веществе будет в пределах от одного до трех ато-
280С происходит с первых секунд. За одну минуту
мов. При более высоких температурах включение
образовывалось около 80% этого изотопомера отно-
дейтерия будет более равномерным, с образованием
сительно максимально возможного его количества
большого количества полностью меченного дейте-
при проведении эксперимента в этих условиях. По-
рием продукта (табл. 20, рис. 9) [243].
видимому, среднее содержание атомов дейтерия со
Из данных, приведенных на рис. 9, следует, что
временем возрастает лишь за счет уменьшения ко-
личества вещества, ранее еще не успевшего всту-
при температурах выше 200°С доминирующим ста-
пить в реакцию.
новится процесс неспецифического изотопного об-
мена с дейтерием в обоих ароматических кольцах
Кривая изменения количества изотопомера с
молекулы 4-фенилбензоата натрия [243].
девятью атомами дейтерия от времени имеет мак-
При уменьшении давления газообразного дейте-
рия количество кластеров с необходимым количе-
ством активированных частиц дейтерия для замены
всех девяти атомов протия в 4-фенилбензоате натрия
на дейтерий будет уменьшаться. Следовательно, ко-
личество изотопомеров, содержащих менее девяти
атомов дейтерия в 4-циклогексилбензоате натрия,
будет возрастать (табл. 21).
Как видно из приведенных данных, действи-
тельно, содержание изотопомеров с 9 и 8 атомами
дейтерия при более низких давлениях газообразно-
го дейтерия заметно упало, а вклад изотопомеров
с меньшим содержанием атомов дейтерия за счет
этого вырос.
Рис.
9.
Эффективность включения дейтерия в
4-фенилбензоат натрия (■) (давление трития 400 гПа,
Кинетические кривые и при 200, и при 280С
10 мин); содержание изотопомера с девятью атомами
имеют отчетливые максимумы (рис. 10).
дейтерия (♦).
РАДИОХИМИЯ том 64 № 4 2022
338
ШЕВЧЕНКО и др.
много легче, чем процесс дегалогенирования [227].
Это связано с тем, что в отличие от введения трития
в молекулы алифатических соединений изотопным
обменом твердофазное дегалогенирование не мо-
жет происходить за счет электрофильного замеще-
ния [227].
Галогены могут быть удалены из молекулы ор-
ганического соединения только в виде HHal. Такой
процесс возможен при взаимодействии с кластера-
ми, содержащими несколько ионных пар [(3Н+)(ē)],
которые будут генерировать атомарный тритий или
сделают возможным участие газообразного трития
Рис.
10. Содержания дейтерия в
4-фенилбензоате
при дегалогенировании. В ряде случаев при синте-
натрия (а) и изотопомере с 9 атомами дейтерия (б) при
зе меченых соединений удавалось селективно заме-
температурах 200 (▲) и 280°С (♦) в зависимости от
нить галоген на дейтерий или тритий [253-255].
времени проведения реакции.
Выявленные закономерности твердофазного
симум при 200С в районе 30 мин, при 280С - в
дегалогенирования алифатических соединений
районе 15 мин (рис. 10). Из этого следует, что вре-
позволили отработать условия введения изотопов
мя реакции нужно строго регулировать, так как в
водорода и в более сложные молекулы (табл. 22),
первую очередь разрушаются изотопомеры с более
например, получить меченый ацетилхолин дега-
высоким содержанием изотопа. Проведенные экс-
логенированием СН2ClСОOСН2-СН2N+(CH3)3Cl-,
перименты еще раз подтверждают, что в тех зонах,
СНBr2СОOСН2СН2N+(CH3)3Cl- и СCl3СОOСН2-
где эффективность изотопного обмена максималь-
СН2N+(CH3)3Cl- (85% метки в дейтериевом аналоге
на, создаются и наиболее благоприятные условия
ацетилхолина содержалось в ацетильном фрагмен-
для деградации молекул вещества.
те, 15% - в холиновом) [256].
Таким образом, можно констатировать, что при
Как видно из полученных данных, при 190С
работе с 4-фенилбензоатом натрия при температу-
(катализатор Линдлара) при замене одного гало-
рах выше 200С происходит качественное измене-
гена в метильной группе включалось в среднем
ние процесса изотопного обмена. Включение изо-
1.24 атома, двух - 1.55 атома, трех - 2.36 атома
топа становится менее избирательным, в результате
дейтерия. Та же закономерность наблюдалась при
чего вероятность замещения всех атомов протия,
введении трития. При 190С при замене одного га-
содержащихся в молекулах вещества, на дейтерий
логена в метильную группу включалось в среднем
или тритий возрастает.
70%, двух - 50%, трех - 43% от расчетного количе-
По-видимому, при повышении температуры
ства трития. Очевидно, что при дегалогенировании
кластеры из сольватированных активированных
СН2ClСОOСН2-СН2N+(CH3)3Cl- часть дейтерия
частиц водорода и электронов перестраиваются.
включается в метильную группу за счет изотопного
Эффективность взаимодействия молекул вещества
обмена.
с газообразными тритием или дейтерием значитель-
По-видимому, атом галогена вызывает сдвиг
но возрастает [25]. Кроме того, катионы изотопов
электронной плотности в метильной группе мо-
водорода, сольватированные полярными группами
нохлоруксусной кислоты, что увеличивает подвиж-
органических соединений, приобретают способ-
ность атомов протия в ней.
ность взаимодействовать с электронами и становят-
ся активными участниками обменного процесса в
6. ЗАКЛЮЧЕНИЕ
атомарном виде.
5.2. Твердофазное дегалогенирование
Главный вывод из приведенных данных кратко
Известно, что изотопный обмен в алифатических
можно сформулировать следующим образом. Как
галогенсодержащих соединениях происходит на-
правило, введение изотопов водорода химическими
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
339
Таблица 22. Набор изотопомеров, образующихся при введении дейтерия в ацетилхолин при дегалогенировании
(палладиевый катализатор, соотношение катализатор : вещество 40 : 1)
СН2ClСОOСН2-
СНBr2СОOСН2-
Вклад
СCl3СОOСН2-СН2N+(CH3)3Cl-
СН2N+(CH3)3Cl-
СН2N+(CH3)3Cl-
изотопомеров
190°С
120°С
190°С
120°С
190°С
2Н0
0.138
0.139
0.081
0.066
0.035
2Н1
0.616
0.456
0.409
0.214
0.161
2Н2
0.163
0.353
0.388
0.310
0.244
2Н3
0.099
0.053
0.119
0.398
0.543
2Н4
0
0
0.002
0.014
0.019
Σ2Н
1.239
1.315
1.551
2.085
2.355
методами или твердофазным изотопным обменом
Введение тритиевой метки изотопным обменом
предпочтительнее. Если вещество или его предше-
в растворе может оказаться перспективным в при-
сутствии катализаторов на основе родия, рутения,
ственник лабильны при нагревании в присутствии
железа, никеля, кобальта и иридия (табл. 23).
катализатора и газообразного трития (или дейте-
Катализаторы на основе кобальта лучше стиму-
рия), а химический синтез нерентабелен, то нуж-
лируют изотопный обмен протия при углеродных
ного результата можно достичь при использовании
атомах в α-положениях к бензольному кольцу. При
тритиевой или дейтериевой воды. В последнем
сравнении трех катализаторов, активирующих изо-
случае активизацию этого процесса осуществляют
топный обмен в МК-7622, 2-(п-толил)пиридине и
не только за счет проведения реакции при высо-
(-)никотине, лучшие результаты демонстрирует ни-
ких температурах, но и за счет посредников, кото-
келевый катализатор [(iPrDI)Ni(μ2-H)]2. В некоторых
рые облегчают изотопный обмен между дейтерием
случаях небольшое преимущество имеет родиевая
(тритием) и протонами органического соединения.
чернь. Худшие результаты показал никель Ренея.
Таблица 23. Распределение дейтерия в молекулах органических соединений при использовании различных методов
введения метки [153, 257, 258]
Распределение дейтерия в ароматическом и алкильном фрагментах вещества при использовании (iPrDI)
Co(CH2SiMe3)2
i
Распределение дейтерия при использовании [(iPrDI)Ni(μ2-H)]2 (значения без скобок), никеля Ренея (значения в
круглых скобках), родиевой черни (значения в квадратных скобках)
(-)Никотин
2-(п-Толил)пиридин
МК-7622
РАДИОХИМИЯ том 64 № 4 2022
340
ШЕВЧЕНКО и др.
Проведено сканирование по основным методам
(H4-iPrCNC)Fe(N2)2
-
[2,6-(2,6-iPr2-C6H3-4,5-H2-имида-
введения дейтериевой и тритиевой метки в раз-
зол-2-илиден)2C5H3N)]Fe(N2)2
личные биологически активные соединения. Даны
ABT-578 - (42S)-42-дезокси-42-(1Н-тетразол-1-ил)рапа-
мицин
основные подходы, на основе которых удается зна-
чительно повысить выход и содержание изотопа во-
AR-C67085MX - [[[[(2R,3S,4R,5R)-5-(6-амино-2-пропил-
сульфанилпурин-9-ил)-3,4-дигидроксиоксолан-2-ил]
дорода в меченных дейтерием и тритием препара-
метоксигидроксифосфорил]оксигидроксифосфорил]
тах. Показано, как можно влиять на распределение
дихлорметил]фосфоновая кислота
дейтерия и трития в молекулах органических сое-
AZD5248 - (S)-4-амино-N-(1-циано-2-(4ꞌ-циано-[1,1ꞌ-би-
динений. Показано, что разработанные методики
фенил]-4-ил)этил)тетрагидро-2Н-пиран-4-карбоксамид
позволяют получать меченые препараты с характе-
AZD6642
- (R)-5-(2-(4-(5-аминопиразин-2-ил)фенил)
ристиками, достаточными для решения самых раз-
тетрагидрофуран-2-ил)-N-(2-гидрокси-2-метилпропил)
нообразных задач, стоящих перед теоретической и
пиколамид
прикладной биологией и медициной.
BIBN
4096
-
1-[3,5-дибром-N-[[4-(1,4-дигидро-2-ок-
В последние годы достигнуты определенные те-
со-3(2H)-хиназолил)-1-пиперидинил] карбонил]-D-тиро-
оретические достижения при получении меченых
зил-L-лизил]-4-(4-пиридинил)пиперазин
соединений при нагревании без использования рас-
BIIL260 - 4-[(3-{4-[2-(4-гидроксифенил)пропан-2-ил]фе-
творителей [1, 11]. Современные представления о
ноксиметил}фенил)метокси]бензол-1-карбоксимидамид
процессах, происходящих при введении дейтерия
CNS-5161
-
(Е)-2-[2-хлор-5-(метилсульфанил)фе-
или трития этим методом, позволили связать эф-
нил]-1-метил-1-[3-(метилсульфанил)фенил] гуанидин
фективность введения дейтерия или трития с раз-
COD - 1,5-циклооктадиен
ной вероятностью сольватации изотопов водорода и
D - дейтерий
электронов на носителе и в пуле вещества. Заметно
DBU - 1,8-диазабицикло[5.4.0]ундец-7-ен
влияют на включение дейтерия и трития стериче-
DMAP - 4-диметиламинопиридин
ские факторы.
DMF - диметилформамид
Осмысленное манипулирование самыми незна-
DMSO - диметилсульфоксид
чительными изменениями в ходе экспериментов
Dmt - 2-(1H-индол-3-ил)-N,N-диметилэтанамин
часто оказывалось решающим при получении иско-
DOFA - 3,4-дигидроксифенилаланин
мого меченого продукта [16, 22, 255, 259-268].
DPA-714 - N,N-диэтил-2-[4-(2-фторэтокси)фенил]-5,7-ди-
метилпиразоло[1,5-а]пиримидин-3-ацетамид
В обзоре приведены примеры использования
dtbbpy - 4,4ꞌ-ди-трет-бутил-2,2ꞌ-дипиридил
новых катализаторов. С их использованием можно
DTBHN - ди-трет-бутилгипонитрит
ожидать введения дейтерия и трития в самые слож-
Et3B - триэтилборан
ные и лабильные соединения. Интересны и новые
i-Pr2EtN - диизопропилэтиламин
подходы к повышению их селективности.
i-PrOH - изопропанол
Рисунки и таблицы сделаны авторами обзора на
KRED - кеторедуктаза
основании публикаций, ссылки на которые указаны
m-CPBA - м-хлорнадбензойная кислота
в подписях к рисункам и таблицам, или после упо-
Mes - 2-(N-морфолино)этансульфоновая кислота
минания об этих рисунках и таблицах в тексте.
Ms- SO2CH3
NMP - N-метил-2-пирролидон
КОНФЛИКТ ИНТЕРЕСОВ
OTf - трифторметансульфонат
Pd(dppf)2Cl2
-
[1,1ꞌ-бис (дифенилфосфино)ферроцен]
Авторы заявляют об отсутствии конфликта ин-
дихлорпалладий(II)
тересов.
ppy - фенилпиридил
T - тритий
СПИСОК СОКРАЩЕНИЙ
TEA - триэтиламин
Tf2O - трифторметансульфоновый ангидрид
(+)-S-145 - (+)-[1R-[1α,2α(Z),3β,4α]]-7-[3-[(фенилсульфо-
нил)амино]бицикло[2.2.1]гепт-2-ил]-5-гептеновая кис-
TFA - трифторуксусная кислота
лота
THF - тетрагидрофуран
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
341
TMP - 2,2,6,6-тетраметилпиперидин
16. Шевченко В.П., Нагаев И.Ю., Шевченко К.В.,
TMS - тетраметилсилан
Мясоедов Н.Ф. // Докл. AH. 2016. Т. 466, № 6. C. 683-
687.
мио-Инозит - цис-1,2,3,5-транс-4,6-циклогексангексаол
17. Chiesa M., Paganini M.C., Spoto G., Giamello E.,
Реактив Бёрджесса - метил-N-(трииэтиламмонийсуль-
Di Valentin C., Del Vitto A., Pacchioni G. // J. Phys.
фонил)карбамат
Chem. B. 2005. Vol. 109, N 15. P. 7314-7322.
https://doi.org/10.1021/jp044783c
СПИСОК ЛИТЕРАТУРЫ
18. Chiesa M., Paganini M.C., Giamello E., Murphy D.M.,
Di Valentin C., Pacchioni G. // Acc. Chem. Res. 2006.
1.
Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Vol. 39, N 11. P. 861-867.
Радиохимия. 2019. Т. 61, № 3 С. 183-216.
https://doi.org/10.1021/ar068144r
19. Napoli F., Chiesa M., Giamello E., Finazzi E.,
https://doi.org/10.1134/S0033831119030018
Di Valentin C., Pacchioni G. // J. Am. Chem. Soc. 2007.
2.
Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Vol. 129, N 34. P. 10575-10581.
Успехи химии. 2003. Т. 72, № 5. C. 471-497.
https://doi.org/10.1021/ja073114k
3.
Розанов В.В., Крылов О.В. // Успехи химии. 1997.
20. Zhao J., Li B., Onda K., Feng M., Petek H. // Chem. Rev.
Т. 66, № 2. C. 117-130.
2006. Vol. 106, N 10. P. 4402-4427.
https://doi.org/10.1070/RC1997v066n02ABEH000308
https://doi.org/10.1021/cr050173c
4.
Voges R., Heys J.R., Moenius T. Preparation of
21. Шевченко В.П., Нагаев И.Ю., Шевченко К.В.,
Compounds Labeled with Tritium and Carbon-14.
Мясоедов Н.Ф. // Радиохимия. 2013. Т. 55, № 5.
Chichester: Wiley, 2009. 664 p.
C. 461-466.
5.
Shu A.Y.L., Heys J.R. // Tetrahedron Lett. 2000. Vol. 41,
22. Shevchenko V.P., Nagaev I.Yu., Myasoedov N.F. //
N 47. P. 9015-9019.
J. Label. Compd. Radiopharm. 2010. Vol. 53, N 11-12.
https://doi.org/10.1016/S0040-4039(00)01652-X
P. 693-703.
6.
Борисов Ю.А., Золотарев Ю.А. // ЖФХ. 2002. Т. 76,
https://doi.org/10.1002/jlcr.1828
№ 4. C. 727-731.
23. Шевченко В.П., Нагаев И.Ю., Шевченко К.В.,
7.
Lockley W.J.S. // J. Label. Compd. Radiopharm. 2010.
Мясоедов Н.Ф. Изотопы в медицине. М.: Физматлит,
Vol. 53, N 5-6. P. 227-230.
2005. Т. 2. С. 484-538.
https://doi.org/10.1002/jlcr.1761
24. Zolotarev Yu.A., Dadayan A.K., Borisov Yu.A.,
8.
Brandt B., Fischer J.-H., Ludwig W., Libuda J., Zaera F.,
Kozik V.S. // Chem. Rev. 2010. Vol. 110, N 9. P. 5425-
Schauermann S., Freund H.-J. // J. Phys. Chem. C. 2008.
5446.
Vol. 112, N 30. P. 11408-11420.
25. Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф.
https://doi.org/10.1021/jp800205j
Меченные тритием липофильные соединения.
М.: Наука, 2003. 246 c.
9.
Doyle A.M., Shaikhutdinov S.K., Freund H.-J. // J. Catal.
2004. Vol. 223, N 2. P. 444-453.
26. Юхимчук А.А. // Успехи в химии и хим. технологии.
2019. Т. 33, № 1. C. 19-21.
https://doi.org/10.1016/j.jcat.2004.02.020
27. Постановление главного гос. санитарного вра-
10. Zaera F. // Catal. Lett. 2003. Vol. 91, N 1-2. P. 1-10.
ча РФ от 07.06.2009 № 47. СанПиН 2.6.1.2523-09.
https://doi.org/10.1023/B:CATL.0000006310.50290.ba
Нормы радиационной безопасности НРБ-99/2009.
11. Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Федеральная служба по надзору в сфере защи-
Радиохимия. 2018. Т. 60, № 2. С. 97-127.
ты прав потребителей и благополучия человека.
12. Prins R., Palfi V.K., Reiher M. // J. Phys. Chem. C. 2012.
Зарегистрировано в Министерстве юстиции РФ
Vol. 116, N 27. P. 14274-14283.
14.08.2009. № 14534.
https://doi.org/10.1021/jp212274y
28. Технологические и организационные аспекты обра-
13. Prins R. // Chem. Rev. 2012. Vol. 112, N 5. P. 2714-
щения с радиоактивными отходами: Серия учебных
2738.
курсов № 27. Вена: МАГАТЭ, 2005. IAEA-TCS-27.
https://doi.org/10.1021/cr200346z
221 c.
14. Prasittichai C., Avila J.R., Farha O.K., Hupp J.T. // J.
29. Постановление главного гос. санитарного врача РФ
Am. Chem. Soc. 2013. Vol. 135, N 44. P. 16328-16331.
от 26.04.2010 № 40 (ред. от 16.09.2013). Об утверж-
https://doi.org/10.1021/ja4089555
дении СП
2.6.1.2612-10
«Основные санитарные
15. Шевченко В.П., Нагаев И.Ю., Андреева Л.А.,
правила обеспечения радиационной безопасности
Мясоедов Н.Ф. // Докл. AH. 2017. Т. 473, № 5. C. 564-
(ОСПОРБ-99/2010)» (вместе с СП
2.6.1.2612-10
567.
«ОСПОРБ-99/2010. Санитарные правила и нормати-
РАДИОХИМИЯ том 64 № 4 2022
342
ШЕВЧЕНКО и др.
вы...»). Зарегистрировано в Министерстве юстиции
43. Fiorio J.L., Goncalves R.V., Teixeira-Neto E.,
РФ 11.08.2010. № 18115.
Ortuno M.A., Lopez N., Rossi L.M. // ACS Catal. 2018.
30. Приказ Федеральной службы по экологическому, тех-
Vol. 8, N 4. P. 3516-3524.
нологическому и атомному надзору от 15.12.2014 №
https://doi.org/10.1021/acscatal.8b00806
572 [Об утверждении федеральных норм и правил в
44. Kahsar K.R., Schwartz D.K., Medlin J.W. // J. Am.
области использования атомной энергии «Критерии
Chem. Soc. 2014. Vol. 136, N 1. P. 520-526.
приемлемости радиоактивных отходов для захоро-
https://doi.org/10.1021/ja411973p
нения» (вместе с НП-093-14. Федеральные нормы
45. Marshall S.T., OꞌBrien M., Oetter B., Corpuz A.,
и правила)] (с изменением от 17.11.2017 № 481).
Richards R.M., Schwartz D.K., Medlin J.W. // Nature
Зарегистрирован в Министерстве юстиции РФ
Mater. 2010. Vol. 9. P. 853-858.
27.03.2015. № 36592.
https://doi.org/10.1038/nmat2849
31. Linnemann E., Fels G. // J. Label. Compd. Radiopharm.
46. Ananikov V.P., Orlov N.V., Zalesskiy S.S., Beletskaya I.P.,
2001. Vol. 44, N 9. P. 661-669.
Khrustalev V.N., Morokuma K., Musaev D.G. // J. Am.
https://doi.org/10.1002/jlcr.500
Chem. Soc. 2012. Vol. 134, N 15. P. 6637-6649.
32. Okuno K., Kobayashi M., Yamanishi T., Oya Y. // Fusion
https://doi.org/10.1021/ja210596w
Eng. Des. 2013. Vol. 88, N 9-10. P. 2328-2331.
47. Deng D., Yang Y., Gong Y., Li Y., Xu X., Wang Y. // Green
https://doi.org/10.1016/j.fusengdes.2013.03.064
Chem. 2013. Vol. 15, N 9. P. 2525-2531.
33. Chen T., Rodionov V.O. // ACS Catal. 2016. Vol. 6, N 6.
https://doi.org/10.1039/C3GC40779A
P. 4025-4033.
48. Mitsudome T., Takahashi Y., Ichikawa S., Mizugaki T.,
https://doi.org/10.1021/acscatal.6b00714
Jitsukawa K., Kaneda K. // Angew. Chem. Int. Ed. 2013.
34. Albani D., Shahrokhi M., Chen Z., Mitchell S., Hauert R.,
Vol. 52, N 5. P. 1481-1485.
Lopez N., Perez-Ramirez J. // Nat. Commun. 2018.
https://doi.org/10.1002/anie.201207845
Vol. 9, N 2634. P. 1-11.
49. Mitsudome T., Urayama T., Yamazaki K., Maehara Y.,
https://doi.org/10.1038/s41467-018-05052-4
Yamasaki J., Gohara K., Maeno Z., Mizugaki T.,
35. Zhao X., Zhou L., Zhang W., Hu C., Dai L., Ren L.,
Jitsukawa K., Kaneda K. // ACS Catal. 2016. Vol. 6, N 2.
Wu B., Fu G., Zheng N. // Chem. 2018. Vol. 4, N 5.
P. 666-670.
P. 1080-1091.
https://doi.org/10.1021/acscatal.5b02518
https://doi.org/10.1016/j.chempr.2018.02.011
50. Mitsudome T., Yamamoto M., Maeno Z., Mizugaki T.,
36. Vile G., Albani D., Almora-Barrios N., Lopez N., Perez-
Jitsukawa K., Kaneda K. // J. Am. Chem. Soc. 2015.
Ramirez J. // ChemCatChem. 2016. Vol. 8, N 1. P. 21-
Vol. 137, N 42. P. 13452-13455.
33.
https://doi.org/10.1021/jacs.5b07521
https://doi.org/10.1002/cctc.201501269
51. Song S., Li K., Pan J., Wang F., Li J., Feng J., Yao S.,
37. Crespo-Quesada M., Cardenas-Lizana F., Dessi-
Ge X., Wang X., Zhang H. // Adv. Mater. 2016. Vol. 29.
moz A.-L., Kiwi-Minsker L. // ACS Catal. 2012. Vol. 2,
N 8. Article 1605332.
N 8. P. 1773-1786.
https://doi.org/10.1002/adma.201605332
52. Niu W., Gao Y., Zhang W., Yan N., Lu X. // Angew. Chem.
38. Guan E., Gates B.C. // ACS Catal. 2018. Vol. 8, N 1.
2015. Vol. 127, N 28. P. 8389-8392.
P. 482-487.
https://doi.org/10.1002/ange.201503148
https://doi.org/10.1021/acscatal.7b03549
53. Huang F., Deng Y., Chen Y., Cai X., Peng M., Jia Z.,
39. Fedorov A., Liu H.-J., Lo H.-K., Coperet C. // J. Am.
Ren P., Xiao D., Wen X., Wang N., Liu H., Ma D. // J.
Chem. Soc. 2016. Vol. 138, N 50. P. 16502-16507.
Am. Chem. Soc. 2018. Vol. 140, N 41. P. 13142-13146.
https://doi.org/10.1021/jacs.6b10817
https://doi.org/10.1021/jacs.8b07476
40. Yan M., Jin T., Ishikawa Y., Minato T., Fujita T.,
54. Riley C., Zhou S., Kunwar D., De La Riva A., Peterson E.,
Chen L.-Y., Bao M., Asao N., Chen M.-W.,
Yamamoto Y. // J. Am. Chem. Soc. 2012. Vol. 134, N 42.
Payne R., Gao L., Lin S., Guo H., Datye A. // J. Am.
P. 17536-17542.
Chem. Soc. 2018. Vol. 140, N 40. P. 12964-12973.
https://doi.org/10.1021/ja3087592
https://doi.org/10.1021/jacs.8b07789
41. Hauwert P., Maestri G., Sprengers J.W., Catellani M.,
55. Ji G., Duan Y., Zhang S., Fei B., Chen X., Yang Y. //
Elsevier C.J. // Angew. Chem. 2008. Vol. 120, N 17.
ChemSusChem. 2017. Vol. 10, N 17. P. 3427-3434.
P. 3267-3270.
https://doi.org/10.1002/cssc.201701127
https://doi.org/10.1002/ange.200705638
56. Chen F., Kreyenschulte C., Radnik J., Lund H.,
42. Gavia D.J., Shon Y.-S. // ChemCatChem. 2015. Vol. 7,
Surkus A.-E., Junge K., Beller M. // ACS Catal. 2017.
N 6. P. 892-900.
Vol. 7, N 3. P. 1526-1532.
https://doi.org/10.1002/cctc.201402865
https://doi.org/10.1021/acscatal.6b03140
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
343
57. Zhang P., Yuan J., Fellinger T.-P., Antonietti M., Li H.,
69. Aich P., Wei H., Basan B., Kropf A.J., Schweitzer N.M.,
Wang Y. // Angew. Chem. Int. Ed. 2013. Vol. 52, N 24.
Marshall C.L., Miller J.T., Meyer R. // J. Phys. Chem. C.
P. 6028-6032.
2015. Vol. 119, N 32. P. 18140-18148.
https://doi.org/10.1002/anie.201301069
https://doi.org/10.1021/acs.jpcc.5b01357
58. Shen L., Mao S., Li J., Li M., Chen P., Li H., Chen Z.,
70. Yan H., Cheng H., Yi H., Lin Y., Yao T., Wang C., Li J.,
Wei S., Lu J. // J. Am. Chem. Soc. 2015. Vol. 137, N 33.
Wang Y. // J. Catal. 2017. Vol. 350. P. 13-20.
P. 10484-10487.
https://doi.org/10.1016/j.jcat.2017.01.021
https://doi.org/10.1021/jacs.5b06485
59. Studt F., Abild-Pedersen F., Bligaard T., Sorensen R.Z.,
71. Delley M.F., Silaghi M.-C., Nunez-Zarur F.,
Christensen C.H., Norskov J.K.
// Science.
2008.
Kovtunov K.V., Salnikov O.G., Estes D.P., Koptyug I.V.,
Vol. 320, N 5881. P. 1320-1322.
Comas-Vives A., Coperet C. // Organometallics. 2017.
https://doi.org/10.1126/science.1156660
Vol. 36, N 1. P. 234-244.
60. Yan N., Chen X. // Nature. 2015. Vol. 524, N 7564.
https://doi.org/10.1021/acs.organomet.6b00744
P. 155-157.
72. Estes D.P., Siddiqi G., Allouche F., Kovtunov K.V.,
https://doi.org/10.1038/524155a
Safonova O.V., Trigub A.L., Koptyug I.V., Coperet C. // J.
61. Fang Y., Duan B., Lu A., Liu M., Liu H., Xu X., Zhang L. //
Am. Chem. Soc. 2016. Vol. 138, N 45. P. 14987-14997.
Biomacromolecules. 2015. Vol. 16, N 4. P. 1410-1417.
https://doi.org/10.1021/jacs.6b08705
https://doi.org/10.1021/acs.biomac.5b00195
73. Zhang B., Asakura H., Zhang J., Zhang J., De S.,
62. Duan B., Zheng X., Xia Z., Fan X., Guo L., Liu J.,
Yan N. // Angew. Chem. Int. Ed. 2016. Vol. 55, N 29.
Wang Y., Ye Q., Zhang L. // Angew. Chem. Int. Ed. 2015.
P. 8319-8323.
Vol. 54, N 17. P. 5152-5156.
https://doi.org/10.1002/anie.201602801
https://doi.org/10.1002/anie.201412129
74. Аксенов И.А. Наноструктурированные катализато-
63. Duan B., Gao X., Yao X., Fang Y., Huang L., Zhou J.,
ры селективного гидрирования ацетиленовых и ди-
Zhang L. // Nano Energy. 2016. Vol. 27. P. 482-491.
еновых углеводородов. Дис. … к.х.н. М.: МГУ, 2014.
https://doi.org/10.1016/j.nanoen.2016.07.034
150 c.
64. Zhao L., He R., Rim K.T., Schiros T., Kim K.S.,
75. Liu B., Liu L., Wang Z., Chai Y., Liu H., Yin C., Liu C. //
Zhou H., Gutierrez C.,
Chockalingam S.P.,
Catal. Today. 2017. Vol. 282, Part. 2. P. 214-221.
Arguello C.J., Palova L., Nordlund D., Hybertsen M.S.,
https://doi.org/10.1016/j.cattod.2016.08.020
Reichman D.R., Heinz T.F., Kim P., Pinczuk A.,
76. Wichmann J., Huguenin P., Adam G. // J. Label. Compd.
Flynn G.W., Pasupathy A.N. // Science. 2011. Vol. 333,
N 6045. P. 999-1003.
org/10.1002/(SICI)1099-1344(200001)43:1<1::AID-
JLCR285>3.0.CO;2-N
https://doi.org/10.1126/science.1208759
77. Atwell G.J., Denny W.A. // J. Label. Compd. Radiopharm.
65. Schiros T., Nordlund D., Palova L., Prezzi D.,
2007. Vol. 50, N 1. P. 7-12.
Zhao L., Kim K.S., Wurstbauer U., Gutierrez C.,
https://doi.org/10.1002/jlcr.1147
Delongchamp D., Jaye C., Fischer D., Ogasawara H.,
78. Szammer J., Simon-Trompler E., Banka Z., Szunyog J.,
Pettersson L.G.M.,
Reichman D.R., Kim P.,
Klebovich I. // J. Label. Compd. Radiopharm. 2005.
Hybertsen M.S., Pasupathy A.N. // Nano Lett.
2012.
Vol. 48, N 9. P. 693-700.
Vol. 12, N 8. P. 4025-4031.
https://doi.org/10.1002/jlcr.964
https://doi.org/10.1021/nl301409h
79. Egan J.A., Filer C.N., Pounds S., Connors T., Knight E. jr.,
66. Li X., Pan Y., Yi H., Hu J., Yang D., Lv F., Li W., Zhou J.,
Hudkins R.L. // J. Label. Compd. Radiopharm. 2005.
Wu X., Lei A., Zhang L. // ACS Catal. 2019. Vol. 9, N 5.
Vol. 48, N 5. P. 331-335.
P. 4632-4641.
https://doi.org/10.1021/acscatal.9b01001
80. Larsen U.S., Begtrup M., Martiny L. // J. Label. Compd.
67. Liu B., Wang Y., Peng H.-Q., Yang R., Jiang Z., Zhou X.,
Radiopharm. 2005. Vol. 48, N 6. P. 429-434.
Lee C.-S., Zhao H., Zhang W. // Adv. Mater. 2018.
Vol. 30, N 36. Article 1803144.
81. Seltzman H.H., Foster M.C., Wyrick C.D., Burgess J.P.,
https://doi.org/10.1002/adma.201803144
Carroll F.I. // J. Label. Compd. Radiopharm. 2005.
68. Boucher M.B., Zugic B., Cladaras G., Kammert J.,
Vol. 48, N 8. P. 589-596.
Marcinkowski M.D., Lawton T.J., Sykes E.C.H., Flytzani-
Stephanopoulos M. // Phys. Chem. Chem. Phys. 2013.
82. Kingston L.P., Lockley W.J.S., Mather A.N.,
Vol. 15, N 29. P. 12187-12196.
Coombs M.E., Wilkinson D.J. // Seventh Int. Symp.
https://doi.org/10.1039/C3CP51538A
on: The Synthesis and Applications of Isotopes and
РАДИОХИМИЯ том 64 № 4 2022
344
ШЕВЧЕНКО и др.
Isotopically Labelled Compounds: Abstracts. Dresden,
99. Malmquist J., Bernlind A., Sandell J., Strom P.,
Germany, 2000. P. 50.
Waldman M. // J. Label. Compd. Radiopharm. 2012.
83. Marek A., Pedersen M.H.F., Vogensen S.B., Clausen R.P.,
Vol. 55, N 2. P. 80-83.
Frolund B., Elbert T. // J. Label. Compd. Radiopharm.
2016. Vol. 59, N 12. P. 476-483.
100.Filer C.N. // J. Label. Compd. Radiopharm.
2010.
Vol. 53, N 3. P. 120-129.
84. Bragg R.A., Bushby N., Ericsson C., Kingston L.P., Ji H.,
101.Meisterhans C., Nuckel S.C., Zeller A. // J. Label.
Elmore C.S. // J. Label. Compd. Radiopharm. 2016.
Compd. Radiopharm. 2010. Vol. 53, N 5-6. P. 253-257.
Vol. 59, N 11. P. 454-461.
102.Pounds S.
// Presentation at the IIS-CED Symp.
85. Filer C.N. // J. Label. Compd. Radiopharm.
2017.
Bad Soden, Germany,
2003. Р.
25-26. Personal
Vol. 60, N 2. P. 96-109.
communication.
103.Ahern D.G., Seguin R.J., Filer C.N. // J. Label. Compd.
86. Marek A., Pedersen M.H.F. // Tetrahedron. 2015. Vol. 71,
Radiopharm. 2002. Vol. 45, N 5. P. 401-405.
N 6. P. 917-921.
104.Tu Y., Zhong J., Wang H., Pan J., Xu Z., Yang W.,
87. Gibbs A.R., Morimoto H., VanBrocklin H.F.,
Luo Y. // J. Label. Compd. Radiopharm. 2016. Vol. 59,
Williams P.G., Biegon A. // J. Label. Compd. Radiopharm.
N 13. P. 546-551.
2002. Vol. 45, N 5. P. 395-400.
105.Sun K., Fang C., Yang W., Xu Z., Wang H., Sun W.,
88. Eriksson J., Ulin J., Langstrom B. // J. Labelled Compd.
Luo Y., Xu Y. // J. Label. Compd. Radiopharm. 2016.
Radiopharm. 2006. Vol. 49, N 13. P. 1177-1186.
Vol. 59, N 13. P. 552-556.
89. Pounds S. Synthesis and Applications of Isotopically
106.Шевченко В.П., Нагаев И.Ю., Шевченко К.В.,
Labeled Compounds / Eds D.C. Dean, C.N. Filer, K.E.
Мясоедов Н.Ф., Бочкарева Н.В., Коломиец Л.А.,
McCarthy. Chichester: Wiley, 2004. Vol. 8. P. 63-66.
Кондакова И.В. // Радиохимия. 2004. Т. 46, № 5.
90. Pounds J.S. Patent WO 2004108636. 2003.
C. 458-463.
91. Pounds J.S. Patent US 8329934B2. 2012.
107.Бочкарева Н.В., Кондакова И.В., Коломиец Л.А.,
92. Шевченко В.П., Нагаев И.Ю., Алфеева Л.Ю.,
Мунтян А.Б., Шевченко В.П., Нагаев И.Ю.,
Андреева Л.А., Шевченко К.В., Мясоедов Н.Ф. //
Шевченко К.В., Мясоедов Н.Ф., Дорофеева Н.Н. //
Радиохимия. 2006. Т. 48, № 3. C. 267-271.
Бюл. эксперим. биологии и медицины. 2006. Т. 142,
№ 10. C. 462-464.
93. Шевченко К.В., Нагаев И.Ю., Алфеева Л.Ю.,
108.Безуглов В.В., Грецкая Н.М., Блаженова А.В.,
Андреева Л.А., Каменский А.А., Левицкая Н.Г.,
Андрианова Е.Л., Акимов М.Г., Бобров М.Ю.,
Шевченко В.П., Гривенников И.А., Мясоедов Н.Ф. //
Назимов И.В., Кисель М.А.,Шарко О.Л., Новиков А.В.,
Биоорганическая химия. 2006. Т. 32, № 1. C. 64-70.
Краснов Н.В., Шевченко В.П., Шевченко К.В.,
94. Maxwell B.D., Bronstein J.C. // J. Label. Compd.
Вьюнова Т.В., Мясоедов Н.Ф. // Биоорганическая хи-
Radiopharm. 2005. Vol. 48, N 14. P. 1049-1054.
мия. 2006. Т. 32, № 3. C. 258-267.
109.Шевченко В.П., Нагаев И.Ю., Алфеева Л.Ю.,
95. Latli B., Stiasni M., Hrapchak M., Li Z., Grinberg N.,
Андреева Л.А., Климова П.А., Малкин А.В.,
Lee H., Busacca C.A., Senanayake C.H. // J. Label.
Шевченко К.В., Мясоедов Н.Ф. // Радиохимия. 2006.
Compd. Radiopharm. 2016. Vol. 59, N 13. P. 557-564.
Т. 48, № 3. C. 260-266.
110. Lindelof A., Ericsson C., Simonsson R., Nilsson G.,
96. Sonnenberger S., Lange S., Langner A., Neubert R.H.H.,
Gronberg G., Elmore C.S.
// J. Label. Compd.
Dobner B. // J. Label. Compd. Radiopharm.
2016.
Radiopharm. 2016. Vol. 59, N 9. P. 340-345.
Vol. 59, N 12. P. 531-542.
111. Allen P., Bragg R.A., Caffrey M., Ericsson C.,
97. Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Hickey M.J., Kingston L.P., Elmore C.S. // J. Label.
Радиохимия. 2009. Т. 51, № 2. C. 156-160.
Compd. Radiopharm. 2017. Vol. 60, N 2. P. 124-129.
98. Bupp J.E., Tanga M.J. // J. Label. Compd. Radiopharm.
2016. Vol. 59, N 7. P. 291-293.
112. Shevchenko V.P., Nagaev I.Yu., Myasoedov N.F.,
Susan A.B., Switek K.-H., Braunger H.
// J. Label.
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
345
Compd. Radiopharm. 2006. Vol. 49, N 5. P. 421-427.
127. Shchepina N.E., Avrorin V.V., Badun G.A., Lewis S.B.,
Shurov S.N. // ISRN Org. Chem. 2012. Vol. 2012. Artcle
113. Rotert G., Fan L., Shevchenko V.P., Nagaev I.Yu.,
ID 526867.
Myasoedov N.F., Susan A.B. // J. Label. Compd.
Radiopharm. 2006. Vol. 49, N 10. P. 849-856.
128. Shchepina N.E., Avrorin V.V., Badun G.A., Shurov S.N.,
Shchepin R.V. // Radiochim. Acta. 2020. Vol. 108, N 2.
114. Shevchenko V.P., Nagaev I.Yu., Myasoedov N.F.,
P. 105-111.
Susan A.B., Anderskewitz R., Birke F.W., Switek K.H.
129.Щепина Н.Е., Аврорин В.В., Бадун Г.А., Уханов С.Е. //
// J. Label. Compd. Radiopharm. 2003. Vol. 46. N 3.
Радиохимия. 2016. Т. 58, № 1. С. 84-86.
130.Щепина Н.Е., Аврорин В.В., Бадун Г.А., Шуров С.Н.,
P. 221-230.
Щепин Р.В. // Радиохимия. 2017. Т. 59, № 3. С. 260-
263.
115. Oba M., Ohkuma K., Hitokawa H., Shirai A.,
131.Щепина Н.Е., Аврорин В.В., Бадун Г.А., Шуров С.Н.,
Nishiyama K. // J. Label. Compd. Radiopharm. 2006.
Щепин Р.В. // Радиохимия. 2020. Т. 62, № 1. С. 73-78.
Vol. 49, N 3. P. 229-235.
132.Hickey M.J., Kingston L.P., Lockley W.J.S., Allen P.,
Mather A., Wilkinson D.J.
// J. Label. Compd.
116. Oba M., Kobayashi M., Oikawa F., Nishiyama K.,
Radiopharm. 2007. Vol. 50, N 5-6. P. 286-289.
Kainosho M. // J. Org. Chem. 2001. Vol. 66. N 17.
P. 5919-5922.
133.Wan Y., Jakway J.P., Qiu H., Shah H., Garlisi C.G.,
Tian F., Ting P., Hesk D., Egan R.W., Billah M.M.,
117. Pajak M., Palka K., Winnicka E., Kanska M. // J. Label.
Umland S.P. // Eur. J. Pharmacol. 2002. Vol. 456, N 1-3.
Compd. Radiopharm. 2016. Vol. 59,. N 1. P. 4-8.
P. 1-10.
118. Wolf J.R. // J. Label. Compd. Radiopharm. 2016. Vol. 59,
134.Hesk D., Cesarz D., Magatti C., Voronin K., Lavey C.,
N 2. P. 38-47.
McNamara P., Koharski D., Saluja S., Hendershot S.,
Pham H., Truong V. // J. Label. Compd. Radiopharm.
119. Arai K., Wakabayashi K., Soga K., Hoson T.
//
2005. Vol. 48, N 1. P. 11-23.
Physiology. 2010. Vol. 167, N 10. P. 800-804.
135.Ray T., Wu A., Allentoff A.J. // J. Label. Compd.
120.Rivera N.R., Moore J., Schenk D.J., Wang H., Hesk D.,
Radiopharm. 2007. Vol. 50, N 5-6. P. 433-434.
Mergelsberg I. // Tetrahedron Lett. 2016. Vol. 57, N 10.
P. 1090-1092.
136.Klei S.R., Golden J.T., Tilley T.D., Bergman R.G. // J.
Am. Chem. Soc. 2002. Vol. 124, N 10. P. 2092-2093.
121.Pajak M., Palka K., Winnicka E., Kanska M. // J.
Radioanal. Nucl. Chem. 2018. Vol. 317, N 2. P. 643-
137.Klei S.R., Golden J.T., Burger P., Bergman R.G. // J.
666.
Mol. Catal. A: Chem. 2002. Vol. 189, N 1. P. 79-94.
122.Щепина Н.Е., Аврорин В.В., Бадун Г.А., Федосеев В.М.,
138.Hesk D., Bignan G., Lee J., Yang J., Voronin K.,
Уханов С.Е., Льюис С.Б. // Радиохимия. 2007. Т. 49.
Magatti C., McNamara P., Koharski D., Hendershot S.,
№ 6. C. 551-553.
Saluja S., Wang S. // J. Label. Compd. Radiopharm.
123.Щепина Н.Е., Аврорин В.В., Бадун Г.А., Федосеев В.М.,
2002. Vol. 45, N 2. P. 145-155.
Уханов С.Е., Льюис С.Б. // Радиохимия. 2009. Т. 51,
139.Hesk D., Borges S., Hendershot S., Koharski D.,
№ 2. C. 167-169.
McNamara P., Ren S., Saluja S., Truong V., Voronin
124.Щепина Н.Е., Аврорин В.В., Бадун Г.А., Федосеев В.М.,
K. // J. Label. Compd. Radiopharm. 2016. Vol. 59, N 5.
Уханов С.Е., Льюис С.Б. // Радиохимия. 2010. Т. 52,
P. 190-196.
№ 3. C. 274-276.
125.Щепина Н.Е., Бойко И.И., Александрова Г.А. //
140. Skaddan M.B., Bergman R.G. // J. Label. Compd.
Химико-фармацевтический журнал. 2011. Т. 45, № 3.
Radiopharm. 2006. Vol. 49, N 7. P. 623-634.
C. 30-32.
126. Shchepina N.E., Avrorin V.V., Badun G.A., Bumagin N.A.,
141. Simonsson R., Stenhagen G., Ericsson C., Elmore C.S. //
Lewis S.B., Shurov S.N. // Org. Med. Chem. Lett. 2012.
J. Label. Compd. Radiopharm. 2013. Vol. 56, N 6.
Vol. 2, N 14. P. 1-5.
P. 334-337.
РАДИОХИМИЯ том 64 № 4 2022
346
ШЕВЧЕНКО и др.
142.Nilsson G.N., Kerr W.J. // J. Label. Compd. Radiopharm.
157.Ellames G.J., Gibson J.S., Herbert J.M., McNeill A.H. //
2010. Vol. 53, N 11-12. P. 662-667.
Tetrahedron. 2001. Vol. 57, N 46. P. 9487-9497.
143.Brown J.A., Cochrane A.R., Irvine S., Kerr W.J., Mondal
158. Johansen S.K., Wagner S.H., Jessen C.U., Valsborg J.S.,
B., Parkinson J.A., Paterson L.C., Reid M., Tuttle T.,
Martiny L. // J. Label. Compd. Radiopharm.
2007.
Andersson S., Nilsson G.N. // Adv. Synth. Catal. 2014.
Vol. 50, N 5-6. P. 466-467.
Vol. 356, N 17. P. 3551-3562.
159.Bushby N., Killick D.A. // J. Label. Compd. Radiopharm.
2007. Vol. 50, N 5-6. P. 519-520.
144.Parmentier M., Hartung T., Pfaltz A., Muri D. // Chem.
Eur. J. 2014. Vol. 20, N 36. P. 11496-11504.
160.Allen P.H., Hickey M.J., Kingston L.P., Wilkinson D.J. //
J. Label. Compd. Radiopharm. 2010. Vol. 53, N 11-12.
145.Powell M.E., Elmore C.S., Dorff P.N., Heys J.R. // J.
P. 731-738.
Label. Compd. Radiopharm. 2007. Vol. 50, N 5-6.
P. 523-525.
161.Herbert J.M. // J. Label. Compd. Radiopharm. 2010.
Vol. 53, N 11-12. P. 658-661.
146.Brown J.A., Irvine S., Kennedy A.R., Kerr W.J.,
Andersson S., Nilsson G.N. // Chem. Commun. 2008.
162.Chappelle M.R., Morgan A.D. // J. Label. Compd.
N 9. P. 1115-1117.
Radiopharm. 2004. Vol. 47, N 5. P. 317.
147.Brown J.A., Irvine S., Kennedy A.R., Kerr W.J.,
163.Chappelle M.R., Hawes C.R. // J. Label. Compd.
Andersson S., Nilsson G.N. // J. Labelled Compd.
Radiopharm. 2010. Vol. 53, N 11-12. P. 745-751.
Radiopharm. 2008. Vol. 51, N 6. P. 257.
164.Palmer W.N., Zarate C., Chirik P.J. // J. Am. Chem. Soc.
148.Prechtl M.H.G., Holscher M., Ben-David Y., Theyssen N.,
2017. Vol. 139, N 7. P. 2589-2592.
Loschen R., Milstein D., Leitner W. // Angew.Chem. Int.
Ed. 2007. Vol. 46, N 13. P. 2269-2272.
165.Kerr W.J., Mudd R.J., Reid M., Atzrodt J., Derdau V. //
ACS Catal. 2018. Vol. 8, N 11. P. 10895-10900.
149.Traff A., Nilsson G.N., Szabo K.J., Eriksson L. // J.
Organomet. Chem. 2007. Vol. 692, N 25. P. 5529-5531.
166.Valero M., Weck R., Gussregen S., Atzrodt J., Derdau V. //
Angew. Chem. Int. Ed. 2018. Vol. 57, N 27. P. 8159-
150.Herbert J.M., Kohler A.D., McNeill A.H. // J. Label.
8163.
Compd. Radiopharm. 2005. Vol. 48, N 4. P. 285-294.
167.Danopoulos A.A., Wright J.A., Motherwell W.B. // Chem.
Commun. 2005. N 6. P. 784-786.
151.Yu R.P., Hesk D., Rivera N., Pelczer I., Chirik P.J. //
Nature. 2016. Vol. 529, N 7585. P. 195-199.
168.Loh Y.Y., Nagao K., Hoover A.J., Hesk D., Rivera N.R.,
Colletti S.L., Davies I.W., Macmillan D.W.C. // Science.
152.Yu R.P., Darmon J.M., Hoyt J.M., Margulieux G.W.,
2017. Vol. 358, N 6367. P. 1182-1187.
Turner Z.R., Chirik P.J. // ACS Catal. 2012. Vol. 2, N 8.
P. 1760-1764.
169.Yamakawa M., Ito H., Noyori R. // J. Am. Chem. Soc.
2000. Vol. 122, N 7. P. 1466-1478.
153.Yang H., Zarate C., Palmer W.N., Rivera N., Hesk D.,
Chirik P.J. // ACS Catal. 2018. Vol. 8, N 11. P. 10210-
170. Soni R., Cheung F.K., Clarkson G.C., Martins J.E.D.,
10218.
Graham M.A., Wills M. // Org. Biomol. Chem. 2011.
N 9. P. 3290-3294.
154.Palmer W.N., Diao T., Pappas I., Chirik P.J. // ACS
Catal. 2015. Vol. 5, N 2. P. 622-626.
171.Keczer S.A. de, Lane T.S.,Voronin T.,Masjedizadeh M.R. //
J. Label. Compd. Radiopharm. 2005. Vol. 48, N 14.
155.Palmer W.N., Obligacion J.V., Pappas I., Chirik P.J. //
P. 1013-1023.
J. Am. Chem. Soc. 2016. Vol. 138, N 3. P. 766-769.
172. Scheigetz J., Berthelette C., Li C., Zamboni R.J. //
156.Wencel-Delord J., Droge T., Liu F., Glorius F. // Chem.
J. Label. Compd. Radiopharm. 2004. Vol. 47, N 12.
Soc. Rev. 2011. Vol. 40, N 9. P. 4740-4761.
P. 881-889.
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
347
173.Ariza X., Asins G., Garcia J., Hegardt F.G., Makowski K.,
Label. Compd. Radiopharm. 2015. Vol. 58, N 2. P. 36-
Serra D., Velasco J. // J. Label. Compd. Radiopharm.
41.
2010. Vol. 53, N 8. P. 556-558.
189.Hesk D., Calvert M., McNamara P. Synthesis and
174.Шевченко В.П., Нагаев И.Ю., Шапошников А.И.,
Applications of Isotopically Labeled Compounds /
Шевченко К.В., Белимов А.А., Баташева С.Н.,
Eds D.C. Dean, C.N. Filer, K.E. McCarthy. Chichester:
Гоголева Н.Е., Гоголев Ю.В., Мясоедов Н.Ф. // Докл.
Wiley, 2004. Vol. 8. P. 51-54.
АН. 2018. Т. 483, № 3. C. 274-278.
190.Maegawa T., Hirota K., Tatematsu K., Mori Y., Sajiki H. //
175.Pajak M., Kanska M. // J. Label. Compd. Radiopharm.
J. Org. Chem. 2005. Vol. 70, N 25. P. 10581-10583.
2006. Vol. 49, N 12. P. 1061-1067.
191.Ito N., Watahiki T., Maesawa T., Maegawa T., Sajiki H. //
176.Kozlowska M., Kanski R., Kanska M. // J. Label. Compd.
Adv. Synth. Catal. 2006. Vol. 348, N 9. P. 1025-1028.
Radiopharm. 2005. Vol. 48, N 3. P. 235-240.
192.Anto S., Getvoldsen G.S., Harding J.R., Jones J.R.,
177.Kingston L.P., Lockley W.J.S., Mather A.N.,
Lu S.-Y., Russell J.C. // J. Chem. Soc., Perkin Trans. 2.
Wilkinson D.J. Synthesis and Applications of
2000. N 11. P. 2208-2211.
Isotopically Labeled Compounds / Eds U. Pleiss, R.
Voges. Chichester: Wiley, 2001. Vol. 7. P. 105-108.
193.Koniarczyk J.L., Hesk D., Overgard A., Davies I.W.,
178.Tanga M.J., Bupp J.E., Bradford W.W. // J. Label.
McNally A. // J. Am. Chem. Soc. 2018. Vol. 140, N 6.
Compd. Radiopharm. 2001. Vol. 44, N 6. P. 405-411.
P. 1990-1993.
179.Leppala E., Wahala K. // J. Label. Compd. Radiopharm.
194.Alexakis E., Hickey M.J., Jones J.R., Kingston L.P.,
2004. Vol. 47, N 1. P. 25-30.
Lockley W.J.S., Mather A.N., Smith T., Wilkinson D.J. //
Tetrahedron Lett. 2005. Vol. 46, N 25. P. 4291-4293.
180. Stack D.E., Eastman R. // J. Label. Comp. Radiopharm.
2016. Vol. 59, N 12. P. 500-505.
195.Kruger J., Manmontri B., Fels G. // Eur. J. Org. Chem.
2005. Vol. 2005, N 7. P. 1402-1408.
181. Shevchenko V.P., Nagaev I.Yu., Bezuglov V.V.,
Myasoedov N.F., Susan A.B.
// J. Label. Compd.
196.Lockley W.J.S. // J. Label. Compd. Radiopharm. 2007.
Radiopharm. 2007. Vol. 50, N 5-6. P. 448-449.
Vol. 50, N 9-10. P. 779-788.
182.Myasoedov N.F., Nagaev I.Yu., Shevchenko V.P.,
197.Atzrodt J., Derdau V., Kerr W.J., Reid M. // Angew.
Susan A.B., Rotert G. // J. Label. Compd. Radiopharm.
Chem. Int. Ed. 2018. Vol. 57, N 12. P. 3022-3047.
2004. Vol. 47, N 4. P. 255-257.
198.Moozeh K., So S.M., Chin J. // Angew. Chem. Int. Ed.
183.Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Радиохимия. 2005. Т. 47, № 4. C. 368-373.
2015. Vol. 54, N 32. P. 9381-9385.
184.Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Радиохимия. 2012. Т. 54, № 1. C. 75-81.
199. Sakamoto T., Mori K., Akiyama T. // Org. Lett. 2012.
185. Shevchenko V.P., Nagaev I.Yu., Myasoedov N.F.,
Vol. 14, N 13. P. 3312-3315.
Susan A.B. // J. Label. Compd. Radiopharm.
2007.
Vol. 50, N 5-6. P. 416-417.
200.Arns S., Moreau A., Young R.N. // J. Label. Compd.
Radiopharm. 2010. Vol. 53, N 4. P. 205-207.
186.Hesk D., Voronin K., McNamara P., Royster P.,
Koharski D., Hendershot S., Saluja S., Truong V.,
201.Okano K., Murata K., Ikariya T. // Tetrahedron Lett.
Chan T.M. // J. Label. Compd. Radiopharm.
2007.
2000. Vol. 41, N 48. P. 9277-9280.
Vol. 50, N 2. P. 131-137.
202.Koike T., Murata K., Ikariya T. // Org. Lett. 2000. Vol. 2,
187.Petros R.A., Shah J. // J. Label. Compd. Radiopharm.
N 24. P. 3833-3836.
2014. Vol. 57, N 1. P. 53-56.
203.Hilton M.C., Dolewski R.D., McNally A. // J. Am. Chem.
188.Hesk D., Borges S., Dumpit R., Hendershot S.,
Soc. 2016. Vol. 138, N 42. P. 13806-13809.
Koharski D., Lavey C., McNamara P., Voronin K. // J.
РАДИОХИМИЯ том 64 № 4 2022
348
ШЕВЧЕНКО и др.
204. Shimada M., Sugimoto O., Sato A., Tanji K.-I. //
221.Vityuk A., Khivantsev K., Aleksandrov H.A.,
Heterocycles. 2011. Vol. 83, N 4. P. 837-847.
Vayssilov G.N., Alexeev O.S., Amiridis M.D. // ACS
Catal. 2019. Vol. 9, N 2. P. 839-847.
205.Deng Z., Lin J.-H., Xiao J.-C. // Nat. Commun. 2016.
Vol. 7. Article 10337.
222.Zheng A., Li S., Liu S.-B., Deng F. // Acc. Chem. Res.
2016. Vol. 49, N 4. P. 655-663.
206.Gerdes G., Chen P. // Organometallics. 2004. Vol. 23,
223.Grimme S. // J. Comput. Chem. 2006. Vol. 27, N 15.
N 12. P. 3031-3036.
P. 1787-1799.
207.Ziatdinov V.R., Oxgaard J., Mironov O.A., Young K.J.H.,
224.Bhering D.L., Ramirez-Solis A., Mota C.J.A. // J. Phys.
Goddard W.A., Periana R.A. // J. Am. Chem. Soc. 2006.
Chem. B. 2003. Vol. 107, N 18. P. 4342-4347.
Vol. 128, N 23. P. 7404-7405.
225.Khivantsev K., Vityuk A., Aleksandrov H.A.,
208.Hickman A.J., Villalobos J.M., Sanford M.S.
//
Vayssilov G.N., Alexeev O.S., Amiridis M.D. // J. Phys.
Organometallics. 2009. Vol. 28, N 18. P. 5316-5322.
Chem. C. 2015. Vol. 119, N 30. P. 17166-17181.
209.Allais F., Boivin J., Nguyen V.T. // Beilstein J. Org.
226.Khivantsev K., Vityuk A., Aleksandrov H.A.,
Chem. 2007. Vol. 3, N 46. P. 1-7.
Vayssilov G.N., Blom D., Alexeev O.S., Amiridis M.D. //
ACS Catal. 2017. Vol. 7, N 9. P. 5965-5982.
210.Boivin J., Nguyen V.T. // Beilstein J. Org. Chem. 2007.
Vol. 3, N 45. P. 1-7.
227.Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Радиохимия. 2018. Т. 60, № 2. C. 97-127.
211. Hioe J., Karton A., Martin J.M.L., Zipse H. // Chem. Eur.
228.Atzrodt J., Derdau V., Kerr W.J., Reid M. // Angew.
J. 2010. Vol. 16, N 23. P. 6861-6865.
Chem. Int. Ed. 2018. Vol. 57, N 7. P. 1758-1784.
212.Povie G., Marzorati M., Bigler P., Renaud P. // J. Org.
229.Chen L., Cooper A.C., Pez G.P., Cheng H. // J. Phys.
Chem. 2013. Vol. 78, N 4. P. 1553-1558.
Chem. C. 2007. Vol. 111, N 51. P. 18995-19000.
213.Povie G., Renaud P. // Chimia. 2013. Vol. 67, N 4.
230.Chen L., Cooper A.C., Pez G.P., Cheng H. // J. Phys.
P. 250-252.
Chem. C. 2008. Vol. 112, N 6. P. 1755-1758.
231.Pyle D.S., Gray E.M., Webb C.J. // Int. J. Hydrogen
214. Jin J., Newcomb M. // J. Org. Chem. 2007. Vol. 72, N 14.
Energy. 2016. Vol. 41, N 42. P. 19098-19113.
P. 5098-5103.
232.Deka R.C., Baishya S. // Catal. Today. 2012. Vol. 198,
215. Jin J., Newcomb M. // J. Org. Chem. 2008. Vol. 73, N 12.
N 1. P. 110-115.
P. 4740-4742.
233.Ensafi A.A., Jafari-Asl M., Nabiyan A., Rezaei B.,
216.Denes F., Pichowicz M., Povie G., Renaud P. // Chem.
Dinari M. // Energy. 2016. Vol. 99. P. 103-114.
Rev. 2014. Vol. 114, N 5. P. 2587-2693.
234.Han L., Qin W., Jian J., Liu J., Wu X., Gao P., Hultman B.,
217.Katsnelson A. // Nat. Med. 2013. Vol. 19. P. 656.
Wu G. // J. Power Sources. 2017. Vol. 358. P. 93-100.
218.Mullard A. // Nat. Rev. Drug Discov. 2016. Vol. 15.
235.Mukherjee S., Ramalingam B., Gangopadhyay S. //
P. 219-221.
J. Mater. Chem. A. 2014. Vol. 2, N 11. P. 3954-3960.
219. Schmidt C. // Nat. Biotechnol. 2017. Vol. 35. P. 493-494.
236.Nguyen H.T., Huynh L.K., Truong T.N. // Carbon. 2017.
Vol. 121. P. 248-256.
220.Povie G., Ford L., Pozzi D., Soulard V., Villa G.,
Renaud P. // Angew. Chem. 2016. Vol. 128, N 37.
237.Tierney H.L., Baber A.E., Kitchin J.R., Sykes E.C.H. //
P. 11387-11391.
Phys. Rev. Lett. 2009. Vol. 103. Article 246102.
РАДИОХИМИЯ том 64 № 4 2022
ВВЕДЕНИЕ ИЗОТОПОВ ВОДОРОДА В БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ
349
238.Zhou H., Zhang J., Ji D., Yuan A., Shen X. // Micropor.
Вестн. МГУ. Сер. 2: Химия. 2011. Т. 52, № 3. C. 220-
Mesopor. Mater. 2016. Vol. 229. P. 68-75.
223.
254.Шевченко В.П., Нагаев И.Ю., Шевченко К.В.,
239.Шевченко В.П., Радилов А.С., Нагаев И.Ю.,
Чернышева М.Г., Бадун Г.А. // Перспективные мате-
Шевченко К.В., Рембовский В.Р., Мясоедов Н.Ф. //
риалы. Спец. вып. 2010. № 8. C. 263-269.
Радиохимия. 2015. Т. 57, № 5. C. 463-465.
255.Шевченко В.П., Нагаев И.Ю., Шевченко К.В.,
240.Шевченко В.П., Нагаев И.Ю., Петунов С.Г.,
Чернышева М.Г., Бадун Г.А., Федосеев В.М.,
Шевченко К.В., Радилов А.С., Мясоедов Н.Ф. // Докл.
Мясоедов Н.Ф. // Радиохимия. 2011. Т. 53, № 3.
AH. 2015. Т. 464, № 5. C. 562-567.
C. 285-288.
241.Баитов А.А., Сидоров Г.В., Мясоедов Н.Ф.
//
256.Шевченко В.П., Вьюнова Т.В., Нагаев И.Ю.,
Радиохимия. 2007. Т. 49, № 1. C. 89-90.
Шевченко К.В., Мясоедов Н.Ф. // Радиохимия. 2017.
242.Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Т. 59, № 5. C. 456-460.
Радиохимия. 2014. Т. 56, № 3. C. 249-251.
257.Palmer W.N., Chirik P.J. // ACS Catal. 2017. Vol. 7, N 9.
243.Шевченко В.П., Нагаев И.Ю., Шевченко К.В.,
P. 5674-5678.
Мясоедов Н.Ф. // Радиохимия. 2015. Т. 57, № 4.
https://doi.org/10.1021/acscatal.7b02051
C. 366-372.
258.Alexakis E., Jones J.R., Lockley W.J.S. // Tetrahedron
244.Nagaev I.Yu., Shevchenko K.V., Shevchenko V.P.,
Lett. 2006. Vol. 47, N 29. P. 5025-5028.
Myasoedov N.F., Grigoriev V.V., Lavrov M.I.,
https://doi.org/10.1016/j.tetlet.2006.05.106
Bondarenko E.V., Kalashnikova E.E.
// Mendeleev
Commun. 2018. Vol. 28, N 1. P. 64-65.
259.Шевченко В.П., Нагаев И.Ю., Пронина Т.С.,
Шевченко К.В., Муртазина А.Р., Сурков С.А.,
245.Сидоров Г.В., Мясоедов Н.Ф. // Радиохимия. 2011.
Угрюмов М.В., Мясоедов Н.Ф. // Докл. AH. 2018.
Т. 53, № 6. C. 545-546.
Т. 480, № 5. C. 551-554.
246.Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
260.Шевченко В.П., Андреева Л.А., Нагаев И.Ю.,
Радиохимия. 2002. Т. 44, № 1. C. 72-77.
Шевченко К.В., Мясоедов Н.Ф. // Докл. AH. 2018.
247.Мясоедов Н.Ф., Шевченко В.П., Нагаев И.Ю.,
Т. 483. № 1. C. 53-54.
Сусан А. Патент RU 2277097. 2004.
261.Шевченко В.П., Андреева Л.А., Нагаев И.Ю., Мясо-
248.Myasoedov N.F., Shevchenko V.P., Nagaev I.Yu.,
едов Н.Ф. // Докл. AH. 2018. Т. 483, № 2. C. 159-161.
Susan A. Patent WO 2006071138A1. 2004.
262.Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. // Докл.
249.Мясоедов Н.Ф., Шевченко В.П., Нагаев И.Ю. Патент
AH. 2013. Т. 448, № 6. C. 668-669.
RU 2291147. 2004.
263.Шевченко В.П., Нагаев И.Ю., Шевченко К.В.,
250. Золотарев Ю.А., Фирстова Ю.Ю., Абаимов Д.А.,
Мясоедов Н.Ф. // Радиохимия. 2013. Т. 55, № 3.
Дадаян А.К., Козик В.С., Новиков А.В., Краснов Н.В.,
C.284-288.
Васьковский Б.В., Назимов И.В., Ковалев Г.И.,
264.Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Мясоедов Н.Ф. // Биоорганическая химия.
2009.
Радиохимия. 2009. Т. 51, № 2. C. 153-155.
Т. 35, № 3. C. 323-333.
265.Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
251.Разживина И.А., Бадун Г.А., Артемкина С.Б.,
Радиохимия. 2009. Т. 51, № 5. C. 452-455.
Чернышева М.Г., Ксенофонтов А.Л., Грайфер Е.Д.,
266.Шевченко В.П., Нагаев И.Ю., Мясоедов Н.Ф. //
Гаршев А.В. // Радиохимия. 2019. Т. 61, № 1. C. 56-
Радиохимия. 2010. Т. 52, № 1. C. 84-86.
62.
https://doi.org/10.1134/S003383111901009X
267.Шевченко В.П., Нагаев И.Ю., Андреева Л.А.,
Мясоедов Н.Ф. // Радиохимия. 2010. Т. 52, № 3.
252.Чернышева М.Г., Буняев В.А., Бадун Г.А.
//
Радиохимия. 2020. Т. 62, № 2. C. 169-174.
C. 277-280.
https://doi.org/10.31857/S0033831120020112
268.Шевченко В.П., Безуглов В.В., Бобров М.Ю.,
253.Шевченко В.П., Бадун Г.А., Нагаев И.Ю.,
Нагаев И.Ю., Мясоедов Н.Ф. // Радиохимия. 2010.
Чернышева М.Г., Шевченко К.В., Федосеев В.М. //
Т. 52, № 3. C. 281-284.
РАДИОХИМИЯ том 64 № 4 2022